

Keywords: colitis, hypoxia, hypoxia-inducible factor-2α
Abstract
Hypoxia is a notable feature of inflammatory bowel disease and chronic induction of hypoxia-inducible factor (HIF)-1α and HIF-2α (endothelial PAS domain protein 1, EPAS1) play important, but opposing, roles in its pathogenesis. While activation of HIF-1α decreases intestinal inflammation and is beneficial in colitis, activation of HIF-2α exacerbates colitis and increases colon carcinogenesis in animal models, primarily due to the role of epithelial HIF-2α in mounting a potent inflammatory response. Previous work from our laboratory showed that mice overexpressing intestinal epithelial HIF-2α led to massive intestinal inflammation and decreased survival. As oxygen homeostasis and HIFs are critical in embryonic development, it is not clear whether the observed intestinal inflammatory response was secondary to developmental defects. To address this question, the present study used a mouse model to temporally modulate expression of intestinal epithelial HIF-2α to assess its role in mediating inflammatory response. Remarkably, activation of HIF-2α in intestinal epithelial cells in adult mice increased expression of proinflammatory mediators; however, no decrease in survival was observed. Furthermore, in an acute model of colitis, activation of HIF-2α was sufficient to exacerbate colitis. These data confirm our previous finding that epithelial HIF-2α mediates inflammatory response and demonstrates that activation of HIF-2α is sufficient to exacerbate colitis.
NEW & NOTEWORTHY Inflammatory bowel disease (IBD) is a chronic relapsing inflammatory disease of the intestinal tract. Hypoxia and activation of its downstream transcription factors hypoxia-inducible factor (HIF)-1α and HIF-2α are notable features of IBD. HIF-1α has well-characterized protective roles in IBD; however, the role of HIF-2α has been less studied. Using novel HIF-2α mouse models, we show that activation of HIF-2α in intestinal epithelial cells is sufficient to exacerbate colitis.
INTRODUCTION
Inflammatory bowel disease (IBD) affects more than 70,000 patients in the United States every year, and ~1.6 million Americans currently have IBD (5, 24). IBD comprises two major subgroups: ulcerative colitis and Crohn’s disease (13, 23). Although the etiology of IBD is not completely understood, a complex interplay of decreased intestinal epithelial barrier integrity, dysregulated mucosal immune response, and gut microbial dysbiosis contribute to IBD pathogenesis (7, 12, 16, 22, 25, 32). These processes ultimately lead to chronic inflammatory response and subsequent tissue injury (15, 35). Hypoxia is a hallmark of the inflammatory foci in IBD (12, 32, 38). Previous studies have shown a robust induction of hypoxia along the intestinal mucosa in a mouse model of colitis, further suggesting that hypoxia participates in the pathogenesis of colitis (17, 33).
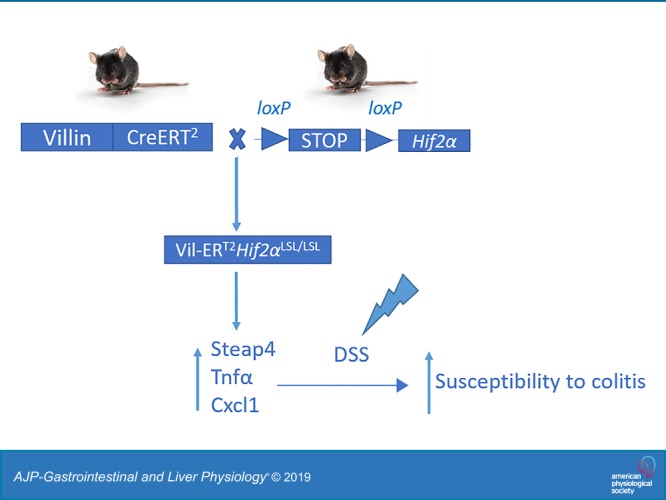
Hypoxic responses are primarily mediated by hypoxia-inducible factor (HIF)-1α and (HIF)-2α. HIF-1α is ubiquitously expressed, while HIF-2α expression is more restricted (31). HIF-1α and HIF-2α share extensive homology in structure and heterodimerize with HIF-1β (ARNT) to execute their transcriptional activities. Although HIF-1α and HIF-2α bind to the same canonical hypoxia-responsive elements (HREs), numerous studies have shown that HIF-1α and HIF-2α have distinct and nonredundant functions (9, 32). Activation of intestinal epithelial HIF-1α regulates genes involved in maintaining the epithelial barrier function and decreases the expression of proinflammatory mediators (1, 17, 19, 20). Moreover, stable expression of intestinal epithelial HIF-1α or pharmacological activation of HIFs by prolyl hydroxylase domain enzymes (PHDs) inhibitors is protective in experimental models of acute colitis (6, 19, 27, 42). The latter protective effects were mainly attributed to the activation of HIF-1α in these models (17, 19). However, activation of intestinal epithelial HIF-2α induces a potent epithelial proinflammatory response by regulating the expression of inflammatory cytokines and chemokines (33, 37, 41). Accordingly, mice with intestinal epithelial HIF-2α deficiency exhibit attenuated inflammatory responses (41). These findings suggest that in setting of colitis, HIF-1α and HIF-2α play important, but distinct, roles in resolution and progression of inflammatory responses, respectively. HIF-2α expression was further shown to be important in regulating the barrier function (11, 39). As both HIF-1α and HIF-2α impact colonic inflammatory responses, a critical factor in colon cancer, their activation was shown to distinctly impact colon cancer development. While HIF-1α had no effect on tumorigenesis (42), activation of HIF-2α resulted in increased tumor burden in mouse models of colon cancer (44). These findings suggested that targeting HIF-2α might be optimal, not only in colitis, but also in colitis-associated colon cancer. One interesting finding from mice with constitutive overexpression of HIF-2α is that these mice fail to survive past 6 wk of age as a result of massive intestinal inflammation. These observations hinted a possible role of HIF-2α in embryonic intestinal development, which could have impacted the observed inflammatory phenotype. Thus, to address this question, we generated a mouse model with a tamoxifen-inducible expression of an oxygen-stable HIF-2α, specifically in the intestinal epithelial cells. Temporal activation of intestinal HIF-2α did not lead to any basal intestinal injury; however, it is sufficient to increase the expression of proinflammatory mediators, six-transmembrane epithelial antigen of prostate 4 (Steap4), tumor necrosis factor-α (Tnf-α), and chemokine (C-X-C motif) ligand 1 (Cxcl1). Importantly, temporal activation of intestinal HIF-2α did not result in spontaneous colitis, as previously reported in mice with constitutive embryonic overexpression of HIF-2α. However, these mice were highly susceptible to inflammatory injury in a colitis model. These findings confirm the proinflammatory role of intestinal epithelial HIF-2α and support the notion that intestinal epithelial HIF-2α expression exacerbates colitis.
