

Abstract
Objective:
Immune complex (IC) deposition activates neutrophils (PMN), increases vascular permeability and leads to organ damage in SLE and RA. The bioactive lipid sphingosine-1-phosphate (S1P), acting via S1P receptor 1 (S1P1), is a key regulator of endothelial cell (EC) barrier function. We hypothesized that augmenting EC integrity via S1P1 signaling would attenuate inflammatory injury mediated by ICs.
Methods:
In vitro barrier function was assessed in human umbilical vein endothelial cells (HUVECs) by Electric Cell-substrate Impedance Sensing (ECIS). Phosphorylation of myosin light chain2 (p-MLC2) and VE-Cadherin staining in HUVECs was assessed by immunofluorescence. Reverse Arthus reaction (RAR) in skin and lung was performed in mice with S1P1 deleted from ECs (ECKO) and mice treated with S1P1 agonists and antagonists.
Results:
S1P1 agonists prevented loss of barrier function in HUVEC treated with IC-activated PMN. S1P1 ECKO and WT mice treated with S1P1 antagonists had amplified RAR, whereas specific S1P1 agonists attenuated skin and lung RAR in WT mice. ApoM-Fc, a novel S1P chaperone, mitigated EC cell barrier dysfunction induced by activated PMN in vitro and attenuated lung RAR. S1P1 agonists and ApoM-Fc markedly reduced p-MLC2 and disruption VE-Cadherin, manifestations of cell contraction and destabilization of adherence junctions, respectively, induced by activated PMN.
Conclusion:
S1P1 signaling in ECs modulates vascular responses to IC deposition. S1P1 agonists and ApoM-Fc enhance the EC barrier, limit leukocyte escape from capillaries, and provide protection from inflammatory injury. The S1P/S1P1 axis is a new target to attenuate tissue responses to IC deposition and mitigate end organ damage.
INTRODUCTION
Systemic lupus erythematosus and rheumatoid arthritis, though complex and heterogeneous, share the basic pathophysiologic mechanisms of immune complex (IC) deposition in tissues and neutrophil activation that cause end organ damage. Circulating ICs induce neutrophil activation by both Fc and complement receptors which trigger release of pro-inflammatory chemokines and cytokines that lead to endothelial cell (EC) barrier dysfunction and increase vascular permeability [1] [2]. Loss of EC barrier integrity has been implicated in inflammatory injury in mouse models of rheumatoid arthritis (RA) [3] and systemic lupus erythematosus (SLE) [4]. When the EC integrity is compromised, plasma proteins extravasate and neutrophils transmigrate via paracellular (inter-endothelial) routes orchestrated by activated adhesion molecules [5]. Paracellular transmigration is mediated in part by phosphorylation of VE-cadherin and transient separation of adherens junctions which creates intercellular gaps [6, 7]. Enhancing barrier integrity with pharmacological or genetic approaches has been shown to impede both vascular leak and neutrophil transmigration in animals treated with LPS or histamine, respectively, to induce vascular permeability [8, 9]. We hypothesized that augmenting EC barrier function would attenuate inflammatory injury mediated by ICs. This approach has the potential to limit IC deposition and leukocyte transmigration into tissues, yet EC integrity has not been pharmacologically targeted in either SLE or RA.
Sphingosine-1-phosphate (S1P), a bioactive lipid secreted mainly by RBCs, ECs, and activated platelets, is a key regulator of EC barrier function and lymphocyte egress out of lymph nodes and thymus [10, 11]. S1P interacts with five broadly expressed GPCRs, S1P1–5. When S1P bind EC S1P1, it stabilizes adherens junctions by increasing the translocation of GTPase Rac and VE-cadherin to the cell periphery, and resultant homotypic VE-cadherin interactions cause cell spreading and closure of intercellular gaps [12–14]. Blocking S1P degradation or administration of S1P1 agonists have been shown to diminish lung inflammation in response to LPS and acute lung injury with bleomycin [15, 16]. We asked whether S1P1 agonists could decrease inflammation in an animal model of IC-mediated vascular injury, the reverse Arthus reaction (RAR). In this model, ICs activate neutrophils via Fc receptors to release reactive oxygen species, proteolytic enzymes, cytokines and chemokines. Complement receptors are required in some animal models, depending upon the strain and organ in which the RAR is performed 2[17, 18]. Although vascular injury is a critical pathophysiologic component of the RAR, pharmacologic targeting of the EC barrier has not been studied. In this study, we show that S1P1 agonists, including a novel engineered S1P chaperone - ApoM-Fc [19], prevent barrier dysfunction in response to activated neutrophil-induced injury in vitro and diminish neutrophil transmigration and red blood cell (RBC) extravasation in vivo. These data support a novel approach to limiting inflammation by enhancing EC barrier integrity and thus, have implications for the treatment of patients with IC-mediated injury.
MATERIALS AND METHODS
Reagents
Specific S1P1 agonists, CYM-5442 (Santa Cruz Biotechnology) and SEW-2871 and a specific S1P1 antagonist, W146 (Cayman Chemical), were dissolved in DMSO . A novel engineered S1P chaperone, ApoM-Fc loaded with S1P- [19], was used in vitro (10 μg/ml) and in vivo (4 mg/kg IP). Human recombinant C5a (R&D Systems; 100 ng/ml) was used to activate PMNs. The following antibodies were used for western blot and/or immunofluorescence: ApoM (ab91656, Abcam), anti-VE-Cadherin (C19, Santa Cruz Biotechnology), and anti-p-MLC2 (T18/S19, Cell Signaling).
In vitro formation of Immune complexes
Goat anti-ovalbumin IgG (500 μg in 300 μl PBS, MP Biomedicals) was incubated with 50 μg ovalbumin for 60 min at 370C. ICs were kept at 4oC overnight, and then centrifuged for 5 min at 10,000 g. After washing in cold PBS, the pellet was resuspended in 300 μl PBS. Previous work by other laboratories has shown that goat IgG binds human and mouse Fc receptors [20, 21].
Human umbilical vein endothelial cell (HUVEC) culture and Electrical Cell Substrate Impedance (ECIS) assays
HUVECs (passages 2 to 8, 1.5X105,) were cultured on fibronectin-coated 8 well ECIS plates (8W10E, Applied BioPhysics) in M199 supplemented with HEPES, 10% heat-inactivated FBS, 10 mM Hepes, penicillin and streptomycin (50 mg/ml), and EC growth factors (Sigma). Cells were plated 18 hrs before use and were serum starved for 2 hrs. In experiments using PMNs, ICs (1:20 dilution) and C5a (100ng/ml) were added to HUVECs 15 min prior to the addition of PMNs. HUVECs treated with unactivated neutrophils were used as a control to validate each experimental run. A AC current (4 kHz) was applied (ECIS Theta, Applied Biophysics), and resistance measurements were obtained every 5 min.
Neutrophil isolation and activation
10 ml of human blood from healthy donors was collected in EDTA. Neutrophils (PMN) were isolated using Polymorphoprep™ (Axis-Shield), washed in HBSS without cations, resuspended at 1–5X105, and used within 1 hr of isolation. Protocols for human subjects were approved by Institutional IRB at HSS and written informed consent was obtained.
