

Abstract
Reactive oxygen species such as H2O2 are believed to play a prominent role in the injury and loss of transport function that affect the intestinal epithelium in inflammatory conditions such as inflammatory bowel diseases. Defects in intestinal epithelial ion transport regulation contribute to dysbiosis and inflammatory phenotypes. We previously showed that H2O2 inhibits Ca2+-dependent Cl− secretion across intestinal epithelial cells (IECs) via a phosphatidylinositol 3-kinase (PI3K)- and extracellular signal-regulated kinase (ERK)-dependent mechanism that occurs, at least in part, through inhibition of the basolateral Na+-K+-2Cl− cotransporter NKCC1. NKCC1 governs Cl− entry into crypt IECs and thus plays a critical role in maintaining the driving force for Cl− secretion. Electrolyte transport consumes large amounts of cellular energy, and direct pharmacological activation of the cellular energy sensor AMP-activated protein kinase (AMPK) has been shown to inhibit a number of ion transport proteins. Here, we show that H2O2 activates AMPK in human IEC lines and ex vivo human colon. Moreover, we demonstrate that the inhibitory effect of H2O2 on Ca2+-dependent Cl− secretion and NKCC1 activity is AMPK-dependent. This inhibitory effect is associated with a physical interaction between AMPK and NKCC1, as well as increased phosphorylation (Thr212,217) of NKCC1, without causing NKCC1 internalization. These data identify a key role for AMPK-NKCC1 interaction as a point of convergence for suppression of colonic epithelial ion transport by inflammatory reactive oxygen species.
NEW & NOTEWORTHY H2O2 inhibition of intestinal epithelial Ca2+-dependent Cl− secretion involves recruitment of AMP-activated protein kinase (AMPK) downstream of ERK and phosphatidylinositol 3-kinase signaling pathways, physical interaction of AMPK with the Na+-K+-2Cl− cotransporter NKCC1, and AMPK-dependent suppression of NKCC1-mediated electrolyte influx without causing NKCC1 internalization. It is intriguing that, in human intestinal epithelial cell lines and human colon, H2O2 activation of AMPK increased phosphorylation of NKCC1 residues required for promoting, not inhibiting, NKCC1 activity. These data identify an elevated complexity of AMPK regulation of NKCC1 in the setting of an inflammatory stimulus.
Keywords: carbachol, chloride, epithelial, HT-29, hydrogen peroxide, T84
INTRODUCTION
The overproduction of reactive oxygen species (ROS) during active episodes of Crohn’s disease and ulcerative colitis is thought to play an important role in the pathophysiology of inflammatory bowel disease (IBD) (6, 51). A number of studies have shown that chronically inflamed colonic tissue is subject to significant oxidative stress due, in part, to decreased expression of endogenous antioxidants (12, 27, 33, 41). Therapeutic approaches using broad-spectrum antioxidants, such as vitamin E, or immunomodulatory agents that show antioxidant activity, such as 5-aminosalicylic acid, have proven effective to differing degrees in treating IBD and reducing markers of oxidative stress (52). In addition, antioxidants have proven effective in delaying the onset of, and enhancing recovery from, colitis including a protective effect against the onset of diarrhea in mouse models (11, 37, 50).
H2O2 is among the most important ROS believed to have a role in IBD, as it is a relatively stable and cell-permeable precursor of numerous more-toxic oxygen radicals such as the hydroxyl radical (38, 51, 69). Furthermore, the large influx of activated phagocytes in ulcerative colitis may secrete millimolar concentrations of H2O2 in close proximity to colonocytes (64). Although pathophysiological concentrations of H2O2 can cause damage, both direct and indirect, to cultured epithelial cells and cells isolated from IBD patients via generation of more ROS, H2O2 is also capable of more subtle influences on cell function. H2O2 has emerged as an important cell-signaling molecule involved in mediating responses to epidermal growth factor and other growth factors (51), as well as participating in MAPK and NF-κB signaling networks (20, 70). H2O2 has also been shown to exert discreet, and often opposing, effects on ion transport functions in intestinal tissue and colonic epithelial cell lines. High concentrations (i.e., 5.5 mmol/l) of H2O2 have been shown to induce a small increase in short-circuit current (Isc), accounted for by Cl− secretion, across T84 colonic epithelial cells (48). Other studies have shown that lower concentrations (500 μM) of H2O2 inhibit cAMP-dependent Cl− secretion through specific effects on transport proteins involved in Cl− transport, without evidence of toxic or injurious effects on cell monolayers (20, 67). The overall effect of H2O2 on Cl− secretion appears to be one of suppression based on studies of colonic epithelial cell lines and isolated mouse colonic mucosa, consistent with the reduced ion-transporting capability of intestinal tissues in the setting of inflammation (9, 35, 58). Indeed, we previously demonstrated that in vivo administration of the H2O2-degrading antioxidant catalase to mice alleviated ion transport dysfunction in dextran sulfate sodium-induced colitis and that this effect was associated with reduced H2O2 levels in the colonic mucosa (3).
In the intestinal tract, Ca2+-dependent Cl− secretion is triggered by neuronal reflexes responding to changes in luminal composition, contact between luminal contents and the epithelium, and physical distension (13). Cl− secretion serves useful “housekeeping” functions, such as hydration of the mucosal surface, dispersal of mucus and antimicrobial peptides from the crypt, and flushing of noxious substances and pathogenic bacteria. Thus, crypt fluid secretion is an important component of intestinal homeostasis and protection of the host epithelial barrier (47). Indeed, compromised secretory function likely contributes to a decrease in the overall effectiveness of the barrier, facilitating bacterial colonization of the intestinal crypt and subsequent excessive inflammatory responses that are a feature of diseases such as IBD. This hypothesis was supported by studies demonstrating that CFTR-knockout mice exhibit inflammation similar to that observed in IBD patients, as well as pronounced changes in richness and diversity of the intestinal microbiome (43, 49).
We previously demonstrated that H2O2 pretreatment is also capable of inhibiting Ca2+-dependent Cl− secretion across colonic epithelial cells via Ca2+-dependent activation of MAPKs, phosphatidylinositol 3-kinase (PI3K), and functional inhibition of the Na+-K+-2Cl− cotransporter NKCC1 (9). NKCC1 is located on the basolateral membrane of colonic epithelial cells at the crypt base and is the principal basolateral uptake mechanism for secreted Cl− in crypt epithelium (2). Therefore, inhibition of NKCC1 compromises the normal entry of electrolytes into crypt epithelial cells and restricts secretory capability (21). Decreased electrogenic secretory capacity is associated with IBD and may be particularly relevant to loss of crypt flushing and dispersal of antimicrobial peptides associated with inflammatory conditions (1, 4, 26, 34, 43, 65).
In our earlier work we established that the inhibitory effect of H2O2 on Ca2+-dependent Cl− secretion across colonic epithelial cells is mediated through activation of a number of signaling kinases previously shown to negatively regulate intestinal Cl− secretion (9, 36) and subsequent inhibition of NKCC1 activity. However, the mechanism by which H2O2-activated kinases inhibit NKCC1 and the signaling effector(s) mediating this inhibition has not been identified. Therefore, in the present study we set out to identify the mechanism by which H2O2 inhibits NKCC1 by focusing on the cellular energy sensor AMP-activated protein kinase (AMPK). AMPK responds to metabolic stress, manifesting as depletion of ATP levels and elevation of the AMP-to-ATP ratio (10, 31, 32) by stimulating energy-producing pathways, such as glucose uptake and glycolysis, while inhibiting energy-consuming processes, such as protein and fatty acid synthesis (25, 28, 32). In the intestinal epithelium, energy-consuming processes also include electrolyte transport and maintenance of the epithelial barrier. Indeed, previous studies have demonstrated that activation of AMPK suppresses intestinal ion transport and synthesis of various proteins (28, 53). In the Cl−-secreting epithelial cells within the colonic crypt, AMPK has been shown to physically interact with and inhibit the CFTR Cl− channel (67). In absorptive intestinal cells, AMPK can promote removal of the epithelial Na+ channel from the apical membrane via increased activity of the ubiquitin ligase enzyme Nedd4-2 (5, 29, 30, 68). These effects would reduce the overall ion-transporting capability of the intestinal epithelium, as demonstrated in colonic tissues from the IL-10−/− mouse model of colitis (67) and intestinal tissues from IBD patients (26, 54, 57).
Therefore, the goal of this study was to determine whether AMPK mediates H2O2-induced alterations in intestinal epithelial Ca2+-dependent Cl− secretion and NKCC1 function. We found that AMPK is activated by H2O2 in human colon and intestinal epithelial cell (IEC) lines. H2O2 activation of AMPK leads to physical interaction of AMPK with NKCC1 and suppression of NKCC1 activity, and inhibition of AMPK partially protects NKCC1 from inhibition by H2O2. Our data indicate a novel role for the cellular energy sensor AMPK in the regulation of a critical epithelial transport protein in a cell model of inflammation. These findings may have implications for suppression of ion transport function in the setting of inflammatory conditions such as IBD.
