

Abstract
Background
Many people with schizophrenia do not achieve a satisfactory treatment response with ordinary antipsychotic drug treatment. In these cases, various add‐on medications are used, and valproate is one of these.
Objectives
To examine whether:
1. valproate alone is an effective treatment for schizophrenia and schizoaffective psychoses; and
2. valproate augmentation of antipsychotic medication is an effective treatment for the same illnesses.
Search methods
We searched the Cochrane Schizophrenia Group’s Study‐Based Register of Trials (July 2002; February 2007; July 2012; March 04, 2016). We also contacted pharmaceutical companies and authors of relevant studies in order to identify further trials.
Selection criteria
We included all randomised controlled trials comparing valproate to antipsychotics or to placebo (or no intervention), whether as the sole agent or as an adjunct to antipsychotic medication for the treatment of people with schizophrenia or schizophrenia‐like psychoses.
Data collection and analysis
We independently inspected citations and, where possible, abstracts, ordered papers, and re‐inspected and quality‐assessed these. At least two review authors independently extracted data. We analysed dichotomous data using risk ratio (RR) and its 95% confidence intervals (CI). We analysed continuous data using mean differences (MD) and their 95% CI. We assessed risk of bias for included studies and used GRADE (Grading of Recommendations Assessment, Development and Evaluation) to create a 'Summary of findings' table.
Main results
The 2012 update search identified 19 further relevant studies, most of which were from China. Thus the review currently includes 26 studies with a total of 2184 participants. All trials examined the effectiveness of valproate as an adjunct to antipsychotics. With the exception of two studies, the studies were small, the participants and personnel were not blinded (neither was outcome assessment), and most were short‐term and incompletely reported.
For this update we prespecified seven main outcomes of interest: clinical response (clinically significant response, aggression/agitation), leaving the study early (acceptability of treatment, overall tolerability), adverse events (sedation, weight gain) and quality of life.
Adding valproate to antipsychotic treatment resulted in more clinically significant response than adding placebo to antipsychotic drugs (14 RCTs, n = 1049, RR 1.31, 95% CI 1.16 to 1.47, I2 = 12%, low‐quality evidence). However, this effect was removed after excluding open RCTs in a sensitivity analysis. In terms of acceptability of treatment (measured by the number of participants leaving the study early due to any reason) valproate was just as acceptable as placebo (11 RCTs, n = 951, RR 0.76, 95% CI 0.47 to 1.24, I2 = 55%). Also overall tolerability (measured by the number of participants leaving the study early for adverse events) between valproate and placebo was similar (6 RCTs, n = 974, RR 1.33, 95% CI 0.90 to 1.97, I2 = 0).
Participants in the valproate group were found to be less aggressive than the control group based on the Modified Overt Aggression Scale (3 RCTs, n = 186, MD ‐2.55, 95% CI ‐3.92 to ‐1.19, I2 = 82%, very low‐quality evidence). Participants receiving valproate more frequently experienced sedation (8 RCTs, n = 770, RR 1.38, 95% CI 1.07 to 1.79, I2 = 0, low‐quality evidence) but were no more likely to gain weight than those receiving placebo (4 RCTs, n = 427, RR 1.17, 95% CI 0.76 to 1.82, I2 = 0, low‐quality evidence). No study reported on the important outcome of quality of life.
Authors' conclusions
There is limited evidence, based on a number of trials, that the augmentation of antipsychotics with valproate may be effective for overall clinical response, and also for specific symptoms, especially in terms of excitement and aggression. However, this evidence was entirely based on open RCTs. Moreover, valproate was associated with a number of adverse events among which sedation and dizziness appeared significantly more frequently than in the control groups. Further randomised studies which are blinded are necessary before any clear recommendation can be made. Ideally these would focus on people with schizophrenia and aggression, on those with treatment‐resistant forms of the illness and on those with schizoaffective disorders.
Plain language summary
Valproate for schizophrenia
Review question
To review the effects of adding valproate to an anitpyschotic for the treatment of schizophrenia and schizophrenia‐like illnesses.
Background
The main treatment for schizophrenia is antipsychotic medication. Despite this treatment, about 30% of people will continue to experience some signs of illness. Other drugs are sometimes added to antipsychotic medication to attempt to reduce the symptoms that people experience. Valproate is one such drug and is typically used to treat epilepsy, to stabilise mood in people who have bipolar disorder and for people who have both schizophrenia and mood disorder (schizoaffective disorder).
Study characteristics
The review includes 26 studies, found through electronic searching of relevant databases, with a total of 2184 participants. All trials examined the effectiveness of valproate as an add on to antipsychotics. With the exception of two studies, the studies were small, and most of them were short‐term and poorly reported.
Key results
Data from the included trials showed that participants receiving valproate plus an antipsychotic had better clinical response, compared to those taking an antipsychotic with a placebo. However, this advantage was lost when lower‐quality trials were taken out of the analysis. Valproate was also indicated to be effective in controlling excitement and aggression. Acceptability and overall tolerability of the combined treatment was similar between treatment groups and did not cause more weight gain, however, adding valproate did cause greater sedation and dizziness. No trial reported effect on quality of life.
Quality of the evidence
Evidence is limited and firm conclusions cannot be made. For the main outcomes of interest, the review authors judged the quality of evidence to be low or very low quality, due to methodological issues in the reviewed studies. Most of them were small, short‐term and did not blind the participants or personnel. Large, double‐blind and long‐term randomised trials should be undertaken to properly determine the clinical effects of adding valproate to antipsychotic treatment for people with schizophrenia.
This summary was written by Ben Gray, Senior Peer Researcher, McPin Foundation. mcpin.org/
Summary of findings
Summary of findings for the main comparison. VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone.
| Valproate plus antipsychotics versus antipsychotics plus placebo or antipsychotics alone | ||||||
| Patient or population: people with schizophrenia Settings: inpatient/outpatient Intervention: Valproate plus antipsychotics versus antipsychotics plus placebo or antipsychotics alone | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Antipsychotics and valproate versus antipsychotics and placebo/no treatment | |||||
| Clinically significant response: important change As defined by each of the studies Follow‐up: short term | 550 per 1000 | 721 per 1000 (638 to 809) | RR 1.31 (1.16 to 1.47) | 1049 (14 studies) | ⊕⊕⊝⊝ low1 | |
| Leaving the study early: Acceptability of treatment Leaving the study early for any reason Follow‐up: short term | 364 per 1000 | 327 per 1000 (284 to 378) | RR 0.76 (0.47 to 1.24) | 951 (11 studies) | ⊕⊝⊝⊝ very low2,3,4,5 | |
| Leaving the study early: Overall tolerability Leaving the study early due adverse events Follow‐up: short term | 77 per 1000 | 102 per 1000 (69 to 150) | RR 1.33 (0.90 to 1.97) | 974 (6 studies) | ⊕⊕⊝⊝ low2,6 | |
| Clinical response: Aggression/agitation Mean Modified Overt Aggression Rating Scale. Scale from: 0‐16 Follow‐up: short term | The mean aggression in the control groups was 5.74 points | The mean aggression in the intervention groups was 2.55 lower (3.92 to 1.19 lower) | 186 (3 studies) | ⊕⊝⊝⊝ very low7,8 | ||
| Adverse events: Sedation Number of participants with sedation/somnolence/drowsiness Follow‐up: short term | 186 per 1000 | 276 per 1000 (214 to 356) | RR 1.38 (1.07 to 1.79) | 770 (8 studies) | ⊕⊕⊝⊝ low1 | |
| Adverse events: Weight gain Number of participants with weight gain Follow‐up: short term | 143 per 1000 | 169 per 1000 (109 to 260) | RR 1.17 (0.76 to 1.82) | 427 (4 studies) | ⊕⊕⊝⊝ low9,10 | |
| Quality of life: Clinically important change in quality of life ‐ as defined by individual studies | See comment | See comment | Not estimable | 0 | See comment | No study reported on the predefined outcome quality of life so it could not be presented in this summary of findings table |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Risk of bias: almost all studies were open RCTs, there was no blinding. Moreover, several studies did not report rates. 2 Risk of bias: half of the studies were open RCTs without blinding. 3 Inconsistency: considerable heterogeneity based on both visual inspection, Chi2 and I2 statistics. 4 Indirectness: acceptability of treatment was measured by the number of participants leaving the study early for any reason which comprised efficacy and tolerability. Nevertheless, this is an indirect measure. 5 Imprecision: overall sample size was too small to provide a precise estimate for the small RR found. 6 Imprecision: too few events for a precise estimate and relatively broad confidence interval. 7 Risk of bias: all open RCTs, no blinding. Unclear whether ITT results were presented. 8 Inconsistency: results were significantly heterogeneous. However, all studies showed a statistically significant superiority of valproate augmentation therefore, the heterogeneity expressed a difference in the degree of the effect, but not of the direction of the effect. 9 Risk of bias: weight gain is an objective outcome. Therefore, we did not downgrade for lack of blinding. 10Imprecision: relatively few events, relatively small overall sample size for a relatively small effect size the confidence interval of which overlaps widely with zero.
Background
Despite the introduction of antipsychotic (neuroleptic) medications in the 1950s, there is still a sizeable minority (at least 30% of people with schizophrenia and related conditions), who do not achieve remission of symptoms (Schooler 1993). Over the last 40 years a variety of adjunctive treatments have been used to treat schizophrenia (Christison 1991). These are often used in addition to antipsychotics, in an attempt to alleviate the symptoms of schizophrenia such as hallucinations and delusional beliefs, although they have been used instead of antipsychotics. Treatments such as lithium (Leucht 2007b, indicated for bipolar affective disorder), carbamazepine (Leucht 2007c), valproate (Casey 2003), benzodiazepine (Dold 2012), beta‐blockers (Cheine 2003) and electroconvulsive therapy (Tharyan 2005) have all been used for people whose psychosis did not respond to traditional therapy. The situation has improved somewhat in recent years, with the re‐introduction of clozapine, which has proven efficacy for those that have not responded to traditional antipsychotic medications (Essali 2010). Whether the other second‐generation ('atypical') antipsychotics are more effective for the treatment of those with treatment‐resistant schizophrenia is unclear (Bagnall 2000, Gilbody 2000, Srisurapanont 2004).
Description of the condition
Schizophrenia is often a chronic and disabling psychiatric disorder. It afflicts approximately one per cent of the population world‐wide with few gender differences. Its typical manifestations are 'positive' symptoms such as fixed, false beliefs (delusions) and perceptions without stimuli (hallucinations), and 'negative' symptoms such as apathy and lack of drive, disorganisation of behaviour and thought, and catatonic symptoms such as mannerisms and bizarre posturing (Carpenter 1994). The degree of suffering and disability is considerable with 80% to 90% unemployed (Marvaha 2004) and 10% going on to commit suicide (Tsuang 1978, Palmer 2005).
Description of the intervention
Valproate (valproic acid) is traditionally used as an anticonvulsant drug and is also used for affective disorders, especially for the treatment of acute mania. Furthermore it is thought to have anti‐aggressive effects and it may reduce impulsive behaviour, which might be useful for some people with schizophrenia (Citrome 2000).
How the intervention might work
It is assumed that GABA‐ergic drugs such as valproate have a potential role in the treatment of schizophrenia as they down‐regulate dopamine (Wassef 2000). Mesolimbic dopamine hyperactivity is considered one of the main reasons for the development of positive symptoms in schizophrenia.
Why it is important to do this review
In this review we examined the role of valproate in the treatment of schizophrenia and schizophrenia‐like psychoses. The importance of performing such a review is emphasised by the fact that valproate is already frequently used for schizophrenia. For example, between 1994 and 1998 the use of valproate almost tripled among inpatients in the New York State psychiatric hospital system, with 43.4% of 4922 participants with a diagnosis of schizophrenia receiving valproate (Citrome 2000).
Objectives
To examine whether:
1. valproate alone is an effective treatment for schizophrenia and schizoaffective psychoses; and
2. valproate augmentation of antipsychotic medication is an effective treatment for the same illnesses.
Methods
Criteria for considering studies for this review
Types of studies
All relevant randomised controlled trials (RCT). If a trial was described as 'double‐blind' but implies randomisation, we included such trials in a sensitivity analysis (see Sensitivity analysis). If their inclusion did not result in a substantive difference, they remained in the analyses. If their inclusion did result in statistically significant differences, we did not add the data from these lower‐quality studies to the results of the better trials, but presented such data within a subcategory. We excluded quasi‐randomised studies, such as those allocating by alternate days of the week. Where people were given additional treatments within valproate, we only included data if the adjunct treatment was evenly distributed between groups and it was only the valproate that was randomised.
Types of participants
Adults, however defined, with schizophrenia or related disorders including schizophreniform disorder, schizoaffective disorder and delusional disorder, again, by any means of diagnosis.
We are interested in making sure that information is as relevant to the current care of people with schizophrenia as possible and planned to clearly highlight the current clinical state (acute, early post‐acute, partial remission, remission) as well as the stage (prodromal, first episode, early illness, persistent) and as to whether the studies primarily focused on people with particular problems (for example, negative symptoms, treatment‐resistant illnesses etc.).
Types of interventions
1. Valproate alone
Any dose; versus
i. Placebo
Includes no intervention
2. Valproate combination
In combination with any antipsychotic treatment: any dose; versus
i. Placebo
Includes no intervention in combination with any antipsychotic treatment, or
ii. Antipsychotics alone
Any dose
Types of outcome measures
We aimed to group outcomes by time: short‐term (up to 12 weeks), medium‐term (13 to 26 weeks) and long‐term (over 26 weeks).
Primary outcomes
1. Clincally significant response: important change ‐ as defined by each of the studies
Secondary outcomes
1. Leaving the study early: acceptability/tolerability of treatment
1.1 Acceptability ‐ leaving the study early for any reason 1.2 Overall tolerability ‐ leaving the study early due to adverse events 1.3 Leaving the study early due to poor clinical effect
2. Service utilisation
2.1 Hospital admission 2.2 Days in hospital 2.3 Change in hospital status
3. Clinical response
3.1 Any change global state ‐ as defined by each of the studies 3.2 Average endpoint/change score on global state scale 3.3 Relapse ‐ as defined by each of the studies 3.4 Clinically important change mental state ‐ as defined by each of the studies 3.5 Average endpoint/change score on mental state scale 3.6 Clinically important change in positive symptoms ‐ as defined by each of the studies 3.7 Average endpoint/change score on positive symptoms scale 3.8 Clinically important change in negative symptoms ‐ as defined by each of the studies 3.9 Average endpoint/change score on negative symptoms scale 3.10 Clinically important change in depressive symptoms ‐ as defined by each of the studies 3.11 Average endpoint/change score on depressive symptoms scale 3.12 Clinically important change in manic symptoms ‐ as defined by each of the studies 3.13 Average endpoint/change score on manic symptoms scale 3.14 Clinically important change in aggressive symptoms/agitation ‐ as defined by each of the studies 3.15 Average endpoint/change score on aggression/agitation symptoms scale
4. Behaviour
4.1 Clincally important change general behaviour ‐ as defined by each of the studies 4.2 Specific behaviour change ‐ as defined by each of the studies 4.2.1 Clinically important change social functioning ‐ as defined by each of the studies 4.2.2 Employment status during trial (employed/unemployed) 4.2.3 Occurrence of violent incidents (to self, others, or property)
5. Adverse events
5.1 General adverse events 5.2 Specific adverse events 5.2.1 Allergic reactions 5.2.2 Reversible thrombocytopenia 5.2.3 Central nervous system (ataxia, nystagmus, drowsiness, fits, diplopia, tremor) 5.2.4 Gastrointestinal (nausea, vomiting, diarrhoea) 5.2.5 Pancreatitis 5.2.6 Weight gain
6. Use of additional medication
6.1 Antipsychotics 6.2 Benzodiazepines
7. Economic (cost of care)
7.1 Direct cost of care 7.2 Indirect cost of care
8. Quality of life
8.1 Clincically important change in quality of life ‐ as defined by individual studies 8.2 Any change in quality of life ‐ as defined by individual studies 8.3 Average endpoint/change score on quality of life scale
'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2011) and used GRADE profiler (GRADEPRO) to import data from RevMan 5 (Review Manager) to create a 'Summary of findings' table. These tables provided outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes we rated as important to participant care and decision making. We selected the following main outcomes for inclusion in the 'Summary of findings' table.
Clinically significant response: important change ‐ as defined by each of the studies
Leaving the study early: acceptability of treatment ‐ leaving the study early for any reason
Leaving the study early: overall tolerability ‐ leaving the study early due to adverse events
Clinical response: aggression/agitation
Adverse events: sedation
Adverse events: weight gain
Quality of life: clincially important change in quality of life ‐ as defined by each of the studies
Search methods for identification of studies
Electronic searches
Cochrane Schizophrenia Group’s Study‐Based Register of Trials
On 4 March 2016, the Information Specialist searched the register using the following search strategy:
*Valproate* in Intervention Field of STUDY
In such study‐based registers, searching the major concept retrieves all the synonyms and relevant studies because all the studies have already been organised based on their interventions and linked to the relevant topics.
This Register is compiled by systematic searches of major resources (including MEDLINE, Embase, AMED, BIOSIS, CINAHL, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates, handsearches, grey literature, and conference proceedings (see Group’s Module). There is no language, date, document type, or publication status limitations for inclusion of records into the Register.
For previous searches, please see Appendix 1.
Searching other resources
1. Reference searching
We inspected references of all identified studies for further relevant studies.
2. Personal contact
We contacted the first author of each included study for information regarding unpublished trials (not done for 2012 update) and we sent requests to all authors for missing data.
3. Pharmaceutical companies
We contacted Sanofi‐Synthelabo, France, and Abbott Laboratories, USA, as the main manufacturers of valproic acid drugs to obtain data on unpublished trials (not done again for 2012 update).
Data collection and analysis
Methods used in data collection and analysis for this update are below, for previous methods please see Appendix 2.
Selection of studies
Two out of four reviewers (YW, JX) independently inspected all citations from the 2012 update searches and identified relevant abstracts. Where disputes arose, we acquired the full report for more detailed scrutiny. If citations met inclusion criteria, we obtained full reports of the papers for more detailed, independent inspection by two out of four reviewers (YW, JX). Where it was not possible to resolve disagreement by discussion, we attempted to contact the authors of the study for clarification.
Data extraction and management
1. Extraction
Two reviewers (YW, JX) extracted data from all newly included studies of the 2012 update search. Again, we discussed any disagreement, documented decisions and, if necessary, contacted authors of studies for clarification. With remaining problems SL helped clarify issues and we documented these final decisions. We planned to extract data presented only in graphs and figures whenever possible, with magnified images and ruler, and by two reviewers independently. Since there were no data to be extracted from graphs and figures, this was not performed. We attempted to contact study authors through an open‐ended request in order to obtain missing information or for clarification whenever necessary. If studies were multi‐centre, where possible, we extracted data relevant to each component centre separately.
2. Management
2.1 Forms
We extracted data onto standard, simple forms.
2.2 Scale‐derived data
We included continuous data from rating scales only if: a. the psychometric properties of the measuring instrument had been described in a peer‐reviewed journal (Marshall 2000); and b. the measuring instrument had not been written or modified by one of the trialists for that particular trial.
Ideally the measuring instrument should either be a self‐report, or completed by an independent rater or relative (not the therapist).
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand calculation of change needs two assessments (baseline and endpoint) which can be difficult in unstable and difficult‐to‐measure conditions such as schizophrenia. We decided to use primarily endpoint data, and only use change data if the former were not available. We combined endpoint and change data in the analysis as we aimed to use mean differences (MD) rather than standardised mean differences throughout (Deeks 2011).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we applied the following standards:
a. We entered skewed data from studies of at least 200 participants in the analysis irrespective of the following rules, because skewed data poses less of a problem in large studies.
b. Endpoint data: when a scale started from the finite number zero, we subtracted the lowest possible value from the mean and divided this by the standard deviation. If this value was lower than one, it strongly suggested a skew and we excluded these data. If this ratio was higher than one but below two, there was suggestion of a skew. We entered these data and tested whether their inclusion or exclusion substantially changed the results. If the ratio was larger than two we included such data because skew was less likely (Altman 1996).
c. When continuous data were presented on a scale which included the possibility of negative values (such as change data), it was difficult to tell whether data were skewed or not. We entered such data because change data tend to be less skewed and because excluding data would also lead to bias, as not all the available information would be used.
2.5 Common measure
To facilitate comparison between trials, where possible, we intended to convert variables that could be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary
Where possible, we made efforts to convert outcome measures to dichotomous data. This was done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It was generally assumed that if there was a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the Positive and Negative Syndrome Scale (PANSS, Kay 1986), this could be considered as a clinically significant response (Leucht 2005a, Leucht 2005b). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for valproate. Where keeping to this made it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'Not un‐improved') we planned to report data where the left of the line indicated an unfavourable outcome and to note this in the relevant graphs.
Assessment of risk of bias in included studies
YW and JX worked independently to assess risk of bias by using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a) to assess trial quality of Chinese studies. BH alone assessed the non‐Chinese articles and SL supervised this task. This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article, such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting.
If the raters disagreed, we made the final rating by consensus, with the involvement of another member of the review group. Where inadequate details of randomisation and other characteristics of trials were provided, we contacted authors of the studies in order to obtain further information. We reported non‐concurrence in quality assessment, but if disputes arose as to which category a trial was to be allocated, again, we resolved by discussion.
We noted the level of risk of bias in both the text of the review (Risk of bias in included studies) and in the 'Summary of findings' table (Characteristics of included studies).
Measures of treatment effect
1. Binary data
For binary outcomes we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). For statistically significant results we had planned to calculate the number needed to treat for an additional beneficial outcome/harmful outcome statistic (NNTB/NNTH), and its 95% CI using Visual Rx (http://www.nntonline.net/) taking account of the event rate in the control group. This, however, has been superseded by Table 1 and calculations therein.
2. Continuous data
For continuous outcomes we estimated mean difference (MD) between groups. We preferred not to calculate effect size measures (standardised mean difference (SMD)). However, if scales of very considerable similarity were used, we would have presumed there was a small difference in measurement, and we would have calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra‐class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997, Gulliford 1999).
If we had included cluster trials and clustering was not accounted for in primary studies, we planned to present data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error and planned to contact first authors of studies to obtain intra‐class correlation coefficients for their clustered data and to adjust for this by using accepted methods (Gulliford 1999).
If clustering had been incorporated into the analysis of primary studies, we planned to present these data as if from a non‐cluster randomised study, but adjust for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the intra‐class correlation coefficient (ICC) (Design effect = 1 + (m ‐ 1)*ICC) (Donner 2002). If the ICC is not reported we would have assumed it to be 0.1 (Ukoumunne 1999).
If cluster studies have been appropriately analysed taking into account intra‐class correlation coefficients and relevant data documented in the report, synthesis with other studies would have been possible using the generic inverse variance technique.
Since we don't have clustered randomised trials in our review, the adjustment for unit of analysis error was not necessary.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we only used data of the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Where a study involved more than two treatment arms, if relevant, we presented the additional treatment arms in comparisons. If data were binary we simply added these and combined within the two‐by‐two table. If data were continuous we combined data following the formula in section 7.7.3.8 (Combining groups) of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b). Where the additional treatment arms were not relevant, we did not reproduce these data.
Dealing with missing data
1. Overall loss of credibility
We share the concern that at some degree of loss of follow‐up data must lose credibility (Xia 2009). However, from which degree of attrition onward this is a problem is unclear. We did therefore not exclude studies on the basis of degree of attrition, but attrition was taken into account in the risk of bias assessment.
2. Binary
We presented data on a 'once‐randomised‐always‐analyse' basis (an intention to treat analysis). We assumed all those leaving the study early to not have changed in the given outcome. This rule is conservative for response, because it assumes that those who left the studies early would not have responded to treatment. It is not conservative concerning side‐effects. But it would often have been an overestimation of the frequency of side‐effects if all participants who discontinued had been assumed to experience rare side‐effects.
3. Continuous
3.1 Attrition
We used intention‐to‐treat data sets when possible, but included completer data if only these were available.
3.2 Standard deviations
If standard deviations were not reported, we first tried to obtain the missing values from the authors. If not available, where there were missing measures of variance for continuous data, but an exact standard error and confidence intervals available for group means, and either P value or 't' value available for differences in mean, we would have calculated them according to the rules described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b). When only the standard error (SE) was reported, standard deviations (SDs) were calculated by the formula SD = SE* square root (n). Chapters 7.7.3 (Higgins 2011b) and 16.1.3 (Higgins 2011c) of the Cochrane Handbook for Systematic Reviews of Interventions presented detailed formulas for estimating SDs from P values, t or F values, confidence intervals, ranges or other statistics. If these formulas did not apply, we would have calculated the SDs according to a validated imputation method which was based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies could introduce error, the alternative would be to exclude a given study’s outcome and thus to lose information. We nevertheless examined the validity of the imputations in a sensitivity analysis excluding imputed values.
3.3 Last observation carried forward
For a long time the method of last observation carried forward (LOCF) was employed to use participants' last assessments before leaving a study in the endpoint calculations (Leucht 2007a). Analyses have shown that more sophisticated statistical methods such as multiple imputation or mixed‐model repeated measures analysis (MMRM, also called 'mixed models') overall lead to more valid results, but they have other limitations. LOCF does also not necessarily overestimate effects, it can both overestimate or underestimate them (Mallinckrodt 2004). We therefore decided to use more sophisticated approaches such as multiple imputation or MMRM if available, but would use LOCF results if the former were not available.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations which we had not predicted would arise, and discussed.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods which we had not predicted would arise, and discussed.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
We investigated heterogeneity between studies by considering the I2 method alongside the Chi2 P value. The I2 provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I2 depends on the magnitude and direction of effects, and the strength of evidence for heterogeneity (e.g. P value from Chi2 test, or a confidence interval for I2). We interpreted an I2 estimate greater than or equal to around 50%, accompanied by a statistically significant Chi2 statistic, as evidence of substantial levels of heterogeneity (Deeks 2011). When there were substantial levels of heterogeneity in the primary outcome, we explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Sterne 2011). We were aware that funnel plots might be useful in investigating reporting biases but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar size. In other cases, where funnel plots were possible, we sought statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. There is, however, a disadvantage to the random effects model. It puts added weight onto small studies which often are the most biased ones. Depending on the direction of effect these studies can either inflate or deflate the effect size. We chose random‐effects model for all analyses.
Subgroup analysis and investigation of heterogeneity
We reported if inconsistency was high. We first investigated whether data had been entered correctly. Second, if data were correct, we re‐inspected outlier studies to find out whether methodological differences were likely to explain the differences. Such reasons were also explored by subgroup analyses. We had a priori decided that the examination of studies in first‐episode participants and treatment‐resistant participants versus the rest of the studies would be conducted in any case for the primary outcome, but others could arise and would be clearly described as post‐hoc. Decisions as to whether the studies should nevertheless be pooled, whether single outlier studies should be removed or at least removed from a sensitivity analysis, or whether studies would not be pooled at all would depend on factors such as whether heterogeneity could be explained by subgroup effects or whether studies differed in direction of effects or just in the degree of differences between intervention and control (Deeks 2011).
Sensitivity analysis
1. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way as to imply randomisation. For the primary outcomes we included these studies and if there was no substantive difference when the implied randomised studies were added to those with better description of randomisation, then we employed all data from these studies.
2. Assumptions for lost binary data
Where assumptions had to be made regarding people lost to follow‐up (see Dealing with missing data) we compared the findings of the primary outcomes when we used our assumption compared with completer data only. If there was a substantial difference, we reported results and discussed them but continued to employ our assumption.
Where assumptions had to be made regarding missing SDs data (see Dealing with missing data), we compared the findings on the primary outcome when we used our assumption compared with completer data only. A sensitivity analysis was undertaken testing how prone results were to change when 'completer' data only were compared to the imputed data using the above assumption. If there was a substantial difference, we reported results and discussed them but continued to employ our assumption.
3. Risk of bias
We analysed the effects of excluding trials that we judged to be at high risk of bias across one or more of the domains of randomisation (implied as randomised with no further details available, allocation concealment, blinding and outcome reporting) for the meta‐analysis of the primary outcome. If the exclusion of trials at high risk of bias did not substantially alter the direction of effect or the precision of the effect estimates, then we included data from these trials in the analysis.
4. Imputed values
We planned to undertake a sensitivity analysis to assess the effects of including data from trials if we used imputed values for ICC in calculating the design effect in cluster randomised trials.
If substantial differences were noted in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we did not pool data from the excluded trials with the other trials contributing to the outcome, but presented them separately.
5. Fixed and random effects
We synthesised all data using a random‐effects model, however, we also synthesised data for the primary outcome using a fixed‐effect model to evaluate whether the greater weights assigned to larger trials with greater event rates altered the significance of the results, compared to the more evenly distributed weights in the random‐effects model.
6. Impact of people with schizoaffective disorder
We followed a reviewer request, and post‐hoc added a sensitivity analysis excluding studies in participants with schizoaffective disorder for the outcome 'behavior ‐ aggression/agitation'.
Results
Description of studies
For substantive description of studies please see Characteristics of included studies and Characteristics of excluded studies tables.
Results of the search
The original search in 2004 identified 34 citations of which we ordered and inspected 12 full‐text articles. We excluded four studies and a further three studies we added to awaiting assessment. We included five studies.
The update search in 2007 yielded 28 citations of which we inspected five closely. Of the latter, one report provided additional usable information on Casey 2003. Two studies were included (Wang 2005a, Yin 2004) and the other two studies (Liang 2004, Zhu 2005) were listed in the 'excluded studies table'.
The update search in 2012 yielded 46 citations of which we ordered and inspected 41 full reports. We added 19 studies to the included studies table. We excluded seven studies (see Excluded studies). There were two identical studies published (Liang 2004 and Ma 2010) from two different author groups and from different hospitals. We assumed that the publications were on the same study and sent a letter to the authors for clarification.
In 2016 we did a scoping search. We identified 81 new references. Eight of these turned out to be additional publications of included studies and nine could be immediately excluded based on the title or abstract. The remaining references had to be classified as 'studies awaiting assessment'. This section now contains 69 reports. They will be examined in detail in the next update.
In summary, after searches carried out in 2004, 2007, 2012 and 2016, the total number of included studies is now 26, 11 are excluded and 69 are awaiting assessment. Please see Figure 1 which presents the search up to 2012.
1.
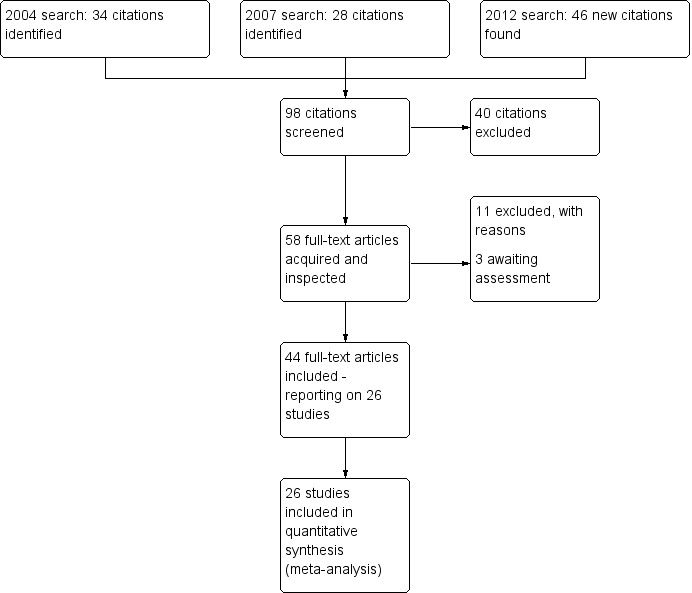
Study flow diagram.
Included studies
The current version of the review includes 26 studies.
1. Length of trials
All studies fell in the short‐term category. The mean duration of the trials was six weeks, with the longest study lasting twelve weeks (Casey 2009; Fisk 1987), and the shortest only two weeks (Guo 2007; Li 2008; Shi 2010).
