

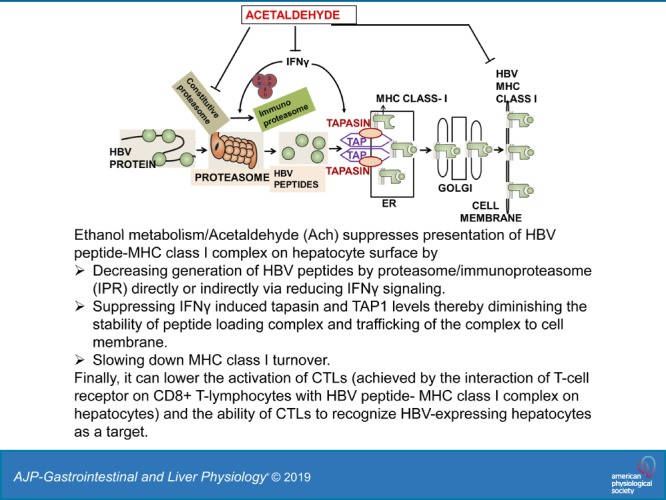
Keywords: acetaldehyde, CTL, HBV, IFNγ, proteasome
Abstract
Hepatitis B virus (HBV) infection and alcoholism are major public health problems worldwide, contributing to the development of end-stage liver disease. Alcohol intake affects HBV infection pathogenesis and treatment outcomes. HBV-specific cytotoxic T lymphocytes (CTLs) play an important role in HBV clearance. Many previous studies have focused on alcohol-induced impairments of the immune response. However, it is not clear whether alcohol alters the presentation of HBV peptide-major histocompatibility complex (MHC) class I complexes on infected hepatocytes resulting in escape of its recognition by CTLs. Hence, the focus of this study was to investigate the mechanisms by which ethanol metabolism affects the presentation of CTL epitope on HBV-infected hepatocytes. As demonstrated here, although continuous cell exposure to acetaldehyde-generating system (AGS) increased HBV load in HepG2.2.15 cells, it decreased the expression of HBV core peptide 18–27-human leukocyte antigen-A2complex (CTL epitope) on the cell surface. Moreover, we observed AGS-induced suppression of chymotrypsin- and trypsin-like proteasome activities necessary for peptide processing by proteasome as well as a decline in IFNγ-stimulated immunoproteasome (IPR) function and expression of PA28 activator and immunoproteasome subunits LMP7 and LMP2. Furthermore, IFNγ-induced activation of peptide-loading complex (PLC) components, such as transporter associated with antigen processing (TAP1) and tapasin, were suppressed by AGS. The attenuation of IPR and PLC activation was attributed to AGS-triggered impairment of IFNγ signaling in HepG2.2.15 cells. Collectively, all these downstream events reduced the display of HBV peptide-MHC class I complexes on the hepatocyte surface, which may suppress CTL activation and the recognition of CTL epitopes on HBV-expressing hepatocytes by immune cells, thereby leading to persistence of liver inflammation.
NEW & NOTEWORTHY Our study shows that in HBV-expressing HepG2.2.15 cells, acetaldehyde alters HBV peptide processing by suppressing chymotrypsin- and trypsin-like proteasome activities and decreases IFNγ-stimulated immunoproteasome function and expression of PA28 activator and immunoproteasome subunits. It also suppresses IFNγ-induced activation of peptide-loading complex (PLC) components due to impairment of IFNγ signaling via the JAK-STAT1 pathway. These acetaldehyde-induced dysfunctions reduced the display of HBV peptide-MHC class I complexes on the hepatocyte surface, thereby leading to persistence of HBV infection.
INTRODUCTION
Alcohol and hepatitis viruses are synergies in the development of liver disease (12). According to a World Health Organization report of 2018, worldwide, ~257 million people are chronically infected with the hepatitis B v irus (HBV), which results in 887,000 deaths from HBV complications (70). Alcohol exacerbates the clinical course of HBV infection by increasing viral load (44) and by suppressing elimination of the infected hepatocytes. The prevalence of hepatocellular carcinoma (HCC) and liver-related mortality is higher in people with chronic HBV infection and concurrent heavy alcohol consumption (29, 47). Importantly, the association of alcohol consumption with chronic hepatitis B in the progression of liver disease has been less extensively studied than that with chronic hepatitis C (30).
In HBV infection, viral clearance and disease pathogenesis are largely mediated by the adaptive immune response (26). Since HBV is not a cytopathogenic virus, the clearance of HBV-expressing hepatocytes is performed by an immune response mediated mainly by cytotoxic CD8+ T lymphocytes (CTLs) (27, 31). Being a major effector component of antiviral immunity, CD8+ T cells are able to recognize and target the HBV+ hepatocytes to terminate viral spread in the host via direct lysis or inhibition of viral replication by cytokine release. Whereas in self-limiting (acute hepatitis) there is a vigorous CD8+ T cell response against viral epitopes presented on HBV-infected hepatocytes, chronic hepatitis B is associated with a defective CTL response (7). Since activation of CTLs is programmed by the presentation of viral peptide-major histocompatibility complex I (MHC class I) complex on HBV-infected hepatocytes (the target cells), the impaired presentation of HBV peptide-MHC class I complexes on the infected hepatocyte surface reduces activation of CTLs, leading to ineffective control on HBV-expressing cells. This may cause chronic liver inflammation without infected cell clearance.
Alcohol can interfere with clearance of HBV-expressing hepatocytes and establishment of chronic hepatitis by evading clearance of HBV-infected cells by CTLs (29). Meanwhile, exact mechanisms how it happens are not well established (23). In other hepatotropic infections, like hepatitis C, alcohol metabolism interferes with effective immune response (53) and, partially, with impaired presentation of antigenic peptides in the context of MHC class I (50). However, whether this is the case for HBV infection, has not been investigated yet.
For presentation of viral peptide-MHC class I complex on the hepatocyte surface, viral antigens need to be processed to peptides by proteasomes. Proteasomes, the major cytosolic proteinase complexes, are essential for the generation of most MHC class I-presented peptides. Both 26S and 20S proteasomes degrade antigenic proteins up to the size of 9–11 amino acids that allows MHC class I folding. The chymotrypsin-like (Cht-L) and trypsin-like (T-L) activities of the proteasome are related to antigen presentation due to their respective abilities to cleave peptide bonds after hydrophobic and basic amino acids (25, 58). Cleavage of antigenic peptides becomes more effective when the constitutive β-subunits of the proteasome are replaced by immunoproteasome (IPR) subunits. Expression of IPR subunits is tissue specific, and their incorporation into 20S proteasome is driven by IFNγ (2). Our previous studies indicated the role of an IPR activator, PA28α, in IFNγ-driven induction of IPR as a key regulator of viral peptide cleavage in infected hepatocytes (50, 55, 56).
Proteasome-generated peptides of certain sizes are transported to the endoplasmic reticulum, where they bind to transporters associated with antigen processing (TAPs, TAP1 and TAP2) and assemble in a complex with β2-microglobulin and the heavy chain of MHC class I. Assembly of peptide-MHC class I with TAPs is stabilized by tapasin and chaperones, allowing optimal MHC class I loading (9, 11). These complexes are trafficked via the trans-Golgi to the plasma membrane, where they are recognized by the T lymphocyte cell receptor (TCR) (13, 18). The importance of TAPs and tapasin for expression of HBV CTL epitopes as well as IPR for IFNγ-activated MHC class I-restricted antigen presentation on hepatocytes has been previously reported (11, 39), and TAP polymorphism plays a significant role in infection outcomes (59). Particularly, HBV core peptide 18–27 (FLPSDEFPSV) has been characterized as a CTL epitope (4, 5, 43, 57) with therapeutic potential (8, 63). This peptide generated by proteasome-mediated cleavage is presented in the context of human leukocyte antigen-A2 (HLA-A2).
Because the hepatocyte is a primary site of both HBV replication and ethanol metabolism altering proteasome function (14, 52, 56), we hypothesized that ethanol metabolism decreases the display of HBV peptide-MHC class I complexes on the hepatocyte surface by impairing HBV peptide proteasomal cleavage and by disrupting peptide-MHC class I traffic to the cell surface.
To mimic ethanol metabolism in stably HBV-transfected ethanol nonmetabolizing HepG2.2.15 cells, which express HBV core peptide 18–27 in the context of HLA-A2 (36), we employed an acetaldehyde (Ach)-generating system (AGS) (19–22) providing a continuous enzymatic generation of physiologically relevant amounts of Ach without causing necrotic cell death (22). Since liver contains mixed proteasomes (constitutive and IPRs), the most efficient generation of antigenic peptides requires hepatocyte exposure to IFNγ (36, 60). Thus, HBV-transfected cells were exposed to IFNγ for activation of IPR/peptide cleavage/trafficking of peptide complex to the cell surface. Then, the effects of ethanol metabolism and the mechanisms behind the observed changes in the display of CTL epitope on hepatocyte surface were elucidated.
MATERIALS AND METHODS
Reagents and media.