MATERIALS AND METHODS
Materials.
Carbachol (CCh), compound C (CC), and metformin hydrochloride were obtained from Sigma (St. Louis, MO); wortmannin, LY294002, PD98059, 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR), and H2O2 from Calbiochem (San Diego, CA); mouse anti-phosphotyrosine antibodies from Upstate Biotechnology (Lake Placid, NY); anti-phosphorylated ERK antibodies from New England Biolabs (Ipswich, MA); goat polyclonal anti-total NKCC1 (N-16) and mouse monoclonal anti-total human NKCC1 (T4) antibodies from Dr. C. Lytle (46); rabbit anti-lamin A/C antibody from Santa Cruz Biotechnology (Santa Cruz, CA); rabbit anti-phosphorylated (Thr172) AMPKα antibody, rabbit anti-AMPKα antibody, rabbit anti-phosphorylated (Ser473) Akt antibody, and rabbit anti-Akt antibody from Cell Signaling Technology (Danvers, MA); and Tris-glycine electrophoresis gels from Bio-Rad. Rabbit polyclonal anti-human ERK1 (K-23; Santa Cruz Biotechnology) was used to measure total ERK. The R5 anti-phosphorylated (Thr212,217) NKCC1 antibody was a kind gift from Dr. Biff Forbush (22). All other reagents were of analytical grade and obtained commercially.
Cell culture.
Methods for maintenance of T84 cells (obtained from primary stocks of Dr. Kim Barrett, University of California, San Diego) in culture were described previously (9). Briefly, T84 cells were grown in DMEM/F-12 medium (Mediatech, Herndon, VA) supplemented with 5% newborn calf serum. For Ussing chamber/voltage-clamp experiments, 5 × 105 cells were seeded onto 12-mm Millicell polycarbonate filters. For Western blot experiments, 106 cells were seeded onto 30-mm filters. Cells were cultured for 10–15 days before use. When grown on polycarbonate filters, T84 cells are known to acquire the polarized phenotype of native colonic epithelia. In accordance with muscarinic M3 receptor distribution on intestinal epithelia, CCh was added basolaterally in all experiments. HT-29.cl19A cells (from Dr. Kim Barrett) were grown to confluence (∼5 days) in DMEM supplemented with 10% FBS, 1% l-glutamine, and streptomycin (100 μg/ml) in 75-cm2 flasks. Thereafter, the cells were seeded onto 12-mm Millicell polycarbonate filters for Ussing chamber experiments or onto 30-mm filters for Western blotting, as described above for T84 cells.
Physiological solutions.
Composition of the Ringer solution used for Ussing chamber, Western blot, and 86Rb+ uptake studies was as follows (in mM): 140 Na+, 120 Cl−, 5.2 K+, 25 , 0.4 , 2.4 , 1.2 Ca2+, 1.2 Mg2+, and 10 d-glucose.
Electrophysiological studies.
T84 cells grown to confluence on permeable 12-mm filters were mounted in Ussing chambers (0.6 cm2 window area) and bathed in oxygenated (95% O2-5% CO2) Ringer solution at 37°C. Monolayers were voltage-clamped to zero potential difference by application of Isc. Under these conditions, changes in Isc in response to agonists are wholly reflective of electrogenic Cl− secretion (8). Transepithelial resistance (TER) was measured using a “chopstick” voltohmmeter (World Precision Instruments, Sarasota, FL) (55, 56). The MEK inhibitor PD98059 was solubilized in DMSO. In control studies, the final concentration of DMSO (0.1%) had no effect on Isc responses to CCh in the presence or absence of H2O2 (data not shown). This finding is consistent with our previous unpublished findings and with studies by other groups on MAPK phosphorylation using IEC lines (39).
Human subjects.
Control patients [n = 3 (2 female and 1 male), 61 ± 5 (range 52–71) yr old] presenting for colon cancer screening were asymptomatic and showed no clinical or macroscopic signs of acute or chronic inflammation. Individuals with a history of IBD or intestinal inflammation at the time of the study were excluded. After informed consent was obtained from the patients, a reusable round-cup jumbo biopsy forceps (model no. FB-25 K-1, Olympus) was used to obtain mucosal punch biopsies (~2 mm2 surface area) from macroscopically normal-appearing regions of the sigmoid colon. Tissue biopsies were placed in cold, oxygenated Ringer solution for transport from the endoscopy suite to the laboratory. Tissues were allowed to equilibrate at 37°C in Ringer solution for 20 min before incubation in test solutions. Biopsies were then washed three times in ice-cold Ringer solution before tissue homogenization in lysis buffer for Western blot analysis (see below). The protocol was evaluated and approved by the University of California, San Diego, Institutional Review Board.
Immunoprecipitation and Western blotting.
T84 cell monolayers and tissue biopsies were washed three times with Ringer solution, allowed to equilibrate for 30 min at 37°C, and then stimulated with agonists (in the presence or absence of antagonists) as appropriate. The reaction was stopped by washing in ice-cold PBS, and the cells were lysed in ice-cold lysis buffer (1% Triton X-100, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 μg/ml antipain, 100 μg/ml phenylmethylsulfonyl fluoride, 1 mM sodium vanadate, 1 mM sodium fluoride, and 1 mM EDTA in PBS) for 45 min. Cell lysate supernatants were assayed for protein content (Bio-Rad protein assay kit) and adjusted so that each sample contained an equal amount of protein. For immunoprecipitation studies, lysates were incubated with immunoprecipitating antibody according to the manufacturer’s instructions for 1 h at 4°C and then with protein A-Sepharose for 1 h at 4°C. For NKCC1 immunoprecipitation, the goat polyclonal anti-total NKCC1 antibody (N-16) was used. Lysates were centrifuged for 3 min at 21,000 g, and the supernatant was discarded. The pellets were washed three times in ice-cold PBS and resuspended in 2× gel loading buffer [50 mM Tris (pH 6.8), 2% SDS, 200 mM dithiothreitol, 40% glycerol, and 0.2% bromophenol blue] and boiled before separation by SDS-polyacrylamide gel electrophoresis. Biopsy lysates were passed through a needle, and insoluble material was removed by centrifugation at 350 g for 5 min at 4°C. The supernatant was centrifuged at 43,000 g for 30 min at 4°C, and pellets were resuspended in lysate buffer. Lysate supernatants were assayed for protein content (Bio-Rad protein assay kit) and adjusted so that each sample contained an equal amount of protein. Aliquots of 20 μg of protein were separated by polyacrylamide gel electrophoresis. Resolved proteins were transferred onto polyvinylidene difluoride membranes (NEN Life Science Products, Boston, MA). Thereafter, the membrane was preblocked with a 1% solution of blocking buffer (Upstate Biotechnology) for 30 min and then incubated for 1 h with the appropriate concentration of primary antibody in 1% blocking buffer. After five 10-min washes in Tris-buffered saline with 1% Tween, membranes were incubated for 30 min with a horseradish peroxidase-conjugated secondary antibody (anti-mouse or anti-rabbit IgG; BD Biosciences, San Jose, CA) in 1% blocking buffer. After five 10-min washes in Tris-buffered saline with 1% Tween, immunoreactive proteins were detected using an enhanced chemiluminescence detection kit (Roche Molecular Biochemicals, Indianapolis, IN) on film. National Institutes of Health Image software was used for densitometric analysis of bands.
Basolateral 86Rb+ uptake studies.
Basolateral K+ uptake was measured using 86Rb+ as a tracer and an adaptation of a previously described method (9). Confluent monolayers of T84 cells grown on 12-mm Transwell inserts were rinsed three times with warm Ringer solution and incubated for 1 h at 37°C. After 1 h of preincubation, cells were exposed bilaterally to H2O2 (500 μM) or vehicle control for 30 min on a warming plate at 37°C. Inserts were transferred to wells containing Ringer solution with or without 100 μM CCh for 1 min and then to wells containing Ringer solution with 1 μCi/ml 86Rb+ and CCh and maintained at 37°C for 3 min. Inserts were immersed several times in ice-cold 100 mM MgCl2 containing 10 mM Tris·HCl (pH 7.5) to terminate 86Rb+ uptake. Filters were immediately excised from the inserts and placed directly into 5 ml of scintillation fluid, and 86Rb+ content was measured by standard scintillation methods. K+ influx (μmol K+·g protein−1·min−1) was calculated as (cpm·g protein−1·min−1)/SA, where SA is the specific activity of the uptake buffer (cpm/μmol K+).
siRNA transfection.
T84 cells (2 × 106) were seeded 3 days before transfection and grown to 50–70% confluence in T75 flasks. Three different annealed Silencer predesigned siRNA oligonucleotides targeting AMPKα1 were obtained from Applied Biosystems (Foster City, CA). For transfection reactions, 100 pmol of each of the three gene-specific siRNA oligonucleotides were transfected into T84 cells using the Nucleofector system (Amaxa, Gaithersburg, MD) according to the manufacturer’s instructions. After transfection, T84 cells were cultured on filter membranes for 48 h before further treatment. The nonspecific control siRNA SMARTpool (100 pmol; Upstate Biotechnology/Dharmacon, Chicago, IL) was used as a negative control.
