

Abstract
Background
This is an update of the original Cochrane review published in 2013 (Issue 6). Pruritus occurs in patients with disparate underlying diseases and is caused by different pathologic mechanisms. In palliative care patients, pruritus is not the most prevalent but is one of the most puzzling symptoms. It can cause considerable discomfort and affects patients' quality of life.
Objectives
To assess the effects of different pharmacological treatments for preventing or treating pruritus in adult palliative care patients.
Search methods
For this update, we searched CENTRAL (the Cochrane Library), and MEDLINE (OVID) up to 9 June 2016 and Embase (OVID) up to 7 June 2016. In addition, we searched trial registries and checked the reference lists of all relevant studies, key textbooks, reviews and websites, and we contacted investigators and specialists in pruritus and palliative care regarding unpublished data.
Selection criteria
We included randomised controlled trials (RCTs) assessing the effects of different pharmacological treatments, compared with a placebo, no treatment, or an alternative treatment, for preventing or treating pruritus in palliative care patients.
Data collection and analysis
Two review authors independently assessed the identified titles and abstracts, performed data extraction and assessed the risk of bias and methodological quality. We summarised the results descriptively and quantitatively (meta‐analyses) according to the different pharmacological interventions and the diseases associated with pruritus. We assessed the evidence using GRADE (Grading of Recommendations Assessment, Development and Evaluation) and created 10 'Summary of findings' tables.
Main results
In total, we included 50 studies and 1916 participants in the review. We added 10 studies with 627 participants for this update. Altogether, we included 39 different treatments for pruritus in four different patient groups.
The overall risk of bias profile was heterogeneous and ranged from high to low risk. However, 48 studies (96%) had a high risk of bias due to low sample size (i.e. fewer than 50 participants per treatment arm). Using GRADE criteria, we downgraded our judgement on the quality of evidence to moderate in seven and to low in three comparisons for our primary outcome (pruritus), mainly due to imprecision and risk of bias.
In palliative care participants with pruritus of different nature, the treatment with the drug paroxetine, a selective serotonin reuptake inhibitor, reduced pruritus by 0.78 points (numerical analogue scale from 0 to 10; 95% confidence interval (CI) −1.19 to −0.37; one RCT, N = 48, quality of evidence: moderate) compared to placebo.
For participants suffering from uraemic pruritus (UP), gabapentin was more effective than placebo (visual analogue scale (VAS): 0 to 10), mean difference (MD) −5.91, 95% CI −6.87 to −4.96; two RCTs, N = 118, quality of evidence: moderate). The κ‐opioid receptor agonist nalfurafine showed amelioration of UP (VAS 0 to 10, MD −0.95, 95% CI −1.32 to −0.58; three RCTs, N = 422, quality of evidence: moderate) and only few adverse events. Moreover, cromolyn sodium relieved UP participants from pruritus by 2.94 points on the VAS (0 to 10) (95% CI −4.04 to −1.83; two RCTs, N = 100, quality of evidence: moderate) compared to placebo.
In participants with cholestatic pruritus (CP), data favoured rifampin (VAS: 0 to 100, MD −24.64, 95% CI −31.08 to −18.21; two RCTs, N = 42, quality of evidence: low) and flumecinol (RR > 1 favours treatment group; RR 1.89, 95% CI 1.05 to 3.39; two RCTs, N = 69, quality of evidence: low) and showed a low incidence of adverse events in comparison with placebo. The opioid antagonist naltrexone reduced pruritus for participants with CP (VAS: 0 to 10, MD −2.26, 95% CI −3.19 to −1.33; two RCTs, N = 52, quality of evidence: moderate) compared to placebo. However, effects in participants with UP were inconclusive (percentage difference −12.30%, 95% CI −25.82% to 1.22%, one RCT, N = 32). Furthermore, large doses of opioid antagonists (e.g. naltrexone) could be inappropriate in palliative care patients because of the risk of reducing analgesia.
For participants with HIV‐associated pruritus, it is uncertain whether drug treatment with hydroxyzine hydrochloride, pentoxifylline, triamcinolone or indomethacin reduces pruritus because the evidence was of very low quality (e.g. small sample size, lack of blinding).
Authors' conclusions
Different interventions tended to be effective for CP and UP. However, therapies for patients with malignancies are still lacking. Due to the small sample sizes in most meta‐analyses and the heterogeneous methodological quality of the included trials, the results should be interpreted cautiously in terms of generalisability.
Plain language summary
Drugs for itching in adult palliative care patients
Background
Pruritus is the medical term for itching. This symptom can be a problem in palliative care settings where treatments for cancer or severe kidney disease are given at the same time. In this updated review, we searched for high quality clinical trials of drugs for preventing or treating itch in palliative care.
Key findings and quality of evidence
In June 2016, we found 50 studies that tested 39 different drugs in 1916 people with itch. An ideal antipruritic (anti‐itch) therapy is currently lacking. However, there was enough evidence to point out some possibly useful treatments for particular causes of the itch. These included gabapentin, nalfurafine and cromolyn sodium for itch associated with chronic kidney disease, and rifampicin and flumecinol for itch associated with liver problems. Paroxetine may be useful for palliative care patients whatever the cause of the itching, although evidence was only available from one study. Overall, most of the drugs caused few and mild side effects. Naltrexone showed by far the most side effects. Overall the evidence ranged from very low to moderate quality.
Further research
Research in palliative care is challenging and often limited to a restricted period of time at the end of life. More high‐quality studies on preventing and treating itch (pruritus) are needed.
Summary of findings
Background
This is an updated version of the original Cochrane review published in 2013 (Issue 6), on "pharmacological interventions for pruritus in adult palliative care patients". Pruritus, derived from the Latin word prurire, which means 'to itch', is defined as "an unpleasant sensation associated with the desire to scratch". This definition of pruritus was introduced in 1660 by the German physician Samuel Hafenreffer (Haffenreffer 1660; Misery 2010; Proske 2010). In modern medicine, the term pruritus is generally used to refer to a pathological condition in which the sensations of itch are intense and often generalised and trigger repeated scratching in an attempt to relieve the discomfort. Pruritus is not a disease, but rather a common and still poorly understood symptom of both localised and systemic disorders that may accompany many conditions (Bernhard 2005; Summey 2009; Zylicz 2004). Pruritus is a prevalent symptom in many skin conditions. However, much less is known about pruritus that is not associated with primary skin disease. This latter problem is of major relevance to many medical specialities, and notably to palliative care. Pruritus or itch is not the most prevalent but is one of the most puzzling symptoms in advanced incurable diseases, and it can cause considerable discomfort in patients receiving treatment for cancer or other non‐malignant terminal illnesses. In addition to social embarrassment, the itch‐scratch‐itch cycle damages skin integrity, decreases resistance to infections, and impairs quality of life in a similar way to pain.
Prevalence of pruritus
Pruritus and malignant diseases
Pruritus may be associated with virtually any malignancy (Chiang 2011). Some neoplasms, particularly haematologic malignancies, are frequently associated with pruritus. Among patients with polycythaemia vera, 48% to 70% have aquagenic pruritus. About 30% of people with Hodgkin's disease also suffer from pruritus (Krajnik 2001b). The incidence and significance of pruritus in other lymphomas and leukaemia are unknown, but investigators have reported its presence in approximately 3% of patients with non‐Hodgkin's lymphoma (Lober 1988). Solid tumours can be associated with paraneoplastic pruritus, which in fact might be a presenting symptom that precedes the diagnosis by months or years. The pathophysiology is not well understood, but it appears to involve an immunologic reaction to tumour‐specific antigens (Seccareccia 2011). Pruritus is also frequent in cutaneous lymphomas (Ahern 2012). Additionally, it is a common symptom in malignancies of the biliary tract. Retrospective studies have revealed that malignant diseases are present in 2% to 11% of chronic itch cases (Weisshaar 2009).
Pruritus and non‐malignant internal diseases
Many internal diseases other than cancer may be associated with pruritus. Pruritus has been reported to herald the onset of thyroid disease, renal insufficiency, liver disease, iron deficiency, diabetes mellitus, paraproteinaemia, Sjögren's syndrome, and other conditions. In internal diseases, itch has been best studied in cholestatic pruritus (CP) and uraemic pruritus (UP) (Metz 2010; Wang 2010; Weisshaar 2009). About one third of uraemic patients treated without dialysis exhibit UP, and on maintenance haemodialysis, the incidence of uraemic itching increases to 70% to 80% (Manenti 2009; Narita 2006). CP affects 100% of patients with biliary cirrhosis and is the initial symptom in almost half of the patients with this disease (Bergasa 2008). Furthermore, the prevalence of pruritus in patients with end‐stage HIV is over 20% (Smith 1997b; Uthayakumar 1997).
Pruritus in palliative care in general
In advanced diseases, as seen in palliative care units, the prevalence of severe pruritus is not too high, but pruritus is a distressing symptom for palliative care patients and may be difficult to manage. A specific problem in palliative medicine involves systemic pruritus in terminal illnesses because pruritus is often a result of changing organ functions in this phase of illness (Twycross 2001; Twycross 2004). In this case, the itch is multifactorial, associated with both liver and kidney function deterioration and increased anxiety (Yosipovitch 2003). Additionally, in the field of palliative care, pruritus is a well‐known adverse effect of opioid administration. Even though the incidence is low (approximately 1% after systemic administration), pruritus as an adverse effect must be kept in mind (Krajnik 2001a).
Description of the condition
In the field of palliative care, pruritus is a symptom occurring in patients with disparate underlying diseases and is caused by different pathologic mechanisms. The pathogenesis of pruritus is complex and not fully elucidated, but it is known that central and peripheral nerves and specific brain regions are involved (Langner 2009). For a long time, itch was regarded as a variant of pain; however, the neural transmission associated with pruritus follows distinct neuronal pathways and causes unique sensations (Ikoma 2006; Schmelz 1997). The pathogenesis of itch is diverse and involves a complex network of cutaneous and neuronal cells. Mediators of pruritus presumably act on nerve fibres or lead to a cascade of mediator release, resulting in nerve stimulation and the sensation of pruritus. The group of potential chemical mediators is large and is steadily increasing. It contains amines (e.g. histamine, serotonin), proteases (e.g. tryptases), neuropeptides (e.g. substance P (SP), calcitonin gene‐related peptide (CGRP), bradykinin), opioids (e.g. morphine, beta‐, met‐, leu‐enkephalin), eicosanoids, growth factors and cytokines (Weisshaar 2003). The identification of different itch‐specific mediators and receptors, such as interleukin‐31, gastrin‐releasing peptide receptor or histamine H4 receptor, is increasing, and the characterisation of itch‐specific neurons is taking shape. The physiological basis of pruritus includes multiple mechanisms that are quite variable. Pruritus is initiated by the stimulation of unmyelinated C‐fibers in the dermal‐epidermal junction (Krajnik 2001a; Steinhoff 2006). Mediators of pruritus include histamine through H1 receptors and serotonin through 5‐HT2 and 5‐HT3 receptors (Jones 1999; Krajnik 2001a; Yamaguchi 1999). The actual sensation may depend on special temporal patterns of neural excitation and location of receptors (Seiz 1999). The perception of pruritus leads to a motor response to scratch, which stimulates myelinated A‐delta sensory fibres and temporarily blocks the sensation.
Description of the intervention
Due to the complex physiology of pruritus, many of the underlying mechanisms are still poorly understood (Krajnik 2001b). Modulations by serotonergic and enkephalinergic systems take place on all levels of the 'pruritus tract'. Additionally, opioid receptors seem to be of particular importance, which is not surprising considering the involvement of almost identical mediators for pain and itch (Moore 2009). This implies that various completely different pathologic mechanisms may form the basis of pruritus, making it difficult to find an effective medication for managing the symptom. Until now, no universally valid therapeutic concept has been developed. For the treatment of pruritus, researchers have tested the efficacy of several different substance classes. Among these, our review includes trials on antiphlogistic substances, psychotropic drugs, antagonistic drugs, anaesthetics, adsorbent substances and topical treatments.
How the intervention might work
For this update, we identified 50 trials studying 39 drugs from different classes. Below we have listed each drug class and individual pharmacological intervention included in this review.
Histaminergic drugs
Of the mediators that trigger pruritus, histamine is the best known and most thoroughly researched. Preformed histamine is present in large amounts in mast cell granules. For this reason, after cell activation, it can be immediately released into the surrounding area, where it may induce pruritus via H1 receptors on nerve fibres. Antihistamines act via prevention of the histamine fixation on the surface of the histamine receptors (Gaudy‐Marqueste 2010; Ständer 2008). Some of the antihistamines mentioned in this review are hydroxyzine and ketotifen.
Opioid receptor antagonists and (partial) agonists
Opioid receptor antagonists were originally developed for the treatment of heroin addiction and for symptom reversal of postanaesthetic depression, narcotic overdose, and opioid intoxication (Gowing 2010; Rösner 2010). Clinical and experimental observations have demonstrated that endogenous or exogenous opioids can evoke or intensify pruritus (Metze 1999a; Metze 1999b). This phenomenon can be explained by the activation of spinal opioid receptors, mainly μ‐opioid receptors on pain transmitting neurons, which often induce analgesia in combination with pruritus. Thus, reversing this effect through μ‐opioid antagonists inhibits pruritus (Ständer 2008).
Serotonergic drugs
Pruritus sensations may arise from the superficial layers of the skin, which contain clustered nerve endings at 'itch points' close to the dermoepidermal junction, as well as the mucous membranes and conjunctiva (Krajnik 2001a; Yosipovitch 2003). These receptors may be acted upon directly by physical or chemical stimuli, or indirectly via histamine release. Itch impulses are transmitted through the C fibres of polymodal nociceptors to the dorsal root ganglia, where a synapse occurs with secondary neurons. Efferents traverse to the contralateral spinothalamic tract and pass to the posterolateral spinothalamic tract, the posterolateral ventral thalamic nucleus, and then to the somatosensory cortex of the postcentral gyrus (Mela 2003). One important neurotransmitter in these pathways is 5‐hydroxytryptamine (5‐HT; serotonin) (O'Donohue 2005). Ondansetron is one of a group of drugs that act as antagonists at 5‐HT3 serotonin subtype receptors. Properties of drugs within this group differ with respect to the selectivity of receptor binding, potency, duration of action and dose‐response relationships.
Serotonin reuptake inhibitors and antidepressants
Selective serotonin reuptake inhibitors (SSRIs) like sertraline and paroxetine play an increasingly important role in the management of pruritus (Balaskas 1998; Larijani 1996; Raap 2012; Schworer 1995; Tandon 2007; Tennyson 2001; Wilde 1996; Ye 2001; Zylicz 1998). Experts believe that they raise the extracellular level of the neurotransmitter serotonin by inhibiting its reuptake into the presynaptic cell and increasing the level of serotonin in the synaptic cleft that is available to bind to the postsynaptic receptor. They have varying degrees of selectivity for the other monoamine transporters, with pure SSRIs having only weak affinity for the noradrenaline and dopamine transporters.
Antiepileptics
Antiepileptics are used to prevent or reduce the severity and frequency of seizures (Duley 2010; Ratilal 2005; Wiffen 2011). Gabapentin and pregabalin, which are γ‐aminobutyric acid analogues, were originally developed as antiepileptics and may hinder the transmission of nociceptive sensations to the brain, thereby also suppressing pruritus (Ständer 2008).
Rifampicin
Rifampicin, or rifampin in the USA, is an antibiotic that induces detoxicating hepatic enzymes and competitively inhibits the reuptake of bile acids by hepatocytic transporters (Trauner 2005). Some hypothesise that rifampicin might influence pruritus by changing the bacterial growth in the intestines, which can influence the reabsorption of pruritrogens.
Thalidomide
Thalidomide is a drug that modifies or regulates the immune system and has anti‐inflammatory properties. It is used as an immunomodulator to treat graft versus host reactions. It suppresses tumour necrosis factor alpha (TNF‐α) production and leads to a predominant differentiation of Th2 lymphocytes with suppression of interleukin‐2 (IL‐2) producing Th1 cells (McHugh 1995; Mettang 2010). The antipruritic action of this drug may be secondary to inhibition of TNF‐α. Another possibility is that thalidomide can act as both a peripheral and a central nerve depressant (Moretti 2010).
Flumecinol
There are reports that flumecinol (3‐trifluoromethyl‐alpha‐ethylbenzhydrol), a benzhydrol derivative, induces microsomal drug metabolising enzymes (Turner 1990). Flumecinol also lowers serum bilirubin in Gilbert's syndrome, possibly by inducing bilirubin undine diphosphate (UDP)‐glucuronyltransferase. Therefore, it may induce a range of enzymes, similar to phenobarbitone and rifampicin.
Colestyramine
Colestyramine is an intestinally active anion exchange resin. It interrupts the enterohepatic circulation of bile acids and has been used for many years to relieve pruritus in cholestatic disorders (Datta 1966; Sharp 1967). Another bile acid sequestrant also used for the treatment of pruritus is colesevelam.
Cromolyn sodium
Cromolyn sodium is a drug that blocks mast cell degranulation in response to antigens, leading to decreased release of histamine, leukotrienes and other inflammatory mast cell products. It is hypothesised that mediators released from mast cells are most likely to be responsible for UP. Another hypothesis is that cromolyn sodium may decrease the severity of pruritus via reducing serum tryptase levels. Both oral and topical administration is possible.
Leukotriene antagonists
Leukotriene antagonists prevent the inflammatory response produced by leukotrienes (Watts 2012).
Erythropoietin
Erythropoietin is a hormone produced naturally by the kidneys that stimulates the production of red blood cells in the bone marrow. Studies have hypothesised that erythropoietin may have an antipruritic effect related to a lowering effect of the hormone on plasma histamine concentrations (Bohlius 2009).
Activated charcoal
Activated charcoal is an agent that can bind many poisons in the stomach and therefore prevent them from being absorbed. Charcoal has also been shown to be effective in UP (Giovanetti 1995; Yatzidis 1972).
Topical capsaicin
Capsaicin is the prototype of topical antipruritic agents that target the transient receptor potential (TRP) gene family of ion channels, which respond to physical activation (heat, cold), protons (pH changes) or biological mediators (for example prostanoids) and counteract itch via activating pain neurons (Derry 2012; Derry 2013; Steinhoff 2011). Capsaicin (trans‐8‐methyl‐N‐vanillyl‐6‐nonenamide) is an alkaloid naturally found in many botanical species of the nightshade plant family (Solanacea).
Tacrolimus
Tacrolimus is an immunosuppressant used for the prevention of transplant rejection. It suppresses the differentiation of Th1 lymphocytes and the ensuing IL‐2 production (Suthanthiran 1994; Webster 2005).
Pramoxine hydrochloride
Pramoxine hydrochloride is a local anaesthetic. Pramoxine stabilises the neuronal membrane by an uncertain mechanism (Elmariah 2011; Hedayati 2005).
Ergocalciferol
Patients under haemodialysis often experience pruritus and may have an impaired metabolism of vitamin D. It is supposed that the administration of ergocalciferol (vitamin D2) may have an antipruritic effect (Shirazian 2013).
Nicotinamide
Nicotinamide (which is the amid of nicotinic acid, i.e. vitamin B3/niacin) may have an antipruritic effect, mediated by its anti‐inflammatory and histamine‐release blocking characteristics (Omidian 2013).
Omega‐3 fatty acids
Fish oil tends to support the immune system and reduces inflammation, free radicals and leukotriene B‐4. Hence, omega‐3 fatty acids may be effective in UP (Ghanei 2012).
Turmeric
Turmeric is the powder of Curcuma longa L. (Zingiberaceae) containing the active component curcumin (diferuloylmethane), which has an anti‐inflammatory effect and may be beneficial for UP (Pakfetrat 2014).
Zinc sulphate
Pruritus patients with UP may benefit from zinc sulphate, as it is an antagonist of calcium (releases histamine) and prevents degranulation of mast cells (Mapar 2015; Najafabadi 2012).
Other treatments
Photodynamic therapy, transcutaneous electrical nerve stimulation (TENS), and other non‐pharmaceutical therapies for pruritus may be assessed in a separate systematic review.
Summary of interventions
In conclusion, pruritus is a frequent and distressing symptom. The medical literature is full of recommendations for its management, but it contains only a few clinical trials and evidence‐based data. There are some reviews about pruritus in general (Winkelmann 1964; Winkelmann 1982), dermatologic causes of pruritus (Fransway 1988), pruritus in systemic diseases (Kantor 1983; Summey 2005), and pruritus related to specific causes like cholestasis or uraemia (Khandelwal 1994; Szepietowski 2004). There is also some literature on the management of pruritus in palliative care patients (Krajnik 2001b). There have also been two recent systematic reviews that assessed the effectiveness of different medical interventions on pruritus in the field of palliative care (Siemens 2014; Xander 2013). This is an updated version of the original Cochrane review by Xander and colleagues.
Why it is important to do this review
Pruritus or itch is one of the most puzzling symptoms in advanced incurable diseases and can cause considerable discomfort in patients. Nevertheless, it is a kind of Cinderella symptom, tucked away and hidden behind more 'fashionable' symptoms such as pain. As already explained, pruritus is multifactorial in origin and can be a symptom of diverse pathophysiologies. Particularly over the last decade, clinical observation and controlled trials have done much to aid the understanding and treatment of pruritus, especially in liver disease, uraemia and other kinds of chronic pruritus. Therefore, this review aimed to systematically collect and evaluate the evidence for adequate treatment of pruritus in the field of palliative care, to put this symptom into perspective, and to make new therapeutic strategies accessible for clinicians and patients (Wee 2008).
Objectives
To assess the effects of different pharmacological treatments for preventing or treating pruritus in adult palliative care patients.
Methods
Criteria for considering studies for this review
Types of studies
We considered full reports concerning pruritus in patients with advanced diseases with a focus on pharmacological treatment. The primary outcome of the studies had to be subjective measures of pruritus. We only included randomised controlled trials (RCTs) in adults. We defined 'randomised' as studies described as such by the authors anywhere in the manuscripts. Both published and unpublished studies were eligible for inclusion.
Contrary to our initial considerations in the protocol, we did not include controlled clinical trials (CCTs) (Differences between protocol and review).
Types of participants
Previous reviews have cited problems defining the population for systematic reviews in palliative care. Therefore, we drew upon the definition that other Cochrane reviews have used, "adult patients in any setting, receiving palliative care or suffering an incurable progressive medical condition" (Dorman 2010; Perkins 2009). Studies eligible for this review included participants:
suffering from pruritus combined with an incurable advanced malignant or non‐malignant disease such as advanced cancer, HIV/AIDS, renal failure, liver failure or others;
aged 18 years or older; and
of both sexes.
Since many of the studies we considered also included participants who were not necessarily in advanced stages of their disease and were not palliative care patients, we decided to define comprehensible criteria for the patients included in this systematic review. Concretely, we included all patients who were described as palliative care patients or as patients in advanced stages of malignant or non‐malignant diseases.
If no detailed information on the stages of the underlying disease was available, we considered the following patients to have palliative care needs.
UP (also known as chronic kidney disease (CKD)‐associated pruritus, renal pruritus or end‐stage renal disease (ESRD) pruritus) in need of haemodialysis.
CP or hepatogenic pruritus: all patients suffering from primary biliary cholestasis or primary sclerosing cholestasis and all patients who were described as being in an advanced stage of the disease. If patients with different kinds of CP were included in the studies, only studies that included more than 75% of patients with primary biliary cholestasis, primary sclerosing cholangitis or advanced‐stage disease were eligible for the systematic review.
HIV‐associated pruritus: all patients with pruritus associated with HIV.
Pruritus associated with malignancies: all patients in advanced stages of cancer (with metastases or described as in an advanced stage of the disease).
We excluded studies in people with pruritus related to acute or chronic cholestasis, acute or chronic dermatological diseases, or acute medical or surgical interventions. Furthermore, we did not include participants with primarily dermatological diseases or infections.
Types of interventions
We included studies using any pharmacological medication to treat pruritus, regardless of dosage, route of administration or duration of follow‐up. Interventions with both internal and external application of the treatment were eligible for inclusion in the review. We did not focus on pharmacological interventions targeting the treatment of underlying diseases but rather on pharmacological interventions for treating pruritus as an accompanying symptom of advanced diseases. We excluded complementary medical interventions and non‐pharmaceutical treatments such as photodynamic therapy or TENS, but a separate review may evaluate them.
Types of outcome measures
Given the heterogeneity of included trials, in the Effects of interventions we organise the reporting of primary and secondary outcomes according to types of participants and pharmacological interventions.
Primary outcomes
Primary outcomes were subjective measurements of pruritus.
Scores on validated and reliable scales, such as unidimensional scales (e.g. visual analogue scales (VAS), numeric rating scales (NRS), categorical scales).
Patient‐reported pruritus according to non‐validated pruritus scores (e.g. 1 to 3 or 1 to 4), which were substituted by estimations by nursing or medical staff if self‐assessment was not possible.
Secondary outcomes
Secondary outcomes included:
quality of life;
patient satisfaction;
depression;
adverse events.
Search methods for identification of studies
There were no language restrictions for either the searching strategies or study inclusion.
Electronic searches
For this update, we searched the following databases using slightly revised search strategies (see Appendix 1; Appendix 2; Appendix 3) after consultation with the Cochrane Pain, Palliative Care and Supportive Care Cochrane Review Group.
Cochrane Central Register of Controlled Trials (CENTRAL; 2016, Issue 5) in the Cochrane Library (searched 2012 to 9 June 2016)
MEDLINE Ovid (2012 to 9 June 2016);
Embase Ovid (2012 to 7 June 2016).
We used the Cochrane highly sensitive search strategy (CHSSS) for identifying RCTs in MEDLINE, a sensitivity maximising version as referenced in Chapter 6.4.11.1 and detailed in box 6.4.c of the Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (Higgins 2011). We used a similar study design filter for other databases, as appropriate.
We decided not to search the other databases used for the original review as they did not yield any useful records.
In the original review, we searched the following databases (see Appendix 4, Appendix 5, Appendix 6, Appendix 7, Appendix 8 and Appendix 9).
Cochrane Pain, Palliative and Supportive Care Trials Register (searched August 2012).
The Cochrane Library via Wiley, including the Cochrane Database of Systematic Reviews (CDSR), the Cochrane Central Register of Controlled Trials (CENTRAL) and the Database of Abstracts of Reviews of Effects (DARE) (searched August 2012);
MEDLINE Ovid (including MEDLINE In‐process and other non‐indexed citations) (1950 to August 2012);
Embase Ovid, including Embase Alert (1980 to August 2012);
BIOSIS previews Ovid and Web of Knowledge (1969 to August 2012);
CINAHL EBSCO (Cumulative Index to Nursing and Allied Health Literature; 1982 to August 2012);
PsycINFO EBSCO (1806 to August 2012).
In addition, we performed Internet searches using Scirus (www.scirus.com) and Google Scholar (scholar.google.de) in the original review.
Searching other resources
We searched the following trial registers.
Current Controlled Trials (www.controlled‐trials.com; searched 9 June 2016).
WHO International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch; searched 9 June 2016).
US National Institutes of Health Ongoing Trials Register ClinicalTrials.gov (www.clinicaltrials.gov; searched 9 June 2016).
Data collection and analysis
Selection of studies
For this update, two review authors (WS, CX) screened all titles and abstracts of studies identified by the search strategies for relevance. We resolved disagreement by consensus and after discussion with a third review author (GB). If it was not possible to accept or reject a study with certainty, we obtained the full text of the study for further evaluation. Two review authors (WS, CX) independently assessed the full text of all potentially relevant studies in accordance with the above inclusion criteria. We resolved any differences in opinion at this stage by consensus and discussion with a third review author (GB). We kept a record of all excluded studies and the reasons for exclusion.
Data extraction and management
Two review authors (WS, CX) independently extracted data from the selected studies using a standardised coding form. We discussed differences in data extraction and sought the input of a third review author (GB) as necessary. The data extraction form, specifically designed for the review, included the following.
Study ID and publication details
Study aim
Study design (randomised, not randomised, controlled, prospective etc.)
Primary and secondary outcomes
Type of control group
Number of participants in each group
Quality of the study
Randomisation procedure
Concealment of treatment allocation
Details of blinding
Per protocol analysis or intention‐to‐treat analysis
Number of withdrawals described
Management of missing data
Follow‐up data
Details of analysis
Patient characteristics
Demographics
Diagnosis
Status or course of disease
Type and stage of treatment
Type of pruritus
Pharmacological interventions
Drug characteristics
Duration of therapy
Pharmacological regimen of drug treatment with the drug of interest (dose, frequency of application)
Description of placebo
Description of alternative treatment
Description of additional non‐pharmacological techniques if additionally used during similar regimens
Outcome measures
Primary outcome, including the measurement of pruritus (mean, standard deviation (SD)) and the change in level of pruritus
Secondary outcomes, including the measurement of quality of life, patient satisfaction, depression and adverse events of treatments
Additional information
Patient narrative comments, etc.
We contacted authors of studies to, if possible, provide unpublished data if required for analysis.
Assessment of risk of bias in included studies
We performed the 'Risk of bias' assessment for RCTs as recommended by the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011; RevMan 2014). Two review authors (WS, CX) independently assessed the quality of included studies using the Cochrane 'Risk of bias' tool (Higgins 2011; RevMan 2014).
Random sequence generation
Low risk: every participant had an equal chance to be selected for either treatment, and the investigator was unable to predict which treatment the participant would be assigned to.
Unclear risk: no information given.
High risk: for example, randomisation by date of birth or date of admission.
Allocation concealment
Low risk: methods to conceal allocation included central randomisation, serially numbered, opaque, sealed envelopes, or other descriptions with convincing concealment.
Unclear risk: authors did not adequately report the method of concealment.
High risk: investigators enrolling participants could possibly foresee assignments because of the use of high risk methods to conceal allocation, such as an open random allocation schedule (e.g. a list of random numbers), assignment envelopes without appropriate safeguards (e.g. if envelopes were unsealed, nonopaque, or not sequentially numbered), alternation or rotation, date of birth.
Blinding of participants and personnel
Low risk: blinding of participants and providers stated and unlikely that the blinding could have been broken.
Unclear risk: blinding not adequate, but the outcome measurement is not likely to have been influenced by lack of blinding.
High risk: no blinding or incomplete blinding, and the outcome or outcome measurement is likely to have been influenced by lack of blinding.
Blinding of outcome assessors
Low risk: blinding of providers and outcome assessor stated and unlikely that the blinding could have been broken.
Unclear risk: blinding of outcome assessment not adequate, but the outcome measurement is not likely to have been influenced by lack of blinding.
High risk: no blinding or incomplete blinding of outcome assessment, and the outcome or outcome measurement is likely to have been influenced by lack of blinding.
Incomplete outcome data
Low risk: no missing outcome data, or reasons for missing outcome data are unlikely to be related to true outcome.
Unclear risk: insufficient information to permit judgement.
High risk: reasons for missing outcome data likely to be related to true outcome, with either imbalance in numbers or reasons for missing data across intervention groups, or 'as‐treated' analysis done with substantial departure of the intervention received from that assigned at randomisation.
Selective outcome reporting
Low risk: reports of the study free of selective outcome reporting.
Unclear risk: insufficient information to permit judgement.
High risk: reports of the study suggest selective outcome reporting.
Size of study
Low risk: 200 participants or more per treatment arm.
Unclear risk: 50 to 199 participants per treatment arm.
High risk: fewer than 50 participants per treatment arm.
Other sources of bias
Low risk of bias: the trial appears to be free of other components that could put it at risk of bias.
Unclear risk of bias: the trial may or may not be free of other components that could put it at risk of bias.
High risk of bias: there are other factors in the trial that could put it at risk of bias, e.g. for‐profit involvement, authors have conducted trials on the same topic, etc.
The review authors were blinded to each others' assessments. We resolved any disagreements by discussion. We did not automatically exclude any study as a result of a rating of 'unclear' or 'high' risk or based on a low quality score. We considered trials assessed as being at low risk of bias in all of the specified individual domains to be trials with overall low risk of bias. We considered studies at unclear risk of bias in one or more of the specified individual domains to be trials with unclear risk of bias. Finally, we considered studies assessed at high risk of bias in one or more of the specified individual domains to be trials with high risk of bias.
Measures of treatment effect
Primary outcome: measurement of itch
Pruritus measurement is problematic because of its subjective nature and poor localisation. In addition, itch has multidimensional aspects (for example severity, duration, frequency, spatial distribution, and quality). Although several authors have suggested that VAS is subjective and represents an inadequate and unreliable method of assessing pruritus (Jones 1999), more sophisticated and objective methods pose several practical difficulties since the main goal of pruritus treatment is to improve patients' well‐being and quality of life. It is only possible to measure these aspects subjectively, as pruritus is primarily based on the subjective perception of the patient. Therefore, for the direct evaluation of itch, we have to rely on the patients' own ratings of their subjective symptoms and on the assumption that the participant is able to relate their experiences accurately.
Studies commonly employ categorical research scales that consist of discrete divisions of the frequency or intensity of pruritus (e.g. none, mild, moderate severe). Moreover, the Duo scale is used by some researches (Duo 1987; Mettang 2002), which is a sum score covering areas like pruritus severity, distribution, frequency and sleep disturbance. The instrument is used in different ways resulting in ranges from 0‐36 to 0‐48.
Continuous scales like the VAS or numeric rating scales (NRS) consist of a line with a specific length (e.g. 100 mm or 10 cm) with descriptive anchors at the extremes, for example 'no pruritus' and 'pruritus as bad as it can be imagined'. Since the VAS is validated (Reich 2008), simple, accurate, and supposedly the most sensitive approach to measuring pruritus intensity, it is probably the most commonly used scale in pruritus research (Wallengren 2010; Weisshaar 2003).
Another approach to measurement of pruritus is scratching behaviour measurement. In contrast to patient‐reported measures of pruritus, it is possible to objectively quantify scratching activity by report of scratching behaviour, for example with hand‐activated counters to record scratching (Melin 1986). This method is not suitable for recording nocturnal scratching, but other methods are, for example nocturnal bed movement measured by a vibration transducer on one of the legs of the bed, and limb or forearm activity measured by movement‐sensitive meters. Researchers can also observe nocturnal scratching by infrared videotaping or by direct observation during the night.
One instrument for recording daily and nocturnal scratching is the pruritometer, which processes the signals of a piezoelectric vibration sensor fixed on the middle finger of the patient's dominant hand and sent to a counter worn by the patient like a wristwatch (Wallengren 2010).
Scores and measurement of scratching may also be determined through questionnaires asking for more information regarding the pruritus, for example the 'Worcester Itch Index' or the 'Eppendorf Itch Questionnaire' (Weisshaar 2003).
In this systematic review, investigators evaluated treatment effect by estimations of nursing or medical staff if self‐assessment was not possible.
Since no gold standard concerning treatment or improvement of pruritus exists, we considered a reduction of pruritus symptoms by 30% as moderate and a reduction by 50% as substantial, assuming that there were no other specifications given in the studies. This is consistent with the IMMPACT recommendations introduced in Turk 2008.
Secondary outcomes
Quality of life, patient satisfaction, depression, and adverse events were recorded as secondary outcomes.
For measuring quality of life, we considered the following scales or methods.
Short Form 36 (8 dimensions that can be transformed on a 0‐100 scale, higher values = better status) and Liver Disease Symptom Index 2.0 (18 items, 0‐4, higher score = worse status) (Kuiper 2010).
VAS (0 to 100 mm), 0 = able to cope with normal activities, 100 = completely incapacitated (Turner 1994a; Turner 1994b).
Other investigators assessed patient satisfaction using a seven‐point scale, where 0 meant indifferent, a value of −3 meant extremely poor, and a value +3 meant excellent (Zylicz 2003).
We considered depression using:
the Hamilton depression rating scale (includes items intrinsic to medical conditions (i.e. fatigue, sleep) and concern about health) (Bergasa 2006);
the Structured Clinical Interview Questionnaire (SCID) for the Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM‐IV), Axis I Disorders (a measure for the diagnosis of depression and anxiety syndromes) (Bergasa 2006); and
the 30‐item Inventory of Depressive Symptomatology‐Self‐Report (IDS‐SR30) (Mayo 2007).
Most included studies reported adverse events.
Unit of analysis issues
We evaluated the data of the RCTs. Identified studies had to evaluate and report the effect of a pharmacological treatment versus placebo, no treatment, or an alternative treatment on pruritus in individuals. Our results did not contain studies with multiple observations or cluster RCTs. However, there were cross‐over studies, and we considered specific challenges, such as possible carryover effects.
Cross‐over trials may be combined with parallel‐group trials in principle (Higgins 2011, chapter 16.4). We included properly reported cross‐over trials (i.e. analysed with paired t‐test and without a carryover or period effect) in meta‐analyses using the generic inverse variance (GIV) method Higgins 2011. When authors reported or analysed results in an inappropriate way that did not allow calculation of the standard error (SE) of the mean difference in a paired analysis, we tried to approximate the SE by estimating the correlation within participants. In case there were insufficient data to calculate the correlation coefficient, we assumed a correlation of zero, which results in a conservative scenario, i.e. SE is slightly overestimated (Gunal 2004; Murphy 2003). Each comparison includes a subgroup analysis by study design when both cross‐over and parallel‐group trials were included in a meta‐analysis.
In addition, the meta‐analyses and 'Summary of findings' tables show the number of patients for parallel‐group trials and the number of cases for cross‐over trials, since there are two post‐treatment values for each patient in a cross‐over trial. However, the number of participants included in this review and also shown in the tables refers to participants and not to cases of the cross‐over RCTs.
Dealing with missing data
We did not impute missing outcome data. We analysed them on an endpoint basis, including only participants for whom final data were available. We did not assume that participants who dropped out after randomisation had a negative outcome.
Assessment of heterogeneity
We investigated heterogeneity using visual inspection of the forest plots as well as the I2 statistic (Higgins 2002).
Assessment of reporting biases
There were insufficient studies in each of the meta‐analyses to assess reporting bias. We had planned funnel plots corresponding to meta‐analyses of the primary outcome to assess the potential for small study effects, such as publication bias.
Data synthesis
We used Review Manager 5 (RevMan) and R statistical software for data entry, statistical analysis, and creation of graphs (R Foundation 2015; RevMan 2014; Schwarzer 2015). We analysed each drug class separately and compared it with its respective control group or alternative intervention. We presented most outcomes in this review as continuous variables. We presented continuous outcomes, including the mean change in pruritus score between treatment and placebo, either as mean difference (MD) or standardised mean difference (SMD; 0.2 = small effect, 0.5 = moderate effect, 0.8 = large effect, Cohen 1988) with 95% confidence intervals (CI), depending on whether trials reported results on the same or different scales. We anticipated that some individual studies would have used final scores and others would use change scores and even analysis of covariance in their statistical analyses of the results. In this case, we combined these different types of analysis as MDs. We used the fixed‐effect model in all meta‐analyses.
We decided not to pool the results in cases of significant clinical heterogeneity. We calculated the 95% CI for each effect size estimate.
We made the following treatment comparisons.
Naltrexone versus placebo.
Nalfurafine versus placebo.
Ondansetron versus placebo.
Gabapentin versus placebo.
Rifampicin versus placebo.
Flumecinol versus placebo.
Cromolyn sodium versus placebo.
Capsaicin versus placebo.
Zinc sulphate versus placebo.
We included studies with parallel‐group and cross‐over designs in the review, handling data from cross‐over trials according to the recommendations in section 16.4 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). If all necessary data were provided in the publications of cross‐over trials and if no carryover effect or periodic effect was apparent, we included the results of a paired analysis in the meta‐analyses. If the required data were available, we included only data from the first period of the cross‐over trial (if available) and thus treated this trial as a parallel‐group trial.
Summary of findings table
We prepared 'Summary of findings' tables with GRADEpro GDT software and in accordance with the latest recommendations of the GRADE working group (GRADEproGDT 2015; Guyatt 2013a; Guyatt 2013b). The 'Summary of findings' tables include each comparison and the primary outcome (pruritus) as well as all secondary outcomes. We included the number of participants who experienced at least one adverse event as a binary outcome in our meta‐analyses and 'Summary of findings' tables.
We assessed the overall quality of the evidence for each outcome using the GRADE system and presented it along with the main findings of the review in 'Summary of findings' tables, following a transparent and simple format (GRADE Handbook; GRADEproGDT 2015). In particular, we included key information concerning the quality of evidence, the magnitude of effect of the interventions examined, and the sum of available data on the main outcomes.
The GRADE system uses the following criteria for assigning grade of evidence.
High: further research is very unlikely to change our confidence in the estimate of effect.
Moderate: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate.
Low: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate.
Very low: any estimate of effect is very uncertain.
We downgraded the quality of evidence under the following conditions.
Serious (−1) or very serious (−2) limitation to study design or execution (risk of bias).
Serious (−1) or very serious (−2) inconsistency of results.
Serious (−1) or very serious (−2) indirectness of evidence.
Serious (−1) or very serious (−2) imprecision.
Serious (−1) or very serious (−2) publication bias.
Subgroup analysis and investigation of heterogeneity
We planned the following subgroup analyses a priori and performed them when possible.
UP versus CP.
Parallel‐group versus cross‐over study design.
We conducted subgroup analyses as recommended in the Cochrane Handbook for Systematic Reviews of Interventions section 9.6 (Higgins 2011).
Sensitivity analysis
We performed sensitivity analyses to assess whether the quality of the chosen trials influenced the results of the meta‐analysis or whether the analysis by the fixed‐effect or the random‐effects model changed the results.
Due to the small numbers of studies for a single comparison, we did not conduct sensitivity analyses based on quality criteria.
Results
Description of studies
Please see the Characteristics of included studies table for full information on the included studies.
Results of the search
In total, we identified 50 studies with 1916 participants for this update. See Figure 1 for a flowchart of the study selection process. We included 10 new studies with 627 additional participants: Amirkhanlou 2016 (N = 52), Feily 2012 (N = 60), Ghanei 2012 (N = 22), Mapar 2015 (N = 40), Najafabadi 2012 (N = 40), Nakhaee 2015 (N = 25), Omidian 2013 (N = 50), Pakfetrat 2014 (N = 100), Shirazian 2013 (N = 50)and Yue 2015 (N = 188). We updated the study flow diagram (Figure 1) according to the latest recommendations (Stovold 2014). The 50 identified studies contained different assessment scales (Table 11) and a total of 39 different drugs for the treatment of pruritus associated with different underlying diseases (Table 12). The drugs assessed were antiphlogistic substances, psychotropic drugs, antagonistic drugs, anaesthetics, adsorbent substances and topical treatments. The participants suffered from UP (1574 participants, 82%), CP caused by hepatobiliary diseases (276 participants, 14%), pruritus associated with malignancies (26 participants, 1%) (Zylicz 2003), and pruritus as a symptom associated with HIV (40 participants, 2%) (Smith 1997a). Among the included studies, 20 were cross‐over studies, while the remaining 30 studies had a parallel‐group design. Two studies were already pooled in a meta‐analysis (Wikström 2005a; Wikström 2005b). The studies took place in 25 different countries in Europe, North America and Asia. Fourteen studies were multicentre trials. A few studies assessed quality of life (Kuiper 2010; Turner 1994a; Turner 1994b; Yue 2015), depression (Zylicz 2003), and patient satisfaction (Bergasa 2006; Mayo 2007), and most of them reported adverse events (see Table 13; Table 14; Table 15).
1.
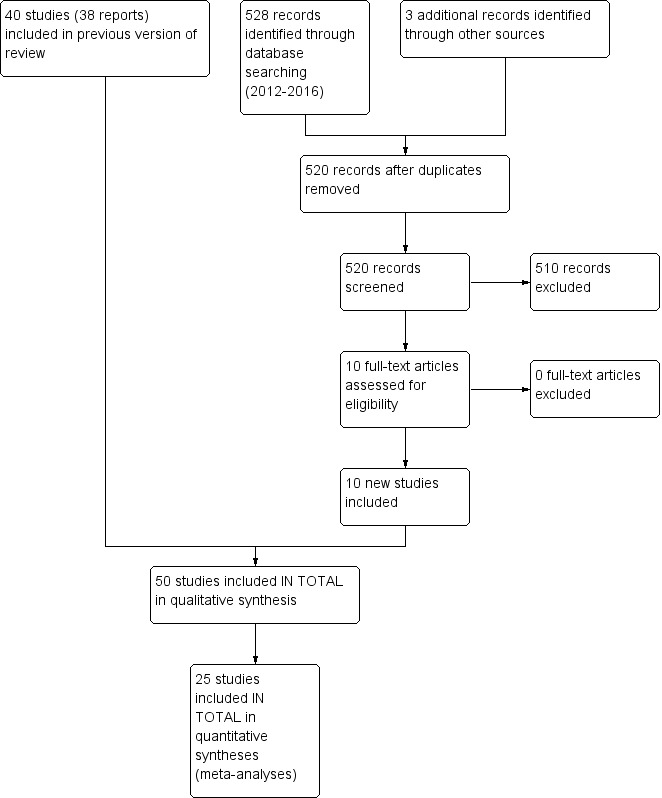
Study flow diagram.
1. Measurement of pruritus.
| Study | Pruritus scale | Determination of Score | Description of scale | Description of partial resolution | |
| Amirkhanlou 2016 | Clinical response | Number and percentage post treatment | 1 | Complete response (no itching or minimal itching after treatment) | See description of scale |
| 2 | Partial response (mild or moderate severity of itching after treatment) | ||||
| 3 | No response (severe pruritus after treatment) | ||||
| Ashmore 2000 | 0‐10 cm VAS | Median daily pruritus score and interquartile range for each period | 0 | No pruritus | Score reduction of 40% to 50% was chosen as desired improvement |
| 10 | Maximum pruritus | ||||
| Bachs 1989 | 0‐3 score | Mean and standard deviation for pruritus 7 days before, and daily during treatment | 0 | No itching | Not available |
| 1 | Mild intermittent pruritus which did not affect the patient's routine or disturb sleep pattern | ||||
| 2 | Moderate pruritus present most of the time but tolerable and not interfering with sleep pattern | ||||
| 3 | Continuous pruritus disturbing sleep pattern | ||||
| Bergasa 2006 | 0‐10 cm VAS | Mean difference of VAS: "Mean differences in measurements were determined by subtracting each on‐treatment value from each pretreatment value for each subject and calculating the mean of all the differences." | — | Not stated | Not available |
| Borgeat 1993 | 0‐10 verbal rating score | Evaluation 10 minutes after administration and then every 10 minutes during the first hour | 0 | No pruritus | Decrease of at least 4 points |
| 10 | Most severe pruritus imaginable | ||||
| Breneman 1992a | 4‐point scale | Follow‐up evaluations at weeks 1, 2, 3, 4 and 6 | 1 | No itching | Not available |
| 2 | Mild itching, occasionally noticeable | ||||
| 3 | Moderate itching, not interfering with daily life and/or sleep | ||||
| 4 | Severe itching, disturbing daily life and/or sleep | ||||
| Cho 1997 | 4‐point scale | Mean values and standard error of the mean | 1 | None | Not available |
| 2 | Mild | ||||
| 3 | Moderate | ||||
| 4 | Severe | ||||
| De Marchi 1992 | Scoring system proposed by Duo and modified by Mettang (Duo 1987; Mettang 1990) | Daily mean values and standard error | Scoring of severity: | Not available | |
| 1 | Pruritus without the need to scratch | ||||
| 2 | Pruritus with the need to scratch but without excoriations | ||||
| 3 | Pruritus that was unrelieved by scratching | ||||
| 4 | Pruritus accompanied by excoriations | ||||
| 5 | Total restlessness | ||||
| Scoring of distribution: | |||||
| 1 | Pruritus at a single location | ||||
| 2 | Scattered pruritus | ||||
| 3 | Generalised pruritus | ||||
| Scoring of frequency: each four short episodes (< 10 min) or one long episode (> 10 min) received 1 point, max. 5 points | |||||
| Scoring sleep disturbance: each episode of awaking due to pruritus received 2 points, max. 14 points | |||||
| The highest possible score for a 24‐hour period: 40 (with 35% of the points attributed to the night‐time period); total range: 0‐26 | |||||
| Duncan 1984 | 0‐3 score | Mean cumulative pruritus score (over 10 days) | 0 | No pruritus | Not available |
| 1 | Mild | ||||
| 2 | Moderate | ||||
| 3 | Severe | ||||
| Duque 2005 | 0‐10 cm VAS | Mean pruritus score; VAS was measured every visit for the intensity of itch in the last 24 hours | 0 | No itch | Not available |
| 10 | Very strong itch | ||||
| Feily 2012 | 0‐5 VAS | Mean weekly score | 0 | No pruritus | Not available |
| 5 | The worst pruritus | ||||
| Ghanei 2012 | Detailed pruritus score introduced by Duo (Duo 1987) | Mean scores, 95% confidence interval | Scoring of severity: | Not available | |
| 1 | Itching sensation without necessity of scratching | ||||
| 2 | Itching that necessitates scratching, but without excoriations | ||||
| 3 | Itching that necessitates frequent scratching | ||||
| 4 | Itching that necessitates scratching accompanied by excoriation | ||||
| 5 | Pruritus causing total restlessness | ||||
| Scoring of distribution: | |||||
| 1 | Pruritus in 2 areas of the body or less | ||||
| 2 | Pruritus in more than 2 areas of the body | ||||
| 3 | Generalised pruritus | ||||
| Scoring sleep disturbance: | |||||
| Every waking up due to pruritus received 2 points (max. 10 points) and every scratching due to pruritus received 1 point (max. 5 points); total range: 0‐45 | |||||
| Ghent 1988 | 0‐100 VAS | Summed 7‐day pruritus score on mean VAS | 0 | None | Preference of rifampicin |
| 100 | Severe | ||||
| Gunal 2004 | 0‐10 cm VAS | Mean and standard deviation; measurement once a day for each period | 0 | No itch | Not available |
| 10 | Worst possible imaginable itch | ||||
| Kuiper 2010 | 0‐10 cm VAS | Morning and evening VAS; proportion of responders | 0 | No pruritus | 40% reduction in pruritus visual analogue scale |
| 10 | Severe pruritus | ||||
| Kumagai 2010 | 0‐100 mm VAS | Mean VAS values of previous 12 hours (morning and evening) were calculated for the last 7 days of the pre‐observation period, the first and latter 7 days of the treatment period and the 8‐day post‐observation period; 95% confidence interval | 0 | No itch | "[A]ssuming an expected difference of 10.0 mm [with a common standard deviation (SD) of 25 mm]" |
| 100 | Strongest possible itch | ||||
| Legroux‐Crespel 2004 | 0‐10 cm VAS | Mean VAS scores; pruritus was evaluated on days 0, 7 and 14. | 0 | Not pruritus or sleep disorders | A marked improvement of pruritus was assessed by a decrease in the VAS score > 3. |
| 10 | Maximum intense of these disorders | ||||
| Makhlough 2010 | Detailed pruritus score introduced by Duo (Duo 1987) | Mean scores ± standard deviation; at the beginning of the study and at the end of weeks 1, 2, 3, and 4 of each study period | Scoring of severity: | Not available | |
| 1 | Itching sensation without necessity of scratching | ||||
| 2 | Itching that necessitates scratching, but without excoriations | ||||
| 4 | Itching that necessitates scratching accompanied by excoriation | ||||
| 5 | Pruritus causing total restlessness | ||||
| Scoring of distribution: | |||||
| 1 | Pruritus in 2 areas of the body or less | ||||
| 2 | Pruritus in more than 2 areas of the body | ||||
| 3 | Generalised pruritus | ||||
| Scoring sleep disturbance | |||||
| Every waking up due to pruritus received 2 points (max. 10 points) and every scratching due to pruritus received 1 point (max. 5 points); total range: 0‐30 | |||||
| Mapar 2015 | Modified Duo Score (0‐45) | Mean scores ± standard deviation; baseline, week 1, 2, 3, and 4 | 0‐45 | higher scores indicating more severe symptoms; (calculation of the final score is based on severity, distribution and sleep disturbance of pruritus) | Discontinuation of pruritus: zinc sulphate: 4 (20%); placebo: 1 (5%) |
| Mayo 2007 | 0‐10 cm VAS | Daily VAS scores were averaged | 0 | No pruritus | Clinically significant improvement defined as a 20% reduction in pruritus from baseline |
| 10 | Worst pruritus imaginable | ||||
| Murphy 2003 | 0‐10 cm VAS | Composite mean VAS score (morning and evening); mean score of the last 5 days of each week was calculated and referred to as the composite mean VAS score | 0 | No itch | Effect size of d = 0.8 |
| 10 | Maximum itch | ||||
| Naini 2007 | 0‐10 cm VAS | Mean pruritus score ± standard deviation; mean score of the last 5 days of each week was calculated and referred to as the composite mean VAS score | 0 | No itch | Not available |
| 10 | Worst possible itch | ||||
| Najafabadi 2012 | 0‐10 VAS | Mean pruritus score ± standard deviation; every two weeks | 0 | No itching | Not available |
| 10 | Worst pruritis | ||||
| Nakhaee 2015 | 0‐10 cm VAS | Mean pruritus score ± standard deviation at baseline and after 2 weeks | 0 | No pruritus | Not available |
| 10 | Worst pruritus imaginable | ||||
| Nasrollahi 2007 | Detailed pruritus score introduced by Duo (Duo 1987) | Not stated | "Assessment of pruritus was done using Detailed Pruritus Score introduced by Duo. The scores for sleep disturbances and intensity, area of pruritus were added and the final score at the beginning and at the end of the study were calculated (maximum score: 45)" | Not available | |
| O'Donohue 2005 | 0‐10 cm VAS; additional measurement of scratching activity by piezo‐electric vibration transducer |
Mean pruritus score standard error of the mean; On day 0 and day 1: every 15 min during the first hour, and hourly thereafter during waking hours. On days 2–5 recordings were made at 3 h intervals from 09:00 to 24:00 hours. | 0 | No pruritus | > 50% reduction in the severity of pruritus |
| 10 | Worst imaginable pruritus | ||||
| Omidian 2013 | 0‐5 VAS | Weekly mean score ± standard deviation | 0 | No itching | Not available |
| 5 | Worst pruritus | ||||
| Pakfetrat 2014 | Detailed pruritus score proposed by Duo (Duo 1987) | Mean scores ± standard deviation; the pruritus score was calculated before and at the end of the study. | Scoring of severity: | Not available | |
| 1 | Itching sensation without necessity of scratching | ||||
| 2 | Itching that necessitates scratching, but without excoriations | ||||
| 3 | Itching that necessitates frequent scratching | ||||
| 4 | Itching that necessitates scratching accompanied by excoriation | ||||
| 5 | Pruritus causing total restlessness | ||||
| Scoring of distribution: | |||||
| 1 | Pruritus in 2 areas of the body or less | ||||
| 2 | Pruritus in more than 2 areas of the body | ||||
| 3 | Generalised pruritus | ||||
| Scoring sleep disturbance: | |||||
| Every waking up due to pruritus received 2 points (max. 10 points) and every scratching due to pruritus received 1 point (max. 5 points); range: 0‐45 | |||||
| Pauli‐Magnus 2000 | 0‐10 cm VAS | Mean weekly pruritus score in percent of initial pruritus score; 95% confidence interval | 0 | No pruritus | Not available |
| 10 | Unbearable pruritus | ||||
| Detailed pruritus score proposed by Duo (Duo 1987) | Mean detailed score and 95% confidence interval | Scoring of severity: | |||
| 1 | Itching sensation without necessity of scratching | ||||
| 2 | Itching that necessitates scratching, but without excoriations | ||||
| 3 | Itching that necessitates frequent scratching | ||||
| 4 | Itching that necessitates scratching accompanied by excoriation | ||||
| 5 | Pruritus causing total restlessness | ||||
| Scoring of distribution: | |||||
| 1 | Pruritus in 2 areas of the body or less | ||||
| 2 | Pruritus in more than 2 areas of the body | ||||
| 3 | Generalised pruritus | ||||
| Scoring sleep disturbance: | |||||
| Every waking up due to pruritus received 2 points (max. 10 points) and every scratching due to pruritus received 1 point (max. 5 points); range: 0‐45 | |||||
| Pederson 1980 | Questionnaire as suggested by Lowrie and Ingham (Lowrie 1975) | Pruritus scores in each individual are presented at week 0, 8 and 16; few information | 1 | I never itch | Not available |
| 2 | I itch rarely but never complain | ||||
| 3 | I itch occasionally with mild annoyance | ||||
| 4 | I itch often; it may be severe but I can be active or rest easily | ||||
| 5 | I itch often; it may be severe and interferes with rest but not activity | ||||
| 6 | I itch always; it is severe and interferes both with rest and activity | ||||
| Statements were arranged in nine paired response alternatives, allowing the ranking of the itching as a severity continuum on a scale of one (no itching) to 10 (severe constant itching) | |||||
| Peer 1996 | 0‐10 cm VAS | Mean daily scores (recorded every 6 hours); medians and interquartile ranges are reported | 0 | No pruritus | Not available |
| 10 | Maximum intensity of pruritus | ||||
|
Podesta 1991a |
0‐100 cm VAS | Mean pruritus score; pruritus was evaluated 15 days before and daily (between 8 and 12 AM, 12‐8 PM, and 8‐8 AM) during treatment | 100 | Pruritus that interfered with sleep, altered daily activities, or resulted in self‐inflicted skin‐breakdown | Full response to treatment was defined as the complete lack of pruritus and a partial response as a 50% reduction in the pruritus score |
| Pour‐Reza‐Gholi 2007 | Not stated | Not stated | Response to treatment was recorded as: complete improvement (no more itching) relative improvement (reduction of the symptom) no effect (symptom remained unchanged or worsened) | Not available | |
| Shirazian 2013 | Pruritus Severity Questionnaire | Biweekly mean scores ± standard deviation | Maximum pruritus score on the survey: 21 points | Not available | |
| 5 | Active itching (yes = 5) | ||||
| 4 | Itching affecting sleep or other activities in the past few days (yes = 4) | ||||
| In the past few days, how would you describe your itching? | |||||
| 0 | None | ||||
| 1 | Mild itching | ||||
| 3 | Moderate itching | ||||
| 4 | Severe itching | ||||
| In the past few days, what part of your body has felt itchy? | |||||
| 1 | Localised itching | ||||
| 2 | Itching in most of the body | ||||
| 3 | Itching in all of the body | ||||
| 5 | Use of medications for itching (yes = 5) | ||||
| Silva 1994 | 0‐3 score | Depicted as a percent of maximum score possible ± standard error; pruritus intensity was scored three times daily | 0 | Absent | Reduction of at least 50% |
| 1 | Pruritus at rest or during usual tasks but not interfering with its accomplishment | ||||
| 2 | Pruritus perturbing but not interrupting performance of regular tasks | ||||
| 3 | Pruritus causing interruption of tasks or sleep | ||||
| Silverberg 1977 | 0‐3 score | Mean of all daily scores; score for the 3 weeks before treatment was the mean of 21 days' values and the score during treatment was the mean of 28 days' values. | 0 | None | Not available |
| 1 | Slight | ||||
| 2 | Moderate | ||||
| 3 | Great | ||||
| Smith 1997a | 1‐4 score | Not stated | 1 | Periodic at night only | Not available |
| 2 | Periodic during the day and night | ||||
| 3 | Periodic during the day, but interferes with sleep at night | ||||
| 4 | Interferes with daily activities as well as sleep at night | ||||
| 0‐4 score | Categorial evaluation; median improvement; 25th and 75th percentile | Overall changes in pruritus after 4‐6 weeks of therapy were graded as follows: | Not available | ||
| 0 | Increased pruritus | ||||
| 1 | No decrease | ||||
| 2 | Slight but definite decrease in pruritus | ||||
| 3 | Moderate decrease in pruritus | ||||
| 4 | Complete resolution of pruritus | ||||
| Tarng 1996 | 4‐point scale | Weekly mean values ± standard deviation | 1 | No itching | Not available |
| 2 | Mild itching, occasionally noticeable | ||||
| 3 | Moderate itching, not interfering with daily life and/or sleep | ||||
| 4 | Severe itching, disturbing daily life and/or sleep | ||||
| Terg 2002 | 0‐10 cm VAS | Mean values ± standard deviation of each period; assessment 1 week before starting treatment and during the 5 weeks of the study; Daytime pruritus was assessed before retiring to sleep while night‐time pruritus was assessed at wake‐up. | 0 | Absence of pruritus | Complete response was defined as disappearance of pruritus and partial response as 50% reduction in the pruritus score |
| 10 | Were pruritus that interfered with sleep, altered daily activities or resulted in self‐inflicted skin breakdown | ||||
|
Turner 1994a Turner 1994b |
0‐100 mm VAS | Mean VAS scores for the last 7 days (daily assessment reflecting the preceding 24 hours); 95% confidence interval | 0 | No itch | Subjective itch improvement (yes or no) |
| 100 | Severe | ||||
| Vessal 2010 | 0‐10 cm VAS | Weekly mean scores ± standard deviation for 12 weeks | 0 | Absence of pruritus | Not available |
| 10 | Greatest severity of symptoms | ||||
| Villamil 2005 | 0‐100 mm VAS | Daily mean VAS scores for the 7 days (measurement at baseline and every 12 hours before going to bed and after awakening); 95% confidence interval | 0 | Absence of pruritus | Difference of 30% |
| 100 | Unbearable intensity | ||||
|
Wikström 2005a Wikström 2005b |
0‐100 mm VAS | Mean worst itching VAS from run‐in to the end of week 4 (study 1) and week 2 (study 2) during the previous 12 hours; assessment every 12 hours during the run‐in period and throughout the studies; 95% confidence interval and standard deviation |
0 | No itching | Patient responders as defined by a reduction from run‐in of at least 50% in "worst itching" VAS |
| 100 | Worst itching ever | ||||
| Wolfhagen 1997 | 0‐100 mm VAS | Mean daytime/night‐time scores each day; ± standard error of the mean and 95% confidence interval | 0 | No itching | Not available |
| 100 | Unbearable itching | ||||
| Young 2009 | 0‐10 cm VAS | [1 − (mean VAS at the end of the study)/(mean VAS at baseline)*100] | Itch intensity after a mosquito bite | Not available | |
| Individual itch on its best intensity | |||||
| Individual itch on its worst intensity | |||||
| Yue 2015 | 0‐10 cm VAS | Biweekly mean scores ± standard deviation (12 weeks) | 0 | No pruritus | Not available |
| 10 | Worst pruritus imaginable | ||||
| Modified Duo's VAG: 0 to 40 | mean scores ± standard deviation at baseline and week 12 | 0 | Higher scores indicating more severe symptoms; (based on criteria such as scratching, severity, frequency, distribution of pruritus, number of sleeping hours, and frequency of waking‐up during the night for scratching) | ||
| 40 | |||||
| Zylicz 2003 | 0 ‐10 numerical analogue scale | Mean value of 7 days ± standard error and 95% confidence interval | 0 | No symptoms | Proportion of clinical responses defined as a pruritus reduction of at least 50% in the last 3 days of each period as compared to the last 3 days of the run‐in period |
| 10 | Worst possible symptoms | ||||
| Özaykan 2001 | Scoring system proposed by Duo and modified by Mettang (Duo 1987; Mettang 1990) | Weekly mean scores | 0‐48 | Period: 0‐3 | Not available |
| Intensity: 0‐10 | |||||
| Allocation: 0‐10 | |||||
| Frequency: 0‐10 | |||||
| Sleeping‐time: 0‐10 | |||||
| Wake‐up time: 0‐5 | |||||
NRS: numerical rating scale; VAS: visual analogue scale.