METHODS
Animals and treatment conditions.
All animal studies were carried out in accordance with Institute of Laboratory Animal Resources guidelines and approved by the University Committee on the Use and Care of Animals at the University of Michigan (IACUC protocol number: PRO00008292). Hif2α LSL/LSL mice, as previously described (41), were crossed with Vil-ERT2 Cre to make Vil-ERT2 Hif2α LSL/LSL mice. All mice are on C57BL/6 background, maintained in standard cages in a light- and temperature-controlled room, and were allowed a standard chow diet and water ad libitum. For basal characterization, 6-wk-old Vil-ERT2 Hif2α LSL/LSL and littermate controls were injected with tamoxifen in corn oil (100 mg/kg) for three consecutive days and were euthanized 1 wk or 4 wk following the last tamoxifen treatments. For dextran sulfate sodium (DSS) studies, Hif2α+/+ and Vil-ERT2 Hif2α LSL/LSL mice were injected with tamoxifen in corn oil (100 mg/kg), respectively, for three consecutive days, and a week later were placed on 2.5% DSS in drinking water for 5 days. For RNA-sequencing (RNA-seq) studies, Hif2αΔIE and age-matched wild-type littermates (Hif2αFlx/Flx) were subjected to 2.5% DSS in drinking water for 5 days, and colonic mucosal cells were scraped.
Protein isolation and immunoblotting.
Colonic mucosal cells were scraped and lysed in radioimmunoprecipitation assay buffer with added protease (1:100 dilution; Sigma) and phosphatase (1:100 dilution; ThermoFisher Scientific) inhibitors using TissueLyser II (Qiagen) to obtain whole cell extracts, and immunoblotting was performed as previously described (41, 42, 44). Briefly, after cell lysis, solubilized proteins were resolved on 10% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membrane, blocked with 3% dry milk, and were immunoblotted with the indicated primary antibodies for HIF-2α (Novus Biologicals) and actin (Proteintech).
RNA isolation and quantitative reverse transcription-PCR.
Total RNA was isolated using phenol-chloroform extraction method, reverse transcribed using M-MLV reverse transcriptase (ThermoFisher Scientific), and was measured by real-time RT-PCR using SYBR Green (Life Technologies), as previously described (41, 42, 44). The primers used for quantitative PCR are listed in Table 1.
Table 1.
Primers for quantitative PCR
| Forward Primer | Reverse Primer | |
|---|---|---|
| Cxcl1 | TCTCCGTTACTTGGGGACAC | CCACACTCAAGAATGGTCGC |
| Dmt1 | TGTTTGATTGCATTGGGTCTG | CGCTCAGCAGGACTTTCGAG |
| DcytB | CATCCTCGCCATCATCTC | GGCATTGCCTCCATTTAGCTG |
| Fpn | ATGGGAACTGTGGCCTTCAC | TCCAGGCATGAATACGGAGA |
| Glut1 | CAAGTCTGCATTGCCCATGAT | CCAGCTGGGAATCGTCGTT |
| Gapdh | TTGATGGCAACAATCTCCAC | CGTCCCGTAGACAAAATGGT |
| Hif2α | CAGAGCTGAGGAAGGAGAAATC | TCATGAGCCAACTCATAGAAGAC |
| Pgk1 | CAAATTTGATGAGAATGCCAAGACT | TTCTTGCTGCTCTCAGTACCACA |
| Steap4 | GGAAACTCATCTGCATGTGCT | CTAGAAGGCAGAGCCCACC |
| Tnf-α | AGGGTCTGGGCCATAGAACT | CCACCACGCTCTTCTGTCTAC |
| Vegf | CCACGTCAGAGAGCAACATCA | TCATTCTCTCTATGTGCTGGCTTT |
Histology and immunofluorescence staining.