NKCC1 biotinylation assay.
NKCC1 surface expression was determined as previously described (7). Briefly, T84 monolayers were treated with 500 μM H2O2 with or without 30 min of pretreatment with compound C (50 μM), metformin (5 mM) alone, or capsaicin (20 μM) alone. After treatment, monolayers were washed twice with ice-cold 1× PBS (pH 8.0). EZ-Link Sulfo-NHS-SS-biotin (10 μM; Thermo Fisher Scientific) was added basolaterally, and monolayers were incubated for 30 min at 4°C. Fresh biotin was added, and monolayers were incubated for an additional 30 min. Monolayers were washed twice with ice-cold 1× PBS (pH 8.0), incubated in quenching buffer [100 mM glycine in 1× PBS (pH 8.0)] for 15 min, and then aspirated. Radioimmunoprecipitation lysis buffer (500 μl) was added, and monolayers were incubated for 30 min at 4°C with gentle agitation. Cells were scraped and collected into 1.7-ml tubes and then subjected to sonication (QSonica) for 30 s (10 s on/off) at 30% amplitude. Lysates were centrifuged at 4°C for 10 min at 16,200 g, and the supernatant was aspirated into new tubes. Protein was quantified using a bicinchoninic acid assay (Pierce). Pierce streptavidin-agarose resin (50 μl; Thermo Fisher Scientific) was placed in 1.7-ml tubes and washed according to the manufacturer’s instructions. Equal concentrations of protein (~1 mg/ml) were added to respective streptavidin-bead aliquots, which were then incubated overnight at 4°C with rotation. On the following day, samples were centrifuged according to instructions of the manufacturer of the streptavidin-agarose beads. Supernatants were aspirated into new tubes for total NKCC1 samples. Beads were washed twice with a high-salt buffer [0.1% Triton X-100, 500 mM NaCl, 50 mM Tris, and 5 mM EDTA (pH 7.5)] for 10 min and once with a low-salt buffer [0.1% Triton X-100, 100 mM NaCl, 50 mM Tris, and 5 mM EDTA (pH 7.5)] for 10 min. Biotinylated proteins were eluted from streptavidin beads using 50 μl of Laemmli sample buffer (Bio-Rad). Samples were run on 8% SDS-PAGE and transferred to polyvinylidene difluoride membranes.
Statistical analysis.
All data are expressed as means ± SE for a series of n experiments. Student’s t-test or ANOVA with the Student-Newman-Keuls post test was used to compare mean values as appropriate. P < 0.05 was considered to represent significant difference.
RESULTS
H2O2 inhibits CCh-stimulated Cl− secretion through activation of AMPK.
The metabolic-sensing serine-threonine kinase AMPK has been shown to play an important role in restoration of cellular energy levels under conditions of metabolic stress, including after exposure to oxidants such as H2O2 (10, 31, 32). AMPK activation occurs in response to AMP binding or via activation by upstream kinases (14). Moreover, binding of AMP to AMPK makes AMPK a better substrate for kinase activation, resulting in phosphorylation of its catalytic α-subunit on the Thr172 residue, with a 50- to 100-fold increase in enzyme activity (14). Therefore, we measured phosphorylation (Thr172) of AMPKα to investigate whether H2O2 increases activation of AMPK. H2O2 increased phosphorylation transiently, with a maximal effect between 5 and 15 min (P < 0.05–0.01, n = 7; Fig. 1, A and B). The AMPK activator AICAR, administered for 30 min, also significantly increased AMPKα phosphorylation (P < 0.05, n = 4; Fig. 1, A and B). While AMPK has been shown to inhibit cAMP-dependent Cl− secretion through effects on CFTR, its effects on Ca2+-dependent epithelial Cl− secretion have not been determined (29, 67). Consequently, we investigated whether AMPK activation was involved in the inhibitory effect of H2O2 on Ca2+-dependent Cl− secretion stimulated by the acetylcholine analog CCh. Pretreatment of T84 monolayers with the specific AMPK inhibitor compound C (1–100 μM) blocked H2O2-mediated inhibition of CCh-stimulated Cl− secretion, indicating that AMPK is involved in H2O2 inhibition of Ca2+-dependent Cl− secretion (50 μM compound C, P < 0.05, n = 4; Fig. 1C). This finding was also observed in HT-29.cl19a monolayers, where the inhibitory effect of H2O2 was significantly blocked by pretreatment with compound C (P < 0.05; Fig. 1D).
Fig. 1.

H2O2 increases AMP-activated protein kinase (AMPK) phosphorylation in intestinal epithelial cells. T84 and HT-29.cl19a cells grown as monolayers on semipermeable supports were treated bilaterally with H2O2 (500 μM) for 2–30 min or the AMPK activator 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR, 2.5 mM) for 30 min. A: Western blot analysis of phosphorylation of the activating Thr172 residue on the catalytic α-subunit of AMPK. Molecular mass (kDa) of each protein is shown at right. B: densitometric quantitation of Western blot results in A and normalization to total AMPKα levels. C: CCh-induced change in ion transport reported as change in short-circuit current (ΔIsc). T84 monolayers mounted in Ussing chambers were not treated (control) or preincubated with compound C (1–100 μM, bilaterally) for 30 min before administration of H2O2 (500 μM) for 30 min followed by stimulation of electrogenic Cl− secretion with carbachol (CCh, 100 μM, basolaterally, n = 4). D: change in ion transport reported as change in short-circuit current (ΔIsc). HT-29.cl19a monolayers mounted in Ussing chambers were untreated (control) or pretreated with compound C (50 μM, bilaterally) for 30 min before administration of H2O2 (500 μM, bilaterally) for 30 min and subsequent CCh stimulation of ion transport (n = 3). Values are means ± SE. *P < 0.05, **P < 0.01 vs. control (B) and vs. H2O2 + CCh (C and D); #P < 0.05 vs. compound C (1 μM).
H2O2-induced AMPK phosphorylation occurs downstream of PI3K and ERK activation.
We previously demonstrated that the inhibitory effect of H2O2 on CCh-stimulated Isc involves activation of two separate, but Ca2+-dependent, signaling pathways. One pathway involves activation of Src, Pyk-2, and ERK/p38 MAPK while the other involves p38-dependent activation of PI3K (9). We therefore investigated whether these signaling pathways are also involved in activation of AMPK by H2O2. Preincubation of T84 monolayers with the MEK1 inhibitor PD98059 or the PI3K inhibitor wortmannin reduced phosphorylation of AMPKα in response to H2O2 (P < 0.05–0.001, n = 3; Fig. 2). AMPK phosphorylation levels in monolayers treated with PD98059 or wortmannin alone were not statistically different from those in monolayers treated with either agent in the presence of H2O2. Consistent with our previous findings, MEK1 inhibition had no effect on H2O2-stimulated PI3K signaling measured by Akt phosphorylation; similarly, PI3K inhibition by wortmannin had no effect on ERK phosphorylation, whereas it successfully reduced Akt phosphorylation (Fig. 2A). These data indicate that the ERK and PI3K signaling pathways activated by H2O2 converge at the level of AMPK.
Fig. 2.

H2O2 activation of AMP-activated protein kinase (AMPK) is downstream of ERK and phosphatidylinositol 3-kinase (PI3K) pathways. A: Western blot analysis of AMPK, ERK, and Akt phosphorylation. T84 monolayers were preincubated with the MEK inhibitor PD98059 (50 μM) or the PI3K inhibitor wortmannin (50 nM) for 30 min before treatment with H2O2 (500 μM) for 5 min. Molecular mass (kDa) of each protein is shown at right. B: densitometric quantitation of Western blot results in A and normalization to total AMPKα levels (n = 3). Values (arbitrary units) are means ± SE. *P < 0.05 vs. control/untreated; #P < 0.05 vs. H2O2 alone.
AMPK activity mediates H2O2 inhibition of NKCC1.
Our prior work indicated that H2O2 pretreatment inhibits CCh-stimulated NKCC1 activity, as measured by bumetanide-sensitive 86Rb+ influx, and that the inhibitory effect of H2O2 does not involve changes in intracellular Cl− (a major regulator of NKCC1 activity) (9). Further experiments explored whether H2O2 inhibition of CCh-stimulated K+ influx also involves AMPK activation. Preincubation of T84 monolayers with the AMPK inhibitor compound C (50 μM) for 30 min reversed the inhibitory effect of H2O2 on CCh-stimulated 86Rb+ influx (P < 0.01 vs. Ringer solution, n = 4; Fig. 3), while there was no significant difference in 86Rb+ influx between CCh alone and CCh in the presence of compound C. These data indicate that H2O2 inhibition of Ca2+-dependent K+ uptake, likely via NKCC1 based on our prior studies, is mediated, at least in part, through AMPK activation.