2. Study interventions and numbers attached to intervention.
| Substance and participants | Dose |
No. of participants included (with dropouts and placebo) |
Authors |
Total number of participants (with dropouts and placebo) |
|
| Paroxetine: 26 participants | |||||
| Palliative care patients | Paroxetine | 20 mg/d | 26 | Zylicz 2003 | 26 |
| Naltrexone: 100 participants | |||||
| UP | Naltrexone | 50 mg/d | 15 | Peer 1996 | 126 (26 from loratidine group) |
| UP | Naltrexone | 50 mg/d | 23 | Pauli‐Magnus 2000 | |
| UP | Naltrexone vs. loratadine | 50 mg/d 50 mg/d |
26 26 |
Legroux‐Crespel 2004 | |
| CP | Naltrexone | 50 mg/d | 16 | Wolfhagen 1997 | |
| CP | Naltrexone | 50 mg/d | 20 | Terg 2002 | |
| Nalfurafine: 450 participants | |||||
| UP | Nalfurafine | 5 µg 3x/week IV | 79 | Wikström 2005a | 450 |
| UP | Nalfurafine | 5 µg 3x/week IV | 34 | Wikström 2005b | |
| UP | Nalfurafine | 2.5µg/d or 5 µg/d | 337 | Kumagai 2010 | |
| Ondansetron: 270 participants | |||||
| UP | Ondansetron | 8 mg 3x/d | 24 | Murphy 2003 | 270 (10 from cyproheptadine and 67 from pregabalin group) |
| UP | Ondansetron | 8 mg 3x/d | 19 | Ashmore 2000 | |
| CP | Ondansetron | 8 mg 2x/d, 5 days | 19 | O'Donohue 2005 | |
| UP | Ondansetron/cyproheptadine | 8 mg/d, 30 days | 20 (10/10) | Özaykan 2001 | |
| UP | Ondansetron vs. pregabalin vs. placebo | ondansetron: 8 mg/d pregabalin: 75 mg twice‐weekly |
188 (64/67/57) | Yue 2015 | |
| Sertraline: 12 participants | |||||
| CP | Sertraline | 25–100 mg/d | 12 | Mayo 2007 | 12 |
| Gabapentin: 127 participants | |||||
| UP | Gabapentin | 300 mg 3x/week | 25 | Gunal 2004 | 127 (26 from ketotifen group) |
| UP | Gabapentin | 400 mg 2x/week | 34 | Naini 2007 | |
| CP | Gabapentin | 300 mg‐2400 mg/d | 16 | Bergasa 2006 | |
| UP | Gabapentin vs ketotifen | Gabapentin 100 mg daily Ketotifen 1 mg twice daily |
26 26 |
Amirkhanlou 2016 | |
| Rifampicin: 23/22 participants | |||||
| CP | Rifampicin | 300 mg 2x/d | 14 | Podesta 1991a | 45 |
| CP | Rifampicin | 150 mg 2‐3x/d | 9 | Ghent 1988 | |
| CP | Rifampicin | 10 mg/kg/d | 22 | Bachs 1989 | |
| Phenobarbitone | 3 mg/kg/d | ||||
| Doxepin: 24 participants | |||||
| UP | Doxepin | 10 mg 2x/d | 24 | Pour‐Reza‐Gholi 2007 | 24 |
| Cholestyramine: 18 participants | |||||
| UP | Cholestyramine | 5 g 2x/d | 10 | Silverberg 1977 | 18 |
| CP | Cholestyramine | 4 g/d | 8 for 2 weeks each | Duncan 1984 | |
| Terfenadine | 60‐180 mg/d | ||||
| Chlorpheniramine | 4 mg–12 mg/d | ||||
| Colesevelam: 38 participants | |||||
| CP | Colesevelam | 1875 mg 2x/d | 38 | Kuiper 2010 | 38 |
| Thalidomide: 29 participants | |||||
| UP | Thalidomide | 100 mg/d | 29 | Silva 1994 | 29 |
| Montelukast: 16 participants | |||||
| UP | Montelukast | 10 mg/d | 16 | Nasrollahi 2007 | 16 |
| Flumecinol: 69 participants | |||||
| CP | Flumecinol low dose | 600 mg 1x/week | 50 | Turner 1994a | 69 |
| CP | Flumecinol high dose | 300 mg/d | 19 | Turner 1994b | |
| Erythropoietin: 20 participants | |||||
| UP | Erythropoietin | 36 units/kg body weight 3x/week IV | 20 | De Marchi 1992 | 20 |
| Cromolyn Sodium: 122 participants | |||||
| UP | Topical cromolyn sodium (CS) | Topical CS 4% 2x/d | 60 | Feily 2012 | 122 |
| UP | Oral CS | 135 mg 3x/d | 62 | Vessal 2010 | |
| Activated oral charcoal: 11 participants | |||||
| UP | Activated oral charcoal | 6 g/d | 20 | Pederson 1980 | 11 |
| Propofol: 12 participants | |||||
| CP | Propofol | 15 mg (1.5 mL)/d IV | 12 | Borgeat 1993 | 12 |
| Lidocaine: 18 participants | |||||
| CP | Lidocaine | 100 mg/d IV | 18 | Villamil 2005 | 18 |
| Topical capsaicin: 105 participants | |||||
| UP | Capsaicin | 0.03% ointment 4x/d | 34 | Makhlough 2010 | 105 |
| UP | Capsaicin | 0.025% cream 4x/d | 19 | Tarng 1996 | |
| UP | Capsaicin | 0.025% cream 4x/d | 22 | Cho 1997 | |
| UP | Capsaicin | 0.025% cream 4x/d | 7 | Breneman 1992a | |
| Tacrolimus: 22 participants | |||||
| UP | Tacrolimus | 0.1% ointment 2x/d | 22 | Duque 2005 | 22 |
| Pramoxine‐HCl: 28 participants | |||||
| UP | Pramoxine‐HCl | 1% lotion 2x/d | 28 | Young 2009 | 28 |
| Hydroxyzine/Pentoxifylline/Indomethacin/Triamcinolone: 65 participants per intervention | |||||
| HIV‐1 disease patients | Hydroxyzine‐HCl with or without doxepin‐HCl at night | 25 mg 3x/d or 25 mg at bedtime | 10 | Smith 1997a | 65 (10 from pentoxifylline, 10 from indomethacin, 10 from triamcinolone, 8 from avena sativa and 9 from vinegar group) |
| Pentoxifylline | 400 mg 3x/d | 10 | |||
| Indomethacin | 25 mg 3x/d | 10 | |||
| Triamcinolone | 0.025% lotion120 mL/week | 10 | |||
| UP | Hydroxyzine vs avena sativa vs vinegar | Hydroxyzine tablet, 10‐mg tablets every night Avena sativa lotion, twice daily Vinegar solution (30‐mL synthetic white vinegar 5% in 500 mL of water), twice daily |
25 (8/8/9) | Nakhaee 2015 | |
| Ergocalciferol: 50 participants | |||||
| UP | Ergocalciferol | 50.000 IU capsule, 1 pill/week | 50 | Shirazian 2013 | 50 |
| Nicotinamide: 50 participants | |||||
| UP | Nicotinamide | 500 mg 2x/d | 50 | Omidian 2013 | 50 |
| Omega‐3 fatty acids: 22 participants | |||||
| UP | Omega‐3 fatty acids | 1 g omega‐3 capsule 3x/d | 22 | Ghanei 2012 | 22 |
| Turmeric: 100 participants | |||||
| UP | Turmeric | 500 mg 3x/d | 100 | Pakfetrat 2014 | 100 |
| Zinc sulphate: 80 participants | |||||
| UP | Zinc sulphate | 220 mg 2x/d | 40 | Najafabadi 2012 | 80 |
| UP | Zinc sulphate | 220 mg daily | 40 | Mapar 2015 | |
CP: cholestatic pruritus; CS: cromolyn sodium; IU: international unit; UP: uraemic pruritus.
3. Secondary outcomes.
| Quality of life | Method/scale | Results |
| Yue 2015 | Health‐related quality of life: Mental Component Summary scale (MCS) from the 12‐item short‐form (SF‐12; version 2); SF‐12 was scored from 0 to 100, with higher scores indicating better HRQoL | Results: Post‐treatment scores:
Mean change from baseline versus placebo (95% CI): statistically significant for pregabalin and not statistically significant for ondansetron or placebo
|
| Kuiper 2010 | Quality‐of‐life scores: Short Form 36 and Liver Disease Symptom Index 2.0 |
No statistically significant changes were found. Both treatment groups were comparable before and after treatment: Short Form 36 questionnaire in the colesevelam group before and after treatment physical functioning (P = 0.67), role physical functioning (P = 0.50), bodily pain (P = 1.00), general health (P = 0.48), vitality (P = 0.90), social functioning (P = 0.37), emotional functioning (P = 0.17) and mental health (P = 0.26) |
| Turner 1994a | VAS: 0 = able to cope with normal activities 100 = completely incapacitated |
Median improvement in quality of life assessment between flumecinol and placebo was 5.0 mm (95% CI 0.4 to 13.0, P = 0.02), in favour of flumecinol At entry: active = 26, placebo = 11 At completion: active = 21, placebo = 7 Median fall: active = 3.5, placebo = 0.1 |
| Turner 1994b | VAS: 0 = able to cope with normal activities 100 = completely incapacitated |
Quality of life was not significantly improved by the higher dose of flumecinol with the difference in median improvement between the 2 groups being 3.5 mm (95% CI 5.9 to 24.9 mm): At entry: active = 32, placebo = 42 At completion: active = 19, placebo = 44 Median fall: active = 4.4, placebo = 3.0 |
| Patient Satisfaction | Method/scale | Results |
| Zylicz 2003 | 7 point scale, where "0" means indifferent, a negative value of "−3" extremely poor, and a positive value "+3" excellent | On average, patients treated with paroxetine had higher satisfaction scores (mean = 0.41 (SE 0.36)) as compared to patients who received placebo (mean = 0.66 (SE 0.36)). Treatment effect: Placebo: mean ± SE = −0.66 ± 0.36 Paroxetine: mean ± SE = 0.41 ± 0.36 Mean difference (95% CI): −1.08 (−0.19 to 1.96); P = 0.027 Period effect: Placebo: mean ± SE = −0.09 ± 0.36 Paroxetine: mean ± SE = −0.16 ± 0.36 Mean difference (95% CI): 0.08 (−0.81 to 0.96); P = 0.967 |
| Depression | Method/scale | Results |
| Bergasa 2006 | ‐Hamilton depression rating scale: includes items intrinsic to medical conditions (i.e. fatigue, sleep) and concern about health ‐Structured Clinical Interview Questionnaire (SCID) for DSM IV, Axis I Disorders: interview measure for the diagnosis of depression and anxiety syndromes. |
Only measured at baseline: Data on the full psychiatric evaluation were available for 13 participants. Hamilton scale: 8 participants with mild depression, 3 moderate depression, 2 no to minimal depression When items relating to medical conditions were omitted: 7 participants mild depression, no moderate depression, and 6 no to minimal depression SCID: 1 major depressive disorder with atypical features. The remainder of the participants were diagnosed with a mood disorder because of a general medical condition: 8 with depressive features and 4 with major depression‐like episodes. The results of the psychiatric evaluations suggested that liver disease and pruritus might have contributed to the depressive symptomatology of the participants. |
| Mayo 2007 | 30‐item Inventory of Depressive Symptomatology‐Self‐report (IDS‐SR30) | All 4 participants with moderate or severe depression improved with sertraline. One of these subjects also improved with placebo. Subjects with mild depressive symptoms did not reliably improve their IDS‐SR30 score with sertraline. 2 of 9 participants improved on open‐label sertraline, but no subject in the cross‐over study with mild depressive symptoms improved. In order to determine how much of the improvement in pruritus may have been related to improvement in depressive symptoms, an analysis of covariance was performed on the serial IDS‐SR30 and VAS scores obtained in the open‐label dose titration. The improvement in VAS after adjusting for IDS‐SR30 was still highly significant (P = 0.0002). The IDS‐SR30 effect was also significant (P = 0.0011). Thus, both VAS and IDSSR30 improved with increasing doses of sertraline, but the change in IDS‐SR30 did not explain all of the change in VAS. Baseline: none (n = 8), mild (n = 9), moderate (n = 2), severe (n = 2) Post‐Sertraline: none (n = 10), mild (n = 2), moderate (n = 0), severe (n = 0) Post‐Placebo: none (n = 7), mild (n = 4), moderate (n = 1), severe (n = 0) |
DSM IV: Diagnostic and Statistical Manual of Mental Disorders, 4th edition; HRQoL: health‐related quality of life; IDS‐SR30 : 30‐item Inventory of Depressive Symptomatology‐Self‐report.
4. Adverse events according to the different studies.
| Study | Design | Pruritus/Disease | Intervention | Dose | Participants(dropouts included) | Adverse events |
| Paroxetine | ||||||
| Zylicz 2003 | Randomised, double‐blind, placebo‐controlled cross‐over study |
Pruritus in palliative care patients (var.) | Paroxetine vs placebo |
20 mg/d | 26 |
Paroxetine: Major: severe nausea and vomiting (leading to withdrawal: n = 2) Minor: sleepiness |
| Naltrexone | ||||||
| Peer 1996 | Randomised, double‐blind, placebo‐controlled cross‐over study | UP | Naltrexone vs placebo |
50 mg/d | 15 |
Naltrexone: Minor: minor heart burn (n = 2), upper abdominal discomfort (n = 3) Placebo: adverse events not given |
| Pauli‐Magnus 2000 | Randomised, double‐blind, placebo‐controlled cross‐over study | UP | Naltrexone | 50 mg/d | 23 |
Naltrexone: Major: gastrointestinal side effects (leading to withdrawal: n = 3) Minor: gastrointestinal side effects (n = 6) Placebo: Major: gastrointestinal side effects (leading to withdrawal) (n = 1) |
| Legroux‐Crespel 2004 | Randomised, comparative trial | UP | Naltrexone vs loratadine |
50 mg/d 50 mg/d |
52 |
Naltrexone: 30 events in 15 participants: Major: vertigo (n = 4) , nausea (n = 9) , malaise (n = 1) , cramps (n = 2), sleeping disturbances (n = 1), anorexia (n = 1) Minor: vomiting (n = 2), abdominal distention (n = 1), sleep disturbances (n = 4), vertigo (n = 1), headaches (n = 2), somnolence (n = 1), paraesthesia (n = 1) Loratadine: 3 events in 2 participants: Major: vomiting (n = 1), malaise (n = 1) Minor: vomiting (n = 1) Withdrawals: Naltrexone: 12 participants (n = 4 due to vertigo, n = 3 due to nausea, n = 1 due to malaise, n = 2 for cramps, n = 1 due to sleeping disturbances and n = 1 due to anorexia) Loratadine: 3 participants (n = 1 due to vomiting, n = 1 due to malaise, n = 1 without relation with loratadine) |
| Wolfhagen 1997 | Randomised, double‐blind, placebo‐controlled study | CP | Naltrexone | 50 mg/d | 16 |
Naltrexone: nausea (n = 4), dizziness (n = 3), flushing (n = 2), drowsiness (n = 2), headache (n = 1), nightmares (n = 1), tremor (n = 1), abdominal cramps (n = 5), dry mouth (n = 2), peripheral oedema (n = 1), night‐sweating (n = 1) Placebo: abdominal cramps (n = 1), dry mouth (n = 1), irritability (n = 1), epistaxis (n = 1), swelling of the hands (n = 1) |
| Terg 2002 | Randomised, double‐blind, placebo‐controlled cross‐over study | CP | Naltrexone | 50 mg/d | 20 |
Naltrexone: dizziness (n = 10), nausea (n = 8), vomit (n = 6), headache (n = 5), abdominal cramps (n = 5), asthenia (n = 3), drowsiness (n = 3), irritability (n = 3), dry mouth (n = 3), insomnia (n = 2), tremor (n = 1), tachycardia (n = 1), anorexia (n = 1), flushing (n = 1), arterial hypertension (n = 1) Placebo: nausea (n = 1), vomit (n = 2), headache (n = 3), abdominal cramps (n = 1), drowsiness (n = 2), irritability (n = 2) |
| Nalfurafine | ||||||
| Wikström 2005a | Meta‐analysis of 2 randomised, double‐blind, placebo‐controlled studies | UP | Nalfurafine | 5 µg 3x/wk IV | 79 |
Nalfurafine: 17 of 26 participants: headache (n = 3), nausea (n = 3), insomnia (n = 2), vertigo (n = 2), vomiting (n = 2), severe headache (n = 1), severe insomnia (n = 1); Leading to withdrawal: moderate nausea and vomiting (n = 1) Placebo: 13 of 25 participants had adverse drug reactions: no description Serious adverse event (SAE): 2 (8%) participants in the nalfurafine 5 µg group and 6 (24%) participants in the placebo group reported at least one SAE, but none were considered to be drug related |
| Wikström 2005b | Meta‐analysis of 2 RCTs | UP | Nalfurafine | 5 µg 3x/wk IV | 65 (not clearly stated) |
Nalfurafine: 2 of 16 participants: vertigo (n = 1), elevations of aspartate aminotransferase and alanine transaminase (n = 1) Placebo: 2 of 18 participants had adverse drug reactions: no description Serious adverse event (SAE): 3 participants in the nalfurafine 5 µg group and 3 participants in the placebo group reported a SAE, but none were considered to be drug related |
| Kumagai 2010 | Randomised, double‐blind, placebo‐controlled study | UP | Nalfurafine | 2.5µg/d or 5 µg/d | 337 |
Adverse events: Nalfurafine (5 µg): nasopharyngitis (12.3%), insomnia (14.9%), somnolence (3.5%), constipation (7.9%) Nalfurafine (2.5 µg): nasopharyngitis (8.0%), insomnia (7.1%), somnolence (4.5%), diarrhoea (4.5%) Placebo: nasopharyngitis (17.1%), headache (3.6%), vomiting (3.6%) Adverse drug reactions: Nalfurafine (5 µg): incidence 35.1%: insomnia (n = 16), anorexia (n = 1), headache (n = 1), pruritus (n = 1), decreased blood TSH (n = 2), mood altered (n = 1), elevated mood (n = 1), feeling abnormal (n = 1), increases in prolactin (n = 3), decrease in free testosterone (n = 1) Nalfurafine (2.5 µg): incidence 25.0%, insomnia (n = 8), sudden hearing loss (n = 1), hypertension (n = 1), vomiting (n = 1), nausea (n = 1), increased eosinophiles (n = 1), increases in prolactin (n = 3), decrease in free testosterone (n = 1) Placebo: incidence 16.2%, headache (n = 1), increases in prolactin (n = 1), decrease in free testosterone (n = 1) |
| Ondansetron | ||||||
| Ashmore 2000 | Randomised, double‐blind, placebo‐controlled cross‐over study | UP | Ondansetron | 8 mg 3x/d | 16 | Not given |
| Özaykan 2001 | Randomised, comparative, controlled trial | UP | Ondansetron vs Cyproheptadine |
8 mg/d 8 mg/d |
10 10 |
None observed; Ondansetron: nausea (n = 3) but disappeared at the end of therapy |
| Murphy 2003 | Randomised, placebo‐controlled double‐blind cross‐over study | UP | Ondansetron | 8 mg 3x/d | 24 |
Ondansetron: Major (leading to withdrawal): constipation (n = 1) Placebo: none given |
| O'Donohue 2005 | Randomised, double‐blind, placebo‐controlled study | CP | Ondansetron | 8 mg 2x/d | 19 |
Ondansetron: Minor: moderate increases from baseline in serum alkaline phosphatase and bilirubin levels (n = 1), constipation (n = 4) Placebo: Minor: nausea (n = 3), headache (n = 2) |
| Yue 2015 | Randomised, double‐blind, placebo‐controlled 3‐armed comparative trial | UP |
|
|
188 |
Pregabalin: Major (leading to withdrawal): somnolence: n = 3; dizziness: n = 1; loss of balance: n = 1 Ondansetron: Major (leading to withdrawal): nausea and vomiting: n = 2 |
| Sertraline | ||||||
| Mayo 2007 | Randomised, double‐blind, placebo‐controlled cross‐over study |
CP | Sertraline | 25‐100 mg/d | 12 |
Sertraline: Minor (during dose‐escalation trial): increase in bowel frequency (n = 2), visual hallucinations (n = 2), increase in fatigue (n = 2), insomnia (n = 3), nausea (n = 1); Major: (during dose‐escalation trial): dizziness (n = 1) beneficial effect of increased mood stability Placebo: Minor: increase in fatigue (n = 1), insomnia (n = 6), nausea (n = 1) taking no drug (baseline, washout): increase in fatigue (n = 1), insomnia (n = 14), nausea (n = 1) |
| Gabapentin | ||||||
| Gunal 2004 | Randomised, double‐blind, placebo‐controlled cross‐over study | UP | Gabapentin | 300 mg 3x/wk | 25 |
Gabapentin: Minor to moderate: somnolence, fatigue, dizziness (usually occurring after first dose) Placebo: none given |
| Bergasa 2006 | Randomised, double‐blind, placebo‐controlled study | CP | Gabapentin | 300‐2400 mg/d | 16 |
Gabapentin: Minor: fatigue (n = 1), dizziness (n = 1), worsening symptoms of carpal tunnel syndrome (n = 1), dizziness on increasing dose and fluctuating rise in serum creatinine (n = 1); Major: vomiting (n = 1) Placebo: Minor: fatigue and leukopaenia (n = 1), symptoms of carpal tunnel syndrome (n = 1) |
| Naini 2007 | Randomised, double‐blind, placebo‐controlled study | UP | Gabapentin | 400 mg 2x/wk | 34 |
Gabapentin: Minor: somnolence, dizziness, nausea (subsided) Major: attacks of dizziness in 1 participant (subsided gradually) Placebo: none given |
| Amirkhanlou 2016 | Randomised, double‐blind, comparative, placebo‐controlled study | UP |
|
|
52 |
Gabapentin: Drowsiness: 4 (7.7%), dizziness: 1 (1.9%) Ketotifen: Drowsiness: 4 (7.7%), dizziness: 1 (1.9%); same between groups |
| Rifampicin | ||||||
| Ghent 1988 | Randomised double‐blind placebo‐controlled cross‐over study | CP | Rifampicin | 300 mg/d‐450 mg/d | 9 | None reported |
| Bachs 1989 | Randomised, placebo‐controlled cross‐over study | CP | Rifampicin | 10 mg/kg/d | 22 | Haemolytic anaemia and renal failure (n = 1) |
| Phenobarbitone | 3 mg/kg/d | Skin rash after a few days of treatment (n = 3), sedative effect | ||||
| Podesta 1991a | Randomised double‐blind placebo‐controlled cross‐over study | CP | Rifampicin | 600 mg/d | 14 | None reported |
| Doxepin | ||||||
| Pour‐Reza‐Gholi 2007 | Randomised, double‐blind, placebo‐controlled cross‐over study | UP | Doxepin | 20 mg/d | 24 |
Minor: drowsiness (n = 11); Major (leading to withdrawal): drowsiness (n = 1) |
| Cholestyramine | ||||||
| Silverberg 1977 | Randomised, double‐blind, placebo‐controlled study | UP | Cholestyramine | 10 g/d | 10 |
Cholestyramine: constipation (n = 1), nausea 10‐15 min after every dose (n = 1) Placebo: none given |
| Duncan 1984 | Randomised, single‐blind, controlled cross‐over trail | CP | Cholestyramine | 4 g/d | 8 | Diarrhoea and vomiting (n = 4) |
| Terfenadine | 60‐180 mg/d | Emotional lability (n = 1) | ||||
| Chlorpheniramine | 4 mg ? 12 mg/d | Drowsiness (n = 2), headache (n = 1) | ||||
| Placebo | lactose, 200 mg 1‐3/d |
Nausea and cutaneous burning (n = 1) | ||||
| Colesevelam | ||||||
| Kuiper 2010 | Randomised, double‐blind, placebo‐controlled study | CP | Colesevelam | 3750 mg/d | 35 |
Colesevelam: minor: 1 (no more than mild stool changes) Placebo: minor: 4 (no more than mild stool changes) |
| Thalidomide | ||||||
| Silva 1994 | Randomised, double‐blind, placebo‐controlled cross‐over study | UP | Thalidomide | 100 mg/d | 29 | None observed |
| Montelukast | ||||||
| Nasrollahi 2007 | Randomised, single‐blind, placebo‐controlled cross‐over study | UP | Montelukast | 10 mg/d | 16 |
Major (leading to withdrawal): 1 man faced anaemia that was diagnosed as myelodysplastic syndrome during placebo period after receiving montelukast for 20 days; 1 diabetic woman with ischaemic heart disease died during placebo period of myocardial infarction |
| Flumecinol | ||||||
| Turner 1994a | Randomised double blind placebo‐controlled study |
CP | Flumecinol low dose | 600 mg 1x/wk | 50 | None observed |
| Turner 1994b | Randomised, double‐blind, placebo‐controlled study | CP | Flumecinol high dose | 300 mg/d | 19 | None observed |
| Erythropoietin | ||||||
| De Marchi 1992 | Randomised, double‐blind, placebo‐controlled cross‐over study | UP | Erythropoietin | 36 units/kg body weight 3x/wk IV | 20 | None given |
| Cromolyn sodium | ||||||
| Feily 2012 | Randomised, double‐blind, vehicle‐controlled study | UP | Topical Cromolyn sodium 4% | topical CS 4% 2x/d | 60 |
Topical cromolyn sodium: Burning sensation (n = 6); gradually subsided during treatment Placebo: none |
| Vessal 2010 | Randomised, double‐blind, vehicle‐controlled study | UP | Oral cromolyn sodium | 405 mg/d | 62 |
Oral cromolyn sodium: Minor: flatulence (n = 1) Placebo: Minor: nausea (n = 2), diarrhoea (n = 1), nausea and diarrhoea (n = 3) |
| Activated oral charcoal | ||||||
| Pederson 1980 | Randomised, double‐blind, placebo‐controlled cross‐over study | UP | Activated oral charcoal | 6 g/d | 20 | None given |
| Propofol | ||||||
| Borgeat 1993 | Randomised, double‐blind, placebo‐controlled cross‐over study | CP | Propofol | 15 mg/d IV | 10 |
Propofol: Minor: presence of pain on injection (n = 3), dizziness of 10‐20 seconds' duration (n = 2) Placebo: none observed |
| Lidocaine | ||||||
| Villamil 2005 | Randomised double blind placebo‐controlled study | CP | Lidocaine | 100 mg/d IV | 18 |
Lidocaine: Minor: mild tinnitus associated with lingual paraesthesia during infusion (n = 2) (symptoms subsided 2‐5 min postinfusion) Placebo: none given |
| Topical capsaicin | ||||||
| Makhlough 2010 | Randomised double‐blind, vehicle‐controlled cross‐over study | UP | Capsaicin | 0.03% ointment 4x/d | 34 |
Capsaicin: Minor: skin burning mild (n = 23), moderate (n = 10), severe (n = 1) Placebo: none observed |
| Tarng 1996 | Randomised double‐blind, vehicle‐controlled cross‐over study | UP | Capsaicin | 0.025% cream 4x/d | 19 |
Capsaicin: 75% (12 of 16) adverse events occurred during treatment Placebo: 25% (4 of 16) adverse events occurred on placebo Adverse events: 93.7% were related to local burning and/or stinging sensations and 6.3% to cutaneous erythaema; adverse events were mild, transient and tolerable by the participants |
| Cho 1997 | Randomised double‐blind, vehicle‐controlled cross‐over study | UP | Capsaicin | 0.025% cream 4x/d | 22 |
Capsaicin: Minor: skin burning or stinging sensations (or both) (n = 11), cutaneous erythaema (n = 5); adverse events were mild and tolerable |
| Breneman 1992a | Randomised, double‐blind, vehicle‐controlled study | UP | Capsaicin | 0.025% cream 4x/d | 7 |
Capsaicin: Minor: mild burning sensation in both arms (n = 1) Major: mild burning sensation in the capsaicin‐treated arm (n = 1) Placebo: none observed |
| Tacrolimus | ||||||
| Duque 2005 | Randomised, double‐blind, vehicle‐controlled study | UP | Tacrolimus | 0.1% ointment 2x/d | 22 |
Tacrolimus: At baseline: warm sensations (n = 8) In week 4: warm sensations (n = 6), significant burning sensation (n = 1) Vehicle: At baseline: warm sensations (n = 2) In week 4: warm sensations (n = 3) |
| Pramoxine‐HCl | ||||||
| Young 2009 | Randomised, double‐blind, vehicle‐controlled study | UP | Pramoxine‐HCl | 1% lotion 2x/d | 28 | None observed |
| Hydroxyne/pentoxyfilline/indomethacin/triamcinolone | ||||||
| Smith 1997a | Randomised comparative trial | Pruritus in patients with HIV‐1 disease | Hydroxyzine‐HCl with/without doxepin‐HCl | 25 mg 3x/d or 25 mg q.h.s. |
10 |
Major: tiredness, drowsiness, sleepiness (n = 2) Minor: tiredness, drowsiness, sleepiness (n = 6); dry mouth or eyes (n = 5), headache (n = 1) |
| Pentoxifylline | 400 mg 3x/d | 10 | Minor: headache (n = 2) | |||
| Indomethacin | 25 mg 3x/d | 10 | Minor: abdominal pain (n = 2), headache (n = 1), indigestion (n = 4); major: abdominal pain (n = 1) | |||
| Triamcinolone | 0.025% lotion | 10 | Minor: nausea (n = 1) | |||
| Nakhaee 2015 | Randomised, non blinded, comparative three‐armed trial | UP |
|
|
25 | None observed |
| Ergocalciferol | ||||||
| Shirazian 2013 | Randomised, double‐blind, placebo‐controlled study | UP | Ergocalciferol | 50.000 IU capsule, 1x/week | 50 | None observed |
| Nicotinamide | ||||||
| Omidian 2013 | Randomised, double‐blind, placebo‐controlled study | UP | Nicotinamide | 500 mg 2x/d | 50 | None observed |
| Omega‐3 with active ingredients: Eicosapentaenoic Acid (EPA), Docosahexaenoic Acid (DHA) | ||||||
| Ghanei 2012 | Randomised double‐blind, placebo‐controlled cross‐over study | UP | Omega‐3 | 1g omega‐3 capsule contained 180 mg of eicosapentaenoic acid (EPA) and 120 mg of docosahexaenoic acid (DHA), 1x/8h | 22 | None observed |
| Turmeric | ||||||
| Pakfetrat 2014 | Randomised, double‐blind, placebo‐controlled study | UP | Turmeric | 500 mg turmeric (22.1 mg was the active ingredient curcumin), 3x/d | 100 | None observed |
| Zinc sulphate | ||||||
| Najafabadi 2012 | Randomised, double‐blind, placebo‐controlled study | UP | Zinc sulphate | 220 mg 2x/d | 40 | None observed |
| Mapar 2015 | Randomised, triple, placebo‐controlled study | UP | Zinc sulphate | 220 mg daily | 40 | None observed |
CP: cholestatic pruritus; IV: intravenous; SAE: serious adverse event; TSH: thyroid stimulating hormone; UP: uraemic pruritus.
5. Adverse events according to different interventions.
| Intervention, participants receiving druga (dropouts included) | Adverse events (intervention) | Placebo/standard medication, number of participantsb (dropouts included) | Adverse events (control) | Withdrawals | ||
| Minor adverse events observed | Major adverse events observed | Minor adverse events observed | Major adverse events observed | |||
| Paroxetine (Zylicz 2003) No of participants: 26 |
Sleepiness | Nausea and vomiting (n = 2) | Placebo No of participants: 26 |
— | — | Paroxetine: 2 Placebo: — |
| Naltrexone (Legroux‐Crespel 2004; Pauli‐Magnus 2000;Peer 1996; Wolfhagen 1997; Terg 2002) No of participants: 92 |
Nausea (n = 21) (Abdominal) cramps (n = 12) Vomiting (n = 8) Gastrointestinal side effects (n = 6) Upper abdominal discomfort (n = 3) Malaise (n = 1) Headache (n = 8) Somnolence (n = 1) Dizziness (n = 13) Drowsiness (n = 3) Irritability (n = 3) Tremor (n = 2) Flushing (n = 1) Peripheral oedema (n = 1) Night‐sweating (n = 1) Nightmares (n = 1) Insomnia (n = 2) Asthenia (n = 3) Dry mouth (n = 5) Minor heart burn (n = 2) Abdominal distention (n = 1) Sleep disturbances (n = 5) Vertigos (n = ) Tachycardia (n = 1) Paraesthesia (n = 1) Anorexia (n = 1) Arterial hypertension (n = 1) |
Gastrointestinal side effects (n = 3) | Placebo (Peer 1996; Pauli‐Magnus 2000; Wolfhagen 1997; Terg 2002) No of participants: 66 |
Abdominal cramps (n = 2) Irritability (n = 3) Epistaxis (n = 1) Swelling of the hands (n = 1) Nausea (n = 1) Vomiting (n = 2) Headache (n = 3) Drowsiness (n = 2) |
Gastrointestinal side effect (n = 1) | Naltrexone: 12 (uncertain) Placebo: 1 |
| Loratadine (Legroux‐Crespel 2004) No of participants: 26 |
Vomiting (n = 2) Malaise (n = 1) |
— | ||||
| Nalfurafine (Kumagai 2010; Wikström 2005a; Wikström 2005b) No of participants: 286 |
Sleep disturbance (n = 24) Headache (n = 1) Nausea (n = 4) Insomnia (n = 26) Vomiting (n = 3) Nasopharyngitis (n = ?) Vertigo (n = 3) Anorexia (n = 1) Somnolence (n = ?) Diarrhoea (n = ?) Constipation (n = ?) Pruritus (n = 1) Hypertension (n = 1) Sudden hearing loss (n = 1) Elevations of aspartate Aminotransferase and alanine Transaminase (n = 1) Decreased blood TSH (n = 1) Mood altered (n = 1) Elevated mood (n = 1) Feeling abnormal (n = 1) Increases in prolactin (n = 6) Decrease in free testosterone (n = 2) Increased eosinophiles (n = 1) Elevated mood (n = 1) Feeling abnormal (n = 1) Increases in prolactin (n = 6) Decrease in free testosterone (n = 2) Increased eosinophiles (n = 1) |
Nausea and vomiting (n = 1) Severe headache (n = 1) Severe insomnia (n = 1) |
Placebo (Kumagai 2010; Wikström 2005a; Wikström 2005b) No of participants: 170 |
Adverse drug reactions (not described) (n = 15) (Wikström 2005a; Wikström 2005b) Nasopharyngitis Vomiting Headache (n = 1) Increases in prolactin (n = 1) Decrease in free testosterone (n = 1) |
— | Nalfurafine: 11 Placebo: 3 |
| Ondansetron (Ashmore 2000; Murphy 2003; O'Donohue 2005;Özaykan 2001;Yue 2015) No of participants: 123 |
Constipation (n = 4) Moderate increases from baseline in serum alkaline phosphatase and bilirubin levels (n = 1) |
Constipation (n = 1) Nausea (n = 5) Vomiting (n = 2) |
Cyproheptadine (Özaykan 2001) No of participants: 10 |
— | — | Ondansetron: 9 Pregabalin: 5 Placebo: 3 |
| Pregabalin (Yue 2015) No of participants: 67 |
— | Somnolence: 3 Dizziness: 1 Loss of balance: 1 |
||||
| Placebo (Ashmore 2000; Murphy 2003; O'Donohue 2005) No of participants: 50 |
Nausea (n = 3) Headache (n = 2) |
— | ||||
| Sertraline (Mayo 2007) No of participants: 12 |
Increase in bowel frequency (n = 1) Fatigue (n = 2) Insomnia (n = 3) Visual hallucinations (n = 2) Nausea (n = 1) |
Dizziness (n = 1) | Placebo No of participants |
Increase in fatigue (n = 1) Insomnia (n = 6) Nausea (n = 1) |
— | — |
| Gabapentin (Bergasa 2006; Gunal 2004; Naini 2007; Amirkhanlou 2016) No of participants: 76 |
Somnolence/drowsiness (n > 4) Dizziness (n > 1) Nausea Fatigue worsening Symptoms of carpal tunnel syndrome (n = 1) |
Attacks of dizziness (1) Vomiting |
Placebo: (Bergasa 2006; Gunal 2004; Naini 2007) No of participants: 49 |
Fatigue and leukopenia (n = 1) Symptoms of carpal tunnel syndrome |
— | Gabapentin: — Ketotifen: — |
| Ketotifen (Amirkhanlou 2016): **No of participants: 26 |
Drowsiness (n = 4) Dizziness (n = 1) |
— | ||||
| Rifampicin (Bachs 1989; Ghent 1988; Podesta 1991a) No of participants: 45 |
Haemolytic anaemia and renal failure (n = 1) | Haemolytic anaemia Renal failure |
Placebo (Ghent 1988; Podesta 1991a) No of participants: 23 |
Skin rash (n = 3) Sedative effect |
— | Rifampicin: 1 Phenobarbitone: 4 |
| Phenobarbitone (Bachs 1989) No of participants: 22 |
— | — | ||||
| Doxepin (Pour‐Reza‐Gholi 2007) No of participants: 24 |
Drowsiness (n = 11) | Drowsiness (n = 1) | Placebo No of participants: 24 |
— | — | Doxepin: 1 |
| Cholestyramine (Duncan 1984; Silverberg 1977) No of participants: 13 |
Constipation (n = 1) Nausea (n = 1) Diarrhoea and vomiting (n = 4) |
— | Placebo (Silverberg 1977; Duncan 1984) No of participants: 13 |
Nausea and cutaneous burning (n = 1) | — | Cholestyramine: — Terfenadine: — Chlorpheniramine: — Placebo: 1 |
| Terfenadine (Duncan 1984) No of participants: 8 |
Emotional lability (n = 1) | — | ||||
| Chlorphenirame (Duncan 1984) No of participants: 8 |
Drowsiness (n = 2) Headache (n = 2) |
— | ||||
| Colesevelam (Kuiper 2010) No of participants: 17 |
Mild stool changes (n = 1) | — | Placebo No of participants: 18 |
Mild stool changes (n = 4) | — | 3 withdrawalsc |
| Thalidomide (Silva 1994) No of participants: 29 |
No adverse events observed | Placebo No of participants: 29 |
No adverse events observed | Thalidomide: 11 | ||
| Montelukast (Nasrollahi 2007) No of participants: 16 |
— | Anaemia (n = 1) | Placebo No of participants: 16 |
— | — | Montelukast: 2 |
| Flumecinol (Turner 1994a; Turner 1994b) No of participants: 34 |
No adverse events observed | Placebo No of participants: 35 |
No adverse events observed | — | ||
| Erythropoietin (De Marchi 1992) No of participants: 10 |
Adverse events not given | Placebo No of participants: 10 |
Adverse events not given | Erythropoietin: 1 | ||
| Topical cromolyn sodium 4% (Feily 2012) No of participants: 30 |
— | Burning sensation (n = 6) | Placebo No of participants: 30 |
No other side effects were reported | — | |
| Oral cromolyn sodium (Vessal 2010) No of participants: 21 |
— | Flatulence (n = 1) | Placebo No of participants: 19 |
Nausea (n = 2) Diarrhoea (n = 1) Nausea and diarrhoea (n = 3) |
— | 23 withdrawalsc |
| Activated oral charcoal (Pederson 1980) No of participants: 20 |
Adverse events not given | Placebo No of participants: 16 |
Adverse events not given | — | — | 9 withdrawalsc |
| Propofol (Borgeat 1993) No of participants: 10 |
Dizziness (n = 2) Pain during injection (n = 3) |
— | Placebo No of participants: 10 |
— | — | 2 withdrawalsc |
| Lidocaine (Villamil 2005) No of participants: 12 |
Mild tinnitus associated with lingual paraesthesia during infusion (n = 2) | — | Placebo No of participants: 6 |
— | — | 2 withdrawalsc |
| Topical capsaicin (Breneman 1992a; Cho 1997; Makhlough 2010; Tarng 1996) No of participants: 82 (uncertain) |
Skin burning (n = 52) Stinging (n = ?) Cutaneous erythaema (n = 6) |
— | Placebo No of participants: 82(?) |
— | — | Capsaicin: 5 |
| Tacrolimus (Duque 2005) No of participants: 12 |
Warmth sensations (n = 14) | Burning sensation (n = 1) | Vehicle No of participants: 10 |
Warming sensation (n = 5) | — | Vehicle: 2 |
| Pramoxine‐HCl (Young 2009) No of participants: 14 |
No adverse events observed | Placebo No of participants: 14 |
No adverse events observed | 1 withdrawalc | ||
| Hydroxine HCL (Smith 1997a; Nakhaee 2015; Nakhaee 2015) No of participants: 18 |
Headache (n = 1) Dry mouth or eyes (n = 5) Tiredness/ Drowsiness/sleepiness (n = 6) |
Sleepiness/tiredness/drowsiness (n = 2) | No placebo | — | — | Hydroxyzine‐HCL: 2 Vinegar solution: 2 (kidney transplantation)c |
| Avena sativa (Nakhaee 2015): No of participants: 8 |
— | — | ||||
| Vinegar solution (Nakhaee 2015): No of participants: 7 |
— | — | ||||
| Pentoxifylline (Smith 1997a) No of participants: 10 |
Headache (n = 2) | — | Placebo | — | — | Pentoxifylline: 1 |
| Indomethacin (Smith 1997a) No of participants: 10 |
Abdominal pain (n = 2) Headache (n = 1) Indigestion (n = 4) |
Abdominal pain (n = 1) | Placebo | — | — | Indomethacin: 2 |
| Triamcinolone (Smith 1997a) No of participants: 10 |
Nausea (n = 1) | — | Placebo | — | — | Triamcinolone: 2 |
| Ergocalciferol (Shirazian 2013) No of participants: 25 |
No other side effects, including hypercalcaemia or hyperphosphataemia, were reported | Placebo No of participants: 25 |
No other side effects, including hypercalcaemia or hyperphosphataemia, were reported | 6 withdrawalsc | ||
| Nicotinamide (Omidian 2013) No of participants: 25 |
No adverse events observed | Placebo No of participants: 25 |
No adverse events observed | 1 withdrawal (reason unclear) | ||
| Omega‐3 with active ingredients (Ghanei 2012) No of participants: 11 |
No adverse events (dyspepsia, skin rash, headache) observed | Placebo No of participants: 11 |
No adverse events (dyspepsia, skin rash, headache) observed | — | ||
| Turmeric (Pakfetrat 2014) No of participants: 50 |
No minor or major complaints attributable to the use of turmeric | Placebo No of participants: 50 |
No minor or major complaints were reported | 1 withdrawalc | ||
| Zinc sulphate (Najafabadi 2012; Mapar 2015) No of participants: 40 |
No minor or major complaints attributable to use of zinc supplementation | Placebo No of participants: 40 |
No minor or major complaints were reported | Zinc sulphate: expired because of congestive heart failure (n = 1) and decreased blood sugar (n = 1); placebo: vomiting (n = 1), "itching improvement" (n = 1) |
||
aTotal number of participants receiving the verum examined. bTotal number of participants receiving placebo or vehicle or standard medication examined. cWithdrawals irrespective of adverse events.
Search results for the previous version of the review
The original review (Xander 2013) identified 40 RCTs. Two review authors (Edith Motschall (EM), SB) searched the databases in June 2010, and one review author (CX) updated the search in November 2011 and January 2012. Overall, the literature search yielded a total of 771 citations. Furthermore, we identified one additional study by handsearching the reference lists of the original studies, textbooks and websites. Personal contact with several investigators did not yield any additional studies or unpublished data. We excluded 144 duplicates. Review authors evaluated the titles and abstracts of 628 studies, selecting relevant studies if they met the inclusion criteria. At this stage, we excluded 546 studies. We obtained a full copy of 82 studies that were potentially eligible for more detailed evaluation. After assessing the reports and in some cases contacting the study authors, we found 38 papers (reporting on 40 studies) that met the eligibility criteria.
Included studies
In this update, we report the study results first organised by type of pruritus and then by type of intervention (see Effects of interventions). Most included studies researched the effect of different interventions on pruritus in participants suffering from advanced diseases and which was associated with UP (34 studies) or CP/hepatogenic pruritus (14 studies). Two studies explored pharmacological interventions in participants with HIV‐infection and in participants treated in palliative care wards, respectively (Smith 1997a; Zylicz 2003). The trials explored a total of 39 different interventions. Among these, 21 treatments focused on UP, 9 on CP, 4 on both UP and CP, 4 on HIV‐associated pruritus, and 1 on pruritus of different origins in palliative care patients. For the drugs researched and the total numbers of participants assigned to the drugs, see Table 12. For an overview of the adverse events according to the different studies or interventions, see Table 14 and Table 15.
See the Characteristics of included studies table for detailed information on each trial.
Palliative care patients with pruritus of different origin
Selective serotonin reuptake inhibitor (SSRI) paroxetine
Zylicz 2003: a prospective, double‐blind, randomised, controlled, multicentre cross‐over trial of the SSRI paroxetine versus placebo took place in two hospices. The 26 total participants,who had solid tumours (17/26), haematological malignancies (4/26), and various non‐malignant or idiopathic conditions (5/26), were heterogeneous and representative of people receiving palliative care. After a run‐in period, participants were randomly assigned to treatment with 20 mg paroxetine or placebo. Due to the advanced nature of their disease, the trial had to be short and the cross‐over took place after seven days.
Participants with advanced diseases suffering from UP or CP
Opioid antagonist naltrexone
Three studies researched the antipruritic effect of the opioid antagonist naltrexone in participants suffering from UP (Legroux‐Crespel 2004; Pauli‐Magnus 2000; Peer 1996).
Peer 1996: in a randomised, double‐blind, placebo‐controlled cross‐over trial, investigators administered 50 mg naltrexone per day by mouth to 15 haemodialysis participants with severe, resistant pruritus. The naltrexone or placebo periods lasted seven days, each with a seven‐day washout between treatment regimens.
Pauli‐Magnus 2000: a placebo‐controlled, double‐blind cross‐over study involved 23 uraemic participants with persistent, treatment‐resistant pruritus. Participants were started with either a four‐week naltrexone sequence (50 mg/d) or matched placebo. This was followed by a seven‐day washout, and participants continued with a four‐week sequence of the alternate medication.
Legroux‐Crespel 2004: in a comparative study researching naltrexone versus loratadine, 52 participants with UP received naltrexone (50 mg/d; 26 participants) or loratadine (10 mg/d; 26 participants) for two weeks, after a washout of 48 hours.
Two double‐blind RCTs researched the antipruritic effect of the opioid antagonist naltrexone in participants suffering from CP (Terg 2002; Wolfhagen 1997).
Wolfhagen 1997: in a double‐blind randomised placebo‐controlled study, 16 participants with pruritus associated with chronic cholestasis were randomised to receive 50 mg/d of naltrexone (8 participants) or placebo (8 participants) daily for four weeks.
Terg 2002: in a randomised, double‐blind, placebo‐controlled cross‐over study, 20 participants with pruritus and cholestasis were included and randomised to receive 50 mg/d of naltrexone or placebo for two weeks. Subsequently, there was a one‐week washout period, and participants were crossed over to the other therapy for two additional weeks.
κ‐Receptor agonist nalfurafine hydrochloride
Three included RCTs investigated the effect of the kappa‐receptor agonist nalfurafine hydrochloride on severe itch in haemodialysis participants (Kumagai 2010; Wikström 2005a; Wikström 2005b). Two of these studies were combined in a meta‐analysis (Wikström 2005a; Wikström 2005b).