Immunofluorescence staining
HUVECs (3X105) were cultured on fibronectin-coated culture slides and used when confluent. HUVECs were serum starved for 2 hrs before addition of S1P1 agonists or antagonists or ICs (1:20 dilution) and C5a (100 ng/ml). PMNs (1X105) were added 10 min after the addition of IC and C5a. After a 30 min incubation at 37oC, supernatants were discarded and wells were washed in PBS. Cells were fixed in PFA (4%) at RT and permeabilized with Triton-X100 (0.1%). The cells were blocked with 1% FBS for 1hr at RT. To detect phosphorylated myosin light chain2 (p-MLC2) and VE-cadherin, samples were incubated with anti-p-MLC2 (5 μg/ml) and anti-VE-cadherin (5μg/ml) antibodies overnight. After 3 washes, samples were incubated with fluorescent secondary antibodies (Invitrogen, Alexa 488 and Alexa 568) for 1 hr at RT. Samples were imaged using an Olympus FluoView FV10i confocal microscope.
Mice
Animal experiments were performed under the guidelines set by the Institutional Animal Care and Use Committee at Weill Cornell Medicine, New York. Male C57BL/6J mice (8–10 weeks) were obtained from Jackson Laboratories. EC specific gene deletion of S1PR1: S1pr1f/f mice [22] were crossed with Cdh5-CreERT2 mice [23] to establish Cre positive S1pr1f/f (S1P1 ECKO) mice and Cre negative controls. To induce EC knockdown of S1P1, 8–10-week-old Cre+ S1pr1f/f mice and control Cre- S1pr1f/f mice were treated with tamoxifen (10mg/ml, 200 μl IP) for 5 days. Deletion of S1P1 exon 2, containing the entire coding region, was confirmed by PCR (supplemental Figure 1). Mice were used 2–4 weeks after tamoxifen administration. Apom−/− mice were a gift from L. B. Nielsen and C. Christeoffersen, Copenhagen, Denmark [24]. Absence of apolipoprotein M was confirmed on western blot of mouse plasma (supplemental Figure 1).
Reverse Arthus Reaction in skin
Anesthetized mice were shaved and injected with intradermal goat anti-ovalbumin IgG (60 ug/30 ul) or PBS alone. Immediately thereafter, ovalbumin (400 μg; Sigma) and Evans Blue (EB; 0.5% in 150 μl PBS) were injected via tail vein. Some mice received intradermal W146 (10μg) or vehicle concomitantly with anti-ovalbumin IgG. After 4 hrs, mice were sacrificed and the diameter of EB leakage was measured. 6 mm punch skin samples were weighed and treated with formamide (500 μl at 60oC overnight) to extract EB and supernatants were measured at 610 nm. In preliminary experiments, we found that nonspecific goat IgG injected intradermally did not elicit a reaction after IV injection with ovalbumin and EB and that injection of goat anti-ovalbumin IgG intradermally did not elicit a reaction after IV injection of EB without ovalbumin.
Reverse Arthus Reaction in lung
Mice were anesthetized and goat anti-ovalbumin IgG (250 μg) was administered intranasally; immediately thereafter, mice were injected with IV 0.5% EB and ovalbumin (750 μg in 150 μl PBS). CYM-5442 (0.5mg/kg) or SEW-2874 (10 mg/kg) was administered IP (in 25% tween: 75% water) starting 2 hrs prior to RAR and then every 5 hrs. In some experiments, W146 (10 μg) or vehicle was administered intranasally concomitantly with anti-ovalbumin IgG. 24 hrs after RAR, mice were anesthetized and broncho-alveolar lavage (BAL) was performed. 500 μl of PBS with 1mM EDTA was administered and collected 3 times. Samples (300 μl) were stained with Wright-Giemsa after cytospin. Cell counts were obtained with a hemocytometer (ADVIA) and the number of PMN was calculated by multiplying the total number of WBCs by the percentage of PMN assessed on cytospin.
Western Blotting
Plasma samples (1 μl) from ApoM −/− and WT mice were denatured at 95°C for 5 min after addition of 10% β-mercaptoethanol. Proteins were electrophoresed and transferred onto nitrocellulose. Transferred proteins were blocked for 1hr and then probed with anti-ApoM antibody (10 μg/ml).
Statistics
All data were presented as the mean ± standard error of mean (S.E.M). Comparisons between different groups were made using the unpaired T-test. Statistical significance was set at P<0.05.
RESULTS
Mice deficient in EC specific S1P1 show amplified inflammation and hemorrhage in response to lung RAR
The RAR is a well-established model of acute IC-mediated vascular injury and inflammation characterized by neutrophil activation and vascular leak [25, 26]. Because IC deposition in microvessels correlates with vascular permeability [27], we posited that decreased EC barrier function and the consequent increase in IC deposition would enhance inflammation. To test this hypothesis, we performed lung RAR in mice with ECs lacking S1P1 (EC knock out, ECKO). At baseline, S1P1 ECKO mice injected with IV EB alone showed increased pulmonary extravasation of EB compared to controls, as previously reported [28]; notably, BAL fluid from S1P1 ECKO mice injected with EB did not show increased numbers of RBCs or WBCs (data not shown). In contrast, after RAR, S1P1 ECKO mice demonstrated marked increase in EB extravasation and lung weight (Figures 1A and B) indicative of vascular leak. BAL from S1P1 ECKO mice exposed to RAR also showed a greater number of WBCs and RBCs compared to Cre-negative littermate controls (Figures 1C and D). WBCs in BAL fluid were comprised mostly of neutrophils (range: 50–80%) and alveolar macrophages (Figures 1E and F). Taken together, these findings demonstrate impaired barrier function and increased inflammation in response to RAR in S1P1 ECKO mice.
Figure 1. S1P1 ECKO mice show accentuated lung RAR compared to Cre-negative littermate controls similarly treated with tamoxifen.
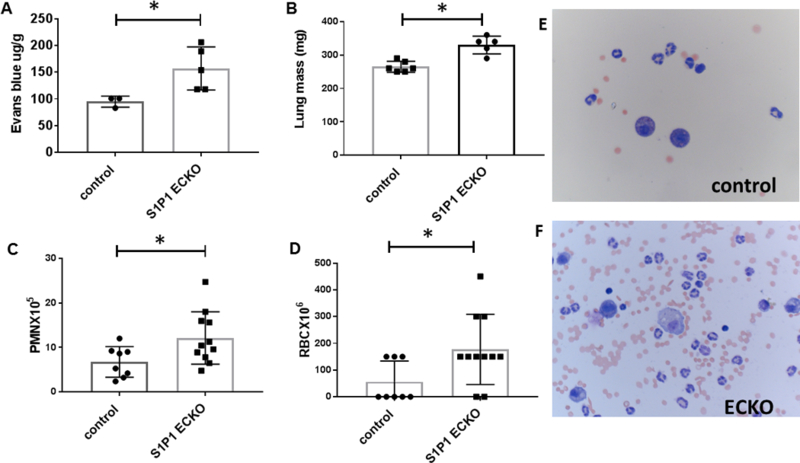
(A) After RAR, Evans blue leakage into lung was quantified by formamide extraction (n=3–5 mice/group, *p=0.04). (B) Lung mass, a surrogate marker of edema, was measured after RAR (n=5–6 mice/group, *p=0.0007). BAL fluid was assessed for (C) total numbers of PMNs (n=8–11 mice/group; *p=0.03) and (D) RBCs (n=8–11 mice/group; *p=0.03). Representative Wright-Giemsa stain of BAL fluid after lung RAR in control (upper panel, E) and ECKO mice (lower panel, F).
