

Abstract
Epithelial cells that line lung airways produce and secrete proteins with important roles in barrier function and host defense. Secretion of airway goblet cells is controlled by autophagy proteins during inflammatory conditions, resulting in accumulation of mucin proteins. We hypothesized that autophagy proteins would also be important in the function of club cells, dominant secretory airway epithelial cells that are dysregulated in chronic lung disease. We found that in the absence of an inflammatory stimulus, mice with club cells deficient for the autophagy protein Atg5 had a markedly diminished expression of secreted host defense proteins secretoglobulin family 1A, member 1 (Scgb1a1) and surfactant proteins A1 and D (Sftpa1 and Sftpd), as well as abnormal club cell morphology. Adult mice with targeted loss of Atg5 also showed diminished levels of host defense proteins in regenerating cells following ablation with naphthalene. A mouse strain with global deficiency of Atg16-like 1 (Atg16l1), an Atg5 binding partner, had a similar loss of host defense proteins and abnormal club cell morphology. Cigarette smoke exposure reduced levels of Scgb1a1 in wild-type mice as expected. Smoke exposure was not required to trigger club cell abnormalities in mice bearing the human ATG16 variant Atg16l1T300A/T300A, which had low Scgb1a1 levels independent of this environmental stress. Evaluation of lung tissues from former smokers with severe chronic obstructive pulmonary disease showed evidence of reduced autophagy and SCGB1A1 expression in club cells. Thus, autophagy proteins are required for the function of club cells, independent of the cellular stress of cigarette smoke, with roles that appear to be distinct from those of other secretory cell types.
Keywords: autophagy, club cell
INTRODUCTION
Airway epithelium is composed of differentiated ciliated and secretory epithelial cell types, and in the normal lung, the club cell is the most prominent secretory cell type. In the rodent lung, particularly distal to the bronchi, including the terminal bronchioles, the club cell is the most abundant epithelial cell type (40). Club cells are unique to the airway and have protective roles, including secretion of anti-inflammatory, antimicrobial, and immune-regulating proteins (18). The major secretory protein of the club cell is secretoglobulin (Scgb) family 1A member 1 [Scgb1a1, club cell secretory protein (CCSP), and Clara cell 10- and 16-kDa proteins (CC10 and CC16)]. Scgb1a1 is an immunomodulator and protects the lung from toxicant injury while maintaining respiratory homeostasis (6). Club cells also secrete protein members of the collectin family, surfactant proteins A (Sftpa1), B (Sftpb), and D (Sftpd), and Scgb family 2A member 3 and cytochrome P-450 (Cyp) family members (40). Collectins are components of the acquired immune response, with functions controlling respiratory bacterial and viral infection and gas exchange, whereas the Cyp proteins are mainly involved in the metabolism of lung xenobiotics and other toxins (6, 15, 20, 47, 49). Multiple studies have linked abnormal Clara cell 16-kDa (CC16) protein (SCGB1A1 protein product) and collectin levels in the lung and serum with abnormal lung function associated with cigarette smoking, chronic obstructive pulmonary disease (COPD), asthma, and other lung diseases (14, 20, 23–25, 28, 51).
Previous reports by our laboratory and others have shown that autophagy proteins play an important role in packaging and secretion of proteins in multiple cell types (3, 10, 34, 37). Goblet cells of the intestine at homeostasis (34) and the inflamed airway (10) require downstream autophagy genes Atg5 (as part of the Atg12-Atg5 complex) or oligomeric Atg16l1 for proper packaging and secretion of mucus granules by mechanisms that have yet to be uncovered. Disruption of the Atg16l1 gene in intestinal Paneth cells results in aberrant packaging of antimicrobial proteins and diffuse protein localization (3, 4). Similarly, loss of Atg7 or Atg5 or inhibition of autophagy-impaired secretion of von Willebrand factor from endothelial cells can induce protein accumulation. The defect is shown to be impaired protein processing and maturation, although the precise mechanism remains undefined (45). Each of these cell types accumulates proteins, as secretion requires either Atg7, the Atg12-Atg5 complex, or Atg16l1, all of which are involved in maturation of the autophagosome.