Wikström 2005a and Wikström 2005b: two multicentre, randomised, double‐blind, placebo‐controlled studies enrolled 144 participants with UP to postdialysis intravenous treatment with either nalfurafine or placebo for two to four weeks. The first study (study one) used a parallel‐group design with treatment lasting for four weeks. Seventy‐nine participants were randomly assigned in this study, and 74 completed the four weeks of treatment. After the run‐in period, participants were randomly assigned to receive nalfurafine 5 g (n = 26) or placebo (n = 25) three times weekly by intravenous infusion, immediately after completion of each haemodialysis session during the four weeks. A follow‐up visit was performed two weeks after administration of the final dose of study medication.
The second study (study 2) used a cross‐over design in which participants were randomly chosen to receive an intravenous infusion of either nalfurafine 5 g or placebo three times weekly for two weeks. At the completion of the first treatment period, participants underwent a three‐week washout period followed by another one‐week run‐in period. Participants were then crossed over to the other study medication for an additional two weeks of therapy. Thirty‐four participants were randomly assigned to this study, and 31 completed the four weeks of treatment.
Kumagai 2010: in a phase III, randomised, double‐blind, placebo‐controlled study the efficacy and safety of nalfurafine hydrochloride were prospectively investigated by randomly (1:1:1) administering 5 μg (n = 114) or 2.5 μg (n = 112) of the drug or a placebo (n = 111) orally for 14 days, using a double‐blind design, in 337 haemodialysis participants with itch.
Serotonin 5‐HT3 antagonist ondansetron
Three RCTs researched the effect of the emetic agent ondansetron, a peripherally and centrally acting selective serotonin 5‐HT3‐receptor antagonist, on haemodialysis participants suffering UP (Ashmore 2000; Murphy 2003; Özaykan 2001). One RCT examined the effect of ondansetron in participants with CP (O'Donohue 2005).
Ashmore 2000: in a prospective, randomised, double‐blind, placebo‐controlled cross‐over study, 16 haemodialysis participants with persistent pruritus were randomly assigned to treatment with ondansetron (8 mg) or placebo three times daily for two weeks. The study period consisted of two washout periods of seven days and two treatment periods of 14 days.
Murphy 2003: in a double‐blind, randomised, placebo‐controlled trial, 24 participants on haemodialysis were enrolled and blindly allocated on a random basis to the ondansetron–placebo sequence (14 participants) or to the placebo–ondansetron sequence (10 participants). During the treatment, participants received either 8 mg of ondansetron three times a day or a placebo tablet three times a day for two weeks. The washout period between the cross‐over treatment periods was seven days.
Özaykan 2001: an open, randomised, comparative trial investigated the antipruritic effects of ondansetron and cyproheptadine in 20 haemodialysis participants. Ten participants were given 8 mg/d ondansetron, and the other 10 participants were given 8 mg/d cyproheptadine orally, for 30 days. The study was published in Turkish.
O'Donohue 2005: in a double‐blind, placebo‐controlled study, a total of 19 participants with resistant pruritus were randomised to receive either ondansetron 8 mg or placebo as a single intravenous bolus, followed by oral ondansetron 8 mg or placebo twice daily for two days.
Yue 2015: in a double‐blind, comparative three‐armed trial the efficacy of two drugs was evaluated in 188 UP participants. The treatment period was 12 weeks. The authors compared pregabalin (75 mg twice weekly), ondansetron (8 mg/d) and placebo.
Selective serotonin reuptake inhibitor (SSRI) sertraline
Mayo 2007: initially, 21 participants with chronic pruritus due to liver disease underwent an open‐label, dose escalation to determine the dose with optimal efficacy and tolerability. After a washout period, 12 of the participants entered a randomised, double‐blind, placebo‐controlled cross‐over trial. Participants were treated for six weeks, then had a washout period of four weeks, and crossed over to the other therapy for six weeks.
Antiepileptic gabapentin
Two RCTs examined the effect of the antiepileptic gabapentin in participants with UP (Gunal 2004; Naini 2007). One study researched gabapentin in participants with CP (Bergasa 2006).
Gunal 2004: in a double‐blind, placebo‐controlled, cross‐over study, the effectiveness of gabapentin against renal itch was assessed in 25 adult participants on haemodialysis. The participants were randomly assigned to receive gabapentin for four weeks followed by placebo for four weeks, or vice versa. Gabapentin (300 mg) or placebo was administered three times weekly at the end of the haemodialysis sessions.
Naini 2007: in a double‐blind, placebo‐controlled trial to evaluate the efficacy of gabapentin in controlling uraemic itch, 34 adult participants on maintenance haemodialysis were enrolled and assigned to receive four weeks of treatment with either gabapentin (400 mg) or placebo administered twice weekly after haemodialysis sessions.
Bergasa 2006: in a double‐blind, randomised, placebo‐controlled trial, the effect of gabapentin on the perception of pruritus and its behavioural manifestation, scratching, in cholestasis was studied in 16 inpatient women with chronic liver disease and chronic pruritus. Participants were randomised to gabapentin or placebo, starting with a divided dose of 300 mg gabapentin orally per day and increasing to a maximum of 2400 mg per day until relief from pruritus.
Amirkhanlou 2016: in a randomised, double‐blind, comparative trial, 52 UP participants received either 100 mg gabapentin daily or 1 mg ketotifen twice daily for two weeks.
Semiantibiotic rifampicin
Three included studies investigated the treatment of CP with semi‐antibiotic rifampicin (Bachs 1989; Ghent 1988; Podesta 1991a).
Ghent 1988: a double‐blind, randomised cross‐over trial studied nine participants with primary biliary cirrhosis receiving 300 mg to 450 mg/d of rifampicin and placebo sequentially, in random order. Investigators administered each treatment for 14 days, with a 14‐day washout between treatments.
Bachs 1989: in a randomised cross‐over trial, investigators assessed the antipruritic effects of rifampicin (10 mg/kg) and phenobarbitone (3 mg/kg) in 22 participants with primary biliary cirrhosis. Participants received each agent for 14 days, with a 30‐day washout period between treatments.
Podesta 1991a: in a randomised, double‐blind, placebo‐controlled cross‐over study, researchers studied 14 participants with CP for three weeks after a 15‐day washout period. During the first and third week, participants received 600 mg of rifampicin or placebo. No treatment was administered during the second week (washout period and cross‐over). Subsequently, an open study evaluated the persistence of antipruritic effect and safety of rifampicin over an eight‐month period.
Antidepressant doxepin
Pour‐Reza‐Gholi 2007: an RCT with a cross‐over design assigned 24 participants to two groups, who received either placebo or oral doxepin, 10 mg, twice a day for one week. After a one‐week washout period, the two groups switched treatments.
Cholestyramine
Two included RCTs investigated the effect of cholestyramine on participants with CP or UP, respectively (Duncan 1984; Silverberg 1977).
Silverberg 1977: 10 participants with UP were examined in a double‐blind, randomised, placebo‐controlled trial during four weeks of treatment with either 5 g cholestyramine twice daily (5 participants) or placebo (5 participants).
Duncan 1984: a single‐blind, randomised, controlled, cross‐over trial compared the antipruritic activity of cholestyramine (4 g twice daily), chlorpheniramine (4 mg up to three times daily), and placebo (lactose 200 mg up to three times daily) versus terfenadine (60 mg up to three times daily) in eight participants with CP over a treatment period of two weeks for each drug.
Colesevelam
Kuiper 2010: in a randomised, double‐blind, investigator‐initiated, multicentre trial, 38 participants with CP, both treatment‐naive and previously treated, received 1875 mg of colesevelam or an identical placebo twice daily for three weeks.
Immunsuppressant thalidomide
Silva 1994: in a randomised, double‐blind, placebo‐controlled cross‐over trial, 29 participants with UP were treated with 100 mg/d of thalidomide or placebo for seven days. After a washout period of one week, drugs were crossed over for another treatment period of one week.
Leukotriene receptor antagonist montelukast
Nasrollahi 2007: a randomised, single‐blind, placebo‐controlled cross‐over multicentre trial involved participants with refractory UP. The 16 participants were divided into two groups to first receive montelukast 10 mg daily for 20 days and then placebo, or vice versa. The washout period was 14 days.
Flumecinol
Turner 1994a: the initial trial was a double‐blind, randomised, placebo‐controlled study investigating the effect of low‐dose flumecinol (600 mg once weekly) in 50 participants with CP for three weeks. After a seven‐day baseline period, participants were randomised to flumecinol 600 mg or the identical placebo once weekly for three weeks. Participants took the medication on days 0, 7, and 14.
Turner 1994b: at least one month after completing the initial low dose trial, another double‐blind, randomised, placebo‐controlled study investigated the effect of high dose flumecinol in 19 participants with CP, 2 of whom had not participated in the low dose trial. The participants completed a seven‐day baseline VAS assessment of pruritus and quality of life and then were randomised to take either the treatment with 300 mg of flumecinol or identical placebo daily for 21 days.
Erythropoietin
De Marchi 1992: in a 10‐week double‐blind, randomised, placebo‐controlled cross‐over study, participants with severe UP were enrolled to investigate the effects of recombinant human erythropoietin on pruritus and plasma histamine levels; 20 participants with uraemia (10 with severe pruritus and 10 without) were randomised to receive either erythropoietin intravenously (36 U/kg of body weight three times weekly) or placebo for five weeks and then crossed over.
Oral and topical mast cell stabilizer cromolyn sodium
Vessal 2010: in a double‐blind, randomised, placebo‐controlled study, 62 haemodialysis participants with pruritus were randomly assigned to receive cromolyn sodium or placebo (135 mg, three times daily) for eight weeks. Participants were asked to record the severity of their pruritus during each dialysis session on a VAS, during the eight weeks of treatment and four weeks following discontinuation of treatment.
Feily 2012: in a double‐blind, randomised, vehicle‐controlled trial, 60 participants with UP received topical cromolyn sodium (4 %) twice a day or a vehicle for 4 weeks.
Activated oral charcoal
Pederson 1980: in a randomised, double‐blind, placebo‐controlled, cross‐over trial, 11 participants with UP were treated with oral charcoal 6 g daily or placebo and then vice versa in two consecutive eight‐week treatment periods. Authors did not mention a washout period between the treatments.
Anaesthetic propofol
Borgeat 1993: in a prospective, randomised, double‐blind, cross‐over, placebo‐controlled study, 10 participants with CP received two doses of propofol (1.5 mL) and two doses of placebo (1.5 mL of intralipid) during a four‐day study period.
Local anaesthetic lidocaine
Villamil 2005: in a double‐blind, placebo‐controlled trial, 18 participants with CP were randomised (2:1) to receive 100 mg lidocaine (5 mL saline) intravenously over five minutes or placebo (5 mL saline). Investigators performed electrocardiographic monitoring during infusion and recorded vital signs every 15 minutes for the first hour after infusion.
Topical capsaicin
Breneman 1992a: in a randomised, double‐blind, vehicle‐controlled trial conducted to evaluate the efficacy and safety of capsaicin 0.025% cream in the treatment of localised areas of pruritus in participants with UP, seven participants were treated with either capsaicin 0.025% cream or the vehicle for six weeks. Each participant was provided with two sets of tubes: one contained capsaicin 0.025% cream and the other contained the vehicle. Participants were assigned on a random basis to either arm in a double‐blinded fashion. The tubes were identical except for the designations of right and left. Participants were instructed to apply the cream four times daily and to apply medication from one set of tubes only, and specifically to one arm, and medication from the other set of tubes likewise to the other arm for six weeks.
Tarng 1996: to assess the efficacy and safety of capsaicin 0.025% cream in the treatment of UP and to further explore the underlying pathomechanism in a double‐blind, vehicle‐controlled, cross‐over, single‐centre study, 19 haemodialysis participants with UP were treated with capsaicin 0.025% cream or vehicle during two four‐week treatment periods, with a two‐week washout phase between treatments and a follow‐up of eight weeks without treatment. Treatment was applied to a selected area four times daily.
Cho 1997: a randomised, double‐blind, vehicle‐controlled, cross‐over single‐centre study of capsaicin 0.025% cream with two, four‐week treatment periods and a 14 day washout period was conducted with 22 participants with UP to evaluate the role of parathyroid hormone (PTH) and substance P in UP and to elucidate the underlying mechanisms. For this purpose, in the first phase of the study the correlation between the intensity of itching and serum levels of intact PTH was tested. For the second phase, participants were further stratified into two subgroups with low intact PTH (≤ 35 pg/mL) and high intact PTH (> 35 pg/mL). Subsequently, the double‐blind cross‐over trial with topical capsaicin was conducted in the two subgroups. Participants applied capsaicin or vehicle creams four times daily to a pre‐selected area of skin throughout each treatment period.
Makhlough 2010: the randomised, double‐blinded, cross‐over clinical trial was performed on 34 participants receiving haemodialysis with UP to research the effect of topical capsaicin versus vehicle in a single‐centre clinical setting. The participants were divided into two groups: one group received capsaicin 0.03% and the other vehicle for four weeks. Treatment was stopped for two weeks during the washout period and was then continued following a cross‐over.
Tacrolimus
Duque 2005: in a randomised, double‐blind, vehicle‐controlled, multicentre study in 22 participants with UP, investigators assessed the efficacy of tacrolimus ointment 0.1% for a treatment period of four weeks. Participants used one to eight 30 g tubes, with an average of four per participant. Medication was applied only on pruritic areas three times weekly by one of the investigators and by the participants at home twice a day for four weeks.
Pramoxine hydrochloride
Young 2009: a randomised, double‐blind, controlled comparative trial assessed the efficacy of a commercially available anti‐itch lotion containing pramoxine hydrochloride versus control lotion in the treatment of UP in 28 adult haemodialysis participants, recruited from a community haemodialysis centre. Fourteen participants were randomised to receive the 1% pramoxine hydrochloride lotion and 14 participants received the bland emollient for a one‐week treatment period.
Hydroxyzine
Nakhaee 2015: in a non‐blinded, three‐armed cross‐over RCT, 25 participants with UP were randomised and treated for two weeks with avena sativa (twice daily), vinegar solution (twice daily) and hydroxyzine (10 mg tablet every night). Although we did not consider avena sativa and vinegar solution as pharmacological interventions, we included the study because the treatments were compared to hydroxyzine.
Participants with HIV‐associated pruritus
Hydroxyzine hydrochloride, pentoxifylline, triamcinolone, and indomethacin
Smith 1997a: a prospective, randomised, controlled trial with four arms investigated the antipruritic effect of hydroxyzine hydrochloride with or without doxepin hydrochloride, pentoxifylline, triamcinolone, and indomethacin in patients with advancing HIV disease. Altogether, 40 participants (10 participants in each treatment group) took part in the study. Duration of the treatment was four to six weeks.
New drugs identified in this update
All new included drugs focused on patients with UP.
Ergocalciferol
Shirazian 2013: in this double‐blind RCT, 50 participants with UP were randomised to the ergocalciferol group (vitamin D2; 50,000 IU, one pill per week) or the placebo group for 12 weeks.
Nicotinamide
Omidian 2013: in a double‐blind RCT, 50 participants with UP were randomised to the nicotinamide group or placebo group. The duration of the treatment was four weeks and nicotinamide was administered orally (500 mg twice daily).
Omega‐3 fatty acids
Ghanei 2012: in a double‐blind cross‐over RCT, 22 participants with UP were randomised and treated with omega‐3 fatty acids or placebo. The 1 g capsules of the study drug and placebo had to be taken every eight hours for 20 days.
Turmeric
Pakfetrat 2014: this double‐blind RCT investigated, the antipruritic effect of turmeric. One hundred participants with UP were randomised to receive three 500 mg turmeric or placebo capsules a day for eight weeks.
Zinc sulphate
Najafabadi 2012: in a double‐blind RCT, 40 participants suffering under UP were randomised to receive 220 mg zinc sulphate (oral) twice daily for eight weeks or placebo. The placebo capsule was similarly shaped and coloured.
Mapar 2015: in this double‐blind RCT, 40 participants with UP were randomised to the zinc sulphate group (single dose of 220 mg daily) or the placebo group for 4 weeks.
Ketotifen
Amirkhanlou 2016: in a randomised, double‐blind, comparative trial, 52 UP participants received either 100 mg gabapentin daily or 1 mg ketotifen twice daily for 2 weeks.
Pregabalin
Yue 2015: a double‐blind, comparative three‐armed trial evaluated the efficacy of two treatments in 188 UP participants. The treatment period was 12 weeks. The authors compared pregabalin (75 mg twice a week), ondansetron (8 mg/d) and placebo.
Funding sources
Pharmaceutical companies sponsored 9 of the 50 studies (18%) (Ashmore 2000; Duque 2005; Ghanei 2012; Kuiper 2010; O'Donohue 2005; Peer 1996; Smith 1997a; Wolfhagen 1997; Young 2009). In Breneman 1992a, industry provided only capsaicin and vehicle creams. Another 12 studies (24%) were funded by the related university hospital (Ghent 1988; Pakfetrat 2014), the Robert Bosch Foundation (Pauli‐Magnus 2000) or other independent sources (Bergasa 2006; Ghent 1988; Mayo 2007; Murphy 2003; Pakfetrat 2014; Pauli‐Magnus 2000; Shirazian 2013; Terg 2002; Vessal 2010). In one study (2%), the authors declared that they did not receive any funding. However, most studies (n = 27; 54%) provided no information on financial resources.
Ongoing studies
The original review included six ongoing studies. Two have since been completed, and we include their results in this update (Kuiper 2010; Shirazian 2013). We initially found 70 ongoing studies for this update using the following search terms: (itch OR pruritus) AND (palliative OR progressive OR advanced OR terminal OR dialysis OR hemodialysis OR "end stage") AND (randomised OR randomized OR random) NOT animal | Adult). After the screening (WS, CX), we identified and included 12 additional ongoing studies resulting in a total of 16 studies classified as ongoing (see Ongoing studies).
Excluded studies
We did not assess the full text of any study that we subsequently excluded in this update.
In the original review, we excluded 44 of 82 studies in the final screening because they did not meet the inclusion criteria: 32 were not RCTs; 7 did not meet the inclusion criteria concerning palliative care patients; one study intervention targeted the treatment of underlying disease; one showed doubly published data; and one did not focus on a pharmacological intervention. Two studies could not be included because only the abstract was available and the data were reported inadequately. For both of these studies, we did not receive a response from the authors despite multiple requests. Details regarding reasons for exclusions are provided in the Characteristics of excluded studies table.
Risk of bias in included studies
We present the risk of bias of the included studies graphically (Figure 2; Figure 3) and report details justifying our decisions in the Characteristics of included studies table. The main reason for giving a high risk of bias rating was a small sample size. Forty‐eight of 50 studies (96%) had fewer than 50 participants per treatment arm. Without the consideration of the sample size bias, four studies (8%) would have a low risk of bias (Kuiper 2010; O'Donohue 2005; Pakfetrat 2014; Vessal 2010). The remaining studies would have an unclear risk of bias (34 studies, 68%) or a high risk of bias (12 studies, 24%).
2.
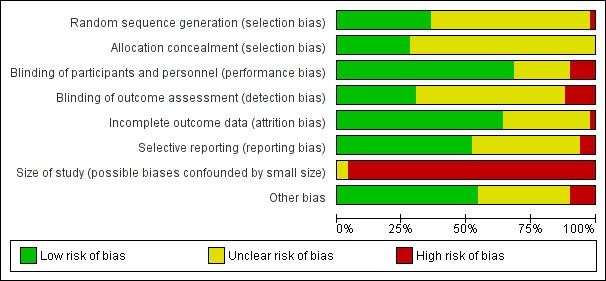
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.
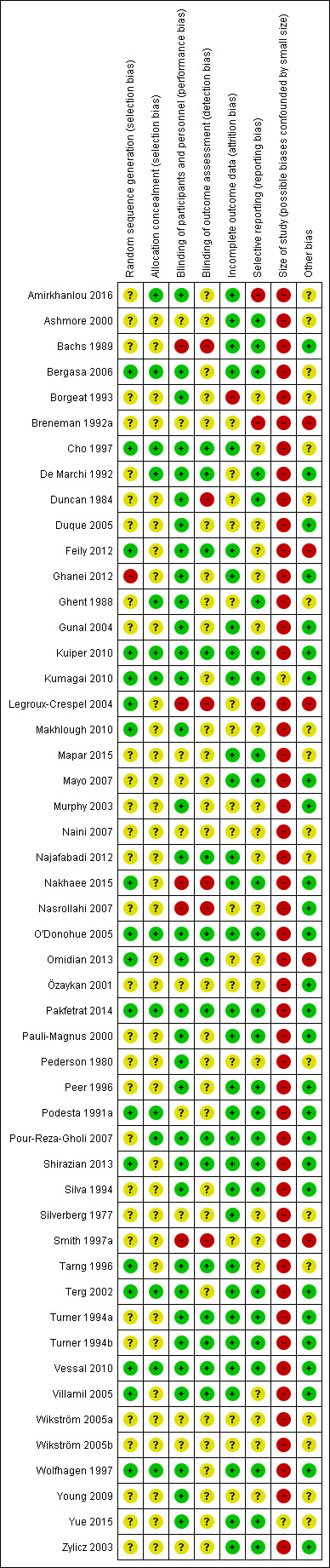
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Random sequence generation (selection bias)
We included 18 studies (36%) that used appropriate methods of randomisation (e.g. drawing lots, flipping coins, computer‐generated table of random numbers) (low risk of bias) and 31 trials (62%) that reported using randomisation but failed to state the method of randomisation (unclear risk of bias). We rated the risk of bias of the random sequence generation in one study (2%) as high because investigators used an alternation method (Ghanei 2012).
Allocation concealment (selection bias)
Fourteen trials (28%) described allocation concealment (low risk), whereas the remainder of the trials (36 studies, 72%) did not mention the method of allocation concealment (unclear risk).
Blinding
Authors described 43 trials (86%) as double‐blind. Thirty‐four studies (68%) were at low risk of performance bias concerning blinding of participants and personnel, but five studies (10%) were at high risk of bias. The remaining 11 studies (22%) were at unclear risk.
Ten studies (20%) described blinding of the outcome assessment. The risk for detection bias was high in six studies (12%), unclear in 29 studies (58%) and low in 15 studies (30%). Two of the included studies (4%) were single‐blind (Duncan 1984; Nasrollahi 2007), three studies (6%) were open trials (Bachs 1989; Nakhaee 2015; Özaykan 2001), and in another two studies (4%) the use of blinding was unclear (Legroux‐Crespel 2004; Smith 1997a). To our knowledge, the included studies did not carry out any analyses of the efficacy of blinding.
Incomplete outcome data
With regard to incomplete outcome data, the quality of the included studies was quite heterogeneous. The dropout rate was low in most trials. In 17 studies (34%) there were no withdrawals. We judged 32 studies (64%) to be at low risk of bias and 17 studies (34%) to be at an unclear risk of bias for this domain. Five trials (10%) reported dropout rates of more than 25% (judged to be at unclear risk), and we judged one study with a maximum dropout rate of 57% to be at high risk of bias (Breneman 1992a). This high dropout rate may be due to the small number of participants included in the trials (only seven participants were included in the trial with the highest dropout rate). On the other hand, it may also be related to the advanced stage of disease of the participants included in the studies. Most trials reported reasons for dropout. Detailed information on dropouts and reasons for dropout are included in the Characteristics of included studies and in Table 15.
Selective reporting
Risk of bias concerning selective reporting was unclear in 21 of 50 studies (42%), and we judged the risk of bias to be low in 26 studies (52%). Since we did not have access to all protocols of the included studies, there was not enough information to assess selective reporting bias in detail. Nevertheless, three studies (6%) showed a high risk of selective reporting bias (Amirkhanlou 2016; Breneman 1992a; Legroux‐Crespel 2004). Please see the Characteristics of included studies table for more details.
Size of study (biases confounded by small size)
The risk of bias concerning the sample size was high in 48 of 50 studies (96%). We rated only two studies that had between 50 and 199 participants per treatment arm as being at unclear risk of bias (Kumagai 2010; Yue 2015).
Other potential sources of bias
Other potential sources of bias may have related to missing washout periods leading to carryover effects in cross‐over studies (Cho 1997; Tarng 1996), inadequate study designs (Smith 1997a; Tarng 1996), and conflicting data (Feily 2012; Omidian 2013). Therefore, we rated the bias of five studies (10%) as high, of 18 studies (36%) as unclear and of 27 studies (54%) as low.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9; Table 10
Summary of findings for the main comparison. Paroxetine versus placebo for pruritus.
| Paroxetine versus placebo | ||||||
| Patient or population: palliative care patients with pruritus Setting: inpatient Intervention: paroxetine Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with paroxetine | |||||
| Pruritus
assessed with: NAS
Scale from: 0 to 10 Follow‐up: 1 week |
The mean pruritus was 6.0 | The mean pruritus in the intervention group was 0.78 lower (1.19 lower to 0.37 lower) | — | 48 (1 RCT) | ⊕⊕⊕⊝ Moderatea | Zylicz 2003: Numerical analogue scale (NAS). Score 0 reflected no symptoms, while score 10 reflected worst possible symptoms. |
| Quality of life ‐ not measured | — | — | — | — | — | Not measured |
| Patient satisfaction
assessed with: 7 point scale
Scale from: −3 to 3 Follow‐up: 1 week |
The mean patient satisfaction was −0.66 | The mean patient satisfaction in the intervention group was 1.08 higher (1.98 higher to 0.18 higher) | — | 48 (1 RCT) | ⊕⊕⊝⊝ Lowa,b | "0" means indifferent, a negative value of "−3" extremely poor, and a positive value "+3" excellent" " (Zylicz 2003) |
| Depression ‐ not measured | — | — | — | — | — | Not measured |
| Nausea
assessed with: NAS
Scale from: 0 to 10 Follow‐up: 1 week |
The mean nausea score was 0.47 | The mean nausea in the intervention group was 0.46 higher (0.05 higher to 0.87 higher) | — | 52 (1 RCT) | ⊕⊕⊕⊝ Moderatec | Score 0 reflected no symptoms, while score 10 reflected worst possible symptoms. |
| Vomiting
assessed with: NAS
Scale from: 0 to 10 Follow‐up: 1 week |
The mean vomiting was 0.25 | The mean vomiting in the intervention group was 0.18 lower (0.44 lower to 0.08 higher) | — | 52 (1 RCT) | ⊕⊕⊕⊝ Moderated | Score 0 reflected no symptoms, while score 10 reflected worst possible symptoms. |
| Sleepiness
assessed with: NAS
Scale from: 0 to 10 follow up: 1 week |
The mean sleepiness was 1.09 | The mean sleepiness in the intervention group was 0.7 higher (0.18 higher to 1.22 higher) | — | 52 (1 RCT) | ⊕⊕⊕⊝ Moderatea | Score 0 reflected no symptoms, while score 10 reflected worst possible symptoms. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; NAS: numerical analogue scale; RCT: randomised controlled trial; RR: risk ratio; OR: odds ratio. | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aQuality of evidence downgraded by one level because of serious imprecision: the 95% CI has a wide range. Parts of the 95% CI are lower than 1 and can be considered as not clinically meaningful whereas some parts are clearly above 1 or −1. bQuality of evidence downgraded by one level because of serious risk of bias: no precise information about randomisation, allocation concealment or blinding. cQuality of evidence downgraded by one level because of serious imprecision: wide 95% CI. dQuality of evidence downgraded by one level because of serious imprecision: wide 95% CI that crosses 0.
Summary of findings 2. Naltrexone versus placebo for pruritus.
| Naltrexone versus placebo | ||||||
| Patient or population: participants with cholestatic pruritus (CP) Setting: inpatients and outpatients Intervention: naltrexone Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with naltrexone | |||||
| Pruritus
assessed with: 10 cm VAS Follow‐up: range 2 weeks to 4 weeks |
The mean pruritus ranged from 5.18 to 5.34 cm | The mean pruritus in the intervention group was 2.26 cm lower (3.19 lower to 1.33 lower) | — | 52 (2 RCTs) | ⊕⊕⊕⊝ Moderatea | Lower scores on VAS indicate less severe pruritus |
| Quality of life ‐ not measured | — | — | — | — | — | Not measured |
| Patient satisfaction ‐ not measured | — | — | — | — | — | Not measured |
| Depression ‐ not measured | — | — | — | — | — | Not measured |
| Risk for at least one adverse event per participant Follow‐up: range 1 weeks to 4 weeks |
Study population | RR 4.07 (2.07 to 8.00) | 116 (3 RCTs) | ⊕⊕⊕⊝ Moderateb | Also uraemic pruritus patients included (see Analysis 1.8) | |
| 12 per 100 | 49 per 100 (25 to 97) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; RCT: randomised controlled trial; VAS: visual analogue scale. | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aQuality of evidence downgraded by one level because of serious inconsistency: meta‐analysis shows I2 = 55%, and mean differences vary seriously. However, 95% CIs overlap and do not cross 0. bQuality of evidence downgraded by one level because of serious imprecision: wide 95% CI.
Summary of findings 3. Nalfurafine versus placebo for pruritus.
| Nalfurafine versus placebo | ||||||
| Patient or population: participants with uraemic pruritus (UP) Setting: inpatient or outpatient Intervention: nalfurafine Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with nalfurafine | |||||
| Pruritus
assessed with: 10 cm VAS Follow‐up: 2 weeks |
The mean pruritus ranged from 4.84 to 5.55 cm | The mean pruritus in the intervention group was 0.95 cm lower (1.32 lower to 0.58 lower) | — | 422** (3 RCTs) | ⊕⊕⊕⊝ Moderatea |
Wikström 2005a and Wikström 2005b: data from study 1 and period 1 of study 2; range of placebo risk only available from Wikström et al; Lower scores on VAS indicate less severe pruritus. |
| Quality of life ‐ not measured | — | — | — | — | — | Not measured |
| Patient satisfaction ‐ not measured | — | — | — | — | — | Not measured |
| Depression ‐ not measured | — | — | — | — | — | Not measured |
| Risk for at least one adverse drug reaction (ADR) per participant Follow‐up: 2 weeks |
Study population | RR 1.62 (1.15 to 2.29) | 422 (3 RCTs) | ⊕⊕⊕⊝ Moderateb | ADR was chosen to enable pooled analysis. | |
| 214 per 1000 | 347 per 1000 (246 to 491) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ADR: adverse drug reaction; CI: confidence interval; RR: risk ratio; RCT: randomised controlled trial; VAS: visual analogue scale. | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aQuality of evidence downgraded by one level because of serious risk of bias: lack of information in study description (randomisation, blinding, withdrawals) of Wikström 2005a and Wikström 2005b. bQuality of evidence downgraded by one level because of serious imprecision: wide 95% CI.
Summary of findings 4. Ondansetron versus placebo for pruritus.
| Ondansetron versus placebo | ||||||
| Patient or population: patients with uraemic pruritus (UP) Setting: inpatient Intervention: ondansetron Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with ondansetron | |||||
| Pruritus assessed with: 10 cm VAS Follow‐up: range 2 weeks to 12 weeks | The mean pruritus was 4.97 cm | The mean pruritus in the intervention group was 0.28 cm lower (0.46 lower to 0.10 lower) | — | 151 (2 RCTs) | ⊕⊕⊕⊝ Moderatea | Lower scores on VAS indicate less severe pruritus; 4.97 is the weighed mean of the placebo groups resulting from Analysis 3.1 |
| Quality of life ‐ not measured | — | — | — | — | — | Not measured |
| Patient satisfaction ‐ not measured | — | — | — | — | — | Not measured |
| Depression ‐ not measured | — | — | — | — | — | Not measured |
| Risk for at least one adverse event per patient Follow‐up: range 2 weeks to 12 weeks | Study population | RR 7.53 (0.97 to 58.51) | 155 (2 RCTs) | ⊕⊝⊝⊝ Very lowa,b | Results for uraemic pruritus (UP) patients from Analysis 3.5 | |
| 0 of 74 | 7 of 81 | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; OR: odds ratio; VAS: visual analogue scale. | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
aQuality of evidence downgraded by one level because of serious risk of bias: little information about blinding; possible adverse events of placebo group not given (Murphy 2003). bQuality of evidence downgraded by two levels because of serious imprecision: the 95% CI has a wide range and crosses 1.
Summary of findings 5. Gabapentin versus placebo for pruritus.
| Gabapentin versus placebo | ||||||
| Patient or population: patients with uraemic pruritus (UP) Setting: inpatient Intervention: gabapentin Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with gabapentin | |||||
| Pruritus
assessed with: 10 cm VAS Follow‐up: 4 weeks |
The mean pruritus ranged from 5.7‐7.6 cm | The mean pruritus in the intervention group was 5.91 cm lower (6.87 lower to 4.96 lower) | — | 118 (2 RCTs) | ⊕⊕⊕⊝ Moderatea | However, Bergasa 2006 concludes that "gabapentin did not provide a significant therapeutic advantage over the placebo"; Lower scores on VAS indicate less severe pruritus. |
| Quality of life ‐ not measured | — | — | — | — | — | Not measured |
| Patient satisfaction ‐ not measured | — | — | — | — | — | Not measured |
| Depression ‐ not reported | — | — | — | — | — | Not measured; Bergasa 2006: Only baseline information given. |
| Risk for at least one adverse event per participant Follow‐up: 4 weeks |
Study population | RR 2.63 (0.76 to 9.05) | 15 (1 RCT) | ⊕⊕⊝⊝ Lowb | Gunal 2004 and Naini 2007 gave no numbers of patients with adverse events; results from Bergasa 2006 | |
| 29 per 100 | 75 per 100 (22 to 100) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; RCT: randomised controlled trial; VAS: visual analogue scale | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
aQuality of evidence downgraded by one level because of serious risk of bias: randomisation and blinding are not well described. Baseline values were pooled for the gabapentin and placebo group. bQuality of evidence downgraded by two levels because of very serious imprecision: very wide 95% CI that, in addition, crosses 1.
Summary of findings 6. Rifampicin versus placebo for pruritus.
| Rifampicin versus placebo | ||||||
| Patient or population: participants with cholestatic pruritus (CP) Setting: outpatient Intervention:rifampicin Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with rifampicin | |||||
| Pruritus
assessed with: 100 mm VAS Follow‐up: range: 2 weeks to 4 weeks |
The mean pruritus was 54.36‐67.50 mm | The mean pruritus in the intervention group was 24.64 mm lower (31.08 lower to 18.21 lower) | — | 42 (2 RCTs) | ⊕⊕⊝⊝ Lowa | Lower scores on VAS indicate less severe pruritus. Only 32 participants could be used for Analysis 5.1 because of carryover effects in the cross‐over design of Podesta 1991a). |
| Quality of life ‐ not measured | — | — | — | — | — | Not measured |
| Patient satisfaction ‐ not measured | — | — | — | — | — | Not measured |
| Depression ‐ not measured | — | — | — | — | — | Not measured |
| Risk for at least one adverse event per participant Follow‐up: 2 weeks |
Study population | RR 0.29 (0.03 to 2.51) | 39 (1 RCT) | ⊕⊝⊝⊝ Very lowb,c | Results from Bachs 1989: rifampicin versus phenobarbitone | |
| 17 per 100 | 5 per 100 (0 to 42) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CP: cholestatic pruritus; RR: risk ratio; RCT: randomised controlled trial; VAS: visual analogue scale | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aQuality of evidence downgraded by two levels because of very serious inconsistency: Meta‐analysis shows I2 of 95% and mean differences vary seriously. However, 95% CIs overlap and do not cross 0. bQuality of evidence downgraded by one level because of serious risk of bias: random sequence generation and allocation concealment unclear; no blinding. cQuality of evidence downgraded by two levels because of very serious imprecision: the 95% CI has a very wide range and crosses 1.
Summary of findings 7. Flumecinol versus placebo for pruritus.
| Flumecinol versus placebo | |||||||
| Patient or population: participants with cholestatic pruritus (CP) Setting: outpatient Intervention: flumecinol Comparison: placebo | |||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | ||
| Risk with placebo | Risk with flumecinol | ||||||
| Pruritus: significant improvement
assessed with: subjective response: yes/no Follow‐up: 3 weeks |
Study population | RR 1.89 (1.05 to 3.39) | 69 (2 RCTs) | ⊕⊕⊝⊝ Lowa,b | An RR > 1 indicates more improvement in participants treated with flumecinol. | ||
| 31 per 100 | 59 per 100 (33 to 100) | ||||||
| Pruritus
assessed with: 100 mm VAS Follow‐up: 3 weeks |
The mean pruritus was 25 mm | The median pruritus in the intervention group was 8 mm lower (2.1 higher to 20.8 lower) | — | 50 (1 RCT) | ⊕⊕⊝⊝ Lowc | Lower scores on VAS indicate less severe pruritus; results from Turner 1994a; Turner 1994b (n = 19): placebo: median: 47 mm, median difference: 19.8 mm, 95% CI 3.3 to 40.7 mm | |
| Quality of life
assessed with: 100 mm VAS Follow‐up: 3 weeks |
The mean quality of life was 7 mm | The mean quality of life in the intervention group was 5 mm lower (0.4 lower to 13 lower) | — | 50 (1 RCT) | ⊕⊕⊕⊝ Moderateb | VAS: 0 = able to cope with normal activities, to 100 = completely incapacitated; Results from Turner 1994a; Turner 1994b (n = 19): placebo: median: 44 mm, median difference: 3.5 mm, 95% CI −5.9 to 24.9 mm |
|
| Patient satisfaction ‐ not measured | — | — | — | — | — | Not measured | ‐ |
| Depression ‐ not measured | — | — | — | — | — | Not measured | |
| Risk for at least one adverse event per participant ‐ not reported Follow‐up: 3 weeks |
See comment | See comment | Not estimable | 69 (2 RCTs) | — | No adverse events attributable to the trial medication" (Turner 1994a; Turner 1994b); no other adverse events stated or documented | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CP: cholestatic pruritus; RR: risk ratio; RCT: randomised controlled trial; VAS: visual analogue scale. | |||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | |||||||
aQuality of evidence downgraded by one level because of serious inconsistency: the meta‐analysis shows an I2 of 59% that implies moderate or substantial inconsistency. bQuality of evidence downgraded by one level because of serious imprecision: the 95% CI has a wide range and covers a large as well as a very small effect. cQuality of evidence downgraded by two levels because of very serious imprecision: the 95% CI has a wide range and crosses zero.
Summary of findings 8. Cromolyn sodium versus placebo for pruritus.
| Cromolyn sodium versus placebo | ||||||
| Patient or population: participants with uraemic pruritus (UP) Setting: inpatient Intervention: cromolyn sodium Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with cromolyn sodium | |||||
| Pruritus
assessed with: 10 cm VAS (values from Feily 2012 multiplied by factor of 2 since presented on VAS 0‐5 scale) Scale from: 0 to 10 Follow‐up: range: 4 weeks to 8 weeks |
The mean pruritus ranged from 2.6 cm to 5.6 cm | The mean pruritus in the intervention group was 2.94 cm lower (4.04 lower to 1.83 lower) | — | 100 (2 RCTs) | ⊕⊕⊕⊝ Moderate a | Risk with placebo: Vessal 2010: 5.6 (scale: VAS 0‐10; '10' represented the greatest severity of symptoms); Feily 2012: 1.3 (scale: 0‐5; "5: the worst pruritus") |
| Quality of life ‐ not measured | — | — | — | — | — | Not measured |
| Patient satisfaction ‐ not measured | — | — | — | — | — | Not measured |
| Depression ‐ not measured | — | — | — | — | — | Not measured |
| Risk for at least one adverse event per participant Follow‐up: range: 4 weeks to 8 weeks |
Study population | RR 1.12 (0.40 to 3.08) | 122 (2 RCTs) | ⊕⊝⊝⊝ Very lowb,c | Oral administration (Vessal 2010); topical use (Feily 2012) | |
| 10 per 100 | 11 per 100 (4 to 31) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; RCT: randomised controlled trial; VAS: visual analogue scale. | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
aQuality of evidence downgraded by one level because of serious inconsistency: meta‐analysis shows an I2 of 81%. The 95% CIs for mean differences do not overlap. bQuality of evidence downgraded by two levels because of very serious inconsistency: meta‐analysis shows I2 of 84% and mean differences are contradictory. cQuality of evidence downgraded by one level because of serious imprecision: the 95% CI has a very wide range and crosses 1.
Summary of findings 9. Topical capsaicin versus vehicle for pruritus.
| Capsaicin versus vehicle | ||||||
| Patient or population: participants with uraemic pruritus (UP) Setting: inpatient Intervention: topical capsaicin Comparison: vehicle | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with topical capsaicin | |||||
| Pruritus
assessed with: different scales Follow‐up: 4 weeks |
Original scale: the mean pruritus was 7.2 (Duo scale 0‐30; Makhlough 2010) and 3.5 (4‐point scale; Cho 1997) | The mean pruritus in the intervention group was 1.02 standard deviations lower (1.35 lower to 0.68 lower) | — | 112 (2 RCTs) | ⊕⊕⊕⊝ Moderate a | SMD calculated from the results of Cho 1997 and Makhlough 2010; Cho uses 4‐point scale (1‐4): −0.88, 95% CI −1.31 to −0.44; Makhlough uses Duo scale (0‐30): −4.70, 95% CI ‐7.57 to −1.83; both favour topical capsaicin; see Analysis 8.1 |
| Quality of life ‐ not measured | — | — | — | — | — | Not measured |
| Patient satisfaction ‐ not measured | — | — | — | — | — | Not measured |
| Depression ‐ not measured | — | — | — | — | — | Not measured |
| Risk for at least one adverse event per participant Follow‐up: range: 2 weeks to 6 weeks |
Study population | RR 4.64 (2.05 to 10.51) | 116 (3 RCTs) | ⊕⊕⊕⊝ Moderate b | Mainly skin burning which is, however, a physiological mechanism of capsaicin. Makhlough 2010: only participants with moderate and severe skin burning included in RR calculation. | |
| 9 per 100 | 40 per 100 (18 to 91) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; RCT: randomised controlled trial | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
aQuality of evidence downgraded by one level because of serious imprecision: the 95% CI has a wide range and includes large and moderate effects. bQuality of evidence downgraded by one level because of serious imprecision: the 95% CI has a very wide range but do not crosses 1.
Summary of findings 10. Zinc sulphate versus placebo for pruritus.
| Zinc sulphate versus placebo | ||||||
| Patient or population: patients with uraemic pruritus (UP) Setting: inpatient Intervention: Zinc sulphate Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with Zinc sulphate | |||||
| Pruritus assessed with: VAS and Duo Scale Follow‐up: range 4 weeks to 12 weeks | Original scale: the mean pruritus was 10.7 (Duo score 0‐45, Mapar 2015) and 4.25 (VAS 0‐10, Najafabadi 2012) | The mean pruritus in the intervention group was 0.13 standard deviations lower (0.58 lower to 0.32 higher) | — | 76 (2 RCTs) | ⊕⊕⊝⊝ Lowa,b | As suggested by Cohen 1988 an SMD of 0.2 is small, 0.5 is moderate and 0.8 is large. |
| Quality of life ‐ not measured | — | — | — | — | — | Not measured |
| Patient satisfaction ‐ not measured | — | — | — | — | — | Not measured |
| Depression ‐ not measured | — | — | — | — | — | Not measured |
| Risk for at least one adverse event per participant Follow‐up: range 4 weeks to 12 weeks | Study population | Not estimable | 160 (2 RCTs) | — | None observed for zinc sulphate; not stated for placebo | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; OR: odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
aQuality of evidence downgraded by one level because of serious risk of bias: no precise information about randomisation, allocation concealment or blinding. bQuality of evidence downgraded by one level because of serious imprecision: wide 95% CI that overlaps the line of no effect.
We researched pruritus as the primary outcome. An overall meta‐analysis was not possible because of the diversity of the different kinds of pruritus and interventions included in this review. Therefore, we considered several different treatment comparisons (see 'Summary of findings' tables and Data and analyses). Most meta‐analyses cover the outcomes 'pruritus' and 'risk for at least one adverse event'. We performed subgroup and sensitivity analyses as stated in the paragraphs Subgroup analysis and investigation of heterogeneity and Sensitivity analysis.
If we were unable to summarise data in meta‐analyses, we reported results descriptively according to the different pharmacological interventions and the underlying kind of pruritus. The description of results focuses on pruritus scores. We present additional outcomes of the studies in the Characteristics of included studies table and Table 13 for secondary outcomes.
Primary outcome: subjective measures of pruritus
Palliative care participants with pruritus of different origin
Selective serotonin reuptake inhibitor (SSRI) paroxetine
One study researched the effect of the selective serotonin reuptake inhibitor paroxetine (SSRI) on pruritus in palliative care patients (Zylicz 2003). In this randomised, controlled, cross‐over study, paroxetine showed an antipruritic effect in palliative care participants with opioid‐induced, paraneoplastic or haematologic pruritus. Twenty‐four of the 26 participants (two dropouts) treated with paroxetine (5.2, SE 0.32) had lower pruritus intensity scores on the 10‐point numerical analogue scale (NAS) over the seven treatment periods when compared to participants receiving placebo (6.0, SE 0.32). The MD between paroxetine and placebo after one week treatment was −0.78 points (95% CI −1.19 to −0.37; one RCT, N = 48, quality of evidence: moderate) (see Table 1) and −1.35 points (95% CI −2.11 to −0.59) on day three. We downgraded the quality of evidence due to serious imprecision. Nine of 24 of participants (37.5%) had a pruritus reduction of at least 50%. Investigators typically observed the onset of antipruritic action after two or three days, irrespective of the order of treatment. This was the only study that specifically researched patients treated in palliative care units or palliative care settings.
Participants with advanced diseases and UP or CP
Opioid antagonist naltrexone
Five trials including 126 participants examined the antipruritic effect of naltrexone in participants suffering from pruritus. Three studies included 90 participants with UP or UP (Legroux‐Crespel 2004; Pauli‐Magnus 2000; Peer 1996), and two studies involving 36 participants evaluated naltrexone in patients with CP (Terg 2002; Wolfhagen 1997).
We conducted separate meta‐analyses, measuring absolute change in pruritus severity in Terg 2002 and Wolfhagen 1997 and relative change in Pauli‐Magnus 2000 and Wolfhagen 1997). In one study, the results concerning pruritus were not interval‐scaled and therefore, we did not include them in the meta‐analysis (Peer 1996). We excluded another study from the meta‐analysis due to poor methodological quality and lack of data (Legroux‐Crespel 2004) (see Risk of bias in included studies).
Naltrexone in UP patients
Results of the RCTs regarding the effects of naltrexone in UP were contradictory (Pauli‐Magnus 2000; Peer 1996). Peer 1996 involved 15 participants and showed that administration of the oral mu‐receptor antagonist naltrexone was associated with a decrease in pruritus perception. At the end of the naltrexone treatment, the median pruritus scores on a 10 cm VAS (10 = maximum intensity of pruritus) were 2.1 cm (interquartile range (IQR) 1.5 to 2.15) for the naltrexone‐placebo sequence and 1.0 (0.4 to 1.15) for the placebo‐naltrexone sequence. The respective baseline values were 9.9 (IQR 9.85 to 9.95) and 9.9 cm (IQR 9.3 to 10.0). The results of this study suggested short‐term efficacy with few side effects for the amelioration of UP with naltrexone (Mettang 2010; Peer 1996). Pauli‐Magnus 2000, involving 23 participants, showed no statistically significant difference between the naltrexone and the placebo treatment periods. During the naltrexone period, pruritus decreased by 29.2% (95% CI 18.7 to 39.6) on the VAS and by 17.6% (95% CI 4.2 to 31.1) on the detailed score (pruritus score proposed by Duo 1987). The percent difference between the naltrexone and the placebo treatment periods was not statistically significant (−12.30%, 95% CI −25.82 to 1.22; P = 0.07; one RCT, N = 32) (Pauli‐Magnus 2000). The third study, researching the effect of naltrexone in participants suffering from UP, was a comparative study comparing naltrexone versus loratadine. In this study, 7 of the 52 participants showed a dramatic improvement when using naltrexone (> 3 cm, marked improvement), whereas the mean VAS score was identical to the alternative medication, the H1 receptor antagonist loratadine (Legroux‐Crespel 2004). The results of the studies are conflicting. Both Pauli‐Magnus 2000 and Peer 1996 were randomised, placebo‐controlled, double‐blind cross‐over trials, so we cannot explain the difference between these two studies by differences in participant compliance, naltrexone dose or study design.
Naltrexone in CP patients
Two studies compared the antipruritic effect of the opioid antagonist naltrexone versus placebo in participants suffering from CP and reported beneficial effects on participants with CP (Terg 2002; Wolfhagen 1997).
Wolfhagen 1997: 16 participants with CP were followed for four weeks. Mean changes in VAS with respect to baseline pruritus scores favoured the naltrexone group for daytime itching (−54% versus 8%; P < 0.001) and night‐time itching (−44% versus 75%; P = 0.003). For five of the eight participants in the naltrexone group, the total pruritus score was reduced by half or more after four weeks of treatment.
Terg 2002: the VAS showed greater and more significant changes with naltrexone than with placebo (P < 0.001). At the end of naltrexone treatment, pruritus decreased significantly compared to baseline (mean daytime pruritus VAS decreased from 6.29 cm (SD 2.28) to 3.55 (SD 2.39); P < 0.001; and night‐time itching improved from 5.89 cm (SD 2.49) to 3.55 (SD 2.42); P = 0.001). These outcomes with naltrexone were seen in both groups separately. In 9 of 20 participants (45%) receiving naltrexone, pruritus decreased 50% compared to the baseline value, including five cases where the pruritus disappeared completely.
Two studies contributed data to the meta‐analysis of naltrexone versus placebo for pruritus on a VAS (Terg 2002; Wolfhagen 1997; Analysis 1.1; Analysis 1.2). The pooled results showed a statistically significant effect, with an MD of −2.26 cm (95% CI −3.19 to −1.33; N = 52, quality of evidence: moderate) (Table 2). There was substantial statistical heterogeneity (I2 = 55%). However, the result of the meta‐analysis did not change substantially when using the random‐effects model, with an MD of −2.42 cm (95% CI −3.90 to −0.94, P = 0.001; Analysis 1.3). We downgraded the quality of evidence due to serious inconsistency.
1.1. Analysis.
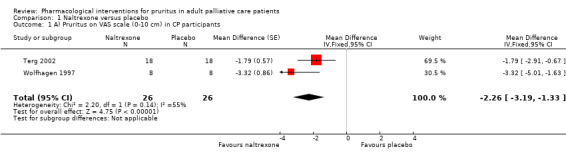
Comparison 1 Naltrexone versus placebo, Outcome 1 A) Pruritus on VAS scale (0‐10 cm) in CP participants.
1.2. Analysis.
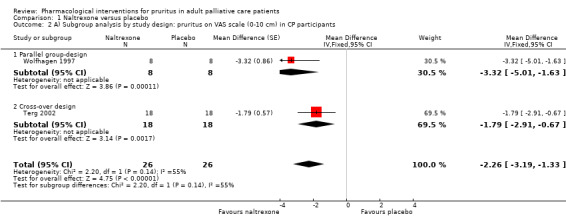
Comparison 1 Naltrexone versus placebo, Outcome 2 A) Subgroup analysis by study design: pruritus on VAS scale (0‐10 cm) in CP participants.
1.3. Analysis.
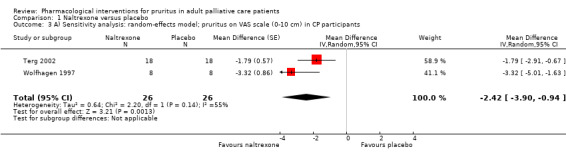
Comparison 1 Naltrexone versus placebo, Outcome 3 A) Sensitivity analysis: random‐effects model; pruritus on VAS scale (0‐10 cm) in CP participants.
The subgroup analysis by nature of pruritus was only possible for group differences in percent change of the two studies (Pauli‐Magnus 2000; N = 16; Wolfhagen 1997; N = 16). However, the sample sizes were small, the studies had different study designs, and CP patients were compared with UP patients, which may have contributed to the different effects (Pauli‐Magnus 2000; −12.30%, 95% CI −25.82 to 1.22; Wolfhagen 1997; −62%, 95% CI −89.42 to −34.58) (Analysis 1.4; Analysis 1.5).
1.4. Analysis.
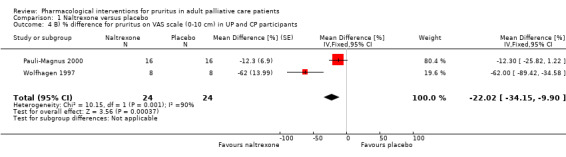
Comparison 1 Naltrexone versus placebo, Outcome 4 B) % difference for pruritus on VAS scale (0‐10 cm) in UP and CP participants.
1.5. Analysis.
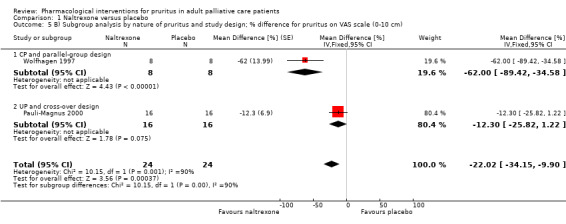
Comparison 1 Naltrexone versus placebo, Outcome 5 B) Subgroup analysis by nature of pruritus and study design; % difference for pruritus on VAS scale (0‐10 cm).
Subgroup analysis by study design (Analysis 1.2) for the studies on CP resulted in an MD of −3.32 cm (95% CI −5.01 to −1.63; N = 16) for Wolfhagen 1997, which had a parallel‐group design, and an MD of −1.79 cm (95% CI −2.91 to −0.67; N = 36) for Terg 2002, a cross‐over study. The difference between the two subgroups was not statistically significant (P = 0.14). For the sensitivity analysis see Analysis 1.3.
κ‐Receptor agonist nalfurafine
A meta‐analysis of two RCTs included 85 participants suffering from UP. Wikström and colleagues showed that treatment with the κ‐receptor agonist nalfurafine could reduce itch in participants on haemodialysis (Wikström 2005a; Wikström 2005b). Data from the first two weeks of both studies showed that participants who received nalfurafine experienced a small but statistically significant (−9.53 mm, 95% CI −1.42 to −17.64; P = 0.0212; two RCTs, N = 85) reduction in worst itching on the VAS score (weighted mean difference (WMD)) from run‐in to week 2 compared to those given placebo. The meta‐analysis combined data from a study with parallel‐group design and a cross‐over design (Wikström 2005a; Wikström 2005b). We considered it appropriate to use only the data from the first two weeks of treatment in both studies because of concerns about carryover effects of the drug and regression to the mean.
We found statistically significant reductions in itching intensity (P = 0.041), and sleep disturbances (P < 0.001) in the nalfurafine group compared with placebo. More nalfurafine‐treated participants responded (defined as 50% reduction in worst itching VAS score) within two weeks of run‐in than placebo‐treated participants (36% versus 14%; P = 0.0226). The number of days with tolerable itching and the number of nights undisturbed by itching increased significantly more in the nalfurafine groups than in the placebo groups during the first two weeks of treatment (+2.2 days versus +1.4 days; P = 0.0410 and +2.5 nights versus +0.9 nights; P < 0.001). There were improvements in itching (P = 0.0025) and excoriations (P = 0.006) for the nalfurafine‐treated participants.