W146, a specific S1P1 antagonist, increases lung and skin RAR
ApoM, a constituent of high density lipoprotein (HDL), carries approximately 65% of circulating S1P while albumin carries 30%. Mice lacking ApoM have a 50% reduction in circulating S1P compared to controls [29], and ApoM delivery of S1P is more protective of barrier function and more anti-inflammatory than albumin delivery [30] [31]. Therefore, we hypothesized that ApoM−/− mice would have an increased inflammatory response to RAR. Surprisingly, ApoM−/− mice and WT mice had similar numbers of PMN and RBCs in BAL fluid after RAR (Figure 2A and B) and similar EB extravasation in their lungs. ApoM deficiency was confirmed by western blot of plasma samples (supplemental Figure 1B). We considered the possibility that local levels of S1P were elevated by platelets and/or EC release during RAR and obscured the effects of ApoM deficiency. At higher levels of S1P (~500nM), albumin and ApoM have been shown to be equally effective at reducing TNF-α induced VCAM-1 in ECs in vitro [32].
Figure 2. S1P1 antagonist W146 increased lung RAR in ApoM−/− mice and skin RAR in WTmice.
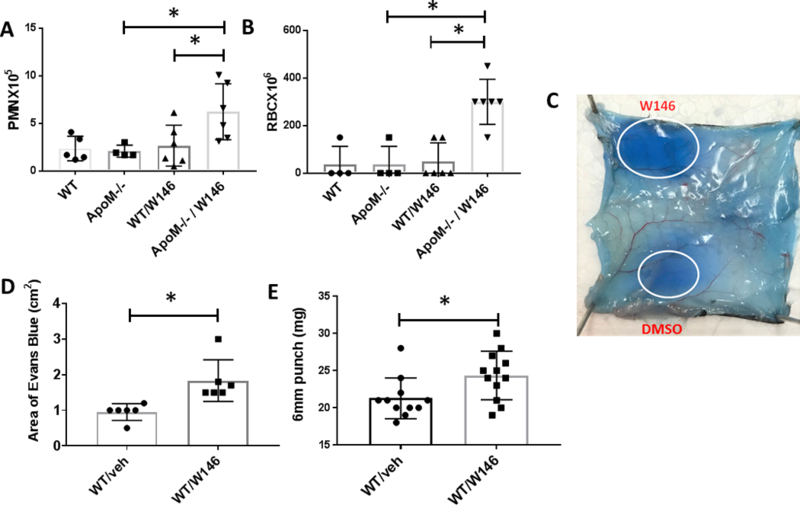
For the lung RAR, mice were treated with intranasal anti-ovalbumin IgG followed by ovalbumin with 0.5% Evans blue IV, and after 24 hrs subjected to bronchoalveolar lavage. Mice received W146 (10 μg) or vehicle intranasally concomitant with anti-ovalbumin IgG. (A) PMN counts (n=6 mice/group *p=0.04) and (B) RBC counts (n=6 mice/group *p=0.005) were performed. (C) For skin RAR, mice were injected intradermally with goat anti-ovalbumin IgG or PBS, followed by ovalbumin and Evan’s blue IV, and mice killed after 4hrs. Some mice received intradermal W146 (10 μg) with anti-ovalbumin IgG. A representative skin sample after RAR with intradermal W146 or vehicle is shown. (D) Skin RAR was assessed by measuring the diameter of the major and minor axis and determining area of EB leakage at the injection site (n=6; *p=0.006), and (E) the weight of 6 mm punch biopsies (n=11–12;*p=0.02).
We hypothesized that partial blockade of S1P1 with a S1P1 antagonist would render ApoM−/− mice more vulnerable to IC-mediated injury than WT mice, in spite of high local levels of albumin S1P. In preliminary studies, systemic administration of W146 IV (10 mg/kg or a total of 250 μg) induced significant pulmonary leakage, as has been published ([33], supplemental Figure 2C) and we verified that W146 decreased baseline HUVEC resistance in ECIS (supplemental Figure 2a). We reasoned that administration lower doses of intranasal W146 (10 μg) would not induce vascular leakage at baseline, but would limit S1P1 availability and unmask the protective effect of ApoM-mediated delivery of S1P during RAR. Indeed, low dose W146 administered intranasally to ApoM−/− concomitant with anti-ovalbumin IgG markedly increased extravasation of RBCs and PMNs from blood vessels into the alveolar space (Figures 2B and C), while treatment of WT mice with W146 at the same dose did not alter RAR. These data demonstrate that decreased signaling of S1P1 (induced by partial blockade of S1P1 with W146) increases vulnerability to endothelial barrier dysfunction during IC-mediated inflammation which can be overcome by delivery of S1P by ApoM.
Because IC-mediated injury in skin is common to patients with SLE, we sought to confirm that modulation of S1P1 signaling also affects skin RAR. Similar to lung RAR, there was no evidence that ApoM deficiency increased skin RAR, and intradermal injection of low doses of W146 alone did not increase EB leak (supplemental figure 3). However, in contrast to our findings in the lung RAR in WT mice, low dose W146 (10 μg) increased the skin RAR when administered concomitantly with anti-ovalbumin IgG, compared to reaction sites (Figures 2C–E). The finding that low dose W146 augmented skin RAR, but not lung RAR in WT mice may be explained by a lower density of S1P1 in skin compared to lung [34]. These data show that low doses of locally administered W146 does not induce skin or lung leakage at baseline but augments vascular leak in IC-mediated injury in skin of WT mice and in lung of ApoM−/− mice. These data further support the concept that blockade of S1P1 receptors increases inflammation in response IC deposition.
S1P1 agonist CYM-5442 increases barrier function, and mitigates vascular injury induced by activated PMN injury in vitro and in vivo.
Given that blockade of S1P1 signaling increases IC-driven injury, we hypothesized that augmentation of EC barrier function would limit inflammation in response to ICs. First, we first tested the ability capacity of CYM-5442, a specific S1P1 agonist, to increase EC barrier function in vitro with ECIS. CYM-5442 induced a rapid increase in EC resistance at doses of 50–200 nM that returned to baseline after 1.5–2 hrs (supplemental figure 2B; n=3). Then we modeled the neutrophil-mediated EC injury characteristic of the RAR in vitro by exposing HUVEC to PMNs activated by IC and C5a and assessed barrier function in the presence or absence of S1P1 agonists. No detectable changes were detected when HUVECs were treated with unstimulated PMN (Figure 3A). C5a or ICs alone did not induce a decrease in HUVEC resistance in ECIS after 1–2 hrs (data not shown) but C5a- and IC- activated PMNs caused a marked decrease in resistance within 30 min that was sustained for 8 hrs (Figure 3A). CYM-5442 (200nM) rescued the PMN-induced drop in resistance (Figure 3A), supporting the possibility that CYM-5442 diminishes inflammatory injury in the RAR by protecting EC barrier function.
Figure 3. S1P1 agonist CYM-5442 protected ECs against IC activated PMN induced injury in vitro and in vivo.