2. Participants
Participants in all included studies were diagnosed with schizophrenia. To diagnose schizophrenia 15 studies used CCMD‐3, three studies used DSM‐IV, two studies used ICD‐10, one study used DSM‐IV‐TR, one study used DSM‐III, and four studies did not specify which criteria they used. In total 2184 participants participated in these 26 trials, with on average 84 participants in each study. The biggest study had 402 participants (Casey 2009) and the smallest only 12 (Wassef 2000). Twenty‐one studies reported gender information, with 1293 male participants and 504 female participants (approximately 13:5); while five studies did not provide gender details. Twenty‐one studies reported the age of participants, with a mean age of around 36 years old (five studies did not provide the participants' mean age).
3. Outcomes
3.1 Leaving the study early
The number of participants leaving the study early was recorded for the categories 'Acceptability of treatment: leaving for any reason', 'Overall tolerability: Leaving due to adverse events' and 'Leaving due to poor clinical effect'.
3.2 Adverse events
Side effects were recorded as the number of participants with an adverse event.
3.3 Scales
Details of scales that provided usable data are shown below.
3.3.1 Clinical Global Impression (CGI)
CGI (Guy 1976) is a rating instrument commonly used in studies on schizophrenia that enables clinicians to quantify severity of illness and overall clinical improvement during therapy. A seven‐point scoring system is usually used with low scores indicating decreased severity or greater recovery, or both.
3.3.2 Brief Psychiatric Rating Scale (BPRS)
BPRS (Overall 1962) is a brief rating scale used to assess the severity of a range of psychiatric symptoms, including psychotic symptoms. The scale has 18 items, and each item can be defined on a seven‐point scale varying from 'not present' (1) to 'extremely severe' (7). Scoring goes from 18‐126.
3.3.3 Positive and Negative Syndrome Scale (PANSS)
PANSS (Kay 1986) is a schizophrenia scale that has 30 items, each of which can be defined on a seven‐point scoring system varying from 1 ‐ absent to 7 ‐ extreme. It can be divided into three subscales for measuring the severity of general psychopathology, positive symptoms (PANSS‐P), and negative symptoms (PANSS‐N). A low score indicates lesser severity.
3.3.4 Abnormal Involuntary Movement Scale (AIMS)
The Abnormal Involuntary Movement Scale (NIMH 1970) has been used to assess tardive dyskinesia, a long‐term, drug‐induced movement disorder. However, using this scale in short‐term trials may also be helpful to assess some rapidly occurring abnormal movement disorders such as tremor.
3.3.5 Treatment Emergent Symptom Scale (TESS)
TESS (NIMH 1991) is a comprehensive scale used to measure adverse events emergent during the antipsychotic therapy. The scale has 33 items, each of which is measured for severity, relationship of the symptom with medication, and treatment taken for the symptom. Severity can be defined on a five‐point scale varying from 'not present' (0) to 'severe' (4). Relationship of the symptom with medication can be defined on a five‐point scale varying from 'not related' (0) to 'definitely related'. Treatment taken for the symptom can be defined on a seven‐point scale varying from 'no treatment required' (0) to 'termination of medication' (6).
3.3.6 Modified Overt Aggression Scale (MOAS)
MOAS (Kay 1986) is a brief rating scale used to assess the severity of aggressive behavior. The scale has four items, and each item can be defined on a five‐point scale varying from 'absent' (0) to 'extreme' (4).
3.3.7 Inpatient Multidimensional Psychiatric Scale (IMPS)
IMPS (Hiller 1986) is a semi‐standardised, multidimensional judgment method for detecting psychopathological findings with 90 operationally defined symptoms (complaints and psychiatrically‐relevant behaviours) from the entire spectrum of psychopathology. The estimates are based on observable behaviour and statements are made immediately after a free exploration call. For differential diagnostic problems, individual findings can be entered on profile sheets for depression, mania, schizophrenia and neuroses.
3.3.8 Scale for Assessment of Negative Symptoms (SANS)
SANS (Andreasen 1989) is a rating scale to measure negative symptoms in schizophrenia. The scale has 26 items, each of which can be defined on a six‐point scale varying from 'absent' (0) to 'severe' (5).
Excluded studies
Eleven studies were excluded from the analysis. Four of them were not randomised or the randomisation was done inappropriately (Centorrino 1994; Ping 1994; Raja 2000; Rifang 2001). One study included no placebo group (Alberti 1999;). One study included no adequate intervention group (Monfort 1991). Two studies included people with mania or bipolar disorder (Bersudsky 2010; NCT00183443). One study only compared valproate with another mood‐stabiliser (Zhu 2005). One study only reported data on metabolic measurements (Haupt 2007). One study (Ma 2010) reported identical data as Liang 2004.
For the comparisons made in excluded studies and suggestions for relevant reviews they could be included in please refer to Table 2 in Additional tables.
1. Excluded studies and suggestions for relevant reviews.
| Excluded study | Comparison | Existing review |
| Alberti 1999 | Valproate + antipsychotics versus gabapentin + antipsychotics in schizoaffective, bipolar and schizophrenic disorders | Carta 2003; Perugi 2004; Goodman 2006 |
| Bersudsky 2010 | Valnoctamide + risperidone versus placebo + risperidone in mania or schizoaffective disorder | Bailer 2012; Konstantinos 2012 |
| Centorrino 1994 | Clozapine + valproate in schizophrenia | Pantelis 1996; Freeman 1998; Balen 1999; Davis 2000; Baethge 2003; Stahl 2004; Besag 2006; Tranulis 2006; Sommer 2012 |
| Haupt 2007 | Valproate + antipsychotics versus antipsychotic monotherapy in schizophrenia | Probably relevant for a new review |
| Ma 2010 | Magnesium valproate + risperidone versus risperidone monotherapy in schizophrenia with impulsive and aggressive symptoms | Probably relevant for a new review |
| Monfort 1991 | Valproate + diazepam versus chlorpromazine in schizophrenia or schizophreniform disorder | Probably relevant for a new review |
| NCT00183443 | Divalproex‐extended release (DV‐ER) + lithium versus DV‐ER + quetiapine versus DV‐ER + placebo in mania | Probably relevant for a new review |
| Ping 1994 | Sodium valproate versus clozapine versus sodium valproate + clozapine in schizophrenia | Probably relevant for a new review |
| Raja 2000 | Antipsychotics versus valproate + antipsychotics in schizophrenia, bipolar, schizoaffective disorder, unipolar depression, personality disorder, mental retardation | Crowhurst 2002; Adams 2014; Pajonk 2005 |
| Rifang 2001 | Sodium valproate + chlorpromazine versus chlorpromazine monotherapy in schizophrenia | Probably relevant for a new review |
| Zhu 2005 | Valproate + antipsychotics versus clonazepam + antipsychotics in schizophrenia | Probably relevant for a new review |
Studies awaiting assessment
There are 69 studies in the Characteristics of studies awaiting classification, 35 are Chinese and awaiting translation, 28 are unclear allocation, six are randomised studies but need the full report for more detailed assessment.
Ongoing studies
We know of four ongoing studies, see Characteristics of ongoing studies.
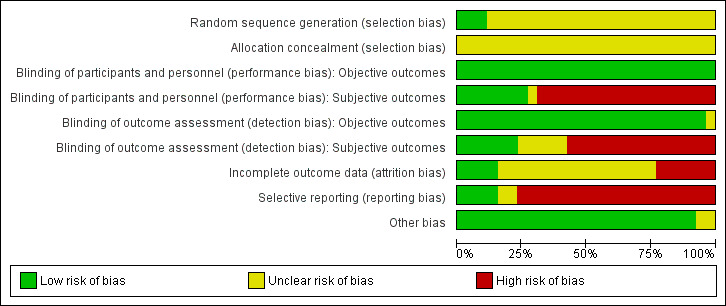
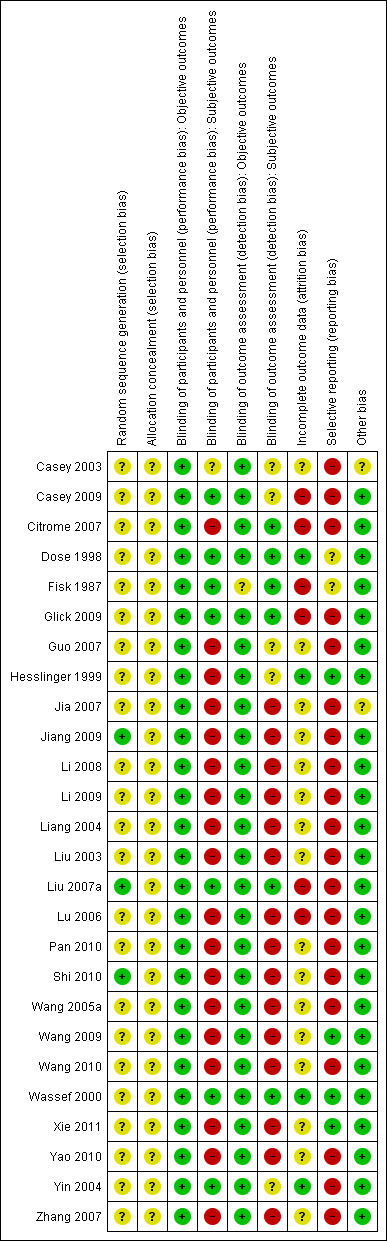
Risk of bias in included studies
For graphical representations of our judgements of risk of bias please refer to Figure 2 and Figure 3. Full details of judgements can be seen in the 'Risk of bias' tables.
2.
3.
Allocation
In the trials, participants were randomly assigned to receive either combinational therapy of antipsychotics and valproate or monotherapy of antipsychotics, with or without placebo. Two studies adopted stratified randomisation (Fisk 1987; Jiang 2009); one study randomised participants by drawing lots (Liu 2007a) and another study used a random number table (Shi 2010). We considered these studies as low risk for randomisation. Other studies failed to specify the process by which allocation to the intervention group was undertaken, therefore we have rated the allocation concealment and allocation for these studies as 'unclear'. Poor reporting of randomisation has been associated with an overestimate of effect (Schulz 1995, Moher 2001).
Blinding
Objective and subjective outcomes were rated separately, because we considered blinding to be less important for objective than for subjective outcomes. We also rated performance and detection bias separately, resulting in four independent assessments of blinding.
Seven studies were described as 'double‐blinded', so we rated them as low risk of performance bias and detection bias (Casey 2003; Casey 2009; Dose 1998; Fisk 1987; Glick 2009; Liu 2007a; Yin 2004). People with schizophrenia and clinicians were not blinded, but raters were blinded in two studies, so we rated them as high risk of performance bias but low risk of detection bias (Citrome 2007; Hesslinger 1999). There was no information about blinding reported in other studies, so we assumed they were open‐label, and at high risk of both performance bias and detection bias.
As for objective outcomes, we judged the vast majority of studies as having a low risk of bias in that respect.
Incomplete outcome data
Twelve studies indicated the numbers of participants who left the study before its completion, but the reasons for leaving the studies early were not consistently indicated. Among these studies, we assessed four as having a low risk of attrition bias, meaning that the problem of incomplete outcome data was addressed in an appropriate way (number of people leaving the study early was not very high and was evenly distributed between groups). We judged three studies as unclear, and five as having high risk of attrition bias.
The rest of the studies did not provide any information about people leaving the study early so we evaluated them as unclear risk of attrition bias.
Selective reporting
Five studies did not selectively report any predefined outcomes. Twenty studies failed to report usable continuous data for some major outcomes, for example BPRS (Casey 2003; Yin 2004), CGI, PANSS (Citrome 2007, Glick 2009), and MOAS (Citrome 2007; Liu 2007a) (many of them did not report results of adverse events and TESS). We gave these studies a 'high risk of bias' rank. The other two studies had only some minor flaws and we judged them as 'unclear'.
Other potential sources of bias
There were no obvious other potential sources of bias in the included studies.
Effects of interventions
See: Table 1
1. Comparison 1: VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone
1.1 Clinically significant response: important change ‐ as defined by each of the studies
We detected a significant difference favouring the valproate group, with 14 studies contributing (14 RCTs, n = 1049, RR 1.31, 95% CI 1.16 to 1.47, I2 = 12%, Analysis 1.1).
1.1. Analysis.
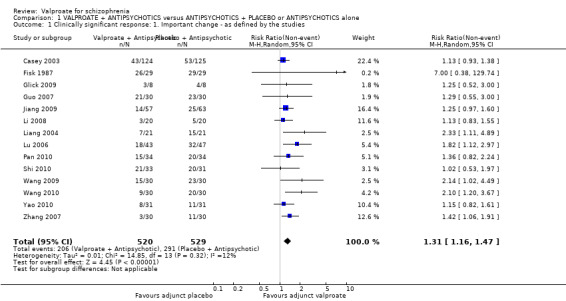
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 1 Clinically significant response: 1. Important change ‐ as defined by the studies.
1.2 Leaving the study early: Acceptability/tolerability of treatment
1.2.1 Acceptability of treatment: leaving for any reason
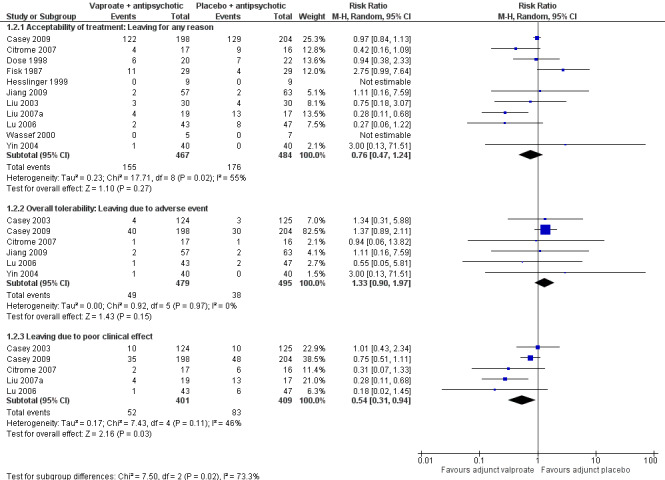
Eleven studies contributed to the outcome of 'number leaving the study early for any reason'. There was no significant difference between participants treated with valproate or placebo (n = 951, RR 0.76, 95% CI 0.47 to 1.24, I2 = 55%, Analysis 1.2).
1.2. Analysis.
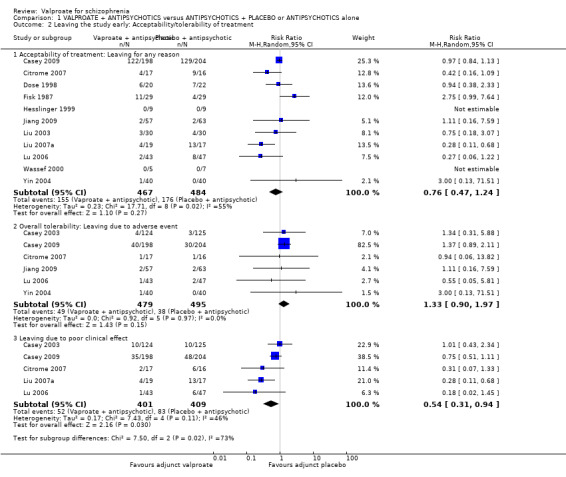
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 2 Leaving the study early: Acceptability/tolerability of treatment.
1.2.2 Overall tolerability: leaving due to adverse event
Six studies contributed to 'leaving the study early due to side effects', again we found no significant difference between the two groups (n = 974, RR 1.33, 95% CI 0.90 to 1.97, I2 = 0, Analysis 1.2).
1.2.3 Leaving due to poor clinical effect
We detected a significant difference favouring the valproate group under the outcome 'leaving the study early due to poor clinical effect', with five studies contributing (n = 810, RR 0.54, 95% CI 0.31 to 0.94, I2 = 46%, Analysis 1.2).
We generated a forest plot of the comparison for visual inspection (Figure 4).
4.
1.3 Clincial response: 2. Global state: 1a. mean change score CGI severity (high = poor)
Casey 2009 reported data on change of CGI severity and improvement.
There were no significant differences between groups in terms of severity (n = 392, MD 0.10, 95% CI ‐0.12 to 0.32, Analysis 1.3).
1.3. Analysis.
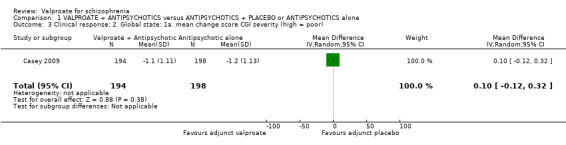
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 3 Clinical response: 2. Global state: 1a. mean change score CGI severity (high = poor).
1.4 Clinical response: 3. Global state: 1b. mean change score CGI Improvement (high = poor)
There were no significant differences between groups in terms of improvement (n = 393, MD 0.00, 95% CI ‐ 0.26 to 0.26, Analysis 1.4).
1.4. Analysis.
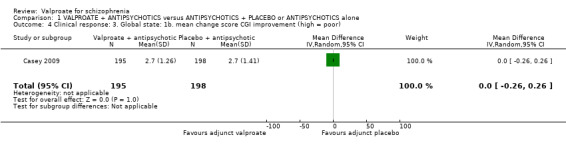
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 4 Clinical response: 3. Global state: 1b. mean change score CGI improvement (high = poor).
1.5 Clinical response: 4. Mental state: 2a mean change score PANSS total (high = poor)
Thirteen studies reported usable data about the mean change score on PANSS. There was a statistically significant difference between the valproate group and the placebo group in favour of the valproate group (n = 1363, MD ‐ 5.85, 95% CI ‐ 7.80 to ‐ 3.91, I2 = 67%, Analysis 1.5). There was significant heterogeneity here, because significant improvement in valproate group was only observed in open Chinese studies, whose quality of evidence was low as described elsewhere in this review. The two blinded Western studies did not show significant difference between the valproate group and the placebo group. Therefore, it may be considered that the open studies exaggerated the effect.
1.5. Analysis.
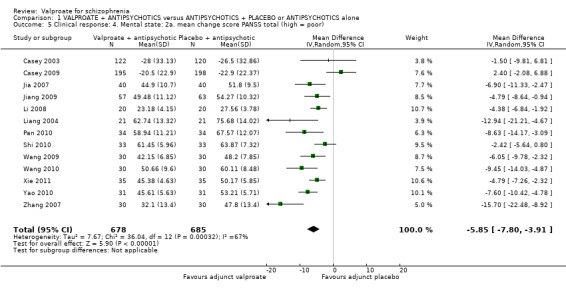
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 5 Clinical response: 4. Mental state: 2a. mean change score PANSS total (high = poor).
1.6 Clinical response 5. Mental sate: 2b. mean endpoint score BPRS total (high = poor)
Seven studies reported usable data about the mean endpoint score on BPRS. There was no statistically significant difference between the valproate group and the placebo group (n = 646, MD ‐ 1.87, 95% CI ‐ 4.46 to 0.73, Z = 1.41, I2 = 73%, Analysis 1.6). There was significant heterogeneity here, but we did not find explanations after looking into individual studies. It may be considered that the heterogeneity was due to natural variation in the data.
1.6. Analysis.
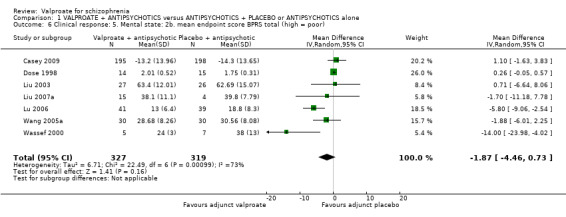
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 6 Clinical response: 5. Mental state: 2b. mean endpoint score BPRS total (high = poor).
1.7 Clinical response: 6. Mental state: 2c. mean change score IMPS total (high = poor)
Hesslinger 1999 reported on the mean change score IMPS . There was no statistically significant difference between the valproate group and the placebo group (n = 18, MD ‐ 5.11, 95% CI ‐ 26.04 to 15.82, Analysis 1.7).
1.7. Analysis.
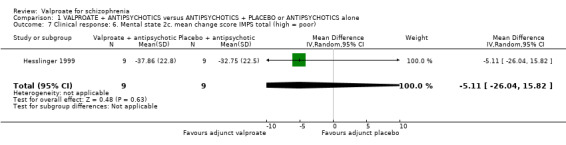
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 7 Clinical response: 6. Mental state 2c. mean change score IMPS total (high = poor).
1.8 Clinical response: 7. Positive symptoms: 3a. mean endpoint score PANSS positive subscale (high = poor)
Nine studies reported usable data on PANSS‐positive subscale. There was a statistically significant difference between the valproate group and the placebo group (n = 1073, MD ‐ 1.72, 95% CI ‐ 3.01 to ‐ 0.43, I2 = 69%, Analysis 1.8). There was significant heterogeneity here, but we did not find explanations after looking into individual studies. It may be considered that the heterogeneity was due to natural variation in the data.
1.8. Analysis.
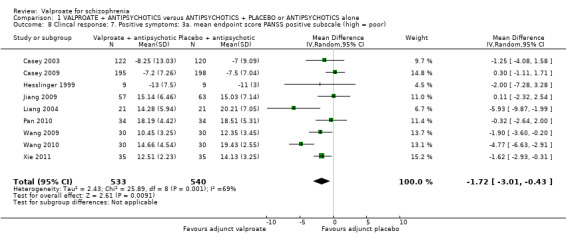
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 8 Clincal response: 7. Positive symptoms: 3a. mean endpoint score PANSS positive subscale (high = poor).
1.9 Clinical response: 8. Positive symptoms: 3b. mean endpoint score PANSS positive subscale (high = poor, skewed data)
Jia 2007 and Zhang 2007 also reported on the mean PANSS‐positive subscale, but their data were skewed and therefore unusable (Analysis 1.9).
1.9. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 9 Clinical response: 8. Positive symptoms: 3b. mean endpoint score PANSS positive subscale (high = poor, skewed data).
| Clinical response: 8. Positive symptoms: 3b. mean endpoint score PANSS positive subscale (high = poor, skewed data) | ||||
|---|---|---|---|---|
| Study | Group | Mean | SD | N |
| Jia 2007 | Valproate | 8.3 | 1.8 | 40 |
| Jia 2007 | Placebo | 9.4 | 2.7 | 40 |
| Zhang 2007 | Valproate + Risperidone | 8.6 | 6.5 | 30 |
| Zhang 2007 | Risperidone monotherapy | 13.5 | 5.3 | 30 |
1.10 Clinical response: 9. Negative symptoms: 4a. mean endpoint score PANSS negative subscale (high = poor)
Five studies reported usable data on the mean PANSS‐negative subscale. There was a statistically significant difference between the valproate group and the placebo group in favour of the valproate group (n = 651, MD ‐ 1.78, 95% CI ‐ 3.13 to ‐ 0.43, I2 = 67% Analysis 1.10). There was significant heterogeneity here, again, because significant improvement in valproate group was only observed in open Chinese studies, whose quality of evidence was low as described elsewhere in this review. Casey 2009 did not show significant difference between the valproate group and the placebo group. Therefore, it may be considered that the open studies exaggerated the effect.
1.10. Analysis.
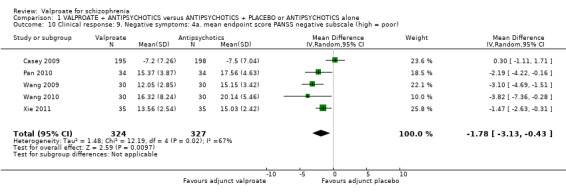
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 10 Clinical response: 9. Negative symptoms: 4a. mean endpoint score PANSS negative subscale (high = poor).
1.11 Clinical Response: 10. Negative symptoms: 4b. mean endpoint score PANSS negative subscale (high = poor, data skewed)
Four other studies also reported on the mean PANSS‐negative subscale, but their data were skewed and therefore unusable (Analysis 1.11).
1.11. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 11 Clinical response: 10. Negative symptoms: 4b. mean endpoint score PANSS negative subscale (high = poor, data skewed).
| Clinical response: 10. Negative symptoms: 4b. mean endpoint score PANSS negative subscale (high = poor, data skewed) | ||||
|---|---|---|---|---|
| Study | Group | Mean | SD | N |
| Jia 2007 | Valproate + Clozapine | 12.9 | 6.9 | 40 |
| Jia 2007 | Clozapine alone | 12.8 | 5.6 | 40 |
| Jiang 2009 | Valproate + Antipsychotics | 11.01 | 6.43 | 57 |
| Jiang 2009 | Antipsychotics monotherapy | 14.19 | 7.28 | 63 |
| Liang 2004 | Valproate | 11.17 | 4.32 | 21 |
| Liang 2004 | Placebo | 13.87 | 4.26 | 21 |
| Zhang 2007 | Valproate + Risperidone | 9.2 | 5.8 | 30 |
| Zhang 2007 | Risperidone monotherapy | 12.9 | 5.7 | 30 |
1.12 Clinical response: 11. Negative symptoms: 4c. mean endpoint score SANS subscale (skewed data)
Wassef 2000 analysed negative symptoms using the SANS total score at endpoint and found a superiority of valproate. The data were skewed and could therefore only be displayed in the other data table (Analysis 1.12).
1.12. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 12 Clinical response: 11. Negative symptoms: 4c. mean endpoint score SANS subscale (skewed data).
| Clinical response: 11. Negative symptoms: 4c. mean endpoint score SANS subscale (skewed data) | ||||
|---|---|---|---|---|
| Study | Interventions | Mean score | SD | N |
| Wassef 2000 | Valproate ‐ mean SANS ‐ high = poor | 2 | 3 | 5 |
| Wassef 2000 | Placebo ‐ mean SANS ‐ high = poor | 16 | 18 | 7 |
1.13 Clinical response: 12. Negative symptoms: 4d. mean endpoint score BPRS lack of energy sub‐score (high = poor)
Three studies reported usable data on the BPRS lack of energy subscore. There was no statistically significant difference between the valproate group and the placebo group (n = 135, MD 0.46, 95% CI ‐ 0.29 to 1.22, I2 = 89% (Analysis 1.13).
1.13. Analysis.
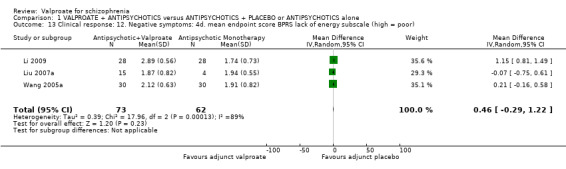
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 13 Clinical response: 12. Negative symptoms: 4d. mean endpoint score BPRS lack of energy subscale (high = poor).
1.14 Clinical response: 13. Aggression/agitation: 5b. clinically important change ‐ MOAS (high = poor)
Liu 2007a reported dichotomous data on the MOAS response. There was a statistically significant difference between the valproate group and the placebo group in favour of the valproate group (n = 36, RR 2.68, 95% CI 1.07 to 6.76, Analysis 1.14).
1.14. Analysis.
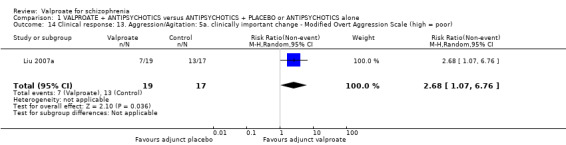
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 14 Clinical response: 13. Aggression/Agitation: 5a. clinically important change ‐ Modified Overt Aggression Scale (high = poor).
1.15 Clinical response: 14. Aggression/agitation 5a. mean endpoint score MOAS (high = poor)
Three studies reported continuous data on the mean MOAS at endpoint. There was a statistically significant difference between the valproate group and the placebo group in favour of the valproate group (n = 186, MD ‐2.55, 95% CI ‐3.92 to ‐1.19, I2 = 82%, Analysis 1.15). There was significant heterogeneity here. As only three studies were involved and all of them reported significant improvement in the valproate group, it may be considered that the heterogeneity was due to natural variation in the data.
1.15. Analysis.
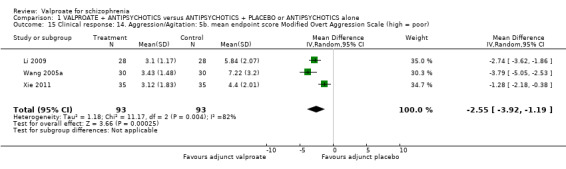
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 15 Clinical response: 14. Aggression/Agitation: 5b. mean endpoint score Modified Overt Aggression Scale (high = poor).
1.16 Clinical response: 15. Aggression/agitation: 5c. mean endpoint PANSS‐EC subscale (high = poor)
Three studies reported usable data on PANSS‐EC sub‐score at endpoint. There was a statistically significant difference between the valproate group and the placebo group in favour of the valproate group (n = 204, MD ‐1.85, 95% CI ‐2.63 to ‐1.08, Analysis 1.16).
1.16. Analysis.
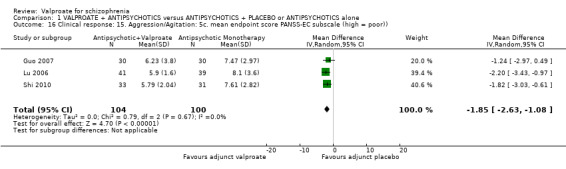
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 16 Clinical response: 15. Aggression/Agitation: 5c. mean endpoint score PANSS‐EC subscale (high = poor)).
1.17 Clinical response: 16. Aggression/agitation: 5d. mean endpoint score PANSS‐EC subscale (skewed data)
Casey 2003 also reported on PANSS‐EC item reduction, but the data were skewed and therefore unusable (Analysis 1.17).
1.17. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 17 Clinical response: 16. Aggression/Agitation: 5d. mean endpoint score PANSS‐EC subscale (skewed data).
| Clinical response: 16. Aggression/Agitation: 5d. mean endpoint score PANSS‐EC subscale (skewed data) | ||||
|---|---|---|---|---|
| Study | Interventions | Mean score | SD | N |
| Casey 2003 | Valproate ‐ mean chlorprothixene dose ‐ high = poor | ‐0.6 mg/d | 1.48 | 122 |
| Casey 2003 | Placebo ‐ mean chlorprothixene dose ‐ high = poor | ‐0.6 mg/d | 1.27 | 120 |
1.18 Clinical response: 17. Aggression/agitation: 5e. mean endpoint score PANSS supplementary subscale (high = poor)
Jiang 2009 reported usable data on PANSS supplementary subscore at endpoint. There was a statistically significant difference between the valproate group and the placebo group in favour of the valproate group (n = 120, MD ‐ 0.85, 95% CI ‐ 1.39 to ‐ 0.31, Analysis 1.18).
1.18. Analysis.
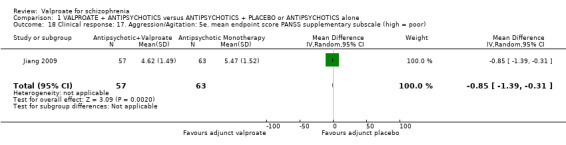
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 18 Clinical response: 17. Aggression/Agitation: 5e. mean endpoint score PANSS supplementary subscale (high = poor).
1.19 Clinical response: 18. Aggression/agitation: 5f. mean endpoint BPRS hostility subscale (high = poor)
Three studies reported usable data on mean BPRS hostility subscore at endpoint. There was no statistically significant difference between the valproate group and the placebo group (n = 135, MD ‐ 0.10, 95% CI ‐ 0.34 to 0.14, Analysis 1.19).
1.19. Analysis.
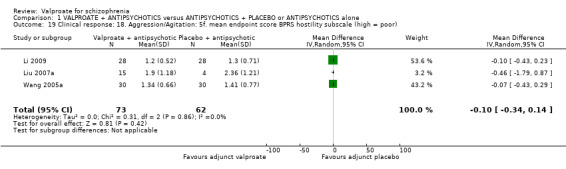
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 19 Clinical response: 18. Aggression/Agitation: 5f. mean endpoint score BPRS hostility subscale (high = poor).
1.20 Clinical response: 19. Depression symptoms: 6a. mild improvement Calgary Depression Scale (high = poor)
Glick 2009 reported a number of people with schizophrenia with mild improvement on the Calgary Depression Scale at endpoint. There was no statistically significant difference between groups (n = 16, RR 1.40, 95% CI 0.77 to 2.54, Analysis 1.20).
1.20. Analysis.
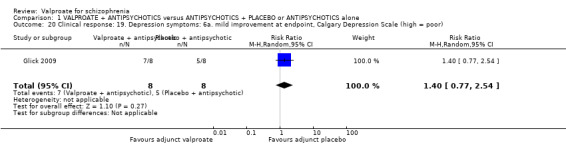
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 20 Clinical response: 19. Depression symptoms: 6a. mild improvement at endpoint, Calgary Depression Scale (high = poor).