High-glucose Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum were purchased from Invitrogen (Carlsbad, CA). TRIzol was from Life Technologies (Carlsbad, CA). PCR and RT-PCR reagents, such as the High Capacity cDNA Reverse Transcription Kit and TaqMan Universal Master Mix II, with UNG were from Applied Biosystems by Thermo Fisher Scientific (Foster city, CA). Human recombinant IFNγ was from PeproTech (Rocky Hill, NJ). C-extended HBV core peptide FLPSDFFPSVRDLLDTA was from Biomatik (Wilmington, DE). Proteasome activity substrates Suc-LLVY-AMC, Boc-LRR-AMC, and ONX-0914 (referred as ONX in this paper) IPR inhibitor were from UBPBio (Aurora, CO). Immunoproteasome substrate Ac-Ala-Asn-Trp-AMC was from BostonBiochem (cat. no. S-320, Cambridge MA). For flow cytometry studies, Human Fc receptor-binding inhibitor, anti-human HLA-A2 allophycocyanin (APC), and mouse isotype control were from Affymetrix-eBioscience (San Diego, CA). Expression of HBV core peptide FLPSDEFPSV-HLA-A2 was measured by flow cytometry using antibody to 18–27 HBV peptide-HLA-A2 purified from supernatant of hybridoma cells at the Department of Internal Medicine, University of Nebraska Medical Center (Omaha, NE). Hybridoma cells were obtained from PHARMEXA (San Diego, CA). LMP2 and LMP7 antibodies were form Santa Cruz Biotechnology (Santa Cruz, CA). Proteasome activator PA28α antibody was from Cell Signaling (Beverly, MA). Anti-TAP-1, anti-tapasin, an danti-HBV core were obtained from Abcam (Cambridge, MA).
Cells and treatments.
In this study, we used HepG2.2.15 cells that are stably transfected with HBV and able to replicate virus and produce viral particles (HBV genotype D) (72). To mimic ethanol metabolism, we employed an AGS, which does not require transfection for generation of Ach. We believe that Ach is a major ethanol metabolite in HBV-infected cells, since HBx protein downregulates cytochrome P-4502E1 (CYP2E1) expression (41). These cells were treated or not with AGS for 72 h. We have characterized the AGS in our recent publications (19–22). Briefly, AGS contains yeast ADH, 50 mM ethanol as a substrate, and nicotinamide adenine dinucleotide (NAD+) as a cofactor and provides continuous enzymatic generation of physiologically relevant amounts of Ach for at least 72 h. This generated Ach easily penetrates the cells and induces biological effects without causing significant cell toxicity (22). For the final 24 h of treatment, cells were exposed (or not) to 2 ng/ml IFNγ. This dose of IFNγ does not affect HBV replication in HepG2.2.15 cells.
Lactate dehydrogenase assay and apoptosis assay.
Cytotoxicity was measured by lactate dehydrogenase (LDH) release to the cell medium as described previously (22). The percentage of apoptotic cells was determined, as previously published (19), by flow cytometry (BD LSR-II-Green) using an Annexin-V Apoptosis Detection kit (BD Biosciences, San Diego, CA) according to the instructions of the manufacturer. Apoptosis was further confirmed by immunoblotting analysis of cleaved caspase-3.
RNA, DNA isolation, real-time PCR, and ddPCR.
Reagents for RNA isolation, cDNA synthesis, and real-time PCR were from Life Technologies and Applied Biosystems by Thermo Fisher Scientific (Carlsbad and Foster City, CA). Total RNA was isolated from cells using TRIzol reagent. A two-step procedure was applied, in which 200 ng of RNA was reverse-transcribed to cDNA using the high-capacity reverse transcription kit. Then, the cDNA was amplified using TaqMan Universal Master Mix-II with fluorescent labeled primers (TaqMan gene expression systems). After incubation in a model 7500 qRT-PCR thermal cycler, the relative quantity of each RNA transcript was calculated by its threshold cycle (CT) after subtraction of that of the reference cDNA (GAPDH). Data were expressed as the quantity of transcript (RQ). HBV infection was confirmed by the measurement of HBV RNA (Applied Biosystems; single vial primer probe).
HBV DNA levels were quantified by the droplet digital (dd)PCR method. Total DNA was prepared using the DNeasy Kit (Qiagen, Germany) according to the manufacturer’s protocol. The concentrations of DNA were quantified using the QX200 Droplet Digital PCR System (Bio-Rad, Hercules, CA) according to the manufacturer’s instructions. Briefly, the 20-μl ddPCR reaction comprised 2× ddPCR Supermix (5 μl), Reverse transcriptase (2 μl), and 300 mM DTT (1 μl; Bio-Rad, Pleasanton, CA) 900 nmol/HBV sense (5′-CGA CGT GCA GAG GTG AAG-3′), and antisense (5′-CAC CTC TCT TTA CGC GGA CT-3′) primers, 250 nmol HBV probe (5′-/56-FAM/ATC TGC CGG/ZEN/ACC GTG TGC AC/3IABkFQ/-3′), and 5 μl of adjusted DNA sample in RNase-free water. Primers and probes were from Integrated DNA Technologies (Coralville, IA). Prepared droplets were transferred to corresponding wells of a Bio-Rad 96-well PCR plate, using an Automated Droplet Generator as described in the instruction manual (no. 10043138). The PCR plate was subsequently heat-sealed with pierceable foil using the PX1 PCR plate sealer (Bio-Rad, Hercules, CA) and then amplified in the C1000 Touch deep-well thermal cycler (Bio-Rad). The cycling conditions were as follows: an initial denaturation cycle of 10 min at 95°C, followed by 45 cycles of denaturation for 30 s at 94°C, annealing for 60 s at 57°C (ramping rate set to 2°C/s), and a final incubation for 10 min at 98°C, ending at 4°C. After amplification, the 96-well plate was fixed in a plate holder and placed in the QX200 Droplet Reader. The ddPCR data were analyzed using QuantaSoft analysis software (Bio-Rad). Fluorescent signals of droplets were manipulated with the QuantaSoft analysis software version 1.8 (Bio-Rad). Positive droplets with higher fluorescent signals and negative droplets with lower fluorescent signals were divided by applying a fluorescence amplitude threshold. The absolute concentration of each sample was automatically reported by the ddPCR software by calculating the ratio of the positive droplets over the total droplets combined with Poisson distribution. Thus, the final concentration of the template was equal to the results, as calculated by the software, multiplied by the dilution factor of the template in the reaction system.
HBV surface antigen sandwich ELISA.
HBV surface antigen (HBsAg) levels were measured in the cell lysates by ELISA using an LSBio Kit (no. LS-F37979; LifeSpan Biosciences, Seattle, WA).
Immunofluorescence.
Large multifunctional proteases LMP2 and LMP7 IPR subunit staining were performed by immunofluorescence. HepG2.2.15 (HBV-transfected) cells were plated on coverslips containing six-well plates. After 48 h of plating, the cells were processed (including washing with PBS, fixation, permeabilization, and blocking) for staining and incubated with LMP2 and LMP7 primary antibodies (1:50 dilution) for 2 h and 1 h with secondary antibody (Alexa fluor 594 donkey anti-goat and goat anti-mouse, 1:200 dilution, Life Technologies). Nuclei were stained with DAPI. The presence of LMP2 and LMP7 was visualized using a ×63 lens in an LSM 710 confocal microscope (Carl Zeiss, Peabody, MA).
HBV core protein staining was performed by immunofluorescence as previously published (19, 22). Briefly, HepG2.2.15 cells (2–3 × 104 per well) were plated on an eight-well chamber slide, and the next day, cells were treated or not with AGS for 72 h. For the final 24 h of treatment, cells were exposed (or not) to 2 ng/ml IFNγ. Then, cells were stained with antibody to HBV core (no. ab8637, Abcam, diluted 1:100) for 2 h at room temperature followed by incubation with secondary antibody (Alexa fluor 555 goat anti-mouse (1:500, Life Technologies) for 1 h. Nuclei were labeled with DAPI. The presence of HBV core protein was visualized using a ×20 lens in the LSM 710 confocal microscope.
HBV 18–27 peptide-HLA-A2 MHC complex was measured by immunofluorescence in HepG2 cells (HBV-negative) and HepG2.2.15 (HBV-transfected) cells plated on coverslips. After 48 h of plating, the cells were processed for staining (including washing with PBS, fixation, permeabilization, and blocking) and incubated with HBV 18–27 peptide antibody (1:50 dilution) for 2 h and 1 h with secondary antibody (Alexa fluor 488 goat anti-mouse,1:200 dilution, Life Technologies). Nuclei were labeled with DAPI. The presence of HBV 18–27 peptide-MHC complex was visualized using the ×63 lens in the LSM 710 confocal microscope.
Proteasome activity.
Proteasome Cht-L and T-L peptidase activities were detected by in vitro fluorometric assay as previously reported by our laboratory (56). IPR activity was detected by in vitro fluorometric assay using the substrate Ac-Ala-Asn-Trp-AMC.
Immunoblotting (Western blot).
Cell lysates prepared in 0.5 M EDTA, 2 M Tris, 20 mM Na3VO4, 200 mM Na4P2O7, 100 mM PMSF, 1 M NaF, 20% Triton X-100, and aprotinin, pH 7, were separated and subjected to immunoblotting technique as previously described (21). Blots were developed using the Odyssey infrared imaging system, and the protein bands were quantified using Li-Cor software (Li-Cor Bioscience, Lincoln, NE).