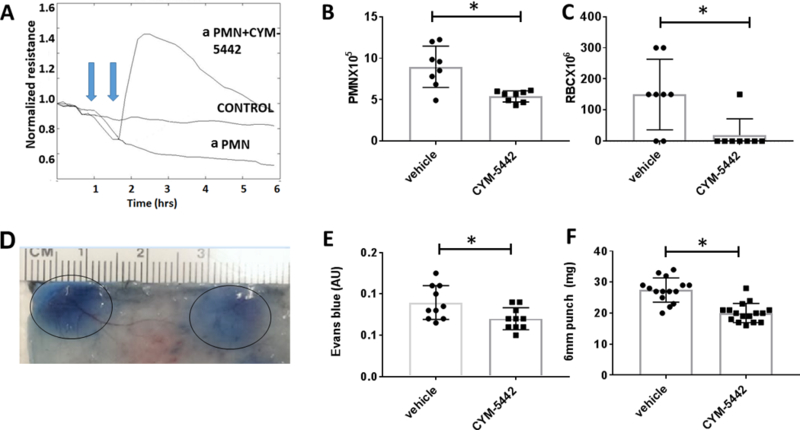
(A) CYM-5442 rescued PMN-induced EC barrier dysfunction. IC and C5a activated PMN (aPMN) (1X105) were added to HUVECs (1st blue arrow) and resistance was measured at 5 min intervals. CYM-5442 was added 30 min (2nd blue arrow) after the addition of activated PMN, and resistance was measured at 5 min intervals. The control tracing (middle) represents ECs treated with unstimulated PMN. ECIS tracing is representative of experiments with 3 different healthy PMN donors. (B, C) Lung RAR was performed in WT C57Bl/B6 mice. CYM-5442 (0.5mg/kg IV) attenuated lung RAR as assessed by BAL PMNs (n=8 mice/group, *p=0.002) and RBCs (n=8 mice/group, *p=0.01). (D-F) Skin RAR was performed in WT mice with CYM-5442 or vehicle control. (D) A representative sample skin injected with CYM-5442 (10 μg) vs. DMSO control is shown. Skin RAR was quantified by measuring (E) area of EB extravasation (n=10 mice, *p=0.02), and (F) weights of 6 mm punch biopsies (n=15 mice; *p<0.0001).
Because CYM-5442 is a S1P1 agonist that can act as a functional antagonist, inducing internalization and degradation of S1P1 in a dose dependent manner, we first determined the dose of CYM-5442 that induced EB leakage. At 10 mg/kg, a dose that has been shown to induce maximal lymphopenia and yield plasma concentrations of approximately 1μM [35], there was significant EB leakage (supplemental figure 2C, 3rd panel). Because our ECIS experiments showed that CYM-5442 was protective of EC barrier at concentrations of 50–200 nM, we hypothesized that administration of CYM-5442 at 0.5 mg/kg to mice would yield plasma concentration such that it would function as an S1P1 agonist (50–200 nM) and increase EC barrier function. In mice treated with CYM-5442 (0.5mg/kg), the lung RAR was blunted. BAL fluid contained fewer PMNs and RBCs in (Figure 3B and C), but EB extravasation or lung weights were not decreased. Treatment with a much weaker S1P1 agonist, SEW-2871 (10 mg/kg; [35]) resulted in a similar attenuation of WBCs and RBCs in BAL after lung RAR, but no significant change in EB extravasation or lung weights, (supplemental Figure 4). We confirmed the protective effect of CYM-5442 for IC-driven inflammation in skin RAR. Treatment with intradermal CYM-5442 reduced vascular leak as demonstrated by a decrease in EB extravasation and tissue edema as assessed by formamide extraction of EB and skin biopsy weights, respectively (Figures 3E and F). The finding that CYM-5442 was effective at blocking EB extravasation in the skin, but not the lung, during RAR is consistent with our observation that W146 exacerbated skin, but not lung RAR, in WT mice.
ApoM-Fc, a novel S1P chaperone, protects ECs from loss of barrier integrity induced by activated PMN and attenuates lung RAR.
A novel biologic chaperone of S1P, ApoM-Fc was recently shown to increase circulating S1P levels by approximately 60% and to attenuate myocardial injury after ischemia-reperfusion injury and reduce brain injury in a mouse model of stroke [19]. In addition, ApoM-Fc increased barrier function to a greater extent than albumin-S1P and did not induce lymphopenia, suggesting specificity to activation of EC S1P receptors, particularly S1P1 [19]. We examined the ability of ApoM-Fc to attenuate IC-mediated injury initiated by activated PMN in vitro and in vivo. ApoM-Fc rescued EC barrier function in HUVECs exposed to IC- and C5a- activated PMN in a sustained manner (Figure 4A). In comparison, albumin-S1P (200 nM) increased resistance, but the effect was transient (Figure 4A).
Figure 4. ApoM-Fc rescued IC activated PMN induced EC injury in vitro and in vivo.
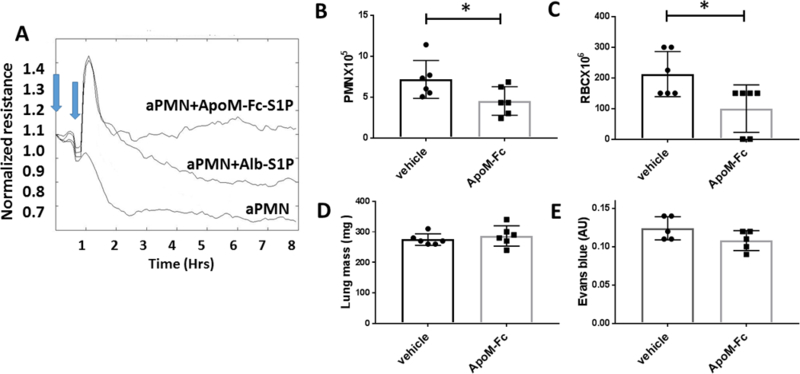
(A) IC and C5a activated PMN (aPMN) (1X105) were added to HUVECs (1st blue arrow) and 30 min later (2nd blue arrow) ApoM-Fc (10 μg/ml) loaded with S1P was added or albumin-S1P (Alb-SIP, 200 nM). EC resistance was measured in ECIS at 5 min intervals. ECIS tracing is representative of experiments with 3 different healthy PMN donors. (B-E) Mice were treated with ApoM-Fc or PBS at the initiation of lung RAR. At 24 hrs inflammation and injury was assessed by (B) PMNs in BAL (n=6 mice/group,* p< 0.05), (C) RBCs in BAL (n=6 mice/group; p=0.03), (D) lung mass (E), and EB extravasation.
To determine whether ApoM-Fc is an effective means to deliver S1P to ECs and mitigate IC-mediated injury in vivo, we treated mice with ApoM-Fc and initiated lung RAR. ApoM-Fc attenuated the inflammatory response in lung RAR indicated by a decrease in PMN and RBCs in BAL fluid (Figure 4B and C), but like CYM-5442 and SEW-2871, there was no effect on EB leakage or lung weights (Figure 4D or E).