1.21 Clinical response: 20. Depression symptoms: 6b. mean endpoint score BPRS anxiety and depression subscale (high = poor)
Three studies reported usable data on mean BPRS anxiety and depression sub‐score at endpoint. There was no statistically significant difference between the valproate group and the placebo group (n = 135, MD ‐0.06, 95% CI ‐0.25 to 0.13, Analysis 1.21).
1.21. Analysis.
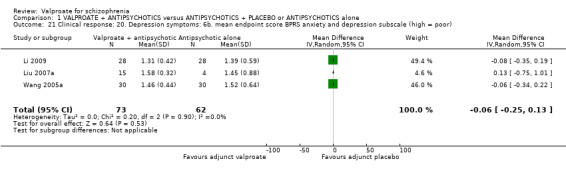
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 21 Clinical response: 20. Depression symptoms: 6b. mean endpoint score BPRS anxiety and depression subscale (high = poor).
1.22 Clinical response: 7. General pathology: 7a. mean change score PANSS general pathology subscale (high = poor)
Eight studies reported usable data. There was a statistically significant difference between the valproate group and the placebo group in favour of the valproate group (n = 873, MD ‐3.05, 95% CI ‐4.30 to ‐1.81, Analysis 1.22).
1.22. Analysis.
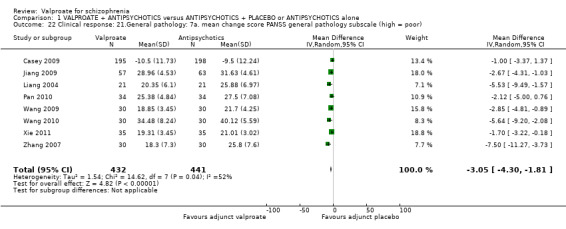
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 22 Clinical response: 21.General pathology: 7a. mean change score PANSS general pathology subscale (high = poor).
1.23 to 1.24 Use of additional medication: mean dose of antiparkinson medication, mean dose chlorprothixene
Dose 1998 and Hesslinger 1999 reported the mean dose of antiparkinson medication that was prescribed. Dose 1998 found a similar dose in both groups, whereas Hesslinger 1999 revealed that in the valproate group more antiparkinson medication was used. The data were skewed in both trials and therefore we could only display the data in the other data table, Analysis 1.23.
1.23. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 23 Use of additional medication: 1. Mean dose of antiparkinson medication (skewed data).
| Use of additional medication: 1. Mean dose of antiparkinson medication (skewed data) | ||||
|---|---|---|---|---|
| Study | Interventions | Mean score | SD | N |
| Dose 1998 | Valproate ‐ mean biperiden dose ‐ high = poor | 3.0 mg/d | 2.4 | 14 |
| Dose 1998 | Placebo ‐ mean biperiden dose ‐ high = poor | 2.4 mg/d | 1.8 | 15 |
| Hesslinger 1999 | Valproate ‐ mean biperiden dose ‐ high = poor | 3.6 mg/ | 3.3 | 9 |
| Hesslinger 1999 | Placebo ‐ mean biperiden dose ‐ high = poor | 2.9 mg/d | 3 | 9 |
Dose 1998 and Hesslinger 1999 analysed the mean dose of chlorprothixene that was used for additional sedation. Dose 1998 found a higher mean chlorprothixene dose in the placebo group whereas Hesslinger 1999 did not find any difference between the two groups The data were skewed and therefore we could only present the data in the other data table (Analysis 1.24).
1.24. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 24 Use of additional medication: 2. Mean chlorprothixene dose (skewed data).
| Use of additional medication: 2. Mean chlorprothixene dose (skewed data) | ||||
|---|---|---|---|---|
| Study | Interventions | Mean score | SD | N |
| Dose 1998 | Valproate ‐ mean chlorprothixene dose ‐ high = poor | 11.8 mg/d | 13.4 | 14 |
| Dose 1998 | Placebo ‐ mean chlorprothixene dose ‐ high = poor | 21.0 mg/d | 25.4 | 15 |
| Hesslinger 1999 | Valproate ‐ mean chlorprothixene dose ‐ high = poor | 21.1 mg/d | 6.7 | 9 |
| Hesslinger 1999 | Placebo ‐ mean chlorprothixene dose ‐ high = poor | 21.6 mg/d | 17.4 | 9 |
1.25 Use of additional medication: 3. Medication for sedation at least once
Two studies provided usable data. The number of participants taking sedative medication at least once was similar between groups (n = 309, RR 3.65, 95% CI 0.11 to 122.31, I2 85%, Analysis 1.25). There was significant heterogeneity here. As only two studies were involved and both of them reported no significant difference between the valproate group and the placebo group, it may be considered that the heterogeneity was due to natural variation in the data.
1.25. Analysis.
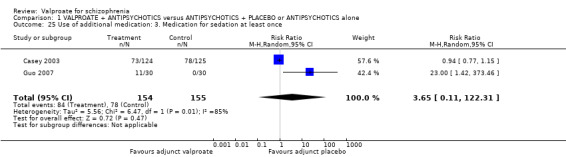
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 25 Use of additional medication: 3. Medication for sedation at least once.
1.26 Adverse events: 1. Abnormal ECG
Citrome 2007 and Jiang 2009 reported the number of participants with abnormal ECG. There was no statistically significant difference between the valproate group and the placebo group (n = 153, RR 0.88, 95% CI 0.35 to 2.18, Analysis 1.26).
1.26. Analysis.
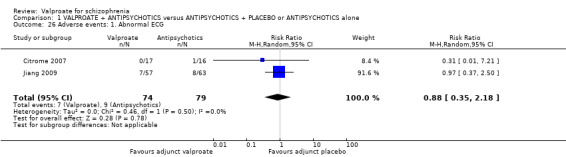
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 26 Adverse events: 1. Abnormal ECG.
1.27 Adverse events: 2. Abnormal liver function/increase in alanine transaminase/gamma glutamyl transpeptidase
Eight studies provided usable data. There was no statistically significant difference between the valproate group and the placebo group (n = 745, RR 1.26, 95% CI 0.72 to 2.22, Analysis 1.27).
1.27. Analysis.
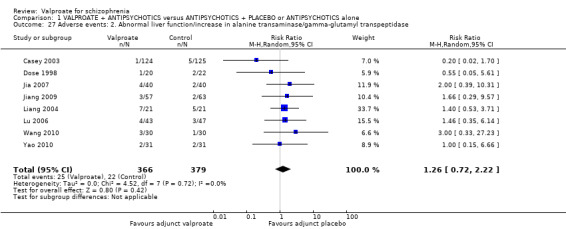
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 27 Adverse events: 2. Abnormal liver function/increase in alanine transaminase/gamma‐glutamyl transpeptidase.
1.28 Adverse events: 3. Akathisia
Li 2009, Wang 2010 and Xie 2011 reported the number of participants with akathisia. There was no statistically significant difference between the valproate group and the placebo group (n = 186, RR 1.06, 95% CI 0.36 to 3.06, Analysis 1.28).
1.28. Analysis.
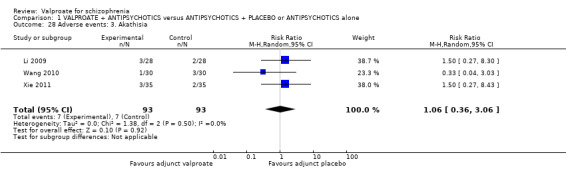
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 28 Adverse events: 3. Akathisia.
1.29 Adverse events: 4. Anxiety
Casey 2003 and Xie 2011 reported the number of participants with anxiety. There was no statistically significant difference between the valproate group and the placebo group (n = 319, RR 0.51, 95% CI 0.21 to 1.24, I2 = 0, Analysis 1.29).
1.29. Analysis.
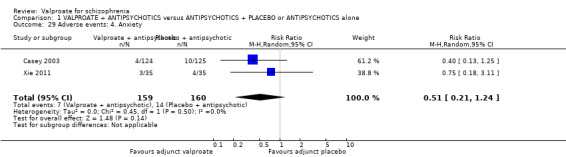
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 29 Adverse events: 4. Anxiety.
1.30 Adverse events: 5. Asthenia
Casey 2003 observed seven participants in the valproate group and 11 participants in the placebo group with asthenia. Again the difference did not reach statistical significance (n = 249, RR 1.58, 95% CI 0.63 to 3.95, Analysis 1.30).
1.30. Analysis.
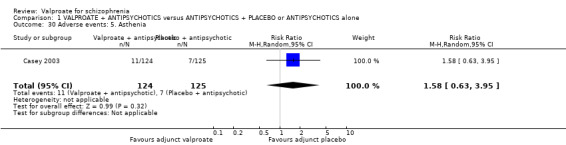
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 30 Adverse events: 5. Asthenia.
1.31 Adverse events: 6. Ataxia
Fisk 1987 and Pan 2010 reported the number of participants with ataxia. There was no statistically significant difference between the valproate group and the placebo group (n = 115, RR 2.42, 95% CI 0.37 to 15.92, Analysis 1.31).
1.31. Analysis.
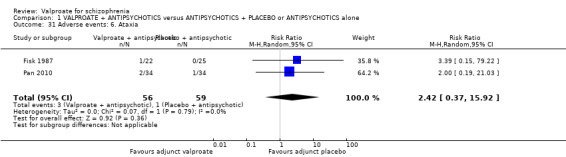
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 31 Adverse events: 6. Ataxia.
1.32 Adverse events: 7. At least one adverse event
There was no statistically significant difference between the valproate group and the placebo group (5 RCTs, n = 493, RR 0.92, 95% CI 0.68 to 1.25, I2 = 74%, Analysis 1.32).
1.32. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 32 Adverse events: 7. At least one adverse event.
1.33 Adverse events: 8. Blood count changes
Dose 1998 reported that four participants in the placebo group and five participants in the valproate group developed eosinophilia (n = 42, RR 1.38, 95% CI 0.43 to 4.42). Monocytosis was found in two participants in the valproate group only (n = 42, RR 5.48, 95% CI 0.28 to 107.62) and transient lymphocystosis was found in three participants of the valproate and one participant of the placebo group (n = 42, RR 3.30, 95% CI 0.37 to 29.21). Five studies reported the number of participants with leukopenia, and there was no statistically significant difference between the valproate group and the placebo group (n = 385, RR 1.18, 95% CI 0.38 to 3.64). Yin 2004 reported the number of participants with thrombocytopenia, and there was a statistically significant difference between the valproate group and the placebo group in favour of the placebo group (n = 79, RR 17.42, 95% CI 1.04 to 291.96) (All in Analysis 1.33).
1.33. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 33 Adverse events: 8. Blood count changes.
1.34 Adverse events: 9. Constipation
Four studies provided usable data. There was no statistically significant difference between the valproate group and the placebo group (n = 515, RR 0.94, 95% CI 0.47 to 1.85, I2 54%, Analysis 1.34).
1.34. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 34 Adverse events: 9. Constipation.
1.35 Adverse events: 10. Convulsion
Jiang 2009 reported one participant with convulsion in the placebo group but no participants with convulsion in the valproate group. This difference did not reach statistical significance (n = 120, RR 0.37, 95% CI 0.02 to 8.85, Analysis 1.35).
1.35. Analysis.
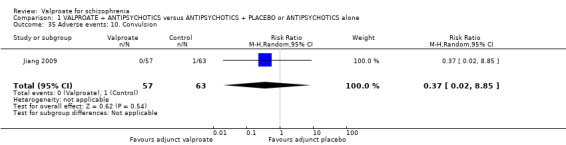
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 35 Adverse events: 10. Convulsion.
1.36 Adverse events: 11. Diarrohea
Three studies provided usable data. There was no statistically significant difference between the valproate group and the placebo group (n = 193, RR 1.03, 95% CI 0.32 to 3.34, Analysis 1.36).
1.36. Analysis.
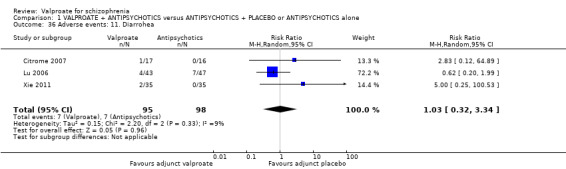
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 36 Adverse events: 11. Diarrohea.
1.37 Adverse events: 12. Dizziness
Seven studies provided usable data. There was no statistically significant difference between the valproate group and the placebo group (n = 662, RR 1.37, 95% CI 0.92 to 2.03, Analysis 1.37).
1.37. Analysis.
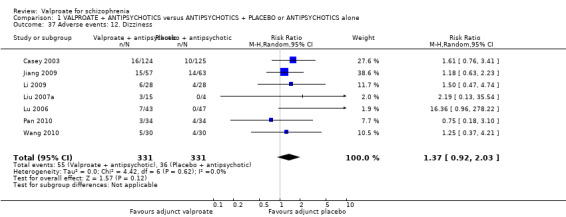
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 37 Adverse events: 12. Dizziness.
1.38 Adverse events: 13. Dry mouth
Citrome 2007, Lu 2006 and Wang 2010 reported the number of participants with dry mouth. There was no statistically significant difference between the valproate group and the placebo group (n = 179, RR 1.19, 95% CI 0.58 to 2.42, Analysis 1.38).
1.38. Analysis.
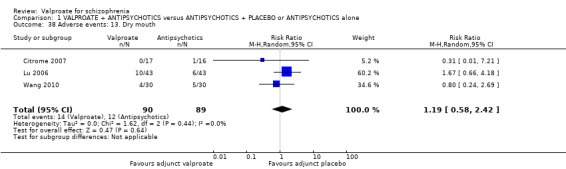
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 38 Adverse events: 13. Dry mouth.
1.39 Adverse events: 14. Dyspepsia
Casey 2003 reported on dyspepsia as an adverse event and found no statistical significant difference between the two treatment groups (n = 249, RR 1.05, 95% CI 0.62 to 1.79, Analysis 1.39).
1.39. Analysis.
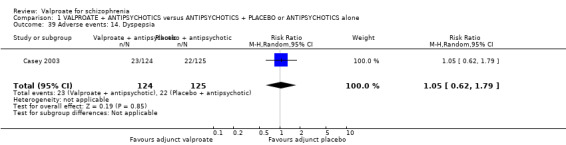
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 39 Adverse events: 14. Dyspepsia.
1.40 Adverse events: 15. Dystonia
Wang 2010 and Xie 2011 reported the number of people with dystonia. There was no statistically significant difference between the valproate group and the placebo group (n = 130, RR 1.00, 95% CI 0.30 to 3.37, Analysis 1.40).
1.40. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 40 Adverse events: 15. Dystonia.
1.41 Adverse events: 16. Extrapyramidal adverse advents
Extrapyramidal adverse advents were recorded by Dose 1998 and Fisk 1987 using the Simpson Angus Scale (Simpson 1970). As the data were skewed they could be displayed in the other data table only (Analysis 1.41).
1.41. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 41 Adverse events: 16. Extrapyramidal adverse events (skewed data).
| Adverse events: 16. Extrapyramidal adverse events (skewed data) | ||||
|---|---|---|---|---|
| Study | Interventions | Mean score | SD | N |
| Dose 1998 | Valproate ‐ (mean EPS) ‐ high = poor | 3.4 | 1.0 | 14 |
| Dose 1998 | Placebo ‐ (mean EPS) ‐ high = poor | 2.4 | 1.7 | 15 |
| Fisk 1987 | Valproate ‐ (mean tardive dyskinesia score) ‐ high = poor | 4.4 | 0.75 | 22 |
| Fisk 1987 | Placebo ‐ (mean tardive dyskinesia score) ‐ high = poor | 3.5 | 0.7 | 25 |
1.42 Adverse events: 17. Headache
Four studies provided usable data. There was no statistically significant difference between the valproate group and the placebo group (n = 469, RR 1.02, 95% CI 0.67 to 1.56, Analysis 1.42).
1.42. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 42 Adverse events: 17. Headache.
1.43 Adverse events: 18. Hypersalivation
Three studies reported the number of people with hypersalivation. The difference between the valproate group and placebo group was not statistically significant (n = 248, RR 1.03, 95% CI 0.62 to 1.70, Analysis 1.43).
1.43. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 43 Adverse events: 18. Hypersalivation.
1.44 Adverse events: 19. Incontinence
In the study by Fisk 1987 one participant who received valproate experienced incontinence but this did not lead to a statistically significant difference between both groups (n = 47, RR 3.39, 95% CI 0.15 to 79.22, Analysis 1.44).
1.44. Analysis.
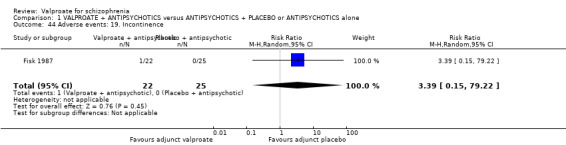
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 44 Adverse events: 19. Incontinence.
1.45 Adverse events: 20. Insomina
Li 2009 and Xie 2011 reported the number of participants with insomnia. There was no statistically significant difference between the valproate group and the placebo group (n = 126, RR 0.83 95% CI 0.39 to 1.78, Analysis 1.45).
1.45. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 45 Adverse events: 20. Insomnia.
1.46 Adverse events: 21. Myocardial ischaemia
Yao 2010 reported one participant in the valproate group and two participants in the placebo group with myocardial ischaemia. There was no statistically significant difference between the valproate group and the placebo group (n = 62, RR 0.50, 95% CI 0.05 to 5.23, Analysis 1.46).
1.46. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 46 Adverse events: 21. Myocardial ischaemia.
1.47 Adverse events: 22. Nausea
Nine studies provided usable data. There was no statistically significant difference between the valproate group and the placebo group (n = 728, RR 1.22, 95% CI 0.80 to 1.86, Analysis 1.47).
1.47. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 47 Adverse events: 22. Nausea.
1.48 Adverse events: 23. Pain
In Casey 2003 more participants in the placebo group (n = 18) experienced pain than in the valproate group (n = 11). This, however, was not statistically significant (n = 249, RR 0.62, 95% CI 0.30 to 1.25, Analysis 1.48).
1.48. Analysis.
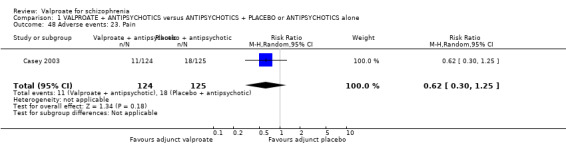
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 48 Adverse events: 23. Pain.
1.49 Adverse events: 24. Rash
Jiang 2009 and Xie 2011 reported the number of participants with a rash. There was no statistically significant difference between the valproate group and the placebo group (n = 190, RR 3.15, 95% CI 0.33 to 29.72, Analysis 1.49).
1.49. Analysis.
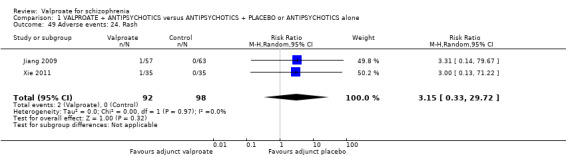
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 49 Adverse events: 24. Rash.
1.50 Adverse events: 25. Rhinitis
In one study (Casey 2003) rhinitis was reported as an adverse event, but without significant findings (n = 249, RR 0.30, 95% CI 0.09 to 1.07, Analysis 1.50).
1.50. Analysis.
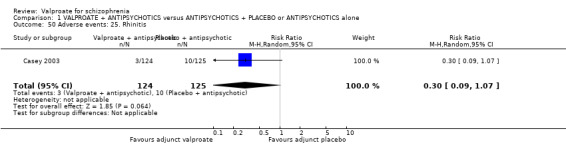
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 50 Adverse events: 25. Rhinitis.
1.51 Adverse events: 26. Rigidity/stiffness
Citrome 2007 reported one participant in the valproate group but no participants in the placebo group with rigidity/stiffness. There was no statistically significant difference between groups (n = 33, RR 2.83, 95% CI 0.12 to 64.89, Analysis 1.51).
1.51. Analysis.
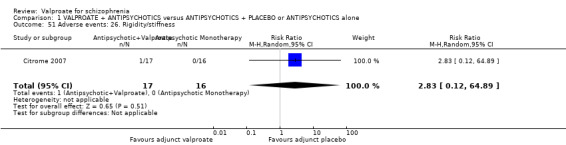
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 51 Adverse events: 26. Rigidity/stiffness.
1.52 Adverse events: 27. Sedation/somnolence/drowsiness
Eight studies provided usable data. Valproate was significantly more likely to cause sedation than placebo (8 RCTs, n = 770, RR 1.38, 95% CI 1.07 to 1.79, Analysis 1.52).
1.52. Analysis.
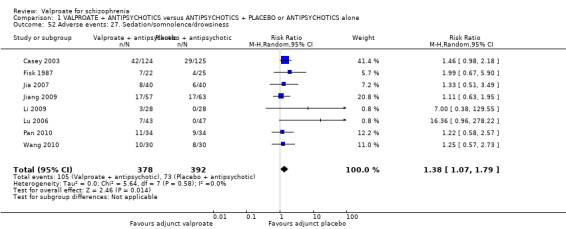
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 52 Adverse events: 27. Sedation/somnolence/drowsiness.
1.53 Adverse events: 28. Sexual dysfunction
Citrome 2007 reported one participant in the placebo group but no participants in the valproate group with sexual dysfunction. There was no statistically significant difference between groups (n = 33, RR 0.31, 95% CI 0.01 to 7.21, Analysis 1.53).
1.53. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 53 Adverse events: 28. Sexual dysfunction.
1.54 Adverse events: 29. Suicidal or depressed
Glick 2009 reported one participant in the placebo group but no participants in the valproate group as suicidal or depressed. There was no statistically significant difference between groups (n = 16, RR 0.33, 95% CI 0.02 to 7.14, Analysis 1.54).
1.54. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 54 Adverse events: 29. Suicidal or depressed.
1.55 Adverse events: 30. Tachycardia
Three studies provided data. There was no statistically significant difference between the valproate group and the placebo group (n = 218, RR 1.28, 95% CI 0.60 to 2.72, Analysis 1.55).
1.55. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 55 Adverse events: 30. Tachycardia.
1.56 Adverse events: 31.Tremor
Four studies reported the number of participants with tremor. There was no statistically significant difference between the valproate group and the placebo group (n = 199, RR 1.17, 95% CI 0.46 to 2.97, Analysis 1.56).
1.56. Analysis.
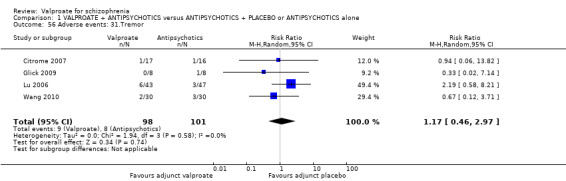
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 56 Adverse events: 31.Tremor.
1.57 Adverse events: 32. Unstable gait
A small study by Liu 2007a reported one participant in the valproate group and one participant in the placebo group with unstable gait. There was no statistically significant difference between the valproate group and the placebo group (n = 19, RR 0.27, 95% CI 0.02 to 3.39, Analysis 1.57).
1.57. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 57 Adverse events: 32. Unstable gait.
1.58 Adverse events: 33. Vegetative adverse events
Fisk 1987 reported six cases of vegetative adverse events which were not further specified in the valproate group and four cases in the placebo group. The difference was not statistically significant (n = 47, RR 1.70, 95% CI 0.55 to 5.27, Analysis 1.58).
1.58. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 58 Adverse events: 33. Vegetative adverse events.
1.59 Adverse events: 34. Vomiting
Casey 2003 and Citrome 2007 found no statistical significance between the two groups regarding vomiting (n = 282, RR 0.99, 95% CI 0.45 to 2.18, Analysis 1.59).
1.59. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 59 Adverse events: 34. Vomiting.
1.60 Adverse events: 35. Weight gain
Four studies provided data. There was no statistically significant difference between groups in the number of participants with weight gain (n = 427, RR 1.17, 95% CI 0.76 to 1.82, Analysis 1.60).
1.60. Analysis.
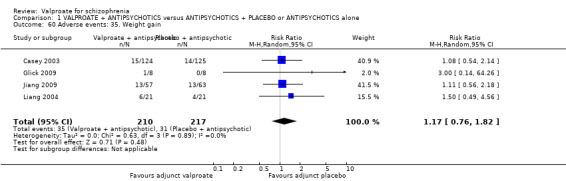
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 60 Adverse events: 35. Weight gain.
1.61 Adverse events: 36. Mean endpoint score TESS (high = poor)
Wang 2009 reported usable continuous data on the mean TESS score at endpoint. There was no statistically significant difference between the valproate group and the placebo group (n = 60, MD 2.06, 95% CI ‐0.41 to 4.53, Analysis 1.61).
1.61. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 61 Adverse events: 36. Mean endpoint score TESS (high = poor).
1.62 Adverse events: 37. Tardive dyskinesia ‐ mean endpoint score AIMS (high = poor)
In the small study by Yin 2004 those in the valproate group had a lower level of tardive dyskinesia than those in the monotherapy group (n = 79, MD ‐3.31, 95% CI ‐4.91 to ‐1.71, Analysis 1.62).
1.62. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 62 Adverse events: 37. Tardive dyskinesia ‐ mean endpoint score AIMS (high = poor).
1.63. Sensitivity analysis: Primary outcome 1. Excluding open studies
(Pimary outcome: Clinical response: Clincally important change ‐ as defined by studies)
After excluding all open studies, only three studies contributed to the primary outcome (Casey 2003, Fisk 1987 and Glick 2009). There was no statistically significant difference between the valproate group and the placebo group (n = 323, RR 1.15, 95% CI 0.95 to 1.39, Analysis 1.63).
1.63. Analysis.
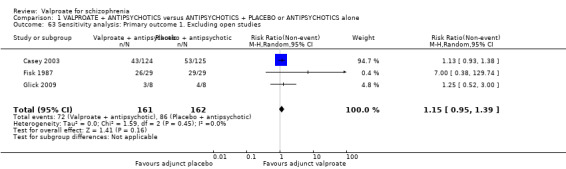
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 63 Sensitivity analysis: Primary outcome 1. Excluding open studies.
1.64 Sensitivity analysis: Primary outcome 2. fixed‐effect model
(Pimary outcome: Clinical response: Clincally important change ‐ as defined by studies)
We also synthesised data for the primary outcome using a fixed‐effect model for all 14 studies to evaluate whether the greater weights assigned to larger trials with greater event rates altered the significance of the results, compared to the more evenly distributed weights in the random‐effects model. There was a statistically significant difference between the valproate group and the placebo group in favour of valproate (n = 1049, RR 1.35, 95% CI 1.21 to 1.50, Analysis 1.64).
1.64. Analysis.
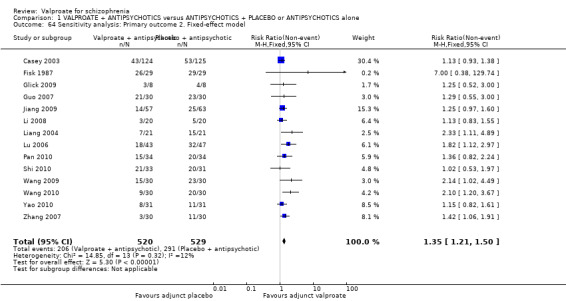
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 64 Sensitivity analysis: Primary outcome 2. Fixed‐effect model.
1.65 Sensitivity analysis: Primary outcome 3. excluding no attrition
(Pimary outcome: Clinical response: Clincally important change ‐ as defined by studies)
After excluding studies which did not report attrition data, only four studies contributed to the primary outcome (Casey 2003; Fisk 1987; Jiang 2009; Lu 2006). There was a statistically significant difference between the valproate group and the placebo group in favour of valproate (n = 517, RR 1.28, 95% CI 1.03 to 1.60, Analysis 1.65).
1.65. Analysis.
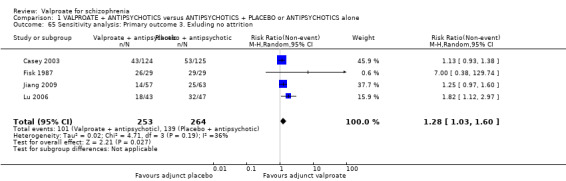
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 65 Sensitivity analysis: Primary outcome 3. Exluding no attrition.
1.66 Sensitivity analysis: Primary outcome 4. Completers
(Pimary outcome: Clinical response: Clincally important change ‐ as defined by studies)
We performed completer analysis for the primary outcome. Ten studies were removed because they did not report on participants leaving early. Three studies were removed because they used LOCF assumptions for missing data. After removing these studies only one study, Lu 2006, reported outcomes of completers only. There was a statistically significant difference between valproate group and placebo group in this single study (n = 80, RR 3.13, 95% CI 1.52 to 6.44, Analysis 1.66).
1.66. Analysis.
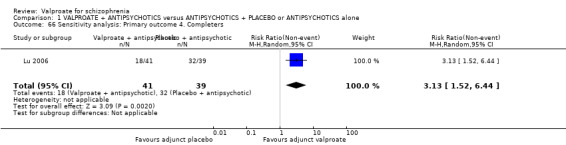
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 66 Sensitivity analysis: Primary outcome 4. Completers.
1.67 Sensitivity analysis: Primary outcome 5. Defined by PANSS reduction
(Pimary outcome: Clinical response: Clincally important change ‐ as defined by studies)
Twelve studies reported number of people with schizophrenia showing significant clinical response to the treatment, by the criteria of more than 50% reduction in PANSS total score. There was a significant difference between participants treated with valproate and participants treated with antipsychotic monotherapy in favour of combination (n = 975, RR 1.31, 95% CI 1.16 to 1.48, Analysis 1.67).
1.67. Analysis.
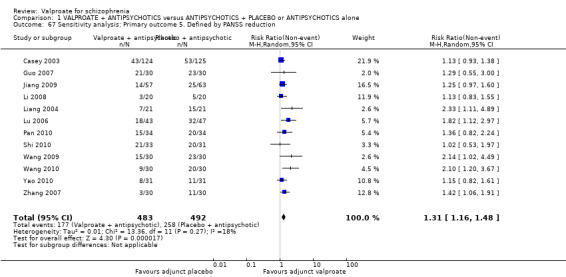
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 67 Sensitivity analysis: Primary outcome 5. Defined by PANSS reduction.
1.68 to 1.71 Sensitivity analysis: excluding schizoaffective studies
Aggression/agitation: 1. MOAS (high = poor)
In the post‐hoc sensitivity analysis on the effects of valproate in managing aggression, we excluded studies with schizoaffective participants, and valproate augmentation was still superior to monotherapy with antipsychotic drugs. This held true for the outcome mean overt aggression scale at endpoint (3 RCTs, n = 186, MD ‐ 2.55, 95% CI ‐ 3.92 to ‐ 1.19, I2 = 82%, Analysis 1.68).
1.68. Analysis.
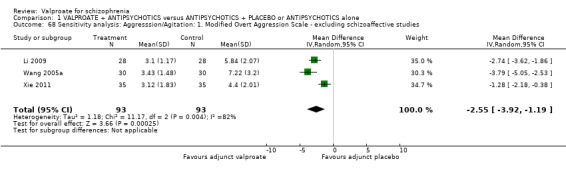
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 68 Sensitivity analysis: Aggresssion/Agitation: 1. Modified Overt Aggression Scale ‐ excluding schizoaffective studies.
Aggression/agitation: 2. PANSS‐EC subscale (high = poor)
Similar results were found for the PANSS‐EC subscore scale (2 RCTs, n = 124, MD ‐ 1.63, 95% CI ‐ 2.62 to ‐ 0.64, Analysis 1.69).
1.69. Analysis.
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 69 Sensitivity analysis: Aggression/Agitation: 2. PANSS‐EC subscale ‐ excluding schizoaffective studies.
Aggression/agitation: 3. PANSS supplementary subscale (high = poor)
However, Jiang 2009 showed no statistically significant difference between the valproate group and the placebo group (n = 120, MD ‐ 0.85, 95% CI ‐ 1.39 to ‐ 0.31, Analysis 1.70).
1.70. Analysis.
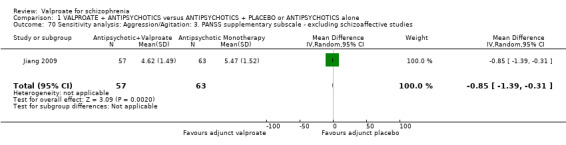
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 70 Sensitivity analysis: Aggression/Agitation: 3. PANSS supplementary subscale ‐ excluding schizoaffective studies.