Peptide cleavage detection.
A peptide cleavage assay was performed using HPLC, as previously reported by our laboratory (56). Cytosol protein (100 µg/ml) derived from cells was mixed with 10 nM C-extended peptide in 50 mM Tris·HCl (pH 8.5) and 5 mM MgCl2 in a total volume of 100 µl. The reaction was stopped by 20% trichloracetic acid. A 50-µl aliquot of each supernatant was subjected to reverse-phase HPLC on a Vydac C18 monomeric column equilibrated with 0.1% trifluoroacetic acid, with a flow rate of 1 ml/min. Peptides were eluted with a linear acetonitrile gradient ranging from 20 to 40% (vol/vol). Peptides were detected by their absorbance at 214 nm and their quantities calculated by integration of the peptide peaks on chromatograms. The percentage of remaining (uncleaved) peptide was calculated as integration units obtained from the intact peptide peak after incubation with cytosol, divided by integration units obtained from identically treated unincubated samples and multiplied by 100.
Flow cytometry analysis.
Cells were exposed to AGS or not for 72 h. IFNγ treatment was done in last 24 h of AGS treatment. The cells were then washed with PBS and detached using accutase (Invitrogen by Thermo Fisher Scientific). Cells were collected by centrifugation and then incubated with human Fc receptor-binding inhibitor for 20 min, followed by HBV peptide 18–27-HLA-A2 primary antibody for 60 min. After a washing, Alexa fluor 647 secondary antibody was added for 30 min, and the cells were fixed in 2% paraformaldehyde for flow cytometry analysis. All incubations were on ice. Data were collected on a BD LSR2 flow cytometer and analyzed using BD FACSDiva software v.6.0.
Peptide pulsed analysis.
HepG2 cells were pulsed with 5 µM HBV core 18–27 peptide for 2 h at room temperature, followed by 2 h at 37°C. Cells were washed three times. Then, flow cytometry analysis of HBV peptide 18–27-HLA-A2 complex was performed as mentioned above. Peptide-unpulsed HepG2 cells were used as a negative control.
Low acid wash.
Cells were treated or not with AGS for 72 h. Eight hours before the end of this experiment, cells were washed with citrate buffer (0.062 M Na2HPO4, 0.132 M citric acid, 0.5% BSA, pH 3) for 2 min, as described by Sugawara et al. (65). The citrate buffer wash was then neutralized by 3× wash with medium. Cells were incubated in fresh medium at 37°C for up to 4 h and analyzed by flow cytometry for expression of HLA-A2, as described above.
Statistical analyses.
Data from at least three duplicate independent experiments are expressed as mean values ± SE. Comparisons among multiple groups were determined by one-way ANOVA, using Tukey’s post hoc test. For comparisons between two groups, we used Student’s t-test. A probability value of 0.05 or less was considered significant.
RESULTS
AGS treatment did not cause toxicity and apoptosis in HepG2.2.15 cells.
To mimic ethanol metabolism, we employed an AGS, which does not require transfection with ethanol-metabolizing enzymes to generate Ach. Since HBx protein downregulates CYP2E1 expression, Ach is a major ethanol metabolite in full viral genome-expressing HepG2.2.15 cells (41). These cells were treated or not with AGS for 72 h. We have characterized the AGS in our recent publications (19–22). Briefly, AGS contains yeast ADH, 50 mM ethanol as a substrate, and NAD+ as a cofactor and provides continuous enzymatic generation of physiologically relevant amounts of Ach for at least 72 h. This generated Ach easily penetrates the cells and induces biological effects without causing significant cell toxicity (22). Here, we provide evidence that 72 h of AGS treatment did not cause apoptosis and necrosis in HBV-transfected HepG2.2.15 cells. Apoptosis was measured by both annexin-V-FITC staining and cleaved caspase-3 immunoblotting analysis (Fig. 1, A–C). Cytotoxicity (necrosis) was measured by LDH release to the cell medium (Fig. 1D).
Fig. 1.

Cytotoxic effect of acetaldehyde-generating system (AGS) on HepG2.2.15 cells. Cells were treated or not with AGS for 72 h and with interferon-γ (IFNγ) for the last 24 h. A: flow cytometric analysis of annexin V-FITC staining: apoptotic cells collected after UV light treatment was used as positive control, and the other 4 treatment cell groups were incubated with annexin V-FITC and analyzed by flow cytometry. B and C: apoptosis was further tested by immunoblotting with antibody to cleaved caspase-3; β-actin was used for normalization. D: cytotoxicity (necrosis) was measured by lactate dehydrogenase (LDH) release to cell medium; media from cells treated with Triton X-100 were used as positive control (100% LDH leakage). Percent cytotoxicity was calculated as (LDH activity in treatment group divided by LDH activity in positive control). Data from 3 independent experiments are presented as means ± SE. Bars marked with the same lowercase letter are not significantly different from each other; bars with different lowercase letters are significantly different (P ≤ 0 0.05).
AGS treatment affects proteasome activities in HepG2.2.15 cells.
Generation of antigenic peptides for MHC class I-restricted antigen presentation is carried out by proteasome. To elucidate whether the effects of Ach on the peptide-MHC class I complex presentation on cell surface are due to proteasome dysfunction, we measured the proteasome ChT-L and T-L activities necessary for processing the peptides for MHC class I loading. ChT-L and T-L proteasome activities were tested in in HepG2.2.15 cells exposed to AGS for 72 h. As shown in Fig. 2, A and B, we observed 18% suppression of ChT-L proteasome activity and 32% suppression of T-L activity by AGS in IFNγ-untreated cells. In IFNγ-treated cells, proteasome was more sensitive to AGS, suppressing both ChT-L and T-L activities in AGS-treated cells by 28 and 39%, respectively. To prove the activation of IPR with IFNγ, we measured IPR (β5i-LMP7 activity) by using an IPR substrate in IFNγ-untreated and -treated cells. Since hepatocytes contain a mixed proteasome (both constitutive and immunoproteasome (16, 17, 28, 37), using an IPR-specific substrate, we found IPR activity even in IFNγ-untreated cells, which was further reduced by 25% upon AGS exposure (Fig. 2C). However, IFNγ treatment enhanced IPR activity by 50% compared with IFNγ-untreated cells. To better characterize the IPR component in IFNγ-treated HepG2.2.15 cells, we also checked whether ChT-L and T-L proteasome activities could be suppressed by a specific IPR inhibitor, ONX (originally known as PR957). Thus, IFNγ-treated cells were exposed to ONX (100 nM, overnight), and then proteasome activities were measured. While ONX almost fully blocked Ch-T-L activity, it was only 40% suppression of T-L proteasome activity. Chronic exposure to AGS was less effective than to ONX for blocking Ch-T-L proteasome activity, but the magnitude of suppression of T-L activity by ONX in IFNγ-treated cells was similar to the effects of AGS (Fig. 2, D and E). Since IPR is known to optimize peptide cleavage for MHC class I-restricted antigen presentation (32) and because hepatocytes are naturally exposed to IFNγ during inflammation (hepatitis), to mimic these natural conditions in our hepatocyte-monoculture studies and to maximize the content of IPR in hepatocyte proteasome, we used IFNγ-treated HepG2.2.15 cells for further proteasome experiments.
Fig. 2.

Effect of acetaldehyde-generating system (AGS) on proteasome activities in HepG2.2.15 cells. Cells were treated or not with AGS for 72 h and with interferon-γ (IFNγ) for the last 24 h. Chymotrypsin-like (Cht-L; A) trypsin-like (T-L; B), and immunoproteasome (IPR)-β5i-LMP7 activities (C) were detected in cytosol fractions by in vitro fluorometric assay using fluorometric substrates Suc-LLVY-AMC, Boc-LRR-AMC, and Ac-ANW-AMC. D and E: Cht-L and T-L activities were measured in IFNγ-induced ONX (100 nM, overnight)-treated HepG2.2.15 cells. Data from 3 independent experiments are presented as means ± SE. Bars marked with the same lowercase letter are not significantly different from each other; bars with different lowercase letters are significantly different (P ≤ 0 0.05).
AGS treatment affects expression of PA28α proteasome activator and IPR subunits in HepG2.2.15 cells.
Here, we exposed cells to AGS in the presence IFNγ and then measured the expression of PA28 and IPR subunits (LMP2 and LMP7) by immunoblotting. IPR-cleaved antigenic peptides possess higher affinity to MHC class I (68), and IFNγ is necessary for maximizing incorporation of IPR subunits to proteasome. AGS suppressed (P ≤ 0 0.05) IFNγ-induced levels of PA28α, LMP2, and LMP7, confirming that Ach mainly targets IPR from IFNγ-stimulated cells (Fig. 3, A–D). These immunoblotting data were further confirmed by immunefluorescence staining of LMP2 and LMP7 (Fig. 4, A and B).
Fig. 3.