The importance of secretion by club cells in the airway lumen and the similarities to the secretory cells of the intestine led us to examine autophagy deficiency in this cell lineage. Using two previously defined models of autophagy deficiency, we examined the effect of reduced expression of Atg16l1 in the entire airway and club cell-specific ablation of Atg5. We initially hypothesized that loss of Atg16l1 or Atg5 would diminish secretion of proteins from club cells. Unexpectedly, we found that, in contrast to the phenotype of autophagy-deficient Paneth or goblet cells in which secretion was impaired and intracellular proteins for host defense were overabundant, autophagy-deficient club cells had reduced intracellular production of collectins and Scgb1a1. In both a human cohort and a mouse model, we found that, during airway injury, expression of autophagy genes and Scgb1a1 was further decreased and could impact disease states.
MATERIALS AND METHODS
Mice.
The Washington University Institutional Animal Care and Use Committee approved all animal experiments. Mice were maintained in a pathogen-free barrier facility under a strict 12:12-h light-dark cycle and fed autoclaved chow diet. Six- to 10-wk-old male and female mice were used for experiments. Microtubule-associated protein Map1lc3b+/− light chain 3B (LC3B−/−) mice (5) were bred to generate Map1lc3b−/− mice and littermate controls (Map1lc3b+/−). Scgb1a1Cre/+ [Clara cell secretory protein (CCSP)-Cre] mice (26) were bred to Atg5fl/fl mice (3, 16) to generate littermate controls. Genotyping and breeding schemes for hypomorphic (hm) Atg16l1 (Atg16l1hm/hm) mice are described elsewhere (3). Male and female Atg16l1T300A /+ mice were bred to generate Atg16l1T300A/T300A mice and littermate controls (Atg16l1+/+) (22).
Human tissues.
The Washington University Institutional Review Board approved the studies with human tissues. Human COPD lung tissue samples were obtained at the time of lung transplantation from individuals who provided consent before surgery. All patients had very severe COPD by pulmonary function testing, all were former cigarette smokers, and four also had α1-antitrypsin deficiency [n = 20 (9 female and 11 male), mean age 61 (range 48–71) yr]. Lung tissue used in the “donor” group was obtained from lungs that were donated for transplantation but were not used [n = 6 (2 female, 2 male, 2 demographics unknown), mean age 38 (range 19–62) yr]. All samples were deidentified before studies were initiated.
Histochemistry and immunohistochemistry.
Lung tissues were inflation-fixed (mouse tissues) or submersion-fixed (human tissues) with 10% neutral buffered formalin (Sigma-Aldrich, St. Louis, MO). The tissue was dehydrated through graded ethanol and embedded in paraffin for 5-μm-thick sectioning. Tissue sections were stained with hematoxylin and eosin (H&E) or periodic acid-Schiff (PAS) for analysis of morphology and goblet cells.