S1P1 agonists CYM-5442 and ApoM-Fc limit endothelial cell phosphorylation of MLC and VE-Cadherin junction disassembly in response to IC and C5a activated neutrophils
Activated neutrophils have been shown to induce the Rho GTPase pathway in EC which contributes to barrier breach, vascular leak [36] and neutrophil migration [37]. Phosphorylation of myosin light chain (p-MLC) of myosin II induces RhoA-mediated assembly of stress fibers and focal adhesions, mediating cell contraction. We hypothesized that S1P protected ECs from injury induced by activated neutrophils and prevented loss of barrier integrity by inhibiting the Rho pathway. To test this experimentally, we compared p-MLC staining in HUVECs exposed to PMN activated with IC and C5a to that of HUVECs exposed to unstimulated PMN (control HUVECs, Figure 5, top panel). Treatment with activated PMNs markedly increased HUVEC p-MLC staining, which appeared to localize to stress fibers (Figure 5, middle panel). p-MLC staining was attenuated in HUVECs pre-incubated with ApoM-Fc and then treated with IC-activated PMN, (Figure 5, bottom panel). Because Rho pathway-mediated endothelial hyper-permeability is associated with disruption of VE-cadherin at adherens junctions [38], we determined whether ApoM-Fc protected VE-cadherin localization in HUVECs treated with IC- and C5a-activated PMNs. HUVECs treated with activated PMN showed marked loss of VE-Cadherin staining, whereas HUVECs pretreated with ApoM-Fc showed mostly intact staining (Figure 5, bottom panel). Similarly, HUVEC pretreated with CYM-5442 were protected from PMN induced injury, demonstrating preserved VE-cadherin and attenuated p-MLC (supplemental Figure 5). Collectively, these data suggest that S1P agonists protect ECs from neutrophil-mediated induction of the Rho pathway.
Figure 5. ApoM-Fc decreases myosin light chain phosphorylation (p-MLC) and attenuates disruption of VE-cadherin in HUVECs treated with activated PMNs.
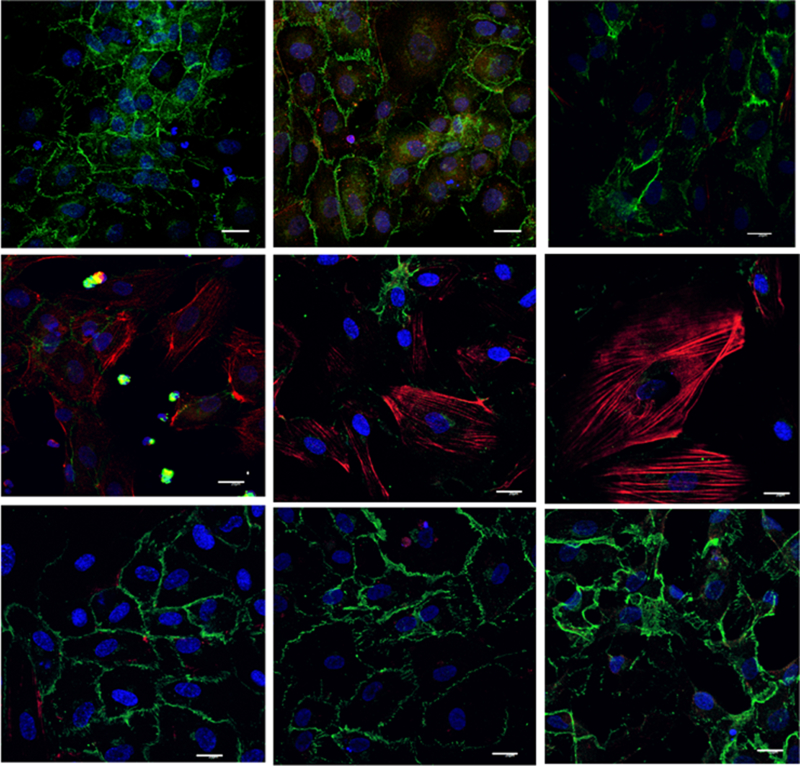
HUVECs were treated for 30 min with IC and C5a activated PMNs (1X105) and stained for VE-Cadherin (green) and p-MLC (red). Nuclei were stained with DAPI. Top panel: unstimulated PMN, middle panel: IC and C5a activated PMN; bottom panel: HUVECs pretreated with ApoM-Fc prior to incubation with IC and C5a activated PMN. Images representative of 3 experiments with 3 different healthy PMN donors. (scale bar = 20 microns)
DISCUSSION
We present genetic and pharmacological evidence that S1P1 signaling in ECs modulates vascular responses to IC deposition. Deletion of S1P1 in ECs augmented vascular leakage and inflammatory induced by the RAR suggesting protective role for S1P1. This was supported by evidence that W146, a specific S1P1 antagonist, decreased EC barrier function in vitro and increased skin and lung RAR. Finally, two S1P1 agonists, CYM-5442, SEW-2871, and a novel biologic chaperone of S1P, ApoM-Fc, protected EC barrier integrity from IC activated PMN induced injury in vitro and in vivo. Collectively, our results demonstrate that S1P1 signaling enhances the EC barrier, limits leukocyte escape from capillaries, and provides protection from inflammatory injury. In addition, we show that S1P1 agonists prevent VE-cadherin disruption and phosphorylation of MLC, a potential mechanism by which they maintain vascular barrier integrity despite pathogenic mediators released by activated PMN.
While the S1P1 agonists were effective in decreasing inflammation, they did not significantly reduce EB extravasation or plasma escape from the microvasculature. Enhanced S1P1 signaling during IC-mediated inflammation may not be sufficient to block transudation of small molecules, such as albumin, but it appears sufficient to prevent transmigration of PMN and leakage of RBC at sites of barrier breaks. It is also possible that S1P1 agonists decrease migration of inflammatory cells at sites of inflammation, at least in part, by influencing the phosphorylation status of VE-cadherin; phosphorylation of T685 on the cytoplasmic domain of VE-Cadherin supports permeability while de-phosphorylation of T731 supports leukocyte transmigration [39]. In contrast to our findings in the lung, CYM-5442 decreased EB extravasation and skin weights in the skin RAR. This may be due to the lower density of S1P1 receptors in the skin compared to the lung. Alternatively, there may be differential expression of S1P family receptors or other receptors on ECs that determine vascular barrier function in different vascular beds. The ability of low dose W146 to augment the skin, but not lung RAR, in WT mice also supports differences in S1P1 density and/or other receptors influencing permeability in the skin and lung.
Increasing barrier function has been shown to limit inflammation in models of ischemia-reperfusion injury and several other models of acute injury [40] [41]. Although the RAR is also an acute model of inflammation, increasing barrier function to limit the number of activated neutrophils transmigrating into tissues may have long term consequences on organ function. PMN influx has been identified as key mediator of renal damage in nephrotoxic serum transfer, a model of IC-initiated glomerulonephritis [42]. Reducing exposure to neutrophil-derived chemokines, cytokines, reactive oxidants and proteolytic enzymes limits acute damage and allows resolution of damage. The critical role of microvascular integrity in limiting joint damage and PMN infiltration is also supported in the K/BxN serum-transfer model of rheumatoid arthritis [3]. Indeed, blood vessels in joints appear to be more susceptible to IC-mediated injury than vasculature in other tissues [43].
Despite the body of evidence demonstrating the key of role microvascular in regulation of tissue injury induced by inflammation, pharmacological targeting of ECs to increase barrier function in RA and SLE has not been pursued. Drug discovery and development for SLE and RA targeting the S1P/S1P1 axis has focused solely on restricting egress of pathogenic lymphocytes from lymph nodes and induction of circulating lymphopenia [44]. Because of their ability to induce lymphopenia, S1P1 antagonists have been studied in mouse models of inflammatory arthritis [45], and while blocking autoreactive T cells from entering target tissues is beneficial for reducing inflammation and organ damage, the effects on ECs may be deleterious. Indeed, prolonged treatment of mice with the S1P1 antagonist NIBR-0213 to treat adjuvant-induced arthritis decreased joint inflammation, but long term administration resulted in pulmonary edema, perivascular fibrosis, and functional lung impairment [46]. Because patients with SLE and RA frequently have lung involvement, they may be more vulnerable to pulmonary vascular leak and inflammation.