Agression/agitation: 4. mean BPRS hostility sub‐score at endpoint or change (high = poor)
There was still no statistically significant difference between the valproate group and the placebo group for the mean BPRS hostility sub‐score at endpoint (n = 135, MD ‐ 0.10, 95% CI ‐ 0.34 to 0.14, Analysis 1.71).
1.71. Analysis.
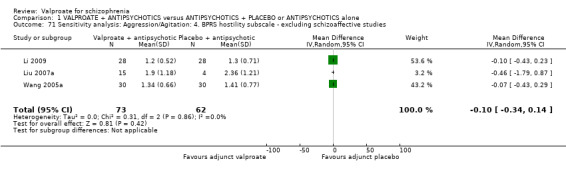
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 71 Sensitivity analysis: Aggression/Agitation: 4. BPRS hostility subscale ‐ excluding schizoaffective studies.
1.72 Subgroup analysis: Primary outcome: Treatment‐resistant participants vs no treatment‐resistant participants
(Pimary outcome: Clinical response: Clincally important change ‐ as defined by studies)
Among the 14 studies contributing to the primary outcome, two studies included treatment‐resistant people with schizophrenia (Pan 2010; Wang 2009); 12 studies did not focus on treatment‐resistant people with schizophrenia. There was no statistically significant difference between the two subgroups (Chi² = 0.77, P = 0.38, I² = 0%, Analysis 1.72).
1.72. Analysis.
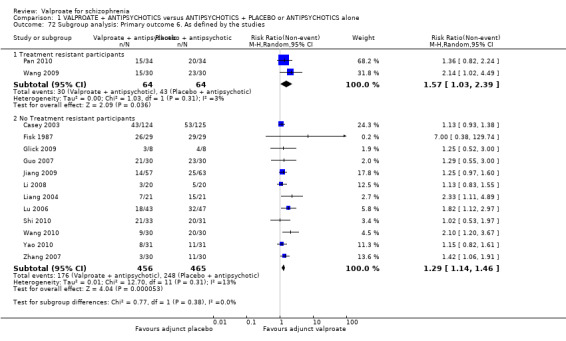
Comparison 1 VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone, Outcome 72 Subgroup analysis: Primary outcome 6. As defined by the studies.
2. Missing outcomes
W found no data for the important 'service utilisation' outcomes such as hospital admission, days in hospital or change in hospital status. Furthermore, there were no data on satisfaction with treatment or costs.
3. Publication bias
We generated a funnel plot from the primary outcome of clinically significant response (Figure 5). With visual inspection, the data distribution was not significantly unsymmetrical. Therefore, we concluded that there was no significant publication bias in the primary outcome.
5.
Funnel plot of comparison: 1 Antipsychotics and valproate versus antipsychotics and placebo/no treatment, outcome: 1.1 Global mental state. 1. Clinically significant response ‐ as defined by the studies
4. Contacts with study authors
In the current update, we contacted all authors of the 18 newly‐included studies (Guo 2007; Jia 2007; Jiang 2009; Li 2008; Li 2009; Liang 2004; Liu 2003; Liu 2007a; Lu 2006; Ma 2010; Pan 2010; Shi 2010; Wang 2009; Wang 2010; Xie 2011; Yao 2010; Yin 2004; Zhang 2007) in an effort to obtain missing data. Unfortunately, we have not received any response from any of these study authors.
Discussion
Summary of main results
1. General
Valproate is widely used in many countries in the treatment of schizophrenia and schizophrenia‐like disorders. Although a considerable number of RCTs were available, the individual trials were usually small and individually lacked sufficient power to detect any small to moderate effect. Only two studies (Casey 2003; Casey 2009) used reasonably large sample sizes involving more than 200 participants. We hope that more large valproate augmentation trials will be undertaken allowing for a better assessment of its effects. Importantly, only seven studies were double‐blinded; approximately 70% of the studies included were open‐label, or only raters were blinded. When the open RCTs were removed, efficacy effects were no longer statistically significant. Furthermore, all included studies were incompletely reported. We sent letters to all first authors of the studies indicating this and hope that this reporting will be improved by the time an updated version of this review is published.
2. Valproate as a sole treatment versus placebo or antipsychotic drugs
No studies for these comparisons were found. Researchers may not feel that use of valproate is indicated as a sole treatment for people with schizophrenia.
3. Valproate plus antipsychotics versus antipsychotics plus placebo or antipsychotics alone
3.1 Clinically significant response: important change ‐ as defined by individual studies
Fourteen studies reported data on the outcome 'clinically important change', with a significant difference favouring combination therapy of valproate and antipsychotics with little heterogeneity noted (n = 1049, RR 1.31, 95% CI 1.16 to 1.47, I2 = 12%). The superiority of valproate augmentation was, however, strongly based on open trials. When such open RCTs were removed, no significant difference remained.
3.2 Leaving the study early: acceptability/tolerability of treatment
About 34.8% (331 out of 951) participants in 11 studies left before completion of the study. This rate is similar to those in recent trials assessing new antipsychotic drugs for schizophrenia. There was no significant difference between valproate group and placebo group in the number of people with schizophrenia leaving the study early for any reason and for adverse events, but significantly fewer people with schizophrenia taking valproate left the studies due to poor clinical effect, compared to people with schizophrenia taking placebo (5 RCTs, n = 810, RR 0.54, 95% CI 0.31 to 0.94, I2 = 45%). Although this finding suggests benefits in terms of overall acceptability for valproate augmentation, the results could be biased by the lack of blinding which limits the conclusion in this regard.
3.3 Clinical response: Mental state
Results on mental state are difficult to interpret as the data were reported using four different scales which made it difficult to synthesize the results. There was a significant difference regarding the general mental state, measured by PANSS total score in 14 studies, in that the valproate group had lower PANSS scores. Valproate was also found to be superior to placebo/no treatment regarding the positive (10 RCTs) and negative (five RCTs) symptoms of schizophrenia, measured by PANSS positive and negative subscaless respectively. However, no significant difference was detected in BPRS total score (seven RCTs). Casey 2009 measured the general mental state by CGI severity and improvement scales, but again identified no significant difference. Hesslinger 1999 measured the general mental state by IMPS change, but still found no statistically significant difference. Overall, it appears that valproate is beneficial for symptoms of schizophrenia, both for the general mental state and for positive and negative symptoms, but these results have to be inspected with caution as they could be largely biased, which will be discussed in the next sections.
3.4 Clinical response: Agitation/aggression
Eleven studies reported measurements on agitation/aggressive behaviour using MOAS, PANSS‐EC subscale, PANSS supplementary subscale and BPRS hostility subscale. MOAS (four RCTs), PANSS‐EC (three RCTs) and PANSS supplementary (Jiang 2009) showed that valproate reduced such symptoms in people with schizophrenia, whereas the BPRS hostility subscale found no significant difference. The participants of these studies were often specifically selected for being aggressive at the beginning of the study. Post‐hoc sensitivity analyses excluding studies in participants with schizoaffective disorder did not change the results, valproate augmentation was still superior. Nevertheless, we can not rule out the possibility that participants with schizophrenia had manic symptoms as well, and that the superiority of valproate on aggression was mediated by improvements of these symptoms. Although we did not formally test this, the information stems almost entirely from open RCTs making replications in blinded trials necessary.
3.5 Use of additional medication
A few study authors reported the mean dose of additional antiparkinson medication and additional use of chlorprothixene. The data were skewed so that meta‐analytic calculations were not possible, but the original publications reported no clear differences between groups. Other data on sedative medicine in general yielded no significant difference between groups either.
3.6 Adverse events
We detected no significant differences between the valproate group and the control group in abnormal ECG, abnormal liver function, akathisia, anxiety, asthenia, ataxia, at least one adverse event, blood count changes, constipation, convulsion, diarrhoea, dry mouth, dyspepsia, dystonia, extrapyramidal adverse events, headache, hypersalivation, incontinence, insomnia, mean TESS score at endpoint, myocardial ischaemia, nausea, pain, rash, rhinitis, rigidity/stiffness, sexual dysfunction, tachycardia, tremor, unstable gait, vegetative adverse events, vomiting, and weight gain. But since many of these adverse events are generally rare, much larger studies would be needed to reveal significant differences between groups.
On the other hand, there were significantly more participants in the valproate groups who experienced dizziness (7 RCTs, n = 662, RR 1.37, 95% CI 0.92 to 2.03, I2 = 0); and sedation (8 RCTs, n = 770, RR 1.38, 95% CI 1.07 to 1.79, I2 = 0). The sedation effect may sometimes be useful for acutely ill people, but it is certainly unwanted in the maintenance phase. There was one study (Yin 2004) that reported that the Abnormal Involuntary Movement Scale (AIMS) score assessing tardive dyskinesia was lower in the valproate group (n = 79, MD ‐3.31, 95% CI ‐4.91 to ‐1.71), but we feel that a study based on only 79 participants is not a sufficient basis for saying that valproate is effective for this condition. Yin 2004 also found that there were significantly more participants with thrombocytopenia in the valproate group (n = 79, RR 17.43, 95% CI 1.04 to 291.96). Again, the study was too small to draw any firm conclusion.
3.7 Missing outcomes
It is hoped that more data on important service outcomes such as duration of hospital stay, satisfaction with treatment or costs will be received for updates of this review.
Overall completeness and applicability of evidence
We believe that the data we gathered is not complete. Firstly, all results were derived from short‐term studies, and long‐term results are missing. Moreover, most of the trials are based on relatively small sample sizes which can be prone to bias, therefore we cannot conclusively judge the potential applicability of the results. The review Davey Smith 1998 suggested that meta‐analyses based on summation of small trials should be interpreted as inconclusive, regardless of whether the combined estimate was significant. Most of the positive findings came from studies conducted in China, while the Western studies were usually negative. This finding also limits generalisability.
Quality of the evidence
The quality of all identified relevant trials has to be judged as moderate at best. The quality of data provided for our main outcomes of interest was low or very low quality. Although all included studies claimed that the treatment allocation was randomised, few of them presented further descriptions about the randomisation procedure. Therefore, it is unclear whether these trials were adequately randomised and whether researchers were concealed from treatment allocation before randomisation.
The positive evidence supporting use of valproate largely stems from the mostly Chinese included studies, which had a problem with methodological biases. They did not provide detailed information regarding randomisation, nor did they provide any details of blinding of clinicians, participants and raters throughout the procedures. Since it is unlikely that a study would not report blinding when blinding was actually performed, we assumed that these studies were open‐label trials, and therefore possibly biased. Many outcomes showed considerable heterogeneity, mainly due to the results from the two, mainly negative, large double‐blinded studies (Casey 2003, Casey 2009) which were different from those of the usually positive, small, open Chinese studies. Only four Chinese studies provided information on people leaving the study early, which made their results even less reliable, as it is quite rare that a study has 100% completion rate. We have sent letters to the first authors of the papers and hope that with their replies we could validate and improve the quality of evidence. We also observed bias in terms of selective reporting in a number of studies. However, this was mainly related to incomplete reporting of the Treatment‐Emergent Symptom Scale (TESS), which is a continuous outcome of side effects. Given that these studies still reported dichotomous data for adverse events, we interpret that this issue should not have severely biased our results.
Potential biases in the review process
The evidence presented here is, to our best knowledge, complete. However one can never be certain whether some additional (unpublished) material exists that was not pooled into the analysis.
Agreements and disagreements with other studies or reviews
We are not aware of any other systematic reviews and meta‐analysis on the question of valproate augmentation for schizophrenia.
Authors' conclusions
Implications for practice.
1. For people with schizophrenia
People with schizophrenia should be informed of the small empirical basis for the use of valproate for their disorder. They should know that valproate might lead to improvement in their general mental state, and also for specific symptoms such as excitement and aggression. But they should also know that this evidence is largely based on open studies without appropriate blinding, and that valproate is associated with sedation, dizziness and other side‐effects.
2. For clinicians
Based on available RCT‐derived evidence there is no randomised evidence to support or refute the use of valproate as a monotherapy for schizophrenia. There is evidence based on a number of trials that the augmentation of antipsychotics with valproate may be effective for overall mental state, and also for specific symptoms, especially in terms of excitement and aggression. However, this evidence was entirely based on open RCTs. Two large double‐blind trials did not find any convincing evidence that valproate augmentation may be beneficial. Moreover, clinicians should take into account that valproate is associated with a number of adverse events among which sedation and dizziness could be documented to appear significantly more frequently than in the control groups of this review.
3. For managers and policy makers
Managers and policy makers should support further trials to assess the impact of valproate augmentation for people with schizophrenia and related disorders.
Implications for research.
1. General
Any future studies should respect standards of measuring outcomes and of reporting data in order to enhance the comparability of study results (Begg 1996). We have written to the first authors of the included studies but we have not received many answers. If study authors consistently provided missing data for meta‐analyses, their quality could be much improved.
2. Specific
Large, double‐blind and also longer‐term randomised trials should be undertaken to examine the clinical effects of valproate as an adjunct to antipsychotic treatment. Open RCTs are available, but there is a dearth of double‐blind studies which should focus on the following groups:
Those with schizophrenia and violent episodes, because valproate is said to have an impact on aggression.
Those with schizoaffective disorder, because valproate might have mood‐stabilising effects.
Those with treatment‐resistant forms of the disorder, because here effective adjuncts to antipsychotic drugs are urgently needed.
For the design of a suggested future trial, please refer to Table 3 in Additional tables.
2. Suggested future trial.
| Methods | Allocation: random Blinding: blind participants, treating team and raters. Duration: minimum one‐year follow‐up |
| Participants | Diagnosis: people with schizophrenia and aggression, on those with treatment‐resistant forms of the disorder and on those with schizoaffective disorders (alternatively, several trials are necessary) Age: Sex: male and female. n = 300 |
| Interventions | 1. Valproate in combination with any antipsychotic treatment: any dose 2. Placebo (or no intervention) in combination with any antipsychotic treatment |
| Outcome | Leaving the study early (due to any reason, inefficacy‐ and tolerability‐related adverse events)
Service utilisation
Global state ‐ clinically important change*
Relapse
Mental state ‐ clinically important change (general and specific)
Behaviour ‐ clinically important change
Aggression ‐ clinically important change
Social functioning ‐ clinically important change Adverse effects ‐ clinically important general adverse effects*; sudden and unexpected death Economic outcomes Satisfaction with treatment Quality of life Pharmacokinetic interactions All outcomes by time ‐ short term (up to 12 weeks), medium term (13‐26 weeks) and long term (over 26 weeks) |
| Notes | *Primary outcomes of interest |
What's new
| Date | Event | Description |
|---|---|---|
| 19 September 2016 | New citation required but conclusions have not changed | New trial data available but overall conclusions regarding effectiveness remain unclear. Large well‐conducted randomised trials needed |
| 4 March 2016 | New search has been performed | Search updated, 81 new references added to 'Classification pending references' section of the review. Eight of these turned out to be additional publications of included studies and nine could be immediately excluded based on the title or abstract. The remaining references had to be classified as 'studies awaiting assessment'. Sixty‐four references were added to those already in 'awaiting classification'. This section now contains 69 reports, 28 of these are unclear allocation, 35 are Chinese and awaiting translation, 6 need more detailed assessment. |
| 15 October 2012 | New search has been performed | Results from July 2012 search added to review. 19 new studies, mostly Chinese, added to included studies table. The risk of bias tables and description of bias added; background information, results and discussion extended; structure of the review updated to meet new standards. |
History
Protocol first published: Issue 3, 2002 Review first published: Issue 1, 2004
| Date | Event | Description |
|---|---|---|
| 14 April 2010 | Amended | Contact details updated |
| 9 April 2009 | Amended | With new Plain Language Summary added |
| 30 October 2008 | Amended | Missing text in Discussion section amended |
| 14 May 2008 | New search has been performed | Update search run Feb 2008 |
| 14 May 2008 | New citation required but conclusions have not changed | Update search run Feb 2008 |
| 4 May 2008 | Amended | Converted to new review format |
| 7 March 2003 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
We would like to thank the editorial team of the Cochrane Schizophrenia Group for its study search and its continuous support. Cochrane Schizophrenia Group holds a standardised methods section which was used for this review and modified were necessary. We would like to thank Anja Volz and Christian Schwarz for their contributions as previous authors on this review.
We would also like to thank Dina Bošnjak for peer reviewing the 2012 version.
Appendices
Appendix 1. Previous searches
1.1 Search in 2002 and 2007
1.1.1 Electronic searches
1.1.1.1 Cochrane Schizophrenia Group's Study‐Based Register of Trials (July 2002 and February 2007)
This register is compiled by methodical searches of BIOSIS, CINAHL, Dissertation abstracts, EMBASE, LILACS, MEDLINE, PSYNDEX, PsycINFO, RUSSMED, Sociofile, supplemented with hand searching of relevant journals and numerous conference proceedings. The following phrase was used: {[(valproat* or *valproat* or valproic acid* or *valproic acid* or sodium valproate* or *sodium valproate* or sodium dipropylacetate* or *sodium dipropylacetate* or calcium valproate* or *calcium valproate* or convulex* or *convulex* or depakene* or *depakene* or ergenyl* or *ergenyl*) in abstract or title or index terms of REFERENCE] or [valproic acid in interventions of STUDY]} All references of articles selected for inclusion were searched for further relevant trials.
1.1.2 Other search methods
1.1.2.1 Contacting pharmaceutical companies
We contacted Sanofi‐Synthelabo, France, and Abbott Laboratories, USA, as the main manufacturers of valproic acid drugs to obtain data on unpublished trials.
1.1.2.2 Personal contact
We contacted the first author of each included study for information regarding unpublished trials.
1.2 Search in 2012
1.2.1 Electronic searches
1.2.1.1 Cochrane Schizophrenia Group's Study‐Based Register of Trials (July 2012)
The Trials Search Co‐ordinator searched the Cochrane Schizophrenia Group’s Trials Register (July 2012).
(*valproate* or *valproic acid* or *sodium valproate* or *sodium dipropylacetate* or *calcium valproate* or *convulex* or *depakene* or *ergenyl*) in Intervention Field of STUDY
The Cochrane Schizophrenia Group's Trials Register was compiled by systematic searches of major databases, handsearches and conference proceedings (see Group's Module). Incoming trials were assigned to existing or new review titles.
1.2.2 Searching other resources
1.2.2.1 Reference searching
We inspected references of all identified studies for further relevant studies.
Appendix 2. Previous data collection and analyses
[For definitions of terms used in this, and other sections, please refer to the Glossary]
1. Study selection
All study citations identified by the searches were independently inspected by two reviewers, and full reports of the studies of agreed relevance were obtained. Where agreement could not be reached, we acquired the full report for more detailed scrutiny. We then independently inspected these articles to assess their relevance to this review. Again, if the disagreement could not be resolved from published information, the article was added to those awaiting assessment and we contacted the authors of the study for further clarification.
2. Quality assessment
We assessed the methodological quality of the trials included in this review using the criteria described in the Cochrane Handbook (Higgins 2005). These criteria are based on the evidence of a strong relationship between allocation concealment and the potential for bias in the results (Schulz 1995) and are defined as below:
A. Low risk of bias (adequate allocation concealment) B. Moderate risk of bias (some doubt about the results) C. High risk of bias (inadequate allocation concealment)
For the purpose of the analysis in this review, we included trials if they meet the criteria A or B.
3. Data extraction
We independently extracted the data from included studies. Again, any disagreement was discussed, the decisions documented and, if necessary, we contacted the authors of the studies for clarification. We documented justification for excluding references from the review.
4. Data management
4.1 Intention to treat
For studies that did not specify the reasons for people leaving the study early, we assumed that these people had no change in the clinical outcome variables. Wahlbeck highlighted the problem of high rate of people leaving the study early in randomised controlled trials of antipsychotics (Wahlbeck 2001). As there is no consensus on the level of people leaving the study early that makes results meaningless, all trials were included in the main analyses. We performed a sensitivity analysis on those trials with a greater than 50% drop‐out rate to test whether including them significantly changed the results of the primary outcome parameters. When insufficient data were provided to identify the original group size (prior to drop outs), we contacted the authors and allocated the trials to the 'awaiting assessment' list.
4.2 Crossover design
We expected that some trials would use a crossover design. In order to exclude the potential additive effect in the second or later stages on these trials, we only analysed data from the first stage.
4.3 Data types
Outcomes are assessed using continuous (for example changes on a behaviour scale), categorical (for example, one of three categories on a behaviour scale, such as 'little change', 'moderate change' or 'much change') or dichotomous measures (for example, either 'no important changes' or 'important changes' in a person's behaviour). Currently RevMan does not support categorical data so they could not be analysed as such.
4.3.1 Dichotomous data
Where possible we made efforts to convert outcome measures to dichotomous data. This may be done by identifying cut off points on rating scales and dividing subjects accordingly into 'clinically improved' or 'not clinically improved'. If the authors of a study had used a predefined cut off point for determining clinical effectiveness we used this where appropriate. Otherwise we generally assumed that a 50% reduction of a scale (e.g. the Brief Psychiatric Rating Scale ‐ Overall 1962) or a rating of 'at least much improved' according to the Clinical Global Impression Scale (Guy 1976) could be considered as a clinically significant response.
For dichotomous outcomes, we estimated a relative risk (RR) with the 95% confidence interval (CI) based on a fixed effects model in case of homogeneous outcomes, and based on a random effects model in the case of heterogeneous outcomes. If overall results were significant, the Number Needed to Treat (NNT) and/or the Number Needed to Harm (NNH) was calculated as the inverse of the risk reduction. It has been shown that RR is more intuitive than odds ratios (Boissel 1999) and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). This misinterpretation then leads to an overestimate of the impression of the effect. Data were inspected to see if an analysis using a Mantel‐Haenszel odds ratio and a random effects model made a substantive difference.
4.3.2 Continuous data
4.3.2.1 Normal distribution
Continuous data on outcomes in trials relevant to mental health issues are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data the following standards were applied to data derived from continuous measures of endpoint ('state' data).
When a scale starts from zero, the standard deviation, when multiplied by two, is less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution ‐ Altman 1996). Endpoint scores on scales often have a finite start and end point and this rule can be applied. When continuous data are presented on a scale which includes a possibility of negative values (such as change on a scale) it is impossible to tell whether data are non‐normally distributed (skewed) or not. It is thus preferable to use scale end point data, which typically cannot have negative values. If end point data were not available, we chose to use change data, because the statistics used in Metaview are rather robust towards skewness. If a scale starts from a positive value (such as PANSS, which can have values from 30‐210) the calculation described above should be modified to take the scale starting point into account. In these cases skewness is present if 2SD>(S‐Smin), where S is the mean score and Smin is the minimum score.
4.3.2.2 Intention‐to‐treat versus completer analyses
In the case of continuous data we assumed that in many cases an intention‐to‐treat analysis would not be available, so the data had to be analysed as they were presented in the original publications.
4.3.2.3 Summary statistic
For continuous outcomes, we estimated a weighted mean difference (WMD) between groups. Again, a fixed effects model was used for homogeneous outcomes and a random effects model for heterogeneous outcomes. Whenever possible we took the opportunity to make direct comparisons between trials that used the same measurement instrument to quantify specific outcomes. Where continuous data was presented from different scales rating the same effect, we presented both sets of data and inspected the general direction of effect.
4.3.2.4 Rating scales
A wide range of instruments is available to measure mental health outcomes. These instruments vary in quality and many are not valid, or even ad hoc. For outcome instruments some minimum standards have to be set. We only included continuous data from rating scales if the measuring instrument had been described in a peer‐reviewed journal (Marshall 2000), the instrument was either a self report or completed by an independent rater or relative (not the therapist), and the instrument could be considered a global assessment of an area of functioning. However, as it was expected that therapists would frequently also be the rater, we commented on such data as 'prone to bias'.
4.4 Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra class correlation in clustered studies, leading to a "unit of analysis" error (Divine 1992) whereby p values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type 1 errors (Bland 1997, Gulliford 1999).
Where clustering was not accounted for in primary studies, we presented the data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies to obtain intra‐class correlation co‐efficients of their clustered data and to adjust for this using accepted methods (Gulliford 1999). Where clustering has been incorporated into the analysis of primary studies, we will also present these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a "design effect". This is calculated using the mean number of participants per cluster (m) and the intraclass correlation co‐efficient (ICC) [Design effect = 1+(m‐1)*ICC] (Donner 2002). If the ICC was not reported it was assumed to be 0.1 (Ukoumunne 1999). If cluster studies had been appropriately analysed taking into account intra‐class correlation coefficients and relevant data documented in the report, synthesis with other studies would have been possible using the generic inverse variance technique.
4.5 Data display
We entered data into RevMan in such a way that the area to the left of the line of no effect indicated a favourable outcome for valproate alone or valproate augmentation.
5. Heterogeneity
Firstly, we undertook consideration of all the included studies within any comparison to judge clinical heterogeneity. Then we visually inspected graphs to investigate the possibility of statistical heterogeneity. We supplemented this using, primarily, the I‐squared statistic. This provides an estimate of the percentage of variability due to heterogeneity rather than chance alone. Where the I‐squared estimate was greater than or equal to 50% we interpreted this as indicating the presence of considerable levels of heterogeneity Higgins 2003. If inconsistency was high and clear reasons explaining the heterogeneity were found we presented the data separately. If not, we commented upon the heterogeneity of the data.
6. Publication bias
We entered data from all included trials into a funnel graph (trial effect versus trial size or 'precision') in an attempt to investigate the likelihood of overt publication bias. We undertook a formal test of funnel plot asymmetry (suggesting potential publication bias) where appropriate (Egger 1997). Significance levels of p < 0.1 were set a priori to accept the presence of asymmetry.
7. Sensitivity analysis
We planned to carry out a sensitivity analysis to investigate whether the exclusion of schizoaffective participants significantly changed the results.
Appendix 3. Previous plain language summary
Most people with schizophrenia or schizophrenia‐like conditions who are in contact with medical services will be treated with antipsychotic medication. Despite this, about 30% will continue to experience some signs of illness. Various other drugs have been added to the antipsychotic medication to try and reduce the symptoms these people experience. One such group of drugs are sodium and magnesium valproate, medication usually used to treat epilepsy or to stabilize mood in people who have bipolar disorder and those who have symptoms of schizophrenia and mood disorder together (schizoaffective disorder). This review looks at trials which attempt to compare valproate with placebo and also looks at valproate in combination with an antipsychotic compared to the antipsychotic alone. The studies included 2184 people in 26 trials, with the largest trial containing 402 people and the smallest, 12 people. All of the trials used an antipsychotic, and compared it to the antipsychotic plus valproate. There were no trials comparing just valproate with placebo. Risperidone was used in 8 trials, haloperidol, olanzapine and clozapine were used in 6 trials, quetiapine was used in 4 trials, aripiprazole in 3 trials, chlopromazine and perphenazine in only one trial. 6 other trials used more than one type of antipsychotic and did not specify the drugs used. For 14 studies reported measures including becoming more well and improvement in mental state there was a significant advantage for those on valproate plus antipsychotic compared to antipsychotic alone. Valproate was also reported to be effective in controlling excitement and aggression. However, such positive evidences were largely based on open Chinese studies. Moreover, it was also found that valproate increased sedation and dizziness. None of the studies were longer than 12 weeks so it is not known whether there would be a difference between the groups in the longer term. In addition, it was difficult to compare one trial with another because they recruited people with different groups of symptoms, used several different antipsychotics and looked at a diverse range of outcomes. The use of valproate for schizophrenia would benefit from some bigger and longer trials, with proper randomisation and double‐blindness.