Effect of acetaldehyde-generating system (AGS) and hepatitis B virus (HBV) on proteasome subunit protein expression. Cells were treated or not with AGS for 72 h and with interferon-γ (IFNγ) for the last 24 h. A: proteasome activator PA28α and immunoproteasome subunits low-molecular-mass poylpeptide (LMP)2 and LMP7 protein expressions were detected by immunoblotting in cell lysates. Equal (20 µg) amounts of protein were loaded in each lane; β-actin was used as internal control. B, C, and D: quantification of immunoblotting bands from 3 independent experiments are presented as means ± SE. Bars marked with the same lowercase letter are not significantly different from each other; bars with different lowercase letters are significantly different (P ≤ 0 0.05).
Fig. 4.

Effects of acetaldehyde-generating system (AGS) on interferon-γ (IFNγ)-induced expression of immunoproteasome (IPR) subunits LMP2 and LMP7 protein expression. Cells were treated or not with AGS for 72 h and with IFNγ for last 24 h. A and B: immunofluorescence staining of low-molecular-mass poylpeptide (LMP)2 and LMP 7 expression. Staining was visualized using a ×63 lens in a LSM 710 confocal microscope. Pictures shown are representative of 3 independent experiments with similar results.
AGS treatment delays IFNγ-induced cleavage of HBV core C-extended peptide.
To confirm that AGS decreases proteasome-dependent cleavage of peptides presented in the context of HLA-A2 on the surface of HBV+ cells, we ran these experiments in IFNγ-treated cells exposed AGS. Here, C-extended HBV peptide FLPSDEFPSV-RDLLDTA was incubated with cell cytosols as source of a crude proteasomal preparation, and then the peptide cleavage was measured by HPLC. This C-extended HBV peptide was chosen because, according to the prediction algorithm, after removal of C-extension (RDLLDTA), FLPSDEFPSV peptide fits in the HLA-A2 groove to serve as a CTL-recognizing epitope on HBV-expressing hepatocytes. We found that the HBV control (AGS unexposed) group showed ~80% of C-extension cleavage at the interval of 30 to 90 min of extended peptide incubation with cytosol. However, when cytosol from AGS-exposed HBV cells was incubated with extended HBV peptide, there was no peptide cleavage for up to 60 min, but then only 20% of antigenic peptide was cleaved, indicating that the peptide cleavage was delayed and suppressed by AGS in HBV-expressing cells (Fig. 5).
Fig. 5.

Effects of acetaldehyde-generating system (AGS) on interferon-γ (IFNγ)-induced cleavage of hepatitis B virus (HBV) core C-extended peptide. Cells were treated or not with AGS for 72 h and with IFNγ for the last 24 h. Cytosolic protein (100 μg/ml) derived from cell lysates were mixed with 10 nM of corresponding C-extended peptide, as described in materials and methods. Percentage of remaining (uncleaved) peptide was calculated as integration units obtained from intact peptide peak after incubation with cytosol divided by integration units obtained from identically treated unincubated samples × 100. Data are shown as representative kinetics of C-extended peptide cleavage repeated in 3 independent experiments with similar results.
Antibody to HBV core peptide 18–27-HLA-A2 in recognizing HBV core 18–27-HLA-A2 complex in HepG2.2.15 cells.
Here, we provide evidence that antibody to HBV core peptide 18–27-HLA-A2 used in this study, specifically recognizes the HBV peptide-HLA-A2 MHC complex in HBV-transfected HepG2.2.15 cells. Immunofluorescent staining with this antibody was positive only in HBV+ HepG2.2.15 cells, but not in HBV− HepG2 cells (Fig. 6A). When HepG2 cells were pulsed with 5 µM HBV 18–27 core peptide, there was positive staining of these pulsed cells with HLA-A2-HBc 18–27 antibody (flow cytometry), whereas peptide-unpulsed HepG2 cells were negative (Fig. 6B); These data clearly demonstrate that the HBV core 18–27-HLA-A2 antibody used for this study specifically recognizes HBV peptide-HLA-A2 complex in HepG2.2.15 cells, but not empty HLA-A2, on control HepG2 cells.
Fig. 6.

Specificity of hepatitis B virus (HBV) core peptide18–27-human leukocyte antigen-A2 (HLA-A2) in recognizing the major histocompatibility complex I (MHC) complex of HBV-transfected HepG2.2.15 cells. A: HepG2 cells and HBV-transfected HepG2.2.15 cells were used for immunoflourescece staining of HBV core peptide (18–27)-HLA-A2 MHC complex. Green shows staining of HBV peptide-HLA-A2 Alexa fluor 488 staining. Staining was visualized using a ×63 lens in a LSM 710 confocal microscope. B: HepG2 cells unpulsed (without HBV 18–27 peptide) and HepG2 cells pulsed with 5 µM HBV 18–27 peptide for 4 h, and then HBV core peptide (18–27)-HLA-A2 MHC complex staining was measured by flow cytometry with antibody to HBV peptide-HLA-A2 followed by exposure to Alexa fluor 647 secondary antibody. Alexa fluor 647 secondary antibody was used as control. Data were collected on a BD LSR2 flow cytometer and analyzed using BD FACSDiva software v.6.0.
Suppression of HBV-HLA-A2 complex presentation on HepG2.2.15 cells by AGS and IPR inhibitor.
We studied whether the presentation of proteasome-processed HBV peptide FLPSDEFPSV (HBV core antigen peptide 18–27), which makes the complex with HLA-A2 (a potential CTL target on HBV-expressing hepatocytes) is altered by exposure to major ethanol metabolite Ach released by AGS. For this purpose, HBV+ HepG2.2.15 cells were exposed to AGS for 72 h in the presence or absence of IFNγ as described above in Cells and treatments). The display of HBV peptide-MHC class I complex was assessed by flow cytometry using anti- FLPSDEFPSV-HLA-A2 (designated as HBV+ HLA-A2) antibody. Figure 7A demonstrates that the presentation of HBV-HLA-A2 complex was higher in IFNγ-stimulated cells and it was 61% suppressed upon AGS exposure. We compared the effects of AGS with the effects of IPR inhibitor on the HBV peptide complex expression measured by flow cytometry. As shown in Fig. 7B, the presentation of HBV peptide-MHC class I complex was reduced by ONX by ~30%, providing evidence of the contribution of IPR activity suppression by AGS/Ach to overall display of the HBV peptide complex in HepG2.2.15 cells. This indicates that AGS provides broader effects on the HBV peptide complex display than just suppression of the peptide processing by IPR.
Fig. 7.

Effect of acetaldehyde-generating system (AGS) on expression of hepatitis B virus (HBV) peptide-human leukocyte antigen-A2 (HLA-A2) complex on the surface of HepG2.2.15 cells. A: HepG2.2.15 cells were treated or not with AGS for 72 h in the presence of interferon-γ (IFNγ), and expression of HBV core peptide (18–27) FLPSDEFPSV-HLA-A2 was measured by flow cytometry with antibody to HBV peptide-HLA-A2, followed by exposure to Alexa fluor 647 secondary antibody. Mouse IgG2b K isotype control was used. B: expression of HBV core peptide (18–27) FLPSDEFPSV-HLA-A2 measured by flow cytometry in IFNγ-induced ONX (ONX 0914, 100 nM, overnight)-treated HepG2.2.15 cells. Data were collected on a BD LSR2 flow cytometer and analyzed using BD FACSDiva software v.6.0. Data are shown as representative expression from 3 independent experiments with similar results.
AGS treatment affects TAP1 and tapasin expression in HepG2.2.15 cells.
The expressions of the transporter for the peptide-MHC class I complex (TAP1) and the protein to stabilize the peptide-MHC class I complex (tapasin) were measured in HepG2.2.15 cells exposed to IFNγ and AGS. We observed the suppression of IFNγ-induced TAP1 and tapasin by AGS (Fig. 8, A–D), indicating that, in addition to IPR, the decreased expression of TAP1 and tapasin may have an impact on delivery of cleaved HBV peptide-HLA-A2 complex to the surface. To test whether the AGS-induced defect in transporter induction affects the turnover of HLA-A2 and the delivery of HLA-A2 to the cell surface, the membrane-expressed structures on HepG2.2.15 cells were removed by low acid wash (as described in materials and methods), and after 8 h incubation of cells in fresh medium at 37C, the restoration of HLA-A2 expression was measured in IFNγ-stimulated cells treated or not with AGS. The expression of HLA-A2 was quantified by flow cytometry with anti-HLA-A2 antibody (Fig. 9, A and B). As it appeared, AGS delayed the restoration of HLA-A2 expression on the cell surface threefold.
Fig. 8.

Effect of acetaldehyde-generating system (AGS) on interferon-γ (IFNγ)-induced expression of transporter associated with antigen processing 1 (TAP1) and tapasin. HepG2.2.15 cells cells were treated or not with AGS for 72 h and with IFNγ for the last 24 h. A and C: TAP1 and tapsin protein expressions were detected by immunoblotting in cell lysates. Equal (20 µg) amounts of protein were loaded in each lane; β-actin was used as internal control. B and D: quantification of immunoblotting bands from 3 independent experiments presented as means ± SE. Bars marked with the same lowercase letter are not significantly different from each other; bars with different lowercase are significantly different (P ≤ 0 0.05).