Fig. 3.
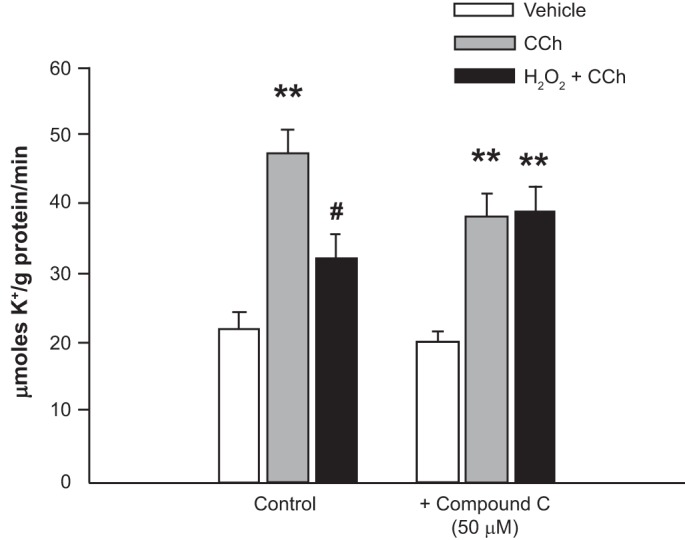
H2O2 inhibits basolateral K+ influx and activity of the Na+-K+-2Cl− cotransporter NKCC1. T84 monolayers were pretreated with the AMP-activated protein kinase (AMPK) inhibitor compound C (50 μM) or Ringer solution (control) and then incubated with vehicle control or H2O2 (500 μM) for 30 min. Cells were subsequently transferred to Ringer solution or Ringer solution + carbachol (CCh, 100 μM, basolaterally) for 1 min and then to a basolateral 86Rb+-containing buffer for 3 min (n = 4). Values are means ± SE. **P < 0.01 vs. control/untreated cells; #P < 0.05 vs. CCh alone.
AMPK physically associates with NKCC1 in IECs.
AMPK activators such as AICAR and A-769662 have been shown to have no effect or to inhibit NKCC1 activity, with the latter effect due, in part, to phosphorylation at the NH2 terminus (23, 60). To determine whether AMPK inhibits NKCC1 through a physical association, we used lysates from T84 cells treated with H2O2 to carry out coimmunoprecipitation experiments (Fig. 4). We observed a significant level of coimmunoprecipitation of AMPK and NKCC1 (detected with anti-NKCC1 T4 antibody), regardless of whether AMPK or NKCC1 was initially immunoprecipitated [P < 0.01, n = 3 (Fig. 4A); P < 0.05, n = 3 (Fig. 4B)]. Thus, inhibition of NKCC1 by AMPK could involve a direct association between the two proteins.
Fig. 4.

H2O2 induces AMP-activated protein kinase (AMPK)-Na+-K+-2Cl− cotransporter type 1 (NKCC1) association. T84 monolayers were treated with H2O2 (500 μM) for 2–30 min or the AMPK activator 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR, 2.5 mM) for 30 min. Cell lysates were immunoprecipitated with anti-AMPKα (A) or anti-NKCC1 (N-16) antibody (B) and subsequently probed for NKCC1 and/or AMPKα binding by Western blotting. The catalytic α-subunit of AMPK was immunoprecipitated (IP) and immunoblotted (Blot) for NKCC1 (T4 antibody). Results from blots were quantified by densitometric analysis (n = 4). Molecular mass of each protein (kDa) is shown at right. Values (arbitrary units) are means ± SE and were normalized to total levels of the primary immunoprecipitate (AMPKα or NKCC1). *P < 0.05, **P < 0.01, ***P < 0.001 vs. control/untreated cells.
H2O2 modifies the phosphorylation status of NKCC1 through AMPK.
Full activity of human NKCC1 requires phosphorylation of Thr residues at positions 212 and 217 (16). We therefore explored whether H2O2 affects NKCC1 phosphorylation and whether this involves AMPK activation. We were surprised to find that treatment of T84 monolayers with H2O2 (500 μM) induced a rapid increase in phosphorylation (Thr212,217) of NKCC1 that peaked at 15 min (P < 0.05, n = 4; Fig. 5A). The AMPK activator AICAR also significantly increased NKCC1 phosphorylation (P < 0.05, n = 4; Fig. 5A). These findings were reproduced using human colonic mucosal biopsies isolated from subjects who were undergoing routine colorectal cancer screening but showed no evidence of intestinal inflammation. Ex vivo treatment with H2O2 (1 mM) and subsequent Western blotting for phosphorylated and total forms of NKCC1 and AMPK showed that H2O2 increased NKCC1 (Thr212,217), as well as AMPK (Thr172), phosphorylation (n = 3; Fig. 5B). Follow-up studies in T84 cells showed that H2O2-stimulated phosphorylation of NKCC1 was significantly reduced by preincubation with the AMPK inhibitor compound C (P < 0.01, n = 4; Fig. 5C). These data indicate that H2O2 actually increases phosphorylation of NKCC1 residues required for NKCC1 activity.
Fig. 5.

H2O2 increases Na+-K+-2Cl− cotransporter type 1 (NKCC1) phosphorylation in human colon and in cell lines in an AMP-activated protein kinase (AMPK)-dependent manner. A: H2O2 treatment of T84 monolayers significantly increased phosphorylation (Thr212,217) of NKCC1 (500 μM, P < 0.05, n = 4). Treatment with 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR, 2.5 mM) for 30 min also increased NKCC1 (Thr212,217) phosphorylation (P < 0.05, n = 4). B: experiment described in A was repeated in ex vivo human colonic biopsies from control subjects without active inflammatory intestinal disease (n = 3). H2O2 (1 mM) also increased AMPKα (Thr172) phosphorylation in human colonic tissue. C: preincubation of T84 monolayers with compound C (50 μM, bilaterally) for 30 min significantly reduced H2O2 induction of NKCC1 phosphorylation (P < 0.01, n = 4). Densitometric data (arbitrary units) are presented as means ± SE for levels of phosphorylation and normalized to total NKCC1 levels. *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle control; &P < 0.05 vs. compound C control; ##P < 0.01 vs. vehicle H2O2.
H2O2-activated AMPK does not promote internalization of membrane-bound NKCC1.
NKCC1 has also been shown to be regulated via internalization of membrane-bound NKCC1 in colonic epithelial cells (7). Therefore, we employed a surface biotinylation assay to determine if surface expression of NKCC1 is altered and if NKCC1 internalization is AMPK-dependent. Monolayers of T84 cells were treated with H2O2 with or without pretreatment with the AMPK inhibitor compound C (50 μM), metformin (5 mM) alone as a positive control for AMPK activation, or capsaicin (20 μM) alone as a positive control for NKCC1 internalization (7, 67). After treatment, proteins expressed in the basolateral membrane were biotinylated and pulled down by immunoprecipitation with streptavidin-coated agarose beads. We observed no significant change in NKCC1 surface expression in response to H2O2 treatment, as well as no effect of compound C pretreatment, although there appeared to be an increasing trend of NKCC1 surface expression with H2O2 treatment (Fig. 6). Furthermore, AMPK activation by metformin (5 mM) did not alter NKCC1 internalization. Capsaicin (20 μM) also did not induce NKCC1 internalization, despite our use of a previously established incubation time and concentration of capsaicin that were shown to promote NKCC1 internalization in T84 cells (7). Our data suggest that H2O2 inhibition of Cl− secretion is independent of NKCC1 internalization and that AMPK does not mediate distinct patterns of membrane turnover of NKCC1.
Fig. 6.

AMP-activated protein kinase (AMPK) inhibition of the Na+-K+-2Cl− cotransporter NKCC1 is independent of internalization. NKCC1 surface expression was detected using a biotinylation assay, where biotinylation indicated membrane insertion. H2O2 treatment of T84 monolayers slightly, but not statistically significantly, increased surface expression of NKCC1 (500 μM; n = 3). Pretreatment with the AMPK inhibitor compound C (CC, 50 μM, bilaterally) for 30 min did not decrease NKCC1 internalization. Neither metformin (Metf, 5 mM, bilaterally; n = 3) nor capsaicin (Cap, 20 μM, bilaterally, n = 3) treatment significantly altered surface expression of NKCC1. Densitometric data (arbitrary units) are presented as means ± SE for levels of biotinylated NKCC1 and normalized to total NKCC1 levels.