Kumagai 2010 confirmed the results of the studies reported by Wikström and colleagues in a randomised, double‐blind, placebo‐controlled, parallel‐group study in 337 haemodialysis participants with itch that was resistant to currently available treatments. Based on the hypothesis that the activation of ĸ‐receptors expressed by dermal cells and lymphocytes might lead to the suppression of pruritus, Kumagai 2010 tested whether κ‐receptor agonists (nalfurafine) were able to reduce UP (Mettang 2010). Of the 337 participants, 114 received nalfurafine 5 μg daily, 112 received 2.5 μg nalfurafine daily, and 111 received placebo daily. Pooled data for all morning and evening VAS values (0 ‐ 100 mm) showed that decreases in the 5 μg nalfurafine group (mean 16 mm, 95% CI 13 to 18 during the first seven days of the treatment period and mean 22 mm, 95% CI 18 to 26 during the latter seven days of the treatment period) were significantly greater than those in the placebo group (mean 8 mm, 95% CI 6 to 11 and mean 13 mm, 95% CI 10 to 16, respectively). In the 2.5 μg nalfurafine group, decreases in the VAS values (mean 16 mm, 95% CI 13 to 19 during the first seven days of the treatment period and mean 23 mm, 95% CI 19 to 27 during the latter seven days of the treatment period) were also significantly larger than those in the placebo group.
We were able to pool the data from the meta‐analysis by Wikström and the study by Kumagai and colleagues in a meta‐analysis with 422 participants (Analysis 2.1; Table 3). We divided the results of Kumagai and Wikström by 10 in order to show meta‐analysis on a 10 point scale (better comparability with other analyses). We pooled the 5 μg and the 2.5 μg groups for the meta‐analysis because they showed similar results (Kumagai 2010). Meta‐analysis of the studies researching ĸ‐receptor‐agonist nalfurafine versus placebo resulted in a small but statistically significant effect in favour of nalfurafine (MD −0.95, 95% CI −1.32 to −0.58; I2 = 0%; three RCTs, N = 422, quality of evidence: moderate) (Analysis 2.1; Table 3). As all studies examined UP, and all studies used a parallel‐group design (only first period of Wikström 2005b was used), subgroup analysis according to underlying kind of pruritus or according to different study design was not necessary. Furthermore, fixed‐effect and random‐effects models yielded the same results (Analysis 2.2).
2.1. Analysis.
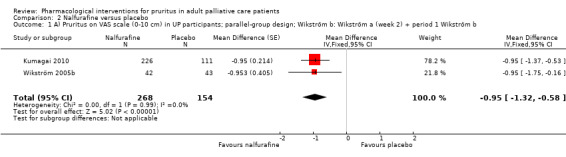
Comparison 2 Nalfurafine versus placebo, Outcome 1 A) Pruritus on VAS scale (0‐10 cm) in UP participants; parallel‐group design; Wikström b: Wikström a (week 2) + period 1 Wikström b.
2.2. Analysis.
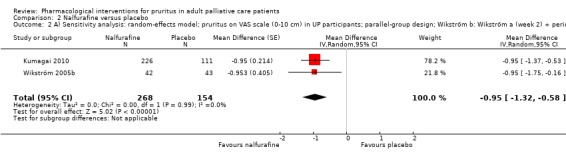
Comparison 2 Nalfurafine versus placebo, Outcome 2 A) Sensitivity analysis: random‐effects model; pruritus on VAS scale (0‐10 cm) in UP participants; parallel‐group design; Wikström b: Wikström a (week 2) + period 1 Wikström b.
Serotonin 5‐HT3 antagonist ondansetron
Five studies including 270 participants examined the effect of ondansetron for treatment of pruritus. Four studies compared ondansetron to placebo (Ashmore 2000; Murphy 2003; O'Donohue 2005; Yue 2015), and one trial used cyproheptadine as a standard medication in comparison with ondansetron (Özaykan 2001). O'Donohue 2005 involved 19 participants with CP, while four other studies in 241 participants researched participants with UP (Ashmore 2000; Murphy 2003; Özaykan 2001; Yue 2015). The administered doses ranged from 8 mg per day to 8 mg three times per day.
In a placebo‐controlled cross‐over study in 19 participants (three dropouts) (Ashmore 2000), the median daily pruritus score measured via a VAS did not change significantly (P = 0.9) during active or placebo treatment (pre‐ondansetron 5.3 cm; IQR 3.4 to 6.3; during ondansetron 3.9 cm; IQR 2.7 to 5.0; P = 0.02; pre‐placebo 3.7 cm; IQR 3.0 to 4.6; during placebo 3.6 cm; IQR 2.4 to 4.8; P = 0.03).
Another placebo‐controlled, cross‐over study examined 24 participants with UP and recorded the severity of pruritus on a VAS (Murphy 2003). In this study, pruritus decreased by 16% (95% CI 0.5 to 32) during active treatment and by 25% (95% CI 9 to 41) during treatment with the placebo. The changes in VAS scores during treatment with ondansetron (P = 0.04) and placebo (P = 0.01) were both statistically significant. However, the between group differences were not significant (N = 24).
Twenty participants with UP were enrolled in a prospective, parallel‐group study researching ondansetron versus cyproheptadine, which served as a standard medication (Özaykan 2001). Pruritus was measured by the 0 to 48 unit pruritus scale introduced in Duo 1987. We found no significant difference among the groups in itching scores at the end of the first and second weeks of the therapy. At the end of the third and fourth weeks, participants receiving ondansetron had statistically significant lower itching scores (week three: 4 units versus 12.6 units; week four: 3.2 units versus 11 units).
Yue 2015 conducted a fully powered (N = 188), placebo‐controlled, three‐armed RCT with an intervention period of 12 weeks, examining the effect of ondansetron and pregabalin on UP. Investigators described pregabalin as more effective than placebo (mean change difference ‐4.6, 95% CI ‐5.2 to ‐4.0), whereas the difference between ondansetron and placebo was not significant (VAS 0 to 10: pregabalin: 1.4 cm (SD 0.2), ondansetron: 5.4 cm (SD 0.6), placebo: 5.7 cm (SD 0.4)).
O'Donohue 2005 investigated the effect of ondansetron in 19 participants with CP in a placebo‐controlled, parallel‐group study. Mean pruritus score using a VAS (0‐10) and scratching activity were reduced on the first treatment day compared with baseline in both the ondansetron and placebo groups (P < 0.05). However, there were no significant differences in mean pruritus perception (ondansetron: ‐21%, placebo ‐22%) or scratching activity (ondansetron: ‐‐6%, placebo ‐16%) between the two groups over the 5‐day treatment period in this study.
There was a very small but statistically significant effect in favour of the ondansetron group compared to the placebo group in patients with UP (MD −0.28 cm, 95% CI −0.46 to −0.09, two RCTs, N = 151, quality of evidence: moderate) (see also Analysis 3.1; Analysis 3.2; Table 4). We downgraded the quality of evidence due to risk of bias. The effect disappeared and 95% CIs increased under the random effects model (Analysis 3.3).
3.1. Analysis.
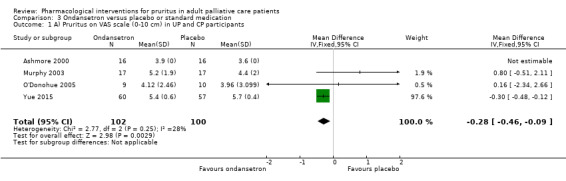
Comparison 3 Ondansetron versus placebo or standard medication, Outcome 1 A) Pruritus on VAS scale (0‐10 cm) in UP and CP participants.
3.2. Analysis.
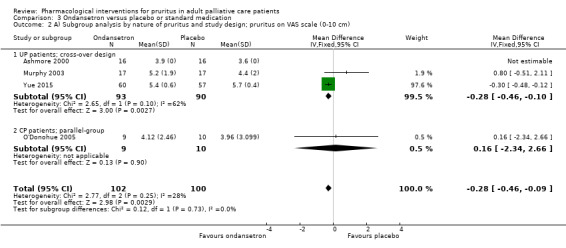
Comparison 3 Ondansetron versus placebo or standard medication, Outcome 2 A) Subgroup analysis by nature of pruritus and study design; pruritus on VAS scale (0‐10 cm).
3.3. Analysis.
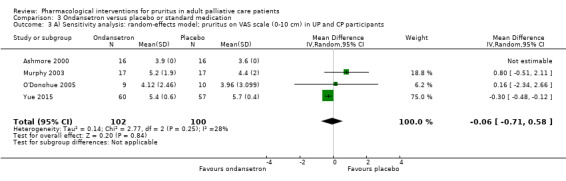
Comparison 3 Ondansetron versus placebo or standard medication, Outcome 3 A) Sensitivity analysis: random‐effects model; pruritus on VAS scale (0‐10 cm) in UP and CP participants.
In principle, four out of five studies evaluating ondansetron were suitable for meta‐analysis. However, we did not include one study (Analysis 3.1) because it used a different measurement (Duo scale) to assess the intensity of pruritus. In addition, it compared ondansetron to cyproheptadine and not to placebo (Özaykan 2001). Interestingly, UP participants on ondansetron had a lower pruritus score (MD 7.80 units, 95% CI −14.27 to −1.33; measured with the 0 to 48 unit Duo scale; one RCT; N = 20) than participants treated with cyproheptadine (Özaykan 2001). Further, it was not possible to include Ashmore 2000 in the pooled analysis because it used IQRs but did not supply information on the SE.
Selective serotonin reuptake inhibitor (SSRI) sertraline
Mayo 2007 enrolled 12 participants suffering from CP in a randomised, double‐blind, placebo‐controlled trial. Participants taking sertraline improved by a mean of 1.86 cm on the 0 to 10 VAS, whereas participants taking placebo worsened by 0.38 cm. This resulted in a difference of 2.24 cm in favour of the intervention group (P = 0.009).
Antiepileptic gabapentin
Three RCTs examined the effect of the antiepileptic gabapentin in 111 participants with UP (Gunal 2004; Naini 2007), and one study researched gabapentin in 16 participants with CP (Bergasa 2006).
In a double‐blind, placebo‐controlled, cross‐over study, Gunal 2004 studied the effectiveness of gabapentin against renal itch in 25 participants. The treatment period was four weeks. The mean pruritus score (VAS) of the cohort before the study was 8.4 cm (SD 0.94). After placebo intake, it decreased to 7.6 cm (SD 2.6, P = 0.098). After gabapentin administration, the mean score decreased to 1.2 cm (SD 1.8, P < 0.001).
An RCT in 34 participants with UP confirmed the effect of gabapentin. Participants received treatment for four weeks, and the mean decrease in pruritus score (VAS) in the gabapentin and placebo groups was 6.7 cm (SD 2.6 and 1.5 cm (SD 1.8, respectively, P < 0.001) (Naini 2007).
We were able to pool data from these two studies in participants with UP (Gunal 2004; Naini 2007), and the result indicated a large effect in favour of gabapentin (−5.91 cm, 95% CI −6.87 to −4.96; two RCTs, N = 118, quality of evidence: moderate) (Analysis 4.1; Analysis 4.2; Table 5). The pooled estimate remained robust in the sensitivity analysis under the random‐effects model (−5.88 cm, 95% CI −7.04 to −4.71) (Analysis 4.3). We downgraded the quality of evidence due to risk of bias.
4.1. Analysis.
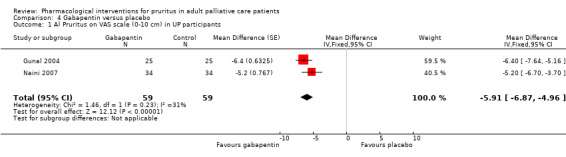
Comparison 4 Gabapentin versus placebo, Outcome 1 A) Pruritus on VAS scale (0‐10 cm) in UP participants.
4.2. Analysis.
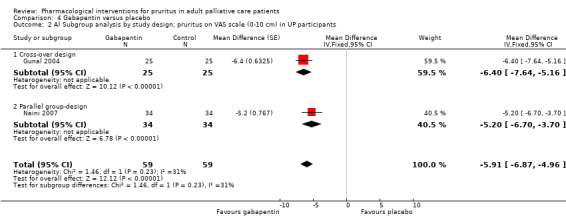
Comparison 4 Gabapentin versus placebo, Outcome 2 A) Subgroup analysis by study design; pruritus on VAS scale (0‐10 cm) in UP participants.
4.3. Analysis.
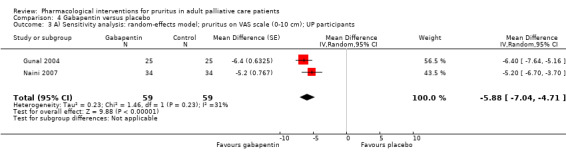
Comparison 4 Gabapentin versus placebo, Outcome 3 A) Sensitivity analysis: random‐effects model; pruritus on VAS scale (0‐10 cm); UP participants.
We did not include the results from Amirkhanlou 2016 in the meta‐analyses because the trial compared gabapentin to ketotifen instead of placebo. The authors assessed the clinical response (complete, partial and no response) of the drugs in 52 participants after a two‐week intervention period. Both drugs tended to be effective (complete response > 50%), and investigators observed only slight differences between the groups (gabapentin: no response: 3 (11.5%), partial response: 9 (34.6%); ketotifen: no response: 6 (23.1%), partial response: 7 (26.9%)).
We could not include data from the study researching the effect of gabapentin on CP because we could not calculate MD or SMD (Bergasa 2006). The study researching the effect of gabapentin on CP did not find any significant therapeutic advantage over the placebo (Bergasa 2006). On the contrary, these data in 15 participants suggested a placebo effect. The data given in the study did not allow us to calculate an estimate of the effect.
Semiantibiotic rifampicin
Three studies including 45 participants researched the treatment of CP with the semiantibiotic rifampicin, comparing it to placebo in Ghent 1988 and Podesta 1991a or to phenobarbitone as a standard treatment in Bachs 1989.
Ghent 1988 conducted a double‐blind, placebo‐controlled, cross‐over clinical trial of rifampicin in nine participants for a treatment period of 14 days. VAS pruritus scores showed no significant placebo response or any effect from the order of the treatment. Rather, there was a difference in favour of the intervention group of −19.86 mm (VAS 0 to 100; 95% CI −26.66 to −13.06) (Analysis 5.1; Analysis 5.2). The SMD was −1.46 (95% CI −2.79 to −0.13) (Analysis 5.3; Analysis 5.4).
5.1. Analysis.
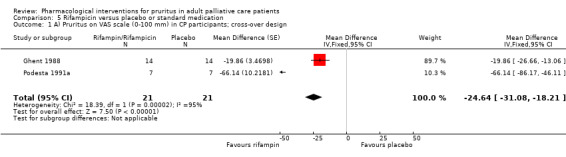
Comparison 5 Rifampicin versus placebo or standard medication, Outcome 1 A) Pruritus on VAS scale (0‐100 mm) in CP participants; cross‐over design.
5.2. Analysis.
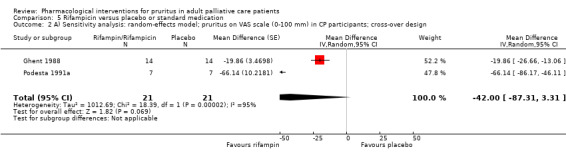
Comparison 5 Rifampicin versus placebo or standard medication, Outcome 2 A) Sensitivity analysis: random‐effects model; pruritus on VAS scale (0‐100 mm) in CP participants; cross‐over design.
5.3. Analysis.
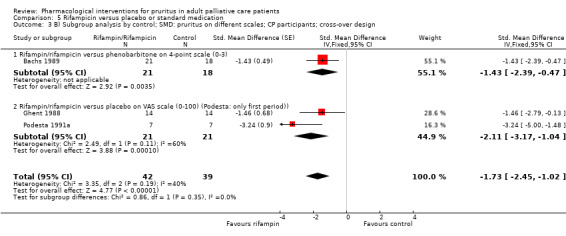
Comparison 5 Rifampicin versus placebo or standard medication, Outcome 3 B) Subgroup analysis by control; SMD: pruritus on different scales; CP participants; cross‐over design.
5.4. Analysis.
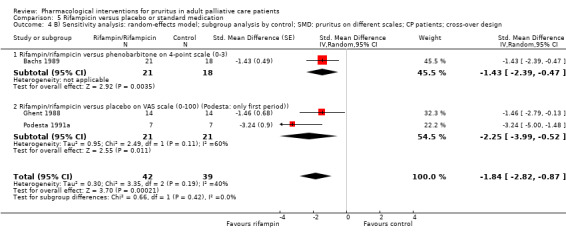
Comparison 5 Rifampicin versus placebo or standard medication, Outcome 4 B) Sensitivity analysis: random‐effects model; subgroup analysis by control; SMD: pruritus on different scales; CP patients; cross‐over design.
Podesta 1991a was a randomised, controlled cross‐over trial in 14 participants with CP that were treated with rifampicin and placebo for seven days, or vice versa. Since the figures provided in the publication suggested a carryover effect, we analysed the data and found an interdependence effect (P = 0.0063) (periodical effect P =< 0.001; therapeutical effect < 0.001). Therefore, in the meta‐analysis we only included the results of the first treatment period and treated the study as a parallel‐group trial. The data of Podesta 1991a showed a statistically significant effect of rifampicin (SMD −3.24, 95% CI −5.00 to −1.48; one RCT, N = 14) (Analysis 5.3).
Bachs 1989 assessed the antipruritic effects of rifampicin and phenobarbitone in 22 participants with CP in a cross‐over RCT. In contrast to the above study, we did not find any interaction between therapy effect and treatment period or carryover effect. Pruritus improved in 19 participants taking rifampicin compared to 8 taking phenobarbitone. Improvement was greater with rifampicin than with phenobarbitone (P < 0.001). We included this study in the meta‐analysis, which showed an SMD of −1.43 (95% CI −2.39 to −0.47; one RCT, N = 39) (Analysis 5.3).
The pooled estimate of the three studies researching rifampicin indicated that it may improve pruritus in participants suffering from CP (SMD −1.73, 95% CI −2.45 to −1.02; three RCTs, N = 71) (Analysis 5.3). The results were consistent, but the 95% CI increased considerably when using the random‐effects model for sensitivity analysis (SMD −2.25, 95% CI −3.99 to −0.52) (Analysis 5.4). We could only estimate the effect on the VAS (0 to 100) for the studies from Ghent 1988 and Podesta 1991a. An effect of −24.64 mm (95% CI −31.08 to −18.21; two RCTs, N = 42, quality of evidence: low) was apparent in the pooled analysis (Analysis 5.1; Table 6), but the effect did not remain statistically significant when using the random‐effects model (MD −42.00 mm, 95% CI −87.31 to 3.31) (Analysis 5.2). We downgraded the quality of evidence two levels due to very serious inconsistency.
Antidepressant doxepin
In a cross‐over RCT by Pour‐Reza‐Gholi 2007, performed on 24 participants with UP, investigators described complete improvement in 58.3% of participants treated with doxepin. This was significantly higher than improvements with placebo (P < 0.001). Overall, doxepin was effective (complete improvement or relative improvement according to the patient) in 21 (87.5%) of the participants (P < 0.001). Since no raw data were available, further statistical analysis was not possible (Pour‐Reza‐Gholi 2007).
Cholestyramine
Two RCTs researched the effect of cholestyramine in participants with UP and CP: Silverberg 1977 was a parallel‐group trial including 10 participants with UP, and Duncan 1984 used a cross‐over design and researched eight participants with CP, comparing the effect of cholestyramine, terfenadine, chlorpheniramine and placebo. Both studies reported some positive effect for cholestyramine and Duncan 1984 for terfenadine as well. However, the very small samples limited the informative value of these results.
Colesevelam
The randomised, double‐blind, investigator‐initiated, multicentre trial by Kuiper 2010 aimed to assess the efficacy of colesevelam versus placebo in participants with CP. Data showed no difference in pruritus score between participants treated with colesevelam and participants receiving placebo (P = 1.00 for the VAS day score and P = 0.74 for the VAS evening score; predefined primary endpoint = proportion of participants with at least a 40% reduction in pruritus VAS scores).
Immunsuppressant thalidomide
There was evaluable data from 18 of 29 participants in a double‐blind, randomised cross‐over trial on thalidomide against UP. Silva 1994 found a similar proportion of participants in phase 1 and 2 responding to thalidomide (responders in phase 1: 55% and responders in phase 2: 57%). The reduction on the three‐item pruritus score was 78% (SE 6%) in phase 1 and 81% (SE 10%) in phase 2 and differed significantly from placebo (54%, SE 1, P < 0.05). The authors found a positive effect of thalidomide in 67% of the study population, with a mean reduction of pruritus scoring of approximately 80% (Silva 1994).
Leukotriene receptor antagonist montelukast
Nasrollahi 2007 researched the effect of the leukotriene receptor antagonist montelukast in a multicentre, randomised, single‐blind, placebo‐controlled cross‐over study in 16 participants with UP. Results at the end of the treatment with montelukast showed a reduction of pruritus by 35% (95% CI 9.5 to 62.5) compared to a reduction of 7% (95% CI 0.5 to 15.9) with placebo (P = 0.002). The mean change in pruritus score was 16.1 units (95% CI 9.5 to 22.5) with montelukast and 7.1 units (95% CI 0.5 to 13.7) with placebo, using the 0 to 45 Detailed Pruritus Score (Duo 1987).
Flumecinol
Two randomised, double‐blind, placebo‐controlled studies examined the effects of low and high doses of flumecinol in participants with CP during a treatment period of three weeks. In the low dose study (Turner 1994a), 50 participants were enrolled and scored with the VAS daily for a seven‐day baseline and for a further 21 days. Subjectively, pruritus improved in 13 of 24 participants (54%) taking flumecinol and in 10 of 26 participants taking placebo (Chi2 = 1.24, P = 0.27). The median difference in the reduction of VAS pruritus score between the baseline week (mean score for each individual used) and the last week was 8.0 mm (95% CI −2.1 to 20.81) and favoured flumecinol over the placebo group. In the subsequent study with high dose flumecinol (Turner 1994b), 19 participants were included. The median difference in reduction of the VAS pruritus score was 19.8 mm (95% CI 3.3 to 40.7), in favour of flumecinol over the placebo.
In addition, participants were asked if their itch had improved significantly (Analysis 6.1). Our subgroup analysis (Analysis 6.2) reveals that the high dose administration of flumecinol (300 mg/d; n = 19, Turner 1994b) could be more effective than the low dose administration (600 mg/week; n = 50) (Turner 1994a). The pooled RR of 1.89 (95% CI 1.05 to 3.39; two RCTs, N = 69, quality of evidence: low; Analysis 6.1; Table 7) shows that flumecinol tends to be effective. However, when using the random‐effects model for sensitivity analysis, the 95% CI increased considerably (RR 2.32, 95% CI 0.54 to 10.10), resulting in no significant effect (P = 0.26) (Analysis 6.3). We downgraded the quality of evidence due to serious inconsistency and imprecision.
6.1. Analysis.
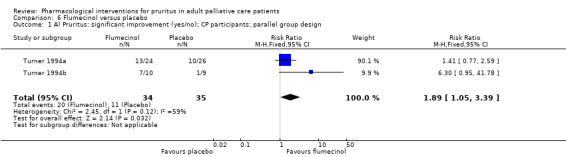
Comparison 6 Flumecinol versus placebo, Outcome 1 A) Pruritus: significant improvement (yes/no); CP participants; parallel group design.
6.2. Analysis.
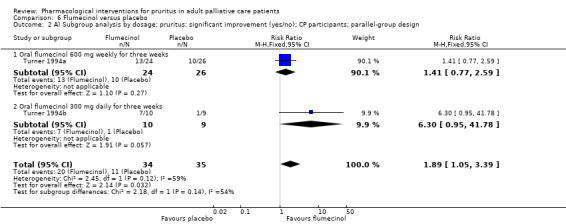
Comparison 6 Flumecinol versus placebo, Outcome 2 A) Subgroup analysis by dosage; pruritus: significant improvement (yes/no); CP participants; parallel‐group design.
6.3. Analysis.
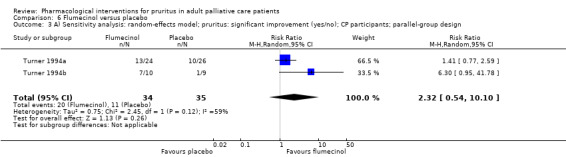
Comparison 6 Flumecinol versus placebo, Outcome 3 A) Sensitivity analysis: random‐effects model; pruritus: significant improvement (yes/no); CP participants; parallel‐group design.
Erythropoietin
A 10‐week, randomised, controlled cross‐over trial in 10 participants with UP and 10 participants without pruritus treated both groups with erythropoietin and placebo, respectively. Eight of the 10 participants suffering from pruritus showed reductions in their mean pruritus score (Duo 1987; Mettang 2002; Duo scale, range: 0 to 40), which decreased from 25 units (SE 3) to 11 units (SE 6) in group one and from 27 units (SE 4) to 9 units (SE 4) in group two during treatment with erythropoietin (De Marchi 1992). The authors concluded a positive effect of erythropoietin. However, the small sample size prohibits the generalisation of the results.
Oral and topical mast cell stabilizer cromolyn sodium
In a double‐blind, placebo‐controlled trial researching the effect of oral cromolyn sodium in participants with UP, Vessal 2010 analysed the data from 40 participants (of 62 participants originally enrolled). Investigators recorded pruritus on a 10 cm VAS. The mean pruritus score was 8.48 cm (SD 2.2, range 4 to 10, median 10) in the placebo group (19 participants evaluated). After eight weeks of therapy, the pruritus score decreased to 5.58 cm (SD 3.8, range 0 to 10, median 6; P = 0.004). The mean pruritus score was 8.68 cm (SD 1.8, range 4 to 10, median 10) in the cromolyn sodium group (21 participants evaluated) and decreased to 0.9 cm (SD 1.8, range 0 to 6, median 0) after eight weeks (P < 0.001). The mean difference between the two groups was −4.70 cm (95% CI −6.57 to −2.83; one RCT, N = 40).
Feily 2012 conducted a double‐blind, randomised, vehicle‐controlled trial with 60 UP participants. They received topical cromolyn sodium (4%) twice a day or a vehicle for four weeks. Pruritus was assessed on a VAS (0 to 5, 0: no pruritus and 5: the worst pruritus). At the end of week four, the vehicle group decreased to 1.3 points (SD 1.4) and the cromolyn sodium group to 0.3 points (SD 1.3) (P = 0.038). Our calculation revealed a mean between‐group difference of −1.00 (95% CI −1.68 to −0.32; one RCTs, N = 60).
We multiplied the values on the VAS (0 to 5) by a factor of 2 in order to enable a pooled analysis with Vessal 2010 Both studies demonstrated a statistically significant and clinically relevant effect (MD −2.94 points, 95% CI −4.04 to −1.83; two RCTs, N = 100, quality of evidence: moderate) (Analysis 7.1; Table 8). However, the subgroup analysis emphasises that the effect could depend on the route of administration (oral versus topical) (Analysis 7.2). The oral administration (MD −4.70 points, 95% CI −6.57 to −2.83; one RCT, N = 40) seemed to be more effective than the topical (MD −2.00 points, 95% CI −3.37 to −0.63; one RCT, N = 60). The 95% CI increased (MD −3.27 points, 95% CI −5.91 to −0.63; two RCTs, N = 100) under the random‐effects model, but the result remained statistically significant (Analysis 7.3). We downgraded the quality of evidence due to serious inconsistency.
7.1. Analysis.
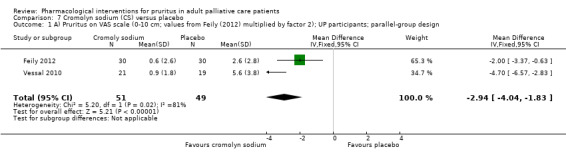
Comparison 7 Cromolyn sodium (CS) versus placebo, Outcome 1 A) Pruritus on VAS scale (0‐10 cm; values from Feily (2012) multiplied by factor 2); UP participants; parallel‐group design.
7.2. Analysis.
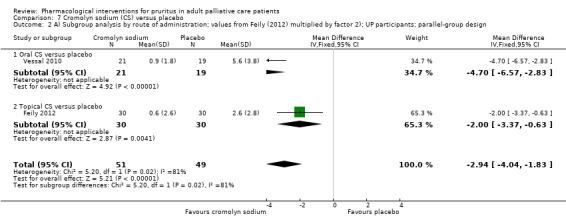
Comparison 7 Cromolyn sodium (CS) versus placebo, Outcome 2 A) Subgroup analysis by route of administration; values from Feily (2012) multiplied by factor 2); UP participants; parallel‐group design.
7.3. Analysis.
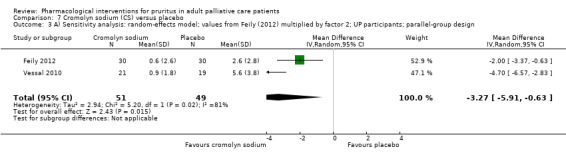
Comparison 7 Cromolyn sodium (CS) versus placebo, Outcome 3 A) Sensitivity analysis: random‐effects model; values from Feily (2012) multiplied by factor 2; UP participants; parallel‐group design.
Activated oral charcoal
Pederson 1980 contained two consecutive eight‐week treatment periods involving 11 participants with UP. This randomised, placebo‐controlled, cross‐over study showed a statistically significant difference favouring charcoal over placebo during the first study period (P = 0.01) and a tendency during the second study period (P = 0.05). Missing data did not enable further statistical analysis. The small sample size affects the generalisation of the results.
Anaesthetic propofol
In a randomised, double‐blind, placebo‐controlled cross‐over trial including 10 participants with CP (Borgeat 1993), investigators described treatment with propofol as successful (defined by authors as a decrease of pruritus of at least 4 points on a verbal rating scale from 0 to 10) in 17 of 20 (85%) doses of propofol, compared to 2 of 20 (10%) doses in the placebo group (P < 0.01). Data did not allow further statistical analysis.
Local anaesthetic lidocaine
A randomised, double‐blind, placebo‐controlled study investigated the efficacy of lidocaine on treatment‐resistant pruritus in 18 participants with chronic cholestatic liver disease (Villamil 2005). Lidocaine administration resulted in a statistically significant reduction of pruritus severity only at day two (mean VAS 39.1 mm (95% CI 15.7 to 62.5) versus 70.8 mm (95% CI 62.7 to 78.9) and three (mean VAS 48.7 mm (95% CI 25.4 to 72) versus 72.0 mm (95% CI 60.3 to 83.7) when compared with placebo administration (P < 0.05). The treatment group, but not the placebo group, improved from baseline (MD of treatment group about 26; P < 0.05).
Topical capsaicin
Four RCTs tested the efficacy of the topical agent capsaicin in treating pruritus in UP. Two of the studies did not provide sufficient data or appropriate statistical analyses for their findings to be evaluated in a meta‐analysis for efficacy (Breneman 1992a; Tarng 1996).
Breneman 1992a compared topical capsaicin and vehicle applied four times daily to either the right or left arm over six weeks. The sample was very small (N = 7), and only five participants were evaluable. Authors presented findings descriptively for these five participants (two capsaicin‐treated participants reported complete resolution of itching), and no statistical analysis was available, but the report states that there was an improvement in the participants treated with capsaicin.
Tarng 1996 carried out a similar cross‐over study with two four‐week treatment periods of capsaicin versus vehicle. The study involved 19 participants and also reported a significant effect of capsaicin. Despite a 14‐day washout period between the treatments, we must assume a carryover effect according to the graphical data presented. Data did not allow inter‐group statistical comparisons to be made for the first phase of the cross‐over.
Cho 1997 also researched the effect of topical capsaicin versus vehicle applied four times daily for four weeks to a pre‐selected area of skin in a cross‐over study involving 22 participants. The authors reported a significant effect of capsaicin in reducing itch (7 with complete and 12 with significant resolution of pruritus). Investigators presented the findings for the individual participants graphically, which indicated a carryover effect of the capsaicin treatment that could impede the interpretability from the second treatment phase. Groups were divided and analysed according to the intact parathyroid hormone (iPTH) ≤ 35 pg/mL and > 35 pg/mL. Both groups showed an improvement in pruritus on the 4‐point scale (1 = none, 4 = severe) (pooled effect: MD −0.88 points, 95% CI −1.31 to −0.44). The iPTH ≤ 35 pg/mL group had a greater (and statistically significant) effect (MD −1.50 points, 95% CI −2.21 to −0.79; one RCT, N = 20) than the iPTH > 35 pg/mL group (MD −0.50 points, 95% CI −1.05 to 0.05).
The fourth cross‐over study researched 34 participants on haemodialysis with UP (Makhlough 2010). Participants received capsaicin 0.03% and vehicle for four weeks each, with a two‐week washout period between the treatment phases. There was no significant difference in pruritus scores between the two groups before the treatment. However, the difference became more statistically significant with each week during treatment (P < 0.001). Repeated measurement tests showed that decreases in pruritus severity in the capsaicin group were more than those in the vehicle group during the treatment period (P < 0.001). Analysis of the data in week 4 of treatment showed a statistically significant effect for topical capsaicin on the Duo scale (0 to 30) with an MD of −4.70 units (95% CI −7.57 to −1.83; one RCT, N = 34).
We calculated the pooled estimate of the results from Cho 1997 and Makhlough 2010and received an SMD of −1.02 (95% CI −1.35 to −0.68; two RCTs, N = 112, quality of evidence: moderate) (Analysis 8.1; Analysis 8.2; Table 9). The result remained stable in the sensitivity analysis (Analysis 8.3). We downgraded the quality of evidence due to imprecision.
8.1. Analysis.
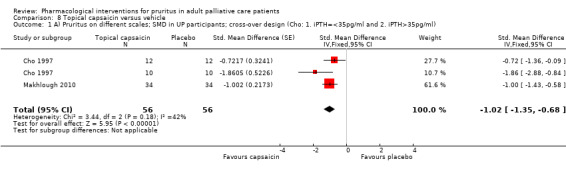
Comparison 8 Topical capsaicin versus vehicle, Outcome 1 A) Pruritus on different scales; SMD in UP participants; cross‐over design (Cho: 1. iPTH=<35pg/ml and 2. iPTH>35pg/ml).
8.2. Analysis.
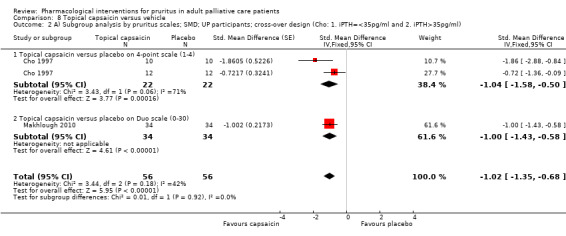
Comparison 8 Topical capsaicin versus vehicle, Outcome 2 A) Subgroup analysis by pruritus scales; SMD; UP participants; cross‐over design (Cho: 1. iPTH=<35pg/ml and 2. iPTH>35pg/ml).
8.3. Analysis.
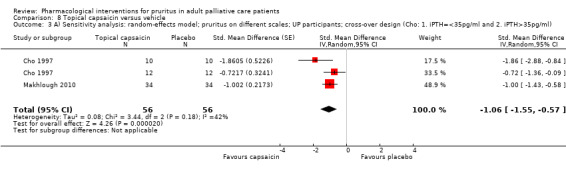
Comparison 8 Topical capsaicin versus vehicle, Outcome 3 A) Sensitivity analysis: random‐effects model; pruritus on different scales; UP participants; cross‐over design (Cho: 1. iPTH=<35pg/ml and 2. iPTH>35pg/ml).
Tacrolimus
A randomised, double‐blind, vehicle‐controlled study assessed the efficacy of tacrolimus ointment 0.1% for the treatment of pruritus in participants undergoing haemodialysis. The study found a similar effect for both tacrolimus ointment and vehicle in regard to the reduction of itch (range for both groups: tacrolimus: 72% to 77%, vehicle: 79% to 81%) (Duque 2005).
Pramoxine hydrochloride
In a randomised, double‐blind, controlled, comparative trial, Young 2009 found a 61% decrease in the average reported VAS in UP participants treated with pramoxine hydrochloride as compared to participants treated with placebo. Since authors presented results graphically with no raw data, statistical analysis was not possible.
Hydroxyzine
Vinegar solution and avena sativa were compared with hydroxyzine in a non blinded cross‐over RCT (Nakhaee 2015). All three treatments reduced pruritus statistically significant by more than 1.1 points on the 10 cm VAS. However, the post treatment scores differed only slightly (Vinegar: 3.73 cm (SD 2.41), Avena sativa: 4.10 cm (SD 2.34), Hydroxyzine: 3.56 cm (SD 2.52)).
Participants with HIV‐associated pruritus
A randomised parallel‐group study in 40 participants examined the four different therapies: hydroxyzine hydrochloride, pentoxifylline, triamcinolone and indomethacin in HIV participants suffering from pruritus (Smith 1997a). Results showed that participants placed on indomethacin obtained a median relief of 2.5 points on a 5‐point verbal rating scale. However, it is uncertain whether drug treatment with hydroxyzine hydrochloride, pentoxifylline, triamcinolone or indomethacin reduces pruritus because the evidence was of very low quality due to a small sample size and lack of blinding.
New drugs identified in this update
Ergocalciferol
Fifty participants with UP were randomised to ergocalciferol (vitamin D2; 50,000 IU, one pill per week) or placebo for 12 weeks in one double‐blind RCT (Shirazian 2013). The groups did not differ significantly at baseline regarding the pruritus score (assessed with the 0 to 21 point Pruritus Severity Questionnaire). None of the biweekly measured pruritus values showed a statistically significant difference between groups. The authors concluded that ergocalciferol was not effective for participants with UP.
Nicotinamide
Omidian 2013 researched 50 participants with UP in a double‐blind RCT. Participants received oral nicotinamide (500 mg twice a day) for four weeks, and pruritus was measured on a VAS (0 to 5: 0 = no pruritus and 5 = the worst pruritus). The pruritus decreased to a score of 1.52 (SD 1.61) in the nicotinamide group and to 1.29 (SD 1.08) in the placebo group. However, the group differences were not statistically significant (P = 0.167).
Omega‐3 fatty acids
In a double‐blind cross‐over RCT, 22 participants with UP received omega‐3 fatty acids or placebo (Ghanei 2012). Investigators assessed pruritus on the Duo pruritus scale (0 to 45, higher scores indicate a more severe pruritus). The duration of the treatment was 20 days, and participants had to take a 1 g capsule (verum or placebo) every eight hours. After 20 days, the pruritus in the fish oil group decreased from a score of 20.3 units (95% CI 16.7 to 23.8) to 6.4 units (95% CI 2.9 to 9.8), a 65% decrease. The placebo group reported a mean score of 14.4 units (95% CI 10.5 to 18.2), from a baseline level of 17.0 units (95% CI 12.4 to 21.6), a 15% decrease.
Turmeric
One hundred participants with UP were randomised in order to investigate the antipruritic effect of turmeric in a double‐blind RCT (Pakfetrat 2014). The participants received 500 mg turmeric or placebo capsules three times a day for eight weeks. Similar to Ghanei 2012, investigators assessed pruritus on the 0 to 45 unit Duo pruritus scale. Groups did not differ at baseline (P = 0.168). After the intervention period, the score of the turmeric group decreased to 10.3 units (SD 1.6) and to 15.9 units (SD 2.1) in the placebo group (P < 0.001). Consequently, the reduction of pruritus was greater in the turmeric than the placebo group (13.6 (SD 2.6) versus 7.2 (SD 2.6), P = 0.001) (Pakfetrat 2014).
Zinc sulphate
Najafabadi 2012 conducted a double‐blind RCT that examined the effect of zinc sulphate on UP. Forty participants were randomised to receive 220 mg zinc sulphate (oral) or placebo twice daily for eight weeks. The baseline values (10 cm VAS) differed slightly between groups (7.3 cm, SD 1.92 versus 6.3 cm, SD 1.62; P = 0.08). Thus, the change score from baseline to week 12 was greater in the zinc sulphate group (MD = 4.5 cm; P = 0.018). However, there were no differences between groups regarding week 2, 4, 6, 8 and 12. The authors still judged zinc sulphate to be more effective than placebo. The findings of another RCT with 40 participants also suggested no effect of zinc sulphate (Mapar 2015). None of the weekly comparisons (baseline to week 4) reached statistical significance (Characteristics of included studies).
We pooled the results in a meta‐analysis and used the SMD since pruritus was measured on different scales (10 cm VAS and Duo scale 0 to 45). The group difference were not significant (SMD −0.13, 95% CI −0.58 to 0.32, two RCTs, N = 76, quality of evidence: low) (Analysis 9.1; Table 10), and the random‐effects model did not change this result (Analysis 9.2). The quality of evidence was downgraded because of risk of bias and imprecision.
9.1. Analysis.
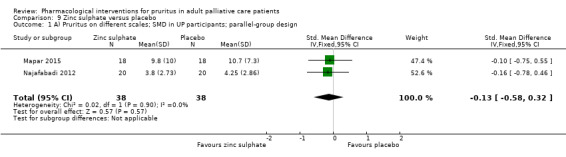
Comparison 9 Zinc sulphate versus placebo, Outcome 1 A) Pruritus on different scales; SMD in UP participants; parallel‐group design.
9.2. Analysis.
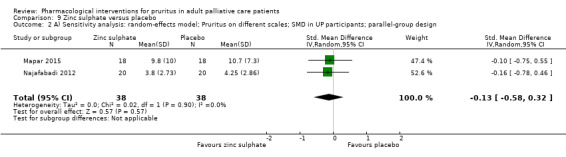
Comparison 9 Zinc sulphate versus placebo, Outcome 2 A) Sensitivity analysis: random‐effects model; Pruritus on different scales; SMD in UP participants; parallel‐group design.
Ketotifen
Amirkhanlou 2016 assessed the clinical response (complete, partial and no response) of ketotifen versus gabapentin in 52 participants after a two‐week intervention period. The effectiveness of the drugs was comparable (complete response > 50%; gabapentin: no response: 3 (11.5%), partial response: 9 (34.6%) versus ketotifen: no response: 6 (23.1%), partial response: 7 (26.9%)).
Pregabalin
A fully powered, three‐armed, placebo‐controlled RCT with 188 participants examined ondansetron and pregabalin in UP patients. Pregabalin, which has almost the same properties as gabapentin in that they are both γ‐aminobutyric acid analogues, was considerably more effective than placebo (10 cm VAS: pregabalin: 1.4 cm (SD 0.2), placebo: 5.7 cm (SD 0.4), Yue 2015). Using even lower doses of pregabalin (75 mg twice weekly), the reported effect was comparable to the results of gabapentin (300 mg 3x/week and 400 mg 2x/week) (Gunal 2004; Naini 2007; Analysis 4.1).
Secondary outcomes
Only a few of the included studies examined the secondary outcomes quality of life, patient satisfaction and depression (Table 13).
Quality of life
Four studies measured quality of life as a secondary outcome (Kuiper 2010; Turner 1994a; Turner 1994b; Yue 2015).
In participants receiving colesevelam for treatment of CP, Kuiper 2010 found no significant changes with respect to the domains of physical functioning (P = 0.67), role physical functioning (P = 0.50), bodily pain (P = 1.00), general health (P = 0.48), vitality (P = 0.90), social functioning (P = 0.37), emotional functioning (P = 0.17) or mental health (P = 0.26). Results were based on the Short‐Form 36 Health Survey in the colesevelam group before and after treatment. The authors reported only p‐values.
For participants receiving low‐dose flumecinol (Turner 1994a), the difference in median improvement in quality of life for flumecinol versus placebo, measured via the 100 mm VAS (lower score = better), was 5.0 mm (95% CI 0.4 to 13.0, P = 0.02; one RCT, N = 50, quality of evidence: moderate), in favour of flumecinol. The higher dose of flumecinol did not significantly improve quality of life. The difference in median improvement between the two groups was 3.5 mm (95% CI −5.9 to 24.9) (Turner 1994b).
Yue 2015 assessed health‐related quality of life with the Mental Component Summary scale (MCS) from the Short‐Form 12 Health Survey (SF‐12; version 2, 0 to 100, higher scores = better quality of life). At week 12, the health‐related quality of life in UP participants were 47.3 (SD 11.6), 42.8 (SD 13.1) and 42.5 (SD 8.7) for pregabalin, ondansetron and placebo, respectively. The baseline values were similar between groups and the mean change from baseline versus placebo was larger in pregabalin (4.1, 95% CI 2.9–5.3) than in ondansetron (1.2, 95% CI ‐0.1 to 2.5).
Patient satisfaction
Only one of the 50 included studies assessed patient satisfaction with the treatment regimen (Zylicz 2003). Using a non‐validated seven‐point scale, where 0 meant indifferent, −3 meant extremely poor, and 3 meant excellent, participants treated with paroxetine had, on average, higher satisfaction scores (mean 0.41, SE = 0.36) when compared to participants who received placebo (mean −0.66, SE = 0.36), regardless of the order in which they were received. The MD was −1.08 points (95% CI 0.18 to 1.98; one RCT, N = 48, quality of evidence: low) in favour of paroxetine. We downgraded the quality of evidence due to serious imprecision and serious risk of bias (Table 1).
Depression
Two studies examined depression as a secondary outcome (Bergasa 2006; Mayo 2007), measuring it with the Hamilton Depression Rating Scale, the Structured Clinical Interview Questionnaire (SCID) (Bergasa 2006), and the 30‐item Inventory of Depressive Symptomatology‐Self‐Report (IDS‐SR30) (Mayo 2007). Whereas Bergasa 2006 only evaluated the psychiatric state of the study participants, particularly with regard to depression, Mayo 2007 aimed to evaluate the effect of sertraline on depression symptoms. Using to the 17‐item Hamilton Rating Scale, Bergasa 2006l found eight participants scoring in the range of mild depression, three in the range of moderate depression, two in the range of none to minimal depression. Data on the full psychiatric evaluation were available for 13 of the 16 participants (Bergasa 2006).
Mayo 2007 found that all four participants with moderate or severe depression improved with sertraline (12 participants included). Participants with mild depression symptoms, however, did not reliably improve their IDS‐SR30 score with sertraline. One of these participants also improved with placebo. Both the VAS and IDS‐SR30 improved with increasing doses of sertraline, but the change in IDS‐SR30 did not completely explain the change in VAS.
Due to the absence of satisfactory data, the feasibility of pooling the secondary outcomes data was limited. For detailed results please see the 'Secondary outcomes' table (Table 13).
Adverse events
All but three included studies collected data on adverse events (Ashmore 2000; De Marchi 1992; Pederson 1980). Fourteen studies (28%) did not observe any adverse events (Ghanei 2012; Ghent 1988; Mapar 2015; Najafabadi 2012; Nakhaee 2015; Omidian 2013; Özaykan 2001; Pakfetrat 2014; Podesta 1991a; Shirazian 2013; Silva 1994; Turner 1994a; Turner 1994b; Young 2009).
Ten (20%) studies described adverse events in the intervention group leading to withdrawal (Kumagai 2010; Legroux‐Crespel 2004; Murphy 2003; Nasrollahi 2007; Pauli‐Magnus 2000; Pour‐Reza‐Gholi 2007; Terg 2002; Wikström 2005a; Wikström 2005b; Zylicz 2003). In contrast, only six studies found adverse events in the placebo groups or the standard medication group (Amirkhanlou 2016; Kumagai 2010; Legroux‐Crespel 2004; Pauli‐Magnus 2000; Wikström 2005a; Yue 2015).
Most adverse events were mild or moderate. Two interventions also showed multiple major adverse events (naltrexone and nalfurafine). We have summarised the different adverse events and the number of withdrawals for each study and intervention in two tables (see Table 14: 'Adverse events according to the different studies', and Table 15: 'Adverse events according to different interventions'). In the following, we will focus on adverse events for the drugs that we included in the 'Summary of findings' tables. We chose 'risk for at least one adverse event per participant' as a pragmatic outcome for our meta‐analyses.
Selective serotonin reuptake inhibitor (SSRI) paroxetine
On a 0 to 10 numerical analogue scale (NAS), participants treated with paroxetine suffered slightly more from nausea (0.46 points, 95% CI 0.05 to 0.87) and sleepiness (0.70 points, 95% CI 0.18 to 1.22) but not from vomiting (−0.18 points, 95% CI −0.44 to 0.08) (Table 1) (N = 52, quality of evidence: moderate). We downgraded the quality of evidence for nausea, sleepiness and vomiting due to serious imprecision. Two participants discontinued treatment because of severe adverse events (nausea and vomiting), presumably because there was no opportunity to titrate the dose to the effect of the medication in this trial.
Opioid antagonist naltrexone
The overall effect of RR 4.07 (95% CI 2.07 to 8.00; three RCTs, N = 116, quality of evidence: moderate) emphasises the increased risk for at least one adverse event for participants under naltrexone (Analysis 1.7; Analysis 1.8; Analysis 1.9; Table 1). We downgraded the quality of evidence due to serious imprecision.
1.7. Analysis.
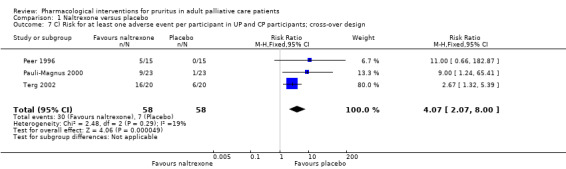
Comparison 1 Naltrexone versus placebo, Outcome 7 C) Risk for at least one adverse event per participant in UP and CP participants; cross‐over design.
1.8. Analysis.
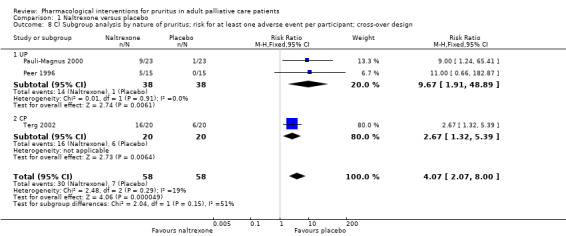
Comparison 1 Naltrexone versus placebo, Outcome 8 C) Subgroup analysis by nature of pruritus; risk for at least one adverse event per participant; cross‐over design.
1.9. Analysis.
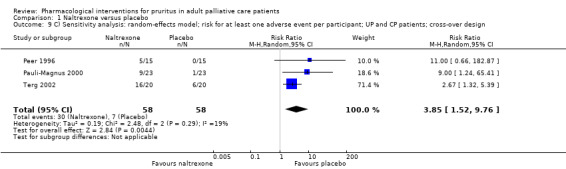
Comparison 1 Naltrexone versus placebo, Outcome 9 C) Sensitivity analysis: random‐effects model; risk for at least one adverse event per participant; UP and CP patients; cross‐over design.
Naltrexone in UP patients
The number of participants with at least one adverse event was higher in the naltrexone group in participants with UP (RR 9.67; two RCTs, N = 76). However, this result was very imprecise as the 95% CI was very wide (1.91 to 48.89) (Pauli‐Magnus 2000; Peer 1996; Analysis 1.8).
Naltrexone in CP patients
We could only integrate the adverse events outcome of Terg 2002 in our meta‐analysis, showing an RR of 2.67 (95% CI 1.32 to 5.39; N = 40) in favour of the placebo group (Analysis 1.8).
κ‐Receptor agonist nalfurafine
The RR for experiencing at least one adverse event per participant was 1.62 (95% CI 1.15 to 2.29; three RCTs, N = 422, quality of evidence: moderate) to the disadvantage of nalfurafine (Analysis 2.3). We downgraded the quality of evidence for sleepiness due to serious imprecision. The results slightly changed when using the random‐effects model (RR 1.51, 95% CI 1.09 to 2.09; Analysis 2.4).
2.3. Analysis.
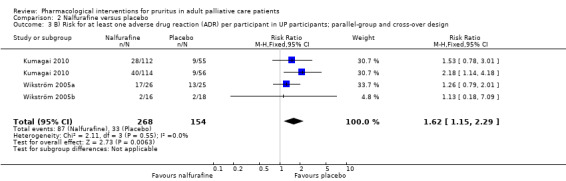
Comparison 2 Nalfurafine versus placebo, Outcome 3 B) Risk for at least one adverse drug reaction (ADR) per participant in UP participants; parallel‐group and cross‐over design.
2.4. Analysis.
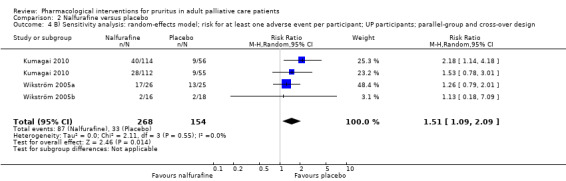
Comparison 2 Nalfurafine versus placebo, Outcome 4 B) Sensitivity analysis: random‐effects model; risk for at least one adverse event per participant; UP participants; parallel‐group and cross‐over design.
Serotonin 5‐HT3 antagonist ondansetron
We included three studies for the analysis of adverse events (Murphy 2003; O'Donohue 2005; Yue 2015). The meta‐analysis showed no significant difference between the groups (RR 2.07, 95% CI 0.87 to 4.93; three RCTs, N = 174) (Analysis 3.4; Analysis 3.5), and there was a noticeable increase of the 95% CI when using the random‐effects model for the sensitivity analysis (RR 2.54, 95% CI 0.38 to 16.78) (Analysis 3.6). Very few adverse events were reported for UP patients resulting in a RR of 7.53 and a wide 95% CI (0.97 to 58.51, two RCTs, N = 155, quality of evidence: very low) (Analysis 3.5; Table 4). We downgraded the quality of evidence due to risk of bias and very serious imprecision.
3.4. Analysis.
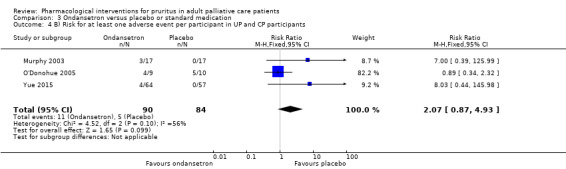
Comparison 3 Ondansetron versus placebo or standard medication, Outcome 4 B) Risk for at least one adverse event per participant in UP and CP participants.
3.5. Analysis.
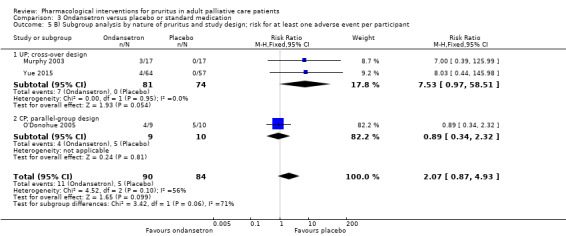
Comparison 3 Ondansetron versus placebo or standard medication, Outcome 5 B) Subgroup analysis by nature of pruritus and study design; risk for at least one adverse event per participant.
3.6. Analysis.
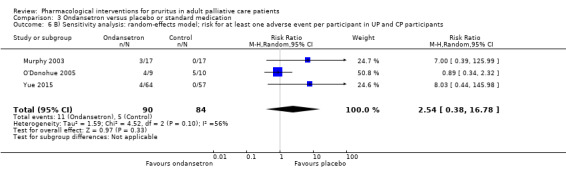
Comparison 3 Ondansetron versus placebo or standard medication, Outcome 6 B) Sensitivity analysis: random‐effects model; risk for at least one adverse event per participant in UP and CP participants.
Antiepileptic gabapentin
Bergasa 2006 was the only study that was appropriate for the analysis of adverse events. The RR for experiencing at least one adverse event was 2.63 (95% CI 0.76 to 9.05; one RCT, N = 15, quality of evidence: low) in favour of the placebo group (P = 0.13). We downgraded the quality of evidence by two levels due very serious imprecision. Common adverse events were somnolence, fatigue, dizziness and nausea (Gunal 2004; Naini 2007).