Fig. 9.

Effects of acetaldehyde-generating system (AGS) on interferon-γ (IFNγ)-induced surface major histocompatibility complex I (MHC class I) levels and signal transducer and activator of transcription 1 (STAT1) signaling. A and B: HepG2.2.15 cells were treated or not with AGS for 72 h in the presence of IFNγ. Then, surface MHC class I were removed from cell surface by low acid wash, followed by measuring restoration of human MHC class I by flow cytometry using anti-human leukocyte antigen-A2 (HLA-A2) antibody. Mouse IgG2b K isotype control (allophycocyanin, APC) was used. C and D: HepG2.2.15 cells were treated or not with AGS for 72 h and with IFNγ for the last 1 h, and STAT1 phosphorylation (pSTAT1) was measured. Total STAT1 was used to normalize the data, and β-actin was also used as internal control. Data are from 3 independent experiments presented as means ± SE. Bars marked with the same lowercase letter are not significantly different from each other; bars with different lowercase letters are significantly different (P ≤ 0 0.05).
AGS suppresses IFNγ-induced signaling via the JAK-STAT1 pathway in HepG2.2.15 cells.
Since AGS-induced suppression of IPR/TAP1/tapasin was observed mainly in IFNγ-treated HepG2.2.15 cells, we next tested whether the mechanism of these suppressive effects are related to AGS-induced impairment of IFNγ-signaling via the Janus kinase (JAK)/signal transducer and activator of transcription 1 (STAT1) pathway. In this regard, in the untreated (control) and AGS-exposed cells, we measured STAT1 phosphorylation (p) in response to IFNγ (immunoblotting). We found that cell exposure to AGS reduced the pSTAT1/STAT1 ratio more than twofold (Fig. 9, C and D), suggesting that Ach suppresses the events downstream from IFNγ signaling in HBV-expressing cells.
AGS increases expression of HBV in HepG2.2.15 cells.
To exclude the suppressive effects of AGS on HBV expression/replication in HepG2.2.15 cells as a reason for decreased presentation of HBV peptide-HLA-A2 complex, we measured HBV RNA and HBV DNA by RT-PCR and ddPCR, respectively, HBsAg expression by quantitative ELISA, and immunofluorescent staining of HBV core protein in cells exposed to IFNγ and AGS (Fig. 10, A–D). While the dose of IFNγ used in this study provided no effects on HBV expression, AGS treatment even increased HBV content in HepG2.2.15 cells.
Fig. 10.

Effect of acetaldehyde-generating system (AGS) on hepatitis B virus (HBV) RNA, HBV DNA, HBV surface antigen (HBsAg), and core protein levels. HepG2.2.15 cells were treated or not with AGS for 72 h and with interferon-γ (IFNγ) for the last 24 h. Then, cells were harvested for RNA and DNA isolation. HBV RNA (A) and HBV DNA (B) levels were measured by RT-PCR and ddPCR, respectively. GAPDH was used as internal control for RT-PCR experiments. C: HBsAg levels were measured by sandwich ELISA. D: representative image of immunostaining of HBV core protein (red) and nuclear staining (blue). Data are from 3 independent experiments presented as means ± SE. Bars marked with the same lowercase letter are not significantly different from each other; bars with different lowercase letters are significantly different (P ≤ 0 0.05).
DISCUSSION
Chronic HBV infection is a major cause of cirrhosis, liver failure, and hepatocellular carcinoma (HCC) (38, 46). HBV-induced immune response (via hepatocyte-CTL receptor interactions or mediated by IFNγ release from activated lymphocytes) is multispecific, polyclonal, and vigorous during acute hepatitis B and plays a vital role in the disease pathogenesis and clearance of virally infected hepatocytes (64, 67, 69). In chronic hepatitis B (CHB), HBV-specific CTL response is suppressed, causing persistence of HBV-expressing hepatocytes (6, 71). While heavy alcohol consumption negatively affects disease outcomes and increases the incidence of HCC in HBV-related cirrhosis (40), the mechanisms, by which alcohol affects chronic persistence of HBV-infection are not fully understood and are linked to enhanced viral replication, enhanced oxidative stress, and a weakened immune response (30). The suppression of immune defense and acceleration of liver inflammation by alcohol exposure (66) is a common feature of hepatitis induced by hepatotropic viruses.
In this study, we hypothesized that, in HBV-infected hepatocytes, the ethanol metabolite Ach suppresses the HBV peptide-MHC class I complexes (CTL epitopes) presentation on hepatocyte surface, which may potentially decrease the clearance of HBV-expressing cells. As shown by others (36), positive immunofluorescent staining with HLA-A2-HBV core 18–27 antibody was found in three of eight liver biopsy samples obtained from CHB patients (36). The same study demonstrated that not viral replication but viral protein synthesis is related to efficient peptide-HLA-A2 complex presentation on hepatocyte surface. Obviously, HepG2.2.15 cells, the HLA-A2+ cell line transfected with HBV, serves an an excellent model to test the effects of ethanol on the peptide-HLA-A2 complex display. In these cells, there is an integration of full HBV genome into host DNA, which allows HepG2.2.15 cells to sensitize not only HBsAg (as happens in chronic asymptomatic HBsAg carriers), but other HBV antigens, including HBcAg, which might be relevant to liver inflammation development. HepG2.2.15 cells, however, do not metabolize ethanol. To mimic continuous generation of the most toxic product, Ach, we exposed cells to AGS. As shown here, AGS by itself induces neither apoptosis nor necrosis in HepG2.2.15 cells. We measured HLA-A2-restricted presentation of HBV core peptide 18–27, a known T cell epitope (4, 8, 43), by using a specific antibody that recognizes HBV peptide-HLA-A2 complex on the surface of HepG2.2.15 cells. Thus, in these cells, HBV antigens, including HBV core protein, are naturally cleaved by proteasome, loaded to MHC class I, and delivered to the cell membrane by protein loading complex (PLC) transporters, TAPs. Here, we tested the major steps critical for the peptide-MHC class I complex presentation that can be affected by chronic exposure to Ach. We used AGS as a tool to establish chronic cell exposure to Ach, because HBV-transfected HepG2.2.15 cells do not express ethanol-metabolizing enzymes and, thus, do not generate Ach by ethanol treatment. Previously, our laboratory demonstrated that alcohol metabolism suppresses antigen processing by proteasome, thereby altering MHC class I-restricted antigen presentation in liver cells (55, 56). However, the contribution of Ach to the regulation of proteasome function and peptide cleavage has not been addressed in the settings of HBV infection.
It is known that the proteasome system is the central proteolytic system of the eukaryotic cells (61). The first indication that IPR plays an important role in the generation of MHC class I-presented peptides came from the identification of the two IFNγ-inducible proteasome subunits LMP2 and LMP7 (10, 24, 34, 49). Stimulation of cells with IFNγ also induces the synthesis of subunits of the 20S proteasome activator PA28 (45).
Hepatocytes do not produce IFNγ. However, being one of the most important proinflammatory cytokines, IFNγ is released by immune cells that infiltrate inflamed liver. Hence, in vivo, hepatocytes are constantly exposed to an IFNγ-rich environment, which activates IFNγ-regulated components of antigen presentation machinery in these cells. To maximize incorporation of IPR subunits into proteasome for the most efficient cleavage of antigenic peptides, we treated cells with IFNγ. In our hands, in these HepG2.2.15 cells, Ach efficiently suppressed IFNγ-induced ChT-L and T-L proteasome activities, IPR activity, and expression of PA28 proteasome activator and LMP7 and LMP2 IPR subunits. Finally, IPR suppression delayed the cleavage of C-extended HBV core peptide FLPSDFFPSV-RDLLDTA, chosen for in vitro studies based on proteasomal cleavage predictions (PAProC program, www.paproc.de). In fact, 18–27 core decapeptideFLPSDFFPSV presented on the cell surface, is generated by IPR by cleavage of C-extension, RDLLDTA.
We asked how Ach suppresses proteasome-mediated antigenic peptide processing. In fact, the decrease in proteasome catalytic core activity by ethanol-induced oxidative stress has been shown before (3, 15, 35, 54). This can be explained, in part, by oxidant-mediated adduct formation that blocks the ability of proteasome regulators (19S particle for 26S proteasome and PA28 for 20S proteasome) to open the catalytic core gate for proteasomal substrates (48, 62). Thus, the suppression of catalytic proteasome activities by AGS observed in this study may be attributed to enhanced adduct formation (4-HNE) as a result of AGS exposure to cultured hepatoma cells, as we reported before (19). In addition, in our study, major suppressive effects of AGS on IPR were observed in IFNγ-stimulated cells. The latter allows hypothesizing that Ach suppresses IFNγ signaling, thereby preventing IPR activation by IFNγ. In this regard, we measured the effects of AGS on STAT1 phosphorylation and observed the decrease in pSTAT1/STAT1 ratio in AGS-treated cells. These results were in agreement with ethanol treatment-induced suppression of IFNγ-activated STAT1 phosphorylation and STAT1 attachment to DNA seen before in HBV-uninfected ethanol-metabolizing hepatoma (VL-17A) cells (51, 55). Therefore, besides direct Ach-induced inhibition of proteasomal catalytic activity via oxidative stress induction, there is the additional level of negative IPR activity regulation via AGS-mediated suppression of IFNγ signaling, which reduces expression of both PA28 and IPR subunits LMP2 and LMP7 in IFNγ-treated HepG2.2.15 cells. This suppression may be crucial for viral peptide processing, as the reduction of IPR function/expression by ~25% has been reported to significantly reduce MHC class I-restricted antigen presentation (32). To further support the crucial role of Ach-impaired IFNγ signaling in the regulation of viral peptide-MHC class I complex presentation on hepatocytes, we tested the the effects of AGS on the other IFNγ-dependent parameters, tapasin and TAP1, which are involved in stabilization and trafficking of peptide-MHC class I complex to the cell surface (1, 33, 42). As in the case of IPR, induction of tapasin and TAP1 by IFNγ was suppressed by AGS treatment and thus represented the downstream changes attributed to disrupted IFNγ signaling in AGS-exposed HepG2.2.15 cells.