DISCUSSION
Regulation of epithelial transport is a complex and multifaceted physiological process that requires careful orchestration of acute vs. chronic and transient vs. sustained events. In the acute regulation of electrolyte transport in response to exposure by ROS, we investigated whether the cellular energy sensor AMPK, itself capable of acute and/or sustained regulation of many metabolic events, was involved in the H2O2 regulation of Ca2+-dependent ion transport. We previously showed that H2O2 inhibits CCh-stimulated Cl− secretion (9). CCh is a more stable analog of the neurotransmitter acetylcholine that plays a prominent role in the minute-to-minute regulation of fluid secretion in intestinal crypts through activation of Ca2+-dependent signals following binding to muscarinic receptors on IECs (2). We further found that the inhibitory effect of H2O2 occurred by virtue of its ability to reduce the activity of the basolateral NKCC1, which is responsible for basolateral Cl− entry into IECs and is critically involved in overall epithelial homeostasis (9). This finding was supported by in vivo studies showing that administration of the antioxidant enzyme catalase, which degrades H2O2, restricted the suppression of colonic ion transport responsiveness and reduced levels of colonic epithelial NKCC1 caused by dextran sulfate sodium-induced colitis (3). In the present study we showed that AMPK activity is a critical mediator of the inhibitory effect of H2O2 on Cl− secretion and NKCC1 activity. H2O2 increased phosphorylation (Thr172) of AMPKα in human colonic epithelial cell lines and in ex vivo human colonic mucosal biopsies. While there was some variability between human samples from different patients with respect to the magnitude and kinetics of H2O2 induction of AMPKα and NKCC1 phosphorylation, the overall effect of H2O2 as a stimulus of phosphorylation of these proteins was consistent and, thus, broadly aligned with our cell line data. The AMPK inhibitor compound C dramatically reduced the inhibitory effect of H2O2 on overall ion transport and CCh-stimulated NKCC1 activity. The inhibitory effect of H2O2 on NKCC1 activity was associated with an increased physical association between AMPK and NKCC1 based on reciprocal coimmunoprecipitation studies. Since NKCC1 activity is dependent on phosphorylation of regulatory Thr212 and Thr217 residues (16, 22, 44, 45), we investigated if H2O2 had an effect on NKCC1 phosphorylation. Using the R5 antibody, which recognizes phosphorylation of these residues, we were surprised to find that H2O2 actually increases NKCC1 phosphorylation in both IEC lines and human colonic biopsies, even while H2O2 exerted an inhibitory effect on overall NKCC1 activity. Phosphorylation of NKCC1 at its regulatory Thr212 and Thr217 residues in response to H2O2 appears, ultimately, to require AMPK activity, since both AICAR and H2O2 elicited NKCC1 phosphorylation in a manner antagonized by compound C.
AMPK activity has been linked to inhibition of renal-specific NKCC2 and, recently, to NKCC1 activity in in vitro model systems. This was shown to occur in response to selective activators of AMPK, such as AICAR and A-769662, and also in response to osmotic shrinkage (23, 24, 60). However, in these studies, AMPK phosphorylation of NKCC1 at Ser38 and Ser214 (dogfish sequence), corresponding to Ser77 and Ser242 in human NKCC1, was, in fact, associated with reduced NKCC1 activity. The consensus amino acid sequence that is associated with AMPK binding and Ser/Thr phosphorylation involves the following recognition motif: φ(Xβ)XXS/TXXXφ, where φ is a hydrophobic residue (M, V, L, I, or F), β is a basic residue (R, K, or H), and the parentheses indicate that the order of residues at the P-4 and P-3 positions is not essential (15, 60). The sequence surrounding Ser242 of human NKCC1 loosely fits the AMPK consensus recognition motif, with an Arg residue at the P-2, rather than P-3, position (60). Additional studies with cell-free assays showed increased phosphorylation of Ser77 following AMPK activation by phenformin, while a NKCC1 construct with a point mutation at Ser77 showed less phosphorylation and a 16% decrease in the inhibitory effect of phenformin on NKCC1 transport of 86Rb+, thus indicating a partial role for this residue in AMPK regulation of NKCC1 (23). One caveat is that phenformin can exert AMPK-independent signaling events (66).
Further studies indicate that AMPK can inhibit activity of Ste-related proline-alanine-rich kinase (SPAK)/oxidative stress-responsive kinase (OSR1), which phosphorylates and activates NKCC1 in response to several stimuli (17, 60). However, there does appear to be a context- and cell type-dependent pattern of regulation of NKCC1 by AMPK that is not always consistent with NKCC1 phosphorylation levels or kinetics. Indeed, this is not an unusual feature of AMPK, as it has been shown to play context-specific roles in tumorigenesis (42). Interestingly, the cell type-specific effects of individual AMPK activators were emphasized by the use of A-769662: Sid et al. observed that A-769662, while increasing phosphorylation of Ser242, had no effect on NKCC1 activity in erythrocytes (60), whereas Fraser et al. observed inhibition of NKCC1 activity by A-769662 in Madin-Darby canine kidney cells (23). There also appear to be important differences between the effects of basal and stimulated AMPK activity in the regulation of NKCC1: Davies et al. observed that in erythrocytes the constitutive absence or suppression of AMPK activity could regulate SPAK/OSR1 through mechanisms different from those invoked by acute changes in AMPK activity (17). Furthermore, the nuanced regulation of NKCC1 activity was also emphasized by findings that the extent of NKCC1 phosphorylation did not directly correlate with NKCC1 activation by hyperosmolarity in mouse erythrocytes; thus, Sid et al. concluded that other factors are likely involved (60).
Even though we observed an increase in H2O2-stimulated phosphorylation of two key residues (Thr212 and Thr217) that are required for increasing NKCC1 activity, increased AMPK activity in Madin-Darby canine kidney cells treated with the AMPK activator A-769662 correlated with reduced NKCC1 phosphorylation on Thr212 and Thr217 (23). Our studies indicate a clear functional role for H2O2-activated AMPK in inhibiting Ca2+-dependent electrolyte transport and NKCC1 activity (cf. Figs. 1A, 2, and 4A). We also observed a partial, although not statistically significant, increase in NKCC1 phosphorylation in cells incubated with compound C alone (cf. Fig. 5), with no effect on NKCC1 activity (cf. Fig. 3), which is consistent with the findings of Fraser et al. (23). Our results suggest that AMPK may have a dual role with respect to NKCC1 phosphorylation in unstimulated/resting conditions vs. that in response to an inflammatory stimulus. Moreover, we did not see an increase in 86Rb+ flux in colonic epithelial cells treated with compound C alone, which is also in agreement with the findings of Fraser et al. (23). AMPK activity is also stimulus-specific, as well-established activators such as AICAR and A-769662 act via different mechanisms. AICAR is phosphorylated by adenylate kinase to generate the AMP analog ZMP, which directly binds to the α-subunit of AMPK, while A-769662 allosterically activates AMPK through binding to its regulatory β1-subunit (14, 59). Indeed, coadministration of these agents has been shown to have a synergistic effect with respect to AMPK activation in mouse hepatocytes (19). Another important consideration of select activators of AMPK is their potential for off-target effects. AICAR and metformin have been shown to inhibit proliferation by AMPK-independent mechanisms, while A-769662 can effectively inhibit proteasomal activity, which is relevant to the study of NKCC1 function, since NKCC1 can be regulated by proteasomal degradation (18, 42).
With respect to the stimulus used to activate AMPK in these studies, several groups have reported that oxidative stress can activate AMPK in a cell type-specific manner independently of alterations in the AMP-to-ATP ratio (reviewed in Refs. 40 and 61). We previously demonstrated that H2O2, at doses used in the present study and in the same cell lines, did not cause disruption of barrier function in T84 cells, thus indicating that cell integrity is maintained (9). Our previous work demonstrated that H2O2 caused a sustained increase in intracellular Ca2+ levels that triggered activation of two signaling pathways, leading to increased phosphorylation of MEK and Akt downstream of PI3K. Our data suggest that these pathways feed into AMPK phosphorylation, as phosphorylated AMPK levels were reduced by ERK or PI3K inhibition (cf. Fig. 3). Our biotinylation-based internalization assays did not detect a clear effect of H2O2 on membrane localization of NKCC1, although there was a counterintuitive trend toward increased membrane localization. This pattern was also observed in response to capsaicin treatment, which was included as a positive control to promote internalization based on a previous study (7). Given the lack of a significant effect of H2O2 on membrane NKCC1 levels, we interpret our findings as indicating that H2O2-AMPK signaling primarily inhibits NKCC1 by altering its activation status, rather than by causing a substantial internalization of NKCC1 away from the basolateral membrane.
In conclusion, we have demonstrated that pathophysiologically relevant concentrations of H2O2 can inhibit Ca2+-dependent Cl− secretion by activating kinase-signaling pathways, leading to activation of AMPK and inhibition of NKCC1 activity and, overall, ion transport responsiveness (Fig. 7). Our findings emphasize the context-dependent nature of the relationship between AMPK and NKCC1 but are consistent with an overall inhibitory effect of H2O2 and AMPK on the ion-transporting capability of IECs that aligns with the loss of transport capacity in intestinal inflammation (35). In agreement with the findings of others, it appears that regulation of NKCC1 activity is more complex than a linear and uniform series of protein kinase signals and may involve multiple protein kinase complexes acting in different cell types and resulting in varied outcomes determined by specific stimuli (62, 63). The findings in our study serve to further expand our understanding of how AMPK may act to negatively regulate energy-consuming cellular activities in epithelia in the setting of inflammation.