Duodenum and colonic tissues were rolled and fixed with PBS-buffered formalin. Hematoxylin-and-eosin analysis was performed in paraffin-embedded tissue sections (5 µm) as described by Xue et al. (41, 42). For immunofluorescence staining, paraffin-embedded tissue sections were subjected to antigen retrieval, followed by blocking with 5% goat serum and probed with primary antibody against Ki67 (1:200 dilution; D3B5; Cell Signaling Technology). Sections were then washed three times with PBST and were incubated with anti-rabbit IgG Alexa Fluor-488 (1:500 dilution) for 1 h (ThermoFisher Scientific). Sections were then washed three times with PBST, and slides were mounted using the Prolong Gold Antifade Reagent with DAPI (ThermoFisher Scientific).
RNA-sequencing analysis.
RNA sequencing was done on the Illumina HiSeq 4000 platform using single-end 50-cycle reads, and analysis was done, as previously described (44). RNA sequencing quality was assessed using FastQC. Reads were aligned to the mouse genome (mm10) using STAR (8). Options out FilterMultimapNmax 10 and sjdbScore 2 were used. Annotation of the mm10 library was completed using the University of California-Santa Cruz annotation Gene Transfer Format (GTF) (mm10.fa) (18). Differential expression was tested first between Hif2αFlx/Flx water versus DSS and Hif2αΔIE water versus DSS. Genes were considered differentially expressed at a P value of <0.05.
The full data set of upregulated and downregulated genes in Hif2αΔIE and Hif2αFlx/Flx is available on Figshare (https://doi.org/10.6084/m9.figshare.8309660). A select number of genes were verified by using the Qiagen RT2 profiler PCR array.
Statistics.
Results are expressed as means ± SE. P values were calculated by independent t-test and one-way ANOVA. P < 0.05 was considered significant.
RESULTS
Generation of mice with temporal overexpression of HIF-2α in intestinal epithelial cells (Vil-ERT2Hif2αLSL/LSL).
To examine the effects of temporal activation of HIF-2α on intestinal homeostasis, we generated Vil-ERT2Hif2αLSL/LSL mice by crossing Vil-ERT2 mice with Hif2α LSL/LSL that harbor an oxygen-stable HIF-2α flanked by loxP-Stop-loxP cassette (Fig. 1A). Tamoxifen injection results in a robust induction of HIF-2α specifically in the intestinal epithelial cells of Vil-ERT2Hif2αLSL/LSL mice (Fig. 1B). Gene expression analysis confirmed the induction of HIF-2α target genes, such as divalent metal transporter 1 (Dmt1; also known as Slc11a2), duodenal cytochrome b (DcytB), and ferroportin [Fpn; also, known as Slc40a1 (34, 36)] in the small intestine (Fig. 1C) and colon (Fig. 1D) of Vil-ERT2Hif2αLSL/LSL mice compared with their littermate controls. However, the expression levels of HIF-1α target genes, such as vascular endothelial growth factor (Vegf), glucose transporter 1 (Glut1), and phosphoglycerate kinase 1 (Pgk1) (42) were unaltered in the small intestine or colon of Vil-ERT2Hif2αLSL/LSL compared with littermate controls (not shown). These results provided the basis to assess the utility of this model in studying the impact of temporal activation of HIF-2α in intestinal homeostasis.
Fig. 1.

Temporal model of intestinal hypoxia-inducible factor-2α (HIF-2α) activation. A: schematic of a temporal HIF-2α-overexpressing mouse model (Vil-ERT2Hif2αLSL/LSL). B: Western blot analysis of HIF-2α from scraped intestinal mucosal cells of Vil-ERT2Hif2αLSL/LSL or littermate control Hif-2α+/+ mice. Quantitative PCR analysis for the expression of divalent metal transporter 1 (Dmt1), duodenal cytochrome b (DcytB), ferroportin (Fpn) in scraped mucosal cells from the small intestine (C) and colon (D) of Vil-ERT2Hif2αLSL/LSL or littermate control, Hif-2α+/+ mice. *P < 0.05, **P < 0.01, ***P < 0.001 for the difference between Vil-ERT2Hif2αLSL/LSL compared with littermate Hif-2α+/+ mice. Four mice were assessed from each group.
Temporal activation of intestinal HIF-2α does not lead to histological changes at steady state.
Previously, we demonstrated that mice with constitutive overexpression of HIF-2α did not survive past 6 wk of age because of massive intestinal inflammation (41). However, Vil-ERT2Hif2αLSL/LSL mice were healthy and survived normally (not shown). Furthermore, histological analysis of Vil-ERT2Hif2αLSL/LSL mice at 1 wk following tamoxifen treatments did not reveal any major differences in the small intestine (Fig. 2A) and colon (Fig. 2B), when compared with their littermate controls. In addition, proliferation in the small intestine (Fig. 2C) and colon (Fig. 2D) were not different between the groups, as assessed by Ki67 staining. We also performed histological analysis of Vil-ERT2Hif2αLSL/LSL mice at 4 wk following tamoxifen treatments and found no major differences in the small intestine (Fig. 2E) and colon (Fig. 2F). Similar to our 1-wk time frame of HIF-2α induction, the proliferation in the small intestine (Fig. 2G) and colon (Fig. 2H) was not different between the groups. Altogether, these data indicate that induction of intestinal HIF-2α either at 7 days or a month does not lead to histological changes in the intestine.
Fig. 2.