To study the input of IPR suppression to the presentation of HBV peptide-HLA-A2 complex, we blocked IPR activity by the specific IPR inhibitor ONX and then quantified the expression of FLPSDFFPSV-HLA-A2 on the cell surface. This experiment demonstrated that ONX suppressed the complex presentation by 30%, whereas the corresponding effect of AGS was twice more potent. However, the ability to block proteasome activity (especially, ChT-L activity) by ONX was stronger than by AGS. These data indicate that Ach suppresses the presentation of the complex not only by blocking of the IPR-mediated peptide processing but also by suppression of some other components regulating the complex presentation, which are beyond the peptide processing. These components may be related to impaired trafficking of FLPSDFFPSV-HLA-A2 to the cell surface.
To ensure the AGS-induced impairment of antigen presentation in HepG2.2.15 cells, we excluded the possibility of AGS-associated reduction of HBV-expression in HepG2.2.15 cells as a reason for decreased HBV antigen supply in these cells. In contrast, we observed an increase in HBV RNA, HBV DNA, HBsAg, and HBV core protein content in AGS-exposed HepG2.2.15 cells attributing the suppression of FLPSDFFPSV-HLA-A2 complex presentation to Ach-impaired HBV-peptide processing/delivery machinery in hepatocytes.
We conclude that, in HBV-expressing hepatocytes, ethanol metabolism impairs proteasome function and IFNγ signaling via the JAK/STAT1 pathway in hepatocytes, thereby decreasing HBV peptide cleavage by IPR and activation of protein loading complex (PLC) components TAP and tapasin, necessary for HBV peptide-MHC class I trafficking to the membrane. Ach-induced defects in both HBV peptide processing and PLC finally decrease the display of HBV core peptide 18–27-MHC class I (FLPSDFFPSV-HLA-A2) complex on the cell surface. All these events may negatively affect the activation of CTLs, ultimately reducing their ability to recognize/eliminate HBV-expressing hepatocytes, which negatively affects HBV infection pathogenesis. (Fig. 11).
Fig. 11.
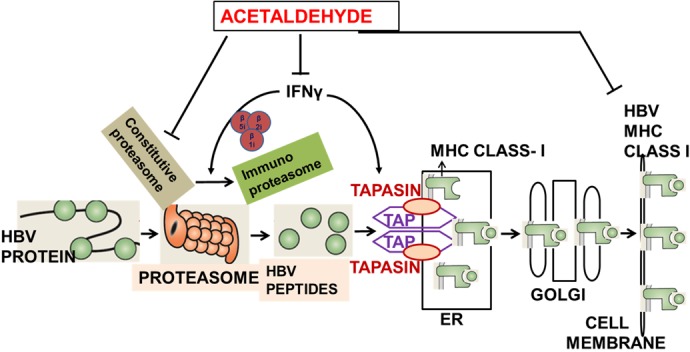
Schema of the possible mechanism of ethanol metabolism/acetaldehyde (Ach) affects presentation of hepatitis B virus (HBV)-peptide-major histocompatibility complex I (MHC class I) [cytotoxic T lymphocyte (CTL) epitopes] on the surface of hepatocytes, which are targets for immune response. Although Ach increases HBV replication, it directly suppresses processing of HBV peptides by proteasome and indirectly affects immunoproteasome function and induction of peptide-loading complex components tapasin and transporter associated with antigen processing 1 (TAP1), by impairing interferon-γ (IFNγ) signaling in hepatocytes. Finally, Ach suppresses presentation of HBV peptide-MHC class I complexes on the surface of hepatocytes, which may lower activation of CTLs (achieved by interaction of T cell receptor on CD8+ T lymphocytes with HBV peptide-MHC class I complex on hepatocytes) and ability of CTLs to recognize HBV-expressing hepatocytes as a target. ER, endoplasmic reticulum.
GRANTS
This work was supported by K01-Mentored Research Scientist Development Award- 1K01 AA-026864-01 from the National Institute on Alcohol Abuse and Alcoholism.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.G. conceived and designed research; M.G., V.M.K., E.M., and S.M. performed experiments; M.G., V.M.K., E.M., and S.M. analyzed data; M.G. interpreted results of experiments; M.G., E.M., and S.M. prepared figures; M.G. drafted manuscript; K.K.K., L.Y.P., C.A.C., and N.A.O. edited and revised manuscript; M.G. and N.A.O. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. George Acs, Baruch S. Blumberg Institute (Doylestown, PA) for providing HepG2.2.15-HBV transfected cells. We also thank Dr. Geoffrey M. Thiele (University of Nebraska Medical Center, Omaha, NE) for the purification of HBV peptide-HLA-A2 antibody from hybridoma cells supernatant.
REFERENCES
- 1.Abarca-Heidemann K, Friederichs S, Klamp T, Boehm U, Guethlein LA, Ortmann B. Regulation of the expression of mouse TAP-associated glycoprotein (tapasin) by cytokines. Immunol Lett 83: 197–207, 2002. doi: 10.1016/S0165-2478(02)00104-9. [DOI] [PubMed] [Google Scholar]
- 2.Arellano-Garcia ME, Misuno K, Tran SD, Hu S. Interferon-γ induces immunoproteasomes and the presentation of MHC I-associated peptides on human salivary gland cells. PLoS One 9: e102878, 2014. doi: 10.1371/journal.pone.0102878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bardag-Gorce F, Li J, French BA, French SW. The effect of ethanol-induced CYP2E1 on proteasome activity: the role of 4-hydroxynonenal. Exp Mol Pathol 78: 109–115, 2005. doi: 10.1016/j.yexmp.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Bertoletti A, Costanzo A, Chisari FV, Levrero M, Artini M, Sette A, Penna A, Giuberti T, Fiaccadori F, Ferrari C. Cytotoxic T lymphocyte response to a wild type hepatitis B virus epitope in patients chronically infected by variant viruses carrying substitutions within the epitope. J Exp Med 180: 933–943, 1994. doi: 10.1084/jem.180.3.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertoletti A, Ferrari C, Fiaccadori F, Penna A, Margolskee R, Schlicht HJ, Fowler P, Guilhot S, Chisari FV. HLA class I-restricted human cytotoxic T cells recognize endogenously synthesized hepatitis B virus nucleocapsid antigen. Proc Natl Acad Sci USA 88: 10445–10449, 1991. doi: 10.1073/pnas.88.23.10445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bertoletti A, Gehring AJ. The immune response during hepatitis B virus infection. J Gen Virol 87: 1439–1449, 2006. doi: 10.1099/vir.0.81920-0. [DOI] [PubMed] [Google Scholar]
- 7.Bertoletti A, Kennedy PTF. HBV antiviral immunity: not all CD8 T cells are born equal. Gut 68: 770–773, 2019. doi: 10.1136/gutjnl-2018-317959. [DOI] [PubMed] [Google Scholar]
- 8.Bertoletti A, Southwood S, Chesnut R, Sette A, Falco M, Ferrara GB, Penna A, Boni C, Fiaccadori F, Ferrari C. Molecular features of the hepatitis B virus nucleocapsid T-cell epitope 18-27: interaction with HLA and T-cell receptor. Hepatology 26: 1027–1034, 1997. doi: 10.1002/hep.510260435. [DOI] [PubMed] [Google Scholar]
- 9.Bitzer A, Basler M, Groettrup M. Chaperone BAG6 is dispensable for MHC class I antigen processing and presentation. Mol Immunol 69: 99–105, 2016. doi: 10.1016/j.molimm.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 10.Brown MG, Driscoll J, Monaco JJ. Structural and serological similarity of MHC-linked LMP and proteasome (multicatalytic proteinase) complexes. Nature 353: 355–357, 1991. doi: 10.1038/353355a0. [DOI] [PubMed] [Google Scholar]
- 11.Chen X, Tang Y, Zhang Y, Zhuo M, Tang Z, Yu Y, Zang G. Tapasin modification on the intracellular epitope HBcAg18-27 enhances HBV-specific CTL immune response and inhibits hepatitis B virus replication in vivo. Lab Invest 94: 478–490, 2014. doi: 10.1038/labinvest.2014.6. [DOI] [PubMed] [Google Scholar]
- 12.Dolganiuc A. Alcohol and viral hepatitis: role of lipid rafts. Alcohol Res 37: 299–309, 2015. [PMC free article] [PubMed] [Google Scholar]
- 13.Donaldson JG, Williams DB. Intracellular assembly and trafficking of MHC class I molecules. Traffic 10: 1745–1752, 2009. doi: 10.1111/j.1600-0854.2009.00979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donohue TM Jr, Zetterman RK, Zhang-Gouillon ZQ, French SW. Peptidase activities of the multicatalytic protease in rat liver after voluntary and intragastric ethanol administration. Hepatology 28: 486–491, 1998. doi: 10.1002/hep.510280228. [DOI] [PubMed] [Google Scholar]