The clinical efficacy of FTY-720, a nonspecific S1P receptor agonist/functional antagonist that also induces lymphopenia, for the treatment of multiple sclerosis has encouraged development of several S1P1 specific agonists/antagonists for the treatment of other autoimmune diseases [44]. In animal models of lupus nephritis, both FTY-720 and a specific S1P1 receptor KRP-203 induced lymphopenia, reduced renal injury, and increased survival [47, 48]. A problem with this approach is that depending on the dose and frequency of its delivery, FTY-720 has been shown to induce and/or exacerbate vascular leak in the lung and brain [49, 50]; the mechanism by which FTY-720 can impair vascular integrity involves S1P1 phosphorylation, internalization, and proteasomal degradation [33]. CYM-5442, while efficacious at low doses in our studies, had a limited therapeutic window because it, too, increased vascular leakage at high doses consistent with its ability to induce S1P1 internalization and proteasome degradation [35]. ApoM-Fc has the potential to effectively enhance delivery S1P without leading to excessive S1P1 internalization. An additional advantage of ApoM-Fc is that its half-life is 4 days compared to 4–5 hrs for CYM-5442 or SEW-2871. Finally, ApoM-Fc does not induce lymphopenia, but formal studies on the effects of ApoM-Fc on leukocyte function are ongoing to determine whether it is immunosuppressive.
Novel approaches to target the S1P/S1P1 axis, particularly in systemic diseases that put patients at risk for pulmonary vascular leak, are needed to augment barrier function without causing degradation of S1P. Such interventions could be used as adjuncts to immunosuppressive therapy, allowing simultaneous targeting to decrease production of autoantibodies and to attenuate tissue responses to IC deposition. Our data support further studies in models of chronic inflammatory injury to determine whether S1P1 augmented EC barrier function will mitigate end organ damage.
Supplementary Material
A. PCR confirmation of S1P1 deletion. PCR confirmation of S1P1 deletion. Mice 3,7,8, and 11 show the 200 bp PCR product corresponding to S1P1 after exon 2 deletion induced by treatment with tamoxifen. 2 bands are present corresponding to WT and deleted S1P1 because the deletion occurs only in ECs. B. Mouse plasmas from ApoM-/- mice (lanes 1-4) confirmed the deletion of ApoM compared to WT plasmas (lanes 5-8).
A. W146 decreased HUVEC resistance ECIS in dose dependent manner (0.1 -10 μM, n=3). B. CYM-5442 increase HUVEC resistance on ECIS in a dose dependent manner (10-200 nM, n=3). C. Mice were injected with 0.5% EB IV and then injected immediately afterward with IP CYM-5442 0.5mg/kg, CYM-5442 10 mg/kg, or W146 10 mg/kg.
Intradermal W146 alone did not induce vascular leak but amplified leak in response to IC vascular injury. W146 (10 μg in 30 μl PBS) was administered intradermally either alone (bottom half of skin sample) or concomitantly with anti-ova IgG (60 ug/30 ul, top half) followed by IV injection of Evans blue (0.5% in 150 μl PBS) and ovalbumin (400μg). 4 hrs post injection skin was assessed for EB extravasation.
SEW 2871 decreases lung RAR. Lung injury after RAR was quantified by assessment of PMN (A) and RBC (B) counts in BAL fluid after 24 hrs (n=4 mice per group, *p=0.01 and *p=0.03, respectively). Lung weights (mg) after RAR are shown in C, n=4, p=ns).
CYM-5442 diminishes p-MLC and preserves VE-cadherin staining in IC and C5a activated HUVECs. HUVECs were treated for 30 min with IC and C5a (100 ng/ml) activated PMN (1X105) and then fixed and solubilized prior to staining with anti-pMLC (in red) and anti-VE-Cadherin (in green). Nuclei were stained with DAPI. 1st row: HUVEC with unstimulated PMN; 2nd row HUVEC with CYM-5442 (no PMNs); 3rd row HUVEC with activated PMN; 4th row HUVEC pretreated with CYM-5442 prior to treatment with activated PMN.
Acknowledgments
This study was supported in part by grants NIH HL135821 (TH and JS), the Empire Clinical Research Investigator Program, NY State Department of Health (JS and NB) and the Clinical and Translational Science Center, Weill Cornell Medical Center (JS and TH).
Footnotes
The authors Nathalie Burg, Stefan Worgall, and Jane Salmon have no competing financial interests. Steven Swendeman and Timothy Hla are the inventors of ApoM-Fc and have filed a patent application through Weill Cornell Medicine [P33974 (7512–01-US)] that describes the development of ApoM-Fc and its uses.
References
- 1.Atehortua L, Rojas M, Vasquez GM, and Castano D, Endothelial Alterations in Systemic Lupus Erythematosus and Rheumatoid Arthritis: Potential Effect of Monocyte Interaction. Mediators Inflamm, 2017. 2017: p. 9680729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodrigues SF and Granger DN, Blood cells and endothelial barrier function. Tissue Barriers, 2015. 3(1–2): p. e978720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stangenberg L, Burzyn D, Binstadt BA, Weissleder R, Mahmood U, Benoist C, and Mathis D, Denervation protects limbs from inflammatory arthritis via an impact on the microvasculature. Proc Natl Acad Sci U S A, 2014. 111(31): p. 11419–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xia Y, Herlitz LC, Gindea S, Wen J, Pawar RD, Misharin A, Perlman H, Wu L, Wu P, Michaelson JS, Burkly LC, and Putterman C, Deficiency of fibroblast growth factor-inducible 14 (Fn14) preserves the filtration barrier and ameliorates lupus nephritis. J Am Soc Nephrol, 2015. 26(5): p. 1053–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woodfin A, Voisin MB, Imhof BA, Dejana E, Engelhardt B, and Nourshargh S, Endothelial cell activation leads to neutrophil transmigration as supported by the sequential roles of ICAM-2, JAM-A, and PECAM-1. Blood, 2009. 113(24): p. 6246–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alcaide P, Newton G, Auerbach S, Sehrawat S, Mayadas TN, Golan DE, Yacono P, Vincent P, Kowalczyk A, and Luscinskas FW, p120-Catenin regulates leukocyte transmigration through an effect on VE-cadherin phosphorylation. Blood, 2008. 112(7): p. 2770–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Su WH, Chen HI, and Jen CJ, Differential movements of VE-cadherin and PECAM-1 during transmigration of polymorphonuclear leukocytes through human umbilical vein endothelium. Blood, 2002. 100(10): p. 3597–603. [DOI] [PubMed] [Google Scholar]