Temporal activation of intestinal hypoxia-inducible factor-2α (HIF-2α) does not lead to any basal histological changes. Representative hematoxylin-and-eosin staining of small intestine (duodenum) (A, E) and colon (B, F) from littermate control Hif2α+/+ and Vil-ERT2Hif2αLSL/LSL mice at 1 wk and 4 wk, respectively. Representative Ki67 staining and quantitation of small intestine (C, G) and colon (D, H) from Hif2α+/+ and Vil-ERT2Hif2αLSL/LSL mice at 1 wk and 4 wk, respectively. Four mice were assessed from each group.
HIF-2α activates proinflammatory targets in the intestine.
Previously, we have characterized the inflammatory genes, such as six-transmembrane epithelial antigen of prostate 4 (Steap4), tumor necrosis factor-α (Tnf-α), and chemokine (C-X-C motif) ligand 1 (Cxcl1) as HIF-2α targets (37, 40, 41). Steap4 has been shown to be highly induced in mouse models of colitis and IBD patients and was regulated by HIF-2α (40). Moreover, HIF-2α has been shown to regulate the Cxcl1 expression through the HRE- and Myc-associated zinc finger (MAZ)-dependent mechanisms, which, in turn, facilitated the progression of colon cancer (37). Thus, we determined the expression levels of these HIF-2α targets in our inducible model and found that these were robustly increased in the small intestine (Fig. 3A) and colon (Fig. 3B) of Vil-ERT2Hif2αLSL/LSL mice. These findings indicated that the temporal activation of intestinal HIF-2α increases expression of proinflammatory mediators, but were not sufficient to cause any basal intestinal injury.
Fig. 3.

Temporal activation of intestinal hypoxia-inducible factor-2α (HIF-2α) activates proinflammatory targets in the intestine. Quantitative PCR analysis of Steap4, Tnf-α, and Cxcl1 in the small intestine (A) and colon (B) from Vil-ERT2Hif2αLSL/LSL or Hif2α+/+ mice. *P < 0.05, **P < 0.01, ***P < 0.001 for the difference between Vil-ERT2Hif2αLSL/LSL and littermate control Hif2α+/+ mice. Four mice were assessed from each group.
Temporal activation of intestinal HIF-2α exacerbates dextran sulfate sodium -induced colitis.
To further characterize the role of temporally induced HIF-2α activation in colitis, an acute model of colitis was assessed using DSS. Vil-ERT2Hif2αLSL/LSL were injected with tamoxifen to temporally induce HIF-2α and then subjected to 2.5% dextran sodium sulfate (DSS) treatment for a period of 5 days (Fig. 4A). Colon length was significantly reduced in Vil-ERT2Hif2αLSL/LSL mice as compared with their littermate controls (Fig. 4B). Histological analysis revealed severe tissue damage with massive loss of crypt architecture, and inflammatory cell infiltrations in the Vil-ERT2Hif2αLSL/LSL mice, while littermate mice showed some crypt damage, erosion, and moderate inflammation as expected under DSS conditions (Fig. 4C). We also assessed the proliferation by Ki67 staining but did not find any differences between the groups (Fig. 4D). Finally, we observed a significant increase in the expression of inflammatory mediators Steap4 and Tnf-α (Fig. 4E). These findings suggest that the temporal activation of HIF-2α is sufficient to exacerbate the development of DSS-induced colitis and confirm our previous findings (41) that intestinal HIF-2α drives experimental colitis.
Fig. 4.

Temporal activation of intestinal hypoxia-inducible factor-2α (HIF-2α) exacerbates colitis. A: schematic of DSS-induced colitis model. Colon length (n = 6; B), representative hematoxylin-and-eosin staining of colons and inflammation score from Hif2α+/+ and Vil-ERT2Hif2αLSL/LSL mice (n = 6; C), Ki67 staining and quantitation of colon from Hif2α+/+ and Vil-ERT2Hif2αLSL/LSL mice (n = 4; D), and qPCR analysis for the expression of Steap4 and Tnf-α in scraped colonic mucosal cells from Vil-ERT2Hif2αLSL/LSL or Hif2α+/+ mice (n = 5; E). *P < 0.05, **P < 0.01 for the differences between Vil-ERT2Hif2αLSL/LSL compared with littermate control Hif2α+/+ mice.
Whole genome mRNA analysis of the colons of Hif2αΔIE mice.