Fig. 7.
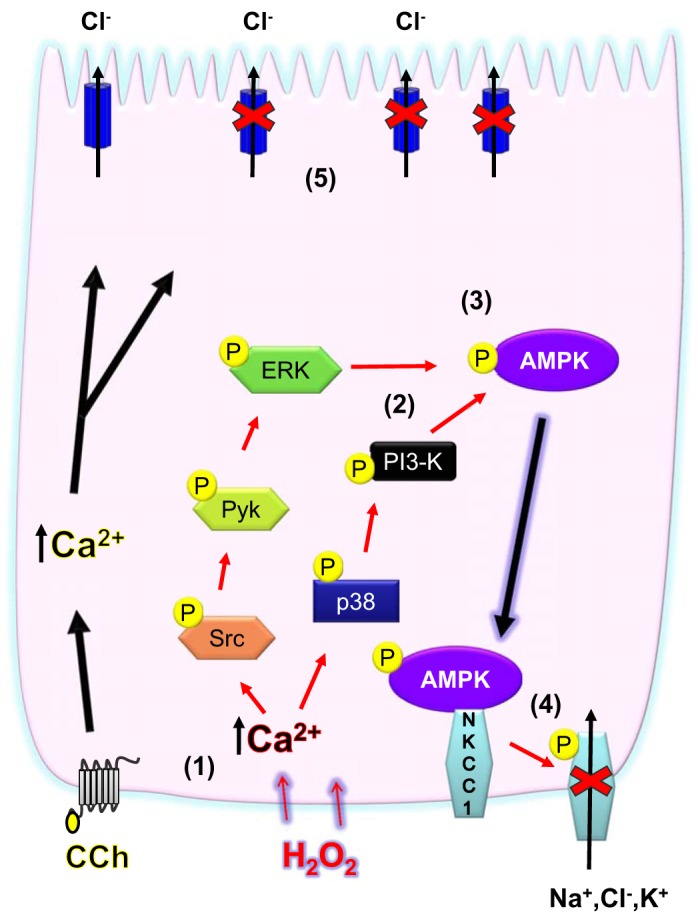
Signaling pathways activated by H2O2 to inhibit Na+-K+-2Cl− cotransporter NKCC1 converge at AMP-activated protein kinase (AMPK). H2O2 stimulates an increase in cytosolic Ca2+ concentration (1). Two separate Ca2+-stimulated signaling pathways [Src-Pyk2-ERK and p38- phosphatidylinositol 3-kinase (PI3K)] are activated (2). These pathways converge to increase AMPK activation (3). H2O2 also inhibits basolateral Ca2+-responsive K+ influx mediated by NKCC1 in an AMPK-dependent manner, and H2O2 increases physical association of AMPK with NKCC1 and increases NKCC1 phosphorylation via AMPK activity (4). Suppression of NKCC1 activity likely contributes to the overall inhibitory effect of H2O2 on Ca2+-dependent ion transport in colonic epithelial cells (5).
GRANTS
This work was supported by a Pease-Burden Fellowship (to S. J. King), a Crohn’s and Colitis Foundation Career Development Award (to D. F. McCole), and National Institute of Diabetes and Digestive and Kidney Diseases Grant 2R01 DK-091281 (to D. F. McCole).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.J.K., M.B., C.L., and D.F.M. conceived and designed research; S.J.K., M.B., A.C., M.S., M.D., B.J., and D.F.M. performed experiments; S.J.K., M.B., A.C., M.S., and D.F.M. analyzed data; S.J.K., M.B., C.L., and D.F.M. interpreted results of experiments; S.J.K., M.B., A.C., and D.F.M. prepared figures; S.J.K., M.B., B.J., C.L., and D.F.M. edited and revised manuscript; S.J.K., M.B., A.C., M.S., M.D., B.J., C.L., and D.F.M. approved final version of manuscript; D.F.M. drafted manuscript.
ACKNOWLEDGMENTS
We are grateful to Merrie Mosedale (University of California, San Diego) and Kristian Lucas (University of California, Riverside) for technical assistance. We are grateful to Dr. Kim E. Barrett (University of California, San Diego) for providing T84 and HT-29.cl19a cell lines and to Dr. Biff Forbush (Yale University) for providing the R5 anti-phosphorylated (Thr212, Thr217) NKCC1 antibody.
Current addresses: A. Chappell, Ionis Pharmaceuticals, 2855 Gazelle Ct., Carlsbad, CA 92010; B. Jung, Div. of Gastroenterology and Hepatology, University of Illinois at Chicago, Chicago, IL 60612.
REFERENCES
- 1.Amasheh S, Barmeyer C, Koch CS, Tavalali S, Mankertz J, Epple HJ, Gehring MM, Florian P, Kroesen AJ, Zeitz M, Fromm M, Schulzke JD. Cytokine-dependent transcriptional down-regulation of epithelial sodium channel in ulcerative colitis. Gastroenterology 126: 1711–1720, 2004. doi: 10.1053/j.gastro.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 2.Barrett KE, Keely SJ. Chloride secretion by the intestinal epithelium: molecular basis and regulatory aspects. Annu Rev Physiol 62: 535–572, 2000. doi: 10.1146/annurev.physiol.62.1.535. [DOI] [PubMed] [Google Scholar]
- 3.Barrett KE, McCole DF. Hydrogen peroxide scavenger, catalase, alleviates ion transport dysfunction in murine colitis. Clin Exp Pharmacol Physiol 43: 1097–1106, 2016. doi: 10.1111/1440-1681.12646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bevins CL, Salzman NH. The potter’s wheel: the host’s role in sculpting its microbiota. Cell Mol Life Sci 68: 3675–3685, 2011. doi: 10.1007/s00018-011-0830-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhalla V, Oyster NM, Fitch AC, Wijngaarden MA, Neumann D, Schlattner U, Pearce D, Hallows KR. AMP-activated kinase inhibits the epithelial Na+ channel through functional regulation of the ubiquitin ligase Nedd4-2. J Biol Chem 281: 26159–26169, 2006. doi: 10.1074/jbc.M606045200. [DOI] [PubMed] [Google Scholar]
- 6.Biasi F, Leonarduzzi G, Oteiza PI, Poli G. Inflammatory bowel disease: mechanisms, redox considerations, and therapeutic targets. Antioxid Redox Signal 19: 1711–1747, 2013. doi: 10.1089/ars.2012.4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouyer PG, Tang X, Weber CR, Shen L, Turner JR, Matthews JB. Capsaicin induces NKCC1 internalization and inhibits chloride secretion in colonic epithelial cells independently of TRPV1. Am J Physiol Gastrointest Liver Physiol 304: G142–G156, 2013. doi: 10.1152/ajpgi.00483.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cartwright CA, McRoberts JA, Mandel KG, Dharmsathaphorn K. Synergistic action of cyclic adenosine monophosphate- and calcium-mediated chloride secretion in a colonic epithelial cell line. J Clin Invest 76: 1837–1842, 1985. doi: 10.1172/JCI112176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chappell AE, Bunz M, Smoll E, Dong H, Lytle C, Barrett KE, McCole DF. Hydrogen peroxide inhibits Ca2+-dependent chloride secretion across colonic epithelial cells via distinct kinase signaling pathways and ion transport proteins. FASEB J 22: 2023–2036, 2008. doi: 10.1096/fj.07-099697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi SL, Kim SJ, Lee KT, Kim J, Mu J, Birnbaum MJ, Soo Kim S, Ha J. The regulation of AMP-activated protein kinase by H2O2. Biochem Biophys Res Commun 287: 92–97, 2001. doi: 10.1006/bbrc.2001.5544. [DOI] [PubMed] [Google Scholar]
- 11.Choudhary S, Keshavarzian A, Yong S, Wade M, Bocckino S, Day BJ, Banan A. Novel antioxidants zolimid and AEOL11201 ameliorate colitis in rats. Dig Dis Sci 46: 2222–2230, 2001. doi: 10.1023/A:1011975218006. [DOI] [PubMed] [Google Scholar]
- 12.Conner EM, Brand SJ, Davis JM, Kang DY, Grisham MB. Role of reactive metabolites of oxygen and nitrogen in inflammatory bowel disease: toxins, mediators, and modulators of gene expression. Inflamm Bowel Dis 2: 133–147, 1996. doi: 10.1097/00054725-199606000-00011. [DOI] [PubMed] [Google Scholar]
- 13.Cooke HJ. “Enteric tears”: chloride secretion and its neural regulation. News Physiol Sci 13: 269–274, 1998. doi: 10.1152/physiologyonline.1998.13.6.269. [DOI] [PubMed] [Google Scholar]
- 14.Corton JM, Gillespie JG, Hardie DG. Role of the AMP-activated protein kinase in the cellular stress response. Curr Biol 4: 315–324, 1994. doi: 10.1016/S0960-9822(00)00070-1. [DOI] [PubMed] [Google Scholar]
- 15.Dale S, Wilson WA, Edelman AM, Hardie DG. Similar substrate recognition motifs for mammalian AMP-activated protein kinase, higher plant HMG-CoA reductase kinase-A, yeast SNF1, and mammalian calmodulin-dependent protein kinase I. FEBS Lett 361: 191–195, 1995. doi: 10.1016/0014-5793(95)00172-6. [DOI] [PubMed] [Google Scholar]
- 16.Darman RB, Forbush B. A regulatory locus of phosphorylation in the N terminus of the Na-K-Cl cotransporter, NKCC1. J Biol Chem 277: 37542–37550, 2002. doi: 10.1074/jbc.M206293200. [DOI] [PubMed] [Google Scholar]
- 17.Davies M, Fraser SA, Galic S, Choy SW, Katerelos M, Gleich K, Kemp BE, Mount PF, Power DA. Novel mechanisms of Na+ retention in obesity: phosphorylation of NKCC2 and regulation of SPAK/OSR1 by AMPK. Am J Physiol Renal Physiol 307: F96–F106, 2014. doi: 10.1152/ajprenal.00524.2013. [DOI] [PubMed] [Google Scholar]