(Plain language summary prepared for this review by Janey Antoniou of RETHINK, UK www.rethink.org)
Data and analyses
Comparison 1. VALPROATE + ANTIPSYCHOTICS versus ANTIPSYCHOTICS + PLACEBO or ANTIPSYCHOTICS alone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically significant response: 1. Important change ‐ as defined by the studies | 14 | 1049 | Risk Ratio (M‐H, Random, 95% CI) | 1.31 [1.16, 1.47] |
| 2 Leaving the study early: Acceptability/tolerability of treatment | 12 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Acceptability of treatment: Leaving for any reason | 11 | 951 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.47, 1.24] |
| 2.2 Overall tolerability: Leaving due to adverse event | 6 | 974 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.90, 1.97] |
| 2.3 Leaving due to poor clinical effect | 5 | 810 | Risk Ratio (M‐H, Random, 95% CI) | 0.54 [0.31, 0.94] |
| 3 Clinical response: 2. Global state: 1a. mean change score CGI severity (high = poor) | 1 | 392 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐0.12, 0.32] |
| 4 Clinical response: 3. Global state: 1b. mean change score CGI improvement (high = poor) | 1 | 393 | Mean Difference (IV, Random, 95% CI) | 0.0 [‐0.26, 0.26] |
| 5 Clinical response: 4. Mental state: 2a. mean change score PANSS total (high = poor) | 13 | 1363 | Mean Difference (IV, Random, 95% CI) | ‐5.85 [‐7.80, ‐3.91] |
| 6 Clinical response: 5. Mental state: 2b. mean endpoint score BPRS total (high = poor) | 7 | 646 | Mean Difference (IV, Random, 95% CI) | ‐1.87 [‐4.46, 0.73] |
| 7 Clinical response: 6. Mental state 2c. mean change score IMPS total (high = poor) | 1 | 18 | Mean Difference (IV, Random, 95% CI) | ‐5.11 [‐26.04, 15.82] |
| 8 Clincal response: 7. Positive symptoms: 3a. mean endpoint score PANSS positive subscale (high = poor) | 9 | 1073 | Mean Difference (IV, Random, 95% CI) | ‐1.72 [‐3.01, ‐0.43] |
| 9 Clinical response: 8. Positive symptoms: 3b. mean endpoint score PANSS positive subscale (high = poor, skewed data) | Other data | No numeric data | ||
| 10 Clinical response: 9. Negative symptoms: 4a. mean endpoint score PANSS negative subscale (high = poor) | 5 | 651 | Mean Difference (IV, Random, 95% CI) | ‐1.78 [‐3.13, ‐0.43] |
| 11 Clinical response: 10. Negative symptoms: 4b. mean endpoint score PANSS negative subscale (high = poor, data skewed) | Other data | No numeric data | ||
| 12 Clinical response: 11. Negative symptoms: 4c. mean endpoint score SANS subscale (skewed data) | Other data | No numeric data | ||
| 13 Clinical response: 12. Negative symptoms: 4d. mean endpoint score BPRS lack of energy subscale (high = poor) | 3 | 135 | Mean Difference (IV, Random, 95% CI) | 0.46 [‐0.29, 1.22] |
| 14 Clinical response: 13. Aggression/Agitation: 5a. clinically important change ‐ Modified Overt Aggression Scale (high = poor) | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 2.68 [1.07, 6.76] |
| 15 Clinical response: 14. Aggression/Agitation: 5b. mean endpoint score Modified Overt Aggression Scale (high = poor) | 3 | 186 | Mean Difference (IV, Random, 95% CI) | ‐2.55 [‐3.92, ‐1.19] |
| 16 Clinical response: 15. Aggression/Agitation: 5c. mean endpoint score PANSS‐EC subscale (high = poor)) | 3 | 204 | Mean Difference (IV, Random, 95% CI) | ‐1.85 [‐2.63, ‐1.08] |
| 17 Clinical response: 16. Aggression/Agitation: 5d. mean endpoint score PANSS‐EC subscale (skewed data) | Other data | No numeric data | ||
| 18 Clinical response: 17. Aggression/Agitation: 5e. mean endpoint score PANSS supplementary subscale (high = poor) | 1 | 120 | Mean Difference (IV, Random, 95% CI) | ‐0.85 [‐1.39, ‐0.31] |
| 19 Clinical response: 18. Aggression/Agitation: 5f. mean endpoint score BPRS hostility subscale (high = poor) | 3 | 135 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.34, 0.14] |
| 20 Clinical response: 19. Depression symptoms: 6a. mild improvement at endpoint, Calgary Depression Scale (high = poor) | 1 | 16 | Risk Ratio (M‐H, Random, 95% CI) | 1.4 [0.77, 2.54] |
| 21 Clinical response: 20. Depression symptoms: 6b. mean endpoint score BPRS anxiety and depression subscale (high = poor) | 3 | 135 | Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐0.25, 0.13] |
| 22 Clinical response: 21.General pathology: 7a. mean change score PANSS general pathology subscale (high = poor) | 8 | 873 | Mean Difference (IV, Random, 95% CI) | ‐3.05 [‐4.30, ‐1.81] |
| 23 Use of additional medication: 1. Mean dose of antiparkinson medication (skewed data) | Other data | No numeric data | ||
| 24 Use of additional medication: 2. Mean chlorprothixene dose (skewed data) | Other data | No numeric data | ||
| 25 Use of additional medication: 3. Medication for sedation at least once | 2 | 309 | Risk Ratio (M‐H, Random, 95% CI) | 3.65 [0.11, 122.31] |
| 26 Adverse events: 1. Abnormal ECG | 2 | 153 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.35, 2.18] |
| 27 Adverse events: 2. Abnormal liver function/increase in alanine transaminase/gamma‐glutamyl transpeptidase | 8 | 745 | Risk Ratio (M‐H, Random, 95% CI) | 1.26 [0.72, 2.22] |
| 28 Adverse events: 3. Akathisia | 3 | 186 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.36, 3.06] |
| 29 Adverse events: 4. Anxiety | 2 | 319 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.21, 1.24] |
| 30 Adverse events: 5. Asthenia | 1 | 249 | Risk Ratio (M‐H, Random, 95% CI) | 1.58 [0.63, 3.95] |
| 31 Adverse events: 6. Ataxia | 2 | 115 | Risk Ratio (M‐H, Random, 95% CI) | 2.42 [0.37, 15.92] |
| 32 Adverse events: 7. At least one adverse event | 5 | 493 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.68, 1.25] |
| 33 Adverse events: 8. Blood count changes | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 33.1 Eosinophilia | 1 | 42 | Risk Ratio (M‐H, Random, 95% CI) | 1.38 [0.43, 4.42] |
| 33.2 Monocytosis | 1 | 42 | Risk Ratio (M‐H, Random, 95% CI) | 5.48 [0.28, 107.62] |
| 33.3 Transient lymphocytosis | 1 | 42 | Risk Ratio (M‐H, Random, 95% CI) | 3.3 [0.37, 29.21] |
| 33.4 Leukopenia | 5 | 385 | Risk Ratio (M‐H, Random, 95% CI) | 1.18 [0.38, 3.64] |
| 33.5 Thrombocytopenia | 1 | 79 | Risk Ratio (M‐H, Random, 95% CI) | 17.42 [1.04, 291.96] |
| 34 Adverse events: 9. Constipation | 4 | 515 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.47, 1.85] |
| 35 Adverse events: 10. Convulsion | 1 | 120 | Risk Ratio (M‐H, Random, 95% CI) | 0.37 [0.02, 8.85] |
| 36 Adverse events: 11. Diarrohea | 3 | 193 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.32, 3.34] |
| 37 Adverse events: 12. Dizziness | 7 | 662 | Risk Ratio (M‐H, Random, 95% CI) | 1.37 [0.92, 2.03] |
| 38 Adverse events: 13. Dry mouth | 3 | 179 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.58, 2.42] |
| 39 Adverse events: 14. Dyspepsia | 1 | 249 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.62, 1.79] |
| 40 Adverse events: 15. Dystonia | 2 | 130 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.30, 3.37] |
| 41 Adverse events: 16. Extrapyramidal adverse events (skewed data) | Other data | No numeric data | ||
| 42 Adverse events: 17. Headache | 4 | 469 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.67, 1.56] |
| 43 Adverse events: 18. Hypersalivation | 3 | 248 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.62, 1.70] |
| 44 Adverse events: 19. Incontinence | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 3.39 [0.15, 79.22] |
| 45 Adverse events: 20. Insomnia | 2 | 126 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.39, 1.78] |
| 46 Adverse events: 21. Myocardial ischaemia | 1 | 62 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.05, 5.23] |
| 47 Adverse events: 22. Nausea | 9 | 728 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.80, 1.86] |
| 48 Adverse events: 23. Pain | 1 | 249 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.30, 1.25] |
| 49 Adverse events: 24. Rash | 2 | 190 | Risk Ratio (M‐H, Random, 95% CI) | 3.15 [0.33, 29.72] |
| 50 Adverse events: 25. Rhinitis | 1 | 249 | Risk Ratio (M‐H, Random, 95% CI) | 0.30 [0.09, 1.07] |
| 51 Adverse events: 26. Rigidity/stiffness | 1 | 33 | Risk Ratio (M‐H, Random, 95% CI) | 2.83 [0.12, 64.89] |
| 52 Adverse events: 27. Sedation/somnolence/drowsiness | 8 | 770 | Risk Ratio (M‐H, Random, 95% CI) | 1.38 [1.07, 1.79] |
| 53 Adverse events: 28. Sexual dysfunction | 1 | 33 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.01, 7.21] |
| 54 Adverse events: 29. Suicidal or depressed | 1 | 16 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.14] |
| 55 Adverse events: 30. Tachycardia | 3 | 218 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [0.60, 2.72] |
| 56 Adverse events: 31.Tremor | 4 | 199 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.46, 2.97] |
| 57 Adverse events: 32. Unstable gait | 1 | 19 | Risk Ratio (M‐H, Random, 95% CI) | 0.27 [0.02, 3.39] |
| 58 Adverse events: 33. Vegetative adverse events | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.70 [0.55, 5.27] |
| 59 Adverse events: 34. Vomiting | 2 | 282 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.45, 2.18] |
| 60 Adverse events: 35. Weight gain | 4 | 427 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.76, 1.82] |
| 61 Adverse events: 36. Mean endpoint score TESS (high = poor) | 1 | 60 | Mean Difference (IV, Random, 95% CI) | 2.06 [‐0.41, 4.53] |
| 62 Adverse events: 37. Tardive dyskinesia ‐ mean endpoint score AIMS (high = poor) | 1 | 79 | Mean Difference (IV, Random, 95% CI) | ‐3.31 [‐4.91, ‐1.71] |
| 63 Sensitivity analysis: Primary outcome 1. Excluding open studies | 3 | 323 | Risk Ratio (M‐H, Random, 95% CI) | 1.15 [0.95, 1.39] |
| 64 Sensitivity analysis: Primary outcome 2. Fixed‐effect model | 14 | 1049 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.35 [1.21, 1.50] |
| 65 Sensitivity analysis: Primary outcome 3. Exluding no attrition | 4 | 517 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [1.03, 1.60] |
| 66 Sensitivity analysis: Primary outcome 4. Completers | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 3.13 [1.52, 6.44] |
| 67 Sensitivity analysis: Primary outcome 5. Defined by PANSS reduction | 12 | 975 | Risk Ratio (M‐H, Random, 95% CI) | 1.31 [1.16, 1.48] |
| 68 Sensitivity analysis: Aggresssion/Agitation: 1. Modified Overt Aggression Scale ‐ excluding schizoaffective studies | 3 | 186 | Mean Difference (IV, Random, 95% CI) | ‐2.55 [‐3.92, ‐1.19] |
| 69 Sensitivity analysis: Aggression/Agitation: 2. PANSS‐EC subscale ‐ excluding schizoaffective studies | 2 | 124 | Mean Difference (IV, Random, 95% CI) | ‐1.63 [‐2.62, ‐0.64] |
| 70 Sensitivity analysis: Aggression/Agitation: 3. PANSS supplementary subscale ‐ excluding schizoaffective studies | 1 | 120 | Mean Difference (IV, Random, 95% CI) | ‐0.85 [‐1.39, ‐0.31] |
| 71 Sensitivity analysis: Aggression/Agitation: 4. BPRS hostility subscale ‐ excluding schizoaffective studies | 3 | 135 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.34, 0.14] |
| 72 Subgroup analysis: Primary outcome 6. As defined by the studies | 14 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 72.1 Treatment resistant participants | 2 | 128 | Risk Ratio (M‐H, Random, 95% CI) | 1.57 [1.03, 2.39] |
| 72.2 No Treatment resistant participants | 12 | 921 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [1.14, 1.46] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Casey 2003.
| Methods | Allocation: randomised Blinding: double Duration: 28 days (preceded by wash‐out period of 3 times the mean elimination half‐life of the antipsychotic) Design: parallel Country: USA |
|
| Participants | Diagnosis: DSM‐IV schizophrenia n = 249* Sex: 184 M, 58 F* Age: ˜ 39 years History: treated with antipsychotic at the time of enrolment (n = 214) |
|
| Interventions | 1. Olanzapine: starting dose 5 mg/d, increased to 10mg/d on day 3 and to target dose of 15 mg/d on day 6; n = 65* 2. Risperidone: starting dose 2 mg/d, increased to 4 mg/don day 3 and to target dose of 6 mg/d on day 6; n = 60* 3. Olanzapine + divalproex: olanzapine was titrated as described in 1. Divalproex starting dose 15 mg/d, titrated to clinical response (maximum 30 mg/d); n = 66* 4. Risperidone + divalproex: risperidone was titrated according to 2. and divalproex according to 3; n = 58* | |
| Outcomes | Clincally significant response: important change (PANSS reduction > 20%)
Leaving the study early: acceptability/tolerability of treatment Clinical response: mental state (PANSS total), positive symptoms (PANSS subscale) Adverse events: at least one event, various events Unable to use: Clinical response: mental state ‐ BPRS (no data), aggression/agitation ‐ PANSS‐EC (data skewed) Adverse events: SAS, AIMS, BAS scores (no data) |
|
| Notes | *The 2 groups with combination therapy and the 2 groups with monotherapy were considered to be one (p 185, Table 1) NB. In the original report 7 participants were not included in the ITT analyses due to missing on‐treatment PANSS score (n = 4) and randomisation at 2 sites (only the second randomisation was excluded; n = 3) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomized" (p 182) no further description |
| Allocation concealment (selection bias) | Unclear risk | Not indicated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "double‐blind" (p 182) |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | "double‐blind" (p 182) |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | "double‐blind" (p 182) |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | "double‐blind" (p 182) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Attrition data for different treatment groups for the reasons of treatment‐emergent adverse events and lack of efficacy were reported and distributed relatively even between groups, but there were no data for the total number of people leaving the study in different treatment groups. |
| Selective reporting (reporting bias) | High risk | No data for those leaving the study early for the different treatment groups; BPRS and movement adverse effect were not reported |
| Other bias | Unclear risk | The 2 groups with combination therapy and the 2 groups with monotherapy were considered to be 1 In the original report 7 people with schizophrenia were not included in the ITT analyses due to missing on‐treatment PANSS score and randomisation at 2 sites (only the second randomisation was excluded) |
Casey 2009.
| Methods | Allocation: randomised Blinding: double Duration: 12 weeks (preceded by wash‐out period: 1‐5 days) Design: parallel Country: USA |
|
| Participants | Diagnosis: current DSM‐IV‐TR diagnosis of schizophrenia, confirmed by a Structured Clinical Interview for DSM‐IV‐TR (SCID)* n = 402 Sex: 304 M, 89 F***. Age: 18 – 65 years (mean: 40.0 ± 10.52 years)** History: mean age of first diagnosis 24.2 ± 8.3 years Setting: multi‐centre, people with schizophrenia hospitalised for a minimum of 14 d during the acute phase of the study and could be discharged (per investigator discretion) anytime after day 14 |
|
| Interventions | 1. Olanzapine + placebo: fixed dose of olanzapine, mean dose = 15 mg/d; n = 103 2. Olanzapine + divalproex ER: fixed dose of olanzapine, mean dose = 15 mg/d; mean dose of divalproex ER = 2828 mg with a flexible dose not exceeding 35 mg/d; n = 99 3. Risperidone + placebo: fixed dose of risperidone, mean dose = 6 mg/d; n = 101 4. Risperidone + divalproex ER: fixed dose of risperidone, mean dose = 6 mg/day, mean dose of divalproex ER = 2712 mg with a flexible dose not exceeding 35 mg/day; n = 99 Co‐medication: lorazepam, propranolol hydrochloride, and benztropine mesylate could be used as adjunctive medications, but were not to be used prophylactically |
|
| Outcomes | Leaving the study early: acceptability/tolerability of treatment Clinical response: global state (CGI severity and improvement scales), mental state (PANSS total, BPRS total), positive symptoms, negative symptoms, general pathology (PANSS subscales) Unable to use: Adverse events: EPS, SAS, BAS, and AIMS (no data) |
|
| Notes | * In the antipsychotic monotherapy group, 172 out of 198 participants (87%) were diagnosed as paranoid type. In the valproate group, 166 out of 195 participants (85%) were diagnosed as paranoid type ** Monotherapy: mean = 39.9 years (SD = 10.49 yrs). Combination: mean = 40.1 years (SD = 10.54 years) *** Monotherapy: 156 M, 42 F. Combination: 148 M, 47 F |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomized", no further details provided (p 1331) |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes. |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | "double‐blind" |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes. |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | "raters completed a rater‐training program", no further details provided (p 1331) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Used technique of LOCF but reported data for people leaving the study early very high (62%) (p 1332) |
| Selective reporting (reporting bias) | High risk | No data reported for extrapyramidal symptoms, measured by SAS, BAS and AIMS |
| Other bias | Low risk | No obvious other bias |
Citrome 2007.
| Methods | Allocation: randomised Blinding: single Duration: 8 weeks Design: parallel Country: not reported |
|
| Participants | Diagnosis: schizophrenia with poor impulse control, aggressive behaviour and/or hostility n = 33 Sex: 31 M, 2 F Age: mean 39.6 yrs ± 10.5 years History: mean age of onset 16.4 ± 4.6 years Settings: inpatient |
|
| Interventions | 1. Risperidone monotherapy: target dose range of 4–6 mg/d within 7–14 d; n = 16 2. Risperidone + valproate: target risperidone dose 4–6 mg/d within 7–14 d; target valproate plasma level 50‐100 mg/ml, usually achieved with dose 1000–1500 mg/d; n = 17 Co‐medication: no other antipsychotics or mood stabilisers permitted. Benztropine (if needed). Lorazepame (if needed) for agitation and insomnia: for weeks 1‐3, max 6 mg/d, for weeks 4–8, up to 4 doses of lorazepam in any week ‐ evidence of treatment failure and study participation discontinued |
|
| Outcomes | Leaving the study early: acceptability/tolerability of treatment Adverse events: various events Unable to use: Clinical response: PANSS, PANSS Hostility, BIS and BDHI (no usable mean and SDs), CGI, NOSIE (results not reported) Overt aggression scale (OAS) (comparison between completers and non‐completers, not between study arms) Adverse events: ESRS score (results not reported) |
|
| Notes | NB. Initially, participants were eligible only if they had at least 2 aggressive incidents during the 2 weeks preceding their transfer to research unit and continued to exhibit aggression during the first 2 weeks after transfer. Aggressive behaviour was defined as at least 2 additional aggressive incidents or ≥ 4 on PANSS ‘Hostility’ item or ‘Poor Impulse Control’ item.” Because of problematic study enrolment (only 16 participants): study entry criteria revised to include participants not exhibiting aggression but scored ≥ 3 on at least one of the PANSS ‘Hostility’, 'Impulsivity’, ‘Excitement’, or ‘Uncooperativeness’ items For participants already receiving valproate and antipsychotic drugs other than risperidone, risperidone and valproate were given for 4 weeks before randomisation For participants already receiving valproate at study entry who were randomised to combination treatment continued valproate at the same dose/plasma level for the remainder of the study Participants already receiving valproate at study entry who were randomised to RMon, valproate tapered down by 50% every 48 hours All participants received valproate delayed release except one who received extended release |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomized", no details provided (p 357) |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcome. |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | Open label, “study participants, treating psychiatrists and other staff were aware” (p 358) |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcome. Raters were blind. |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Low risk | Blinded raters (p 358) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | People leaving the study early 13/33 = 39.4% (p 359) AND reasons for missing outcome data likely to be related to true outcome, with imbalance in numbers: significantly fewer participants randomised to risperidone‐alone completed the study |
| Selective reporting (reporting bias) | High risk | Endpoint data of CGI, NOISE, ESRS not reported, PANSS, PANSS hostility, BIS, BDHI no endpoint mean and SDs reported; Overt aggression scale comparison between completers and non‐completers, not between study arms |
| Other bias | Low risk | No obvious other bias |
Dose 1998.
| Methods | Allocation: randomised Blinding: double Duration: 28 d (followed by 1 week continuation of antipsychotic medication alone) Design: not reported Country: Germany |
|
| Participants | Diagnosis: DSM‐III schizophrenia and schizoaffective disorder n = 42 Sex: no information Age: no information History: no history of epileptic seizures, no ECG abnormalities Setting: inpatient |
|
| Interventions | 1. Haloperidol + valproate: haloperidol starting dose 3 mg/d with possible increase by 3 mg/d every fifth day, valproate starting dose 300 mg/d, target plasma level 60‐90 µg/ml; n = 20 2. Haloperidol + placebo: haloperidol starting dose 3 mg/d with possible increase by 3 mg/d every fifth day; n = 22. Valproate and placebo discontinued abruptly after 28 days | |
| Outcomes | Leaving the study early: acceptability/tolerability of treatment Clinical response: mental state (BPRS total) Adverse events: various events Unable to use: Use of additional medication: mean dose of antiparkinsonian and chlorprothixene medication (data skewed) Adverse events: SAS (data skewed) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned" (p 122) |
| Allocation concealment (selection bias) | Unclear risk | Not indicated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "double‐blind"; "Plasma VPA levels were (...) communicated to the project leader only in order to maintain the blind conditions" (p 122) |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | "double‐blind"; "psychiatrist, who was not aware whether the participant was receiving an anticonvulsant or placebo"; "VPA tablets and placebo of identical appearance" (p 122) |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | "double‐blind"; "Plasma VPA levels were (...) communicated to the project leader only in order to maintain the blind conditions" (p 122) |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Low risk | "double‐blind"; "psychiatrist, who was not aware whether the participant was receiving an anticonvulsant or placebo"; "VPA tablets and placebo of identical appearance" (p 122) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 31% rate for people leaving the study early, evenly distributed between groups (6/20 and 7/22) |
| Selective reporting (reporting bias) | Unclear risk | No numerical values for EEG and lab tests provided |
| Other bias | Low risk | No other bias |
Fisk 1987.
| Methods | Allocation: randomised Blinding: double Duration: 12 weeks (6 week baseline period) Design: parallel Country: UK |
|
| Participants | Diagnosis: chronic functional psychotic illness, all with at least 6 months of pre‐trial tardive dyskinesia according to Barnes and Kidger Rating Scale, maintenance antipsychotic treatment > 1 year n = 62* Sex: no information Age: < 70 years History: physically fit, no liver disease, and no acute or chronic organic brain syndromes Settings: multi‐centre |
|
| Interventions | 1. Antipsychotic medication + valproate: constant antipsychotic dose, valproate dose stepwise increased to 1500 mg/d or 1200 mg/d according to weight; n = 29* 2. Antipsychotic medication + placebo: constant antipsychotic dose; n = 29* | |
| Outcomes | Clinically significant response: important change (Krawiecka scale) Leaving the study early: acceptability/tolerability of treatment Adverse events: various events Unable to use: Adverse events: SAS score (data skewed) |
|
| Notes | *During a 6‐week baseline period all participants received a stable dose of antipsychotic drugs only. 4 participants left the study during this phase and were not considered in the analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised (...) stratified for age, sex, setting and pretrial tardive dyskinesia score (p 542) ‐ no further details |
| Allocation concealment (selection bias) | Unclear risk | Not indicated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "double‐blind" (p 542); "double‐blind procedures were used throughout (...) Hospital pharmacists acted as coordinators of treatment"; "further blind ratings by experienced independent raters" (p 543) |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | "double‐blind" (p 542); "double‐blind procedures were used throughout (...) Hospital pharmacists acted as coordinators of treatment"; "further blind ratings by experienced independent raters" (p 543) |
| Blinding of outcome assessment (detection bias) Objective outcomes | Unclear risk | "double‐blind" (p 542); "double‐blind procedures were used throughout (...) Hospital pharmacists acted as coordinators of treatment"; "further blind ratings by experienced independent raters" (p 543) |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Low risk | "double‐blind" (p 542); "double‐blind procedures were used throughout (...) Hospital pharmacists acted as coordinators of treatment"; "further blind ratings by experienced independent raters" (p 543) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 26% rate for people leaving the study early, not evenly distributed between groups (11/29 and 4/29) |
| Selective reporting (reporting bias) | Unclear risk | Data from physiological monitoring not reported |
| Other bias | Low risk | No other bias |
Glick 2009.
| Methods | Allocation: randomised Blinding: double Duration: 12 weeks Design: not reported Country: USA |
|
| Participants | Diagnosis: DSM‐IV schizophrenia or schizoaffective disorder n = 24* Sex: no information Age: adults (no further details provided) Settings: outpatient |
|
| Interventions | 1. Antipsychotic drugs + lamotrigine: flexible dose of lamotrigine, weeks 1–6: gradually increase dose from 25 mg/d to target dose of 150 mg/d. Weeks 7‐9: between 100 mg/d and 400 mg/d according to clinical judgement of the clinician. Weeks 9–12: study medications discontinued in a stepwise reduction; n = 8* 2. Antipsychotic drugs + divalproex sodium: flexible dose of divalproex sodium, weeks 1–6: gradually increase dose from 250 mg/d to target dose of 1500 mg/d. Weeks 7‐9: between 1000 mg/d and 4000 mg/d according to clinical judgement of the clinician. Weeks 9‐12, study medications discontinued in a stepwise reduction; n = 8 3. Antipsychotic drugs + placebo: n = 8 |
|
| Outcomes | Clincally significant response: important change Clinical response: depression (CDS) Adverse events: various events Unable to use: Leaving the study early: acceptability/tolerability of treatment (no data for each study arm) Clinical response: demoralisation (no usable data) Adverse events: EPS: SAS, BAS, AIMS scores (no data) Quality of life: Lehman quality of life improvement scale (no data) |
|
| Notes | *Data from intervention group 1 were not used for this review | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised" (...) randomly assigned in a 1:1: ratio. No further details provided (p 268) |
| Allocation concealment (selection bias) | Unclear risk | "all drugs identically encapsulated by an independent pharmacy", not sure if the pharmacy ran the randomisation (p 268) |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes. |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | Clinicians, participants and family were blind to study assignment (p 268) |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes. |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Low risk | Raters were blind to study assignment (p 268) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 7 out of 25 (28%) participants left early, no data for people leaving the study early in each group (p 269) |
| Selective reporting (reporting bias) | High risk | Almost no usable data reported apart from dichotomous data for PANSS, CGI, CDS and adverse events |
| Other bias | Low risk | No obvious other bias |
Guo 2007.
| Methods | Allocation: randomised Blinding: open Duration: 2 weeks Design: Parallel Country: China |
|
| Participants | Diagnosis: CCMD‐3 schizophrenia, with excitement and agitation symptoms n = 60 Sex: 40 M, 20 F Age: range 18‐60 years, mean 34.5 ± 2.4 years History: duration of illness (total) ‐ mean 4.75 ± 0.6 years Setting: inpatient |
|
| Interventions | 1. Magnesium valproate + quetiapine: valproate 0.5‐1 g/d, mean dose 0.8 g ± 5.7 mg/d, maximum 1.0 g/d, quetiapine 100‐800 mg (517 ± 4 mg/d); n = 30 2. Haloperidol: IM. injection, 5‐30 mg/d (15.8 ± 3.8 mg/d); n = 30 Co‐medication: 11 participants in valproate group given IM injection of clonazepam 1‐2 mg/d |
|
| Outcomes | Clinically significant response: important change (PANSS reduction) Clinical response: aggression/agitation (PANSS subscale) Use of additional medication Unable to use: Leaving the study early: (no information provided) Clinical response: global state ‐ CGI (data not reported) Adverse events: TESS (data not reported) |
|
| Notes | Valproate group & monotherapy group do not differ significantly in demographic data (P > 0.05) Quetiapine initial dose 100 mg/day; valproate initial dose 0.5 g/day; haloperidol days 1‐3, 5‐10 mg/d, and days 4‐7, 10‐30 mg/d |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned", p183 |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes. |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | "opening", personnel and participants not blinded |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes. |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated |
| Selective reporting (reporting bias) | High risk | CGI, TESS score were measured, but not reported |
| Other bias | Low risk | None obvious |
Hesslinger 1999.
| Methods | Allocation: randomised Blinding: single (rater blind) Duration: 4 weeks (4‐day wash‐out) Design: not reported Country: Germany |
|
| Participants | Diagnosis: ICG‐10 schizophrenia and schizoaffective disorder, acute exacerbation within the last 7 days n = 27* Sex: no information Age: no information History: no evidence of serious medical conditions, substance abuse, organic psychosis, use of depot neuroleptics, antiepileptic drugs or serotonin reuptake inhibitors 3 months prior to trial Setting: inpatient |
|
| Interventions | 1. Haloperidol: mean dose 15.5 mg/d; n = 9 2. Haloperidol + valproate: haloperidol mean dose 15.5 mg/d, valproate dose 757.1 mg/d, target plasma level 50‐100 µg/ml; n = 9 3. Haloperidol + carbamazepine: haloperidol mean dose 15.5 mg/day, carbamazepine mean dose 567.3 mg/day, target plasma level 6‐12 µg/ml; n = 9* | |
| Outcomes | Leaving the study early: acceptability/tolerability of treatment Clinical response: mental state (IMPS), positive symptoms (PANSS subscale) Unable to use: Use of additional medication: (skewed data) |
|
| Notes | *Data from intervention group 3 were not used for this review | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned" (p 311) |
| Allocation concealment (selection bias) | Unclear risk | "sealed envelopes" |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "psychiatrist (...) was blind to medication and drug levels. However, he had access to the nursing charts" (p 311). People with schizophrenia were not aware of the treatment (information received by letter) |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | "psychiatrist (...) was blind to medication and drug levels. However, he had access to the nursing charts" (p 311). People with schizophrenia were not aware of the treatment (information received by letter) |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | "psychiatrist (...) was blind to medication and drug levels. However, he had access to the nursing charts" (p 311). People with schizophrenia were not aware of the treatment (information received by letter) |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | "psychiatrist (...) was blind to medication and drug levels. However, he had access to the nursing charts" (p 311). People with schizophrenia were not aware of the treatment (information received by letter) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participants left the study early. |
| Selective reporting (reporting bias) | Low risk | No selective reporting |
| Other bias | Low risk | No other bias |
Jia 2007.
| Methods | Allocation: randomised Blinding: open label Duration: 4 weeks Design: parallel Country: China |
|
| Participants | Diagnosis: ICD‐10 schizophrenia n = 80 Sex: male only Age: 18‐50 years, mean 38.9 ± 11.0 years History: duration of illness total, range 10‐25 years, mean 15.3 ± 9.1 years Setting: inpatient. |
|
| Interventions | 1. Sodium valproate + clozapine: valproate 400‐800 mg/d, clozapine dosage unclear; n = 40 2. Clozapine alone: dosage not reported; n = 40 |
|
| Outcomes | Clinical response: mental state (PANSS total)
Adverse events: various events Unable to use: Clinical response: positive symptoms, negative symptoms ‐ PANSS subscales (skewed data) Leaving the study early (no information provided) Adverse events: TESS scores (no means and SDs reported) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned", p 3 "Pairs of participants with no significant differences in age, BMI, duration of illness, smoking, Clozapine dosage taken, co‐medication and PANSS score", were randomly divided into two equal groups" |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Not stated. No information about blinding provided, therefore assuming open‐label |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | Not stated. No information about blinding provided, therefore assuming open‐label |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Not stated |
| Blinding of outcome assessment (detection bias) Subjective outcomes | High risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated |
| Selective reporting (reporting bias) | High risk | TESS scores measured but no means and SDs reported; study authors only stated that no significant difference in TESS score between groups (P > 0.05). Study authors also selectively reported binary outcomes of a few adverse events, but didn't provide numbers for all adverse events |
| Other bias | Unclear risk | None obvious |
Jiang 2009.
| Methods | Allocation: randomised Blinding: open label Duration: 6 weeks Design: parallel Country: China |
|
| Participants | Diagnosis: CCMD‐3 schizophrenia (at least 2 of the 5 PANSS subscale scores > or = 4) n = 120 Sex: 64 M, 56 F Age: 33.92 ± 11.06 years History: mean duration of illness 1.73 ± 0.93 years Setting: inpatient |
|
| Interventions | 1. Magnesium valproate + routine antipsychotics: valproate magnesium flexible dose ≤ 1.5 g/d, mean 1.05 ± 0.57 g/d; antipsychotic drugs titrated to treatment dose within 2 weeks. Clozapine mean 275 ± 56 mg/d, risperidone mean 4.1 ± 0.98 mg/d, quetiapine mean 670 ± 120 mg/d, and olanzapine mean 12.4 ± 7.8 mg/d; n = 57 2. Routine antipsychotics: antipsychotic drugs titrated to treatment dose within 2 weeks. Clozapine mean 328 ± 78 mg/d, risperidone mean 4.9 ± 0.99 mg/d, quetiapine mean 710 ± 150 mg/d, and olanzapine mean 14 ± 8.6 mg/d; n = 63 |
|
| Outcomes | Clinically significant response: important change (PANSS reduction) Leaving the study early: acceptability/tolerability of treatment Clinical response: mental state (PANSS total), positive symptoms, aggression/agitation, general pathology (PANSS subscales) Adverse events: various events Unable to use: Clinical response: negative symptoms ‐ PANSS subscale (data skewed) |
|
| Notes | Before antipsychotic drugs and valproate were given, participants were first given an IM injection of clonazepam Dosage for antipsychotic drugs was adjusted according to clinical conditions, so that in the end the mean dosage used for valproate group was lower than mean dosage used for monotherapy group, but not sure if significant. Valproate magnesium extended‐release tablets were given, initial dose 0.5 g/d, 0.25 g/tablet |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratified randomisation, p176 |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of outcome assessment (detection bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Two participants left early from each group due to leukopenia. It is unclear, if these four people were included in the final analysis. (p 177) |
| Selective reporting (reporting bias) | High risk | TESS scores were measured but no means and SDs were reported |
| Other bias | Low risk | No obvious other bias |
Li 2008.
| Methods | Allocation: randomised Blinding: open label Duration: 2 weeks Design: parallel Country: China |
|
| Participants | Diagnosis: DSM‐IV schizophrenia, with obvious excitement n = 40 Sex: 22 M, 18 F Age: mean 32.5 ± 4.2 years History: mean duration of illness 6.6 ± 0.8 years Settings: inpatient |
|
| Interventions | 1. Olanzapine + valproate magnesium group: olanzapine flexible dose ≥ 10 mg/d and ≤ 20 mg/d. Valproate flexible dose ≥ 0.5 g/d and ≤ 1.5 g/d; n = 20 2. Olanzapine monotherapy group: olanzapine flexible dose ≥ 10 mg/d and ≤ 20 mg/d; n = 20 |
|
| Outcomes | Clinically significant response: important change (PANSS reduction ≥ 50%) Clinical response: mental state (PANSS total) Unable to use: Leaving the study early: (no data reported) Adverse events: (no data reported) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly divided into two groups" (p 178) |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of outcome assessment (detection bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated |
| Selective reporting (reporting bias) | High risk | Adverse events: no information provided |
| Other bias | Low risk | No obvious other bias |
Li 2009.
| Methods | Allocation: randomised Blinding: open label Duration: 4 weeks Design: not reported Country: China |
|
| Participants | Diagnosis: CCMD‐3 schizophrenia, with poor impulse control n = 56 Sex: 34 M 22 F Age: range 16–63 years, mean 31.8 ± 7.3 years History: mean duration of illness 3.6 ± 4.0 years Settings: inpatient |
|
| Interventions | 1. Aripiprazole + valproate group: aripiprazole titrated to target dose within two weeks, ≤ 30 mg/d. Valproate titrated to 800‐1200 mg/d within 1 week; n = 28 2. Aripiprazole monotherapy group: aripiprazole titrated to 20‐30 mg/d; n = 28 |
|
| Outcomes | Clinical response: negative symptoms (BPRS subscale), aggression/agitation (MOAS, BPRS subscale), depression (BPRS subscale) Adverse events: various events Unable to use: Leaving the study early (no information provided) Adverse events: TESS scores (no means and SDs reported) |
|
| Notes | No significant differences in demographic data of valproate group & monotherapy group, P > 0.05 Aripiprazole initial dose 5 mmg/d, valproate initial dose 400 mg/d Valproate group: once aripiprazole titrated to maximum 30 mg/d and valproate titrated to 800‐1200mg/d, they were maintained on these doses as long as possible |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomized", no further details provided (p 788) |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes. |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes. |
| Blinding of outcome assessment (detection bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated |
| Selective reporting (reporting bias) | High risk | TESS scores measured but no means and SDs reported |
| Other bias | Low risk | No obvious other bias |
Liang 2004.