Previous work has shown that activation of HIF-1α and/or HIF-2α is essential and sufficient to activate glycolytic, angiogenic, and iron-absorptive HIF target genes (26, 30, 31). Our work has shown that HIF-2α is sufficient to activate a number of proinflammatory genes (37, 40, 41), but it is not clear whether HIF-2α drives the expression of proinflammatory genes following injury. To better assess gene expression changes impacted by HIF-2α in the colitis model, mice with intestinal epithelium-specific knockout of HIF-2α (Hif2αΔIE) and age-matched wild-type littermates were subjected to 2.5% DSS treatment for a period of 5 days, and whole genome mRNA analysis (RNA-seq) was performed on scraped colonic mucosal cells from both groups. We confirmed the expression levels of HIF-2α to be significantly reduced in Hif2αΔIE mice (Fig. 5A). First, we examined known inflammatory HIF-2α target genes and found expression levels of Steap4, Tnf-a, and Cxcl1 to be significantly induced following DSS treatments; however, only Steap4 (Fig. 5B) and Cxcl1 (Fig. 5C), but not Tnf-α (Fig. 5D), levels were reduced in the Hif2αΔIE mice. These data indicated that HIF-2α is essential in Steap4 and Cxcl1 expression; however, it is dispensable in regulating Tnf-α expression in DSS-induced colitis model. To further identify the essential inflammatory genes either upregulated or downregulated, RNA-seq analyses of littermate controls and Hif2αΔIE mice on DSS and regular drinking were assessed. We observed that 654 genes were significantly upregulated and 847 genes were significantly downregulated in Hif2αΔIE compared with Hif2αFlx/Flx mice following DSS (Supplemental Table S1 and S2 via Figshare: doi.or/10.6084/m9.figshare.8309660). As expected, inflammatory gene signatures were among the most significant pathway altered following HIF-2α disruption, corroborating the role of HIF-2α as a driver of inflammatory mediators (Fig. 5E). Interestingly, expression of numerous oncogenes like Yap1, Pi3kca, Met, Cdkn1b, and Wnt8b was found to be significantly decreased in Hif2αΔIE compared with Hif2α Flx/Flx mice following DSS (Fig. 5E). On the basis of differential transcripts between Hif2αFlx/Flx and Hif2αΔIE following DSS treatment, we examined enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. The top 15 KEGG pathways altered demonstrated a significant association with inflammatory and oncogenic signaling (Fig. 5F). Finally, inflammatory genes that were significantly downregulated in Hif2αΔIE following DSS treatment were assessed in the Vil-ERT2Hif2αLSL/LSL mice. Several inflammatory genes and pathways were significantly increased in the Vil-ERT2Hif2αLSL/LSL assessed by qPCR array analysis (Fig. 5, G and H). These observations suggest HIF-2α drives colon cancer development, not only by modulating iron metabolism (21, 43, 44), but also by regulating oncogenic signaling pathways.
Fig. 5.

RNA-Seq analysis in the colons from Hif2αFlx/Flx and Hif2αΔIE mice. A: expression of HIF-2α in Hif2αFlx/Flx and Hif2αΔIE mice. Quantitative PCR analysis for the expression of Steap4 (B), Cxcl1 (C), and Tnf-α (D) in scraped colonic mucosal cells from Hif2αFlx/Flx and Hif2αΔIE mice. E: heat map generated from RNA-seq data on scrapped colonic mucosal cells isolated from Hif2αFlx/Flx and Hif2αΔIE mice. F: list of top 15 KEGG pathways altered in Hif2αFlx/Flx and Hif2αΔIE mice. G: qPCR array analysis from scraped colonic mucosal cells from Vil-ERT2Hif2αLSL/LSL and Hif2α+/+. H: list of top 10 KEGG pathways with respective altered genes in Hif2α+/+ and Vil-ERT2Hif2αLSL/LSL mice. Three mice were assessed for each group. *P < 0.05, **P < 0.01, ***P < 0.001 for the differences between Hif2αΔIE or Vil-ERT2Hif2αLSL/LSL mice compared with littermate control mice.
DISCUSSION
Hypoxia has emerged as a key feature of many diseases, including IBD, and numerous studies have investigated the role of HIFs in affecting the outcome of these pathologies (29). In models of colitis, activation of HIF-1α has been shown to improve barrier function, limit the expression of proinflammatory cytokines, and protect against colitis (17, 20), while activation of HIF-2α increases the expression of proinflammatory mediators, promoting colitis (41). These studies indicated a complex role of HIF-1α and HIF-2α in affecting the outcome of colitis. These differences in their specificities could be attributed to varying degree of tissue expression, differential coactivators, transcriptional factors, and protein-interactome of HIFα subunits, thereby affecting their function (14, 26, 31). A few notable observations of constitutive HIF-2α-overexpressing mice were that they were smaller in size, body weights were significantly lower, and mice died at less than 6 wk of age, as compared with wild-type littermates (41). Moreover, histological analysis revealed massive intestinal inflammation in these mice (41). The question then arose about whether the observed phenotype of severe colitis in HIF-2α-overexpressing mice is due to a specific effect of epithelial HIF-2α in mounting a potent inflammatory response or due to other roles of HIF-2α in embryonic development, thereby impacting colitis. To answer this question, we used a mouse model, whereby intestinal HIF-2α levels could be temporally modulated (Vil-ERT2Hif2αLSL/LSL). This model allowed us to dissect the role of epithelial HIF-2α expression in colitis without interfering with HIF-2α function in embryonic development. Vil-ERT2Hif2αLSL/LSL mice did not impact HIF-1α target genes, further allowing us to use this model to specifically evaluate role of HIF-2α in colitis. Remarkably, temporal induction of intestinal epithelial-specific HIF-2α in Vil-ERT2Hif2αLSL/LSL mice did not result in spontaneous colitis. Vil-ERT2Hif2αLSL/LSL mice were healthy and survived normally compared with littermate controls. These findings indicate