- 15.Donohue TM, Osna NA, Clemens DL. Recombinant Hep G2 cells that express alcohol dehydrogenase and cytochrome P450 2E1 as a model of ethanol-elicited cytotoxicity. Int J Biochem Cell Biol 38: 92–101, 2006. doi: 10.1016/j.biocel.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 16.Drews O, Wildgruber R, Zong C, Sukop U, Nissum M, Weber G, Gomes AV, Ping P. Mammalian proteasome subpopulations with distinct molecular compositions and proteolytic activities. Mol Cell Proteomics 6: 2021–2031, 2007. doi: 10.1074/mcp.M700187-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Ferrington DA, Gregerson DS. Immunoproteasomes: structure, function, and antigen presentation. Prog Mol Biol Transl Sci 109: 75–112, 2012. doi: 10.1016/B978-0-12-397863-9.00003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fritzsche S, Springer S. Investigating MHC class I folding and trafficking with pulse-chase experiments. Mol Immunol 55: 126–130, 2013. doi: 10.1016/j.molimm.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 19.Ganesan M, Natarajan SK, Zhang J, Mott JL, Poluektova LI, McVicker BL, Kharbanda KK, Tuma DJ, Osna NA. Role of apoptotic hepatocytes in HCV dissemination: regulation by acetaldehyde. Am J Physiol Gastrointest Liver Physiol 310: G930–G940, 2016. doi: 10.1152/ajpgi.00021.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ganesan M, Poluektova LY, Tuma DJ, Kharbanda KK, Osna NA. Acetaldehyde disrupts interferon alpha signaling in hepatitis C virus-infected liver cells by up-regulating USP18. Alcohol Clin Exp Res 40: 2329–2338, 2016. doi: 10.1111/acer.13226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ganesan M, Tikhanovich I, Vangimalla SS, Dagur RS, Wang W, Poluektova LI, Sun Y, Mercer DF, Tuma D, Weinman SA, Kharbanda KK, Osna NA. Demethylase JMJD6 as a new regulator of interferon signaling: effects of HCV and ethanol metabolism. Cell Mol Gastroenterol Hepatol 5: 101–112, 2018. doi: 10.1016/j.jcmgh.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ganesan M, Zhang J, Bronich T, Poluektova LI, Donohue TM Jr, Tuma DJ, Kharbanda KK, Osna NA. Acetaldehyde accelerates HCV-induced impairment of innate immunity by suppressing methylation reactions in liver cells. Am J Physiol Gastrointest Liver Physiol 309: G566–G577, 2015. doi: 10.1152/ajpgi.00183.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gitto S, Vitale G, Villa E, Andreone P. Update on alcohol and viral hepatitis. J Clin Transl Hepatol 2: 228–233, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glynne R, Powis SH, Beck S, Kelly A, Kerr LA, Trowsdale J. A proteasome-related gene between the two ABC transporter loci in the class II region of the human MHC. Nature 353: 357–360, 1991. doi: 10.1038/353357a0. [DOI] [PubMed] [Google Scholar]
- 25.Goldberg AL, Cascio P, Saric T, Rock KL. The importance of the proteasome and subsequent proteolytic steps in the generation of antigenic peptides. Mol Immunol 39: 147–164, 2002. doi: 10.1016/S0161-5890(02)00098-6. [DOI] [PubMed] [Google Scholar]
- 26.Guidotti LG, Chisari FV. Immunobiology and pathogenesis of viral hepatitis. Annu Rev Pathol 1: 23–61, 2006. doi: 10.1146/annurev.pathol.1.110304.100230. [DOI] [PubMed] [Google Scholar]
- 27.Guidotti LG, Isogawa M, Chisari FV. Host-virus interactions in hepatitis B virus infection. Curr Opin Immunol 36: 61–66, 2015. doi: 10.1016/j.coi.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guillaume B, Chapiro J, Stroobant V, Colau D, Van Holle B, Parvizi G, Bousquet-Dubouch MP, Théate I, Parmentier N, Van den Eynde BJ. Two abundant proteasome subtypes that uniquely process some antigens presented by HLA class I molecules. Proc Natl Acad Sci USA 107: 18599–18604, 2010. doi: 10.1073/pnas.1009778107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hughes SA, Wedemeyer H, Harrison PM. Hepatitis delta virus. Lancet 378: 73–85, 2011. doi: 10.1016/S0140-6736(10)61931-9. [DOI] [PubMed] [Google Scholar]
- 30.Iida-Ueno A, Enomoto M, Tamori A, Kawada N. Hepatitis B virus infection and alcohol consumption. World J Gastroenterol 23: 2651–2659, 2017. doi: 10.3748/wjg.v23.i15.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Isogawa M, Tanaka Y. Immunobiology of hepatitis B virus infection. Hepatol Res 45: 179–189, 2015. doi: 10.1111/hepr.12439. [DOI] [PubMed] [Google Scholar]
- 32.Joeris T, Schmidt N, Ermert D, Krienke P, Visekruna A, Kuckelkorn U, Kaufmann SH, Steinhoff U. The proteasome system in infection: impact of β5 and LMP7 on composition, maturation and quantity of active proteasome complexes. PLoS One 7: e39827, 2012. doi: 10.1371/journal.pone.0039827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson DR, Pober JS. Tumor necrosis factor and immune interferon synergistically increase transcription of HLA class I heavy- and light-chain genes in vascular endothelium. Proc Natl Acad Sci USA 87: 5183–5187, 1990. doi: 10.1073/pnas.87.13.5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelly A, Powis SH, Glynne R, Radley E, Beck S, Trowsdale J. Second proteasome-related gene in the human MHC class II region. Nature 353: 667–668, 1991. doi: 10.1038/353667a0. [DOI] [PubMed] [Google Scholar]
- 35.Kessova IG, Cederbaum AI. The effect of CYP2E1-dependent oxidant stress on activity of proteasomes in HepG2 cells. J Pharmacol Exp Ther 315: 304–312, 2005. doi: 10.1124/jpet.105.088047. [DOI] [PubMed] [Google Scholar]
- 36.Khakpoor A, Ni Y, Chen A, Ho ZZ, Oei V, Yang N, Giri R, Chow JX, Tan AT, Kennedy PT, Maini M, Urban S, Bertoletti A. Spatiotemporal differences in presentation of CD8 T cell epitopes during hepatitis B virus infection. J Virol 93: e01457-18, 2019. doi: 10.1128/JVI.01457-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klare N, Seeger M, Janek K, Jungblut PR, Dahlmann B. Intermediate-type 20 S proteasomes in HeLa cells: “asymmetric” subunit composition, diversity and adaptation. J Mol Biol 373: 1–10, 2007. doi: 10.1016/j.jmb.2007.07.038. [DOI] [PubMed] [Google Scholar]
- 38.Lavanchy D. Worldwide epidemiology of HBV infection, disease burden, and vaccine prevention. J Clin Virol 34, Suppl 1: S1–S3, 2005. doi: 10.1016/S1386-6532(05)00384-7. [DOI] [PubMed] [Google Scholar]
- 39.Lee HG, Lim JS, Lee KY, Choi YK, Choe IS, Chung TW, Kim K. Peptide-specific CTL induction in HBV-seropositive PBMC by stimulation with peptides in vitro: novel epitopes identified from chronic carriers. Virus Res 50: 185–194, 1997. doi: 10.1016/S0168-1702(97)00068-3. [DOI] [PubMed] [Google Scholar]
- 40.Lin CW, Lin CC, Mo LR, Chang CY, Perng DS, Hsu CC, Lo GH, Chen YS, Yen YC, Hu JT, Yu ML, Lee PH, Lin JT, Yang SS. Heavy alcohol consumption increases the incidence of hepatocellular carcinoma in hepatitis B virus-related cirrhosis. J Hepatol 58: 730–735, 2013. doi: 10.1016/j.jhep.2012.11.045. [DOI] [PubMed] [Google Scholar]
- 41.Liu H, Lou G, Li C, Wang X, Cederbaum AI, Gan L, Xie B. HBx inhibits CYP2E1 gene expression via downregulating HNF4α in human hepatoma cells. PLoS One 9: e107913, 2014. doi: 10.1371/journal.pone.0107913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma W, Lehner PJ, Cresswell P, Pober JS, Johnson DR. Interferon-γ rapidly increases peptide transporter (TAP) subunit expression and peptide transport capacity in endothelial cells. J Biol Chem 272: 16585–16590, 1997. doi: 10.1074/jbc.272.26.16585. [DOI] [PubMed] [Google Scholar]
- 43.Maini MK, Boni C, Ogg GS, King AS, Reignat S, Lee CK, Larrubia JR, Webster GJ, McMichael AJ, Ferrari C, Williams R, Vergani D, Bertoletti A. Direct ex vivo analysis of hepatitis B virus-specific CD8+ T cells associated with the control of infection. Gastroenterology 117: 1386–1396, 1999. doi: 10.1016/S0016-5085(99)70289-1. [DOI] [PubMed] [Google Scholar]