| Methods | Allocation: randomised Blinding: open label Duration: 4 weeks Design: parallel Country: China |
|
| Participants | Diagnosis: CCMD‐3 schizophrenia n = 42 Sex: 31 M, 11 F Age: mean 37.85 ± 11.8 years History: mean duration of illness 5.3 ± 4.6 years Setting: inpatient |
|
| Interventions | 1. Sodium valproate + routine antipsychotic medication: sodium valproate dosage = 600 mg/d, antipsychotic drugs mean dose 385 ± 119 mg/d; n = 21 2. Routine antipsychotic: antipsychotic drugs mean dose 390 ± 120 mg/d; n = 21 |
|
| Outcomes | Clinically significant response: important change (PANSS reduction) Clinical response: mental state (PANSS total), positive symptoms, general pathology (PANSS subscales) Adverse events: various events Unable to use: Clinical response: mental state ‐ PANSS negative subscale (skewed data) Leaving the study early (no information provided) Adverse events: TESS score no means and SDs reported) |
|
| Notes | Another paper (Ma 2010) with identical data as Liang 2004 was not included in the data analysis, the 2 references were combined and letters for clarification were sent to both study authors Valproate group & monotherapy group were not significantly different in sex, age, duration of illness and PANSS score (P > 0.05) Every participant received only one antipsychotic drug. In total there were 12 chlorpromazine, 10 clozapine, 10 risperidone, 4 perphenazine, and 6 haloperidol; participants continued with the same antipsychotic drug as they were given before study entry Valproate group & monotherapy group did not differ significantly in the mean dose of antipsychotic drugs (P > 0.05) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned", p151 |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcome. |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | No details about blinding provided, assume open‐label |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcome. |
| Blinding of outcome assessment (detection bias) Subjective outcomes | High risk | No details about blinding provided, assume open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated |
| Selective reporting (reporting bias) | High risk | TESS score was measured, but not reported. Study authors only stated that there was no difference between groups on TESS score, but the actual measurements were not reported. Authors also selectively reported binary outcomes of a few adverse events, but didn't provide numbers for all adverse events |
| Other bias | Low risk | None obvious |
Liu 2003.
| Methods | Allocation: randomised Blinding: open label Duration: 8 weeks (wash‐out period: 3‐7 days) Design: parallel Country: China |
|
| Participants | Diagnosis: CCMD‐3 schizophrenia n = 60 Sex: male only Age: not stated, but likely to be adult, as their illness onset age is reported as 25 ± 5.27 years History: mean duration of illness: 14.73 ± 5.59 years Setting: inpatient |
|
| Interventions | 1. Sodium valproate + clozapine: valproate fixed dose of 600 mg/day. n = 30 2. Clozapine: dosage unclear. n = 30 |
|
| Outcomes | Leaving the study early: acceptability/tolerability of treatment Clinical response: mental state (BPRS total) Unable to use: Adverse events: no information provided |
|
| Notes | All participants took clozapine+placebo during wash‐out No information about the dosage of clozapine provided |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned in a 1:1 ratio", p11596 |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of outcome assessment (detection bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Three and four participants left early from the experiment and control groups respectively. Valproate group: 3/30 = 10%, monotherapy group: 4/30 = 13.3%, no reason provided for people leaving the study early. They are not included in the final analysis. (p 11,596) |
| Selective reporting (reporting bias) | High risk | Adverse events not reported |
| Other bias | Low risk | None obvious |
Liu 2007a.
| Methods | Allocation: randomised Blinding: double Duration: 3 weeks Design: not reported Country: China |
|
| Participants | Diagnosis: schizophrenia n = 36 Sex: 29 M, 7 F Age: mean 36.06 ± 11.72 years History: mean duration of illness 8.32 ± 8.21 years Settings: inpatient |
|
| Interventions | 1. Antipsychotic drugs + sodium valproate group: continue with previous antipsychotics; valproate 0.6‐1.2 g/d (0.2 g/capsule, 3‐6 capsules/d); n = 19 2. Antipsychotic + starch placebo monotherapy group: continue with previous antipsychotics; placebo 3‐6 capsules/d; n = 17 |
|
| Outcomes | Leaving the study early Clinical response: mental state (BPRS total, BPRS lack of energy subscale), aggression/agitation (important change MOAS, BPRS hostility subscale), depression (BPRS anxiety and depression subscale) Adverse events: various events Unable to use: aggression/agitation ‐ MOAS endpoint data (no mean and SDs given) |
|
| Notes | Valproate sodium and placebo starch were placed into identical capsules A and B by staff Standard for people leaving the study early: "still high danger of aggression after 10 days of medication and difficult to manage within the ward, stop medication and treat as no effect" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "draw lots" (p 490) |
| Allocation concealment (selection bias) | Unclear risk | Not indicated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | Sodium valproate and placebo starch were placed into identical capsules A and B by staff, clinicians and participants did not know the contents of capsules (p 490) |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Low risk | Raters did not know contents of capsules (p 490) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | People leaving the study early due to inefficacy: group A (monotherapy group) 13/17 = 76.5%, group B (valproate group) 4/19 = 21.1%, not balanced and too high in group A (p 490) |
| Selective reporting (reporting bias) | High risk | Aggression: MOAS, endpoint data unusable, no mean and SDs given |
| Other bias | Low risk | No obvious other bias |
Lu 2006.
| Methods | Allocation: randomised Blinding: open label Duration: six weeks Design: not reported Country: China |
|
| Participants | Diagnosis: CCMD‐3 schizophrenia (CCMD‐3) and schizoaffective disorder with excitement and agitation (schizophrenia: n = 78, schizoaffective disorder: n = 12) n = 90 Sex: 78 M, 12 F Age: mean 34.8 ± 13.8 years Setting: inpatient |
|
| Interventions | 1. Sodium valproate + routine antipsychotic mediation: atypical antipsychotics titrated to target dose within 7‐10 days, mean dose: clozapine 308 ± 54 mg/d, olanzapine 12.6 ± 5.9 mg/d, risperidone 4.4 ± 0.81 mg/d, quetiapine 786 ± 221 mg/d; Depakine titrated to 1‐1.5 g/d within 5 days, mean dose 1.01 ± 0.46 g/d; n = 43 2. Routine antipsychotic medication: atypical antipsychotics titrated to target dose within 7‐10 days, mean dose: clozapine 366 ± 71 mg/d, olanzapine 15.2 ± 8.8 mg/d, risperidone 4.9 ± 0.80 mg/d, quetiapine 842 ± 263 mg/d; n = 47 |
|
| Outcomes | Clinically significant response: important change Leaving the study early: acceptability/tolerability of treatment Clinical response: mental state (BPRS total), aggression/agitation (PANSS‐EC sub scale) Adverse events: various events Unable to use: Adverse events: TESS scores (no means and SDs reported) |
|
| Notes | Valproate group and monotherapy group did not differ significantly in sex, age, schizophrenia subtypes (P > 0.05) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned", p1291 |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of outcome assessment (detection bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition was unaccounted for in the final analysis Valproate group 2 (out of 43), monotherapy group 8 (out of 47), no significant difference (P > 0.05) However, 2/43 = 4.6% BUT 8/47 = 17%. Also, due to inefficacy, it is 1/43 vs 6/47 Authors stated that they would use ITT in methods section but in analysis they used completers not ITT |
| Selective reporting (reporting bias) | High risk | TESS scores measured but no means and SDs reported |
| Other bias | Low risk | None obvious |
Pan 2010.
| Methods | Allocation: randomised Blinding: open label Duration: 8 weeks Design: parallel Country: China |
|
| Participants | Diagnosis: CCMD‐3 schizophrenia n = 68 Sex: 28 M, 40 F Age: range 21‐55 years, mean 32.4 ± 6.5 years History: duration of illness: range: 5‐23 years, mean 11.2 ± 6.4 years Settings: inpatient |
|
| Interventions | 1. Clozapine + sodium valproate group: clozapine: flexible dose of 400–700 mg/d; valproate: fixed dose of 800 mg/d; n = 34 2. Clozapine monotherapy group: clozapine: flexible dose of 400–700 mg/day; n = 34 |
|
| Outcomes | Clinically significant response: important change (PANSS reduction ≥ 50%) Clinical response: mental state (PANSS total) positive symptoms, negative symptoms, general pathology (PANSS subscales) Adverse events: various events Unable to use: Leaving the study early (no information provided) Adverse events: TESS scores (no means and SDs reported) |
|
| Notes | Valproate group & monotherapy group no significant differences in demographic characteristics | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomized", no further details provided (p 203) |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of outcome assessment (detection bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated |
| Selective reporting (reporting bias) | High risk | TESS scores measured but no means and SDs reported; authors only stated that valproate group & monotherapy group TESS scores not significantly different |
| Other bias | Low risk | No obvious other bias |
Shi 2010.
| Methods | Allocation: randomised Blinding: open label Duration: 2 weeks Design: parallel Country: China |
|
| Participants | Diagnosis: CCMD‐3 schizophrenia, with excitement symptoms n = 64 Sex: total, 37 M, 27 F Age: total, range 18–60 years, mean 31.3 ± 4.4 years History: mean duration of illness 0.1–10 years Setting: inpatients |
|
| Interventions | 1. Olanzapine + magnesium valproate group: oral intake, olanzapine mean dose 10 ± 2.5 mg/d, valproate mean dose 1.25 ± 0.25 g/d; n = 33 2. Haloperidol monotherapy group: IM injection, haloperidol mean dose 15 ± 2.5 mg/d; n = 31 Co‐medication: benzhexol used for EPS but not used prophylactically |
|
| Outcomes | Clinically significant response: important change (PANSS reduction ≥ 50%) Clinical response: mental state (PANSS total), aggression/agitation (PANSS‐EC subscale) Adverse events: at least one adverse event Unable to use: Leaving the study early (no information provided) Adverse events: TESS (no means and SDs reported) |
|
| Notes | Olanzapine initial dose 5 mg/d; valproate magnesium initial dose 0.5 g/d, haloperidol initial dose 5 mg/d. Valproate magnesium extended‐release tablets used |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomized "using a random number table into two groups" (p 398) |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes No information about blinding provided, assume open‐label |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of outcome assessment (detection bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated |
| Selective reporting (reporting bias) | High risk | TESS scores measured but no means and SDs reported |
| Other bias | Low risk | No obvious other bias |
Wang 2005a.
| Methods | Allocation: randomised ‐ no further description Blinding: open label Duration: 4 weeks Design: not reported Country: China |
|
| Participants | Diagnosis: CCMN‐3 schizophrenia, with aggressive behaviours according to scores of MOAS > = 4 n = 60. Sex: 35 M, 25 F Age: total range 18‐56 years, mean 32.2 ± 7.3 years History: mean duration of illness Settings: inpatients |
|
| Interventions | 1. Antipychotics + magnesium valproate: magnesium valproate starting dose 400 mg/d, increased to 800‐1200 mg/d after 7 days; antipsychotic drugs same as before study entry; n = 30 2. Antipychotics + placebo: antipsychotic drugs same as before study; n = 30 | |
| Outcomes | Clinical response: mental state (BPRS total) negative symptoms (BPRS subscale), aggression/agitation (MOAS, BPRS subscale), depression (BPRS subscale) Adverse events: at least one adverse event, various events Unable to use: Leaving the study early: acceptability/tolerability of treatment (no information provided) Adverse events: TESS score (not reported) |
|
| Notes | No information about blinding provided, assume open‐label Valproate group & monotherapy group no significant differences in demographic data |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly divided" (p 10) |
| Allocation concealment (selection bias) | Unclear risk | Not indicated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcome. No information provided about blinding provided, assume open‐label. |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | No information provided about blinding provided, assume open‐label |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcome. |
| Blinding of outcome assessment (detection bias) Subjective outcomes | High risk | No information provided about blinding provided, assume open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated |
| Selective reporting (reporting bias) | High risk | TESS score measured but not reported |
| Other bias | Low risk | No other bias |
Wang 2009.
| Methods | Allocation: randomised Blinding: open label Duration: 8 weeks Design: parallel Country: China |
|
| Participants | Diagnosis: CCMD‐3 schizophrenia, treatment‐resistant schizophrenia n = 60 Sex: 35 M, 25 F Age: mean 30.95 ± 7.26 years History: mean duration of illness 8.08 ± 4.63 years Settings: inpatient |
|
| Interventions | 1. Aripiprazole + magnesium valproate group: aripiprazole initial dose 5 mg/d, ≤ 20 mg/day; valproate magnesium extended‐release tablets initial dose 250 mg/d, ≤ 1000 mg/d; n = 30 2. Aripiprazole monotherapy group: aripiprazole initial dose 5 mg/d, ≤ 20 mg/d; n = 30 Co‐medication: benzodiazepine could be given; no other antipyschotics or electroconvulsive therapy given |
|
| Outcomes | Clinically significant response: important change (PANSS reduction ≥ 50%) Clinical response: mental state (PANSS total), positive symptoms, negative symptoms, general pathology (PANSS subscales) Adverse events: TESS Unable to use: Leaving the study early: acceptability/tolerability of treatment (no information provided) |
|
| Notes | Valproate group & monotherapy group had no significant differences in demographic data and PANSS score (P > 0.05) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomized", no further details provided (p 250) |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes. No information about blinding provided, assume open‐label |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes. |
| Blinding of outcome assessment (detection bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated |
| Selective reporting (reporting bias) | Low risk | All outcomes reported |
| Other bias | Low risk | No obvious other bias |
Wang 2010.
| Methods | Allocation: randomised Blinding: open label Duration: 8 weeks Design: parallel Country: China |
|
| Participants | Diagnosis: CCMD‐3 schizophrenia, with impulsive and aggressive behaviours n = 60 Sex: 40 M, 20 F Age: mean 38.35 ± 10.82 years History: mean duration of illness 5.75 ± 10.8 years Settings: inpatients |
|
| Interventions | 1. Antipsychotic drugs + valproate group: antipsychotic drugs: mean 345 ± 108 mg; valproate: 600 mg/d; n = 30 2. Antipsychotic monotherapy group: antipsychotic drugs: mean 360 ± 110 mg; n = 30 |
|
| Outcomes | Clinically significant response: important change (PANSS reduction ≥ 60%) Clinical response: mental state (PANSS total), positive symptoms, negative symptoms, general pathology (PANSS subscales) Adverse events: at least one event, various events Unable to use: Leaving the study early (no information provided) Adverse events: TESS scores measured (no means and SDs reported) |
|
| Notes | Valproate group & monotherapy group not significantly different in sex, age, duration of illness & PANSS total score, P > 0.05 Every participant only received one antipsychotic drug (continued with the drug and dose used before study entry, in total 9 risperidone, 4 quetiapine, 18 chlorpromazine, 12 clozapine, 11 perphenazine, and 6 haloperidol, drug dosage converted to chlorpromazine equivalent dose Valproate group & monotherapy group antipsychotic dosage not significantly different (P > 0.05) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomized", no further details provided (p 548) |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes. No information about blinding provided, assume open‐label |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of outcome assessment (detection bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated |
| Selective reporting (reporting bias) | High risk | TESS scores measured but no means and SDs reported |
| Other bias | Low risk | No obvious other bias |
Wassef 2000.
| Methods | Allocation: randomised Blinding: double Duration: 21 days Design: parallel Country: USA |
|
| Participants | Diagnosis: chronic schizophrenia (Research Diagnostic Criteria) n = 12 Sex: 7 M, 5 F Age: mean ˜ 28,6 years History: discontinued standard oral antipsychotic drugs > 3 weeks prior to trial, discontinued depot neuroleptics > 5 weeks prior to trial, no substance abuse, no seizures, normal EEG Setting: inpatients |
|
| Interventions | 1. Haloperidol + valproate: haloperidol starting dose 10 mg/d, increased to 15 mg/d after 3 days, valproate dose adjusted to target plasma level 75‐100 µg/ml; n = 5 2. Haloperidol + placebo: haloperidol starting dose 10 mg/d, increased to 15 mg/d after 3 days; n = 7 | |
| Outcomes | Leaving the study early: acceptability/tolerability of treatment Clinical response: mental state (BPRS total) Unable to use: Clinical response: global state ‐ CGI (only P values reported); negative symptoms ‐ SANS (skewed data) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomized" (p 357) |
| Allocation concealment (selection bias) | Unclear risk | Not indicated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "double‐blind" (p 357); "identical appearing tablets"; "doses were adjusted by an unblinded investigator who was not involved in medication assignment, participant assessment or clinical care. Dose adjustments were done in a blinded fashion" (p 358) |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | "double‐blind" (p 357); "identical appearing tablets"; "doses were adjusted by an unblinded investigator who was not involved in medication assignment, participant assessment or clinical care. Dose adjustments were done in a blinded fashion" (p 358) |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | "double‐blind" (p 357); "identical appearing tablets"; "doses were adjusted by an unblinded investigator who was not involved in medication assignment, participant assessment or clinical care. Dose adjustments were done in a blinded fashion" (p 358) |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Low risk | "double‐blind" (p 357); "identical appearing tablets"; "doses were adjusted by an unblinded investigator who was not involved in medication assignment, participant assessment or clinical care. Dose adjustments were done in a blinded fashion" (p 358) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No people leaving the study early |
| Selective reporting (reporting bias) | Low risk | No selective reporting |
| Other bias | Low risk | No other bias |
Xie 2011.
| Methods | Allocation: randomised Blinding: open label Duration: six weeks Design: parallel Country: China |
|
| Participants | Diagnosis: CCMD‐3 schizophrenia n = 70 Sex: 41 M, 29 F Age: mean 30.13 ± 5.29 years History: mean duration illness (valproate group: 4.25 ± 4.05 years, monotherapy group: 4.30 ± 3.85 years) Settings: inpatient |
|
| Interventions | 1. Aripiprazole + magnesium valproate group: aripiprazole initial dose 5 mg/d, ≤ 20 mg/d; magnesium valproate extended‐release tablets initial dose 250 mg/d, ≤ 1000 mg/d; n = 35 2. Aripiprazole monotherapy group: aripiprazole initial dose 5 mg/d, ≤ 20 mg/d; n = 35 Co‐medication: benzodiazepine given when necessary, no other neuroleptics and electroconvulsive therapy used |
|
| Outcomes | Clinical response: mental state (PANSS total), positive symptoms, negative symptoms (PANSS subscales), aggression/agitation (MOAS), general pathology (PANSS subscale) Adverse events: various events Unable to use: Leaving the study early: acceptability/tolerability of treatment (no information provided) |
|
| Notes | Valproate group & monotherapy group do not differ significantly in demographic data and PANSS, MOAS scores (P > 0.05) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomized", no further details provided (p 191) |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of outcome assessment (detection bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated |
| Selective reporting (reporting bias) | Low risk | All outcomes reported |
| Other bias | Low risk | No obvious other bias |
Yao 2010.
| Methods | Allocation: randomised Blinding: open label Duration: 8 weeks Design: parallel Country: China |
|
| Participants | Diagnosis: CCMD‐3 and ICD‐10 schizophrenia n = 62 Sex: 33 M, 29 F Age: mean 26.8 ± 9.2 years Settings: inpatient |
|
| Interventions | 1. Risperidone + magnesium valproate group: risperidone initial dose 1 mg/d, titrated to target flexible dose of 4‐6 mg/d within 7‐15 days, mean 4.98 ± 1.07 mg/d; valproate 0.5 g/d; n = 31 2. Risperidone monotherapy group: risperidone initial dose 1 mg/d, titrated to flexible dose of 4‐6 mg/d; n = 31 Co‐medication: benzodiazepine drugs (alprazolam tablets) were given orally to participants with poor sleep quality; no other antipsychotics were used |
|
| Outcomes | Clinical significant response: important change (PANSS reduction > 60%) Clinical response: mental state (PANSS total) Adverse events: various events Unable to use: Leaving the study early: acceptability/tolerability of treatment (no information provided) Adverse events: TESS scores (no means and SDs reported) |
|
| Notes | Alcohol was forbidden during treatment Food intake was the same as before treatment |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly divided", no further details provided (p 2713) |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of outcome assessment (detection bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated |
| Selective reporting (reporting bias) | High risk | TESS scores measured but no means and SDs reported Authors also selectively reported binary outcomes of a few adverse events, but didn't provide numbers for all adverse events (p 2714) |
| Other bias | Low risk | No obvious other bias |
Yin 2004.
| Methods | Allocation: randomised Blinding: double (participants and researchers did not know what kind of medication given, however unclear if raters blind as rating completed by 6 experienced psychiatrists) Duration: 6 weeks Design: not reported Country: China |
|
| Participants | Diagnosis: CCMD‐3 schizophrenia, with tardive dyskinesia according to scores of AIMS great than 3 (> = 3) at least one item, or greater than 2 (> = 2) at least two items n = 80 Sex: all male Age: mean 44.5 ± 7.5 years History: mean duration of illness 22.5 ± 7.5 years Setting: inpatient |
|
| Interventions | 1. Antipsychotics + valproate: antipsychotics mean dose 324 (SD = 141) mg/d, valproate dose 0.6 g per day at first week, it increased to 1.2 g per day after 4 weeks; n = 40 2. Antipsychotics + placebo: antipsychotics mean dose 356 (SD = 120) mg/d, placebo initial dose 0.2 g x 3 times/d, after 4 weeks increased to 0.4 g x 3 times/d for 2 weeks if no severe adverse events; n = 40 | |
| Outcomes | Leaving the study early: acceptability/tolerability of treatment Adverse events: blood count changes, tardive dyskinesia (AIMS) Unable to use: Clinical response: mental state ‐ BPRS (results not reported) Adverse events: TESS score measured but no means and SDs reported |
|
| Notes | This study was purposed for the treatment of tardive dyskinesia Participants received the same type and dosage of antipsychotic drugs as before study entry All antipsychotic drug dosage converted to chlorpromazine equivalent dose Valproate sodium and placebo (starch) were put into identical oblique capsules and labelled as I and II respectively by researchers |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "divided randomly" (p 92) |
| Allocation concealment (selection bias) | Unclear risk | Not indicated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think binding causes significant bias in objective outcomes |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | Double‐blind, "Valproate sodium and placebo (starch) were put into identical oblique capsules and labelled as I and II respectively by researchers" |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think binding causes significant bias in objective outcomes |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | Outcomes measured by "6 experienced psychiatrists", no further information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Only one participant in one group left early (out of 40 participants in each group) due to low blood pressure and disturbances of consciousness |
| Selective reporting (reporting bias) | High risk | BPRS results not reported TESS score measured but no means and SDs reported |
| Other bias | Low risk | No other bias |
Zhang 2007.
| Methods | Allocation: randomised Blinding: open label Duration: 8 weeks Design: parallel Country: China |
|
| Participants | Diagnosis: CCMD‐3 schizophrenia, with psychomotor excitement symptoms n = 60 Sex: no information provided Age: range 17‐55 years, mean: 37.2 ± 12.8 years History: mean duration of illness 10.0 ± 3.5 years Settings: inpatient |
|
| Interventions | 1. Risperidone + Depakine group: risperidone: titrated to flexible dose 4.6 ± 2.4 mg/d; Depakine: titrated to flexible dose 1.6 ± 0.4 g/d, ≤ 2.0 g/d; n = 30 2. Risperidone monotherapy group: risperidone: titrated to flexible dose 4.6 ± 2.4 mg/d; n = 30 Co‐medication: according to the conditions of the participants: zolpidem or benzodiazepines (improve sleep); benzhexol hydrochloride or propranolol (EPS) |
|
| Outcomes | Clinically significant response: important change (PANSS reduction ≥ 50%) Clinical response: mental state (PANSS total), general psychopathology (PANSS subscales) Adverse events: at least one event Unable to use: Leaving the study early: acceptability/tolerability of treatment (no information provided) Clinical response: positive symptoms, negative symptoms ‐ PANSS subscale (skewed data) TESS scores measured but no means and SDs reported |
|
| Notes | Valproate (Depakine) group & monotherapy group not significantly different in age, duration of illness (P > 0.05) and PANSS total score and sub‐scores | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised" no details provided (p 265) |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | We don't think blinding causes significant bias in objective outcomes |
| Blinding of outcome assessment (detection bias) Subjective outcomes | High risk | No information about blinding provided, assume open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated |
| Selective reporting (reporting bias) | High risk | TESS scores measured but no means and SDs reported |
| Other bias | Low risk | No obvious other bias |
General abbreviations: EEG ‐ Electroencephalograph ER ‐ Extended release IM ‐ intramuscular ITT ‐ intention‐to‐treat LOCF ‐ last observation carried forward n.i. ‐ not indicated lab ‐ Laboratory
Diagnostic abbreviations: ICD‐10 ‐ International Classification of Diseases, tenth revision DSM‐III Diagnostic and Statistical Manual of Mental disorders, third edition CCMD‐3 Chinese Classification of Mental Disorders, version 3
Global effect scales: CGI ‐ Clinical Global Impression.
Mental state scales: BPRS ‐ Brief Psychiatric Rating Scale CDS ‐ Calgary Depression Scale IMPS ‐ Inpatient Multidimensional Psychiatric Scale SANS ‐ Scale for Assessment of Negative Symptoms PANSS ‐ Positive and Negative Symptom Scale pPANSS ‐ Positive and Negative Symptom Scale, positive sub‐score MOAS Modified Overt Aggression Scale
Adverse events: AIMS ‐ Abnormal Involuntary Movement Scale BAS ‐ Barnes Akathisia Scale SAS ‐ Simpson Angus ScaleTESS ‐ Treatment Emergent Symptom Scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Alberti 1999 | Allocation: randomised Participants: those with schizoaffective, bipolar and schizophrenic disorders Intervention: Valproate combined with antipsychotics versus gabapentin combined with antipsychotics. No placebo group |
| Bersudsky 2010 | Allocation: "assigned by the control psychiatrist according to random order" Participants: people with mania or schizoaffective disorder |
| Centorrino 1994 | Allocation: not randomised, matched groups |
| Haupt 2007 | Allocation: randomised (open label) Participants: people with schizophrenia Interventions: valproate combined with antipsychotic medications versus antipsychotic monotherapy Outcomes: metabolic processes, no usable data |
| Ma 2010 | As noted above, this study reported identical data as Liang 2004 and we sent mails to the editor and author of both studies to rule out that it was just a copy, but have not received a reply. We therefore decided that it would be more careful to exclude this study at this stage |
| Monfort 1991 | Allocation: possibly randomised (double‐blind) Participants: people with schizophrenia or schizophreniform disorder Interventions: valproate combined with diazepam versus chlorpromazine, no adequate intervention group |
| NCT00183443 | Allocation: randomised Participants: people with mania |
| Ping 1994 | Allocation: quasi‐randomisation, randomised according to hospitalisation order |
| Raja 2000 | Allocation: not randomised, prospective naturalistic study |
| Rifang 2001 | Allocation: quasi‐randomisation, randomised according to hospitalisation order |
| Zhu 2005 | Allocation: randomised, no further details Participants: people with schizophrenia Interventions: antipsychotics combined with valproate versus antipsychotics combined with clonazepam |
.