that overexpressing HIF-2α during embryonic development results in spontaneous colitis and increased lethality. Interestingly, in mice with an intestine-specific disruption of HIF-2α, the intestines are indistinguishable from wild-type littermates. Together, these data suggest that HIF-2α is not essential in intestinal development; however, pathological induction during development may have severe consequences. Currently, we are further assessing the role of HIF-2α in intestinal development. Temporal induction of intestinal epithelium-specific HIF-2α in Vil-ERT2Hif2αLSL/LSL mice did not lead to any overt histological or gross changes in the mice. However, this was sufficient to mount an inflammatory response by increasing mediators like Steap4, Tnf-α, and Cxcl1. These data are in line with previous studies and corroborate that Steap4, Tnf-α, and Cxcl1 are direct targets of HIF-2α (37, 40, 41). These findings suggest that the activation of intestinal epithelial-specific HIF-2α is sufficient to drive basal inflammation but not sufficient to drive experimental colitis. Recently, we reported that intestinal overexpression of Steap4 aggravated experimental colitis by dysregulating mitochondrial iron homeostasis (40). Interestingly, spontaneous colitis did not occur in mice with intestinal epithelium-specific overexpression of Steap4, indicating that dysregulating mitochondrial iron homeostasis alone does not promote colitis. These observations are in line with current findings and further reflect the complexity of IBD as a multihit, multifactorial disease. Although the parameters of intestinal mitochondrial iron and oxidative stress remained to be examined in the current model, it is plausible that Vil-ERT2Hif2αLSL/LSL mice may have dysregulated mitochondrial iron homeostasis via induction of Steap4. Using DSS-induced colitis model, we found that Vil-ERT2Hif2αLSL/LSL mice were highly susceptible to injury, as evidenced by heightened inflammatory response and decreased colon length, suggesting that the activation of intestinal HIF-2α is sufficient to exacerbate experimental colitis under these settings. DSS is known to damage the colonic epithelial monolayer, allowing the dissemination of bacteria into underlying tissue (3). Thus, activation of intestinal HIF-2α drives inflammatory response, and together, in the setting of DSS, exacerbates colitis. Considering the fact that a myriad of cellular processes may be affected, directly or indirectly, by HIF-2α through its transcriptional or nontranscriptional mechanisms in colitis, we performed RNA sequencing to obtain unbiased information on global gene signatures in mice with intestinal epithelium-specific knockout of HIF-2α, (Hif2αΔIE) mice. On assessing global gene expression changes by HIF-2α deficiency, our RNA-sequencing data demonstrated significant changes in numerous previously unknown genes. Although the significance of these changes will require additional work, these findings uncovered underappreciated regulation of genes by HIF-2α. Moreover, canonical HIF-2α targets were found to be significantly decreased in Hif2αΔIE mice, including Dmt1 and Steap4. Interestingly, inflammatory mediators, including IL-1β, chemokines like CXCR6 were found to be increased in Hif2αΔIE mice. This may point to a compensatory upregulation of these mediators impacted by HIF-2α or differential regulation of these mediators. To investigate the biological processes and cellular pathways affected by loss of HIF-2α, we evaluated the differentially expressed genes by KEGG analysis. The KEGG analysis showed enrichment in biological processes associated with signal transduction and regulation of several oncogenes. Although our previous studies focused on the effects of HIF-2α in inflammatory responses and iron regulation in driving colitis (41) and colorectal cancer (43, 44), these signaling pathways impacted by HIF-2α suggest that these are also likely to contribute to these phenotypes. Recent studies from our laboratory showed that HIF-2α potentiated Yes-associated protein 1 (Yap1) expression and activity, and it increased colon cancer (21). Moreover, the role of Pi3kca, Wnt8b, and mTORC1 signaling pathways driving colorectal cancer are well documented (2, 10, 28). These suggest that HIF-2α not only regulates inflammatory processes but also impacts cellular transduction pathways driving cell growth. The present data provide strong evidence that activation of intestinal HIF-2α is sufficient to induce inflammatory response and exacerbate experimental colitis. These findings also provide a unique resource of information for future studies aimed at identifying signaling pathways affected by HIF-2α in driving colorectal cancer. Previously thought to be an undruggable target, several potential inhibitors of HIF-2α have been characterized and are in clinical trials for renal carcinoma (4, 45). Our data suggest that pharmacological inhibition of HIF-2α may be protective in colitis and colitis-associated colon cancers.
GRANTS
This work was supported by the National Institutes of Health, R01CA148828 and R01DK095201 to Y. M. Shah, K99DK110537 to S. K. R Ramakrishnan, T32GM007315 and T32CA140044 to S. N. Devenport by and the American Heart Association (19POST34380588) to S. Solanki.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.S. and Y.M.S. conceived and designed research; S.S., S.K.R., and Y.M.S. performed experiments; S.S., S.N.D., S.K.R., and Y.M.S. analyzed data; S.S., S.N.D., S.K.R., and Y.M.S. interpreted results of experiments; S.S., S.N.D., and Y.M.S. prepared figures; S.S. and Y.M.S. drafted manuscript; S.S. and Y.M.S. edited and revised manuscript; Y.M.S. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address of S. K. Ramakrishnan: Dept. of Medicine, Division of Endocrinology and Metabolism, University of Pittsburgh, Pittsburgh, PA 15261.