Semiantibiotic rifampicin
We could only use Bachs 1989 to analyse the RR for experiencing at least one adverse event. The result was not statistically significant (RR 0.29, 95% CI 0.03 to 2.51; one RCT, N = 39 quality of evidence: very low) (Table 6). We downgraded the quality of evidence due to risk of bias and very serious imprecision. Overall, investigators observed very few adverse events for rifampicin (Table 14).
Flumecinol
No adverse events were attributable to the trial medication according to the authors (Turner 1994a; Turner 1994b), who reported no adverse events in the publication.
Oral and topical mast cell stabilizer cromolyn sodium
Adverse events were rare in both groups, but they showed a conflicting pattern (I2 = 84%) that indicated a non‐significantly (P = 0.08) higher risk of adverse events (burning sensation) for the topical treatment (Feily 2012) compared with vehicle (RR 13.00, 95% 0.76 to 220.96; one RCT, N = 60). Interestingly, the risk for at least one adverse event per participant was lower (but not statistically significant, P = 0.08) for the oral administration (Vessal 2010) compared with placebo (RR 0.16, 95% CI 0.02 to 1.22; one RCT, N = 62) (Analysis 7.4; Analysis 7.5; Table 8: two RCTs, N = 122, quality of evidence: very low). We downgraded the quality of evidence due very serious inconsistency and serious imprecision. The random‐effects model considerably increased the 95% CI (Analysis 7.6).
7.4. Analysis.
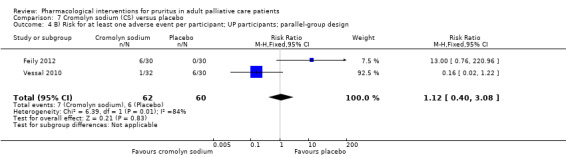
Comparison 7 Cromolyn sodium (CS) versus placebo, Outcome 4 B) Risk for at least one adverse event per participant; UP participants; parallel‐group design.
7.5. Analysis.
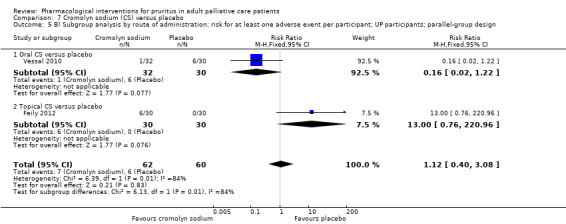
Comparison 7 Cromolyn sodium (CS) versus placebo, Outcome 5 B) Subgroup analysis by route of administration; risk for at least one adverse event per participant; UP participants; parallel‐group design.
7.6. Analysis.
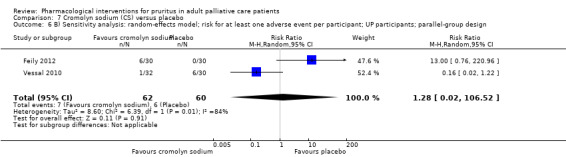
Comparison 7 Cromolyn sodium (CS) versus placebo, Outcome 6 B) Sensitivity analysis: random‐effects model; risk for at least one adverse event per participant; UP participants; parallel‐group design.
Topical capsaicin
The pooled RR for experiencing at least one adverse event was 4.64 (95% CI 2.05 to 10.51; three RCTs, N = 116, quality of evidence: moderate) in favour of the vehicle group (Analysis 8.4). However, participants mostly reported mild skin burning, which is part of the intended mechanism. The results changed slightly when using the random‐effects model but remained statistically significant (RR 3.69, 95% CI 1.17 to 11.67) (Analysis 8.5). We downgraded the quality of evidence due serious imprecision.
8.4. Analysis.
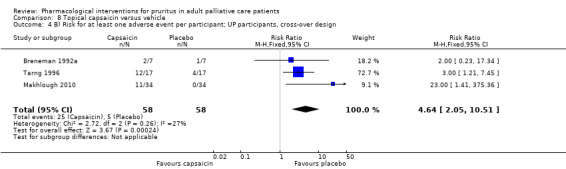
Comparison 8 Topical capsaicin versus vehicle, Outcome 4 B) Risk for at least one adverse event per participant; UP participants, cross‐over design.
8.5. Analysis.
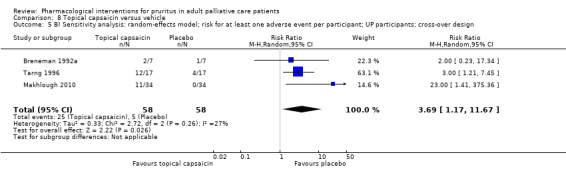
Comparison 8 Topical capsaicin versus vehicle, Outcome 5 B) Sensitivity analysis: random‐effects model; risk for at least one adverse event per participant; UP participants; cross‐over design.
Zinc sulphate
Studies reported no adverse events for zinc sulphate or placebo during a 4‐week or an 8‐week intervention period (Mapar 2015; Najafabadi 2012, respectively).
Discussion
Summary of main results
We identified 50 studies assessing the effects of 39 different interventions for pruritus of multiple origin in participants with advanced disease. Ten studies with 627 participants were new to this update. Eight investigated new pharmacological interventions: ergocalciferol (Shirazian 2013; N = 50), nicotinamide (Omidian 2013; N = 50), omega‐3 fatty acids (Ghanei 2012; N = 22), turmeric (Pakfetrat 2014; N = 100), zinc sulphate (Najafabadi 2012; N = 40; Mapar 2015; N = 40), pregabalin (Yue 2015; N = 188) and ketotifen (Amirkhanlou 2016; N = 52).
For now, only one RCT has researched pruritus as a symptom in inpatients of two palliative care centres (Zylicz 2003). All other studies focused on different kinds of pruritus based on the underlying disease of the participants. The kind of pruritus most frequently researched was UP (N = 1574 participants, 82%), followed by CP (N = 276, 14%). Furthermore, one included study focused on HIV‐related pruritus (Smith 1997a). The main results of this review were reported according to the structure resulting from these findings.
Palliative care patients with pruritus of different origin
Most 'palliative care' participants, as defined in the Methods, were treated on normal wards, perhaps because many hospitals still do not have a palliative care ward or patients did not have palliative care needs in the early phase of their disease. There were only 26 participants including 2 dropouts who received treatment on palliative care wards (Zylicz 2003). The study indicates that paroxetine may be effective but also may increase nausea and sleepiness (quality of evidence: moderate) (Table 1). Still, the number of participants included in this study was quite small, the treatment period was brief, and there was no follow‐up.
Participants with advanced diseases and UP or CP
Fifteen studies (N = 387) assessed four interventions for treating both UP and CP.
Three naltrexone studies focused on UP (Legroux‐Crespel 2004; Peer 1996; Pauli‐Magnus 2000), and two evaluated naltrexone for CP (Terg 2002; Wolfhagen 1997). We analysed the percent change of pruritus in CP and UP participants in a subgroup analysis, finding a larger effect in CP participants. However, the results of the studies pertaining to UP are conflicting. For at least Pauli‐Magnus 2000 and Peer 1996, we cannot explain differences by variations in participant compliance, naltrexone dose, study design or methodological quality. Moreover, readers should interpret the results of Legroux‐Crespel 2004 cautiously because of missing data, high dropout rates and high risk of bias. Concerning CP, meta‐analysis favoured naltrexone over placebo. Still, the study population included was quite small (16 and 20 participants in Wolfhagen 1997 and Terg 2002, respectively), and participants on naltrexone had the highest incidence of adverse events (moderate‐quality evidence) (Table 2).
Ondansetron was applied for either UP or CP. Three of five included studies reported no advantage over placebo, either for use in CP (O'Donohue 2005) or for UP (Ashmore 2000; Murphy 2003). One study researching ondansetron for treatment of UP found a benefit for participants treated with ondansetron compared to participants treated with the standard medication cyproheptadine (Özaykan 2001). In another RCT, the differences were marginal between ondansetron and placebo and indicated a minimal effect in favour of ondansetron (Yue 2015). Interestingly, in all studies comparing ondansetron with placebo, the placebo group showed an improvement in pruritus, which was even statistically significant in one study (Murphy 2003). Overall, ondansetron may be not effective for treatment of UP or CP. Adverse events seem hardly to differ between groups (quality of evidence: low) (Table 4).
Four studies evaluated gabapentin: three for the treatment of UP (Amirkhanlou 2016; Gunal 2004; Naini 2007) and one for CP (Bergasa 2006). For UP, gabapentin showed a clinically relevant improvement for pruritus in an analysis with 118 participants (quality of evidence: moderate) (Table 5). Authors reported some adverse events like dizziness or somnolence for gabapentin, but the quality of evidence was low and the results were not statistically significant (Table 14; Table 5). In contrast, the findings in CP patients even indicate that gabapentin appears to worsen pruritus (Bergasa 2006). On the other hand, the authors also observed a strong placebo effect, prompting them to discuss whether gabapentin possibly interferes with the placebo effect (possibly an association of the placebo intervention with dopamine release).
Duncan 1984 and Silverberg 1977 studied the bile acid sequestrant cholestyramine for CP and UP, respectively. The results tended to favour the cholestyramine group for UP and CP. However, both studies suffered from very small sample sizes (8 and 10 participants), and they were of low methodological quality. Thus, the validity of the results is very limited.
Participants with UP
Three studies examined nalfurafine for UP in 422 participants (Kumagai 2010; Wikström 2005a; Wikström 2005b). The results indicated that nalfurafine could have a small benefit compared to placebo, but the risk for at least one adverse event (most frequently insomnia) was also slightly higher (quality of evidence: moderate) (Table 3). In addition, the statistically significant benefit did not persist after four weeks of treatment in a subgroup of participants (Wikström 2005a).
Additional studies investigated systemic interventions like erythropoietin (De Marchi 1992), montelukast (Nasrollahi 2007), thalidomide (Silva 1994), activated oral charcoal (Pederson 1980), and doxepin (Pour‐Reza‐Gholi 2007). All of these interventions could have a positive effect on UP. However, the sample sizes were small (16 to 29 participants), and in four of the six studies methodological quality was limited (De Marchi 1992; Nasrollahi 2007; Pederson 1980; Silva 1994). The results for cromolyn sodium suggest that the oral administration that Vessal 2010 tested may be more effective than the topical use featured in Feily 2012 (quality evidence: moderate) (Table 8) and that the topical use is associated with more adverse events (skin burning).
Seven studies investigated the effect of topical agents on UP. Whereas tacrolimus ointment was not more effective than the vehicle in relieving UP (Duque 2005), pramoxine lotion tended to reduce pruritus to a greater degree than the control lotion (Young 2009). Adverse events for tacrolimus were minor, and there were none for pramoxine hydrochloride. However, both studies were at high risk of bias. Thus, evidence for the use of these topical applications is limited. Another study examined avena sativa and vinegar solution as topical agents (Nakhaee 2015). Both treatments, as well as placebo, reduced pruritus. The evidence of the study is questionable because of the small sample size (N = 25) and the impossibility of blinding.
Four studies compared the efficacy of topical capsaicin in treating UP (Breneman 1992a; Cho 1997; Makhlough 2010; Tarng 1996). A common minor adverse event of topical capsaicin was a transient burning sensation and local erythaema with initial application, and the risk for at least one adverse event per participant was considerably increased (moderate quality of evidence) (Table 9; Table 14). Three of the studies demonstrated methodological flaws such as inappropriate designs leading to carryover effects, inadequate depiction of data and failure to provide appropriate statistical analyses (Breneman 1992a; Cho 1997; Tarng 1996). Nevertheless, our analysis suggests a moderate quality of evidence.
Nicotinamide and ergocalciferol tended to be ineffective for pruritus treatment (Omidian 2013; Shirazian 2013). The effect of zinc sulphate was neither statistically nor clinically significant. We did not observe any dose response (220 mg twice daily in Najafabadi 2012 versus 220 mg daily in Mapar 2015). Thus, the effectiveness of zinc sulphate is questionable. One RCT with low risk of bias found that turmeric may be effective for participants with UP, but the authors advised caution because of the small sample size (N = 100), the short treatment phase (eight weeks) and the lack of a follow‐up (Pakfetrat 2014). Ghanei 2012 evaluated the efficacy of Omega‐3 fatty acid. Though there were statistically significant group differences, the wide confidence intervals due to the small sample size and the low methodological quality of the trial confer some uncertainty on the findings. These (rather small) new included studies did not report adverse events, except skin burning in patients receiving topical cromolyn sodium (Feily 2012). Pregabalin might reduce pruritus in a similar way to gabapentin because both drugs are γ‐aminobutyric acid analogues (Yue 2015). In addition, pregabalin improved health‐related quality of life. However, the effect was small and of questionable clinical relevance.
Participants with CP
Six interventions targeted participants with advanced disease and CP in nine studies with 194 participants. Three studies explored the effect of rifampicin (Bachs 1989; Ghent 1988; Podesta 1991a). The quantitative results indicate that rifampicin could be effective for treating CP when compared with a placebo or phenobarbitone. However, the heterogeneity was very high (I2 = 95%) when using the random‐effects model, leading to an extremely wide 95% CI that reversed statistical significance and impeded the interpretability of results (though the mean effect was large) (Analysis 5.2). The quality of evidence was judged as low for pruritus, and very low for the adverse event outcome because only Bachs 1989 contributed to the meta‐analysis (Table 6).
Two studies researched flumecinol with different dosages in CP participants (Turner 1994a; Turner 1994b). The results of both studies only reached statistical significance in our analysis when we combined them. However, the quality of evidence remains low for this comparison (Table 7). The results suggested that there could be a dose‐response‐relationship, but the small sample sizes impeded generalisation and led to a wide 95% CI. Authors reported no adverse events for flumecinol.
Two studies investigated the effect of anaesthetics: Borgeat 1993 assessed propofol in 12 participants, and Villamil 2005 studied lidocaine in 18. Both trials reported an amelioration of pruritus for participants treated with propofol and lidocaine, but we cannot draw conclusions since the sample sizes were small and the studies were not free of bias. Adverse events reported were minor. However, the applicability of lidocaine as an alternative therapy for CP is limited because of the necessity of hospital or inpatient treatment of the patients concerned.
Mayo 2007 described the SSRI sertraline as effective and well‐tolerated in participants with CP. However, the sample size of 12 participants was very small, and the blinding of outcome assessment was somewhat precarious. Additional studies confirming or disagreeing with these results are lacking.
Participants with HIV‐associated pruritus
Smith 1997a compared four different interventions to a placebo for pruritus in 40 participants with HIV. Since authors provided no information about blinding and additional data were missing, we assessed the risk of bias to be high, and we cannot make conclusive recommendations based on the study results.
Overall completeness and applicability of evidence
This review included 50 RCTs. Only one study researched pruritus in palliative care patients who received palliative care. According to the presumed definition of palliative care patients as 'adult patients in any setting, receiving palliative care or suffering an incurable progressive medical condition', we also included studies researching pruritus in participants with advanced and incurable diseases in different settings. Thus, included studies explored a total of 39 different interventions for treatment of four different groups of patients suffering from pruritus of different origins (Table 12). Due to the diversity of the interventions and the small number of studies per intervention, we could not compare the effectiveness of all interventions. As the overall quality of studies varied, missing data in several studies did not allow for including data from all studies in a single meta‐analysis. We had to describe the results of several studies as a narrative summary. Due to the diversity and different character of pruritus and the patient groups being researched, the applicability of evidence is restricted to the particular patient group targeted by the intervention.
Quality of the evidence
The summary of findings tables, structured according to the GRADE system (GRADE Handbook), show that the quality of evidence for the primary outcome (i.e. pruritus) was moderate for paroxetine, naltrexone, ondansetron, nalfurafine, gabapentin, cromolyn sodium and capsaicin. This means we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. The quality of evidence was low for rifampicin and flumecinol. This means that our confidence in the effect estimate is limited and the true effect may be substantially different from the estimate of the effect. Risk of bias and imprecision were the main factors for downgrading the quality of evidence for the primary outcome (Table 1; Table 3; Table 4; Table 5; Table 7; Table 9; Table 10).
Concerning the risk of experiencing at least one adverse event per participant, the quality of evidence was moderate for naltrexone, nalfurafine and capsaicin, low for gabapentin and very low for ondansetron, rifampicin and cromolyn sodium. For paroxetine, we rated the quality of evidence as moderate for nausea, vomiting and sleepiness. We mostly downgraded the quality of evidence because of serious imprecision (Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 8; Table 9).
Potential biases in the review process
Previous reviews have reported difficulties in defining the population of palliative care patients. Therefore, we used the definition 'adult patients in any setting, receiving palliative care or suffering an incurable progressive medical condition', which has previously been used in other Cochrane reviews (Dorman 2010; Perkins 2009). However, it is still difficult to identify the patients or patient groups who are appropriate to be included in a systematic review on palliative care topics, as there is still a lack of original studies, particularly RCTs, in palliative care. In a survey of 25 Cochrane reviews in palliative care (Wee 2008), the authors concluded that "Cochrane reviews in palliative care ... fail to provide good evidence for clinical practice because the primary studies are few in number, small, clinically heterogeneous, and of poor quality and external validity". Nevertheless, data on palliative care patients may be hidden in studies not described as palliative care topics. Therefore, it is possible that we failed to identify some published or unpublished trials including palliative care patients for this review.
In this update, we performed a comprehensive literature search, specified inclusion and exclusion criteria and conducted meta‐analysis where possible. We revised the search strategies, updated MEDLINE, Embase and CENTRAL (August 2012 to June 2016), checked reference lists of all relevant trials and searched registers of ongoing trials.
In the original review, we identified two studies meeting all inclusion criteria (Ghorbani 2011; Sja'bani 1997), but we could not retrieve data, and we were unable to make contact with the authors. Inability to include this data could have led to publication bias or changed the results concerning a special intervention.
In order to avoid duplicate publication bias, it was crucial to identify all redundant and multiple publications, which can lead to overestimation of intervention effects. We found one study whose data overlapped substantially with an included study and therefore excluded it (Borgeat 1994).
In the original review and in this update, we imposed no language restriction. As a result, we identified a Turkish language study that we had translated before data extraction (Özaykan 2001). We accept possible lack of clarity due to translation.
A limitation of this review is that, with the exception of two studies (Kumagai 2010, N = 337; Yue 2015, N = 188), all included studies were at high risk of bias concerning sample sizes (i.e. fewer than 50 per treatment arm; Figure 3). Consequently, the evidence for most studies was limited.
The title of this review suggests that a variety of patients with different underlying diseases were included. However, the vast majority of participants suffered from UP (n = 1574, 82%) pruritus, and only 26 participants were treated in palliative care settings.
Choosing the adverse event outcome 'risk for at least one adverse event per participant' for meta‐analysis was a pragmatic decision in order to give a quantitative overview. In many cases, the number of adverse events exceeded the number of participants, making the results of statistical analyses incoherent. On the one hand, one participant may experience several adverse events simultaneously. On the other hand, especially in the cross‐over studies, the data do not allow us to identify which participant in which group of the study experienced the adverse event. Therefore, it was not possible to allocate the adverse events described in the studies to an individual or to adequately calculate the number of adverse events in relation to the factual number of participants included. Table 14 and Table 15 describe all different types of adverse events more precisely.
The risk of bias at the study level was high in 48 of the 50 (96%) included studies in this review (Figure 3), mostly because of the small sample size. This result emphasises the challenge and difficulty of conducting large high‐quality RCTs with 200 or more participants with advanced disease per treatment arm. The latter was our criterion for a low risk of bias (see Methods). Though other Cochrane Reviews have used this cutoff, which seems reasonable, it may be somewhat arbitrary. Moreover, the sample size is also indirectly addressed by the 'imprecision' item of GRADE.
It is difficult to decide on the extent of bias at the outcome level. On one hand, participants and personnel in 36 (72%) studies were adequately blinded. However, the outcome assessment was only blinded in 15 studies (30%). The latter in particular could have contributed to bias when assessing the intensity of pruritus or adverse events. The quality of evidence at the outcome level can be found in the 'Summary of findings' tables for the all comparisons.
Agreements and disagreements with other studies or reviews
The results of this review are generally consistent with the findings of another recent review by our working group that was conducted as an intermediate non‐Cochrane update between the original and the updated Cochrane review (Siemens 2014).
We found reviews and systematic reviews on the following interventions for pruritus, and we compared the results to the results of this review.
Nalfurafine (Inui 2015).
Ondansetron (To 2012).
Gabapentin (Anand 2013).
Rifampicin (Khurana 2006; Tandon 2007).
Systemic μ‐opioid receptor antagonists (Phan 2010).
Topical capsaicin (Gooding 2010).
Inui 2015 included the same studies featured in this review. In addition, the authors included a long‐term study that showed efficacy of nalfurafine hydrochloride over one year without resulting in abuse liability of nalfurafine (Ueno 2013). However, our meta‐analysis suggests that the effect of nalfurafine should not be overestimated (Analysis 2.1).
Due to different inclusion criteria concerning palliative care patients, To 2012 included some studies that we excluded in our review (Jones 2007; Müller 1998a; Müller 1998b; Characteristics of excluded studies). The authors concluded, similarly to our working group, that ondansetron has negligible effects on UP or CP (To 2012).
According to Anand 2013, gabapentin is safe and effective for UP and some other indications. Although the authors did not include Naini 2007 or conduct a meta‐analysis, their conclusions are comparable to those of our working group.
In accordance with the results of our review, recent meta‐analyses of prospective RCTs revealed that rifampicin might be effective and has tolerable adverse events as short‐term treatment for pruritus (Khurana 2006; Tandon 2007).
Phan 2010 reviewed the use of systemic μ‐opioid receptor antagonists in the treatment of various forms of chronic pruritus; the included RCTs in participants with advanced diseases are similar to the RCTs included in this review.
Gooding 2010 explored the effect of topical capsaicin . Since it was conducted in 2010, the review did not include the Makhlough 2010 study on topical capsaicin; however, the overall results are comparable with our findings.
Authors' conclusions
Implications for practice.
Since the last version of this review, none of the new relevant studies have provided additional information to change the conclusions.
For people with pruritus and advanced disease
An ideal or general antipruritic therapy for patients with advanced disease is currently lacking. However, we identified some possibly useful treatments for particular patients. Gabapentin (and maybe pregabalin), nalfurafine and cromolyn sodium may be useful for itch associated with UP, and rifampicin and flumecinol could be effective for itch associated with liver problems. Paroxetine showed promise in palliative care patients, although evidence was only available from one study. Overall, most of the drugs were associated with few and mild adverse events. Naltrexone showed the most adverse events.
For clinicians
The varying pathogenesis of pruritus in different disorders means that a universally accepted therapy is difficult to establish. Therapy for pruritus is challenging and requires an individualistic approach. Therefore, identifying the underlying cause of pruritus is still of prime importance in order to develop tailored treatment plans. Especially in palliative care, patients with pruritus may have more than one origin for their pruritus. The fact that itch affects the skin, immune system, and the peripheral and central nervous system means that complex and combinatory pathways are likely to be more effective than a single‐line approach.
Palliative care participants with pruritus of different origin
For participants in palliative care settings who mainly suffered from pruritus related to neoplasms, paroxetine tended to be effective (quality of evidence: moderate).
Participants with advanced diseases suffering from UP or CP
The opioid antagonist naltrexone offered a therapeutic alternative for participants suffering from UP or CP. However, these drugs are sometimes inappropriate in the palliative care population that suffers from pain because of the risk of loss of analgesia at higher doses of naltrexone (Higginson 1997; Potter 2003; Walsh 2000).
For patients suffering from UP, gabapentin, nalfurafine, cromolyn sodium (oral rather than topical) and capsaicin could be effective for pruritus relief (quality of evidence: moderate), and in CP participants with advanced disease, rifampicin and flumecinol (low quality of evidence) may be beneficial.
Ondansetron tended to have only very small or no effect for treatment of UP or CP, and the results for cholestyramine, thalidomide, lidocaine and sertraline are very limited due to the small sample sizes.
Participants with HIV‐associated pruritus
For patients suffering from pruritus associated with HIV infection, we could not draw any distinct conclusions, as the evidence was very low quality. Indomethacin was described as the most effective drug, but the results cannot be generalised.
For policy‐makers
Two factors could be taken into account for policy‐making. First, many drugs presented here were used off‐label. The off‐label use of drugs is a typical and well‐known phenomenon in palliative care. Second, treatments for pruritus are not necessarily bound to the pharmaceutical law (e.g. omega‐3 fatty acid). This influences time and costs of RCTs.
For funders
Funders could consider supporting high‐quality RCTs with 50 or more participants per treatment arm. Most of the investigated drugs showed a positive tendency in reducing pruritus, but these results need to be confirmed in further RCTs (e.g. gabapentin, cromolyn sodium; see also below).
Implications for research.
General
This update revealed that omega‐3 fatty acids and turmeric could be effective in participants with UP (Ghanei 2012; Pakfetrat 2014). These results have to be confirmed in future RCTs.
Gabapentin may have the largest effect of the investigated drugs (Gunal 2004; Naini 2007). However, these results are only valid for UP patients. The conflicting results for CP participants should be clarified in a future RCT with an adequate sample size (at least 50 participants per treatment arm) (Anand 2013). Furthermore, it is necessary to reproduce those large effects for the UP population in a powered and sound RCT.
A future goal is a wide range of topical and systemic therapies that target the various receptors and neural pathways that mediate different types of itch and lead to improved management of all kinds of pruritus.
As already mentioned, opioid antagonists (e.g. naltrexone) are often inappropriate in the palliative care population because of the risk of loss of analgesia at higher doses of naltrexone. Methylnaltrexone, a peripherally acting μ‐opioid receptor antagonist, was approved by the Food and Drug Administration (FDA) in 2008 for the treatment of opioid‐induced constipation in patients with advanced illness. Notably, methylnaltrexone does not cross the blood‐brain barrier, offering the advantage of peripheral action only and thus has significantly fewer adverse events, including addiction. Researching the efficacy of methylnaltrexone in larger, high‐quality studies, especially in the field of palliative care, would be of particular interest. In addition, given the possible role of the opioidergic system in pruritus, the role of the μ‐opioid receptor antagonist naltrexone in a topical 1% form in the treatment of severe pruritus might be of interest for palliative care patients and could also be researched in a future RCT (Bigliardi 2007).
Design
In the future, larger studies would help delineate the efficacy of the available and proposed antipruritics. Studies in the field of palliative care are especially lacking, and the evidence for interventions targeting palliative care patients is low. Therefore, well‐designed treatment studies, where possible placebo‐controlled and randomised, are needed to further verify the effectiveness of many antipruritic agents currently in use. Ideally, these RCTs should at least include 50 participants per treatment arm (depends also on sample size calculation).
Measurement (endpoints)
Most authors used a simple 10 cm or 100 mm VAS to assess pruritus. The VAS should be the minimum standard for assessing pruritus (e.g. in addition to the Duo scale, satisfaction and quality of life). Another advantage of the VAS is that the results can be easily pooled and interpreted in meta‐analyses.
What's new
| Date | Event | Description |
|---|---|---|
| 16 November 2016 | Review declared as stable | See Published notes. |
History
Protocol first published: Issue 1, 2010 Review first published: Issue 6, 2013
| Date | Event | Description |
|---|---|---|
| 9 June 2016 | New search has been performed | This review has been updated. We included results of a new search and updated the 'Risk of bias table, the 'Summary of findings' tables and the meta‐analyses. |
| 9 June 2016 | New citation required but conclusions have not changed | i. Summary of the differences between the update and the current published version.
ii. Date and year of the last search: 9 June 2016. iii. Ten new included studies after reading the full text: Amirkhanlou 2016; Feily 2012; Ghanei 2012; Mapar 2015; Najafabadi 2012; Nakhaee 2015; Omidian 2013; Pakfetrat 2014; Shirazian 2013; Yue 2015. iv. Participants involved in this review update (N = 1916):
v. New meta‐analyses as a result of our new findings and general revision:
vi. Conclusion
|
Notes
A new search within two years is not likely to identify any potentially relevant studies likely to change the conclusions. Therefore, this review has now been stabilised following discussion with the authors and editors. The review will be re‐assessed for updating in 2019. If appropriate, we will update the review before this date if new evidence likely to change the conclusions is published, or if standards change substantially which necessitate major revisions.
Acknowledgements
The authors wish to thank the participants who entered the trials and the investigators who conducted them.
We are very grateful to Anna Erskine (nee Hobson) (Managing Editor), Joanne Abbott (Information Specialist) and Kerry Harding (Assistant Managing Editor) of the Cochrane Pain, Palliative and Supportive Care Review Group and would like to thank them for their help and support during the preparation of this update.
We thank Mayang Mayang, graduate assistant at the Department of Palliative Care, University Medical Center Freiburg, for her help with data extraction and proofreading.
We thank Edith Motschall (librarian at the Center for Medical Biometry and Medical Informatics, Medical Center ‐ University of Freiburg, Freiburg, Germany) for her support concerning the search strategies in the original review.
The original review was funded by the German Ministry for Education and Research (BMBF), Germany Grant No. 01KG0819. The review was conducted independently of funding from any interested party.
This update was not funded.
Cochrane Review Group funding acknowledgement: the National Institute for Health Research (NIHR) is the largest single funder of the Cochrane PaPaS Group. Disclaimer: the views and opinions expressed therein are those of the authors and do not necessarily reflect those of the NIHR, National Health Service (NHS) or the Department of Health.
Appendices
Appendix 1. MEDLINE search strategy via Ovid, 9 June 2016
1. exp Pruritus/
2. (prurit* or itch* or scratch*).ti.
3. ((prurit* or itch* or scratch*) adj10 (prevent* or stop* or alleviat* or relief* or reliev*)).mp.
4. 1 or 2 or 3
5. ((advance* or late or last or end or final) adj4 (stage* or phase*)).mp.
6. (palliat* or terminal* or endstage or end‐stage or hospice* or (end adj3 life) or (care adj3 dying)).mp.
7. ((advance* or progressi* or terminal*) adj6 (ill* or disease* or condition*)).mp.
8. (terminal* adj6 (care or therap* or treat*)).mp.
9. (hospice or (nursing adj3 home*)).mp.
10. exp Palliative Care/ or Palliative Medicine/ or Terminal Care/ or Terminally Ill/ or Hospice Care/ or "Hospice and Palliative Care Nursing"/ or exp Home Care Services/ or exp Hospitals, Special/ or Attitude to Death/ or exp Medicare/ or Patient Care/ or Nursing Homes/ or Homes for the Aged/
11. 5 or 6 or 7 or 8 or 9 or 10
12. 4 and 11
13. randomized controlled trial.pt.
14. controlled clinical trial.pt.
15. randomized.ab.
16. placebo.ab.
17. drug therapy.fs.
18. randomly.ab.
19. trial.ab.
20. groups.ab.
21. 13 or 14 or 15 or 16 or 17 or 18 or 19 or 20
22. exp animals/ not humans.sh.
23. 21 not 22
24. 12 and 23
25. limit 24 to yr="2012 ‐ 2016"
Appendix 2. Embase search strategy via Ovid, 7 June 2016
1. exp Pruritus/
2. pruri$.tw.
3. itch$.tw.
4. scratch.tw.
5. ((pruri$ or itch$ or scratch$) adj5 prevent$).tw.
6. or/1‐5
7. exp Antipruritic Agent/
8. antipruritic$.tw.
9. (Antipruritic Agent or Pruritus or anti‐pruritic$).mp.
10. anti‐pruritic$.tw.
11. exp Drug Therapy/
12. drug therapy combination$.mp.
13. exp Drug Combination/
14. prevention control.mp.
15. (pharmacol$ adj3 (therap$ or treat$ or intervent$)).mp.
16. (pharmaceutic$ adj3 (therap$ or treat$ or intervent$)).mp.
17. (drug$ adj3 prevent$).mp.
18. pharmaceutic aid$.mp.
19. exp "Pharmaceutical Vehicles and Additives"/
20. or/7‐19
21. (endstage or end‐stage).mp.
22. progressive.mp.
23. (terminal$ adj3 ill$).mp.
24. palliative care.mp.
25. exp Palliative Therapy/
26. (palliativ$ adj3 car$).mp.
27. (palliative adj3 therap$).mp.
28. (palliative adj3 treat$).mp.
29. (terminal adj3 care).mp.
30. (terminal adj3 disease).mp.
31. (terminal adj3 treat$).mp.
32. (end adj3 life adj3 care).mp.
33. exp Home Care/
34. hospice care.mp.
35. exp Psychological Aspect/
36. exp Terminal Disease/
37. exp Terminal Care/
38. exp Terminally ill Patient/
39. exp Medicare/
40. exp Hospice/
41. exp Hospice Care/
42. exp Hospice Patient/
43. exp Patient Care/
44. (hospice adj3 care).mp.
45. hospice$.mp.
46. exp hospice nursing/
47. exp hospital patient/
48. exp nursing home/
49. exp nursing home patient/
50. exp home for the aged/
51. ("nursing adj2 home$" or "home for the aged" or "old age home$").mp.
52. or/21‐51
53. 6 and 20 and 52
54. random$.tw.
55. factorial$.tw.
56. crossover$.tw.
57. cross over$.tw.
58. cross‐over$.tw.
59. placebo$.tw.
60. (doubl$ adj blind$).tw.
61. (singl$ adj blind$).tw.
62. assign$.tw.
63. allocat$.tw.
64. volunteer$.tw.
65. Crossover Procedure/
66. double‐blind procedure.tw.
67. Randomized Controlled Trial/
68. Single Blind Procedure/
69. or/54‐68
70. (animal/ or nonhuman/) not human/
71. 69 not 70
72. 53 and 71
73. (201208* or 201209* or 201210* or 201211* or 201212* or 2013* or 2014* or 2015* or 2016*).dd.
74. 72 and 73
Appendix 3. Cochrane Central Register of Controlled Trials (CENTRAL) search strategy via The Cochrane Library, 9 June 2016
#1 [mh pruritus]
#2 (pruri*):ti,ab,kw
#3 (itch*):ti,ab,kw
#4 (scratch*):ti,ab,kw
#5 (antipruritic*):ti,ab,kw
#6 {or #1‐#5}
#7 (drug therap*):ti,ab,kw
#8 (pharmacologic* therap*):ti,ab,kw
#9 (pharmacologic* treat*):ti,ab,kw
#10 (pharmaceutic* therap*):ti,ab,kw
#11 (pharmaceutic* treat*):ti,ab,kw
#12 {or #7‐#11}
#13 (advanced disease*):ti,ab,kw
#14 (palliative care):ti,ab,kw
#15 (hospice care):ti,ab,kw
#16 (terminal care):ti,ab,kw
#17 (terminal* ill*):ti,ab,kw
#18 {or #13‐#17}
#19 (#6 and #12)
#20 (#18 and #19)
#21 (#18 and #19) Publication Year from 2012 to 2016
Appendix 4. MEDLINE search strategy via Ovid, August 2012
mp=title, original title, abstract, name of substance word, subject heading word; ti=title; pt=publication type; ab=abstract; fs=floating subheading; sh=MeSH subject heading
1. exp Pruritus/
2. (prurit* or itch* or scratch*).ti.
3. ((prurit* or itch* or scratch*) adj10 (prevent* or stop* or alleviat* or relief* or reliev*)).mp.
4. 1 or 2 or 3
5. ((advance* or late or last or end or final) adj4 (stage* or phase*)).mp.
6. (palliat* or terminal* or endstage or end‐stage or "end stage" or hospice* or (end adj3 life) or (care adj3 dying)).mp.
7. ((advance* or progressi* or terminal*) adj6 (ill* or disease* or condition*)).mp.
8. (terminal* adj6 (care or therap* or treat*)).mp.
9. exp Palliative Care/ or Terminal Care/ or Terminally Ill/ or Hospice Care/ or exp Home Care Services/ or exp Hospitals, Special/ or Attitude to Death/
10. 5 or 6 or 7 or 8 or 9
11. 4 and 10
For identification of randomised controlled trials (humans or human and animals) the subject search above will be combined with the following search strategy
Cochrane Highly Sensitive Search Strategy for identifying randomised trials in MEDLINE: sensitivity‐maximizing version (2008 revision); Ovid format
1. randomized controlled trial.pt.
2. controlled clinical trial.pt.
3. randomized.ab.
4. placebo.ab.
5. drug therapy.fs.
6. randomly.ab.
7. trial.ab.
8. groups.ab.
9. 1 or 2 or 3 or 4 or 5 or 6 or 7 or 8
10. exp animals/ not humans.sh.
11. 9 not 10
Appendix 5. Biosis search strategy via Ovid, August 2012
1. (prurit* or itch* or scratch*).m_titl.
2. ((prurit* or itch* or scratch*) adj10 (prevent* or stop* or alleviat* or relief* or reliev*)).mp.
3. 1 or 2
4. ((advance* or late or last or end or final) adj4 (stage* or phase*)).mp.
5. (palliat* or terminal* or endstage or end‐stage or "end stage" or hospice or (end adj3 life) or (care adj3 dying)).mp.
6. ((advance* or progressi* or terminal*) adj6 (ill* or disease* or condition)).mp.
7. (terminal* adj6 (care or therap* or treat*)).mp.
8. 4 or 5 or 6 or 7
9. 3 and 8
10. (animals not (humans and animals)).sh.
11. 9 not 10
Appendix 6. CINAHL search strategy via EBSCOhost, August 2012
1. MH pruritus
2. MJ prurit* or MJ itch* or MJ scratch*
3. prurit* N10 prevent* or prurit* N10 stop* or prurit* N10 alleviat* or prurit* N10 relief* or prurit* N10 reliev*
4. itch* N10 prevent* or itch* N10 stop* or itch* N10 alleviat* or itch* N10 relief* or itch* N10 reliev*
5. scratch* N10 prevent* or scratch* N10 stop* or scratch* N10 alleviat* or scratch* N10 relief* or scratch* N10 reliev*
6. S1 or S2 or S3 or S4 or S5
7. advance* N4 stage* or advance* N4 phase* or late N4 stage* or late N4 phase* or last N4 stage* or last N4 phase* or end N4 stage* or end N4 phase* or final N4 stage* or final N4 phase*
8. palliat* or terminal* or endstage or end‐ stage or "end stage" or hospice* or end N3 life or care N3 dying
9. advance* N6 ill* or advance* N6 disease* or advance* N6 condition* or progressi* N6 ill* or progressi* N6 disease* or progressi* N6 condition* or terminal* N6 ill* or terminal* N6 disease* or terminal* N6 condition*
10. terminal* N6 care* or terminal* N6 therap* or terminal* N6 treat*
11. MH Palliative Care or MH Terminal Care or MH Terminally Ill or MH Hospice Care or MH Home Care Services or MH Hospitals, Special or MH Attitude to Death
12. S7 or S8 or S9 or S10 or S11
13. S6 and S12
14. animals not (humans and animals)
15. S13 not S14
Appendix 7. Embase search strategy via Ovid August 2012
1. exp Pruritus/
2. pruri$.tw.
3. itch$.tw.
4. scratch.tw.
5. ((pruri$ or itch$ or scratch$) adj5 prevent$).mp.
6. or/1‐5
7. exp Antipruritic Agent/
8. antipruritic$.tw.
9. (Antipruritic Agent or Pruritus or anti‐pruritic$).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer, device trade name, keyword]
10. anti‐pruritic$.tw.
11. exp Drug Therapy/
12. drug therapy combination$.mp.
13. exp Drug Combination/
14. prevention control.mp.
15. (pharmacol$ adj3 (therap$ or treat$ or intervent$)).mp.
16. (pharmaceutic$ adj3 (therap$ or treat$ or intervent$)).mp.
17. (drug$ adj3 prevent$).mp.
18. pharmaceutic aid$.mp.
19. exp "Pharmaceutical Vehicles and Additives"/
20. or/7‐19
21. (endstage or end‐stage).mp.
22. progressive.mp.
23. (terminal$ adj3 ill$).mp.
24. palliative care.mp.
25. exp Palliative Therapy/
26. (palliativ$ adj3 car$).mp.
27. (palliative adj3 therap$).mp.
28. (palliative adj3 treat$).mp.
29. (terminal adj3 care).mp.
30. (terminal adj3 disease).mp.
31. (terminal adj3 treat$).mp.
32. (end adj3 life adj3 care).mp.
33. exp Home Care/
34. hospice care.mp.
35. exp Psychological Aspect/
36. exp Terminal Disease/
37. exp Terminal Care/
38. exp Terminally ill Patient/
39. exp Medicare/
40. exp Hospice/
41. exp Hospice Care/
42. exp Hospice Patient/
43. exp Patient Care/
44. (hospice adj3 care).mp.
45. hospice$.mp.
46. exp hospice nursing/
47. exp hospital patient/
48. exp nursing home/
49. exp nursing home patient/
50. exp home for the aged/
51. ("nursing adj2 home$" or "home for the aged" or "old age home$").mp.
52. or/21‐51
53. 6 and 20 and 52
Appendix 8. PsycINFO search strategy via EBSCOhost, August 2012
S1 DE "Pruritus"
S2 MJ prurit* or MJ itch* or MJ scratch*
S3 prurit* N10 prevent* or prurit* N10 stop* or prurit* N10 alleviat* or prurit* N10 relief* or prurit* N10 reliev*
S4 itch* N10 prevent* or itch* N10 stop* or itch* N10 alleviat* or itch* N10 relief* or itch* N10 reliev*
S5 scratch* N10 prevent* or scratch* N10 stop* or scratch* N10 alleviat* or scratch* N10 relief* or scratch* N10 reliev*
S6 S1 or S2 or S3 or S4 or S5
S7 advance* N4 stage* or advance* N4 phase* or late N4 stage* or late N4 phase* or last N4 stage* or last N4 phase* or end N4 stage* or end N4 phase* or final N4 stage* or final N4 phase*
S8 palliat* or terminal* or endstage or end‐ stage or "end stage" or hospice* or end N3 life or care N3 dying
S9 advance* N6 ill* or advance* N6 disease* or advance* N6 condition* or progressi* N6 ill* or progressi* N6 disease* or progressi* N6 condition* or terminal* N6 ill* or terminal* N6 disease* or terminal* N6 condition*
S10 terminal* N6 care* or terminal* N6 therap* or terminal* N6 treat*
S11 MH Palliative Care or MH Terminal Care or MH Terminally Ill or MH Hospice Care or MH Home Care Services or MH Hospitals, Special or MH Attitude to Death
S12 Palliative or Palliative care or Palliative treatment or Terminal care or Terminally ill or Hospice or Hospice care or Home care service or Attitude to death
S13 S7 or S8 or S9 or S10 or S11 or S12
S14 S6 and S13
Appendix 9. Search strategy for The Cochrane Library via Wiley, August 2012
#1 MeSH descriptor Pruritus explode all trees #2 (pruri*):ti,ab,kw #3 (itch*):ti,ab,kw #4 (scratch*):ti,ab,kw #5 (antipruritic*):ti,ab,kw #6 (antipruritic* NEXT therap*):ti,ab,kw #7 (#1 OR #2 OR #3 OR #4 OR#5 OR #6) #8 (drug therap*):ti,ab,kw #9 (pharmacologic* therap*):ti,ab,kw #10 (pharmacologic* treat*):ti,ab,kw #11 (pharmaceutic* therap*):ti,ab,kw #12 (pharmaceutic* treat*):ti,ab,kw #13 (#9 OR #10 OR #11OR #12) #14 (advanced disease*):ti,ab,kw #15 (advanced NEXT disease*):ti,ab,kw #16 (palliative care):ti,ab,kw #17 (palliative NEXT care):ti,ab,kw #18 (hospice care):ti,ab,kw #19 (hospice NEXT care):ti,ab,kw #20 (terminal care):ti,ab,kw #21 (terminal NEXT care):ti,ab,kw #22 (terminal* ill*):ti,ab,kw #23 (#14 OR #22) #24 (#7 AND #13) '#25 (#'23 AND #24)
Data and analyses
Comparison 1. Naltrexone versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 A) Pruritus on VAS scale (0‐10 cm) in CP participants | 2 | 52 | Mean Difference (Fixed, 95% CI) | ‐2.26 [‐3.19, ‐1.33] |
| 2 A) Subgroup analysis by study design: pruritus on VAS scale (0‐10 cm) in CP participants | 2 | 52 | Mean Difference (Fixed, 95% CI) | ‐2.26 [‐3.19, ‐1.33] |
| 2.1 Parallel group‐design | 1 | 16 | Mean Difference (Fixed, 95% CI) | ‐3.32 [‐5.01, ‐1.63] |
| 2.2 Cross‐over design | 1 | 36 | Mean Difference (Fixed, 95% CI) | ‐1.79 [‐2.91, ‐0.67] |
| 3 A) Sensitivity analysis: random‐effects model; pruritus on VAS scale (0‐10 cm) in CP participants | 2 | 52 | Mean Difference (Random, 95% CI) | ‐2.42 [‐3.90, ‐0.94] |
| 4 B) % difference for pruritus on VAS scale (0‐10 cm) in UP and CP participants | 2 | 48 | Mean Difference [%] (Fixed, 95% CI) | ‐22.02 [‐34.15, ‐9.90] |
| 5 B) Subgroup analysis by nature of pruritus and study design; % difference for pruritus on VAS scale (0‐10 cm) | 2 | 48 | Mean Difference [%] (Fixed, 95% CI) | ‐22.02 [‐34.15, ‐9.90] |
| 5.1 CP and parallel‐group design | 1 | 16 | Mean Difference [%] (Fixed, 95% CI) | ‐62.0 [‐89.42, ‐34.58] |
| 5.2 UP and cross‐over design | 1 | 32 | Mean Difference [%] (Fixed, 95% CI) | ‐12.3 [‐25.82, 1.22] |
| 6 B) Sensitivity analysis: random‐effects model; % difference for pruritus on VAS scale (0‐10) in UP and CP participants | 2 | 48 | Mean Difference [%] (Random, 95% CI) | ‐35.66 [‐84.28, 12.96] |
| 7 C) Risk for at least one adverse event per participant in UP and CP participants; cross‐over design | 3 | 116 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.07 [2.07, 8.00] |
| 8 C) Subgroup analysis by nature of pruritus; risk for at least one adverse event per participant; cross‐over design | 3 | 116 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.07 [2.07, 8.00] |
| 8.1 UP | 2 | 76 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.67 [1.91, 48.89] |
| 8.2 CP | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.67 [1.32, 5.39] |
| 9 C) Sensitivity analysis: random‐effects model; risk for at least one adverse event per participant; UP and CP patients; cross‐over design | 3 | 116 | Risk Ratio (M‐H, Random, 95% CI) | 3.85 [1.52, 9.76] |
1.6. Analysis.
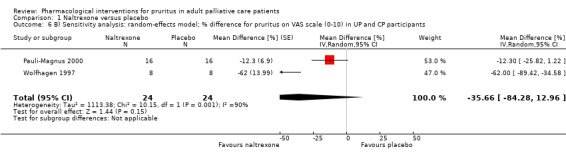
Comparison 1 Naltrexone versus placebo, Outcome 6 B) Sensitivity analysis: random‐effects model; % difference for pruritus on VAS scale (0‐10) in UP and CP participants.
Comparison 2. Nalfurafine versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 A) Pruritus on VAS scale (0‐10 cm) in UP participants; parallel‐group design; Wikström b: Wikström a (week 2) + period 1 Wikström b | 2 | 422 | Mean Difference (Fixed, 95% CI) | ‐0.95 [‐1.32, ‐0.58] |
| 2 A) Sensitivity analysis: random‐effects model; pruritus on VAS scale (0‐10 cm) in UP participants; parallel‐group design; Wikström b: Wikström a (week 2) + period 1 Wikström b | 2 | 422 | Mean Difference (Random, 95% CI) | ‐0.95 [‐1.32, ‐0.58] |
| 3 B) Risk for at least one adverse drug reaction (ADR) per participant in UP participants; parallel‐group and cross‐over design | 3 | 422 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.62 [1.15, 2.29] |
| 4 B) Sensitivity analysis: random‐effects model; risk for at least one adverse event per participant; UP participants; parallel‐group and cross‐over design | 3 | 422 | Risk Ratio (M‐H, Random, 95% CI) | 1.51 [1.09, 2.09] |
Comparison 3. Ondansetron versus placebo or standard medication.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 A) Pruritus on VAS scale (0‐10 cm) in UP and CP participants | 4 | 202 | Mean Difference (IV, Fixed, 95% CI) | ‐0.28 [‐0.46, ‐0.09] |
| 2 A) Subgroup analysis by nature of pruritus and study design; pruritus on VAS scale (0‐10 cm) | 4 | 202 | Mean Difference (IV, Fixed, 95% CI) | ‐0.28 [‐0.46, ‐0.09] |
| 2.1 UP patients; cross‐over design | 3 | 183 | Mean Difference (IV, Fixed, 95% CI) | ‐0.28 [‐0.46, ‐0.10] |
| 2.2 CP patients; parallel‐group | 1 | 19 | Mean Difference (IV, Fixed, 95% CI) | 0.16 [‐2.34, 2.66] |
| 3 A) Sensitivity analysis: random‐effects model; pruritus on VAS scale (0‐10 cm) in UP and CP participants | 4 | 202 | Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐0.71, 0.58] |
| 4 B) Risk for at least one adverse event per participant in UP and CP participants | 3 | 174 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.07 [0.87, 4.93] |
| 5 B) Subgroup analysis by nature of pruritus and study design; risk for at least one adverse event per participant | 3 | 174 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.07 [0.87, 4.93] |
| 5.1 UP; cross‐over design | 2 | 155 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.53 [0.97, 58.51] |
| 5.2 CP; parallel‐group design | 1 | 19 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.34, 2.32] |
| 6 B) Sensitivity analysis: random‐effects model; risk for at least one adverse event per participant in UP and CP participants | 3 | 174 | Risk Ratio (M‐H, Random, 95% CI) | 2.54 [0.38, 16.78] |
Comparison 4. Gabapentin versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 A) Pruritus on VAS scale (0‐10 cm) in UP participants | 2 | 118 | Mean Difference (Fixed, 95% CI) | ‐5.91 [‐6.87, ‐4.96] |
| 2 A) Subgroup analysis by study design; pruritus on VAS scale (0‐10 cm) in UP participants | 2 | 118 | Mean Difference (Fixed, 95% CI) | ‐5.91 [‐6.87, ‐4.96] |
| 2.1 Cross‐over design | 1 | 50 | Mean Difference (Fixed, 95% CI) | ‐6.4 [‐7.64, ‐5.16] |
| 2.2 Parallel group‐design | 1 | 68 | Mean Difference (Fixed, 95% CI) | ‐5.2 [‐6.70, ‐3.70] |
| 3 A) Sensitivity analysis: random‐effects model; pruritus on VAS scale (0‐10 cm); UP participants | 2 | 118 | Mean Difference (Random, 95% CI) | ‐5.88 [‐7.04, ‐4.71] |
Comparison 5. Rifampicin versus placebo or standard medication.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 A) Pruritus on VAS scale (0‐100 mm) in CP participants; cross‐over design | 2 | 42 | Mean Difference (Fixed, 95% CI) | ‐24.64 [‐31.08, ‐18.21] |
| 2 A) Sensitivity analysis: random‐effects model; pruritus on VAS scale (0‐100 mm) in CP participants; cross‐over design | 2 | 42 | Mean Difference (Random, 95% CI) | ‐42.00 [‐87.31, 3.31] |
| 3 B) Subgroup analysis by control; SMD: pruritus on different scales; CP participants; cross‐over design | 3 | 81 | Std. Mean Difference (Fixed, 95% CI) | ‐1.73 [‐2.45, ‐1.02] |
| 3.1 Rifampin/rifampicin versus phenobarbitone on 4‐point scale (0‐3) | 1 | 39 | Std. Mean Difference (Fixed, 95% CI) | ‐1.43 [‐2.39, ‐0.47] |
| 3.2 Rifampin/rifampicin versus placebo on VAS scale (0‐100) (Podesta: only first period)) | 2 | 42 | Std. Mean Difference (Fixed, 95% CI) | ‐2.11 [‐3.17, ‐1.04] |
| 4 B) Sensitivity analysis: random‐effects model; subgroup analysis by control; SMD: pruritus on different scales; CP patients; cross‐over design | 3 | 81 | Std. Mean Difference (Random, 95% CI) | ‐1.84 [‐2.82, ‐0.87] |
| 4.1 Rifampin/rifampicin versus phenobarbitone on 4‐point scale (0‐3) | 1 | 39 | Std. Mean Difference (Random, 95% CI) | ‐1.43 [‐2.39, ‐0.47] |
| 4.2 Rifampin/rifampicin versus placebo on VAS scale (0‐100) (Podesta: only first period)) | 2 | 42 | Std. Mean Difference (Random, 95% CI) | ‐2.25 [‐3.99, ‐0.52] |
Comparison 6. Flumecinol versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 A) Pruritus: significant improvement (yes/no); CP participants; parallel group design | 2 | 69 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.89 [1.05, 3.39] |
| 2 A) Subgroup analysis by dosage; pruritus: significant improvement (yes/no); CP participants; parallel‐group design | 2 | 69 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.89 [1.05, 3.39] |
| 2.1 Oral flumecinol 600 mg weekly for three weeks | 1 | 50 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.41 [0.77, 2.59] |
| 2.2 Oral flumecinol 300 mg daily for three weeks | 1 | 19 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.3 [0.95, 41.78] |
| 3 A) Sensitivity analysis: random‐effects model; pruritus: significant improvement (yes/no); CP participants; parallel‐group design | 2 | 69 | Risk Ratio (M‐H, Random, 95% CI) | 2.32 [0.54, 10.10] |
Comparison 7. Cromolyn sodium (CS) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 A) Pruritus on VAS scale (0‐10 cm; values from Feily (2012) multiplied by factor 2); UP participants; parallel‐group design | 2 | 100 | Mean Difference (IV, Fixed, 95% CI) | ‐2.94 [‐4.04, ‐1.83] |
| 2 A) Subgroup analysis by route of administration; values from Feily (2012) multiplied by factor 2); UP participants; parallel‐group design | 2 | 100 | Mean Difference (IV, Fixed, 95% CI) | ‐2.94 [‐4.04, ‐1.83] |
| 2.1 Oral CS versus placebo | 1 | 40 | Mean Difference (IV, Fixed, 95% CI) | ‐4.70 [‐6.57, ‐2.83] |
| 2.2 Topical CS versus placebo | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐2.0 [‐3.37, ‐0.63] |
| 3 A) Sensitivity analysis: random‐effects model; values from Feily (2012) multiplied by factor 2; UP participants; parallel‐group design | 2 | 100 | Mean Difference (IV, Random, 95% CI) | ‐3.27 [‐5.91, ‐0.63] |
| 4 B) Risk for at least one adverse event per participant; UP participants; parallel‐group design | 2 | 122 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.40, 3.08] |
| 5 B) Subgroup analysis by route of administration; risk for at least one adverse event per participant; UP participants; parallel‐group design | 2 | 122 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.40, 3.08] |
| 5.1 Oral CS versus placebo | 1 | 62 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.16 [0.02, 1.22] |
| 5.2 Topical CS versus placebo | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 13.0 [0.76, 220.96] |
| 6 B) Sensitivity analysis: random‐effects model; risk for at least one adverse event per participant; UP participants; parallel‐group design | 2 | 122 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [0.02, 106.52] |
Comparison 8. Topical capsaicin versus vehicle.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 A) Pruritus on different scales; SMD in UP participants; cross‐over design (Cho: 1. iPTH=<35pg/ml and 2. iPTH>35pg/ml) | 2 | 112 | Std. Mean Difference (Fixed, 95% CI) | ‐1.02 [‐1.35, ‐0.68] |
| 2 A) Subgroup analysis by pruritus scales; SMD; UP participants; cross‐over design (Cho: 1. iPTH=<35pg/ml and 2. iPTH>35pg/ml) | 2 | 112 | Std. Mean Difference (Fixed, 95% CI) | ‐1.02 [‐1.35, ‐0.68] |
| 2.1 Topical capsaicin versus placebo on 4‐point scale (1‐4) | 1 | 44 | Std. Mean Difference (Fixed, 95% CI) | ‐1.04 [‐1.58, ‐0.50] |
| 2.2 Topical capsaicin versus placebo on Duo scale (0‐30) | 1 | 68 | Std. Mean Difference (Fixed, 95% CI) | ‐1.00 [‐1.43, ‐0.58] |
| 3 A) Sensitivity analysis: random‐effects model; pruritus on different scales; UP participants; cross‐over design (Cho: 1. iPTH=<35pg/ml and 2. iPTH>35pg/ml) | 2 | 112 | Std. Mean Difference (Random, 95% CI) | ‐1.06 [‐1.55, ‐0.57] |
| 4 B) Risk for at least one adverse event per participant; UP participants, cross‐over design | 3 | 116 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.64 [2.05, 10.51] |
| 5 B) Sensitivity analysis: random‐effects model; risk for at least one adverse event per participant; UP participants; cross‐over design | 3 | 116 | Risk Ratio (M‐H, Random, 95% CI) | 3.69 [1.17, 11.67] |
Comparison 9. Zinc sulphate versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 A) Pruritus on different scales; SMD in UP participants; parallel‐group design | 2 | 76 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.13 [‐0.58, 0.32] |
| 2 A) Sensitivity analysis: random‐effects model; Pruritus on different scales; SMD in UP participants; parallel‐group design | 2 | 76 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.13 [‐0.58, 0.32] |
Characteristics of studies
Characteristics of included studies [author‐defined order]
Amirkhanlou 2016.