- 8.Frye M, Dierkes M, Kuppers V, Vockel M, Tomm J, Zeuschner D, Rossaint J, Zarbock A, Koh GY, Peters K, Nottebaum AF, and Vestweber D, Interfering with VE-PTP stabilizes endothelial junctions in vivo via Tie-2 in the absence of VE-cadherin. J Exp Med, 2015. 212(13): p. 2267–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Lorenzo A, Fernandez-Hernando C, Cirino G, and Sessa WC, Akt1 is critical for acute inflammation and histamine-mediated vascular leakage. Proc Natl Acad Sci U S A, 2009. 106(34): p. 14552–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwab SR, Pereira JP, Matloubian M, Xu Y, Huang Y, and Cyster JG, Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science, 2005. 309(5741): p. 1735–9. [DOI] [PubMed] [Google Scholar]
- 11.Proia RL and Hla T, Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J Clin Invest, 2015. 125(4): p. 1379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, and English D, Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest, 2001. 108(5): p. 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, Volpi M, Sha’afi RI, and Hla T, Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell, 1999. 99(3): p. 301–12. [DOI] [PubMed] [Google Scholar]
- 14.Camerer E, Regard JB, Cornelissen I, Srinivasan Y, Duong DN, Palmer D, Pham TH, Wong JS, Pappu R, and Coughlin SR, Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest, 2009. 119(7): p. 1871–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao Y, Gorshkova IA, Berdyshev E, He D, Fu P, Ma W, Su Y, Usatyuk PV, Pendyala S, Oskouian B, Saba JD, Garcia JG, and Natarajan V, Protection of LPS-induced murine acute lung injury by sphingosine-1-phosphate lyase suppression. Am J Respir Cell Mol Biol, 2011. 45(2): p. 426–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L, Sammani S, Moreno-Vinasco L, Letsiou E, Wang T, Camp SM, Bittman R, Garcia JG, and Dudek SM, FTY720 (s)-phosphonate preserves sphingosine 1-phosphate receptor 1 expression and exhibits superior barrier protection to FTY720 in acute lung injury. Crit Care Med, 2014. 42(3): p. e189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baumann U, Kohl J, Tschernig T, Schwerter-Strumpf K, Verbeek JS, Schmidt RE, and Gessner JE, A codominant role of Fc gamma RI/III and C5aR in the reverse Arthus reaction. J Immunol, 2000. 164(2): p. 1065–70. [DOI] [PubMed] [Google Scholar]
- 18.Sylvestre D, Clynes R, Ma M, Warren H, Carroll MC, and Ravetch JV, Immunoglobulin G-mediated inflammatory responses develop normally in complement-deficient mice. J Exp Med, 1996. 184(6): p. 2385–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swendeman SL, Xiong Y, Cantalupo A, Yuan H, Burg N, Hisano Y, Cartier A, Liu CH, Engelbrecht E, Blaho V, Zhang Y, Yanagida K, Galvani S, Obinata H, Salmon JE, Sanchez T, Di Lorenzo A, and Hla T, An engineered S1P chaperone attenuates hypertension and ischemic injury. Sci Signal, 2017. 10(492). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelton JG, Sheridan D, Santos A, Smith J, Steeves K, Smith C, Brown C, and Murphy WG, Heparin-induced thrombocytopenia: laboratory studies. Blood, 1988. 72(3): p. 925–30. [PubMed] [Google Scholar]
- 21.Ciraci C, Janczy JR, Jain N, Haasken S, Pecli ESC, Benjamim CF, Sadler JJ, Olivier AK, Iwakura Y, Shayakhmetov DM, Sutterwala FS, and Cassel SL, Immune Complexes Indirectly Suppress the Generation of Th17 Responses In Vivo. PLoS One, 2016. 11(3): p. e0151252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jung B, Obinata H, Galvani S, Mendelson K, Ding BS, Skoura A, Kinzel B, Brinkmann V, Rafii S, Evans T, and Hla T, Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Dev Cell, 2012. 23(3): p. 600–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Luthi U, Barberis A, Benjamin LE, Makinen T, Nobes CD, and Adams RH, Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature, 2010. 465(7297): p. 483–6. [DOI] [PubMed] [Google Scholar]
- 24.Christoffersen C, Jauhiainen M, Moser M, Porse B, Ehnholm C, Boesl M, Dahlback B, and Nielsen LB, Effect of apolipoprotein M on high density lipoprotein metabolism and atherosclerosis in low density lipoprotein receptor knock-out mice. J Biol Chem, 2008. 283(4): p. 1839–47. [DOI] [PubMed] [Google Scholar]
- 25.Kaburagi Y, Hasegawa M, Nagaoka T, Shimada Y, Hamaguchi Y, Komura K, Saito E, Yanaba K, Takehara K, Kadono T, Steeber DA, Tedder TF, and Sato S, The cutaneous reverse Arthus reaction requires intercellular adhesion molecule 1 and L-selectin expression. J Immunol, 2002. 168(6): p. 2970–8. [DOI] [PubMed] [Google Scholar]
- 26.Li JL, Lim CH, Tay FW, Goh CC, Devi S, Malleret B, Lee B, Bakocevic N, Chong SZ, Evrard M, Tanizaki H, Lim HY, Russell B, Renia L, Zolezzi F, Poidinger M, Angeli V, St John AL, Harris JE, Tey HL, Tan SM, Kabashima K, Weninger W, Larbi A, and Ng LG, Neutrophils Self-Regulate Immune Complex-Mediated Cutaneous Inflammation through CXCL2. J Invest Dermatol, 2016. 136(2): p. 416–24. [DOI] [PubMed] [Google Scholar]
- 27.Stokol T, O’Donnell P, Xiao L, Knight S, Stavrakis G, Botto M, von Andrian UH, and Mayadas TN, C1q governs deposition of circulating immune complexes and leukocyte Fcgamma receptors mediate subsequent neutrophil recruitment. J Exp Med, 2004. 200(7): p. 835–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blaho VA, Galvani S, Engelbrecht E, Liu C, Swendeman SL, Kono M, Proia RL, Steinman L, Han MH, and Hla T, HDL-bound sphingosine-1-phosphate restrains lymphopoiesis and neuroinflammation. Nature, 2015. 523(7560): p. 342–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Christoffersen C, Obinata H, Kumaraswamy SB, Galvani S, Ahnstrom J, Sevvana M, Egerer-Sieber C, Muller YA, Hla T, Nielsen LB, and Dahlback B, Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci U S A, 2011. 108(23): p. 9613–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Argraves KM, Gazzolo PJ, Groh EM, Wilkerson BA, Matsuura BS, Twal WO, Hammad SM, and Argraves WS, High density lipoprotein-associated sphingosine 1-phosphate promotes endothelial barrier function. J Biol Chem, 2008. 283(36): p. 25074–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galvani S, Sanson M, Blaho VA, Swendeman SL, Obinata H, Conger H, Dahlback B, Kono M, Proia RL, Smith JD, and Hla T, HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci Signal, 2015. 8(389): p. ra79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruiz M, Frej C, Holmer A, Guo LJ, Tran S, and Dahlback B, High-Density Lipoprotein-Associated Apolipoprotein M Limits Endothelial Inflammation by Delivering Sphingosine-1-Phosphate to the Sphingosine-1-Phosphate Receptor 1. Arterioscler Thromb Vasc Biol, 2017. 37(1): p. 118–129. [DOI] [PubMed] [Google Scholar]
- 33.Oo ML, Chang SH, Thangada S, Wu MT, Rezaul K, Blaho V, Hwang SI, Han DK, and Hla T, Engagement of S1P(1)-degradative mechanisms leads to vascular leak in mice. J Clin Invest, 2011. 121(6): p. 2290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chae SS, Proia RL, and Hla T, Constitutive expression of the S1P1 receptor in adult tissues. Prostaglandins Other Lipid Mediat, 2004. 73(1–2): p. 141–50. [DOI] [PubMed] [Google Scholar]