REFERENCES
- 1.Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, Bayless AJ, Scully M, Saeedi BJ, Golden-Mason L, Ehrentraut SF, Curtis VF, Burgess A, Garvey JF, Sorensen A, Nemenoff R, Jedlicka P, Taylor CT, Kominsky DJ, Colgan SP. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity 40: 66–77, 2014. doi: 10.1016/j.immuni.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cathomas G. PIK3CA in colorectal cancer. Front Oncol 4: 35, 2014. doi: 10.3389/fonc.2014.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr Protoc Immunol 104: 25, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, Homayoun F, Ma Y, Patel N, Yell P, Hao G, Yousuf Q, Joyce A, Pedrosa I, Geiger H, Zhang H, Chang J, Gardner KH, Bruick RK, Reeves C, Hwang TH, Courtney K, Frenkel E, Sun X, Zojwalla N, Wong T, Rizzi JP, Wallace EM, Josey JA, Xie Y, Xie XJ, Kapur P, McKay RM, Brugarolas J. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 539: 112–117, 2016. doi: 10.1038/nature19796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colombel JF, Mahadevan U. Inflammatory Bowel Disease 2017: innovations and changing paradigms. Gastroenterology 152: 309–312, 2017. doi: 10.1053/j.gastro.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 6.Cummins EP, Seeballuck F, Keely SJ, Mangan NE, Callanan JJ, Fallon PG, Taylor CT. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology 134: 156–165.e1, 2008. doi: 10.1053/j.gastro.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 7.de Mattos BR, Garcia MP, Nogueira JB, Paiatto LN, Albuquerque CG, Souza CL, Fernandes LG, Tamashiro WM, Simioni PU. Inflammatory bowel disease: an overview of immune mechanisms and biological treatments. Mediators Inflamm 2015: 493012, 2015. doi: 10.1155/2015/493012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21, 2013. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flück K, Fandrey J. Oxygen sensing in intestinal mucosal inflammation. Pflugers Arch 468: 77–84, 2016. doi: 10.1007/s00424-015-1722-4. [DOI] [PubMed] [Google Scholar]
- 10.Francipane MG, Lagasse E. mTOR pathway in colorectal cancer: an update. Oncotarget 5: 49–66, 2014. doi: 10.18632/oncotarget.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glover LE, Bowers BE, Saeedi B, Ehrentraut SF, Campbell EL, Bayless AJ, Dobrinskikh E, Kendrick AA, Kelly CJ, Burgess A, Miller L, Kominsky DJ, Jedlicka P, Colgan SP. Control of creatine metabolism by HIF is an endogenous mechanism of barrier regulation in colitis. Proc Natl Acad Sci USA 110: 19820–19825, 2013. doi: 10.1073/pnas.1302840110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glover LE, Colgan SP. Hypoxia and metabolic factors that influence inflammatory bowel disease pathogenesis. Gastroenterology 140: 1748–1755, 2011. doi: 10.1053/j.gastro.2011.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gohil K, Carramusa B. Ulcerative colitis and Crohn’s disease. P T 39: 576–577, 2014. [PMC free article] [PubMed] [Google Scholar]
- 14.Greer SN, Metcalf JL, Wang Y, Ohh M. The updated biology of hypoxia-inducible factor. EMBO J 31: 2448–2460, 2012. doi: 10.1038/emboj.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guan Q, Zhang J. Recent advances: the imbalance of cytokines in the pathogenesis of inflammatory bowel disease. Mediators Inflamm 2017: 4810258, 2017. doi: 10.1155/2017/4810258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang Y, Chen Z. Inflammatory bowel disease related innate immunity and adaptive immunity. Am J Transl Res 8: 2490–2497, 2016. [PMC free article] [PubMed] [Google Scholar]
- 17.Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest 114: 1098–1106, 2004. doi: 10.1172/JCI200421086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karolchik D, Hinrichs AS, Kent WJ. The UCSC Genome Browser. Curr Protoc Bioinformatics Unit 1: 4, 2009. doi: 10.1002/0471250953.bi0104s17. [DOI] [PubMed] [Google Scholar]
- 19.Keely S, Campbell EL, Baird AW, Hansbro PM, Shalwitz RA, Kotsakis A, McNamee EN, Eltzschig HK, Kominsky DJ, Colgan SP. Contribution of epithelial innate immunity to systemic protection afforded by prolyl hydroxylase inhibition in murine colitis. Mucosal Immunol 7: 114–123, 2014. doi: 10.1038/mi.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Louis NA, Hamilton KE, Canny G, Shekels LL, Ho SB, Colgan SP. Selective induction of mucin-3 by hypoxia in intestinal epithelia. J Cell Biochem 99: 1616–1627, 2006. doi: 10.1002/jcb.20947. [DOI] [PubMed] [Google Scholar]
- 21.Ma X, Zhang H, Xue X, Shah YM. Hypoxia-inducible factor 2α (HIF-2α) promotes colon cancer growth by potentiating Yes-associated protein 1 (YAP1) activity. J Biol Chem 292: 17046–17056, 2017. doi: 10.1074/jbc.M117.805655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McIlroy J, Ianiro G, Mukhopadhya I, Hansen R, Hold GL. Review article: the gut microbiome in inflammatory bowel disease-avenues for microbial management. Aliment Pharmacol Ther 47: 26–42, 2018. doi: 10.1111/apt.14384. [DOI] [PubMed] [Google Scholar]
- 23.Mulder DJ, Noble AJ, Justinich CJ, Duffin JM. A tale of two diseases: the history of inflammatory bowel disease. J Crohn’s Colitis 8: 341–348, 2014. doi: 10.1016/j.crohns.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 24.Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, Panaccione R, Ghosh S, Wu JCY, Chan FKL, Sung JJY, Kaplan GG. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet 390: 2769–2778, 2018. doi: 10.1016/S0140-6736(17)32448-0. [DOI] [PubMed] [Google Scholar]