| Methods | RCT Comparative trial Parallel‐group design |
|
| Participants | Pruritus: UP Description: UP participants treated with haemodialysis Number of participants randomised: 52 Number of participants evaluable: 52
Withdrawals/dropouts: 0 Reason for dropout: NA Age (mean ± SD): gabapentin (group G): 60.2 years ± 7.4, ketotifen (group K): 57.6 years ± 6.2 Sex (male/female): gabapentin: 12 (46.2%)/14 (53.8%); ketotifen: 13 (50%)/13 (50%) Underlying disease(s): ESRD Participant pool: single‐centre Setting: inpatient Haemodialysis: similar in frequency and method (maximum duration and frequency of haemodialysis) Baseline pruritus assessment: no Duration/severity of pruritus: NA Baseline parameters: NA |
|
| Interventions |
Additional medication: NA Route of administration: oral Duration of treatment: 2 weeks Follow‐up: no information |
|
| Outcomes | Clinical response: complete response (no itching or minimal itching after treatment), partial response (mild or moderate severity of itching after treatment) and no response (severe pruritus after treatment) Adverse events Pruritus severity: Clinical response to treatment: (1) Complete response, (2) Partial response and (3) No response |
|
| Notes | Not reported: before and at the end of study, authors determined pruritus severity based on Shiratori's severity scores (0 = no itching, 1 = minimal, 2 = mild, 3 = moderate and 4 = severe itching) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomly assigned" |
| Allocation concealment (selection bias) | Low risk | "The patients and drug distributors were not aware of the prescribed medications" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The patients and drug distributors were not aware of the prescribed medications" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No attrition |
| Selective reporting (reporting bias) | High risk | Pruritus severity were determined based on Shiratori's severity scores but not determined |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 52 |
| Other bias | Unclear risk | Trial was not registered |
Ashmore 2000.
| Methods | RCT Placebo‐controlled Cross‐over design |
|
| Participants | Pruritus: UP Description: participants on haemodialysis with ESRD and pruritus Number of participants randomised: 19 Number of participants evaluable: 16 Withdrawals/dropouts: 3 Reason for dropout: non‐compliance Age (years): range 28‐77 (mean 60) for the participants completing the study Sex (male/female): 11/8; participants completing the study: 10/6 Underlying disease(s): chronic renal failure of unknown cause (n = 3), hypertensive nephrosclerosis (n = 3), immunoglobulin A nephropathy (n = 2), nephrotic syndrome (n = 2), connective tissue disease (n = 1), diabetic nephropathy (n = 1), Henoch‐Schönlein purpura (n = 1), obstructive uropathy (n = 1), systemic lupus erythaematosus (n = 1), adult polycystic kidney disease (n = 1) Participant pool: single‐centre Setting: inpatient Haemodialysis: median of 20 months (1 to 53 months) Duration/severity of pruritus: no information Baseline parameters: Symptom score (measured by antihistamine escape medication):
Pruritus score (measured by VAS 10 cm):
|
|
| Interventions |
Additional medication: antihistamines as escape medication chlorpheniramine (n = 9), terfenadine (n = 4), hydroxyzine (n = 3), iron supplements (n = 16), erythropoietin (n = 15), crotamiton cream (n = 9), H2‐receptor antagonist (n = 5) Route of administration: oral Duration of treatment: 2 weeks: washout 1 (day 1‐7) ‐ ondansetron/placebo (8‐21) ‐ washout 2 (22‐28) ‐ cross‐over (29‐42) Follow‐up: no information |
|
| Outcomes | Symptom relief (measured by antihistamine escape medication) Pruritus relief (measured by VAS 10 cm) Additional outcomes: serum calcium, phosphate, magnesium, urea, creatinine levels, bilirubin, alanine transaminase, alkaline phosphatase, haemoglobin, ferritin, parathyroid hormone |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "We randomly assigned 16 haemodialysis patients . . ." Participants were randomised to receive active drug or placebo . . ." Method not stated |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Participants were randomised . . . in a double‐blind crossover study." Unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data, (3 participants withdrawn due to protocol violation), then PP analysis; no intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | No indication |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 19 |
| Other bias | Unclear risk | Partly sponsored by Glaxo Smith Kline |
Bachs 1989.
| Methods | RCT Comparative trial Cross‐over design |
|
| Participants | Pruritus: CP Description: participants with clinical, biochemical, immunological, and histological features of primary biliary cirrhosis (PBC) and pruritus Number of participants randomised: 22 Number of participants evaluable: 17
Withdrawals/dropouts:
Reason for dropout:
Age (mean ± SD): 49.7 years ± 8.4 Sex (male/female): 0/22 Underlying disease(s): PBC Participant pool: no information Setting: outpatient Haemodialysis: NA Baseline pruritus assessment: yes Duration/severity of pruritus: no information Baseline parameters: Pruritus score (measured by a 4‐point scale; 0=no itching, 3=continuous pruritus):
|
|
| Interventions |
Additional medication: stopped 1 month prior the study Route of administration: oral Duration of treatment: 2 weeks (2 weeks rifampicin/phenobarbitone ‐ 30 days washout ‐ 2 weeks cross‐over) Follow‐up: no information |
|
| Outcomes | Pruritus assessment: 4‐point scale Adverse events Additional outcomes: fasting serum concentrations of bile acids, alkaline phosphatase, and gamma‐glutamyl‐transpeptidase decreased with rifampicin but not with phenobarbitone |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The order of treatment was randomised." |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No information provided, probably no blinding |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No information provided, probably no blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data; unclear if intention‐to‐treat analysis was used |
| Selective reporting (reporting bias) | Low risk | No indication of selective reporting although no results on cholesterol given |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 22 |
| Other bias | Low risk | No indication |
Bergasa 2006.
| Methods | RCT Placebo‐controlled Parallel‐group design |
|
| Participants | Pruritus: CP Description: participants with cholestasis Number of participants randomised: 16 Number of participants evaluable: 13
Number of participants enrolled: 15 (1 withdrawal prior to randomisation)
Withdrawals/dropouts: 3
Reason for dropout:
Age (years): median: 49 (range 44 to 63) Sex (male/female): 0/16 Underlying disease(s): PBC (n = 9), chronic liver disease secondary to infection with hepatitis C (n = 6), PSC (n = 1) Participant pool: single‐centre Setting: inpatient Haemodialysis: NA Duration/severity of pruritus:1‐12 years, except one participant for whom it was 4 months; antipruritic drugs had not provided satisfactory relief Baseline parameters: no information |
|
| Interventions |
Additional medication: Antipruritic drugs were discontinued 5 days before collecting baseline data. Participants who took antihistamines to sleep were kept on those doses. Route of administration: oral Duration of treatment: 4 weeks Follow‐up: no information |
|
| Outcomes | Pruritus assessment:
Adverse events Additional outcomes:
|
|
| Notes | Gabapentin was discontinued in 5 participants during the study and 2 more after completing the study; 2 participants took gabapentin after completing treatment with placebo;1 dropped because of side effects. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "[R]andomised to receive the placebo or the active drug according to a randomisation code generated and kept at the research pharmacy." |
| Allocation concealment (selection bias) | Low risk | " [A]ccording to a randomisation code generated and kept at the research pharmacy." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The study was a double‐blind . . ." Unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Gabapentin was discontinued in 5 participants; unclear if used intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | No indication |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 16 |
| Other bias | Unclear risk | Washout period only 5 days; co‐medication with antihistamines |
Borgeat 1993.
| Methods | RCT Placebo‐controlled Cross‐over design |
|
| Participants | Pruritus: CP Description: participants with cholestasis Number of participants randomised: 12 Number of participants evaluable: 10 Withdrawals/dropouts: 2 Reason for exclusion: history of skin disease associated with pruritus Age (years): 21‐79 Sex (male/female): 4/6 Underlying disease(s): pancreatic neoplasia (n = 3), cholangitis (n = 2), PBC (n = 2), hepatic metastasis (n = 2), bile duct neoplasia (n = 1) Participant pool: no information Setting: no information Haemodialysis: NA Baseline pruritus assessment: no information Duration/severity of pruritus: no information Baseline parameters: no information |
|
| Interventions |
Additional medication: no information Route of administration: intravenously Duration of treatment: 2 days (2 days propofol/placebo ‐ 2 days cross‐over) Follow‐up: no information |
|
| Outcomes | Pruritus assessment: verbal rating score (0 to 10 cm) Adverse events |
|
| Notes | Presentation of study results is inappropriate. Study period is very short, and the sample size is very small (N =10). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Medication was blinded and randomised by our pharmacy, which delivered a set of four coded vials per patient of either propofol or Intralipid" |
| Allocation concealment (selection bias) | Unclear risk | "[Pharmacy] delivered a set of four coded vials per patient" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Medication was blinded and randomised by our pharmacy" Double‐blind; unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No information on dropouts; no information on response to placebo |
| Selective reporting (reporting bias) | Unclear risk | Inclusion and exclusion criteria inadequately described; no data for hallucinations, mood changes, haemodynamic values |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 12 |
| Other bias | Unclear risk | Poor additional information; small number of participants; very short study period; only two doses of propofol/placebo for each participant; assessment of compliance not stated |
Breneman 1992a.
| Methods | RCT Placebo‐controlled Study design unclear |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number of participants randomised: 7 Number of participants evaluable: 5 (4 completed the entire 6‐week trial; 1 additional withdrawal after 5 weeks of treatment) Withdrawals/dropouts: 2 + 1 Reason for dropout: worsening medical status (1 + 1), insufficient improvement (1) Age (years): 20‐78 Sex (male/female): 3/4 Underlying disease(s): no information Participant pool: no information Setting: inpatient Haemodialysis: at least 1 month Baseline pruritus assessment: yes Duration/severity of pruritus: no information on duration; moderate to severe Baseline parameters: Pruritus score (measured by 4‐point score; 1 = no itching, 4 = severe itching interfering with daily activities and/or sleep):
|
|
| Interventions |
Additional medication: no information Route of administration: topical (cream) Duration of treatment: 6 weeks Follow‐up: no information |
|
| Outcomes | Pruritus assessment: 4‐point score Adverse events |
|
| Notes | Study design is inappropriate. Participants were instructed to apply one cream solely on one arm and the cream only on the other arm; the risk that participants may have mixed up the creams is very high. Sample size was very small. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Therapies were assigned on a random basis" Method of randomisation not stated |
| Allocation concealment (selection bias) | Unclear risk | Not information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | ". . . in a double‐blinded fashion." Unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 2/7 participants not evaluated (1: insufficient improvement), 4/7 participants completed full trial, no intention‐to‐treat analysis |
| Selective reporting (reporting bias) | High risk | Unclear study design; missing participant characteristics |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 7 |
| Other bias | High risk | Very small number of participants included; only 4 of 7 participants completed the trial. Participants were instructed to apply one cream solely on one arm and the cream from the other tube specifically on the other arm; the risk that participants may have mixed up the creams is very high. Assessment of compliance not stated. |
Cho 1997.
| Methods | RCT Placebo‐controlled Cross‐over design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number of participants randomised: 22 Two subgroups: low intact PTH (parathyroid hormone) < 35 pg/mL (n = 10) and high intact PTH > 35 pg/mL (n = 12)
Number of participants evaluable: 22 Withdrawals/dropouts: 0 Reason for dropout: NA Age (mean ± SD): 62 years ± 4 Sex (male/female): 14/8 Underlying disease(s):chronic glomerulonephritis (n = 8), chronic intestinal nephritis (n = 6), ESRD (n = 3), chronic pyelonephritis (n = 2), nephrosclerosis (n = 2), lupus nephritis (n = 1) Participant pool: no information Setting: inpatient Haemodialysis: average duration of 63 ± 14 months; 3x4−4.5 h/week Baseline pruritus assessment: yes Duration/severity: no information Baseline parameters: Pruritus score (measured by 4‐point score; 1 if no itching, 2, 3, or 4 if mild, moderate, or severe, respectively):
|
|
| Interventions |
Additional medication: ongoing medications were continued without alterations in dosage; topical agents were discontinued at least 2 weeks prior the study. Route of administration: topical (cream) Duration of treatment: 4 weeks (4 weeks capsaicin/vehicle ‐ 2 weeks washout ‐ 4 weeks cross‐over) Follow‐up: no information |
|
| Outcomes | Pruritus assessment: 4‐point score Adverse events Additional outcomes:
|
|
| Notes | No raw data given. Carryover effect because there was no wash‐out period between verum and vehicle phases. No between‐group comparisons are reported separately for the two phases of the cross‐over study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Treatment order was arranged from the computer‐generated random numbers . . ." |
| Allocation concealment (selection bias) | Low risk | "Treatment order was arranged from the computer‐generated random numbers by one of the co‐authors who did not participate in the observation." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Treatments with either capsaicin 0.025% cream or vehicle Base creams "were unknown by the observers and patients" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Treatments with either capsaicin 0.025% cream or placebo base cream were unknown by the observers and patients." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data, all 22 participants evaluated; unclear if used intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Unclear risk | No raw data or between‐group comparisons reported for the two phases of the cross‐over‐study |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 22 |
| Other bias | Unclear risk | Indication for carryover effect; self‐assessment of pruritus by participants |
De Marchi 1992.
| Methods | RCT Placebo‐controlled Cross‐over design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number of participants randomised: 20
Number of participants evaluable: 9 (from 10 patients because group 3 and 4 had no pruritus) Withdrawals/dropouts: 1 participant before cross‐over Reason for dropout: participant did not respond to erythropoietin Age (mean ± SD) :
Sex (male/female):
Underlying disease(s): no information Participant pool: no information Setting: inpatient Haemodialysis: 3 times weekly Baseline pruritus assessment: yes Duration/severity of pruritus:
Baseline parameters: Biochemical parameters (measured by blood samples): Group 1 (pruritus, erythropoietin ‐ placebo):
Group 2 (pruritus, placebo ‐ erythropoietin):
Group 3 (no pruritus, erythropoietin ‐ placebo):
Group 4 (no pruritus, placebo ‐ erythropoietin):
Pruritus score (measured by scoring system proposed by Duo, modified by Mettang, maximum score for a 24 h period: 40 (14 of 40 attributed to the night time period)): Group 1 (pruritus, erythropoietin ‐ placebo):
Group 2 (pruritus, placebo ‐ erythropoietin):
|
|
| Interventions |
Additional medication: no information Route of administration: intravenous Duration of treatment: 5 weeks (2 weeks baseline ‐ 5 weeks erythropoietin/placebo ‐ 5 weeks cross‐over) Follow‐up: baseline, weekly blood samples before and at the end of each 5‐week study |
|
| Outcomes | Pruritus assessment:
Additional outcomes: plasma histamine levels |
|
| Notes | Route of administration not clearly stated. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The patients were randomly assigned" |
| Allocation concealment (selection bias) | Low risk | Code broken only after completion |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind including outcome assessor: "a single investigator who was unaware of the treatment assignments evaluated all patients". Participants and personnel were blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessor was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | One withdrawal; unclear if used intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | No indication |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 20 |
| Other bias | Low risk | No conflicts of interest declared |
Duncan 1984.
| Methods | RCT Comparative trial Cross‐over design |
|
| Participants | Pruritus: CP Description: participants with cholestasis Number of participants randomised: 8 Number of participants evaluable: 8 Withdrawals/dropouts: 1 dropout in placebo‐group Reason for dropout: nausea and cutaneous burning Age: no information Sex (male/female): no information Underlying disease(s): primary biliary cirrhosis (7), sclerosing cholangitis (1) Participant pool: single‐centre Setting: outpatient Haemodialysis: NA Baseline pruritus assessment: no information Duration/severity of pruritus: no information Baseline parameters: no information |
|
| Interventions |
Additional medication: all antipruritic drugs were stopped at least 1 week prior to the study Route of administration: oral Duration of treatment: 2 weeks each (cross‐over) Follow‐up: no information |
|
| Outcomes | Pruritus assessment: 4‐point score Adverse events Additional outcomes: psychometric testing |
|
| Notes | Presentation of the results is inappropriate. Some participants stopped taking one of the trial medications, but there is no data on whether they were excluded from the analysis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The order of administration of the drugs was randomised, different for each patient, and concealed from the assessor" |
| Allocation concealment (selection bias) | Unclear risk | "Treatment was supplied in unlabelled bottles by the hospital pharmacy." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Single‐blind; unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Likely not blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not clear if all participants analysed; unclear if used intention‐to‐treat analysis; missing participant data like sex and age |
| Selective reporting (reporting bias) | Low risk | |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 8 |
| Other bias | Unclear risk | No washout period in between the treatments; no conflicts of interest declared |
Duque 2005.
| Methods | RCT Vehicle‐controlled Cross‐over design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number of participants randomised: 22
Number of participants evaluable: 20 Withdrawals/dropouts: vehicle (n = 2) Reason for dropout: 1 received a kidney transplant, 1 dropped out after two weeks because of lack of improvement Age (mean ± SD): 59 years ± 13.2 Sex (male/female): no information Underlying disease(s): no information Participant pool: multicentre (2 dialysis centres) Setting: outpatient Haemodialysis: at least 3 months Baseline pruritus assessment: yes Duration/severity of pruritus: no information on duration; frequent and severe itch that was resistant to conventional therapies, initial VAS between 3 and 10 Baseline parameters: Pruritus score (measured by VAS 10 cm):
|
|
| Interventions |
Additional medication: stopped 2‐4 weeks prior the study Route of administration: topical Duration of treatment: 4 weeks (4 weeks tacrolimus/placebo ‐ 4 weeks cross‐over) Follow‐up: baseline‐ 4 weeks: 3x/week ‐ 2 weeks after treatment completion |
|
| Outcomes | Pruritus assessment: VAS 10 cm Skin conditions: measured by 3‐point Lickert scale (0 = no skin signs, 3 = severe excoriations, scaliness, lichenification) |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study was randomised; method not stated |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind; unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 2 dropouts in the placebo group; unclear if used intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Unclear risk | Pre‐specified outcomes not mentioned |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 22 |
| Other bias | Low risk | Conflicts of interest mentioned but negative study |
Feily 2012.
| Methods | RCT Vehicle‐controlled Parallel‐group design |
|
| Participants | Pruritus: UP Description: participants with ESRD who were treated with haemodialysis Number of participants randomised: 60 Number of participants evaluable: 60
Withdrawals/dropouts: 0 Reason for dropout: NA Age (mean ± SD): 53 years ± 11.4 Sex (male/female): 38 (63%)/22 (37%) Underlying disease(s): ESRD Participant pool: single‐centre Setting: inpatient Haemodialysis: 3 times per week Baseline pruritus assessment: yes Duration/severity of pruritus: for at least 6 weeks without any systemic or topical treatment for the pruritus Baseline parameters: mean VAS ± SD:
|
|
| Interventions |
Additional medication: all other anti‐pruritus treatments were prohibited during the study; other routine medications were allowed Route of administration: topical Duration of treatment: 4 weeks Follow‐up: no information |
|
| Outcomes | Pruritus assessment: VAS (0‐5, 0: no pruritus and 5: the worst pruritus) Adverse events |
|
| Notes | Results in abstract and full text (e.g. Table 1) are conflicting; we worked with results stated in the abstract. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "[S]imple random table" |
| Allocation concealment (selection bias) | Unclear risk | "[P]atients were randomly allocated to one of the two arms of the study" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The medications used were not revealed to their physicians" "A similar tube was used to store CS 4% to make both creams to look physically identical" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The placebo was formulated by a pharmacist to have a similar base with the drug but not containing the active ingredient and stored in a tube without any labelling. A similar tube was used to store CS 4% to make both creams to look physically identical" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts; unclear if intention‐to‐treat analysis was used |
| Selective reporting (reporting bias) | Unclear risk | No indication of selective reporting; no protocol or registration number |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 60 |
| Other bias | High risk | Results in abstract and full text (e.g. Table 1) are conflicting |
Ghanei 2012.
| Methods | RCT Placebo‐controlled Cross‐over design |
|
| Participants | Pruritus: UP Description: participants with ESRD who were under the intermittent haemodialysis Number of participants randomised: 22 Number of participants evaluable: 22 Withdrawals/dropouts: 0 Reason for dropout: NA Age (mean ± SD): omega‐3‐placebo: 59.90 years ± 14.82, placebo‐omega‐3: 53.09±13.08 Sex (male/female): omega‐3‐placebo: 72%/28%, placebo‐omega‐3: 54%/46% Underlying disease(s): ESRD Participant pool: multicentre Setting: inpatient Haemodialysis (mean ± SD): omega‐3‐placebo: 3.81 ± 2.04, placebo‐omega‐3: 5.09 ± 4.88 Baseline pruritus assessment: yes Duration/severity of pruritus: for over 3 months with no response to antipruritic drugs Baseline parameters: mean (95% CI):
|
|
| Interventions |
Additional medication: All other anti‐pruritus treatments were discontinued 1 week before the study Route of administration: oral Duration of treatment: 20 days (20 days omega‐3/placebo ‐ 14 days washout ‐ 20 days cross‐over) Follow‐up: no information |
|
| Outcomes | Pruritus assessment: Duo Pruritus Score (0‐45) Adverse events Additional outcomes: KT/V index, blood pressure, cholesterol, triglyceride, haemoglobin |
|
| Notes | No protocol or registration number | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | "Patients were divided into two groups randomly by alternation method" |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Fish oil and placebo capsules with the same shape and volume" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "The pruritus assessment was carried out throughout the study by the same person at the start, during, and at the end of the study." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts; unclear if intention‐to‐treat analysis was used |
| Selective reporting (reporting bias) | Unclear risk | No indication of selective reporting; no protocol or registration number |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 22 |
| Other bias | Low risk | Small sample size |
Ghent 1988.
| Methods | RCT Placebo‐controlled Cross‐over design |
|
| Participants | Pruritus: CP Description: participants with PBC Number of participants randomised: 9
Number of participants evaluable: 9 Withdrawals/dropouts: 0 Reason for dropout: NA Age (years): 45‐64 Sex (male/female): 1/8 Underlying disease(s): primary biliary cirrhosis Participant pool: single‐centre Setting: outpatient Haemodialysis: NA Baseline pruritus assessment: 1 week baseline record of the pruritus VAS was obtained for 5 subjects prior to the study Duration/severity of pruritus: persistent pruritus Baseline parameters: no information |
|
| Interventions |
Additional medication: cholestyramine (n = 5) Route of administration: oral Duration of treatment: 2 weeks (2 weeks washout ‐ 2 weeks cross‐over) Follow‐up: no information |
|
| Outcomes | Pruritus assessment: VAS (0‐100 mm) Adverse events: none observed Additional outcomes: urine analysis, blood count, serum bilirubin, creatinine, alanine transaminases, aspartate transaminases, alkaline phosphatases, fasting total serum bile acids |
|
| Notes | Presentation of study results is inappropriate | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The order of treatment was random . . .." Method of randomisation not stated |
| Allocation concealment (selection bias) | Low risk | "The coding of the medication order was done by an independent research pharmacist." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ". . . in a double‐blind manner." Unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | All completed and analysed; unclear if used intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | No indication of selective reporting |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 9 |
| Other bias | Unclear risk | Small number of participants |
Gunal 2004.
| Methods | RCT Placebo‐controlled Cross‐over design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number of participants randomised: 25 Number of participants evaluable: 25 Withdrawals/dropouts: 0 Reason for dropout: NA Age (mean ± SD): 55 ± 11, range 32‐77 years, Sex (male/female): 14/11 Underlying disease(s): ESRD, diabetes (n = 8) Participant pool: single‐centre Setting: inpatient Haemodialysis: duration of 42 ± 33 months Baseline pruritus assessment: yes Duration/severity of pruritus: duration > 8 weeks; not relieved by antihistamines, nicergoline, moisturizers Baseline parameters: Pruritus score (measured by VAS 10 cm): 8.4 (SD 0.94) |
|
| Interventions |
Additional medication: any medication with presumed antipruritic effects was discontinued 1 week before the study Route of administration: oral Duration of treatment: 4 weeks (4 weeks gabapentin/placebo ‐ 1 week washout ‐ 4 weeks cross‐over) Follow‐up: daily record of pruritus severity (VAS) |
|
| Outcomes | Pruritus assessment: VAS 10 cm Adverse events Additional outcomes: hematocrit, serum calcium, phosphate, albumin, parathyroid hormone levels |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study was randomised; method not stated |
| Allocation concealment (selection bias) | Unclear risk | Not information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind; unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data, all completed and analysed; unclear if intention‐to‐treat analysis was used |
| Selective reporting (reporting bias) | Unclear risk | No exclusion criteria mentioned; only means of participant groups given; poor raw data given. Pruritus was scored only once a day and the scores were only subjective indications of the severity of itching; number and details of adverse events were not given. |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 25 |
| Other bias | Low risk | No conflict of interests declared |
Kuiper 2010.
| Methods | RCT Placebo‐controlled Parallel‐group design |
|
| Participants | Pruritus: CP Description: participants with cholestasis Number of participants randomised: 38 Number of participants evaluable: 35
Withdrawals/dropouts: 3 Reason for dropout: 1 participant withdrew after randomisation but before treatment, 1 participant stopped the intake of naltrexone during the trial, 1 participant was unable to fill out the questionnaires Age (mean ± SD):
Sex (male/female):
Underlying disease(s):
Participant pool: multicentre Setting: outpatient Haemodialysis: NA Baseline pruritus assessment: yes Duration/severity of pruritus: symptoms present for a median period of 24 months; pruritus most severe in the evening and/or at night, scratch lesions present in 55% of cases Baseline parameters: no information |
|
| Interventions |
Additional medication: treatment with ursodeoxycholic acid was continued and participants were allowed to continue rifampicin and naltrexone at a stable dose; other antipruritic drugs were stopped 3 days prior the study. Route of administration: oral Duration of treatment: 21 days Follow‐up: no information |
|
| Outcomes | Pruritus assessment: VAS 10 cm Adverse events Quality of life |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Participants were assigned to one of the two arms according to a standard randomization schedule (1:1) in blocks of four and were stratified by trial center." |
| Allocation concealment (selection bias) | Low risk | "Randomization was centralized with opaque serial‐numbered envelopes prepared by the trial statistician." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Both participants and investigators were blinded." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Both participants and investigators were blinded." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "We included and randomized 38 patients, 35 of whom were analyzed because one patient withdrew from participation after randomization and before the start of treatment, one patient stopped the intake of naltrexone during the trial period, and one patient was unable to fill out the questionnaires." Per‐protocol analysis with 35 participants Modified intention‐to‐treat analysis with 36 participants (38 were included and randomised) |
| Selective reporting (reporting bias) | Low risk | No indication |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 38 |
| Other bias | Low risk | Broad information about participant characteristics; presentation of data partially unclear; assessment of compliance not stated |
Kumagai 2010.
| Methods | RCT Placebo‐controlled Parallel‐group design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number of participants randomised: 337
Number of participants evaluable: 329
Withdrawals/dropouts:
Reason for dropout:
Number lost to follow‐up:
Age (mean ± SD):
Sex (male/female):
Underlying disease(s): ESRD Participant pool: multicentre (73 centres) Setting: inpatient Haemodialysis: 3x/week Baseline pruritus assessment: yes Duration/severity of pruritus: no information on duration; pruritus resistant to currently available treatments (mean morning or evening VAS > 50 mm, daytime or night‐time VAS > 20 mm on more than 5 days during the 7 day pre‐observation period) Baseline parameters: Pruritus score (mean VAS value 100 mm in the pre‐observation period): (mean ± SD)
|
|
| Interventions |
Additional medication: opioids and phototherapy were prohibited; hypnotics, antidepressants, antipsychotics, antiepileptics and anxiolytics that were likely to affect itch were administered at a consistent dosage and via normal method of administration, as were the antipruritic drugs administered for basic therapy. Route of administration: oral Duration of treatment: 2 weeks (2 weeks pre‐observation ‐ 2 weeks nalfurafine 5µg/2.5µg/placebo ‐ 8 days post observation) Follow‐up: 8 days |
|
| Outcomes | Pruritus assessment: VAS 100 mm Adverse events |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | ". . . were randomized 1:1:1 to receive 5 µg, 2.5 µg nalfurafine or a placebo using a variable size permuted block design stratified by center." |
| Allocation concealment (selection bias) | Low risk | "a variable size permuted block design stratified by center" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind; unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data; intention‐to‐treat‐analysis |
| Selective reporting (reporting bias) | Low risk | No indication |
| Size of study (possible biases confounded by small size) | Unclear risk | Number of participants randomised: 337 |
| Other bias | Low risk | Broad information about participant characteristics; power calculation |
Legroux‐Crespel 2004.
| Methods | RCT Comparative trial Parallel‐group design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number of participants randomised: 52
Number of participants evaluable: approximately 42, number not clearly stated Withdrawals/dropouts: approximately 10/15, number not clearly stated Reason for dropout: adverse events Age: no information Sex (male/female): no information Underlying disease(s): no information Participant pool: multicentre Setting: inpatient Haemodialysis: 3x/week in sessions of 4.2h; mean duration of 82 months Baseline pruritus assessment: yes Duration/severity of pruritus: at least 1 month; substantial pruritus Baseline parameters: Pruritus score (measured by VAS 10 cm):
Sleep parameters (measured by VAS 10 cm):
|
|
| Interventions |
Additional medication: no information Route of administration: oral Duration of treatment: 2 weeks Follow‐up: day 0‐7‐14 |
|
| Outcomes | Pruritus assessment: VAS 10 cm Adverse events Additional outcomes: blood urea, creatinine, creatinine clearance, calcium, phosphate, parathyroid hormone, ASTA, ALAT, alkaline phosphatases, bilirubin, haemoglobin |
|
| Notes | Compliance assessed by collecting drug boxes at the end of the study | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "This was a randomized study (drawing of lots) . . ." |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding not reported; likely not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Likely not blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Missing endpoint data for some participants |
| Selective reporting (reporting bias) | High risk | Missing raw data; conflicting data |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 52 |
| Other bias | High risk | Conflicting data given Only 2 measurements of pruritus over the study period; most results for day 7 instead of day 14; number of participants and of withdrawals confusing; missing endpoint data |
Makhlough 2010.
| Methods | RCT Vehicle‐controlled Cross‐over design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number of participants randomised: 34
Number of participants evaluable: no information Withdrawals/dropouts: no information Reason for dropout: NA Age (mean ± SD): 57 years ± 18.6 Sex (male/female): 14/20 Underlying disease(s): hypertension (n = 14), diabetes mellitus (n = 12), glomerulonephritis (n = 1), urological problems (n = 1), unknown (n = 1) Participant pool: single‐centre Setting: inpatient Haemodialysis: mean duration of 25 months (SD 15) Baseline pruritus assessment: yes Duration/severity of pruritus: no information on duration; pruritus non‐responsive to common treatment options Baseline parameters: Pruritus score (measured by score by Duo): (mean ± SD)
|
|
| Interventions |
Additional medication: no information Route of administration: topical Duration of treatment: 4 weeks (2 weeks washout ‐ 4 weeks cross‐over) Follow‐up: no information |
|
| Outcomes | Pruritus assessment: score by Duo (0‐30) Adverse events Additional outcomes: parathyroid hormone, serum alkaline phosphatase level |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The patients were equally divided and randomly assigned by lottery into two groups" |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind; unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Unclear if all participants were evaluable; unclear if used intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Unclear risk | Number of evaluable participants not given; no raw data given |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 34 |
| Other bias | Unclear risk | Poor additional information (e.g. dermatological, psychological evaluation); no power calculation; assessment of compliance not stated |
Mapar 2015.
| Methods | RCT Placebo‐controlled Parallel‐group design |
|
| Participants | Pruritus: UP Description: participants with ESRD who were treated with haemodialysis Number of participants randomised: 40 Number of participants evaluable: 36
Withdrawals/dropouts: 4 Reason for dropout: zinc group, expired because of congestive heart failure (n = 1) and decreased blood sugar (n = 1); placebo: vomiting (n = 1), "did not continue the study for the itching improvement" (n = 1) Age (range): 23‐79 years Sex (men/women): 27 (67.5%)/13 (32.5%) Underlying disease(s): ESRD Participant pool: single‐centre Setting: inpatient Haemodialysis: average of 3 years; frequency: 2‐3 per week Baseline pruritus assessment: yes Duration/severity of pruritus: pruritus for more than 6 weeks Baseline parameters: Modified Duo Score (mean ± SD):
|
|
| Interventions |
Additional medication: no steroids or opiate analgesics, discontinuation if antipruritic agents Route of administration: oral Duration of treatment: 4 weeks Follow‐up: no information |
|
| Outcomes | Pruritus assessment: Modified Duo Score from 0 to 45, with higher scores indicating more severe symptoms; (based on severity, distribution and sleep disturbance of pruritus) Adverse events |
|
| Notes | Conclusion: "decrease and discontinuation of pruritus in hemodialytic patients" Intention‐to‐treat analysis |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Triple blind; not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Triple blind; not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 2 dropouts per group |
| Selective reporting (reporting bias) | Low risk | No indication when compared with Methods chapter |
| Size of study (possible biases confounded by small size) | High risk | 20 patients per group |
| Other bias | Unclear risk | No information about study registration |
Mayo 2007.
| Methods | RCT Placebo‐controlled Cross‐over design |
|
| Participants | Pruritus: CP Description: participants with cholestasis Number of participants randomised: 12 Number of participants evaluable: 12 Withdrawals/dropouts: 0 Reason for dropout: NA Age: no information Sex (male/female): 2/10 Underlying disease(s): PBC (n = 9), PSC (n = 2), drug‐induced (postnecrotic cirrhosis) (n = 1) Participant pool: single‐centre Setting: outpatient Haemodialysis: NA Baseline pruritus assessment: yes Duration/severity of pruritus: at least 3 months; no information on severity Baseline parameters: no information |
|
| Interventions |
Additional medication: concomitant ursodiol treatment was allowed if participants were on a stable dose; other antipruritic medications were stopped at least 2 weeks prior the study. Route of administration: oral Duration of treatment: 6 weeks (4 weeks washout ‐ 6 weeks cross‐over) Follow‐up: no information |
|
| Outcomes | Pruritus assessment: VAS 10 cm Adverse events Additional outcomes: pruritus course, duration, distribution, pruritus insomnia, tolerability, depression, study drug preference, scratching lesions |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Part B of the study consisted of subjects randomized to double‐blind treatment with either sertraline or placebo." Method not stated |
| Allocation concealment (selection bias) | Unclear risk | Not information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind; no information how blinding was managed and difficulty because of different doses; unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts mentioned; unclear whether intention‐to‐treat analysis was used |
| Selective reporting (reporting bias) | Low risk | No indication |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 12 |
| Other bias | Low risk | Assessment of compliance not stated; power calculation used |
Murphy 2003.
| Methods | RCT Placebo‐controlled Cross‐over design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number of participants randomised: 24
Number of participants evaluable: 17 (Group A: 10, Group B: 7) Withdrawals/dropouts: 7 Reason for dropout:
Age (years, median): 59 Sex (male/female): 20/4 Underlying disease(s): no information Participant pool: multicentre Setting: inpatient Haemodialysis: no information Baseline pruritus assessment: yes Duration/severity of pruritus: duration > 8 weeks; mean VAS at least 5/10 during baseline Baseline parameters: Pruritus score (measured by VAS 10 cm): (mean ± SD)
|
|
| Interventions |
Additional medication: no information Route of administration: oral Duration of treatment: 2 weeks (7 days baseline ‐ 2 weeks ondansetron/placebo ‐ 7 days washout ‐ 2 weeks cross‐over) Follow‐up: pruritus VAS 2x/d, collected weekly |
|
| Outcomes | Pruritus assessment: VAS 10 cm Adverse events |
|
| Notes | No raw data on the participants. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "On a random basis 14 patients were blindly allocated to the ondansetron‐placebo sequence and 10 to the placebo‐ondansetron sequence" Method not stated |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ". . . 14 patients were blindly allocated to the ondansetron‐placebo sequence and 10 to the placebo‐ondansetron sequence." Unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Dropouts and reasons for dropout addressed, per protocol analysis |
| Selective reporting (reporting bias) | Unclear risk | No information provided |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 24 |
| Other bias | Low risk | Power calculation was carried out before the study; per protocol analysis |
Naini 2007.
| Methods | RCT Placebo‐controlled Parallel‐group design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number of participants randomised: 34 Number of participants evaluable: 34 Withdrawals/dropouts: 0 Reason for dropout: NA Age (mean ± SD): 62 ± 10, range 43‐81 years Sex (male/female): 16/18 Underlying disease(s): no information Participant pool: single‐centre Setting: inpatient Haemodialysis: at least twice a week for at least 3 months Baseline pruritus assessment: yes Duration/severity of pruritus: itching > 8 weeks; unresponsive to antihistamines Baseline parameters: Mean pruritus score (measured by VAS 10 cm): (mean ± SD)
|
|
| Interventions |
Additional medication: no information Route of administration: oral Duration of treatment: 4 weeks Follow‐up: no information |
|
| Outcomes | Pruritus assessment: VAS 10 cm Adverse events Additional outcomes: haemoglobin, serum parathormone, serum phosphorus, liver enzymes, alkaline phosphatase, bilirubin |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The patients were randomly allocated to receive either gabapentin 400 mg or placebo." Method not stated |
| Allocation concealment (selection bias) | Unclear risk | Not information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blinding stated in the abstract, but not further mentioned; unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No missing outcome data; no information whether intention‐to‐treat analysis used |
| Selective reporting (reporting bias) | Unclear risk | Number of side effects not stated |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 34 |
| Other bias | Unclear risk | Number of participants of the verum and the placebo group not stated; no power calculation; assessment of compliance not stated |
Najafabadi 2012.
| Methods | RCT Placebo‐controlled Parallel‐group design |
|
| Participants | Pruritus: UP Description: ESRD participants under haemodialysis Number of participants randomised: 40 Number of participants evaluable: 40 Withdrawals/dropouts: 0 Reason for dropout: NA Age (mean ± SD): zinc sulphate group: 53.35 years ± 14.5, placebo group: 57.55 years ± 16.1 Sex (male/female): zinc sulphate group: 15 (75%)/5 (25%), placebo group: 14 (70%)/6 (30%) Underlying disease(s): ESRD (the cause of ESRD was determined to be: diabetes in 37.5%; hypertension in 17.5%; congenital kidney disease in 10%; glomerulonephritis in 7.5%; and other causes in 27.5%) Participant pool: multicentre Setting: no information Haemodialysis (months ± SD): Zinc sulphate group: 45.95 ± 28.8, placebo group: 52.9 ± 33.1; at least 1 month Baseline pruritus assessment: yes Duration/severity of pruritus: pruritis complaints for more than 8 weeks, not taking other oral or local anti‐pruritic drugs Baseline parameters: mean ± SD: (0 = no itching; 10 = worst pruritis)
|
|
| Interventions |
Additional medication: To reduce confounding variables, participants with co‐morbidities were advised on how to take the medications as to not interfere with the effects of zinc Route of administration: oral Duration of treatment: 8 weeks Follow‐up: at week 12 |
|
| Outcomes | Pruritus assessment: VAS (0‐10) Adverse events Additional outcomes: demographic data of patients, haemodialysis duration, cause of renal failure, pruritus score, skin examinations, possible side effects and extra laboratory tests |
|
| Notes | Authors' conclusion questionable: baseline differences; no group differences at all time points | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The patients were then randomly assigned into treatment and placebo groups." |
| Allocation concealment (selection bias) | Unclear risk | "The patients were then randomly assigned into treatment and placebo groups." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ". . . while the other group received a similar shaped and colored capsule which was a placebo" "Neither the patients nor the physicians had any knowledge of the group to which patients were assigned." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The patients were assigned codes, and at the end of the study the drug and placebo groups were determined by decoding." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts; unclear if intention‐to‐treat analysis was used |
| Selective reporting (reporting bias) | Unclear risk | No indication of selective reporting; no protocol or registration number |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 40 |
| Other bias | Unclear risk | Small baseline differences; primary outcome not stated |
Nakhaee 2015.
| Methods | RCT Comparative trial three‐armed trial Cross‐over design |
|
| Participants | Pruritus: UP Description: participants with ESRD who were treated with haemodialysis Number of participants randomised: 25 Number of participants evaluable: 23
Withdrawals/dropouts: 2 (vinegar solution group) Reason for dropout: 2 kidney transplantations Age (mean ± SD): 57.04 years ± 12.20 Sex (male/female): 17 (73.9%)/6 (26.1%) Underlying disease(s): ESRD Participant pool: single‐centre Setting: inpatient Haemodialysis (mean ± SD): duration: 3.55 ± 2.78; frequency (per week): 2.57 ± 0.51 Baseline pruritus assessment: yes Duration/severity of pruritus (mean ± SD): 5.19 years ± 4.85 Baseline parameters:
|
|
| Interventions |
Additional medication: NA Route of administration: intervention 1 and 2: topical; intervention 3: oral Duration of treatment: 2 weeks Follow‐up: no information |
|
| Outcomes | Clinical response to treatment: complete response, partial response and no response VAS: a 10 cm long line on which 0 referred to no pruritus and 10 showed the most severe pruritus the patient could imagine Adverse events Additional outcomes: frequency, surface percentage of total body surface, verbal descriptor, consequences |
|
| Notes | Washout: 72 hours "All the patients performed the interventions completely." |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Patients were assigned by random numbers to 3 groups |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding not possible due to type of interventions |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blinding not possible due to type of interventions |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 2 kidney transplantations |
| Selective reporting (reporting bias) | Low risk | No indication |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 23 |
| Other bias | Low risk | The study protocol was registered in the Iranian Registry of Clinical Trials (IRCT2013021912525N1); http://www.irct.ir/searchresult.php?id=12525&number=1 |
Nasrollahi 2007.
| Methods | RCT Placebo‐controlled Cross‐over design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number of participants randomised: 16
Number of participants evaluable: 14 Withdrawals/dropouts: 2 Reason for dropout:
Age: range 20‐85 years (mean: 65 for male participants, 63 for female participants) Sex (male/female): 10/6 Underlying disease(s): ESRD Participant pool: multicentre Setting: outpatient Haemodialysis: 3x/week Baseline pruritus assessment: yes Duration/severity of pruritus: persistent pruritus >3 months; at least 1 course of unsuccessful treatment Baseline parameters: no information |
|
| Interventions |
Additional medication: Erythropoietin, other antipruritic treatment options (14 participants were receiving antihistamines, naltrexone or doxepin) were discontinued 1 week prior the study. Route of administration: oral Duration of treatment: 20 days (20 days montelukast/placebo ‐ 14 days washout ‐ 20 days cross‐over) Follow‐up: no information |
|
| Outcomes | Pruritus assessment: score by Duo (0‐45) Adverse events Additional outcomes: serum levels of calcium, phosphorus, alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase, bilirubin, urea, creatinine, parathyroid hormone, haemoglobin |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The study was designed as a randomized, single‐blind, placebo‐controlled, crossover clinical trial." Method of randomisation not stated |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Single‐blind; unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Dropouts and reasons for dropout mentioned; unclear if used intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Unclear risk | Only score reduction in percentages; no raw data given |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 16 |
| Other bias | Low risk | No power calculation; no raw data given; per protocol analysis; assessment of compliance stated |
O'Donohue 2005.
| Methods | RCT Placebo‐controlled Parallel‐group design |
|
| Participants | Pruritus: CP Description: participants with cholestasis Number of participants randomised: 19
Number of participants evaluable: 18
Withdrawals/dropouts:
Reason for dropout: participant with cholestasis due to chronic rejection; postorthotopic liver transplantation required rescue antipruritic treatment (oral antihistamines) Age: range 27‐80 years (mean 55)
Sex (male/female):
Underlying disease(s): primary biliary sclerosis (PBC) (n = 17), cirrhosis because of hepatitis C (n = 1), chronic rejection after orthotopic liver transplantation for hepatitis C cirrhosis (n = 1) Participant pool: single‐centre Setting: combination of inpatient and outpatient setting Haemodialysis: NA Baseline pruritus assessment: yes Duration/severity of pruritus: no information on duration; resistant pruritus Baseline parameters: Pruritus score (measured by VAS 10 cm):
|
|
| Interventions |
Additional medication: all antipruritic medications were withdrawn at least 3 days before entry into the study; antipruritic medication before entry into the study was comparable in both groups (cholestyramine (n = 17), ursodeoxycholic acid (n = 12), antihistamines (n = 9), tamoxifen (n = 2), cyclosporin (n = 1), rifampicin (n = 1)). Route of administration:
Duration of treatment: 5 days (day 0: 24 h observation phase) Follow‐up: within 2 weeks of the last dose of study medication; adverse events documented and blood sample taken for routine hepatic and renal biochemical markers |
|
| Outcomes | Pruritus assessment: VAS 10 cm Adverse events Additional outcomes: serum albumin, alkaline phosphatase, serum bilirubin, prothrombin time |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Each patient was consecutively allocated an individual sequential treatment number that corresponded to one of two . . ." |
| Allocation concealment (selection bias) | Low risk | "Each patient was consecutively allocated an individual sequential treatment number that corresponded to one of two, identical in appearance, medication regimes. The study pharmacist held sealed envelopes containing the codes to the treatment regimes, so that the patient and all investigators were unaware which of the regimes was being administered." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "[T]he patient and all investigators were unaware which of the regimes was being administered." Participants and personnel were blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No information provided, but not likely |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropouts and reasons for dropout provided; unclear if used intention‐to‐treat‐analysis |
| Selective reporting (reporting bias) | Low risk | No indication |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 19 |
| Other bias | Low risk | Very transparent treatment regimen; exact instruction for pruritus rating; no carryover effects |
Omidian 2013.
| Methods | RCT Placebo‐controlled Parallel‐group design |
|
| Participants | Pruritus: UP Description: ESRD participants under haemodialysis Number of participants randomised: 50 Number of participants evaluable: 49 Withdrawals/dropouts: 1 Reason for dropout: no information Age (mean ± SD): 49.6 ± 12.7 years (range 18‐60) Sex (male/female): no information Underlying disease(s): ESRD Participant pool: single‐centre Setting: outpatient Haemodialysis: average dialysis time before treatment: 44 months; 3 times per week Baseline pruritus assessment: yes Duration/severity of pruritus: pruritis complaints for more than 8 weeks, not taking other oral or local anti‐pruritic drugs Baseline parameters:
|
|
| Interventions |
Additional medication: All other anti‐pruritus treatments were prohibited during the study; other routine medications were allowed Duration of treatment: 4 weeks Follow‐up: no information |
|
| Outcomes | Pruritus assessment: VAS (0‐5) [0: no pruritus and 5: the worst pruritus] Adverse events |
|
| Notes | Results and conclusion in abstract (e.g. Table 1) conflict with the full text, i.e. different SDs, and nicotinamide and placebo seem to be interchanged | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "[S]imple random table and the study patients were randomly allocated to one of the two arms of the study" |
| Allocation concealment (selection bias) | Unclear risk | "[S]imple random table and the study patients were randomly allocated to one of the two arms of the study" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The placebo was formulated by a pharmacist to have similar base with the drug but not containing the active ingredient" "The used medications were not revealed to the treating physicians" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The used medications were not revealed to the treating physicians" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 1 patient dropped out but a reason was not given |
| Selective reporting (reporting bias) | Unclear risk | No indication of selective reporting; no protocol or registration number |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 50 |
| Other bias | High risk | Results and conclusion in abstract (e.g. Table 1) conflict with the full text, i.e. different SDs, and nicotinamide and placebo seem to be interchanged |
Özaykan 2001.
| Methods | RCT Comparitive trial Parallel‐group design |
|
| Participants | Pruritus: UP Description: participants on haemodialysis Number of participants randomised: 20
Number of participants evaluable: 20 (group A: 10, group B: 10) Withdrawals/dropouts: 0 Reason for dropout: NA Age:
Sex (male/female):
Underlying disease(s): no information Participant pool: single‐centre Setting: no information Haemodialysis: no information Baseline pruritus assessment: yes Duration/severity of pruritus: > 8 weeks Baseline parameters: Pruritus scoring system proposed by Duo and modified by Mettang (mean ± SD)
|
|
| Interventions |
Additional medication: antipruritic medication was discontinued 2 weeks prior the study Route of administration: oral Duration of treatment: 30 days |
|
| Outcomes | Pruritus assessment: Duo scale modified by Mettang 1990 (0‐48) Adverse events |
|
| Notes | Article in Turkish | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised; method not stated |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No missing outcome data; not reported if intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Unclear risk | Only data for the treatment groups given; no data on single participants |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 20 |
| Other bias | Low risk | No indication |
Pakfetrat 2014.
| Methods | RCT Placebo‐controlled Parallel‐group design |
|
| Participants | Pruritus: UP Description: participants on haemodialysis with a refractory pruritus Number of participants randomised: 100 ("One patient before the start of the study dropped out due to renal transplantation") Number of participants evaluable: 100 Withdrawals/dropouts: NA Reason for dropout: due to renal transplantation Age (mean ± SD): turmeric group: 55.6 years ± 14.7, placebo group: 51.0 years ± 16.6 Sex (male/female): turmeric group: 33 (66%)/17 (34%), placebo group: 27 (54%)/23 (46%) Underlying disease(s): ESRD Participant pool: single‐centre Setting: outpatients Haemodialysis in months (mean ± SD): turmeric group: 3.5 ± 2.6, placebo group: 6.4 ± 4.8 (P=0.001) Baseline pruritus assessment: yes Duration/severity of pruritus: for at least 6 weeks without any response to anti‐pruritic drugs Baseline parameters (mean ± SD): pruritus score (measured by Detailed Pruritus Score proposed by Duo, based on severity and distribution of pruritus as well as sleep disturbances caused by pruritus, max. 45 points)
|
|
| Interventions |
Additional medication: any medications with antipruritic effect were discontinued 1 week before the study Route of administration: oral Duration of treatment: 8 weeks Follow‐up: no information |
|
| Outcomes | Pruritus assessment: Detailed Pruritus Score proposed by Duo (max. 45) Adverse events Additional outcomes: levels of serum albumin (Alb), lipid profile, blood urea nitrogen (BUN), serum creatinine (cr), calcium (Ca), phosphorus (P), C‐reactive protein (hs‐CRP) |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Our statistical consultant assigned by a table of random numbers a number 0 to 4 to AB block and 5–9 to BA block" |
| Allocation concealment (selection bias) | Low risk | "The allocation sequence was concealed from the researcher enrolling and assessing participants in sequentially numbered, opaque, sealed envelopes" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Clinical investigators, laboratory personnel, and patients were all masked to the treatment assignment." "All drugs and placebo tablets were similar in size, shape, weight and colour." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Clinical investigators, laboratory personnel, and patients were all masked to the treatment assignment." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "Based on repeated interviews during the study, the patients mentioned no minor or major complaints attributable to the use of turmeric" "One patient dropped out due to renal transplantation" |
| Selective reporting (reporting bias) | Low risk | "The trial was registered at clinicaltrials.gov (NCT01037595)." |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 100 |
| Other bias | Low risk | No baseline differences; detailed study description |
Pauli‐Magnus 2000.
| Methods | RCT Placebo‐controlled Cross‐over design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis HD (n = 18) and peritoneal dialysis (PD) (n = 5) Number of participants randomised: 23 Number of participants evaluable: 16 Withdrawals/dropouts: 7 Reason for dropout:
Age (range): 20‐85 years Sex (male/female): no information Underlying disease(s): no information Participant pool: multicentre Setting: inpatient Haemodialysis: 3x4‐5h/week, Kt/V>1.2, dialysis Peritoneal dialysis: weekly, Kt/V > 2, Hb > 10 g/L Baseline pruritus assessment: yes Duration/severity of pruritus: duration > 6 months; substantial pruritus: persistent, treatment‐resistant, impairing sleep/daytime activities, Baseline parameters: Pruritus score (measured by VAS 10 cm and Duo detailed score, range 0‐45; higher values = more pruritus):
|
|
| Interventions |
Additional medication: no information Route of administration: oral Duration of treatment: 4 weeks (4 weeks naltrexone/placebo ‐ 1 week washout ‐ 4 weeks cross‐over) Follow‐up: no information |
|
| Outcomes | Pruritus assessment: VAS (10 cm) and modified Duo score (comprising severity and distribution of pruritus and sleep disturbance) Adverse events Additional outcomes: plasma haemoglobin concentrations, serum concentrations of creatinine, urea, calcium, phosphate, alkaline phosphatase, bilirubin, transaminase, parathyroid hormone |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study was randomised; method not stated |
| Allocation concealment (selection bias) | Unclear risk | Not information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind; unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat analysis and per protocol analysis performed |
| Selective reporting (reporting bias) | Low risk | No indication |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 23 |
| Other bias | Low risk | Missing participant characteristics; sex and underlying disease not stated; no raw data given Assessment of compliance by collecting drug boxes at the end of each study period and taking blood samples for naltrexone measurement at randomly chosen time |
Pederson 1980.
| Methods | RCT Placebo‐controlled Cross‐over design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number of participants randomised: 20 Number of participants evaluable: 11 Withdrawals/dropouts:
Reason for dropout:
Age (years): 34‐72 (mean 53) Sex (male/female): 16/4 Underlying disease(s): no information Participant pool: no information Setting: inpatient Haemodialysis: duration of 3.5 months to 72 months Baseline pruritus assessment: not clear if conducted Duration/severity of pruritus: 1 to 72 months; no information regarding severity Baseline parameters: no information |
|
| Interventions |
Additional medication: no information Route of administration: oral Duration of treatment: 8 weeks (8 weeks charcoal/placebo ‐ 8 weeks cross‐over) Follow‐up: no information |
|
| Outcomes | Pruritus assessment: questionnaire as suggested by Lowrie and Ingham Additional outcomes:
|
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomly assigned . . ." Method not stated |
| Allocation concealment (selection bias) | Unclear risk | Treatments "administered orally in identical opaque capsules" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind; unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Dropouts and reasons for dropout provided; no intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Unclear risk | Missing participant characteristics |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 20 |
| Other bias | Unclear risk | Small sample size; high number of dropouts; no information on whether there was a washout period between the treatment periods |
Peer 1996.