- 18.Del Castillo IC, Fedor-Chaiken M, Song JC, Starlinger V, Yoo J, Matlin KS, Matthews JB. Dynamic regulation of Na+-K+-2Cl− cotransporter surface expression by PKC-ε in Cl−-secretory epithelia. Am J Physiol Cell Physiol 289: C1332–C1343, 2005. doi: 10.1152/ajpcell.00580.2004. [DOI] [PubMed] [Google Scholar]
- 19.Ducommun S, Ford RJ, Bultot L, Deak M, Bertrand L, Kemp BE, Steinberg GR, Sakamoto K. Enhanced activation of cellular AMPK by dual-small molecule treatment: AICAR and A769662. Am J Physiol Endocrinol Metab 306: E688–E696, 2014. doi: 10.1152/ajpendo.00672.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DuVall MD, Guo Y, Matalon S. Hydrogen peroxide inhibits cAMP-induced Cl− secretion across colonic epithelial cells. Am J Physiol Cell Physiol 275: C1313–C1322, 1998. doi: 10.1152/ajpcell.1998.275.5.C1313. [DOI] [PubMed] [Google Scholar]
- 21.Flagella M, Clarke LL, Miller ML, Erway LC, Giannella RA, Andringa A, Gawenis LR, Kramer J, Duffy JJ, Doetschman T, Lorenz JN, Yamoah EN, Cardell EL, Shull GE. Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J Biol Chem 274: 26946–26955, 1999. doi: 10.1074/jbc.274.38.26946. [DOI] [PubMed] [Google Scholar]
- 22.Flemmer AW, Gimenez I, Dowd BF, Darman RB, Forbush B. Activation of the Na-K-Cl cotransporter NKCC1 detected with a phospho-specific antibody. J Biol Chem 277: 37551–37558, 2002. doi: 10.1074/jbc.M206294200. [DOI] [PubMed] [Google Scholar]
- 23.Fraser SA, Davies M, Katerelos M, Gleich K, Choy SW, Steel R, Galic S, Mount PF, Kemp BE, Power DA. Activation of AMPK reduces the co-transporter activity of NKCC1. Mol Membr Biol 31: 95–102, 2014. doi: 10.3109/09687688.2014.902128. [DOI] [PubMed] [Google Scholar]
- 24.Fraser SA, Gimenez I, Cook N, Jennings I, Katerelos M, Katsis F, Levidiotis V, Kemp BE, Power DA. Regulation of the renal-specific Na+-K+-2Cl− co-transporter NKCC2 by AMP-activated protein kinase (AMPK). Biochem J 405: 85–93, 2007. doi: 10.1042/BJ20061850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fryer LG, Hajduch E, Rencurel F, Salt IP, Hundal HS, Hardie DG, Carling D. Activation of glucose transport by AMP-activated protein kinase via stimulation of nitric oxide synthase. Diabetes 49: 1978–1985, 2000. doi: 10.2337/diabetes.49.12.1978. [DOI] [PubMed] [Google Scholar]
- 26.Greig E, Sandle GI. Diarrhea in ulcerative colitis. The role of altered colonic sodium transport. Ann NY Acad Sci 915: 327–332, 2000. doi: 10.1111/j.1749-6632.2000.tb05260.x. [DOI] [PubMed] [Google Scholar]
- 27.Grisham MB. Oxidants and free radicals in inflammatory bowel disease. Lancet 344: 859–861, 1994. doi: 10.1016/S0140-6736(94)92831-2. [DOI] [PubMed] [Google Scholar]
- 28.Hallows KR. Emerging role of AMP-activated protein kinase in coupling membrane transport to cellular metabolism. Curr Opin Nephrol Hypertens 14: 464–471, 2005. doi: 10.1097/01.mnh.0000174145.14798.64. [DOI] [PubMed] [Google Scholar]
- 29.Hallows KR, Kobinger GP, Wilson JM, Witters LA, Foskett JK. Physiological modulation of CFTR activity by AMP-activated protein kinase in polarized T84 cells. Am J Physiol Cell Physiol 284: C1297–C1308, 2003. doi: 10.1152/ajpcell.00227.2002. [DOI] [PubMed] [Google Scholar]
- 30.Hallows KR, Raghuram V, Kemp BE, Witters LA, Foskett JK. Inhibition of cystic fibrosis transmembrane conductance regulator by novel interaction with the metabolic sensor AMP-activated protein kinase. J Clin Invest 105: 1711–1721, 2000. doi: 10.1172/JCI9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hardie DG, Carling D. The AMP-activated protein kinase—fuel gauge of the mammalian cell? Eur J Biochem 246: 259–273, 1997. doi: 10.1111/j.1432-1033.1997.00259.x. [DOI] [PubMed] [Google Scholar]
- 32.Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem 67: 821–855, 1998. doi: 10.1146/annurev.biochem.67.1.821. [DOI] [PubMed] [Google Scholar]
- 33.Harris ML, Schiller HJ, Reilly PM, Donowitz M, Grisham MB, Bulkley GB. Free radicals and other reactive oxygen metabolites in inflammatory bowel disease: cause, consequence or epiphenomenon? Pharmacol Ther 53: 375–408, 1992. doi: 10.1016/0163-7258(92)90057-7. [DOI] [PubMed] [Google Scholar]
- 34.Hirota CL, McKay DM. Loss of Ca-mediated ion transport during colitis correlates with reduced ion transport responses to a Ca-activated K channel opener. Br J Pharmacol 156: 1085–1097, 2009. doi: 10.1111/j.1476-5381.2009.00122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kachur JF, Keshavarzian A, Sundaresan R, Doria M, Walsh R, de las Alas MM, Gaginella TS. Colitis reduces short-circuit current response to inflammatory mediators in rat colonic mucosa. Inflammation 19: 245–259, 1995. doi: 10.1007/BF01534465. [DOI] [PubMed] [Google Scholar]
- 36.Keely SJ, Calandrella SO, Barrett KE. Carbachol-stimulated transactivation of epidermal growth factor receptor and mitogen-activated protein kinase in T84 cells is mediated by intracellular Ca2+, PYK-2, and p60src. J Biol Chem 275: 12619–12625, 2000. doi: 10.1074/jbc.275.17.12619. [DOI] [PubMed] [Google Scholar]
- 37.Keshavarzian A, Haydek J, Zabihi R, Doria M, D’Astice M, Sorenson JR. Agents capable of eliminating reactive oxygen species. Catalase, WR-2721, or Cu(II)2(3,5-DIPS)4 decrease experimental colitis. Dig Dis Sci 37: 1866–1873, 1992. [DOI] [PubMed] [Google Scholar]
- 38.Kruidenier L, Kuiper I, Lamers CB, Verspaget HW. Intestinal oxidative damage in inflammatory bowel disease: semi-quantification, localization, and association with mucosal antioxidants. J Pathol 201: 28–36, 2003. doi: 10.1002/path.1409. [DOI] [PubMed] [Google Scholar]
- 39.Li W, Duzgun A, Sumpio BE, Basson MD. Integrin and FAK-mediated MAPK activation is required for cyclic strain mitogenic effects in Caco-2 cells. Am J Physiol Gastrointest Liver Physiol 280: G75–G87, 2001. doi: 10.1152/ajpgi.2001.280.1.G75. [DOI] [PubMed] [Google Scholar]
- 40.Liangpunsakul S, Wou SE, Zeng Y, Ross RA, Jayaram HN, Crabb DW. Effect of ethanol on hydrogen peroxide-induced AMPK phosphorylation. Am J Physiol Gastrointest Liver Physiol 295: G1173–G1181, 2008. doi: 10.1152/ajpgi.90349.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lih-Brody L, Powell SR, Collier KP, Reddy GM, Cerchia R, Kahn E, Weissman GS, Katz S, Floyd RA, McKinley MJ, Fisher SE, Mullin GE. Increased oxidative stress and decreased antioxidant defenses in mucosa of inflammatory bowel disease. Dig Dis Sci 41: 2078–2086, 1996. doi: 10.1007/BF02093613. [DOI] [PubMed] [Google Scholar]
- 42.Liu X, Chhipa RR, Pooya S, Wortman M, Yachyshin S, Chow LM, Kumar A, Zhou X, Sun Y, Quinn B, McPherson C, Warnick RE, Kendler A, Giri S, Poels J, Norga K, Viollet B, Grabowski GA, Dasgupta B. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc Natl Acad Sci USA 111: E435–E444, 2014. doi: 10.1073/pnas.1311121111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lynch SV, Goldfarb KC, Wild YK, Kong W, De Lisle RC, Brodie EL. Cystic fibrosis transmembrane conductance regulator knockout mice exhibit aberrant gastrointestinal microbiota. Gut Microbes 4: 41–47, 2013. doi: 10.4161/gmic.22430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lytle C, Forbush B III. Na-K-Cl cotransport in the shark rectal gland. II. Regulation in isolated tubules. Am J Physiol Cell Physiol 262: C1009–C1017, 1992. doi: 10.1152/ajpcell.1992.262.4.C1009. [DOI] [PubMed] [Google Scholar]