- 35.Gonzalez-Cabrera PJ, Jo E, Sanna MG, Brown S, Leaf N, Marsolais D, Schaeffer MT, Chapman J, Cameron M, Guerrero M, Roberts E, and Rosen H, Full pharmacological efficacy of a novel S1P1 agonist that does not require S1P-like headgroup interactions. Mol Pharmacol, 2008. 74(5): p. 1308–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Breslin JW and Yuan SY, Involvement of RhoA and Rho kinase in neutrophil-stimulated endothelial hyperpermeability. Am J Physiol Heart Circ Physiol, 2004. 286(3): p. H1057–62. [DOI] [PubMed] [Google Scholar]
- 37.Saito H, Minamiya Y, Saito S, and Ogawa J, Endothelial Rho and Rho kinase regulate neutrophil migration via endothelial myosin light chain phosphorylation. J Leukoc Biol, 2002. 72(4): p. 829–36. [PubMed] [Google Scholar]
- 38.Sanchez T, Skoura A, Wu MT, Casserly B, Harrington EO, and Hla T, Induction of vascular permeability by the sphingosine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN. Arterioscler Thromb Vasc Biol, 2007. 27(6): p. 1312–8. [DOI] [PubMed] [Google Scholar]
- 39.Wessel F, Winderlich M, Holm M, Frye M, Rivera-Galdos R, Vockel M, Linnepe R, Ipe U, Stadtmann A, Zarbock A, Nottebaum AF, and Vestweber D, Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat Immunol, 2014. 15(3): p. 223–30. [DOI] [PubMed] [Google Scholar]
- 40.Sammani S, Moreno-Vinasco L, Mirzapoiazova T, Singleton PA, Chiang ET, Evenoski CL, Wang T, Mathew B, Husain A, Moitra J, Sun X, Nunez L, Jacobson JR, Dudek SM, Natarajan V, and Garcia JG, Differential effects of sphingosine 1-phosphate receptors on airway and vascular barrier function in the murine lung. Am J Respir Cell Mol Biol, 2010. 43(4): p. 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kannan L, Kis-Toth K, Yoshiya K, Thai TH, Sehrawat S, Mayadas TN, Dalle Lucca JJ, and Tsokos GC, R-spondin3 prevents mesenteric ischemia/reperfusion-induced tissue damage by tightening endothelium and preventing vascular leakage. Proc Natl Acad Sci U S A, 2013. 110(35): p. 14348–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosetti F, Tsuboi N, Chen K, Nishi H, Ernandez T, Sethi S, Croce K, Stavrakis G, Alcocer-Varela J, Gomez-Martin D, van Rooijen N, Kyttaris VC, Lichtman AH, Tsokos GC, and Mayadas TN, Human lupus serum induces neutrophil-mediated organ damage in mice that is enabled by Mac-1 deficiency. J Immunol, 2012. 189(7): p. 3714–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Binstadt BA, Patel PR, Alencar H, Nigrovic PA, Lee DM, Mahmood U, Weissleder R, Mathis D, and Benoist C, Particularities of the vasculature can promote the organ specificity of autoimmune attack. Nat Immunol, 2006. 7(3): p. 284–92. [DOI] [PubMed] [Google Scholar]
- 44.Kunkel GT, Maceyka M, Milstien S, and Spiegel S, Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat Rev Drug Discov, 2013. 12(9): p. 688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fujii Y, Hirayama T, Ohtake H, Ono N, Inoue T, Sakurai T, Takayama T, Matsumoto K, Tsukahara N, Hidano S, Harima N, Nakazawa K, Igarashi Y, and Goitsuka R, Amelioration of collagen-induced arthritis by a novel S1P1 antagonist with immunomodulatory activities. J Immunol, 2012. 188(1): p. 206–15. [DOI] [PubMed] [Google Scholar]
- 46.Bigaud M, Dincer Z, Bollbuck B, Dawson J, Beckmann N, Beerli C, Fishli-Cavelti G, Nahler M, Angst D, Janser P, Otto H, Rosner E, Hersperger R, Bruns C, and Quancard J, Pathophysiological Consequences of a Break in S1P1-Dependent Homeostasis of Vascular Permeability Revealed by S1P1 Competitive Antagonism. PLoS One, 2016. 11(12): p. e0168252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ando S, Amano H, Amano E, Minowa K, Watanabe T, Nakano S, Nakiri Y, Morimoto S, Tokano Y, Lin Q, Hou R, Ohtsuji M, Tsurui H, Hirose S, and Takasaki Y, FTY720 exerts a survival advantage through the prevention of end-stage glomerular inflammation in lupus-prone BXSB mice. Biochem Biophys Res Commun, 2010. 394(3): p. 804–10. [DOI] [PubMed] [Google Scholar]
- 48.Wenderfer SE, Stepkowski SM, and Braun MC, Increased survival and reduced renal injury in MRL/lpr mice treated with a novel sphingosine-1-phosphate receptor agonist. Kidney Int, 2008. 74(10): p. 1319–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shea BS, Brooks SF, Fontaine BA, Chun J, Luster AD, and Tager AM, Prolonged exposure to sphingosine 1-phosphate receptor-1 agonists exacerbates vascular leak, fibrosis, and mortality after lung injury. Am J Respir Cell Mol Biol, 2010. 43(6): p. 662–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yanagida K, Liu CH, Faraco G, Galvani S, Smith HK, Burg N, Anrather J, Sanchez T, Iadecola C, and Hla T, Size-selective opening of the blood-brain barrier by targeting endothelial sphingosine 1-phosphate receptor 1. Proc Natl Acad Sci U S A, 2017. 114(17): p. 4531–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. PCR confirmation of S1P1 deletion. PCR confirmation of S1P1 deletion. Mice 3,7,8, and 11 show the 200 bp PCR product corresponding to S1P1 after exon 2 deletion induced by treatment with tamoxifen. 2 bands are present corresponding to WT and deleted S1P1 because the deletion occurs only in ECs. B. Mouse plasmas from ApoM-/- mice (lanes 1-4) confirmed the deletion of ApoM compared to WT plasmas (lanes 5-8).
A. W146 decreased HUVEC resistance ECIS in dose dependent manner (0.1 -10 μM, n=3). B. CYM-5442 increase HUVEC resistance on ECIS in a dose dependent manner (10-200 nM, n=3). C. Mice were injected with 0.5% EB IV and then injected immediately afterward with IP CYM-5442 0.5mg/kg, CYM-5442 10 mg/kg, or W146 10 mg/kg.
Intradermal W146 alone did not induce vascular leak but amplified leak in response to IC vascular injury. W146 (10 μg in 30 μl PBS) was administered intradermally either alone (bottom half of skin sample) or concomitantly with anti-ova IgG (60 ug/30 ul, top half) followed by IV injection of Evans blue (0.5% in 150 μl PBS) and ovalbumin (400μg). 4 hrs post injection skin was assessed for EB extravasation.
SEW 2871 decreases lung RAR. Lung injury after RAR was quantified by assessment of PMN (A) and RBC (B) counts in BAL fluid after 24 hrs (n=4 mice per group, *p=0.01 and *p=0.03, respectively). Lung weights (mg) after RAR are shown in C, n=4, p=ns).
CYM-5442 diminishes p-MLC and preserves VE-cadherin staining in IC and C5a activated HUVECs. HUVECs were treated for 30 min with IC and C5a (100 ng/ml) activated PMN (1X105) and then fixed and solubilized prior to staining with anti-pMLC (in red) and anti-VE-Cadherin (in green). Nuclei were stained with DAPI. 1st row: HUVEC with unstimulated PMN; 2nd row HUVEC with CYM-5442 (no PMNs); 3rd row HUVEC with activated PMN; 4th row HUVEC pretreated with CYM-5442 prior to treatment with activated PMN.