- 25.Nishida A, Inoue R, Inatomi O, Bamba S, Naito Y, Andoh A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin J Gastroenterol 11: 1–10, 2018. doi: 10.1007/s12328-017-0813-5. [DOI] [PubMed] [Google Scholar]
- 26.Patel SA, Simon MC. Biology of hypoxia-inducible factor-2α in development and disease. Cell Death Differ 15: 628–634, 2008. doi: 10.1038/cdd.2008.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology 134: 145–155, 2008. doi: 10.1053/j.gastro.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saitoh T, Mine T, Katoh M. Up-regulation of WNT8B mRNA in human gastric cancer. Int J Oncol 20: 343–348, 2002. doi: 10.3892/ijo.20.2.343. [DOI] [PubMed] [Google Scholar]
- 29.Schito L, Semenza GL. Hypoxia-inducible factors: master regulators of cancer progression. Trends Cancer 2: 758–770, 2016. doi: 10.1016/j.trecan.2016.10.016. [DOI] [PubMed] [Google Scholar]
- 30.Schwartz AJ, Das NK, Ramakrishnan SK, Jain C, Jurkovic MT, Wu J, Nemeth E, Lakhal-Littleton S, Colacino JA, Shah YM. Hepatic hepcidin/intestinal HIF-2α axis maintains iron absorption during iron deficiency and overload. J Clin Invest 129: 336–348, 2019. doi: 10.1172/JCI122359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell 148: 399–408, 2012. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shah YM. The role of hypoxia in intestinal inflammation. Mol Cell Pediatr 3: 1, 2016. doi: 10.1186/s40348-016-0030-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shah YM, Ito S, Morimura K, Chen C, Yim SH, Haase VH, Gonzalez FJ. Hypoxia-inducible factor augments experimental colitis through an MIF-dependent inflammatory signaling cascade. Gastroenterology 134: 2036–2048, 2008. doi: 10.1053/j.gastro.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shah YM, Matsubara T, Ito S, Yim SH, Gonzalez FJ. Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab 9: 152–164, 2009. doi: 10.1016/j.cmet.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology 140: 1756–1767, 2011. doi: 10.1053/j.gastro.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor M, Qu A, Anderson ER, Matsubara T, Martin A, Gonzalez FJ, Shah YM. Hypoxia-inducible factor-2α mediates the adaptive increase of intestinal ferroportin during iron deficiency in mice. Gastroenterology 140: 2044–2055, 2011. doi: 10.1053/j.gastro.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Triner D, Xue X, Schwartz AJ, Jung I, Colacino JA, Shah YM. Epithelial hypoxia-inducible factor 2alpha facilitates the progression of colon tumors through recruiting neutrophils. Mol Cell Biol 37: e00481-16, 2017. doi: 10.1128/MCB.00481-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Welden S, Selfridge AC, Hindryckx P. Intestinal hypoxia and hypoxia-induced signalling as therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol 14: 596–611, 2017. doi: 10.1038/nrgastro.2017.101. [DOI] [PubMed] [Google Scholar]
- 39.Xie L, Xue X, Taylor M, Ramakrishnan SK, Nagaoka K, Hao C, Gonzalez FJ, Shah YM. Hypoxia-inducible factor/MAZ-dependent induction of caveolin-1 regulates colon permeability through suppression of occludin, leading to hypoxia-induced inflammation. Mol Cell Biol 34: 3013–3023, 2014. doi: 10.1128/MCB.00324-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xue X, Bredell BX, Anderson ER, Martin A, Mays C, Nagao-Kitamoto H, Huang S, Győrffy B, Greenson JK, Hardiman K, Spence JR, Kamada N, Shah YM. Quantitative proteomics identifies STEAP4 as a critical regulator of mitochondrial dysfunction linking inflammation and colon cancer. Proc Natl Acad Sci USA 114: E9608–E9617, 2017. doi: 10.1073/pnas.1712946114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xue X, Ramakrishnan S, Anderson E, Taylor M, Zimmermann EM, Spence JR, Huang S, Greenson JK, Shah YM. Endothelial PAS domain protein 1 activates the inflammatory response in the intestinal epithelium to promote colitis in mice. Gastroenterology 145: 831–841, 2013. doi: 10.1053/j.gastro.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xue X, Ramakrishnan SK, Shah YM. Activation of HIF-1α does not increase intestinal tumorigenesis. Am J Physiol Gastrointest Liver Physiol 307: G187–G195, 2014. doi: 10.1152/ajpgi.00112.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xue X, Shah YM. Hypoxia-inducible factor-2α is essential in activating the COX2/mPGES-1/PGE2 signaling axis in colon cancer. Carcinogenesis 34: 163–169, 2013. doi: 10.1093/carcin/bgs313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xue X, Taylor M, Anderson E, Hao C, Qu A, Greenson JK, Zimmermann EM, Gonzalez FJ, Shah YM. Hypoxia-inducible factor-2α activation promotes colorectal cancer progression by dysregulating iron homeostasis. Cancer Res 72: 2285–2293, 2012. doi: 10.1158/0008-5472.CAN-11-3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu T, Tang B, Sun X. Development of inhibitors targeting hypoxia-inducible factor 1 and 2 for cancer therapy. Yonsei Med J 58: 489–496, 2017. doi: 10.3349/ymj.2017.58.3.489. [DOI] [PMC free article] [PubMed] [Google Scholar]