- 44.Min BY, Kim NY, Jang ES, Shin CM, Lee SH, Park YS, Hwang JH, Jeong SH, Kim N, Lee DH, Kim JW. Ethanol potentiates hepatitis B virus replication through oxidative stress-dependent and -independent transcriptional activation. Biochem Biophys Res Commun 431: 92–97, 2013. doi: 10.1016/j.bbrc.2012.12.081. [DOI] [PubMed] [Google Scholar]
- 45.Nandi D, Jiang H, Monaco JJ. Identification of MECL-1 (LMP-10) as the third IFN-gamma-inducible proteasome subunit. J Immunol 156: 2361–2364, 1996. [PubMed] [Google Scholar]
- 46.Neuveut C, Wei Y, Buendia MA. Mechanisms of HBV-related hepatocarcinogenesis. J Hepatol 52: 594–604, 2010. doi: 10.1016/j.jhep.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 47.Niro GA, Smedile A, Ippolito AM, Ciancio A, Fontana R, Olivero A, Valvano MR, Abate ML, Gioffreda D, Caviglia GP, Rizzetto M, Andriulli A. Outcome of chronic delta hepatitis in Italy: a long-term cohort study. J Hepatol 53: 834–840, 2010. doi: 10.1016/j.jhep.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 48.Okada K, Wangpoengtrakul C, Osawa T, Toyokuni S, Tanaka K, Uchida K. 4-Hydroxy-2-nonenal-mediated impairment of intracellular proteolysis during oxidative stress. Identification of proteasomes as target molecules. J Biol Chem 274: 23787–23793, 1999. doi: 10.1074/jbc.274.34.23787. [DOI] [PubMed] [Google Scholar]
- 49.Ortiz-Navarrete V, Seelig A, Gernold M, Frentzel S, Kloetzel PM, Hämmerling GJ. Subunit of the ‘20S’ proteasome (multicatalytic proteinase) encoded by the major histocompatibility complex. Nature 353: 662–664, 1991. doi: 10.1038/353662a0. [DOI] [PubMed] [Google Scholar]
- 50.Osna NA, Bardag-Gorce F, White RL, Weinman SA, Donohue TM Jr, Kharbanda KK. Ethanol and hepatitis C virus suppress peptide-MHC class I presentation in hepatocytes by altering proteasome function. Alcohol Clin Exp Res 36: 2028–2035, 2012. doi: 10.1111/j.1530-0277.2012.01813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Osna NA, Clemens DL, Donohue TM Jr. Ethanol metabolism alters interferon gamma signaling in recombinant HepG2 cells. Hepatology 42: 1109–1117, 2005. doi: 10.1002/hep.20909. [DOI] [PubMed] [Google Scholar]
- 52.Osna NA, Donohue TM Jr. CYP2E1-catalyzed alcohol metabolism: role of oxidant generation in interferon signaling, antigen presentation and autophagy. Subcell Biochem 67: 177–197, 2013. doi: 10.1007/978-94-007-5881-0_6. [DOI] [PubMed] [Google Scholar]
- 53.Osna NA, Ganesan M, Kharbanda KK. Hepatitis C, innate immunity and alcohol: friends or foes? Biomolecules 5: 76–94, 2015. doi: 10.3390/biom5010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Osna NA, Haorah J, Krutik VM, Donohue TM Jr. Peroxynitrite alters the catalytic activity of rodent liver proteasome in vitro and in vivo. Hepatology 40: 574–582, 2004. doi: 10.1002/hep.20352. [DOI] [PubMed] [Google Scholar]
- 55.Osna NA, White RL, Thiele GM, Donohue TM Jr. Ethanol metabolism alters major histocompatibility complex class I-restricted antigen presentation in liver cells. Hepatology 49: 1308–1315, 2009. doi: 10.1002/hep.22787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Osna NA, White RL, Todero S, McVicker BL, Thiele GM, Clemens DL, Tuma DJ, Donohue TM Jr. Ethanol-induced oxidative stress suppresses generation of peptides for antigen presentation by hepatoma cells. Hepatology 45: 53–61, 2007. doi: 10.1002/hep.21442. [DOI] [PubMed] [Google Scholar]
- 57.Penna A, Chisari FV, Bertoletti A, Missale G, Fowler P, Giuberti T, Fiaccadori F, Ferrari C. Cytotoxic T lymphocytes recognize an HLA-A2-restricted epitope within the hepatitis B virus nucleocapsid antigen. J Exp Med 174: 1565–1570, 1991. doi: 10.1084/jem.174.6.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qian SB, Princiotta MF, Bennink JR, Yewdell JW. Characterization of rapidly degraded polypeptides in mammalian cells reveals a novel layer of nascent protein quality control. J Biol Chem 281: 392–400, 2006. doi: 10.1074/jbc.M509126200. [DOI] [PubMed] [Google Scholar]
- 59.Qiu B, Huang B, Wang X, Liang J, Feng J, Chang Y, Li D. Association of TAP1 and TAP2 polymorphisms with the outcome of persistent HBV infection in a northeast Han Chinese population. Scand J Gastroenterol 47: 1368–1374, 2012. doi: 10.3109/00365521.2012.725090. [DOI] [PubMed] [Google Scholar]
- 60.Robek MD, Garcia ML, Boyd BS, Chisari FV. Role of immunoproteasome catalytic subunits in the immune response to hepatitis B virus. J Virol 81: 483–491, 2007. doi: 10.1128/JVI.01779-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78: 761–771, 1994. doi: 10.1016/S0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 62.Rouach H, Andraud E, Aufrère G, Beaugé F. The effects of acetaldehyde in vitro on proteasome activities and its potential involvement after alcoholization of rats by inhalation of ethanol vapours. Alcohol Alcohol 40: 359–366, 2005. doi: 10.1093/alcalc/agh174. [DOI] [PubMed] [Google Scholar]
- 63.Shi TD, Wu YZ, Jia ZC, Zou LY, Zhou W. Therapeutic polypeptides based on HBV core 18-27 epitope can induce CD8+ CTL-mediated cytotoxicity in HLA-A2+ human PBMCs. World J Gastroenterol 10: 1902–1906, 2004. doi: 10.3748/wjg.v10.i13.1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shimizu Y. T cell immunopathogenesis and immunotherapeutic strategies for chronic hepatitis B virus infection. World J Gastroenterol 18: 2443–2451, 2012. doi: 10.3748/wjg.v18.i20.2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sugawara S, Abo T, Kumagai K. A simple method to eliminate the antigenicity of surface class I MHC molecules from the membrane of viable cells by acid treatment at pH 3. J Immunol Methods 100: 83–90, 1987. doi: 10.1016/0022-1759(87)90175-X. [DOI] [PubMed] [Google Scholar]
- 66.Szabo G, Saha B. Alcohol’s effect on host defense. Alcohol Res 37: 159–170, 2015. [PMC free article] [PubMed] [Google Scholar]
- 67.Tan AT, Loggi E, Boni C, Chia A, Gehring AJ, Sastry KS, Goh V, Fisicaro P, Andreone P, Brander C, Lim SG, Ferrari C, Bihl F, Bertoletti A. Host ethnicity and virus genotype shape the hepatitis B virus-specific T-cell repertoire. J Virol 82: 10986–10997, 2008. doi: 10.1128/JVI.01124-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vigneron N, Van den Eynde BJ. Proteasome subtypes and regulators in the processing of antigenic peptides presented by class I molecules of the major histocompatibility complex. Biomolecules 4: 994–1025, 2014. doi: 10.3390/biom4040994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Westover KM, Hughes AL. Evolution of cytotoxic T-lymphocyte epitopes in hepatitis B virus. Infect Genet Evol 7: 254–262, 2007. doi: 10.1016/j.meegid.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 70.World Health Organization Hepatitis B. https://www.who.int/news-room/fact-sheets/detail/hepatitis-b. 2018. [28 Feburary 2019].
- 71.Zhang Y, Ren Y, Wu Y, Zhao B, Qiu L, Li X, Xu D, Liu J, Gao GF, Meng S. The L60V variation in hepatitis B virus core protein elicits new epitope-specific cytotoxic T lymphocytes and enhances viral replication. J Virol 87: 8075–8084, 2013. doi: 10.1128/JVI.00577-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhao R, Wang TZ, Kong D, Zhang L, Meng HX, Jiang Y, Wu YQ, Yu ZX, Jin XM. Hepatoma cell line HepG2.2.15 demonstrates distinct biological features compared with parental HepG2. World J Gastroenterol 17: 1152–1159, 2011. doi: 10.3748/wjg.v17.i9.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]