| Methods | RCT Placebo‐controlled Cross‐over design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number of participants randomised: 15
Number of participants evaluable: 15 Withdrawals/dropouts: 0 Reason for dropout: NA Age (years): no information Sex (male/female): no information Underlying disease(s): no information Participant pool: no information Setting: inpatient Haemodialysis: NA Baseline pruritus assessment: yes Duration/severity of pruritus: persistent, treatment resistant pruritus Baseline parameters: Pruritus score (measured by VAS 10 cm):
Biochemical parameters:
|
|
| Interventions |
Additional medication: all antipruritic therapy was stopped 1 week before the study; erythropoietin for at least 3 months before the study. Route of administration: oral Duration of treatment: 7 days (7 days naltrexone/placebo ‐ 7 days cross‐over) Follow‐up: 5 participants continued treatment with the same dose of drug for 10 weeks with no pruritus. The other participants discontinued because of the high cost of naltrexone. |
|
| Outcomes | Pruritus assessment: VAS 10 cm Adverse events Additional outcomes: plasma, endorphin, histamine |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study was randomised; method not stated |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind; unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data; no information on intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | No indication |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 15 |
| Other bias | Low risk | No indication |
Podesta 1991a.
| Methods | RCT Placebo‐controlled Cross‐over design |
|
| Participants | Pruritus: CP Description: participants with cholestasis Number of participants randomised: 14 Number of participants evaluable: 14 Withdrawals/dropouts: 0 Reason for dropout: NA Age: range 32‐72 years (mean 43) Sex (male/female): 1/13 Underlying disease(s): PBC stage IV (n = 5), PBC stage III (n = 4), PBC stage II (n = 3), PBC stage I (n = 2) Participant pool: multicentre Setting: outpatient Haemodialysis: NA Baseline pruritus assessment: yes Duration/severity of pruritus: no information regarding duration; 9 participants were receiving cholestyramine with a poor or no response in 6 participants Baseline parameters: no information |
|
| Interventions |
Additional medication: participants stopped treatment 15 days before the study (washout period). Route of administration: oral Duration of treatment: 7 days ‐ 7 days wash‐out period ‐ cross‐over treatment for 7 days Follow‐up: no information |
|
| Outcomes | Pruritus assessment: VAS 100 mm Adverse events Additional outcomes: bilirubin, alkaline phosphatase, aspartate aminotransferase, alanine aminotransferase, anti‐mitochondrial antibody |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation made using a coin toss |
| Allocation concealment (selection bias) | Low risk | "Vials containing a one‐week‐supply of drug or placebo by an independent observer and randomization made with the toss of a coin" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Blinding of participants used but disclosed because of treatment effects |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data; no information on intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | No indication |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 14 |
| Other bias | Low risk | Assessment of compliance using pill count |
Pour‐Reza‐Gholi 2007.
| Methods | RCT Placebo‐controlled Cross‐over design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number of participants randomised: 24 Number of participants evaluable: 23 Withdrawals/dropouts: 1 Reason for dropout: drowsiness Age (mean ± SD): 48 ± 5.6, range: 35 to 65 Sex (male/female): 13/11 Underlying disease(s): ESRD Participant pool: single‐centre Setting: outpatient Haemodialysis: 3x/week, Kt/V> 1.2 Baseline pruritus assessment: no information Duration/severity of pruritus: no information Baseline parameters: no information |
|
| Interventions |
Additional medication: erythropoietin weekly (n = 10); others did not receive erythropoietin regularly Route of administration: oral Duration of treatment: 1 week (1 week doxepin/placebo ‐ 1 week washout ‐ 1 week cross‐over) Follow‐up: no information |
|
| Outcomes | Pruritus assessment: subjective outcome determined by asking the participants to describe their pruritus as completely improved, relatively improved, or remained unchanged/worsened Adverse events Additional outcomes: serum calcium levels, serum phosphate levels, serum intact parathyroid hormone, serum aluminium, serum magnesium, blood haemoglobin |
|
| Notes | No scales or score for rating of pruritus; subjective outcome report of participants | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "They were randomly assigned . . ." Method not stated |
| Allocation concealment (selection bias) | Low risk | "Doxepin was placed in another capsule in order to provide placebo capsules similar in shape, size and colour" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The patients and the physicians involving in their management were blind to the randomization." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The patients and the physicians involving in their management were blind to the randomization." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropouts and reasons for dropout provided; intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | No indication |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 24 |
| Other bias | Low risk | No scales or score for rating of pruritus; subjective outcome report of participants |
Shirazian 2013.
| Methods | RCT Placebo‐controlled Parallel‐group design |
|
| Participants | Pruritus: UP Description: participants with UP on haemodialysis Number of participants randomised: 50 Number of participants evaluable: 44 Withdrawals/dropouts: 6 Reason for dropout: ergocalciferol group: 1 relocated out of town, 1 renal transplantation, 2 withdrew consent; placebo group: 1 death, 1 withdrew consent Age (mean ± SD): ergocalciferol group: 66.1 years ± 14.7, placebo group: 66.2 years ± 13.7 Sex (male/female): ergocalciferol group: 15/10 (60%/40%), placebo group: 14/11 (56%/44%) Underlying disease(s): ESRD; comorbidities in ergocalciferol and placebo groups, respectively: diabetes (n = 11, 44%;, n = 11, 44%); hypertension (n = 20, 80%; n = 23, 92%); coronary artery disease (n = 10, 40%; n = 8, 32%); congestive heart failure (n = 6, 24%; n = 5, 20%; skin condition (n = 6, 24%; n = 4, 20%) Participant pool: single‐centre Setting: outpatient Haemodialysis (mean ± SD): ergocalciferol group: 27.3 months ± 19.6, placebo group: 43.6 months ± 27.5; at least 3 months Baseline pruritus assessment: yes Duration/severity of pruritus: no information/excessive pruritus Baseline parameters:
|
|
| Interventions |
Additional medication: no information Route of administration: oral Duration of treatment: 12 weeks Follow‐up: no information |
|
| Outcomes | Pruritus assessment: Pruritus Severity Questionnaire (0‐21): "Active itching received 5 points whereas itching affecting sleep or other activities in the past few days received 4 points. Itching that was perceived as mild received 1 point, moderate received 3 points, and severe received 4 points. Localised itching received 1 point, itching in most of the body received 2 points, and itching in all of the body received 3 points. Use of medications for itching received 5 points. A maximum pruritus score on the survey was 21 points." Adverse events Additional outcomes: calcium, phosphorus, PTH, and vitamin D levels |
|
| Notes | "From the 50 patients that were dispensed study medication, 6 patients (4 in the ergocalciferol group and 2 in the control group) did not complete all study visits. These patients were included in all analyses by intention to treat." "[A]cceptable compliance was present in 41 of 50 patients, or 82%" Absolute values were only shown in figure and numbers are not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "[U]sing simple randomization procedures (computer‐generated random numbers)" |
| Allocation concealment (selection bias) | Unclear risk | "Study subjects and investigators administering study surveys to the patient were blinded to randomization assignment" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "A research pharmacist prepackaged ergocalciferol and placebo tablets into opaque bottles." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "A research nurse, who did not participate in consent, pruritus surveys, or study analysis assigned patients to the appropriate pill bottle. The research nurse also dispensed the medication to the patient" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "These patients were included in all analyses by intention to treat" |
| Selective reporting (reporting bias) | Low risk | No indication; NCT01114672 ‐ however, number not stated in the publication |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 50 |
| Other bias | Low risk | No indication; "acceptable compliance was present in 41 of 50 patients, or 82%" |
Silva 1994.
| Methods | RCT Placebo‐controlled Cross‐over design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number of participants randomised: 29
Division into 3 subgroups: according to baseline scoring/proportion of responders analysed Number of participants evaluable: 18
Withdrawals/dropouts: 11
Reason for dropout:
Age (mean ± SD):
Sex (male/female):
Underlying disease(s): malignant nephrosclerosis (n = 11), chronic glomerulonephritis (n = 5), polycystic disease (n = 3), others (n = 10) Participant pool: no information Setting: inpatient Haemodialysis: 3x/week for over 6 months Baseline pruritus assessment: yes Duration/severity of pruritus: no information on duration; > 25% of the maximum score during baseline Baseline parameters: Pruritus score as percent of maximum score (measured by 4‐point score; 0 = absence of itching, 1 = not interfering with usual tasks, 2 = perturbing but not interrupting usual tasks, 3 = causing interruptions of usual tasks/sleep): (mean ± SE)
|
|
| Interventions |
Additional medication: phosphate binders (n = 17), vitamins (n = 12), antihypertensives (n = 10), calcitriol (n = 10), iron (n = 6), H2‐blockers (n = 3), EPO (n = 2) Route of administration: oral Duration of treatment: 1 week (1 week baseline ‐ 1 week thalidomide/placebo ‐ 1 week washout ‐ 1 week cross‐over) Follow‐up: pruritus score 3x/d; blood samples at beginning/end of each study period |
|
| Outcomes | Pruritus assessment: 4‐point score Adverse events Additional outcomes: complete blood count, plasma levels of calcium, phosphorus, magnesium, alkaline phosphatase, blood urea nitrogen |
|
| Notes | Response rate higher in participants with low baseline pruritus score; differentiation between responders and non‐responders | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ". . . they were randomly assigned . . ." Method not stated |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ". . . in a double‐blind fashion." Unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropouts and reasons for dropout provided; not reported if intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | No indication |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 29 |
| Other bias | Low risk | Residual effect identified and participants therefore excluded from cross‐over |
Silverberg 1977.
| Methods | RCT Placebo‐controlled Parallel‐group design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number of participants randomised: 10
Number of participants evaluable: 10 Withdrawals/dropouts: 0 Reason for dropout: NA Age (years): no information Sex (male/female): 10/0 Underlying disease(s): no information Participant pool: no information Setting: inpatient Haemodialysis: 3 x 3‐5 h/week Baseline pruritus assessment: yes Duration/severity of pruritus: long lasting; no information regarding severity Baseline parameters: Pruritus score (measured by 4‐point score; 0 = none, 3 = great):
|
|
| Interventions |
Additional medication: no information Route of administration: oral Duration of treatment: 4 weeks Follow‐up: no information |
|
| Outcomes | Pruritus assessment: 4‐point score Adverse events Additional outcomes: prothrombin time, blood urea, serum creatinine, sodium, potassium, chloride, bicarbonate, calcium, phosphate, alkaline phosphatase, albumin, cholesterol, triglyceride concentration |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The 10 patients were randomly assigned to two treatments . . .." Method not stated |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blinding stated in the abstract but not further mentioned; unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | Small number of participants; inclusion and exclusion criteria not mentioned; only means of 21 days before treatment and means of 28 days of treatment were compared |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 10 |
| Other bias | Unclear risk | Missing participant characteristics; assessment of compliance not stated |
Smith 1997a.
| Methods | RCT Comparitive trial Parallel‐group |
|
| Participants | Pruritus: HIV Description: participants with HIV‐1 Number of participants randomised: 40
Number of participants evaluable: 33
Withdrawals/dropouts: 7
Reason for dropout:
Age (years): no information Sex (male/female): no information Underlying disease(s): HIV‐1 Participant pool: no information Setting: no information Haemodialysis: NA Baseline pruritus assessment: yes Duration/severity of pruritus: no information Baseline parameters: Pruritus score (Baseline: measured by 4‐point score; 1 = periodic at night only, 4 = interferes with daily activities and sleep at night):
|
|
| Interventions |
Additional medication: no information Route of administration: oral, oral, oral, topical Duration of treatment: 4‐6 weeks Follow‐up: no information Number lost to follow‐up:
|
|
| Outcomes | Pruritus assessment: 4‐point score (1 = periodic at night only, 4 = interferes with daily activities and sleep at night) Adverse events Additional outcomes: skin lesions |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "[W]e randomly placed patients of four different forms of therapy for their pruritus" |
| Allocation concealment (selection bias) | Unclear risk | "Patients were assigned to one of three treatment groups and a control group based on entry into the study" |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No information |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Reasons for dropouts not clearly stated; per protocol analysis |
| Selective reporting (reporting bias) | Unclear risk | Inclusion and exclusion criteria inadequately described |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 40 |
| Other bias | High risk | Duration of treatment not clearly stated (overall changes in pruritus after 4‐6 weeks of treatment were graded) Missing participant characteristics; poor additional information; study design comparing 4 treatments to placebo is problematic; assessment of compliance by asking the participants the number of pills they had left and if they had received any refills if follow‐up was longer than 4 weeks |
Tarng 1996.
| Methods | RCT Vehicle‐controlled Cross‐over design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number of participants randomised: 19
Number of participants evaluable: 17 Withdrawals/dropouts:
Reason for dropout:
Age (range): 27‐85 years Sex (male/female): 13/6 Underlying disease(s): no information Participant pool: single‐centre Setting: inpatient Haemodialysis: mean duration of 71.4 months (range 4‐219); 3 x 4−4.5 h/week Baseline pruritus assessment: yes Duration/severity of pruritus (mean ± SD): 33.1 ± 39.3 months; 5 had moderate, 12 had severe pruritus Baseline parameters: Pruritus score (measured by a 4‐point score, 1 = no itching, 4 = severe itching disturbing daily life and/or sleep):
|
|
| Interventions |
Additional medication: all topical agents other than moisturisers were discontinued at least 2 weeks prior the study; any ongoing medications were continued Route of administration: topical Duration of treatment: 4 weeks (4 weeks capsaicin/placebo ‐ 2 weeks washout ‐ 4 weeks cross‐over) Follow‐up: 8 weeks (without treatment) |
|
| Outcomes | Pruritus assessment: 4‐point score (self‐assessment): 1 = no itching, 4 = severe itching disturbing daily life and/or sleep Adverse events Additional outcomes:
|
|
| Notes | Carryover effect up to 8 weeks after end of treatment | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Treatment order is block‐randomized with the use of computer‐generated random numbers." |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind; "to which both observers and patients were blind" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropouts and reasons for dropout provided; no intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Unclear risk | No separate data for phase 1 reported |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 19 |
| Other bias | Unclear risk | Poor additional information; not clear whether significant difference in itch improvement is within or between group; assessment of compliance not stated |
Terg 2002.
| Methods | RCT Placebo‐controlled Cross‐over design |
|
| Participants | Pruritus: CP Description: participants with cholestasis Number of participants randomised: 20
Number of participants evaluable: 18 Withdrawals/dropouts: 2 Reason for dropout: 1 because of clinical impairment due to progression of prior hepatocarcinoma, 1 because of nausea and vomiting Age: (mean ± SD)
Sex (male/female):
Underlying disease(s): PBC (n = 15), chronic hepatitis C (n = 2), PSC (n = 1), overlap syndrome (n = 1), cryptogenic cirrhosis (n = 1) Participant pool: 2 centres Setting: inpatient Haemodialysis: NA Baseline pruritus assessment: yes Duration/severity of pruritus: lasting 6 to 11 months; no information on severity Baseline parameter: Pruritus score (VAS 10 cm): (mean ± SD)
|
|
| Interventions |
Additional medication: All participants were instructed to continue with their previous medication throughout the study. Route of administration: oral Duration of treatment: 2 weeks (2 weeks naltrexone/placebo ‐ 1 week washout ‐ 2 weeks cross‐over) Follow‐up: no information |
|
| Outcomes | Pruritus assessment: VAS 10 cm Adverse events Additional outcomes: serum bilirubin, alkaline phosphatase, aspartate aminotransferase, alanine aminotransferase, prothrombin time, serum albumin, platelets, red and white cell count, serum urea, creatinine, sodium, potassium, γ‐glutamyltransferase |
|
| Notes | Additional open trial: 2 additional months naltrexone for participants with at least 50% pruritus decrease (9 participants included) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization was generated by tables with two random numbers for each patient. These were the numbers of the bottles containing medication or placebo" |
| Allocation concealment (selection bias) | Low risk | "Information was placed in sealed, opaque, and numbered envelopes." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "This study was double‐blind . . .. " Unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data; no information on intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | No indication |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 20 |
| Other bias | Low risk | Assessment of pruritus by counting the pills remaining in the box |
Turner 1994a.
| Methods | RCT Placebo‐controlled Parallel‐group design |
|
| Participants | Pruritus: CP Description: participants with cholestasis Number of participants randomised: 50
Number of participants evaluable: 50 Withdrawals/dropouts: 0 Reason for dropout: NA Age (years): no information Sex (male/female): 3/47 Underlying disease(s): primary biliary cirrhosis (n = 46), alcoholic liver disease (n = 2), autoimmune chronic active hepatitis (n = 1), cholestatic phase of hepatitis A (n = 1) Participant pool: no information Setting: outpatient Haemodialysis: NA Baseline pruritus assessment: yes Duration/severity of pruritus: no information Baseline parameters: Pruritus score (measured by VAS 100 mm), median (IQR):
Quality of life (measured by 100 mm scale), median (IQR):
|
|
| Interventions |
Additional medication: antipruritic treatment was stopped at least 1 week prior the study; if unable to stop, they continued medication at an unchanged dose. Cholestyramine was stopped in 6 of 9 actively‐treated and 7 of 8 placebo participants Route of administration: oral Duration of treatment: 3 weeks Follow‐up: no information |
|
| Outcomes | Pruritus assessment: VAS 100 mm Adverse events Additional outcomes:
|
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation not stated. |
| Allocation concealment (selection bias) | Unclear risk | "[D]ouble‐blindly allocated"; not precisely stated |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study was double‐blind, "without the questioner or the patient knowing the treatment arm" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | ". . . without the questioner or the patient knowing the treatment arm" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data; not reported if intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | No indication |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 50 |
| Other bias | Low risk | Poor additional information; imbalance male/female 3/47; assessment of compliance not stated |
Turner 1994b.
| Methods | RCT Placebo‐controlled Parallel‐group design |
|
| Participants | Pruritus: CP Description: participants with cholestasis Number of participants randomised: 19
Number of participants evaluable: 19 Withdrawals/dropouts: 0 Reason for dropout: NA Age (years): no information Sex (male/female): 0/19 Underlying disease(s): primary biliary cirrhosis (19) Participant pool: no information Setting: outpatient Haemodialysis: NA Baseline pruritus assessment: yes Duration/severity of pruritus: no information Baseline parameters: Pruritus relief (measured by VAS 100 mm), median (IQR):
Quality of life (measured by 100 mm scale), median (IQR):
|
|
| Interventions |
Additional medication: antipruritic treatment was stopped at least 1 week prior the study; if unable to stop they continued medication at an unchanged dose. Cholestyramine was stopped in 2 of 10 actively‐treated and 1 of 9 placebo participants but 1 continued using it. Route of administration: oral Duration of treatment: 3 weeks Follow‐up: no information |
|
| Outcomes | Pruritus assessment: VAS 100 mm Adverse events Additional outcomes:
|
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation not stated. |
| Allocation concealment (selection bias) | Unclear risk | "double‐blindly allocated"; not precisely stated |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind " . . . without the questioner or the patient knowing the treatment arm . . ." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | " ... without the questioner or the patient knowing the treatment arm ..." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data; no information on intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | No indication |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 19 |
| Other bias | Low risk | Poor additional information; assessment of compliance not stated |
Vessal 2010.
| Methods | RCT Placebo‐controlled Parallel‐group design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number total (randomised): 62 (+ 20 as negative control)
Number of participants evaluable: 40 (+19 as negative control)
Withdrawals/dropouts: 22 (+1 as negative control)
Reason for dropout:
Age (mean ± SD):
Sex (male/female):
Underlying disease(s): no information Participant pool: multicentre (2 centres) Setting: inpatient Haemodialysis: 4‐5 hours for 2‐3x per week Baseline pruritus assessment: yes Duration/severity of pruritus: pruritus that did not respond to treatment for at least 6 weeks Baseline parameters: Pruritus relief (measured by VAS 10 cm):
|
|
| Interventions |
Additional medication: antipruritic medication was discontinued 1 week prior the study. Route of administration: oral (capsule was dissolved in a minimal amount of water and administered half an hour before each meal) Duration of treatment: 8 weeks Follow‐up: no information |
|
| Outcomes | Pruritus assessment: VAS 10 cm Adverse events Additional outcomes: haemoglobin, calcium, phosphorus, albumin, ferritin, parathyroid hormone, white blood cells, serum tryptase, platelet, hematocrit |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomly assigned into two groups using the stratified randomization method where the prognostic factor was the gender variable." |
| Allocation concealment (selection bias) | Low risk | "Drug packages were prepared by the principal investigator . . . Both the participants and the investigator that administered the interventions and assessed the outcomes were blinded to group assignment. Code breaking was performed at the end of data analysis." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind; participants and personnel blinded "Both the participants and the investigator that administered the interventions and assessed the outcomes were blinded to group assignment. Code breaking was performed at the end of data analysis." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessor blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropouts and reasons for dropout mentioned; per protocol analysis "19 resp. 21 remained and data were analyzed." |
| Selective reporting (reporting bias) | Low risk | No indication |
| Size of study (possible biases confounded by small size) | High risk | Number total (randomised): 62 |
| Other bias | Low risk | No indication |
Villamil 2005.
| Methods | RCT Placebo‐controlled Parallel‐group design |
|
| Participants | Pruritus: CP Description: participants with cholestasis Number of participants randomised: 18
Number of participants evaluable: 16
Withdrawals/dropouts: 2
Reason for dropout:
Age: (mean ± SD)
Sex (male/female):
Underlying disease(s):
Participant pool: single‐centre Setting: no information Haemodialysis: NA Baseline pruritus assessment: yes Duration/severity of pruritus: at least 3 months; resistant to treatment Baseline parameters: no information |
|
| Interventions |
Additional medication: ursodeoxycholic acid Route of administration: intravenous (5 minutes) Duration of treatment: 7 days Follow‐up: no information |
|
| Outcomes | Pruritus assessment: VAS (0‐100 mm) Adverse events Additional outcomes: total serum bilirubin, alkaline phosphatase, albumin |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomised (2:1) to receive . . ." Method not stated |
| Allocation concealment (selection bias) | Unclear risk | Not information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Unclear who was blinded "Drug administration was done under strict double‐blind conditions and the code was not opened until the final analysis of the results." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessor probably blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropouts and reasons for dropout mentioned; not reported if intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Unclear risk | Presentation of study results inappropriate; neither baseline nor endpoint data given |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 18 |
| Other bias | Low risk | Assessment of compliance not stated |
Wikström 2005a.
| Methods | RCT Placebo‐controlled Parallel‐group design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number of participants randomised: 51
Number of participants evaluable: 48 Withdrawals/dropouts:
Reason for dropout:
Age (years): no information Sex (male/female): no information Underlying disease(s): ESRD Participant pool: multicentre Setting: no information Haemodialysis: no information Baseline pruritus assessment: yes Duration/severity of pruritus: severe, uncontrolled pruritus secondary to ESRD; at least 3 "worst itching" VAS measurements during run‐in period of > 50 mm and average worst itching > 25 mm Baseline parameters: Pruritus relief (measured by VAS 100 mm): (mean ± SD)
|
|
| Interventions |
Additional medication: before the run‐in period all antipruritic medications, except for topical neutral agents, were discontinued for at least 7 days Route of administration: intravenous Duration of treatment: 4 weeks (1 week run‐in period ‐ 4 weeks nalfurafine/placebo) Follow‐up: 2 weeks after the administration of the final dose |
|
| Outcomes | Pruritus assessment: VAS 100 mm Adverse events |
|
| Notes | Not clear who conducted the study; not clear if and where study was published; 17 (65%) of 26 participants had adverse drug events in the nalfurafine group, but only 15 were described | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not stated "Seventy‐nine patients were randomly assigned in this study." |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Substantial missing outcome data; number of participants included unclear |
| Selective reporting (reporting bias) | Unclear risk | Conflicting data (number of participants included); combined results from study Wikström 2005b |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 51 |
| Other bias | Unclear risk | Missing participant characteristics; Bergstrom effect; assessment of compliance not stated |
Wikström 2005b.
| Methods | RCT Placebo‐controlled Cross‐over design |
|
| Participants | Pruritus: UP Description: patients with ESRD on haemodialysis Number of participants randomised: 34
Number of participants evaluable: 31 Withdrawals/dropouts: 3 Reason for dropout: no information Age (years): no information Sex (male/female): no information Underlying disease(s): ESRD Participant pool: multicentre Setting: no information Haemodialysis: "routine" Baseline pruritus assessment: yes Duration/severity of pruritus: no information on duration; severe, uncontrolled pruritus secondary to ESRD, at least 3 "worst itching" VAS measurements during run‐in period of > 50 mm and average worst itching > 25 mm Baseline parameters: Pruritus relief (measured by VAS 100 mm): (mean ± SD)
|
|
| Interventions |
Additional medication: before the run‐in period all antipruritic medications, except for topical neutral agents, were discontinued for at least 7 days. Route of administration: intravenous Duration of treatment: 2 weeks (1 week run‐in period ‐ 2 weeks nalfurafine/placebo ‐ 3 weeks washout ‐ 2 weeks cross‐over) Follow‐up: no information |
|
| Outcomes | Pruritus assessment: VAS 100 mm Adverse events |
|
| Notes | Results combined with results of study Wikström 2005a. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ". . . patients were randomly assigned 1:1 . . .." Method not stated |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind; unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information on withdrawals |
| Selective reporting (reporting bias) | Unclear risk | Combined with results of study Wikström 2005a, conflicting information concerning number of included participants |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 34 |
| Other bias | Unclear risk | Missing participant characteristics; assessment of compliance not stated |
Wolfhagen 1997.
| Methods | RCT Placebo‐controlled Parallel‐group design |
|
| Participants | Pruritus: CP Description: participants with cholangitis Number of participants randomised: 16
Number of participants evaluable: 16 Withdrawals/dropouts: 0 Reason for dropout: NA Age:
Sex (male/female):
Underlying disease(s):
Participant pool: single‐centre Setting: inpatient for one day, then outpatient Haemodialysis: NA Baseline pruritus assessment: yes Duration/severity of pruritus: no information Baseline parameters: Pruritus relief (measured by VAS 100 mm):
|
|
| Interventions |
Additional medication: UDCA (n = 8), anion binders (n = 7), antihistamines (n = 3), rifampicin (n = 3), light therapy (n = 2), plasmapheresis (n = 1) Route of administration: oral Duration of treatment: 4 weeks Follow‐up: twice before randomisation ‐ after 2 weeks ‐ after 4 weeks |
|
| Outcomes | Pruritus assessment: VAS 100 mm Adverse events Additional outcomes:
|
|
| Notes | Study describes participants as responders and non‐responders | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomly assigned (using opaque envelopes) . . ." |
| Allocation concealment (selection bias) | Low risk | Opaque envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind; unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts; not reported if intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | No indication |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 16 |
| Other bias | Low risk | "Treatment compliance, assessed by pill counts, was 100%" |
Young 2009.
| Methods | RCT Vehicle‐controlled Parallel‐group design |
|
| Participants | Pruritus: UP Description: participants with ESRD on haemodialysis Number of participants randomised: 28
Number of participants evaluable: 27
Withdrawals/dropouts: 1 Reason for dropout: unrelated subject death Age (range): 18‐70 years Sex (male/female): 14/14 Underlying disease(s): ESRD Participant pool: single‐centre Setting: inpatient Haemodialysis: at least 3 months Baseline pruritus assessment: yes Duration/severity of pruritus: symptoms of itch in a regular pattern over 6 months; at least 2 episodes of itch > 2 minutes within 2 weeks Baseline parameters: (mean ± SD)
|
|
| Interventions |
Additional medication: no information Route of administration: topical to all affected areas of pruritus/2x daily Duration of treatment: 4 weeks Follow‐up: baseline ‐ week 1‐ week 4 |
|
| Outcomes | Pruritus assessment: VAS 10 cm Adverse events Additional outcomes:
|
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised; method not stated |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind; unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | One participant lost because of unrelated subject death; not reported if intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Unclear risk | Inclusion and exclusion criteria inadequately described; insufficient data on pruritus VAS (no confidence interval, only graphical illustration) |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 28 |
| Other bias | Unclear risk | Assessment of compliance not stated; possible carryover effect not mentioned |
Yue 2015.
| Methods | RCT Placebo‐controlled three‐armed trial Parallel‐group design |
|
| Participants | Pruritus: UP Description: participants with ESRD who were treated with haemodialysis Number of participants randomised: 188 Number of participants evaluable: 179
Withdrawals/dropouts: 9 Reason for dropout: pregabalin: somnolence: 3; dizziness: 1; loss of balance: 1; ondansetron: nausea and vomiting: 2; kidney transplantation: 2 Age (mean ± SD): pregabalin: 57.7 ± 16.9, ondansetron: 56.5 ± 12.7, placebo: 57.2 ± 10.8 Sex (% male sex): pregabalin: 62.9%, ondansetron: 60.0%, placebo: 57.9% Underlying disease(s): ESRD Participant pool: single‐centre Setting: inpatient Haemodialysis (mean ± SD): pregabalin: 56.5 months ± 12.2, ondansetron: 57.6 months ± 16.2, placebo: 54.9 months ± 10.7 Duration/severity of pruritus: all patients suffered from persistent pruritus. Baseline parameters:
|
|
| Interventions |
Additional medication: the use of concomitant pruritus medications was not allowed. Route of administration: oral Duration of treatment: 12 weeks Follow‐up: no information |
|
| Outcomes | Pruritus assessment:
Adverse events Additional outcomes: Health related quality of life: Mental Component Summary scale (MCS) from the 12‐item short‐form (SF‐12; version 2); SF‐12 was scored from 0 to 100, with higher scores indicating better HRQoL |
|
| Notes | Imprecise: the primary end point of the study was to compare the effects between pregabalin and ondansetron on UP in dialysis patients. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomly assigned" |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Reason for dropout: pregabalin: somnolence: 3; dizziness: 1; loss of balance: 1; ondansetron: nausea and vomiting: 2; kidney transplantation: 2 |
| Selective reporting (reporting bias) | Low risk | Primary outcomes stated; no information about registration of the study |
| Size of study (possible biases confounded by small size) | Unclear risk | "The 179 patients included 62 cases from the pregabalin group, 60 from the ondansetron group, and 57 from the placebo group" |
| Other bias | Unclear risk | No information about registration of the study |
Zylicz 2003.
| Methods | RCT Placebo‐controlled Cross‐over design |
|
| Participants | Pruritus: various malignancies Description: participants with the following conditions:
Number of participants is higher than total number of participants due to simultaneously existing causes for pruritus. Number of participants randomised: 26
Number of participants evaluable: 24 Withdrawals/dropouts: 2 participants in group B during paroxetine treatment Reason for dropout: severe nausea and vomiting Age (mean ± SD):
Sex (male/female):
Underlying disease(s): solid tumours (n = 17), haematological malignancies (n = 4), osteoporosis (n = 2), Parkinson's (n = 1), skin amyloidosis due to MEN2A (n = 1), rheumatoid arthritis (n = 1) Participant pool: multicentre (2 palliative care centres) Setting: inpatient Haemodialysis: NA Duration/severity of pruritus:
Baseline parameters: Pruritus relief (measured by numerical analogue scale = NAS) (mean ± SD)
|
|
| Interventions |
Additional medication: cisapride (in case of severe nausea) Route of administration: oral Duration of treatment: 1 week (1 week run in ‐ 1 week paroxetine/placebo ‐ 1 week cross‐over) Follow‐up: no information |
|
| Outcomes | Pruritus assessment: NAS (0‐10) Adverse events Participant satisfaction |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not stated "The patients were randomised when the mean NAS of pruritus . . ." |
| Allocation concealment (selection bias) | Unclear risk | Allocation not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind; unclear who was blinded "Blinding and randomisation were in the hands of the same pharmacist as well." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data; planning of sample size; power calculation done, intention‐to‐treat approach for the primary endpoint done |
| Selective reporting (reporting bias) | Low risk | No indication |
| Size of study (possible biases confounded by small size) | High risk | Number of participants randomised: 26 |
| Other bias | Low risk | Assessment of compliance not stated; no washout period but carryover effect evaluated |
ALAT: glutamate‐pyruvat‐transaminase;ASTA: glutamate‐oxalacetat‐transaminase; CI: confidence interval; CP: cholestatic pruritus; CS: cromolyn sodium;EPO: erythropoietin; ESRD: end‐stage renal disease; IQR: interquartile range;HD: haemodialysis; MEN2A: multiple endocrine neoplasia type 2A; NA: not applicable/available; NAS: numerical analogue scale; PBC: primary biliary cholangitis; PD: peritoneal dialysis; PSC: primary sclerosing cholangitis;iPTH: intact parathyroid hormone; RCT: randomised controlled trial; SD: standard deviation; SF‐12 MCS: Short Form 12 Health Survey, mental health composite score; UDCA: ursodeoxycholic acid; UP: uraemic pruritus; VAS: visual analogue scale.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Almasio 2000 | Study intervention targeted on the treatment of underlying disease |
| Aperis 2010 | Not an RCT |
| Aymard 1980 | Not an RCT |
| Balaskas 1998 | Not an RCT |
| Bergasa 1991 | Not an RCT |
| Bergasa 1992 | Not an RCT |
| Bergasa 1995 | Does not meet inclusion criteria concerning palliative care patients |
| Bergasa 1999 | Does not meet inclusion criteria concerning palliative care patients |
| Berman 1998 | Not an RCT |
| Bigliardi 2007 | Does not meet inclusion criteria concerning palliative care patients |
| Borgeat 1994 | Doubly published data |
| Bousquet 1989 | Not an RCT |
| Breneman 1992b | Not an RCT |
| Castello 2011 | Not an RCT |
| Chen 2006 | Not a pharmacological intervention as defined in inclusion criteria, will be enclosed in an additional review |
| Datta 1966 | Not an RCT |
| Davis 2003 | Not an RCT |
| Easton 1978 | Not an RCT |
| Fjellner 1979 | Not an RCT |
| Ghorbani 2011 | Insufficient data provided |
| Giovanetti 1995 | Not an RCT |
| Goicoechea 1999 | Not an RCT |
| Goncalves 2010 | Not an RCT |
| Hellier 1963 | Not an RCT |
| Jones 2005 | Not an RCT |
| Jones 2007 | Does not meet inclusion criteria concerning palliative care patients |
| Juby 1994 | Not an RCT |
| Kato 2001 | Not an RCT |
| Korfitis 2008 | Not an RCT |
| Kuypers 2004 | Not an RCT |
| Lysy 2003 | Does not meet inclusion criteria concerning palliative care patients |
| Mansour‐Ghanaei 2006 | Not an RCT |
| Marquez 2012 | Not an RCT |
| Metze 1999a | Not an RCT |
| Montero 2006 | Not an RCT |
| Müller 1998a | Does not meet inclusion criteria concerning palliative care patients |
| Müller 1998b | Does not meet inclusion criteria concerning palliative care patients |
| Podesta 1991b | Not an RCT |
| Price 1998 | Not an RCT |
| Prieto 2004 | Not an RCT |
| Razeghi 2009 | Not an RCT |
| Rifai 2006 | Not an RCT |
| Sja'bani 1997 | Only abstract available; contact with the authors could not be established |
| Ständer 2009 | Does not meet inclusion criteria concerning palliative care patients |
| Szepietowski 2005 | Not an RCT |
| Tokgöz 2005 | Not an RCT |
RCT: randomised controlled trial.
Characteristics of ongoing studies [ordered by study ID]
2005‐003469‐18.
| Trial name or title | A randomized, double‐blind, placebo‐controlled study of TRK‐820 in haemodialysis patients with uremic pruritus |
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled, parallel assignment |
| Participants | 300 participants |
| Interventions | Group 1: nalfurafine hydrochloride TRK‐820 5 µg IV; Group 2: placebo |
| Outcomes | The change in worst itching recorded on the Visual Analogue Scale (VAS) from baseline |
| Starting date | NA |
| Contact information | NA |
| Notes | Multinational study: LT (completed), BE (completed), CZ (ongoing), IT (completed) |
2009‐010437‐50.
| Trial name or title | A Phase 2, randomized, double‐blind, placebo‐controlled study to evaluate the efficacy of different Gabapentin doses in haemodialysis patients with uremic pruritus |
| Methods | Randomised, double‐blind, placebo‐controlled, parallel assignment, phase 2 |
| Participants | 45 haemodialysis patients |
| Interventions | Group 1: gabapentin 100 mg/HD or 300 mg/HD; Group 2: placebo |
| Outcomes | Change of itch severity using a pruritus score based on a visual analogue scale (VAS) |
| Starting date | NA |
| Contact information | NA |
| Notes | Ongoing |
ISRCTN13971661.
| Trial name or title | Balneum Plus cream for the treatment of itchy skin in renal patients |
| Methods | Single‐centre, randomised, double‐blind, placebo‐controlled, parallel‐group trial, phase IV |
| Participants | 58 participants |
| Interventions | Group 1: balneum Plus cream (active ingredients: 3.0% urea and 5.0% lauromacrogols); Group 1: placebo |
| Outcomes | Reduction in itch intensity as measured by VAS; quality of life as measured by a validated questionnaire for itch in renal disease |
| Starting date | 1 January 2015 |
| Contact information | Dr Jacqueline Nevols (Wessex Kidney Centre, Portsmouth, UK) |
| Notes | Completed |
NCT00577967.
| Trial name or title | Gabapentin ‐ a solution to uremic pruritus? A prospective, randomized, placebo‐controlled, double‐blind Study |
| Methods | Prospective, randomised, placebo‐controlled, double‐blind study; parallel assignment |
| Participants | 80 participants suffering from UP |
| Interventions | Gabapentin |
| Outcomes | Subjective measurement of reduction in pruritus |
| Starting date | October 2005 |
| Contact information | Hospital Authority, Hong Kong |
| Notes | The recruitment status of this study is unknown because the information has not been verified recently. |
NCT00693654.
| Trial name or title | A controlled comparative study of the efficacy of SARNA sensitive lotion for treatment of uremic pruritus in adult hemodialysis patients |
| Methods | Randomised, controlled, double‐blind study, parallel assignment |
| Participants | 30 adult haemodialysis patients |
| Interventions | Group 1: SARNA sensitive lotion against pruritus Group 2: cetaphil |
| Outcomes | Investigator assessment of pruritus score and response to treatment using an itch questionnaire |
| Starting date | November 2006 |
| Contact information | Alan Fleischer, MD, Wake Forest University Health Sciences |
| Notes | The recruitment status of this study is unknown because the information has not been verified recently. |
NCT00793156.
| Trial name or title | A randomized‐withdrawal phase 3 study evaluation the safety and efficacy of oral nalfurafine HCl (AC‐820) in subjects on hemodialysis with uremic pruritus (renal itch) |
| Methods | Multicentre, double‐blind, placebo‐controlled, randomised withdrawal study, cross‐over assignment |
| Participants | 350 participants with moderate to severe itching associated with ESRD and haemodialysis |
| Interventions | Group 1: nalfurafine HCl 2.5 µg Group 2: nalfurafine HCl 5.0 µg Group 1: placebo |
| Outcomes | Primary efficacy endpoint is the change in worst itching intensity from baseline, compared to that in the last 2 weeks of the double blind, placebo‐controlled, randomised withdrawal period. Safety/efficacy study |
| Starting date | December 2009 |
| Contact information | Acologix, Inc |
| Notes | The recruitment status of this study is unknown because the information has not been verified recently. |
NCT01073501.
| Trial name or title | Efficacy of pregabalin in the management of chronic uremic pruritus |
| Methods | Randomised, double‐blind, placebo‐controlled, cross‐over study design |
| Participants | 36 participants |
| Interventions | Group 1: pregabalin 25 mg Group 2: placebo |
| Outcomes | Reduction of UP by more than 50% after pregabalin administration; reduction of chronic pain of various origin and improvement in insomnia after pregabalin administration |
| Starting date | April 2010 |
| Contact information | Dr. Linda Shavit (lshavit@szmc.org.il) |
| Notes | The recruitment status of this study is unknown because the information has not been verified recently. |
NCT01513161.
| Trial name or title | Efficacy and safety study of TRK‐820 to treat conventional‐treatment‐resistant pruritus in patients receiving hemodialysis |
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group, fixed dose, phase III clinical trial |
| Participants | 104 haemodialysis patients |
| Interventions | Group 1: nalfurafine hydrochloride (TRK‐820) Group 2: placebo |
| Outcomes | Change in pruritus degree measured by VAS |
| Starting date | April 2008 |
| Contact information | SK Chemicals Co, Ltd |
| Notes | Completed |
NCT01660243.
| Trial name or title | A Phase 2, randomized, double‐blind, placebo‐controlled, fixed‐dose, parallel‐group, multicenter, efficacy, and safety study of MT‐9938 for treatment of uremic pruritus in subjects with end‐stage renal disease receiving hemodialysis |
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled, parallel assignment, phase 2 |
| Participants | 45 haemodialysis patients |
| Interventions | Group 1: nalfurafine hydrochloride (2.5μg, 5μg, 10μg) Group 2: placebo |
| Outcomes | Change from baseline in: worst‐itching 11‐point numerical rating scale (NRS); worst‐itching VAS; itch severity score; sleep quality assessment; excoriation; QoL assessment; treatment satisfaction (Patient's Global Impression of Change) |
| Starting date | September 2012 |
| Contact information | Mitsubishi Tanabe Pharma Corporation |
| Notes | This study was terminated because of insufficient patient recruitment. There were no safety concerns. |
NCT01852318.
| Trial name or title | A multicenter, double‐blind, randomized, placebo and active‐controlled study of pregabalin for the treatment of uremic pruritus |
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled, parallel assignment, phase 4 |
| Participants | 210 haemodialysis patients |
| Interventions | Group 1: pregabalin 75mg; Drug: fexofenadine 60 mg Group 2: placebo |
| Outcomes | Changes in pruritus symptoms assessed by VAS; changes in pruritus score (PS), Skindex‐10, brief itching inventory and itch medical outcomes study |
| Starting date | April 2014 |
| Contact information | Hsien‐Yi Chiu, MD (extra.owl0430@yahoo.com.tw); Tsen‐Fang Tsai, MD (tftsai@yahoo.com) |
| Notes | The recruitment status of this study is unknown because the information has not been verified recently. |
NCT02008864.
| Trial name or title | Phase II study of the effect of senna alexandrina mill. on uremic pruritus and serum IL‐2, INF‐δ and TNF‐α levels of hemodialysed patients |
| Methods | Randomised, double‐blind, placebo‐controlled, parallel assignment, Phase 2 |
| Participants | 60 haemodialysis patients |
| Interventions | Group 1: senna (7.5 mg of sennosoides A and B) Group 2: placebo |
| Outcomes | Severity of pruritis, as measured by VAS (0‐10); serum IL‐2 level; serum INF‐δ level; serum TNF‐α level |
| Starting date | August 2011 |
| Contact information | Shiraz University of Medical Sciences |
| Notes | Completed |
NCT02032537.
| Trial name or title | Efficacy of Calmmax cream in the management of chronic uremic pruritus |
| Methods | Prospective, double‐blind, placebo‐controlled, randomised trial, single group assessment |
| Participants | 28 participants with UP |
| Interventions | Group 1: Callmax cream application over affected skin Group 2: placebo |
| Outcomes | Improvement of UP measured by reduction of VAS by more than 50 percent from baseline score; quality of life assessed by questionnaire |
| Starting date | November 2014 |
| Contact information | Dr. Linda Shavit (lshavit@szmc.org.il) |
| Notes | The recruitment status of this study is unknown because the information has not been verified recently. |
NCT02143648.
| Trial name or title | A randomized, double‐blind, placebo‐controlled, parallel, 3‐arm study of the safety and anti‐pruritic efficacy of nalbuphine HCl ER tablets in hemodialysis patients with uremic pruritus |
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled, parallel assignment, phase 2 and 3 |
| Participants | 360 haemodialysis patients |
| Interventions | Group 1: nalbuphine HCl ER (60 mg, 120 mg) Group 2: placebo |
| Outcomes | Itch on the 0‐10 itch numerical rating scale; total Skindex‐10 score; sleep on the 12 question Medical Outcomes Study Sleep Survey's Sleep Problems Index II, SLP‐9; depression and anxiety on the 14 question Hospital Anxiety and Depression Scale (HADS); patient's assessment of their pruritus on the Patient Assessed Disease Severity Scale (PADS) |
| Starting date | June 2014 |
| Contact information | Thomas Sciascia, MD (Trevi Therapeutics) |
| Notes | Completed |
NCT02229929.
| Trial name or title | A double‐blind, randomized, placebo‐controlled study to evaluate the safety and pharmacokinetics of intravenous CR845 in hemodialysis patients, and its safety and efficacy in hemodialysis patients with uremic pruritus |
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled, parallel assignment, phase 2 |
| Participants | 84 haemodialysis patients |
| Interventions | Group 1: CR845 (0.5 mcg/kg, 1.0 mcg/kg, 2.5 mcg/kg) Group 2: placebo |
| Outcomes | Pharmacokinetics of Repeated Doses of CR845 in Haemodialysis Patients (half‐life, Cmax, Tmax, AUC, Vd); change in worst itching intensity using a 100 mm VAS; change in quality‐of‐life assessed using the Skindex‐10 survey; sleep disturbance assessed using itch Medical Outcomes Study (MOS) survey |
| Starting date | July 2014 |
| Contact information | Stephen Cincotta (scincotta@clinicalresearchmgt.com) |
| Notes | Completed |
NCT02696499.
| Trial name or title | Treatment of uremic pruritus with inhaled PA101B in patients with end‐stage renal disease requiring hemodialysis |
| Methods | Randomised, double blind, placebo‐controlled, parallel‐arm, multicentre, phase 2, proof‐of‐concept efficacy and safety study |
| Participants | Patients with end‐stage renal disease requiring haemodialysis |
| Interventions | Group 1: 40 mg PA101B administered via inhalation twice daily for 7 weeks; Group 2: matching placebo administered via inhalation twice daily for 7 weeks |
| Outcomes | Primary outcome: itching intensity (numerical rating scale) Secondary outcomes: pruritus‐specific quality of life (Skindex‐10), pruritus‐specific sleep quality (itch MOS), assessment of depression (Beck Depression Inventory‐II), Patient Global Impression of Change |
| Starting date | This study is ongoing, but not recruiting participants |
| Contact information | Patara Pharma |
| Notes | This study is ongoing, but not recruiting participants. First received: 25 February 2016 |
NCT02701166.
| Trial name or title | The effect of bezafibrate on cholestatic itch (FITCH) |
| Methods | Randomised, double blind, placebo‐controlled, parallel‐arm, multi‐centre efficacy study |
| Participants | Primary biliary cholangitis or primary/secondary sclerosing cholangitis |
| Interventions | Group 1: bezafibrate 400 mg per day Group 2: placebo 400 mg per day |
| Outcomes | Proportion of patients with a reduction in itch intensity of 50% or more |
| Starting date | First received 2 March 2016 |
| Contact information | Ulrich Beuers, Prof. Dr. +31205662422 u.h.beuers@amc.uva.nl |
| Notes | This study was recruiting participants at the time of writing. |
ESRD: end‐stage renal disease; IV: intravenous; MOS: medical outcomes scale; NA: not available/applicable; UP; uraemic pruritus; VAS: visual analogue scale.
Differences between protocol and review
In contrast to our preliminary definition in the protocol, the present review only includes randomised controlled trials. We did not include controlled trials or observational studies.
The primary outcome was slightly rephrased/specified in comparison to the protocol. This had no impact on the included studies or on the results.
In addition, in the Background we have made some modifications and improvements concerning the Cochrane Review Management Program. First of all, in Review Manager 5 an updated version of the 'Risk of bias tool' has been implemented. We adapted our assessment criteria as described in the protocol according to the new 'Risk of bias' tool. Hence, we performed the 'Risk of bias' assessment as recommended by the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011; RevMan 2014).
We added 'Size of study' as new risk of bias domain.
In this update, we did not specify a main comparison (original review: naltrexone) in order to avoid misleading conclusions. We deleted the paroxetine and the satisfaction analysis (only one study included). In addition, we added the comparisons flumecinol versus placebo, cromolyn sodium versus placebo and zinc sulphate versus placebo to Data synthesis and Summary of findings tables.
Contributions of authors
WS: revised the search strategies and searched the databases, screened and extracted the new included studies, conducted meta‐analyses, updated and revised the manuscript.
CX: supported the whole update process, extracted and screened data from the new included papers.
SB: engaged in revising the search strategies for CENTRAL, MEDLINE and Embase.
JJM, GA: supported the whole update process, provided guidance in developing the summary of findings tables.
GS: provided methodological and statistical expertise, supervised meta‐analyses.
GB: supported the whole update process, resolved any disagreements.
All review authors critically reviewed the manuscript and gave suggestions for refining the final draft.
WS and CX are responsible for updates.
Sources of support
Internal sources
German Cochrane Centre, Freiburg, Germany.
External sources
-
German Ministry for Education and Research (BMBF), Germany.
Grant No. 01KG0819
Declarations of interest
WS: none known.
CX: none known.
JM: none known.
SB: none known.
GA: none known.
GS: none known.
GB: none known.