- 45.Lytle C, Forbush B III. The Na-K-Cl cotransport protein of shark rectal gland. II. Regulation by direct phosphorylation. J Biol Chem 267: 25438–25443, 1992. [PubMed] [Google Scholar]
- 46.Lytle C, Xu JC, Biemesderfer D, Forbush B III. Distribution and diversity of Na-K-Cl cotransport proteins: a study with monoclonal antibodies. Am J Physiol Cell Physiol 269: C1496–C1505, 1995. doi: 10.1152/ajpcell.1995.269.6.C1496. [DOI] [PubMed] [Google Scholar]
- 47.McCole DF, Barrett KE. Varied role of the gut epithelium in mucosal homeostasis. Curr Opin Gastroenterol 23: 647–654, 2007. doi: 10.1097/MOG.0b013e3282f0153b. [DOI] [PubMed] [Google Scholar]
- 48.Nguyen TD, Canada AT. Modulation of human colonic T84 cell secretion by hydrogen peroxide. Biochem Pharmacol 47: 403–410, 1994. doi: 10.1016/0006-2952(94)90032-9. [DOI] [PubMed] [Google Scholar]
- 49.Norkina O, Kaur S, Ziemer D, De Lisle RC. Inflammation of the cystic fibrosis mouse small intestine. Am J Physiol Gastrointest Liver Physiol 286: G1032–G1041, 2004. doi: 10.1152/ajpgi.00473.2003. [DOI] [PubMed] [Google Scholar]
- 50.Oz HS, Chen TS, McClain CJ, de Villiers WJ. Antioxidants as novel therapy in a murine model of colitis. J Nutr Biochem 16: 297–304, 2005. doi: 10.1016/j.jnutbio.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 51.Pavlick KP, Laroux FS, Fuseler J, Wolf RE, Gray L, Hoffman J, Grisham MB. Role of reactive metabolites of oxygen and nitrogen in inflammatory bowel disease. Free Radic Biol Med 33: 311–322, 2002. doi: 10.1016/S0891-5849(02)00853-5. [DOI] [PubMed] [Google Scholar]
- 52.Rezaie A, Parker RD, Abdollahi M. Oxidative stress and pathogenesis of inflammatory bowel disease: an epiphenomenon or the cause? Dig Dis Sci 52: 2015–2021, 2007. doi: 10.1007/s10620-006-9622-2. [DOI] [PubMed] [Google Scholar]
- 53.Rogers AC, Huetter L, Hoekstra N, Collins D, Collaco A, Baird AW, Winter DC, Ameen N, Geibel JP, Kopic S. Activation of AMPK inhibits cholera toxin stimulated chloride secretion in human and murine intestine. PLoS One 8: e69050, 2013. doi: 10.1371/journal.pone.0069050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sandle GI, Higgs N, Crowe P, Marsh MN, Venkatesan S, Peters TJ. Cellular basis for defective electrolyte transport in inflamed human colon. Gastroenterology 99: 97–105, 1990. doi: 10.1016/0016-5085(90)91235-X. [DOI] [PubMed] [Google Scholar]
- 55.Scharl M, Paul G, Barrett KE, McCole DF. AMP-activated protein kinase mediates the interferon-γ-induced decrease in intestinal epithelial barrier function. J Biol Chem 284: 27952–27963, 2009. doi: 10.1074/jbc.M109.046292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scharl M, Paul G, Weber A, Jung BC, Docherty MJ, Hausmann M, Rogler G, Barrett KE, McCole DF. Protection of epithelial barrier function by the Crohn’s disease associated gene protein tyrosine phosphatase N2. Gastroenterology 137: 2030–2040.e5, 2009. doi: 10.1053/j.gastro.2009.07.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmitz H, Barmeyer C, Gitter AH, Wullstein F, Bentzel CJ, Fromm M, Riecken EO, Schulzke JD. Epithelial barrier and transport function of the colon in ulcerative colitis. Ann NY Acad Sci 915: 312–326, 2000. doi: 10.1111/j.1749-6632.2000.tb05259.x. [DOI] [PubMed] [Google Scholar]
- 58.Schultheiss G, Hennig B, Diener M. Sites of action of hydrogen peroxide on ion transport across rat distal colon. Br J Pharmacol 154: 991–1000, 2008. doi: 10.1038/bjp.2008.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scott JW, van Denderen BJ, Jorgensen SB, Honeyman JE, Steinberg GR, Oakhill JS, Iseli TJ, Koay A, Gooley PR, Stapleton D, Kemp BE. Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chem Biol 15: 1220–1230, 2008. doi: 10.1016/j.chembiol.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 60.Sid B, Miranda L, Vertommen D, Viollet B, Rider MH. Stimulation of human and mouse erythrocyte Na+-K+-2Cl− cotransport by osmotic shrinkage does not involve AMP-activated protein kinase, but is associated with STE20/SPS1-related proline/alanine-rich kinase activation. J Physiol 588: 2315–2328, 2010. doi: 10.1113/jphysiol.2009.185900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sid B, Verrax J, Calderon PB. Role of AMPK activation in oxidative cell damage: implications for alcohol-induced liver disease. Biochem Pharmacol 86: 200–209, 2013. doi: 10.1016/j.bcp.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 62.Smith L, Litman P, Liedtke CM. COMMD1 interacts with the COOH terminus of NKCC1 in Calu-3 airway epithelial cells to modulate NKCC1 ubiquitination. Am J Physiol Cell Physiol 305: C133–C146, 2013. doi: 10.1152/ajpcell.00394.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smith L, Smallwood N, Altman A, Liedtke CM. PKCδ acts upstream of SPAK in the activation of NKCC1 by hyperosmotic stress in human airway epithelial cells. J Biol Chem 283: 22147–22156, 2008. doi: 10.1074/jbc.M801752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Test ST, Weiss SJ. Quantitative and temporal characterization of the extracellular H2O2 pool generated by human neutrophils. J Biol Chem 259: 399–405, 1984. [PubMed] [Google Scholar]
- 65.Tomas J, Mulet C, Saffarian A, Cavin JB, Ducroc R, Regnault B, Kun Tan C, Duszka K, Burcelin R, Wahli W, Sansonetti PJ, Pédron T. High-fat diet modifies the PPAR-γ pathway leading to disruption of microbial and physiological ecosystem in murine small intestine. Proc Natl Acad Sci USA 113: E5934–E5943, 2016. doi: 10.1073/pnas.1612559113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vincent EE, Coelho PP, Blagih J, Griss T, Viollet B, Jones RG. Differential effects of AMPK agonists on cell growth and metabolism. Oncogene 34: 3627–3639, 2015. doi: 10.1038/onc.2014.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Walker J, Jijon HB, Churchill T, Kulka M, Madsen KL. Activation of AMP-activated protein kinase reduces cAMP-mediated epithelial chloride secretion. Am J Physiol Gastrointest Liver Physiol 285: G850–G860, 2003. doi: 10.1152/ajpgi.00077.2003. [DOI] [PubMed] [Google Scholar]
- 68.Walker J, Jijon HB, Diaz H, Salehi P, Churchill T, Madsen KL. 5-Aminoimidazole-4-carboxamide riboside (AICAR) enhances GLUT2-dependent jejunal glucose transport: a possible role for AMPK. Biochem J 385: 485–491, 2005. doi: 10.1042/BJ20040694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Watson AJ, Askew JN, Sandle GI. Characterisation of oxidative injury to an intestinal cell line (HT-29) by hydrogen peroxide. Gut 35: 1575–1581, 1994. doi: 10.1136/gut.35.11.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zheng Y, Shen X. H2O2 directly activates inositol 1,4,5-trisphosphate receptors in endothelial cells. Redox Rep 10: 29–36, 2005. doi: 10.1179/135100005X21660. [DOI] [PubMed] [Google Scholar]