Characteristics of studies awaiting assessment [ordered by study ID]
Boylan 2004.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Unclear |
| Outcomes | Unclear |
| Notes | Needs detailed assessment, probably only a statistical paper |
Chien 1978.
| Methods | Unclear |
| Participants | People with tardive dyskinesia |
| Interventions | Unclear |
| Outcomes | Unclear |
| Notes | Needs detailed assessment |
Cui 2012.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate |
| Outcomes | Weight, insulin levels, details unclear |
| Notes | Needs translation from Chinese |
Ellenor 1995.
| Methods | Unclear |
| Participants | People with bipolar disorder or depression |
| Interventions | Divalproex |
| Outcomes | Unclear |
| Notes | Needs detailed assessment, possibly only about serum levels |
Friis 1983.
| Methods | Allocation: people were treated in three consecutive phases with sodium valproate, biperiden and placebo in randomized orde Blindness: double‐blinded Duration: 2‐week wash‐out period followed by three 4‐week treatment phases |
| Participants | Diagnosis: people with akathisia, unclear whether they had schizophrenia |
| Interventions | Valproate or biperiden or placebo |
| Outcomes | Akathisia, parkinsonism and hyperkinesia Adverse events |
| Notes | Needs detailed assessment |
Gong 2010.
| Methods | Unclear |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes |
Gu 2012.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate, unclear |
| Outcomes | Cognitive function, unclear |
| Notes | Needs translation from Chinese |
Gu 2014.
| Methods | Unclear |
| Participants | Stable people with schizophrenia |
| Interventions | Valproate combined with antipsychotics, control unclear |
| Outcomes | "Prognosis", details unclear |
| Notes | Needs translation from Chinese |
Huang 2013.
| Methods | Unclear |
| Participants | Women with treatment‐resistant schizophrenia |
| Interventions | Small dose of valproate combined with ziprasidone, control unclear |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Jiang 2014.
| Methods | Unclear |
| Participants | Women with schizophrenia |
| Interventions | Valproate combined with quetiapine, control unclear |
| Outcomes | Aggressive and impulsive behaviour |
| Notes | Needs translation from Chinese |
Jin 2013.
| Methods | Unclear |
| Participants | People with schizophrenia and aggression |
| Interventions | Valproate, unclear |
| Outcomes | Aggresssive behaviour, unclear |
| Notes | Needs translation from Chinese |
Lee 1996.
| Methods | Allocation: randomised ‐ divided into 3 groups randomly Blindness: not mentioned, assume open‐label |
| Participants | Diagnosis: people with chronic schizophrenia, residual type according to DSM‐III‐R and had predominant negative symptoms evaluated by PANSS |
| Interventions | Haloperidol + carbamazepine or haloperidol + valproate or haloperidol only |
| Outcomes | Mental states: BPRS, PANSS, CGI Depression: HRDS Adverse events: SAS |
| Notes |
Li 2008a.
| Methods | Unclear. |
| Participants | People with schizophrenia, acutely ill. |
| Interventions | Valproate, unclear. |
| Outcomes | Unclear. |
| Notes | Needs translation from Chinese. |
Li 2012a.
| Methods | Unclear |
| Participants | People with treatment resistant schizophrenia |
| Interventions | Valproate combined with ziprasidone |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Li 2013.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate, rest unclear |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Li 2013a.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate, details unclear |
| Outcomes | Cognitive function, details unclear |
| Notes | Needs translation from Chinese |
Li 2014.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate combined with low dose quetiapine, control unclear |
| Outcomes | Aggressive behaviour |
| Notes | Needs translation from Chinese |
Li 2014a.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Low dose valproate combined with antipsychotics, control unclear |
| Outcomes | Aggressive behaviour |
| Notes | Needs translation from Chinese |
Liang 2004a.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate, other unclear |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Linnoila 1976a.
| Methods | Allocation: not clear Blindness: double‐blind |
| Participants | People with schizophrenia and various forms of psychosis |
| Interventions | Valproate or placebo + neuroleptics |
| Outcomes | Mental states: BPRS Adverse events |
| Notes | Needs detailed assessment |
Linnoila 1976b.
| Methods | Allocation: not clear |
| Participants | People with tardive dyskinesia |
| Interventions | Valproate, other unclear |
| Outcomes | Unclear |
| Notes | Needs detailed assessment, could be part of Linnoila 1976 |
Liu 2007b.
| Methods | Unclear |
| Participants | Psychiatric patients |
| Interventions | Valproate, other unclear |
| Outcomes | Aggression, other unclear |
| Notes | Needs translation from Chinese |
Liu 2012.
| Methods | Unclear |
| Participants | People with schizophrenia and excitement |
| Interventions | Valproate, other unclear |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Liu 2012a.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate, details unclear |
| Outcomes | Agitated behaviour, rest unclear |
| Notes | Needs translation from Chinese |
Liu 2013.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate + risperidone, other unclear |
| Outcomes | Lipids, other unclear |
| Notes | Needs translation from Chinese |
Liu 2014.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate combined with risperidone, details unclear |
| Outcomes | Serum levels, efficacy, details unclear |
| Notes | Needs translation from Chinese |
Lv 2014.
| Methods | Unclear |
| Participants | People with a first episode of schizophrenia |
| Interventions | Valproate, details unclear |
| Outcomes | Cognitive function, details unclear |
| Notes | Needs translation from Chinese |
Ma 2012.
| Methods | Unclear |
| Participants | People with treatment‐resistant schizophrenia |
| Interventions | Valproate combined with ziprasidone, control unclear |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Monteleone 1986.
| Methods | Allocation: probably randomised |
| Participants | People with chronic schizophrenia |
| Interventions | Valproate, rest unclear |
| Outcomes | Prolactin, other unclear |
| Notes | Needs detailed assessment |
Monteleone 1988.
| Methods | Allocation: probably randomised |
| Participants | People with schizophrenia |
| Interventions | Valproate, other unclear |
| Outcomes | Unclear |
| Notes | Needs detailed assessment |
Nair 1980.
| Methods | Unclear |
| Participants | People with tardive dyskinesia |
| Interventions | Valproate, other unclear |
| Outcomes | Unclear |
| Notes | Needs more detailed assessment |
Nasrallah 1986.
| Methods | Unclear |
| Participants | Unclear |
| Interventions | Valproate, unclear |
| Outcomes | Unclear |
| Notes | Needs detailed assessment |
NCT00552500.
| Methods | Unclear |
| Participants | Unclear |
| Interventions | Valproate, other unclear |
| Outcomes | Lipid metabolism, other unclear |
| Notes | Need to contact author |
Peng 2012.
| Methods | Unclear |
| Participants | People with treatment resistant schizophrenia |
| Interventions | Valproate combined with clozapine |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Peng 2013.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate, other unclear |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Pu 2013.
| Methods | Unclear |
| Participants | People with treatment‐resistant schizophrenia |
| Interventions | Valproate combined with olanzapine, control unclear |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Qiu 2014.
| Methods | Unclear |
| Participants | Unclear |
| Interventions | Valproate combined with risperidone, control unclear |
| Outcomes | Efficacy, serum levels, details unclear |
| Notes | Needs translation from Chinese |
Qu 2014.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate combined with olanzapine |
| Outcomes | Aggressive behaviour, details unclear |
| Notes | Needs translation from Chinese |
Ruiz‐Doblado 2010.
| Methods | Unclear |
| Participants | People with schizophrenia resistant to clozapine |
| Interventions | Valproate |
| Outcomes | Unclear |
| Notes | Needs detailed assessment, possibly only review |
Shu 2014.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate combined with clozapine, control unclear |
| Outcomes | Aggressive behaviour |
| Notes | Needs translation from Chinese |
Su 2013.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Chlorpromazine, clozapine, valproate, details are unclear |
| Outcomes | Unclear |
| Notes | Abstract only, more details are necessary. Chinese |
Sun 2007.
| Methods | Allocation: stratified randomisation according to hospitalisation order Blindness: open label Duration: 12 weeks |
| Participants | Diagnosis: schizophrenia (CCMD‐3), used at least two different types of typical antipsychotics, each treated for at least six weeks at the dosage of at least 500mg chlorpromazine equivalents per day, but no significant response; PANSS ≥ 60 History: duration of illness 7.4 ± 4.7 years n = 86 Sex: male and female Age: 26.7 ± 3.6 years |
| Interventions | 1. Magnesium valproate + risperidol: valproate 250‐750 mg/d, mean dose 425 ± 195 mg/d; risperidol 2‐6 mg/d, mean dose 3.25 ± 1.85 mg/d; n = 42 2. Risperidol alone: 2‐6 mg/day, mean dose 3.25 ± 1.85 mg/day, n = 44 |
| Outcomes | Neurocognitive tests: WAIS‐R, WMS, WCST Mental states: PANSS Adverse events Unable to use: Adverse events: TESS ‐ no data reported |
| Notes | We suspect randomisation of this study was not properly done as it is not clear how they did stratified randomisation according to hospitalisation order; we were unable to contact the study author to verify |
Sun 2008.
| Methods | Unclear |
| Participants | Difficult‐to‐treat people with schizophrenia |
| Interventions | Valproate, details unclear |
| Outcomes | Cognitive function, other unclear |
| Notes | Needs translation from Chinese |
Sun 2012.
| Methods | Unclear |
| Participants | People with treatment resistant schizophrenia |
| Interventions | Valproate combined with quetiapine or clozapine, details unclear |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Suzana 2009.
| Methods | Unclear, possibly not randomised |
| Participants | People with schizophrenia |
| Interventions | Mood‐stabilisers, details unclear |
| Outcomes | Hostility, impulsivity, details unclear |
| Notes | Needs detailed assessment and full report from authors |
Tang 2012.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate, details unclear |
| Outcomes | Aggressive behaviour, details unclear |
| Notes | Needs translation from Chinese |
Wang 2005b.
| Methods | Unclear |
| Participants | People with schizophrenia and aggression |
| Interventions | Valproate, antipsychotic drugs |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Wang 2006.
| Methods | Allocation: randomised ‐ no further details Blindness: no further details Duration: 28 days |
| Participants | Diagnosis: schizophrenia (CCMD‐3), with aggressive behaviours according to scores of MOAS > = 4, or BPRS score > 38 History: no information n = 64 Sex: male and female Age: 31.15 years |
| Interventions | 1. Magnesium valproate + risperidol: valproate 800‐1200 mg/d, risperidol 4‐6 mg/d, n = 32 2. Risperidol alone: 4‐6 mg/d, n = 32 |
| Outcomes | Mental state: BPRS scores Behaviour: MOAS scores Adverse events Unable to use Adverse events: TESS ‐ no data reported |
| Notes | Data of this study apears to be very closely related to Wang 2005a, which was conducted by the same author one year earlier. Both studies measured exactly the same outcomes (Wang 2005a, n = 60; Wang 2006, n = 64), had the same inclusion criteria, with the same dosage of drugs. The numbers in the continuous outcomes are very similar. We suspect that Wang 2006 could be a follow up study of 2005, but we were unable to contact the author to verify. |
Wang 2012.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Wang 2013.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate, olanzapine, details unclear |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Wang 2013a.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate combined with risperidone, control unclear |
| Outcomes | Aggressive and impulsive behaviour |
| Notes | Needs translation from Chinese |
Wang 2014.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate combined with risperidone, control unclear |
| Outcomes | Aggressive behaviour |
| Notes | Needs translation from Chinese |
Wang 2014a.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate, control unclear |
| Outcomes | Behaviour disorder, details unclear |
| Notes | Needs translation from Chinese |
Wen 2013.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate, other unclear |
| Outcomes | Aggressive behaviour, details unclear |
| Notes | Needs translation from Chinese |
Wu 2013a.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate, quetiapine, details unclear |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Wu 2013b.
| Methods | Unclear |
| Participants | People with schizophrenia and positive symptoms |
| Interventions | Valproate, olanzapine, details unclear |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Xiao 2013.
| Methods | Unclear, possibly retrospective analysis |
| Participants | People with schizophrenia |
| Interventions | Valproate, risperidone, details unclear |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Xu 2002.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate, control unclear |
| Outcomes | Auditory hallucinations |
| Notes | Needs translation from Chinese |
Xu 2013.
| Methods | Unclear |
| Participants | Women with treatment‐resistant schizophrenia |
| Interventions | Valproate combined with ziprasidone, control unclear |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Xu 2014.
| Methods | Unclear |
| Participants | Women with treatment‐resistant schizophrenia |
| Interventions | Valproate combined with clozapin |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Zhang 2007a.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate, risperidone, details unclear |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Zhang 2007b.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Zhang 2008.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate combined with risperidone, control unclear |
| Outcomes | Negative symptoms |
| Notes | Needs translation from Chinese |
Zhang 2012.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate combined with haloperidol, details unclear |
| Outcomes | Unclear |
| Notes | Needs translation from Chinese |
Zhou 2012.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate combined with ziprasidone, details unclear |
| Outcomes | Aggressive behaviour, rest unclear |
| Notes | Needs translation from Chinese |
Zhou 2013.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate combined with risperidone, details unclear |
| Outcomes | Agitated behaviour, details unclear |
| Notes | Needs translation from Chinese |
Zhou 2014.
| Methods | Unclear |
| Participants | Men with schizophrenia |
| Interventions | Valproate combined with olanzapine, details unclear |
| Outcomes | Agitated behaviour, rest unclear |
| Notes | Needs translation from Chinese |
Zhu 2005a.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Valproate combined with clonazapam |
| Outcomes | Agitation, details unclear |
| Notes | Needs translation from Chinese |
Zhu 2014.
| Methods | Unclear |
| Participants | People with schizophrenia |
| Interventions | Low‐dose valproate combined with antipsychotics |
| Outcomes | Aggressive behaviour |
| Notes | Needs translation from Chinese |
Characteristics of ongoing studies [ordered by study ID]
NCT00167934.
| Trial name or title | Determining metabolic effects of valproate and antipsychotic therapy |
| Methods | Allocation: randomised (single‐blind) |
| Participants | People with schizophrenia |
| Interventions | Valproate combined with antipsychotic medications versus antipsychotic monotherapy |
| Outcomes | Blood glucose, metabolism disorders and weight gain |
| Starting date | December 2004 |
| Contact information | Martha J Hessler, BS 314‐362‐2423 hesslema@psychiatry.wustl.edu |
| Notes | This study is recruiting participants |
NCT00306475.
| Trial name or title | Does the addition of divalproex sodium ER to an atypical antipsychotic drug (APD) improve cognition and psychopathology in outpatients with schizophrenia (SCH) or schizoaffective disorder (SAD)? |
| Methods | Allocation: randomised Blindness: double‐blinded Duration: six weeks |
| Participants | DSM‐IV diagnosis of schizophrenia or schizoaffective disorder Sex: Male or female Age: 18‐65 |
| Interventions | 1. Divalproex sodium + atypical antipsychotics 2. Atypical antipsychotics + placebo |
| Outcomes | Cognition Psychopathology (positive, negative, and mood symptoms) |
| Starting date | March 2006 |
| Contact information | Kara L Watts, M.A. 615‐343‐9717 kara.l.watts@vanderbilt.edu Claudia Diaz‐Byrd, M.S. 615‐322‐2916 claudia.c.diaz‐byrd@vanderbilt.edu |
| Notes |
Norrie 2000.
| Trial name or title | Norrie 2000 |
| Methods | |
| Participants | Participants with schizophrenia treated as inpatients or outpatients n = 50 |
| Interventions | 1. Olanzapine (10‐20 mg/d) + valproate (500‐2000 mg/d) 2.Olanzapine (10‐20 mg/d) + placebo |
| Outcomes | PANSS, REHAB and Life Skills Profile Scales, adverse events, laboratory parameters |
| Starting date | Unclear |
| Contact information | Norrie PD Glenside Hospital, Adelaide, Australia |
| Notes |
Wang 2003.
| Trial name or title | 60 week, randomised, double‐blind, placebo‐controlled trial of valproate added to risperidone in 200 treatment‐naive, first‐episode people with schizophrenia |
| Methods | Allocation: not stated Blindness: double‐blinded Duration: 52 weeks |
| Participants | Those with schizophrenia, n = 200 |
| Interventions | 1. Valproate + antipsychotics 2. Placebo + antipsychotics |
| Outcomes | Global state: CGI‐ HAM‐D Mental state: PANSS Cognitive battery |
| Starting date | 2003 |
| Contact information | Gang Wang MD
Capital University of Medical Sciences Beijing Anding Hospital No.5 An Kang Hutong De Shengmen Wai Street Beijing Xicheng District 100088 China wangg@intra.nimh.nih.gov |
| Notes |
Differences between protocol and review
Compared to original versions, we have adapted the methods to changes made by the Cochrane (e.g. 'Risk of bias' table and 'Summary of findings' table added) and by the Cochrane Schizophrenia Group. This review contains some rewording and reorganisation of the original outcomes noted in the protocol (the original methods can be found in the appendix). The type of outcomes, however, remain the same.
Contributions of authors
Yijun Wang: contacted Chinese authors and extracted data from articles in Chinese, data analysis, data interpretation and writing of the update.
Jun Xia: extracted data from articles in Chinese, data analysis and interpretation.
Bartosz Helfer: searching, data checking, data interpretation, and revision of the report.
Chunbo Li: contacted Chinese authors and extracted data from articles in Chinese.
Stefan Leucht: protocol development, searching, data checking, data interpretation, and revision of the report.
Sources of support
Internal sources
Freistaat Bayern, Germany.
External sources
German Ministry of Health, Network of Competence Schizophrenia, Germany.
Declarations of interest
Yijun Wang: none known.
Jun Xia: none known.
Bartosz Helfer: none known.
Chunbo Li: none known.
Stefan Leucht has received honoraria for lectures from Otsuka, Lundbeck, Janssen, ICON, Lilly, SanofiAventis, AOP Orphan and Servier; for consulting from Janssen, Roche, Lilly, Otsuka, Lundbeck, TEVA and for a publications from Roche.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Casey 2003 {published data only}
- Casey DE, Daniel DG, Wassef AA, Tracy KA, Wolnlak P, Sommerville W. Effect of divalproex combined with olanzapine or risperidone in participants with acute exacerbation of schizophrenia. Neuropsychopharmacology 2003;28:182‐92. [DOI] [PubMed] [Google Scholar]
- Citrome L, Casey DE, Daniel DG, Wozniak P, Kochan LD, Tracy KA. Adjunctive divalproex and hostility among participants with schizophrenia receiving olanzapine or risperidone. Psychiatric Services 2004;55(3):290‐4. [MEDLINE: ; Embase 2004111171] [DOI] [PubMed] [Google Scholar]
- Jafari M, Jiang P, Casey DE. Adjunctive divalproex sodium lowers cholesterol elevation with olanzapine. 157th Annual Meeting of the American Psychiatric Association; 2004 May 1‐6; New York, New York, USA. 2004. [APA Conference Abstracts NR634]
Casey 2009 {published data only}
- Casey DE, Daniel DG, Tamminga C, Kane JM, Tran‐Johnson T, Wozniak P, et al. Divalproex ER combined with olanzapine or risperidone for treatment of acute exacerbations of schizophrenia. Neuropsychopharmacology 2009;34(5):1330‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- M02‐547. A randomized, double‐blind study of the safety and efficacy of Depakote ER plus an atypical antipsychotic vs. an atypical antipsychotic alone in the treatment of schizophrenia. www.abbvie.com/content/dam/abbviecorp/us/desktop/research/clinical‐trials‐data‐and‐information‐sharing/synopses/divalproex%20sodium_M02‐547.pdf [accessed 08 April 2016].
- NCT00073164. A study of the safety and efficacy of Depakote ER plus an atypical antipsychotic vs an atypical antipsychotic alone in the treatment of schizophrenia. clinicaltrials.gov/ct2/show/NCT00073164 [Date Accessed: 17 November 2003].
Citrome 2007 {published data only}
- Citrome L. Schizophrenia and valproate. Psychopharmacology Bulletin. United States of America, 2003; Vol. 37, issue Suppl 2:74‐88. [PubMed]
- Citrome L, Daniel D, Wassef A, Tracy K, Wozniak P, Casey D. Antipsychotic monotherapy versus combination treatment with valproate in hospitalized patients with acute schizophrenia: a double‐blind, multi‐center study. Proceedings of the 155th Annual Meeting of the American Psychiatric Association; 2002 May 18‐23; Philadelphia, Pennsylvania, USA 2002:NR316.
- Citrome L, Shope CB, Nolan KA, Czobor P, Volavka J. Risperidone alone versus risperidone plus valproate in the treatment of participants with schizophrenia and hostility. International Clinical Psychopharmacology 2007;22(6):356‐62. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- NCT00308360. Risperidone alone vs risperidone plus valproate in the treatment of participants with schizophrenia and hostility. clinicaltrials.gov/ct2/show/NCT00308360 [Date Accessed: 28 March 2006].
- Nolan K, Shope C, Citrome L, Volavka J. Staff and patient views of the reasons for aggressive incidents: a prospective, incident‐based study. Psychiatric Quarterly. United States, 2009; Vol. 80, issue 3:167‐72. [DOI] [PubMed]
Dose 1998 {published data only}
- Dose M. Die Bedeutung von Antikonvulsiva und Calciumantagonisten für die Pharmakotherapie von Psychosen. Habilitationsschrift. Technische Universität München, 1991. [Google Scholar]
- Dose M, Hellweg R, Yassouridis A, Theison M, Emrich HM. Combined treatment of schizophrenic psychoses with haloperidol and valproate. Pharmacopsychiatry 1998;31(4):122‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fisk 1987 {published data only}
- Fisk GG, York SM. The effect of sodium valproate on tardive dyskinesia ‐ revisited. British Journal of Psychiatry 1987;150:542‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Glick 2009 {published data only}
- Glick ID, Bosch J, Casey DE. A double‐blind randomized trial of mood stabilizer augmentation using lamotrigine and valproate for participants with schizophrenia who are stabilized and partially responsive. Journal of Clinical Psychopharmacology 2009;29(3):267‐71. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Guo 2007 {published data only}
- Guo CR. Study of quetiapine combined with magnesium valproate release tablets in treatment of schizophrenia with symptoms of elation and agitation [奎硫平合并丙戊酸镁缓释片治疗精神分裂症兴奋激越研究]. Linchuang Jingshen Yixue Zazhi [临床精神医学杂志] 2007;17(3):183‐4. [Google Scholar]
Hesslinger 1999 {published data only}
- Hesslinger B, Normann C, Langosch JM, Klose P, Berger M, Walden J. Effects of carbamazepine and valproate on haloperidol plasma levels and on psychopathologic outcome in schizophrenic participants. Journal of Clinical Psychopharmacology 1999;19(4):310‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Normann C, Klose P, Hesslinger B, Langosch J, Berger M, Walden J. Haloperidol plasma levels and psychopathology in schizophrenic patients with antiepileptic co‐medication: a clinical trial. PharmacoPsychiatry 1997; Vol. 30:204.
- Walden J, Hesslinger B, Normann C, Langosch J, Berger M. Actions of carbamazepine and valproate on haloperidol plasma levels and psychopathological outcome. Proceedings of the 21st Collegium Internationale Neuro‐Psychopharmacologicum Congress; 1998 Jul 12‐16; Glasgow, UK 1998.
Jia 2007 {published data only}
- Jia HX, Bian ZY, Zhu H, Jiang T, Wang LF, Liu HB, et al. Effect of sodium valproate on serum concentration and clinical efficacy of clozapine [丙戊酸钠对氯氮平血药浓度和疗效的影响]. Linchuang Jingshen Yixue Zazhi [临床精神医学杂志] 2007;17(5):310‐1. [Google Scholar]
Jiang 2009 {published data only}
- Jiang CY, Chen JX, He GQ. The effect of adjuvant magnesium valproate sustained ‐ release tablets for schizophrenia with agitation and agitation. [丙戊酸镁缓释片辅助治疗精神分裂症兴奋激越的效果观察]. Chinese Magazine of Drug Abuse Prevention and Treatment [中国药物滥用防治杂志] 2009;15(3):175‐7. [Google Scholar]
Li 2008 {published data only}
- Li WS, Yu XD, Cui Y. A comparative study of olanzapine combined with low‐dose magnesium valproate sustained‐release tablets in the treatment of acute phrase of schizophrenia [奥氮平合并小剂量丙戊酸镁缓释片治疗急性期精神分裂症的对照研究]. Journal of Psychiatry [精神医学杂志] 2008;21(3):178‐9. [Google Scholar]
Li 2009 {published data only}
- Li SS. The effect of aripiprazole combined with magnesium valproate in the treatment of impulse control disorder of schizophrenia [阿立哌唑联合丙戊酸镁对精神分裂症冲动控制障碍的疗效观察]. Chongqing Medical Journal [重庆医学杂志] 2009;38(7):788, 798. [Google Scholar]
Liang 2004 {published data only}
- Liang HX, Du JY, Wu ZJ. A study of additive effect of sodium‐valproate in the treatment of schizophrenia. Journal of Clinical Psychological Medicine 2004;14(3):151‐2. [Google Scholar]
- Ma XW. The effect of adjuvant magnesium valproate on schizophrenia [丙戊酸镁对精神分裂症的辅助治疗作用]. China Practical Medicine [中国实用医药] 2010;5(12):43﹣4. [Google Scholar]
Liu 2003 {published data only}
- Liu C. The effect of sodium valproate in participants with schizophrenia. Chinese Journal of Clinical Medicinal Professional Research 2003;70:11, 596‐7. [Google Scholar]
Liu 2007a {published data only}
- Liu Z. Sodium valproate in treatment of aggression for psychiatric participants [丙戊酸钠治疗精神病患者的攻击行为]. Chongqing Journal of Medicine [重庆医学杂志] 2007;36(6):490‐1. [Google Scholar]
Lu 2006 {published data only}
- Lu W, Jin W, Wang H, Ma Y, Tong Z. Comparing study of sodium valproate xr (Depakine) in the supplementary therapy of excitement state in participants with schizophrenia and schizoaffective disorder. Proceedings of the Zhejiang Annual Psychiatry Conference, Yiwu, China. 2005:75‐7.
- Lu WM, Jin WD, Shen Y, Ma YC, Tong ZH. Clinical evaluation of sodium valproate xr (Depakine) in adjunctive therapy for participants with excitomania associated with schizophrenia and schizoaffective disorder. Chinese New Drugs Journal 2006;15(15):1291‐3. [Google Scholar]
Pan 2010 {published data only}
- Pan ZX, Yin DF, Liu NH. Sodium valproate combined clozapine in the treatment of refractory schizophrenia 34 cases [丙戊酸钠联合氯氮平治疗难治性精神分裂症34例]. Herald of Medicine [医学导报] 2010;29(2):203‐4. [Google Scholar]
Shi 2010 {published data only}
- Shi YX, Dai TG, Yi PC. A study of olanzapine combined with magnesium valproate sustained‐release tablets for the treatment of excitement and agitation in acute phase of schizophrenia [奥氮平合并丙戊酸镁缓释片治疗精神分裂症急性期兴奋激越研究]. Modern Practical Medicine [现代实用医学] 2010;22(4):398‐9. [Google Scholar]
Wang 2005a {published data only}
- Wang H. Combination of magnesium valproate and anti‐psychotics in aggressive behaviors of schizophrenics. Journal of Clinical Psychosomatic Diseases 2005;11(1):10‐1. [113903] [Google Scholar]
Wang 2009 {published data only}
- Wang GP, Xie R, Pei GX, Zhang XJ. The effect of adjuvant magnesium valproate sustained‐release tablets in the treatment of schizophrenia [丙戊酸镁缓释片治疗精神分裂症的辅助作用]. Journal of Clinical Psychiatry [临床精神医学杂志] 2009;19(4):250‐1. [Google Scholar]
Wang 2010 {published data only}
- Wang LJ, Zhang J. A comparative study of sodium valproate combined with other drugs for schizophrenia with impulsive and aggressive behavior [合用丙戊酸钠治疗伴有冲动和攻击行为的精神分裂症的对照研究]. Medical Journal of Chinese People's Health [中国民康医学] 2010;22(5):548‐9. [Google Scholar]
Wassef 2000 {published data only}
- Wassef AA, Dott SG, Harris A, Brown A, O'Boyle M, Meyer WJ 3rd, et al. Randomized, placebo‐controlled pilot study of divalproex sodium in the treatment of acute exacerbations of chronic schizophrenia. Journal of Clinical Psychopharmacology 2000;20(3):357‐61. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Xie 2011 {published data only}
- Xie R, Wang G, Pei G. The effect of adjuvant magnesium valproate sustained‐release tablet in the treatment of schizophrenic participants with aggressive behaviours [丙戊酸镁缓释片辅助治疗精神分裂症攻击行为的疗效观察]. Journal of Clinical Psychiatry [临床精神医学杂志] 2011;21(3):191‐2. [Google Scholar]
Yao 2010 {published data only}
- Yao F, Jiang F. Short‐term efficacy of risperidone combined with magnesium valproate sustained‐release tablets in the treatment of schizophrenia with aggressive behavior [利培酮联合丙戊酸镁缓释片治疗精神分裂症攻击行为的近期疗效]. Occupation and Health [职业与健康] 2010;26(22):2713‐4. [Google Scholar]
Yin 2004 {published data only}
- Yin XR, Xie BQ, Jiang L. A double‐blind comparative study of sodium valproate in treating of TD. Journal of Clinical Psychological Medicine 2004;14(2):92‐3. [113903] [Google Scholar]
Zhang 2007 {published data only}
- Zhang SL, Wang CX, Wang LT, Feng YF. The effect of debakin combined with risperidone in the treatment of schizophrenia with psychmotor excitement [德巴金联合利培酮治疗伴精神运动性兴奋的精神分裂症的效果]. Medical Journal of Qilu [齐鲁医学杂志] 2007;22(3):265‐6. [Google Scholar]
References to studies excluded from this review
Alberti 1999 {published data only}
- Alberti GG, Garini R, Marzolini M, Beltz J, Greco MI, Maresca G. Efficacy and tolerability of gabapentin (GBP) and valproate (VPA) as an adjunct in the neuroleptic treatment of acute manic syndromes. Journal of the European College of Neuropsychopharmacology 1999;9:210. [Google Scholar]
Bersudsky 2010 {published data only}
- Bersudsky Y, Applebaum J, Gaiduk Y, Sharony L, Mishory A, Podberezsky A, et al. Valnoctamide as a valproate substitute with low teratogenic potential in mania: a double‐blind, controlled, add‐on clinical trial. Bipolar Disorders 2010;12(4):376‐82. [DOI] [PubMed] [Google Scholar]
Centorrino 1994 {published data only}
- Centorrino F, Baldessarini RJ, Kando J, Frankenburg FR, Volpicelli SA, Puopolo PR, et al. Serum concentrations of clozapine and its major metabolites: effects of co‐treatment with fluoxetine or valproate. American Journal of Psychiatry 1994;151(1):123‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Haupt 2007 {published data only}
- Haupt DW, Fahnestock PA, Hessler MJ, Flavin K, Bagley S, Yingling M, et al. Changes in direct measures of adiposity in schizophrenia participants during divalproex augmentation of antipsychotic treatment. Schizophrenia Bulletin 2007;33(2):501. [Google Scholar]
Ma 2010 {published data only}
- Ma XW. The effect of adjuvant magnesium valproate in the treatment of schizophrenia [丙戊酸镁对精神分裂症的辅助治疗作用]. China Practical Medicine [中国实用医药] April 2010;5(12):43‐4. [Google Scholar]
Monfort 1991 {published data only}
- Monfort JC, Mager C, Thorpe D, Ropert R, Ferrey G, Mentre F, et al. Valproate‐diazepam combination versus chlorpromazine: a double‐blind study in schizophrenic and schizophreniform disorders [Valproate‐diazépam versus chlorpromazine: étude en double aveugle dans les troubles schizophréniques et schizophréniformes]. Encephale 1991;17(2):101. [MEDLINE: ] [Google Scholar]
NCT00183443 {published data only}
- NCT00183443. Divalproex extended release and placebo, lithium, or quetiapine for mania. clinicaltrials.gov/ct2/show/NCT00183443 [Date Accessed: 13 September 2005].
Ping 1994 {published data only}
- Ping Y, Xinjuan OY, Lin T. A controlled study of sodium valproate and chlorpromazine for schizophrenia. Shanghai Archives of Psychiatry 1994;6(4):208‐9. [Google Scholar]
Raja 2000 {published data only}
- Raja M, Azzoni A. Second generation antipsychotics in the emergency care setting. A prospective naturalistic study. General Hospital Psychiatry 2000;22(2):107‐14. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Rifang 2001 {published data only}
- Rifang C, Fenfang Z. The impact of sodium valproate for social function rehabilitation in schizophrenia participants. Modern Rehabilitation 2001;5(9):100. [Google Scholar]
Zhu 2005 {published data only}
- Zhu C, Fu R, Yang Y. Sodium valproate and clonazepam for the treatment of agitated symptoms of participants with schizophrenia: a controlled trial. Medical Journal of the Chinese People's Armed Police Forces 2005;16(12):934‐5. [CAJ MEDI0602] [Google Scholar]
References to studies awaiting assessment
Boylan 2004 {published data only}
Chien 1978 {published data only}
- Chien C, Jung K, Ross‐Townsend A. Efficacies of agents related to GABA, dopamine, and acetylcholine in the treatment of tardive dyskinesia. Psychopharmacology Bulletin 1978; Vol. 14, issue 2:20‐2. [PubMed]
Cui 2012 {published data only}
- Cui LJ, Shen WM. Effects of magnesium valproate treatment on weight and blood insulin levels in patients with schizophrenia [丙戊酸镁对精神分裂症患者体重及胰岛素水平的影响]. Frontiers of Medicine [医药前沿] 2012; Vol. 22:97.
Ellenor 1995 {published data only}
- Ellenor G, Lohr J, Ito M, Dishman B, Gammariello A. The comparative efficacy of divalproex low and high serum level in bipolar disorder and schizophrenia. Psychopharmacology Bulletin 1995; Vol. 31, issue 3:563.
Friis 1983 {published data only}
- Friis T, Christensen TR, Gerlach J. Sodium valproate and biperiden in neuroleptic‐induced akathisia, parkinsonism and hyperkinesia. A double‐blind cross‐over study with placebo. Acta Psychiatrica Scandinavica 1983;67(3):178‐87. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Gong 2010 {published data only}
- Gong Y, Guo P. Effects of magnesium valproate on cognitive function in patients with refractory schizophrenia [丙戊酸镁对难治性精神分裂症患者认知功能的影响]. Chronic Pathematology Journal [慢性病学杂志] 2010; Vol. 12, issue 2:107‐9.
Gu 2012 {published data only}
- Gu P, Wang J, Cao J, Li B‐z. A comparative study of quetiapine with sustained‐release tablets of magnesium valproate in treatment of old patients with acute phase of schizophrenia [奎硫平合并丙戊酸镁治疗老年精神分裂症急性期的对照研究]. Chinese Journal of Clinical Psychology [中国临床心理学杂志] 2012; Vol. 20, issue 4:520‐2.
Gu 2014 {published data only}
- Gu Q. Prognostic analysis of magnesium valproate combined with antipsychotics for patients with convalescent schizophrenia [丙戊酸镁联合抗精神病药物治疗对恢复期精神分裂症患者的预后分析]. Medical Journal of Chinese People's Health [中国民康医学] 2014; Vol. 26, issue 18:59, 99.
Huang 2013 {published data only}
- Huang WD, Luo HF, Lan L, Li CH, Xu LH, Li ZM. Efficacy of ziprasidone combined with low‐dose sodium valproate in the treatment of female refractory schizophrenia [齐拉西酮联合小剂量丙戊酸钠治疗女性难治性精神分裂症疗效分析]. Youjiang Medical Journal [右江医学] 2013, issue 02:159‐62.
Jiang 2014 {published data only}
- Jiang TC, Xiong Y. Efficacy of quetiapine combined with sodium valproate in the treatment of schizophrenia with impulsive and aggression in female patients [喹硫平联合丙戊酸钠治疗女性精神分裂症患者冲动攻击行为的疗效观察]. World Latest Medicine Information [世界最新医学信息文摘(连续型电子期刊)] 2014; Vol. 14, issue 32:79‐80.
Jin 2013 {published data only}
- Jin G. Efficacy of magnesium valproate for schizophrenia with impulsive and aggressive behavior [丙戊酸镁对精神分裂症冲动攻击行为疗效观察]. Chinese Journal of Modern Drug Application [中国现代药物应用] 2013, issue 14:160‐1.