Stable (no update expected for reasons given in 'What's new')
References
References to studies included in this review
Amirkhanlou 2016 {published data only}
- Amirkhanlou S, Rashedi A, Taherian J, Hafezi AA, Parsaei S. Comparison of gabapentin and ketotifen in treatment of uremic pruritus in hemodialysis patients. Pakistan Journal of Medical Sciences 2016; Vol. 32, issue 1:22‐6. [DOI] [PMC free article] [PubMed]
Ashmore 2000 {published data only}
- Ashmore SD, Jones CH, Newstead CG, Daly MJ, Chrystyn H. Ondansetron therapy for uremic pruritus in hemodialysis patients. American Journal of Kidney Diseases 2000;35(5):827‐31. [DOI] [PubMed] [Google Scholar]
Bachs 1989 {published data only}
- Bachs L, Parés A, Montserrat E, Piera C, Rodés J. Comparison of rifampicin with phenobarbitone for treatment of pruritus in biliary cirrhosis. Lancet 1989;1(8638):574‐6. [DOI] [PubMed] [Google Scholar]
Bergasa 2006 {published data only}
- Bergasa NV, McGee M, Ginsburg IH, Engler D. Gabapentin in patients with the pruritus of cholestasis: a double‐blind, randomized, placebo‐controlled trial. Hepatology 2006;44(5):1317‐23. [DOI] [PubMed] [Google Scholar]
Borgeat 1993 {published data only}
- Borgeat A, Wilder‐Smith OH, Saiah M, Rifat K. Subhypnotic doses of propofol relieve pruritus induced by epidural and intrathecal morphine. Anesthesiology 1992;76(4):510‐2. [DOI] [PubMed] [Google Scholar]
Breneman 1992a {published data only}
- Breneman DL, Cardone JS, Blumsack RF, Lather RM, Searle EA, Pollack VE. Topical capsaicin for treatment of hemodialysis‐related pruritus. Journal of the American Academy of Dermatology 1992;26(1):91‐4. [DOI] [PubMed] [Google Scholar]
Cho 1997 {published data only}
- Cho YL, Liu HN, Huang TP, Tarng DC. Uremic pruritus: roles of parathyroid hormone and substance P. Journal of the American Academy of Dermatology 1997;36(4):538‐43. [DOI] [PubMed] [Google Scholar]
De Marchi 1992 {published data only}
- Marchi S, Cecchin E, Villalta D, Sepiacci G, Santini G, Bartoli E. Relief of pruritus and decreases in plasma histamine concentrations during erythropoietin therapy in patients with uremia. New England Journal of Medicine 1992;326(15):969‐74. [DOI] [PubMed] [Google Scholar]
Duncan 1984 {published data only}
- Duncan JS, Kennedy HJ, Triger DR. Treatment of pruritus due to chronic obstructive liver disease. Brtish Medical Journal (Clinical Research Edition) 1984;289(6436):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Duque 2005 {published data only}
- Duque MI, Yosipovitch G, Fleischer AB Jr, Willard J, Freedman BI. Lack of efficacy of tacrolimus ointment 0.1% for treatment of hemodialysis‐related pruritus: a randomized, double‐blind, vehicle‐controlled study. Journal of the American Academy of Dermatology 2005;52:519‐21. [DOI] [PubMed] [Google Scholar]
Feily 2012 {published data only}
- Feily A, Dormanesh B, Ghorbani AR, Moosavi Z, Kouchak M, Cheraghian B, et al. Efficacy of topical cromolyn sodium 4% on pruritus of uremic nephrogenic patients; a randomized double blind study on 60 patients. International Journal of Clinical Pharmacology and Therapeutics 2012;50(7):510‐3. [DOI] [PubMed] [Google Scholar]
Ghanei 2012 {published data only}
- Ghanei E, Zeinali J, Borghei M, Homayouni M. Efficacy of omega‐3 fatty acids supplementation in treatment of uremic pruritus in hemodialysis patients: a double‐blind randomized controlled trial. Iranian Red Crescent Medical Journal 2012;14(9):515–22. [PMC free article] [PubMed] [Google Scholar]
Ghent 1988 {published data only}
- Ghent CN, Carruthers SG. Treatment of pruritus in primary biliary cirrhosis with rifampin. Results of a double‐blind, crossover, randomized trial. Gastroenterology 1988;94(2):488‐93. [DOI] [PubMed] [Google Scholar]
Gunal 2004 {published data only}
- Gunal AI, Ozalp G, Yoldas TK, Gunal SY, Kirciman E, Celiker H. Gabapentin therapy for pruritus in haemodialysis patients: a randomized, placebo‐controlled, double‐blind trial. Nephrology, Dialysis, Transplantation 2004;19(12):3137‐9. [DOI] [PubMed] [Google Scholar]
Kuiper 2010 {published data only}
- Kuiper EM, Erpecum KJ, Beuers U, Hansen BE, Thio HB, Man RA, et al. The potent bile acid sequestrant colesevelam is not effective in cholestatic pruritus: results of a double‐blind, randomized, placebo‐controlled trial. Hepatology 2010;52(4):1334‐40. [DOI] [PubMed] [Google Scholar]
Kumagai 2010 {published data only}
- Kumagai H, Ebata T, Takamori K, Muramatsu T, Nakamoto H, Suzuki H. Effect of a novel kappa‐receptor agonist, nalfurafine hydrochloride, on severe itch in 337 haemodialysis patients: a phase III, randomized, double‐blind, placebo‐controlled study. Nephrology, Dialysis, Transplantation 2010;25(4):1251‐7. [DOI] [PubMed] [Google Scholar]
Legroux‐Crespel 2004 {published data only}
- Legroux‐Crespel E, Clèdes J, Misery L. A comparative study on the effects of naltrexone and loratadine on uremic pruritus. Dermatology 2004;208(4):326‐30. [DOI] [PubMed] [Google Scholar]
Makhlough 2010 {published data only}
- Makhlough A. Topical capsaicin therapy for uremic pruritus in patients on hemodialysis. Iranian Journal of Kidney Diseases 2010;4(2):137‐40. [PubMed] [Google Scholar]
Mapar 2015 {published data only}
- Mapar MA, Pazyar N, Siahpoosh A, Latifi SM, Beladi Mousavi SS, Khazanee A. Comparison of the efficacy and safety of zinc sulfate vs. placebo in the treatment of pruritus of hemodialytic patients: a pilot randomized, triple‐blind study. Giornale Italiano di Dermatologia e Venereologia [Official Journal of the Italian Society of Dermatology and Sexually Transmitted Diseases] 2015; Vol. 150, issue 4:351‐5. [PubMed]
Mayo 2007 {published data only}
- Mayo MJ, Handem I, Saldana S, Jacobe H, Getachew Y, Rush AJ. Sertraline as a first‐line treatment for cholestatic pruritus. Hepatology 2007;45(3):666‐74. [DOI] [PubMed] [Google Scholar]
Murphy 2003 {published data only}
- Murphy M, Reaich D, Pai P, Finn P, Carmichael AJ. A randomized, placebo‐controlled, double‐blind trial of ondansetron in renal itch. British Journal of Dermatology 2003;148(2):314‐7. [DOI] [PubMed] [Google Scholar]
Naini 2007 {published data only}
- Naini AE, Harandi AA, Khanbabapour S, Shahidi S, Seirafiyan S, Mohseni M. Gabapentin: a promising drug for the treatment of uremic pruritus. Saudi Journal of Kidney Diseases and Transplantation 2007;18(3):378‐81. [PubMed] [Google Scholar]
Najafabadi 2012 {published data only}
- Najafabadi MM, Faghihi G, Emami A, Monghad M, Moeenzadeh F, Sharif N, et al. Zinc sulfate for relief of pruritus in patients on maintenance hemodialysis. Therapeutic Apheresis & Dialysis 2012;16(2):142‐5. [DOI] [PubMed] [Google Scholar]
Nakhaee 2015 {published data only}
- Nakhaee S, Nasiri A, Waghei Y, Morshedi J. Comparison of Avena sativa, vinegar, and hydroxyzine for uremic pruritus of hemodialysis patients: a crossover randomized clinical trial. Iranian Journal of Kidney Diseases 2015; Vol. 9, issue 4:316‐22. [PubMed]
Nasrollahi 2007 {published data only}
- Nasrollahi AR, Miladipour A, Ghanei E, Yavari P, Haghverdi F. Montelukast for treatment of refractory pruritus in patients on hemodialysis. Iranian Journal of Kidney Diseases 2007;1(2):73‐7. [PubMed] [Google Scholar]
O'Donohue 2005 {published data only}
- O'Donohue JW, Pereira SP, Ashdown AC, Haigh CG, Wilkinson JR, Williams R. A controlled trial of ondansetron in the pruritus of cholestasis. Alimentary Pharmacology & Therapeutics 2005;21(8):1041‐5. [DOI] [PubMed] [Google Scholar]
Omidian 2013 {published data only}
- Omidian, M, Khazanee, A, Yaghoobi, R, Ghorbani, A. R, Pazyar, N, Beladimousavi, S. S, Ghadimi, M, Mohebbipour, A, Feily, A. Therapeutic effect of oral nicotinamide on refractory uremic pruritus: a randomized, double‐blind study. Saudi Journal of Kidney Diseases and Transplantation 2013;24(5):995‐9. [DOI] [PubMed] [Google Scholar]
Özaykan 2001 {published data only}
- Özaykan S, Mansur T, Gündüz S, Güney O. Comparison of ondansetron and cyproheptadine in treatment of uremic pruritus [Üremi Kaşintisi Olan Hastlarda Ondansetron ve Siproheptadinin Etkinliğinin Karşılaştırılması]. Türkderm 2001;35:130‐4. [Google Scholar]
Pakfetrat 2014 {published data only}
- Pakfetrat M, Basiri F, Malekmakan L, Roozbeh J. Effects of turmeric on uremic pruritus in end stage renal disease patients: a double‐blind randomized clinical trial. Journal of Nephrology 2014;27(2):203‐7. [DOI] [PubMed] [Google Scholar]
Pauli‐Magnus 2000 {published data only}
- Pauli‐Magnus C, Mikus G, Alscher DM, Kirschner T, Nagel W, Gugeler N, et al. Naltrexone does not relieve uremic pruritus: results of a randomized, double blind, placebo‐controlled crossover study. Journal of the American Society of Nephrology 2000;11:514‐9. [DOI] [PubMed] [Google Scholar]
Pederson 1980 {published data only}
- Pederson JA, Matter BJ, Czerwinski AW, Llach F. Relief of idiopathic generalized pruritus in dialysis patients treated with activated oral charcoal. Annals of Internal Medicine 1980;93(3):446‐8. [DOI] [PubMed] [Google Scholar]
Peer 1996 {published data only}
- Peer G, Kivity S, Agami O, Fireman E, Silverberg D, Blum M, et al. Randomized cross over trial of naltrexone in uraemic pruritus. Lancet 1996;348(9041):1552‐4. [DOI] [PubMed] [Google Scholar]
Podesta 1991a {published data only}
- Podesta A, Lopez P, Terg R, Villamil F, Flores D, Mastai R, et al. Treatment of pruritus of primary biliary cirrhosis with rifampin. Digestive Diseases and Sciences 1991;36(2):216‐20. [DOI] [PubMed] [Google Scholar]
Pour‐Reza‐Gholi 2007 {published data only}
- Pour‐Reza‐Gholi F, Nasrollahi A, Firouzan A, Nasli Esfahani E, Farrokhi F. Low‐dose doxepin for treatment of pruritus in patients on hemodialysis. Iranian Journal of Kidney Diseases 2007;1(1):34‐7. [PubMed] [Google Scholar]
Shirazian 2013 {published data only}
- Shirazian S, Schanler M, Shastry S, Dwivedi S, Kumar M, Rice K, et al. The effect of ergocalciferol on uremic pruritus severity: a randomized controlled trial. Journal of Renal Nutrition 2013;23(4):308‐14. [DOI] [PubMed] [Google Scholar]
Silva 1994 {published data only}
- Silva SR, Viana PC, Lugon NV, Hoette M, Ruzany F, Lugon JR. Thalidomide for the treatment of uremic pruritus: a crossover randomized double‐blind trial. Nephron 1994;67(3):270‐3. [DOI] [PubMed] [Google Scholar]
Silverberg 1977 {published data only}
- Silverberg DS, Iaina A, Reisin E, Rotzak R, Eliahou HE. Cholestyramine in uraemic pruritus. British Medical Journal 1977;1(6063):752‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Smith 1997a {published data only}
- Smith KJ, Skelton HG, Yeager J, Lee RB, Wagner KF. Pruritus in HIV‐1 disease: therapy with drugs which may modulate the pattern of immune dysregulation. Dermatology 1997;195(4):353‐8. [DOI] [PubMed] [Google Scholar]
Tarng 1996 {published data only}
- Tarng DC, Cho YL, Liu HN, Huang TP. Hemodialysis‐related pruritus: a double‐blind, placebo‐controlled, crossover study of capsaicin 0.025% cream. Nephron 1996;72(4):617‐22. [DOI] [PubMed] [Google Scholar]
Terg 2002 {published data only}
- Terg R, Coronel E, Sordá J, Muñoz AE, Findor J. Efficacy and safety of oral naltrexone treatment for pruritus of cholestasis, a crossover, double blind, placebo‐controlled study. Journal of Hepatology 2002;37(6):717‐22. [DOI] [PubMed] [Google Scholar]
Turner 1994a {published data only}
- Turner IB, Rawlins MD, Wood P, James OF. Flumecinol for the treatment of pruritus associated with primary biliary cirrhosis. Alimentary Pharmacology & Therapeutics 1994;8(3):337‐42. [DOI] [PubMed] [Google Scholar]
Turner 1994b {published data only}
- Turner IB, Rawlins MD, Wood P, James OF. Flumecinol for the treatment of pruritus associated with primary biliary cirrhosis. Alimentary Pharmacology & Therapeutics 1994;8(3):337‐42. [DOI] [PubMed] [Google Scholar]
Vessal 2010 {published data only}
- Vessal G, Sagheb MM, Shilian S, Jafari P, Samani SM. Effect of oral cromolyn sodium on CKD‐associated pruritus and serum tryptase level: a double‐blind placebo‐controlled study. Nephrology, Dialysis, Transplantation 2010;25(5):1541‐7. [DOI] [PubMed] [Google Scholar]
Villamil 2005 {published data only}
- Villamil AG, Bandi JC, Galdame OA, Gerona S, Gadano AC. Efficacy of lidocaine in the treatment of pruritus in patients with chronic cholestatic liver diseases. The American Journal of Medicine 2005;118(10):1160‐3. [DOI] [PubMed] [Google Scholar]
Wikström 2005a {published data only}
- Wikström B, Gellert R, Ladefoged SD, Danda Y, Akai M, Ide K, et al. Kappa‐opioid system in uremic pruritus: multicenter, randomized, double‐blind, placebo‐controlled clinical studies. Journal of the American Society of Nephrology 2005;16(12):3742‐7. [DOI] [PubMed] [Google Scholar]
Wikström 2005b {published data only}
- Wikström B, Gellert R, Ladefoged SD, Danda Y, Akai M, Ide K, et al. Kappa‐opioid system in uremic pruritus: multicenter, randomized, double‐blind, placebo‐controlled clinical studies. Journal of the American Society of Nephrology 2005;16(12):3742‐7. [DOI] [PubMed] [Google Scholar]
Wolfhagen 1997 {published data only}
- Wolfhagen FH, Sternieri E, Hop WC, Vitale G, Bertolotti M, Buuren HR. Oral naltrexone treatment for cholestatic pruritus: a double‐blind, placebo‐controlled study. Gastroenterology 1997;113(4):1264‐9. [DOI] [PubMed] [Google Scholar]
Young 2009 {published data only}
- Young TA, Patel TS, Camacho F, Clark A, Freedman BI, Kaur M, et al. A pramoxine‐based anti‐itch lotion is more effective than a control lotion for the treatment of uremic pruritus in adult hemodialysis patients. Journal of Dermatological Treatment 2009;20(2):76‐81. [DOI] [PubMed] [Google Scholar]
Yue 2015 {published data only}
Zylicz 2003 {published data only}
- Zylicz Z, Krajnik M, Sorge AA, Costantini M. Paroxetine in the treatment of severe non‐dermatological pruritus: a randomized, controlled trial. Journal of Pain and Symptom Management 2003;26(6):1105‐12. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Almasio 2000 {published data only}
- Almasio PL, Floreani A, Chiaramonte M, Provenzano G, Battezzati P, Crosignani A, et al. Multicentre randomized placebo‐controlled trial of ursodeoxycholic acid with or without colchicine in symptomatic primary biliary cirrhosis. Alimentary Pharmacology & Therapeutics 2000;14(12):1645‐52. [DOI] [PubMed] [Google Scholar]
Aperis 2010 {published data only}
- Aperis G, Paliouras C, Zervos A, Arvanitis A, Alivanis P. The use of pregabalin in the treatment of uraemic pruritus in haemodialysis patients. Journal of Renal Care 2010;36(4):180‐5. [DOI] [PubMed] [Google Scholar]
Aymard 1980 {published data only}
- Aymard JP, Lederlin P, Witz F, Colomb JN, Herbeuval R, Weber B. Cimetidine for pruritus in Hodgkin's disease. British Medical Journal 1980;280(6208):151‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Balaskas 1998 {published data only}
- Balaskas EV, Bamihas GI, Karamouzis M, Voyiatzis G, Tourkantonis A. Histamine and serotonin in uremic pruritus: effect of ondansetron in CAPD‐pruritic patients. Nephron 1998;78(4):395‐402. [DOI] [PubMed] [Google Scholar]
Bergasa 1991 {published data only}
- Bergasa NV, Jones EA. Management of the pruritus of cholestasis: potential role of opiate antagonists. American Journal of Gastroenterology 1991;86(10):1404‐12. [PubMed] [Google Scholar]
Bergasa 1992 {published data only}
- Bergasa NV, Talbot TL, Alling DW, Schmitt JM, Walker EC, Baker BL, et al. A controlled trial of naloxone infusions for the pruritus of chronic cholestasis. Gastroenterology 1992;102(2):544‐9. [DOI] [PubMed] [Google Scholar]
Bergasa 1995 {published data only}
- Bergasa NV, Alling DW, Talbot TL, Swain MG, Yurdaydin C, Turner ML, et al. Effects of naloxone infusions in patients with the pruritus of cholestasis. A double‐blind, randomized, controlled trial. Annals of Internal Medicine 1995;123(3):161‐7. [DOI] [PubMed] [Google Scholar]
Bergasa 1999 {published data only}
- Bergasa NV, Alling DW, Talbot TL, Wells MC, Jones EA. Oral nalmefene therapy reduces scratching activity due to the pruritus of cholestasis: a controlled study. Journal of the American Academy of Dermatology 1999;41(3 Pt 1):431‐4. [DOI] [PubMed] [Google Scholar]
Berman 1998 {published data only}
- Berman B, Flores F, Burke G 3rd. Efficacy of pentoxifylline in the treatment of pruritic papular eruption of HIV‐infected persons. Journal of the American Academy of Dermatology 1998;38(6):955‐9. [DOI] [PubMed] [Google Scholar]
Bigliardi 2007 {published data only}
- Bigliardi PL, Stammer H, Jost G, Rufli T, Büchner S, Bigliardi‐Qi M. Treatment of pruritus with topically applied opiate receptor antagonist. Journal of the American Academy of Dermatology 2007;56(6):979‐88. [DOI] [PubMed] [Google Scholar]
Borgeat 1994 {published data only}
- Borgeat A, Mentha G, Savioz D, Wilder‐Smith OH. Pruritus associated with liver disease: Propofol, a new therapeutic approach?. Schweizerische Medizinische Wochenschrift [Swiss Medical Weekly] 1994;124(15):649‐50. [PubMed] [Google Scholar]
Bousquet 1989 {published data only}
- Bousquet J, Rivory JP, Maheut M, Michel FB, Mion C. Double‐blind, placebo‐controlled study of nicergoline in the treatment of pruritus in patients receiving maintenance hemodialysis. Journal of Allergy and Clinical Immunology 1989;83(4):825‐8. [DOI] [PubMed] [Google Scholar]
Breneman 1992b {published data only}
- Breneman DL, Cardone JS, Blumsack RF, Lather RM, Searle EA, Pollack VE. Topical capsaicin for treatment of hemodialysis‐related pruritus. Journal of the American Academy of Dermatology 1992;26(1):91‐4. [DOI] [PubMed] [Google Scholar]
Castello 2011 {published data only}
- Castello M, Milani M. Efficacy of topical hydrating and emollient lotion containing 10% urea ISDIN® plus dexpanthenol (Ureadin Rx 10) in the treatment of skin xerosis and pruritus in hemodialyzed patients: an open prospective pilot trial. Giornale Italiano Di Dermatologia e Venereologia [Official Journal of the Italian Society of Dermatology and Sexually Transmitted Diseases] 2011;46(5):321‐5. [PubMed] [Google Scholar]
Chen 2006 {published data only}
- Chen YC, Chiu WT, Wu MS. Therapeutic effect of topical gamma‐linolenic acid on refractory uremic pruritus. American Journal of Kidney Diseases 2006;48(1):69‐76. [DOI] [PubMed] [Google Scholar]
Datta 1966 {published data only}
- Datta DV, Sherlock S. Cholestyramine for long term relief of the pruritus complicating intrahepatic cholestasis. Gastroenterology 1966;50(3):323‐32. [PubMed] [Google Scholar]
Davis 2003 {published data only}
- Davis MP, Frandsen JL, Walsh D, Andresen S, Taylor S. Mirtazapine for pruritus. Journal of Pain and Symptom Management 2003;25(3):288‐91. [DOI] [PubMed] [Google Scholar]
Easton 1978 {published data only}
- Easton P, Galbraith PR. Cimetidine treatment of pruritus in polycythemia vera. New England Journal of Medicine 1978;299(20):1134. [DOI] [PubMed] [Google Scholar]
Fjellner 1979 {published data only}
- Fjellner B, Hagermark O. Pruritus in polycythemia vera: treatment with aspirin and possibility of platelet involvement. Acta Dermato‐Venereologica 1979;59(6):505‐12. [DOI] [PubMed] [Google Scholar]
Ghorbani 2011 {published data only}
- Ghorbani AR, Feily A, Khalili A, Dormanesh B. Lack of efficacy of topical calcineurin inhibitor pimecrolimus 1% on pruritus of severely uremic patients: a randomized double‐blind study in 60 patients. Dermatitis 2011;22(3):167‐8. [PubMed] [Google Scholar]
Giovanetti 1995 {published data only}
- Giovannetti S, Barsotti G, Cupisti A, Dani L, Bandini S, Angelini D, et al. Oral activated charcoal in patients with uremic pruritus. Nephron 1995;70(2):193‐6. [DOI] [PubMed] [Google Scholar]
Goicoechea 1999 {published data only}
- Goicoechea M, Sequera P, Ochando A, Andrea C, Caramelo C. Uremic pruritus: an unresolved problem in hemodialysis patients. Nephron 1999;82(1):73‐4. [DOI] [PubMed] [Google Scholar]
Goncalves 2010 {published data only}
- Goncalves F. Thalidomide for the control of severe paraneoplastic pruritus associated with Hodgkin's disease. American Journal of Hospice & Palliative Medicine 2010;27(7):486‐7. [DOI] [PubMed] [Google Scholar]
Hellier 1963 {published data only}
- Hellier FF. A comparative trial of trimeprazine and amylobarbitone in pruritus. Lancet 1963;1(7279):471‐2. [DOI] [PubMed] [Google Scholar]
Jones 2005 {published data only}
- Jones EA, Zylicz Z. Treatment of pruritus caused by cholestasis with opioid antagonists. Journal of Palliative Medicine 2005;8(6):1290‐4. [DOI] [PubMed] [Google Scholar]
Jones 2007 {published data only}
- Jones EA, Molenaar HA, Oosting J. Ondansetron and pruritus in chronic liver disease: a controlled study. Hepato‐Gastroenterology 2007;54(76):1196‐9. [PubMed] [Google Scholar]
Juby 1994 {published data only}
- Juby LD, Wong VS, Losowsky MS. Buprenorphine and hepatic pruritus. British Journal of Clinical Practice 1994;48(6):331. [PubMed] [Google Scholar]
Kato 2001 {published data only}
- Kato A, Takita T, Furuhashi M, Takahashi T, Watanabe T, Maruyama Y, et al. Polymethylmethacrylate efficacy in reduction of renal itching in hemodialysis patients: crossover study and role of tumor necrosis factor‐alpha. Artificial Organs 2001;25(6):441‐7. [DOI] [PubMed] [Google Scholar]
Korfitis 2008 {published data only}
- Korfitis C, Trafalis DT. Carbamazepine can be effective in alleviating tormenting pruritus in patients with hematologic malignancy. Journal of Pain and Symptom Management 2008;35(6):571‐2. [DOI] [PubMed] [Google Scholar]
Kuypers 2004 {published data only}
- Kuypers DR, Claes K, Evenepoel P, Maes B, Vanrenterghem Y. A prospective proof of concept study of the efficacy of tacrolimus ointment on uraemic pruritus (UP) in patients on chronic dialysis therapy. Nephrology, Dialysis, Transplantation 2004;19(7):1895‐901. [DOI] [PubMed] [Google Scholar]
Lysy 2003 {published data only}
- Lysy J, Sistiery‐Ittah M, Israelit Y, Shmueli A, Strauss‐Liviatan N, Mindrul V, et al. Topical capsaicin‐a novel and effective treatment for idiopathic intractable pruritus ani: a randomised, placebo controlled, crossover study. Gut 2003;52(9):1323‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mansour‐Ghanaei 2006 {published data only}
- Mansour‐Ghanaei F, Taheri A, Froutan H, Ghofrani H, Nasiri‐Toosi M, Bagherzadeh AH, et al. Effect of oral naltrexone on pruritus in cholestatic patients. World Journal of Gastroenterology 2006;12(7):1125‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Marquez 2012 {published data only}
- Marquez D, Ramonda C, Lauxmann JE, Romero CA, Vukelic VL, Martinatto C, et al. Uremic pruritus in hemodialysis patients: treatment with desloratidine versus gabapentin. Jornal Brasileiro Nefrologia 2012;34(2):148‐52. [DOI] [PubMed] [Google Scholar]
Metze 1999a {published data only}
- Metze D, Reimann S, Beissert S, Luger T. Efficacy and safety of naltrexone, an oral opiate receptor antagonist, in the treatment of pruritus in internal and dermatological diseases. Journal of the American Academy of Dermatology 1999;41:533‐9. [PubMed] [Google Scholar]
Montero 2006 {published data only}
- Montero JL, Pozo JC, Barrera P, Fraga E, Costán G, Domínguez JL, et al. Treatment of refractory cholestatic pruritus with molecular adsorbent recirculating system (MARS). Transplantation Proceedings 2006;38(8):2511‐3. [DOI] [PubMed] [Google Scholar]
Müller 1998a {published data only}
- Müller C, Pongratz S, Pidlich J, Penner E, Kaider A, Schemper M, et al. Treatment of pruritus in chronic liver disease with the 5‐hydroxytryptamine receptor type 3 antagonist ondansetron: a randomized, placebo‐controlled, double‐blind cross‐over trial. European Journal of Gastroenterology & Hepatology 1998;10(10):865‐70. [DOI] [PubMed] [Google Scholar]
Müller 1998b {published data only}
- Müller C, Pongratz S, Pidlich J, Penner E, Kaider A, Schemper M, et al. Treatment of pruritus in chronic liver disease with the 5‐hydroxytryptamine receptor type 3 antagonist ondansetron: a randomized, placebo‐controlled, double‐blind cross‐over trial. European Journal of Gastroenterology & Hepatology 1998;10:865‐70. [DOI] [PubMed] [Google Scholar]
Podesta 1991b {published data only}
- Podesta A, Lopez P, Terg R, Villamil F, Flores D, Mastai R, et al. Treatment of pruritus of primary biliary cirrhosis with rifampin. Digestive Diseases and Sciences 1991;36(2):216‐20. [DOI] [PubMed] [Google Scholar]
Price 1998 {published data only}
- Price TJ, Patterson WK, Olver IN. Rifampicin as treatment for pruritus in malignant cholestasis. Supportive Care in Cancer 1998;6(6):533‐5. [DOI] [PubMed] [Google Scholar]
Prieto 2004 {published data only}
- Prieto LN. The use of midazolam to treat itching in a terminally ill patient with biliary obstruction. Journal of Pain and Symptom Management 2004;28(6):531‐2. [DOI] [PubMed] [Google Scholar]
Razeghi 2009 {published data only}
- Razeghi E, Eskandari D, Ganji MR, Meysamie AP, Togha M, Khashayar P. Gabapentin and uremic pruritus in hemodialysis patients. Renal Failure 2009;31(2):85‐90. [DOI] [PubMed] [Google Scholar]
Rifai 2006 {published data only}
- Rifai K, Hafer C, Rosenau J, Athmann C, Haller H, Peter Manns M, et al. Treatment of severe refractory pruritus with fractionated plasma separation and adsorption (Prometheus ®). Scandinavian Journal of Gastroenterology 2006;41(10):1212‐7. [DOI] [PubMed] [Google Scholar]
Sja'bani 1997 {published data only}
- Sja'bani M, Asdie AH. Effect of erythropoietin on pruritus and quality of life in chronic hemodialyzed end stage renal disease patients. Journal of Clinical Epidemiology 1997;50(Suppl 1):10S. [Google Scholar]
Ständer 2009 {published data only}
- Ständer S, Böckenholt B, Schürmeyer‐Horst F, Weishaupt C, Heuft G, Luger TA, et al. Treatment of chronic pruritus with the selective serotonin re‐uptake inhibitors paroxetine and fluvoxamine: results of an open‐labelled, two‐arm proof‐of‐concept study. Acta Dermato‐Venereologica 2009;89(1):45‐51. [DOI] [PubMed] [Google Scholar]
Szepietowski 2005 {published data only}
- Szepietowski JC, Szepietowski T, Reich A. Efficacy and tolerance of the cream containing structured physiological lipids with endocannabinoids in the treatment of uremic pruritus: a preliminary study. Acta Dermatovenerologica Croatica 2005;13(2):97‐103. [PubMed] [Google Scholar]
Tokgöz 2005 {published data only}
- Tokgöz B, Ata A, Sİpahİoğlu M, Oymak O, Utaş S, Utaş C. Effects of oral granisetron treatment on uremic pruritus. Turkish Journal of Medical Sciences 2005;35:93‐7. [Google Scholar]
References to ongoing studies
2005‐003469‐18 {unpublished data only}
- 2005‐003469‐18. TRK‐820 UP 5µg [A randomised, double‐blind, placebo‐controlled study of TRK‐820 in haemodyalis patients with uremic pruritus]. clinicaltrialsregister.eu/ctr‐search/trial/2005‐003469‐18/LT 2005‐09‐07.
2009‐010437‐50 {unpublished data only}
- 2009‐010437‐50. NA [A Phase 2, randomized, double‐blind, placebo‐controlled study to evaluate the efficacy of different Gabapentin doses in haemodialysis patients with uremic pruritus]. clinicaltrialsregister.eu/ctr‐search/trial/2009‐010437‐50/IT 2009‐03‐27.
ISRCTN13971661 {unpublished data only}
- ISRCTN13971661. Balneum Plus cream for the treatment of itchy skin in renal patients. isrctn.com/ISRCTN13971661 2014‐12‐17.
NCT00577967 {unpublished data only}
- NCT00577967. Gabapentin ‐ A Solution to Uremic Pruritus? [Gabapentin ‐ A Solution to Uremic Pruritus? A Prospective, Randomized, Placebo‐controlled, Double‐blind Study]. www.clinicaltrials.gov/ct2/show/NCT00577967 2007‐12‐19.
NCT00693654 {unpublished data only}
- NCT00693654. Comparative Study of the Efficacy of SARNA Sensitive Lotion for Treatment of Uremic Itch in Adult Hemodialysis Patients [A Controlled Comparative Study of the Efficacy of SARNA Sensitive Lotion for Treatment of Uremic Pruritus in Adult Hemodialysis Patients]. clinicaltrials.gov/ct2/show/NCT00693654 2008‐06‐05.
NCT00793156 {unpublished data only}
- NCT00793156. A Randomized‐Withdrawal Phase 3 Study Evaluating the Safety and Efficacy of Oral Nalfurafine HCl (AC‐820)in Subjects on Hemodialysis With Uremic Pruritus (Renal Itch) (AC120‐8231) [A Randomized‐Withdrawal Phase 3 Study Evaluation the Safety and Efficacy of Oral Nalfurafine HCl (AC‐820)in Subjects on Hemodialysis With Uremic Pruritus (Renal Itch)]. clinicaltrials.gov/ct2/show/NCT00793156 2008‐11‐17.
NCT01073501 {unpublished data only}
- NCT01073501. Efficacy of Pregabalin in the Management of Chronic Uremic Pruritus. clinicaltrials.gov/ct2/show/NCT01073501 2010‐02‐17.
NCT01513161 {unpublished data only}
- NCT01513161. Efficacy and Safety Study of TRK‐820 to Treat Conventional‐treatment‐resistant Pruritus in Patients Receiving Hemodialysis (TRK‐820) [A 14 Day, Multi‐center, Randomized, Double Blinded, Placebo‐controlled, Parallel Group, Fixed Dose, Phase III Clinical Trial to Assess the Efficacy and Safety of TRK‐820 in Treating Conventional‐treatment‐resistant Pruritus in Patients Receiving Hemodialysis]. clinicaltrials.gov/ct2/show/NCT01513161 2010‐11‐23.
NCT01660243 {published data only}
- NCT01660243. Efficacy and Safety of MT‐9938 for Treatment of Uremic Pruritus in Subjects With End‐stage Renal Disease Receiving Hemodialysis [A Phase 2, Randomized, Double‐blind, Placebo‐controlled, Fixed‐dose, Parallel‐group, Multicenter, Efficacy, and Safety Study of MT‐9938 for Treatment of Uremic Pruritus in Subjects With End‐stage Renal Disease Receiving Hemodialysis]. clinicaltrials.gov/ct2/show/NCT01660243 2012‐08‐02.
NCT01852318 {unpublished data only}
- NCT01852318. Pregabalin for the Treatment of Uremic Pruritus [A Multicenter, Double‐blind, Randomized, Placebo and Active‐controlled Study of Pregabalin for the Treatment of Uremic Pruritus]. clinicaltrials.gov/ct2/show/NCT01852318 2013‐05‐08.
NCT02008864 {unpublished data only}
- NCT02008864. Evaluating the Effect of Senna in Uremic Pruritus [Phase II Study of the Effect of Senna Alexandrina Mill. on Uremic Pruritus and Serum IL‐2, INF‐δ and TNF‐α Levels of Hemodialysed Patients]. clinicaltrials.gov/ct2/show/NCT02008864 2013‐12‐01.
NCT02032537 {unpublished data only}
- NCT02032537. Efficacy of Calmmax Cream in the Management of Chronic Uremic Pruritus. clinicaltrials.gov/ct2/show/NCT02032537 2014‐01‐06.
NCT02143648 {unpublished data only}
- NCT02143648. Study of Nalbuphine HCl ER Tablets in Hemodialysis Patients With Uremic Pruritus [A Randomized, Double‐Blind, Placebo‐Controlled, Parallel, 3‐Arm Study of the Safety and Anti‐Pruritic Efficacy of Nalbuphine HCl ER Tablets in Hemodialysis Patients With Uremic Pruritus]. clinicaltrials.gov/ct2/show/NCT02143648 2014‐05‐17.
NCT02229929 {unpublished data only}
- NCT02229929. Safety and Pharmacokinetics of IV CR845 in Hemodialysis Patients, and Its Efficacy in Patients With Uremic Pruritus [A Double‐Blind, Randomized, Placebo‐Controlled Study to Evaluate the Safety and Pharmacokinetics of Intravenous CR845 in Hemodialysis Patients, and Its Safety and Efficacy in Hemodialysis Patients With Uremic Pruritus]. clinicaltrials.gov/ct2/show/NCT02229929 2014‐08‐27.
NCT02696499 {unpublished data only}
- NCT02696499. Treatment of Uremic Pruritus With PA101B [Treatment of Uremic Pruritus With Inhaled PA101B in Patients With End‐Stage Renal Disease Requiring Hemodialysis]. clinicaltrials.gov/ct2/show/NCT02696499 2016‐02‐25.
NCT02701166 {unpublished data only}
- NCT02701166. The Effect of Bezafibrate on Cholestatic Itch (FITCH) [The Effect of Bezafibrate on Cholestatic Itch]. clinicaltrials.gov/ct2/show/NCT02701166 2016‐03‐02.
Additional references
Ahern 2012
- Ahern K, Gilmore ES, Poligone B. Pruritus in cutaneous T‐cell lymphoma: a review. Journal of the American Academy of Dermatology 2012;67(4):760‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Anand 2013
- Anand S. Gabapentin for pruritus in palliative care. American Journal of Hospice & Palliative Care 2013;30(2):192‐6. [DOI] [PubMed] [Google Scholar]
Bergasa 2008
- Bergasa NV. Pruritus in primary biliary cirrhosis: pathogenesis and therapy. Clinics in Liver Disease 2008;12(2):385‐406. [DOI] [PubMed] [Google Scholar]
Bernhard 2005
- Bernhard JD. Itch and pruritus: what are they, and how should itches be classified?. Dermatologic Therapy 2005;18:288‐91. [DOI] [PubMed] [Google Scholar]
Bohlius 2009
- Bohlius J, Schmidlin K, Brillant C, Schwarzer G, Trelle S, Seidenfeld J, et al. Erythropoietin or darbepoetin for patients with cancer‐‐meta‐analysis based on individual patient data. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD007303.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chiang 2011
- Chaing HC, Huang V, Cornelius LA. Cancer and itch. Seminars in Cutaneous Medicine and Surgery 2011;30(2):107‐12. [DOI] [PubMed] [Google Scholar]
Cohen 1988
- Cohen J. Statistical Power Analysis for the Behavioral Sciences. United States of America: Erlbaum, 1988. [Google Scholar]
Derry 2012
- Derry S, Moore RA. Topical capsaicin (low concentration) for chronic neuropathic pain in adults. Cochrane Database of Systematic Reviews 2012, Issue 9. [DOI: 10.1002/14651858.CD010111] [DOI] [PMC free article] [PubMed] [Google Scholar]
Derry 2013
- Derry S, Rice ASC, Cole P, Tan T, Moore RA. Topical capsaicin (high concentration) for chronic neuropathic pain in adults. Cochrane Database of Systematic Reviews 2013, Issue 2. [DOI: 10.1002/14651858.CD007393.pub3] [DOI] [PubMed] [Google Scholar]
Dorman 2010
- Dorman S, Perkins P. Droperidol for treatment of nausea and vomiting in palliative care patients. Cochrane Database of Systematic Reviews 2010;10:CD006938. [DOI: 10.1002/14651858.CD006938.pub2] [DOI] [PubMed] [Google Scholar]
Duley 2010
- Duley L, Gülmezoglu AM, Henderson‐Smart DJ, Chou D. Magnesium sulphate and other anticonvulsants for women with pre‐eclampsia. Cochrane Database of Systematic Reviews 2010, Issue 11. [DOI: 10.1002/14651858.CD000025.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Duo 1987
- Duo LJ. Electrical needle therapy of uremic pruritus. Nephron 1987;47(3):179‐83. [DOI] [PubMed] [Google Scholar]
Elmariah 2011
- Elmariah SB, Lerner EA. Topical therapies for pruritus. Seminars in Cutaneous Medicine and Surgery 2011;30(2):118‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fransway 1988
- Fransway AF, Winkelmann RK. Treatment of pruritus. Seminars in Dermatology 1988;7(4):310‐25. [PubMed] [Google Scholar]
Gaudy‐Marqueste 2010
- Gaudy‐Marqueste C. Antihistamines. In: Misery L, Ständer S editor(s). Pruritus. London: Springer, 2010:277‐88. [Google Scholar]
Gooding 2010
- Gooding SM, Canter PH, Coelho HF, Boddy K, Ernst E. Systematic review of topical capsaicin in the treatment of pruritus. International Journal of Dermatology 2010;49(8):858‐65. [DOI] [PubMed] [Google Scholar]
Gowing 2010
- Gowing L, Ali R, White JM. Opioid antagonists under heavy sedation or anaesthesia for opioid withdrawal. Cochrane Database of Systematic Reviews 2010, Issue 1. [DOI: 10.1002/14651858.CD002022.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADE Handbook
- Schünemann H, Brożek J, Guyatt G, Oxman A editor(s). GRADE handbook for grading quality of evidence and strength of recommendations (updated October 2013). The GRADE Working Group, 2013. Available from guidelinedevelopment.org/handbook.
GRADEproGDT 2015 [Computer program]
- GRADE Working Group, McMaster University. GRADEproGDT. Version accessed prior to 4 October 2016. Hamilton (ON): GRADE Working Group, McMaster University, 2015.
Guyatt 2013a
- Guyatt GH, Oxman AD, Santesso N, Helfand M, Vist G, Kunz R, et al. GRADE guidelines: 12. Preparing summary of findings tables‐binary outcomes. Journal of Clinical Epidemiology 2013;66:158‐72. [DOI] [PubMed] [Google Scholar]
Guyatt 2013b
- Guyatt GH, Thorlund K, Oxman AD, Walter SD, Patrick D, Furukawa TA, et al. GRADE guidelines: 13. Preparing summary of findings tables and evidence profiles‐continuous outcomes. Journal of Clinical Epidemiology 2013;66:173‐83. [DOI] [PubMed] [Google Scholar]
Haffenreffer 1660
- Haffenreffer S. On pruritus [De pruriti, in Nosodochium, in quo cutis, eique adhaerentium partium, affectus omnes, singulari methodo, et cognoscendi et curandi fidelissime traduntur]. De Pruriti. Ulm: Haffenreffer S, 1660:98‐102. [Google Scholar]
Hedayati 2005
- Hedayati H, Parsons J, Crowther CA. Topically applied anaesthetics for treating perineal pain after childbirth. Cochrane Database of Systematic Reviews 2005, Issue 2. [DOI: 10.1002/14651858.CD004223.pub2] [DOI] [PubMed] [Google Scholar]
Higgins 2002
- Higgins JPT, Thompson SG. Quantifying heterogeneity in a meta‐analysis. Statistics in Medicine 2002;21(11):1539‐58. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S, editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org. The Cochrane Collaboration.
Higginson 1997
- Higginson IJ, Hearn J. A multicenter evaluation of cancer pain control by palliative care teams. Journal of Pain and Symptom Management 1997;14(1):29‐35. [DOI] [PubMed] [Google Scholar]
Ikoma 2006
- Ikoma A, Steinhoff M, Stander S, Yosipovitch G, Schmelz M. The neurobiology of itch. Nature Reviews. Neuroscience 2006;7(7):535‐47. [DOI] [PubMed] [Google Scholar]
Inui 2015
- Inui S. Nalfurafine hydrochloride to treat pruritus: a review. Clinical, Cosmetic and Investigational Dermatology 2015;8:249‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jones 1999
- Jones EA, Bergasa NV. The pruritus of cholestasis. Hepatology 1999;29(4):1003‐6. [DOI] [PubMed] [Google Scholar]
Kantor 1983
- Kantor GR, Lookingbill DP. Generalized pruritus and systemic disease. Journal of American Medical Dermatology 1983;9(3):375‐82. [DOI] [PubMed] [Google Scholar]
Khandelwal 1994
- Khandelwal M, Malet PF. Pruritus associated with cholestasis. A review of pathogenesis and management. Digestive Diseases and Sciences 1994;39(1):1‐8. [DOI] [PubMed] [Google Scholar]
Khurana 2006
- Khurana S, Singh P. Rifampin is safe for treatment of pruritus due to chronic cholestasis: a meta‐analysis of prospective randomized‐controlled trials. Liver International 2006;26(8):943‐8. [DOI] [PubMed] [Google Scholar]
Krajnik 2001a
- Krajnik M, Zylicz Z. Understanding pruritus in systemic disease. Journal of Pain and Symptom Management 2001;21(2):151‐68. [DOI] [PubMed] [Google Scholar]
Krajnik 2001b
- Krajnik M, Zylicz Z. Pruritus in advanced internal diseases. Pathogenesis and treatment. Netherlands Journal of Medicine 2001;58(1):27‐40. [DOI] [PubMed] [Google Scholar]
Langner 2009
- Langner MD, Maibach HI. Pruritus measurement and treatment. Clinical and Experimental Dermatology 2009;34(3):285‐8. [DOI] [PubMed] [Google Scholar]
Larijani 1996
- Larijani GE, Goldberg ME, Rogers KH. Treatment of opioid‐induced pruritus with ondansetron: report of four patients. Pharmacotherapy 1996;16(5):958‐60. [PubMed] [Google Scholar]
Lober 1988
- Lober CW. Should the patient with generalized pruritus be evaluated for malignancy?. Journal of the American Academy of Dermatology 1988;19(2 Pt 1):350‐2. [DOI] [PubMed] [Google Scholar]
Lowrie 1975
- Lowrie EG, Lazarus JM, Hampers CL, Merrill JP. Some statistical methods for use in assessing the adequacy of hemodialysis. Kidney International Supplement 1975;2:231‐42. [PubMed] [Google Scholar]
Manenti 2009
- Manenti L, Tansinda P, Vaglio A. Uraemic pruritus: clinical characteristics, pathophysiology and treatment. Drugs 2009;69(3):251‐63. [DOI] [PubMed] [Google Scholar]
McHugh 1995
- McHugh SM, Rifkin IR, Deighton J, Wilson AB, Lachmann PJ, Lockwood CM, et al. The immunosuppressive drug thalidomide induces T helper cell type 2 (Th2) and concomitantly inhibits Th1 cytokine production in mitogen‐ and antigen‐stimulated human peripheral blood mononuclear cell cultures. Clinical & Experimental Immunology 1995;99(2):160‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mela 2003
- Mela M, Mancuso A, Burroughs AK. Review article: pruritus in cholestatic and other liver diseases. Alimentary Pharmacology & Therapeutics 2003;17(7):857–70. [DOI] [PubMed] [Google Scholar]
Melin 1986
- Melin L, Frederikson T, Noren P, Swebilius BG. Behavioural treatment of scratching in patients with atopic dermatitis. British Journal of Dermatology 1986;115(4):467–74. [DOI] [PubMed] [Google Scholar]
Mettang 1990
- Mettang T, Fritz P, Weber J, Machleidt C, Hubl E, Kuhlmann U. Uremic pruritus in patients on hemodialysis or continuous ambulatory peritoneal dialysis (CAPD): the role of plasma histamine and skin mast cells. Clinical Nephrology 1990;34(3):136‐41. [PubMed] [Google Scholar]
Mettang 2002
- Mettang T, Pauli‐Magnus C, Alscher DM. Uraemic pruritus – new perspectives and insights from recent trials. Nephrology, Dialysis, Transplantation 2002;17(9):1558–63. [DOI] [PubMed] [Google Scholar]
Mettang 2010
- Mettang T. Chronic kidney disease‐associated pruritus. In: Misery L, Ständer S editor(s). Pruritus. London: Springer, 2010:167‐77. [Google Scholar]
Metz 2010
- Metz M, Ständer S. Chronic pruritus ‐ pathogenesis, clinical aspects and treatment. Journal of the European Academy of Dermatology and Venereology 2010;24(11):1249‐60. [DOI] [PubMed] [Google Scholar]
Metze 1999b
- Metze D, Reimann S, Luger TA. Effective treatment of pruritus with naltrexone, an orally active opiate antagonist. Annals of the New York Academy of Sciences 1999;885:430‐2. [DOI] [PubMed] [Google Scholar]
Misery 2010
- Misery L, Städer S. Pruritus. London: Springer, 2010. [Google Scholar]
Moore 2009
- Moore RA, Straube S, Wiffen PJ, Derry S, McQuay HJ. Pregabalin for acute and chronic pain in adults. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD007076.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moretti 2010
- Moretti S, Prignano F, Lotti T. Systemic Immunosuppressants in the treatment of pruritus. In: Misery L, Ständer S editor(s). Pruritus. London: Springer, 2010:307‐10. [Google Scholar]
Narita 2006
- Narita I, Alchi B, Omori K, Sato F, Ajiro J, Saga D, et al. Etiology and prognostic significance of severe uremic pruritus in chronic hemodialysis patients. Kidney International 2006;69(9):1626‐32. [DOI] [PubMed] [Google Scholar]
Perkins 2009
- Perkins P, Dorman S. Haloperidol for the treatment of nausea and vomiting in palliative care patients. Cochrane Database of Systematic Reviews 2009, Issue 2. [DOI: 10.1002/14651858.CD006271.pub2] [DOI] [PubMed] [Google Scholar]
Phan 2010
- Phan NQ, Bernhard JD, Luger TA, Ständer S. Antipruritic treatment with systemic μ‐opioid receptor antagonists: a review. Journal of the American Academy of Dermatology 2010;63(4):680‐8. [DOI] [PubMed] [Google Scholar]
Potter 2003
- Potter J, Hami F, Bryan T, Quigley C. Symptoms in 400 patients referred to palliative care services: prevalence and patterns. Palliative Medicine 2003;17(4):310‐4. [DOI] [PubMed] [Google Scholar]
Proske 2010
- Proske S, Hartschuh W. Anal Pruritus. In: Misery L, Ständer S editor(s). Pruritus. 1st Edition. London: Springer, 2010:125‐8. [Google Scholar]
R Foundation 2015 [Computer program]
- The R Foundation. R. Version 3.2.2. The R Foundation, 2015. Available from www.r‐project.org/.
Raap 2012
- Raap U, Kapp A, Darsow U. Antidepressant drugs: a reasonable therapy for pruritus?. Der Hautarzt 2012;63(7):553‐7. [DOI] [PubMed] [Google Scholar]
Ratilal 2005
- Ratilal B, Costa J, Sampaio C. Anticonvulsants for preventing seizures in patients with chronic subdural haematoma. Cochrane Database of Systematic Reviews 2005, Issue 3. [DOI: 10.1002/14651858.CD004893.pub2] [DOI] [PubMed] [Google Scholar]
Reich 2008
- Reich A, Heisig M, Szepietowski JC. Visual analogue scale as a validated assessment of pruritus intensity (Abstract OP03; 5th International Workshop for the Study of Itch). Acta Dermato‐Venereologica. 2008:688.
RevMan 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Rösner 2010
- Rösner S, Hackl‐Herrwerth A, Leucht S, Vecchi S, Srisurapanont M, Soyka M. Opioid antagonists for alcohol dependence. Cochrane Database of Systematic Reviews 2010, Issue 12. [DOI: 10.1002/14651858.CD001867.pub3] [DOI] [PubMed] [Google Scholar]
Schmelz 1997
- Schmelz M, Schmidt R, Bickel A, Handwerker HO, Torebjork HE. Specific C‐receptors for itch in human skin. Journal of Neuroscience 1997;17(20):8003‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schwarzer 2015
- Schwarzer G, Carpenter JR, Rücker G. Meta‐Analysis with R. Heidelberg: Springer, 2015. [Google Scholar]
Schworer 1995
- Schworer H, Hartmann H, Ramadori G. Relief of cholestatic pruritus by a novel class of drugs: 5‐hydroxytryptamine type 3 (5‐HT3) receptor antagonists: effectiveness of ondansetron. Pain 1995;61(1):33‐7. [DOI] [PubMed] [Google Scholar]
Seccareccia 2011
- Seccareccia D, Gebara N. Pruritus in palliative care. Canadian Family Physician 2011;57(9):1010‐3. [PMC free article] [PubMed] [Google Scholar]
Seiz 1999
- Seiz A, Yarbro C. Pruritus. In: Yarbro CH, Frogge M, Goodman M editor(s). Cancer Symptom Management. Boston: Jones and Bartlett, 1999:148‐60. [Google Scholar]
Sharp 1967
- Sharp HL, Carey JBJ, White JG, Krivit W. Cholestyramine therapy in patients with a paucity of intrahepatic bile ducts. Journal of Pediatrics 1967;71(5):723‐36. [DOI] [PubMed] [Google Scholar]
Siemens 2014
- Siemens W, Xander C, Meerpohl JJ, Antes G, Becker G. Drug treatments for pruritus in adult palliative care. Deutsches Ärzteblatt International 2014;111(50):863‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Smith 1997b
- Smith PF, Corelli RL. Doxepin in the management of pruritus associated with allergic cutaneous reactions. Annals of Pharmacotherapy 1997;31(5):633‐5. [PubMed] [Google Scholar]
Steinhoff 2006
- Steinhoff M, Bienenstock J, Schmelz M, Maurer M, Wei E, Biro T. Neurophysiological, neuroimmunological, and neuroendocrine basis of pruritus. Journal of Investigative Dermatology 2006;126(8):1705‐18. [DOI] [PubMed] [Google Scholar]
Steinhoff 2011
- Steinhoff M, Cevikbas F, Ikoma A, Berger TG. Pruritus: management algorithms and experimental therapies. Seminars in Cutaneous Medicine and Surgery 2011;30(2):127‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Stovold 2014
- Stovold E, Beecher D, Foxlee R, Noel‐Storr A. Study flow diagrams in Cochrane systematic review updates: an adapted PRISMA flow diagram. Systematic Reviews 2014;3:1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ständer 2008
- Ständer S, Weisshaar E, Luger TA. Neurophysiological and neurochemical basis of modern pruritus treatment. Experimental Dermatology 2008;17(3):161‐9. [DOI] [PubMed] [Google Scholar]
Summey 2005
- Summey BTJ, Yosipovitch G. Pharmacologic advances in the systemic treatment of itch. Dermatologic Therapy 2005;18(4):328‐32. [DOI] [PubMed] [Google Scholar]
Summey 2009
- Summey B. Pruritus. In: Walsh DT, Caraceni AT, Fainsinger R, et al. editor(s). Palliative Medicine. Philadelphia: Saunders Elsevier, 2009:910‐3. [Google Scholar]
Suthanthiran 1994
- Suthanthiran M, Strom TB. Renal transplantation. New England Journal of Medicine 1994;331(6):365‐76. [DOI] [PubMed] [Google Scholar]
Szepietowski 2004
- Szepietowski JC, Salomon J. Pruritus: still an important clinical problem. Journal of American Academic Dermatology 2004;51(5):842‐3. [DOI] [PubMed] [Google Scholar]
Tandon 2007
- Tandon P, Rowe BH, Vandermeer B, Bain VG. The efficacy and safety of bile acid binding agents, opioid antagonists, or rifampin in the treatment of cholestasis‐associated pruritus. American Journal of Gastroenterology 2007;102(7):1528‐36. [DOI] [PubMed] [Google Scholar]
Tennyson 2001
- Tennyson H, Levine N. Neurotropic and psychotropic drugs in dermatology. Dermatologic Clinics 2001;19(1):179‐97. [DOI] [PubMed] [Google Scholar]
To 2012
- To TH, Clark K, Lam L, Shelby‐James T, Currow DC. The role of ondansetron in the management of cholestatic or uremic pruritus‐‐a systematic review. Journal of Pain and Symptom Management 2012;44(5):725‐30. [DOI] [PubMed] [Google Scholar]
Trauner 2005
- Trauner M, Wagner M, Fickert P, Zollner G. Molecular regulation of hepatobiliary transport systems: clinical implications for understanding and treating cholestasis. Journal of Clinical Gastroenterology 2005;39(4 Suppl 2):111‐24. [DOI] [PubMed] [Google Scholar]
Turk 2008
- Turk DC, Dworkin RH, McDermott MP, Bellamy N, Kurke LB, Chandler JM, et al. Analyzing multiple endpoints in clinical trials of pain treatments: IMMPACT recommendations. Pain 2008;139(3):485‐93. [DOI] [PubMed] [Google Scholar]
Turner 1990
- Turner IB, James O, Wood P, Ward A, Nagy JT, Kawlins MD. Flumecinol is a powerful inducer of bilirubin conjugation in Gilbert’s syndrome. [Abstract]. Hepatology 1990;12(2):415. [Google Scholar]
Twycross 2001
- Twycross R, Wilcock A. Symptom Management in Advanced Cancer. Oxford: Radcliffe Publishing, 2001. [Google Scholar]
Twycross 2004
- Twycross R. Pruritus: past, present, and future. In: Zylicz Z, Twycross R, Jones EA editor(s). Pruritus in Advanced Disease. Oxford: Oxford University Press, 2004:191‐9. [Google Scholar]
Ueno 2013
- Ueno Y, Mori A, Yanagita T. One year long‐term study on abuse liability of nalfurafine in hemodialysis patients. International Journal of Clinical Pharmacology and Therapeutics 2013;51(11):823‐31. [DOI] [PubMed] [Google Scholar]
Uthayakumar 1997
- Uthayakumar S, Nandwani R, Drinkwater T, Nayagam AT, Darley CR. The prevalence of skin disease in HIV infection and its relationship to the degree of immunosuppression. British Journal of Dermatology 1997;137(4):595‐8. [DOI] [PubMed] [Google Scholar]
Wallengren 2010
- Wallengren J. Measurement of itch. In: Misery L, Ständer S editor(s). Pruritus. London: Springer, 2010:45‐50. [Google Scholar]
Walsh 2000
- Walsh D, Donnelly S, Rybicki L. The symptoms of advanced cancer: relationship to age, gender, and performance status in 1000 patients. Supportive Care in Cancer 2000;8(3):175‐9. [DOI] [PubMed] [Google Scholar]
Wang 2010
- Wang H, Yosipovitch G. New insights into the pathophysiology and treatment of chronic itch in patients with end‐stage renal disease,chronic liver disease, and lymphoma. International Journal of Dermatology 2010;49(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Watts 2012
- Watts K, Chavasse RJ. Leukotriene receptor antagonists in addition to usual care for acute asthma in adults and children. Cochrane Database of Systematic Reviews 2012, Issue 5. [DOI: 10.1002/14651858.CD006100.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Webster 2005
- Webster A, Woodroffe RC, Taylor RS, Chapman JR, Craig JC. Tacrolimus versus cyclosporin as primary immunosuppression for kidney transplant recipients. Cochrane Database of Systematic Reviews 2005, Issue 4. [DOI: 10.1002/14651858.CD003961.pub2] [DOI] [PubMed] [Google Scholar]
Wee 2008
- Wee B, Hadley G, Derry S. How useful are systematic reviews for informing palliative care practice? Survey of 25 Cochrane systematic reviews. BMC Palliative Care 2008;7:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Weisshaar 2003
- Weisshaar E, Kucenic MJ, Fleischer AB. Pruritus ‐ a review. Acta Dermato‐Venereologica 2003;213:5‐32. [PubMed] [Google Scholar]
Weisshaar 2009
- Weisshaar E, Dalgard F. Epidemilogy of itch: adding the burden of skin morbidity. Acta Dermato‐Venereologica 2009;89:339‐50. [DOI] [PubMed] [Google Scholar]
Wiffen 2011
- Wiffen PJ, Derry S, Moore RA, McQuay HJ. Carbamazepine for acute and chronic pain in adults. Cochrane Database of Systematic Reviews 2011, Issue 1. [DOI: 10.1002/14651858.CD005451.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wilde 1996
- Wilde MI, Markham A. Ondansetron. A review of its pharmacology and preliminary clinical findings in novel applications. Drugs 1996;52(5):773‐94. [DOI] [PubMed] [Google Scholar]
Winkelmann 1964
- Winkelmann RK, Müller SA. Pruritus. Annual Review of Medicine 1964;15:53‐64. [DOI] [PubMed] [Google Scholar]
Winkelmann 1982
- Winkelmann RK. Pharmacologic control of pruritus. Medical Clinics of North America 1982;66(5):1119‐33. [DOI] [PubMed] [Google Scholar]
Yamaguchi 1999
- Yamaguchi T, Nagasawa T, Satoh M, Kuraishi Y. Itch‐associated response induced by intradermal serotonin through 5‐HT2 receptors in mice. Neuroscience Research 1999;35(2):77‐83. [DOI] [PubMed] [Google Scholar]
Yatzidis 1972
- Yatzidis H. Activated charcoal rediscovered. British Medical Journal 1972;4(5831):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ye 2001
- Ye JH, Ponnudurai R, Schaefer R. Ondansetron: a selective 5‐HT(3) receptor antagonist and its applications in CNS‐related disorders. CNS Drug Review 2001;7(2):199‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yosipovitch 2003
- Yosipovitch G, Greaves MW, Schmelz M. Itch. Lancet 2003;361(9358):690‐4. [DOI] [PubMed] [Google Scholar]
Zylicz 1998
- Zylicz Z, Smits C, Krajnik M. Paroxetine for pruritus in advanced cancer. Journal of Pain and Symptom Management 1998;16(2):121‐4. [DOI] [PubMed] [Google Scholar]
Zylicz 2004
- Zylicz Z, Twycross R, Jones AE. Pruritus in Advanced Disease. New York: Oxford University Press, 2004. [Google Scholar]
References to other published versions of this review
Xander 2013
- Xander C, Meerpohl JJ, Galandi D, Buroh S, Schwarzer G, Antes G, et al. Pharmacological interventions for pruritus in adult palliative care patients. Cochrane Database of Systematic Reviews 2013, Issue 6. [DOI: 10.1002/14651858.CD008320.pub2] [DOI] [PubMed] [Google Scholar]