Lee 1996 {published data only}
- Lee MS, Choi BH, Kim SH. Combined use of carbamazepine and valproic acid in negative symptom schizophrenia. 9th Congress of the European College of Neuropsychopharmacology; 1996 Sep 21‐25; Amsterdam, The Netherlands. 1996. [MEDLINE: ]
Li 2008a {published data only}
- Li W, Yu X, Cui Y. A comparative study of olanzapine with small‐dose sustained‐release tablets of magnesium valproate in treatment of acute phase of schizophrenia. Shandong Archives of Psychiatry 2008;21(3):178‐9. [Google Scholar]
Li 2012a {published data only}
- Li DM. Clinical efficacy of ziprasidone combined with valproate in the treatment of refractory schizophrenia [齐拉西酮联合丙戊酸钠治疗难治性精神分裂症的临床疗效观察]. China Medical Engineering [中国医学工程] 2012, issue 01:92‐3.
Li 2013 {published data only}
- Li C, Liu Y, Wang M. The effect of adjuvant magnesium valproate sustained‐release tablets combined with risperidone in the treatment of schizophrenia with impulsive aggression [丙戊酸镁缓释片合用利培酮对精神分裂症冲动攻击行为的辅助治疗]. Journal of Practical Medical Techniques [实用医技杂志] 2013, issue 01:69‐71.
Li 2013a {published data only}
- Li J, Chen LS, Chen Wang Y. A comparative study of effect of sodium valproate on cognitive dysfunction in patients with schizophrenia [丙戊酸钠改善精神分裂症患者认知功能障碍的对照研究]. Journal of Psychiatry [精神医学杂志] 2013, issue 04:292‐4.
Li 2014 {published data only}
- Li YM, Sun WB, Yang CQ. A study of low‐dose quetiapine combined with magnesium valproate in the treatment of schizophrenia [低剂量喹硫平合并丙戊酸镁治疗精神分裂症攻击行为疗效研究]. Medical Information [医学信息] 2014; Vol. 27, issue 24:47‐8.
Li 2014a {published data only}
- Li Y. A comparative study of antipsychotic drugs combined with low‐dose sodium valproate in the treatment of schizophrenia [抗精神病药物联合小剂量丙戊酸镁治疗精神分裂症攻击行为的对照研究]. The Chinese and Foreign Health Abstract [中外健康文摘] 2014, issue 11:53.
Liang 2004a {published data only}
- Liang H‐X, Du J‐Y, Wu Z‐J. A study of additive effect of sodium‐valproate in the treatment of schizophrenia. Journal of Clinical Psychological Medicine 2004;14(2):92‐3. [Google Scholar]
Linnoila 1976a {published data only}
- Linnoila M, Viukari M, Kietala O. Effect of sodium valproate on tardive dyskinesia. British Journal of Psychiatry 1976;129:114‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Linnoila 1976b {published data only}
- Linnoila M, Viukari M, Hietala O. Sodium valproate therapy for tardive dyskinesia. International Drug Therapy Newsletter 1976;11(Nov):33‐5. [MEDLINE: ] [Google Scholar]
Liu 2007b {published data only}
- Liu Z. Sodium valproate in treatment of aggression of psychiatric participants [丙戊酸钠治疗精神病患者的攻击行为]. Chongqing Medical Journal [重庆医学] 2007;36(6):490‐1. [Google Scholar]
Liu 2012 {published data only}
- Liu Z, Wang L, Sun Y. Clinical observation of magnesium valproate sustained‐release tablets for schizophrenia with excitement [丙戊酸镁缓释片对精神分裂症兴奋状态的疗效观察]. China Journal of Health Psychology [中国健康心理学杂志] 2012; Vol. 20, issue 11:1630‐1.
Liu 2012a {published data only}
- Liu J. The effect of adjuvant magnesium valproate on schizophrenia with aggressive behavior [丙戊酸镁对精神分裂症兴奋激越行为的辅助疗效观察]. Journal of Community Medicine [社区医学杂志] 2012; Vol. 10, issue 14:31‐2.
Liu 2013 {published data only}
- Liu Z, Song S, Wang L. Effects of magnesium valproate sustained‐release tablets combined with risperdal on lipid metabolism in patients with schizophrenia [丙戍酸镁缓释片合并维思通对精神分裂症患者脂代谢影响]. China Journal of Health Psychology [中国健康心理学杂志] 2013; Vol. 21, issue 11:1631‐2.
Liu 2014 {published data only}
- Liu Z, Li HJ, Pei SY. Effects of risperidone and magnesium valproate on blood concentration in patients with schizophrenia [丙戍酸镁合并利培酮治疗精神分裂症血药浓度变化及临床疗效]. China Journal of Health Psychology [中国健康心理学杂志] 2014; Vol. 22, issue 8:1141‐3.
Lv 2014 {published data only}
- Lv S. Effects of magnesium valproate sustained‐release tablets on cognitive function in first‐episode schizophrenic patients [丙戊酸镁缓释片对首发精神分裂症病人认知功能的影响]. China & Foreign Medical Treatment [中外医疗] 2014, issue 17:120‐1.
Ma 2012 {published data only}
- Ma TB. Analysis of effects of valproic acid sodium jointed with ziprasidone in the treatment of refractory schizophrenia [丙戊酸钠联合齐拉西酮治疗难治性精神分裂症的效果分析]. China Modern Medicine [中国当代医药] 2012, issue 04:73‐4.
Monteleone 1986 {published data only}
Monteleone 1988 {published data only}
Nair 1980 {published data only}
- Nair N, Lal S, Schwartz G. Effect of sodium valproate and baclofen in tardive dyskinesia: clinical and neuroendocrine studies. In: Cattabeni FRG editor(s). Long Term Effects of Neuroleptics. Advances in Biochemistry and Psychopharmacology. Vol. 24, New York, USA: Raven Press, 1980:437‐41. [PubMed] [Google Scholar]
Nasrallah 1986 {published data only}
- Nasrallah H, Dunner F, McCalley Whitters M, Smith R. Pharmacologic probes of neurotransmitter systems in tardive dyskinesia: implications for clinical management. Journal of Clinical Psychological Medicine. United‐States, 1986; Vol. 47, issue 2:56‐9. [PubMed]
NCT00552500 {published data only}
- NCT00552500. Effects of atypical antipsychotic and valproate combination therapy on glucose and lipid metabolism in schizophrenia. clinicaltrials.gov/ct2/show/NCT00552500 [Date Accessed: 1 November 2007].
Peng 2012 {published data only}
- Peng ZY, Xia CL, Liu XM. A comparative study of clozapine and clozapine unite magnesium valproate in the treatment of patients with treatment‐resistant schizophrenia [氯氮平合并丙戊酸镁治疗难治性精神分裂症的对照研究]. World Health Digest Medical Periodieal [中外健康文摘] 2012; Vol. 38:164‐6.
Peng 2013 {published data only}
- Peng H. Effect of magnesium valproate on aggressive behavior in schizophrenia [丙戊酸镁对精神分裂症攻击行为的疗效观察]. Chinese Journal of Trauma and Disability Medicine [中国伤残医学] 2013, issue 07:187‐8.
Pu 2013 {published data only}
- Pu QX, Wu HB, Huang X, Yu GH, Wang DF, Gu ZW, et al. Contrast analysis of olanzapine combined with sodium valproate on refractory schizophrenia [奥氮平合并丙戊酸钠治疗难治性精神分裂症的对照分析]. Progress in Modern Biomedicine [现代生物医学进展] 2013, issue 18:3503‐6.
Qiu 2014 {published data only}
- Qiu LL, Wang JF. Effect of sodium valproate on risperidone therapy and blood concentration [丙戊酸镁对利培酮疗效及血药浓度的影响]. Medical Journal of Liaoning [辽宁医学杂志] 2014; Vol. 28, issue 1:9‐10.
Qu 2014 {published data only}
- Qu CH, Liu XL, Sun XH. A comparative study of valproate combined with olanzapine in the treatment of schizophrenia [丙戊酸钠合用奥氮平治疗精神分裂症患者攻击行为的对照研究]. Medical Information [医学信息] 2014; Vol. 27, issue 21:602.
Ruiz‐Doblado 2010 {published data only}
- Ruiz‐Doblado S, Baena‐Baldomero A, Esparrago‐Llorca G. Pharmacological augmentation strategies in clozapine‐resistant schizophrenia: overcoming the resistance. Psiquiatria Biologica 2010;17(3):96‐101. [Google Scholar]
Shu 2014 {published data only}
- Shu Z, Zhou W. Clinical observation of clozapine combined with magnesium valproate sustained‐release tablets in the treatment of schizophrenia [氯氮平联合丙戊酸镁缓释片治疗精神分裂症攻击行为的疗效观察]. Guide of China Medicine [中国医药指南] 2014; Vol. 12, issue 2:28‐9.
Su 2013 {published data only}
- Su L, Xu Y, Shi S, Dutt A, Bramon E. Effects of chlorpromazine, clozapine and valproate on auditory P300 in schizophrenia: a prospective longitudinal randomized controlled study. Schizophrenia Bulletin 2013; Vol. 39:S51.
Sun 2007 {published data only}
- Sun Q, Quo H, Zhang H, Liu Y, Zhu Y, Zhang A. The effects of magnesium valproate sustained release tablets on the cognitive function in treatment‐resistant schizophrenic participants. Proceedings of the Shanghai Regional Meeting and CSP Annual Congress; 2007 Sep 20‐23; Shanghai. 2007.
Sun 2008 {published data only}
- Sun Q‐X, Guo H, Liu Y. Effect of magnesium valproate sustained release tablets on cognition function in difficult cure sex schizophrenic. Chinese Journal of Modern Drug Application [中国现代药物应用] 2008;2(22):25‐7. [Google Scholar]
Sun 2012 {published data only}
- Sun XG. A comparative study of quetiapine combined with haloperidol, sodium valproate and clozapine in the treatment of refractory schizophrenia [奎硫平合并氟哌啶醇、丙戊酸钠与氯氮平治疗难治性分裂症对照研究]. Medical Journal of Chinese People's Health [中国民康医学] 2012; Vol. 24, issue 13:1566‐7.
Suzana 2009 {published data only}
- Suzana S, Olivera Z, Gordana N, Violeta S, Dragoslava G, Vladica S. Adjunctive mood stabilising treatment: the effects on hostility and impulsivity among patients with schizophrenia. European Neuropsychopharmacology 2009; Vol. 19:S532.
Tang 2012 {published data only}
- Tang SY, Qin CL, Zhang W, Xiong MQ. Clinical observation of sodium valproate sustained‐release tablets on schizophrenia hostile attack [丙戊酸钠治疗精神分裂症敌意攻击行为疗效观察]. J Medical Forum [医药论坛杂志] 2012; Vol. 9:36‐7.
Wang 2005b {published data only}
- Wang H. Combination of magnesium valproate and anti‐psychotics in aggressive behaviors of schizophrenics. Journal of Clinical Psychosomatic Diseases 2005;11(1):10‐1. [Google Scholar]
Wang 2006 {published data only}
- Wang HL. Therapeutic effect of risperidone combined with magnesium valproate in aggressive behaviors of schizophrenics. Chinese Journal of Health Psychology 2006;14(4):441‐2. [Google Scholar]
Wang 2012 {published data only}
- Wang G, Xie R, Pei G. A comparative study of quetiapine combined with magnesium valproate sustained‐release tablets in the treatment of schizophrenia with aggressive behavior [喹硫平合并丙戊酸镁缓释片治疗精神分裂症攻击行为的对照研究]. Journal of Psychiatry [精神医学杂志] 2012, issue 01:59‐60.
Wang 2013 {published data only}
- Wang Y. Efficacy of olanzapine combined with magnesium valproate in the treatment of schizophrenia [奥氮平合用丙戊酸镁缓释片治疗精神分裂症疗效分析]. Chinese Journal of General Practice [中华全科医学] 2013, issue 05:730+749.
Wang 2013a {published data only}
- Wang ML, Jiang TC. Clinical observation of risperidone combined with sodium valproate in the treatment of schizophrenia with impulse and aggressive behavior [利培酮联合丙戊酸钠治疗伴有冲动和攻击行为的精神分裂症患者的临床观察]. Clinical Rational Drug Use [临床合理用药杂志] 2013, issue 01:72‐3.
Wang 2014 {published data only}
- Wang GL, Fu PX, Li XY, Lin JX, Ji QS, Li ZJ. A comparative study of the efficacy of risperidone combined with valproate sodium on the aggressive behavior of the patients with schizophrenia [利培酮合并丙戊酸钠对精神分裂症患者攻击行为疗效的对照研究]. Chinese Journal of Drug Abuse Prevention and Treatment [中国药物滥用防治杂志] 2014; Vol. 20, issue 4:200‐2.
Wang 2014a {published data only}
- Wang ZB. The effect of adjuvant sodium valproate for schizophrenia with behavioral disorders [丙戊酸钠对精神分裂症行为紊乱的辅助治疗作用]. Medical Journal of Chinese People's Health [中国民康医学] 2014; Vol. 26, issue 22:57‐8.
Wen 2013 {published data only}
- Wen J, Li Y, Wang G. Clinical efficacy and safety of quetiapine combined with magnesium valproate in the treatment of schizophrenia with aggressive behavior [喹硫平合并丙戊酸镁缓释片治疗具有攻击行为精神分裂症的临床疗效及安全性探讨]. Medical Journal of Chinese Civil Administration [中国民康医学] 2013, issue 11:55‐6.
Wu 2013a {published data only}
- Wu J, Lin T. Efficacy of magnesium valproate combined with quetiapine in the treatment of refractory schizophrenia [丙戊酸镁联合喹硫平治疗难治性精神分裂症的疗效研究]. Practical Journal of Cardiac Cerebral Pneumal and Vascular Disease [实用心脑肺血管病杂志] 2013, issue 04:140‐1.
Wu 2013b {published data only}
- Wu X, Yang Q, Zhang L, Zhou S. A comparative study of olanzapine combined with magnesium valproate sustained‐release tablets in the treatment of patients with positive symptoms of schizophrenia [奥氮平联合丙戊酸镁缓释片治疗精神分裂症伴阳性症状患者的对照研究]. Medical Journal of West China [西部医学] 2013, issue 12:1842‐4.
Xiao 2013 {published data only}
- Xiao H. A retrospective study of risperidone combined with magnesium valproate in the treatment of schizophrenia [利培酮合并丙戊酸镁治疗精神分裂症用药现状回顾分析]. Nei Mongol Journal of Traditional Chinese Medicine [内蒙古中医药] 2013, issue 06:30‐1.
Xu 2002 {published data only}
- Xu HL, Wang Y. The effect of adjuvant sodium valproate on auditory hallucinations in schizophrenia [丙戊酸钠对精神分裂症幻听症状的辅助疗效]. Journal of Clinical Psychological Medicine [临床精神医学杂志] 2002; Vol. 12, issue 3:157‐8.
Xu 2013 {published data only}
- Xu LH, Huang WD, Li CH, Ban RY, Luo HF, Li ZM. Ziprasidone combined with low‐dose sodium valproate in the treatment and nursing of female refractory schizophrenia [齐拉西酮联合小剂量丙戊酸钠治疗女性难治性精神分裂症的观察及护理]. Journal of Aerospace Medicine [航空航天医学杂志] 2013, issue 09:1133‐5.
Xu 2014 {published data only}
- Xu BF, Lou YM, Cai XH. Effects of magnesium valproate sustained‐release tablets combined with clozapine in the treatment of female refractory schizophrenia [丙戊酸镁缓释片联合氯氮平治疗女性难治性精神分裂症的疗效观察]. Chinese Journal of Trauma and Disability Medicine [中国伤残医学] 2014; Vol. 22, issue 6:148‐9.
Zhang 2007a {published data only}
- Zhang S‐L, Wang C‐X, Wang L‐T. Therapeutic efficacy of combination of risperidone with Depakine for schizophrenia. Medical Journal of Qilu 2007;22(3):265‐6. [Google Scholar]
Zhang 2007b {published data only}
- Zhang DW, Dai J, Xie WF, Chen WT. Sodium valproate combined with EEG as an adjuvant therapy for schizophrenia [丙戊酸钠对伴脑电图异常的精神分裂症的辅助治疗作用]. Medical Journal of Chinese People's Health [中国民康医学] 2007; Vol. 19, issue 12:1049, 1051.
Zhang 2008 {published data only}
- Zhang X, Wang XM. Magnesium valproate combined with risperidone in the treatment of schizophrenia with negative symptoms [丙戊酸镁辅助利培酮治疗精神分裂症阴性症状的疗效观察]. Chinese Journal of Misdiagnostics [中国误诊学杂志] 2008; Vol. 18, issue 18:4361‐2.
Zhang 2012 {published data only}
- Zhang X, Ye CP, Liu WP. Efficacy of sodium valproate combined with haloperidol in the treatment of early schizophrenia [丙戊酸钠联合氟哌啶醇治疗早期精神分裂症的疗效观察]. Chinese and Foreign Medical Research [中外医学研究] 2012; Vol. 23:38‐9.
Zhou 2012 {published data only}
- Zhou YH, Zhong YF, Zeng DZ, Dai LJ. Efficacy of magnesium valproate sustained‐release tablets combined with ziprasidone in the treatment of schizophrenia with aggressive behavior [丙戊酸镁缓释片联合齐拉西酮治疗精神分裂症攻击行为的疗效观察]. The 10th National conference of Psychiatry of Chinese Medical Association 2012:1.
Zhou 2013 {published data only}
- Zhou LC, Nong YX, Chen QM. Effect and safety comparison of risperidone single therapy or combined valproate on excited restless of schizophrenia [利培酮单药或与丙戊酸钠联用对精神分裂症兴奋躁动的作用及安全性比较]. Journal of Clinical Psychological Medicine [临床精神医学杂志] 2013, issue 04:261‐2.
Zhou 2014 {published data only}
- Zhou W, Xie WJ, Hu YZ. Clinical observation of olanzapine combined sustained‐release magnesium valproate tablet treating male schizophrenic patients with aggressive behavior [奥氮平合并丙戊酸镁缓释片治疗伴攻击行为的男性精神分裂症临床观察]. Journal of Clinical Psychiatry [临床精神医学杂志] 2014; Vol. 24, issue 3:194‐6.
Zhu 2005a {published data only}
- Zhu CH, Fu R, Yang Y. A comparative study of sodium valproate combined with clonazepam for schizophrenic agitation [丙戊酸钠与氯硝西泮控制精神分裂症激越症状的对照研究]. Medical Journal of the Chinese People's Armed Police Forces [武警医学] 2005;16(12):934‐5. [Google Scholar]
Zhu 2014 {published data only}
- Zhu YT, Zeng DF. A comparative study of antipsychotic drugs combined with low‐dose sodium valproate in the treatment of schizophrenia [抗精神病药物联合小剂量丙戊酸镁治疗精神分裂症攻击行为的对照研究]. China Practical Medicine [中国实用医药] 2014; Vol. 9, issue 13:140‐1.
References to ongoing studies
NCT00167934 {published data only}
- NCT00167934. Metabolic effects of valproate and antipsychotic therapy. clinicaltrials.gov/ct2/show/NCT00167934 [Date Accessed: 9 September 2005].
NCT00306475 {published data only}
- NCT00306475. Does the addition of divalproex sodium ER to an atypical antipsychotic drug (APD) improve cognition and psychopathology in outpatients with schizophrenia (sch) or schizoaffective disorder (sad)?. clinicaltrials.gov/ct2/show/NCT00306475 [Date Accessed: 22 March 2006].
Norrie 2000 {published data only}
- Norrie PD. Valproate augmentation of antipsychotic therapy. International Journal of Neuropsychopharmacology 2000;3 (Suppl 1):S173. [Google Scholar]
Wang 2003 {published data only}
- Wang G. 60 week, randomized, double‐blind, placebo‐controlled trial of valproate added to risperidone in 200 treatment‐naive, first‐episode participants with schizophrenia. Stanley Foundation Research Programs 2003.
Additional references
Adams 2014
- Adams CE, Awad GA, Rathbone J, Thornley B, Soares‐Weiser K. Chlorpromazine versus placebo for schizophrenia. Cochrane Database of Systematic Reviews 2014, Issue 1. [DOI: 10.1002/14651858.CD000284.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313:1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Andreasen 1989
- Andreasen NC. Scale for the assessment of negative symptoms. British Journal of Psychiatry 1989;155:53‐8. [PubMed] [Google Scholar]
Baethge 2003
- Baethge C. Long‐term treatment of schizoaffective disorder: review and recommendations. Pharmacopsychiatry 2003;36(2):45‐56. [DOI] [PubMed] [Google Scholar]
Bagnall 2000
- Bagnall AM, Lewis RA, Leitner ML. Ziprasidone for schizophrenia and severe mental illness. Cochrane Database of Systematic Reviews 2000, Issue 4. [DOI: 10.1002/14651858.CD001945] [DOI] [PubMed] [Google Scholar]
Bailer 2012
- Bailer M. Chemical properties of antiepileptic drugs (AEDs). Advanced Drug Delivery Reviews 2012;64(10):887‐95. [DOI] [PubMed] [Google Scholar]
Balen 1999
- Balen RM, Procyshyn RM. Valproic acid for seizure prophylaxis during clozapine therapy: where's the evidence?. International Journal of Psychiatry in Clinical Practice 1999;3(4):249‐51. [DOI] [PubMed] [Google Scholar]
Begg 1996
- Begg C, Cho M, Eastwood S, Horton R, Moher D, Olkin I, et al. Improving the quality of reporting of randomized controlled trials: the CONSORT statement. JAMA 1996;276:637‐9. [DOI] [PubMed] [Google Scholar]
Besag 2006
- Besag FMC, Berry D . Interactions between antiepileptic and antipsychotic drugs. Drug Safety 2006;29(2):95‐118. [DOI] [PubMed] [Google Scholar]
Bland 1997
- Bland JM, Kerry SM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use. Therapie 1999;54(4):405‐11. [PubMed] [Google Scholar]
Carpenter 1994
- Carpenter WT, Buchanan RW. Schizophrenia. New England Journal of Medicine 1994;330:681‐90. [DOI] [PubMed] [Google Scholar]
Carta 2003
- Carta, MG, Hardoy MC, Hardoy MJ, Grunze H, Carpiniello B. The clinical use of gabapentin in bipolar spectrum disorders. Journal of Affective Disorders June 2003;75(1):83‐91. [DOI] [PubMed] [Google Scholar]
Cheine 2003
- Cheine M, Ahonen J, Wahlbeck K. Beta‐blocker supplementation of standard drug treatment for schizophrenia (Cochrane Review). Cochrane Database of Systematic Reviews 2003, Issue 1. [DOI: 10.1002/14651858.CD000234] [DOI] [Google Scholar]
Christison 1991
- Christison GW, Kirch DG, Wyatt RJ. When symptoms persist: choosing among alternative somatic treatments for schizophrenia. Schizophrenia Bulletin 1991;17:217‐45. [DOI] [PubMed] [Google Scholar]
Citrome 2000
- Citrome L, Levine L, Allingham B. Changes in use of valproate and other mood stabilizers for participants with schizophrenia from 1994 to 1998. Psychiatric Services 2000;51(5):634‐8. [DOI] [PubMed] [Google Scholar]
Crowhurst 2002
- Crowhurst N. Philosophy, care and treatment on the psychiatric intensive care unit: themes, trends and future practice. Journal of Psychiatric and Mental Health Nursing 2002;9(6):689‐95. [DOI] [PubMed] [Google Scholar]
Davey Smith 1998
- Davey Smith G, Egger M. Meta‐analysis: unresolved issues and future developments. BMJ 1998;16:221‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Davis 2000
- Davis LL, Ryan W, Adinoff B, Petty F. Comprehensive review of the psychiatric uses of valproate. Journal of Clinical Psychopharmacology 2000;20(1):1‐17. [DOI] [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Abstracts of 8th International Cochrane Colloquium; 2000 Oct 25‐28th; Cape Town, South Africa. 2000.
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG (editors). Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Divine 1992
- Divine GW, Brown JT, Frazer LM. The unit of analysis error in studies about physicians' participant care behavior. Journal of General Internal Medicine 1992;7:623‐9. [DOI] [PubMed] [Google Scholar]
Dold 2012
- Dold M, Li C, Tardy M, Khorsand V, Gillies D, Leucht S. Benzodiazepines for schizophrenia. Cochrane Database of Systematic Reviews 2012, Issue 11. [DOI: 10.1002/14651858.CD006391.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Essali 2010
- Essali A, Al‐Haj Haasan N, Li C, Rathbone J. Clozapine versus typical neuroleptic medication for schizophrenia. Cochrane Database of Systematic Reviews 2010, Issue 1. [DOI: 10.1002/14651858.CD000059.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Freeman 1998
- Freeman MP, Stoll AL. Mood stabilizer combinations: a review of safety and efficacy. The American Journal of Psychiatry 1998;155(1):12‐21. [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(7):7‐10. [DOI] [PubMed] [Google Scholar]
Gilbody 2000
- Gilbody SM, Bagnall AM, Duggan L, Tuunainen A. Risperidone versus other atypical antipsychotic medication for schizophrenia. Cochrane Database of Systematic Reviews 2000, Issue 3. [DOI: 10.1002/14651858.CD002306] [DOI] [PubMed] [Google Scholar]
Goodman 2006
- Goodman F, Glassman, P, Maglione M, Suttorp M. Drug Class Review on Antiepileptic Drugs in Bipolar Mood Disorder, Neuropathic Pain, and Fibromyalgia. Oregon Health & Science University, 2006. [Google Scholar]
Gulliford 1999
- Gulliford MC, Ukoumunne OC, Chinn S. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy U. ECDEU assessment manual for psychopharmacology. Early Clinical Drug Evaluation (ECDEU) Assessment Manual for Psychopharmacology. Washington DC: National Institute of Mental Health, 1976. [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JPT, Deeks JJ (editors). Chapter 7: Selecting studies and collecting data. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011c
- Higgins JPT, Deeks JJ, Altman DG (editors). Chapter 16: Special topics in statistics. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Hiller 1986
- Hiller W, Zerssen D, Mombour W, Wittchen HU. Inpatient Multidimensional Psychiatric Scale. Eine multidimensionale Skala zur systematischen Erfassung des psychopathologischen Befundes (Dt. Version). Manual. Weinheim: Beltz Test GmbH, 1986. [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and negative syndrome scale (PANSS) manual. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Konstantinos 2012
- Konstantinos N, Fountoulakis, Siegfried K, Ole A, Pierre B, Ahmed O, et al. Efficacy of pharmacotherapy in bipolar disorder: a report by the WPA section on pharmacopsychiatry. European Archives of Psychiatry and Clinical Neuroscience 2012;262(1):1‐48. [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of Brief Psychiatric Rating Scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2005b
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Leucht 2007a
- Leucht S, Engel RR, Bauml J, Davis JM. Is the superior efficacy of new generation antipsychotics an artifact of LOCF?. Schizophrenia Bulletin 2007;33(1):183‐91. [PUBMED: 16905632] [DOI] [PMC free article] [PubMed] [Google Scholar]
Leucht 2007b
- Leucht S, Kissling W, McGrath J. Lithium for schizophrenia. Cochrane Database of Systematic Reviews 2007, Issue 3. [DOI: 10.1002/14651858.CD003834.pub2] [DOI] [PubMed] [Google Scholar]
Leucht 2007c
- Leucht S, McGrath J, White P, Kissling W. Carbamazepine for schizophrenia. Cochrane Database of Systematic Reviews 2007, Issue 3. [DOI: 10.1002/14651858.CD001258.pub2] [DOI] [PubMed] [Google Scholar]
Mallinckrodt 2004
- Mallinckrodt CH, Watkin JG, Molenberghs G, Carroll RJ. Choice of the primary analysis in longitudinal clinical trials. Pharmaceutical Statistics 2004;3:161‐9. [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Marvaha 2004
- Marvaha S, Johnson S. Schizophrenia and employment? A review. Social Psychiatry and Psychiatric Epidemiology 2004;39:337‐49. [DOI] [PubMed] [Google Scholar]
Moher 2001
- Moher D, Schulz KF, Altman D, CONSORT Group (Consolidated Standards of Reporting Trials). The CONSORT statement: revised recommendations for improving the quality of reports of parallel‐group randomized trials. JAMA 2001;258:1987‐91. [DOI] [PubMed] [Google Scholar]
NIMH 1970
- National Institute of Mental Health. Clinical Global Impression. In: Guy W, Bonato RR editor(s). Manual for the ECDEU Assessment. 2nd Edition. NIMH, 1970. [Google Scholar]
NIMH 1991
- Guy W. ECDEU Assessment Manual for Psychopharmacology. ECDEU Assessment Manual for Psychopharmacology, Revised. National Institute of Mental Health, 1991. [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The brief psychiatric rating scale. Psychological Reports 1962;10:790‐812. [Google Scholar]
Pajonk 2005
- Pajonk FG. Clinical trial design in schizophrenia: implications for clinical decisions. Current Opinion in Psychiatry 2005;18(6):692‐99. [DOI] [PubMed] [Google Scholar]
Palmer 2005
- Palmer BA, Pankratz VS, Bostwick JM. The lifetime risk of suicide in schizophrenia: a reexamination. Archives of General Psychiatry 2005;62(3):247‐53. [DOI] [PubMed] [Google Scholar]
Pantelis 1996
- Pantelis C, Barnes TRE. Drug strategies and treatment‐resistant schizophrenia. Australian and New Zealand Journal of Psychiatry 1996;20(1):20‐37. [DOI] [PubMed] [Google Scholar]
Perugi 2004
- Perugi G, Ruffolo G. The empirical basis of the use of antiepileptics in the treatment of bipolar disorder. Clinical Neuropsychiatry 2004;1(3):165‐74. [Google Scholar]
Schooler 1993
- Schooler NR, Keith SJ. Clinical research for the treatment of schizophrenia. Psychopharmacology Bulletin 1993;29:431‐46. [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias: dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273:408‐12. [DOI] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Simpson 1970
- Simpson M, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatrica Scandinavia 1970;212:11‐9. [DOI] [PubMed] [Google Scholar]
Sommer 2012
- Sommer IE, Begemann MJH, Temmerman A, Leucht S. Pharmacological augmentation strategies for schizophrenia patients With insufficient response to clozapine: a quantitative literature review. Schizophrenia Bulletin 2012;38(5):1003‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Srisurapanont 2004
- Srisurapanont M, Disayavanish C, Taimkaew K. Quetiapine for schizophrenia. Cochrane Database of Systematic Reviews 2004, Issue 2. [DOI: 10.1002/14651858.CD000967.pub2] [DOI] [PubMed] [Google Scholar]
Stahl 2004
- Stahl, SM, Grady, MM. A critical review of atypical antipsychotic utilization: comparing monotherapy with polypharmacy and augmentation. Current Medicinal Chemistry 2004;11(3):313‐27. [DOI] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JAC, Egger M, Moher D (editors). Chapter 10: Addressing reporting biases. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Intervention. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Tharyan 2005
- Tharyan P, Adams CE. Electroconvulsive therapy for schizophrenia. Cochrane Database of Systematic Reviews 2005, Issue 2. [DOI: 10.1002/14651858.CD000076.pub2] [DOI] [PubMed] [Google Scholar]
Tranulis 2006
- Tranulis C, Mouaffak F, Chouchana L, Stip E, Gourevitch R, Poirier MF, et al. Somatic augmentation strategies in clozapine resistance ‐ what facts?. Clinical Neuropharmacology 2006;29(1):34‐44. [DOI] [PubMed] [Google Scholar]
Tsuang 1978
- Tsuang MT. Suicide in schizophrenics, manics, depressives, and surgical controls: a comparison with general population suicide mortality. Archives of General Psychiatry 1978;35:153‐55. [DOI] [PubMed] [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organisation‐based interventions in health and health care: a systematic review. Health Technology Assessment 1999;3(5):3‐92. [MEDLINE: ] [PubMed] [Google Scholar]
Wahlbeck 2001
- Wahlbeck K, Tuunainen A, Ahokas A, Leucht S. Drop‐out rates in randomised antipsychotic drug trials. Psychopharmacology 2001;155:230‐3. [DOI] [PubMed] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]
References to other published versions of this review
Leucht 2004
- Leucht S, White P, McGrath J, Kissling W. Lithium for schizophrenia revisited: a systematic review and meta‐analysis of randomized controlled trials. Journal of Clinical Psychiatry 2004;65:177‐86. [PubMed] [Google Scholar]
Schwarz 2008
- Schwarz C, Volz A, Li C, Leucht S. Valproate for schizophrenia. Cochrane Database of Systematic Reviews 2008, Issue 3. [DOI: 10.1002/14651858.CD004028.pub3] [DOI] [PubMed] [Google Scholar]