Immunohistochemistry was performed in formalin-fixed paraffin-embedded tissue sections that were processed using standard procedures. Tissue sections were subjected to antigen retrieval using Trilogy buffer (Cell Marque, Rocklin, CA), blocked in 1% BSA-0.1% Triton X-100-PBS, and incubated with primary antibodies overnight at 4°C. Primary antibodies included rabbit anti-mouse LC3B (1:1,000 dilution; catalog no. NB600-1384, Novus Biological, Centennial, CO), rabbit anti-human LC3B (1:1,000 dilution; catalog no. 3868S, Cell Signaling Technology, Danvers, MA), guinea pig anti-p62/sequestosome 1 (Sqstm1) COOH terminus (1:500 dilution; catalog no. GP62, Progen, Heidelberg, Germany), rat anti-human Scgb1a1 antibody (1:75 dilution; catalog no. MAB4218, R & D Systems, Minneapolis, MN), rabbit anti-mouse Scgb1a antibody (1:10,000 dilution; provided by Barry Stripp, Cedars-Sinai Medical Center, and catalog no. WRAB-3950, Seven Hills Bioreagents, Cincinnati, OH) or goat anti-mouse Scgb1a antibody (1:5,000 dilution, provided by Barry Stripp), acetylated α-tubulin (1:1,000 dilution; clone 6-11B-1, catalog no. T6793, Sigma-Aldrich), goat anti-Sftpa1 antibody (1:100 dilution; catalog no. SC-7699, Santa Cruz Biotechnology, Dallas, TX), rabbit anti-Sftpd (1:100 dilution; catalog no. BS-1583R, Bioss, Woburn, MA), and rabbit anti-Cyp2f2 (1:1,000 dilution, catalog no. SC-67283, Santa Cruz Biotechnology). Primary antibodies were detected with species-specific Alexa Fluor-labeled secondary antibodies (Life Technologies, Carlsbad, CA) at a 1:500 dilution for 1 h at room temperature. Tissues were then counterstained with bis-benzamide and sealed with Fluoromount (Sigma-Aldrich). Slides were visualized by epifluorescence microscopy (Zeiss, Jena, Germany). Images were globally adjusted for brightness and contrast using Photoshop CS5 (Adobe, San Jose, CA).
Quantification of immunostaining.
The total number of club cells was counted and scored as either normal (>8 distinct granules of Scgb1a) or sparse (<8 distinct granules of Scgb1a) in a cross section of a cell that included the nucleus. Percent staining of each was calculated as the number of normal or sparse cells relative to total club cells per airway. In each mouse, four to six airways were quantified (average 90–100 total cells per airway). For quantification of Sftpa-, Sftpd-, and Cyp2f2-positive cells, the total number of cells positive for the specific marker was divided by the total number of cells in the airway identified by nuclear staining.
Protein analysis.
Protein was isolated from whole nonperfused lung lobes in 1 ml of radioimmunoprecipitation buffer with 1 μl of protease inhibitor cocktail (Sigma-Aldrich). Lung lobes were isolated, flash-frozen in liquid nitrogen, sonicated in radioimmunoprecipitation buffer, and then processed with Laemmli buffer. Proteins were separated by electrophoresis on 18% polyacrylamide gels (Bio-Rad, Hercules, CA), transferred to nitrocellulose membranes, blocked with 5% nonfat milk in Tris-buffered saline-Tween 20 for 1 h, and probed with antibodies overnight at 4°C. Club cell secretory protein polyclonal rabbit anti-Scgb1a antibody (for immunostaining) was used at a 1:5,000 dilution. Sftpa was detected using polyclonal goat anti-SP-A antibody (1:100 dilution; catalog no. SC-7699, Santa Cruz Biotechnology), and Sftpd was detected using monoclonal mouse anti-SP-D (1:5,000 dilution, clone 1A10a9; catalog no. WMAB-1A10A9, Seven Hills Bioreagents). Cyp2f2 was detected using a monoclonal mouse anti-Cyp2f2 (1:1,000 dilution; catalog no. SC-374540, Santa Cruz Biotechnology). Actin was detected using a polyclonal rabbit anti-actin antibody (1:1,000 dilution; catalog no. A-2066, Sigma-Aldrich). The blots were incubated with anti-rabbit horseradish peroxidase (HRP), anti-goat HRP, or anti-mouse HRP at a 1:2,500 dilution for 1 h and then with a SuperSignal West Dura kit (Thermo Scientific, Waltham, MA) and exposed to film. Band intensity was measured using ImageJ to quantify intensity and normalized to actin.
Quantitative RT-PCR analysis.
RNA from whole lung lobes was isolated and purified using the NucleoSpin RNA II extraction kit (Machery-Nagel, Bethlehem, PA) according to the manufacturer's protocol. For cDNA synthesis, SuperScript reverse transcriptase and random primers were used (Invitrogen, Carlsbad, CA). Quantitative RT-PCR was performed in triplicate for each sample using SYBR green master mix (Clontech, Mountain View, CA) with sequence-specific primers (Table 1) and analyzed using a RealPlex Mastercycler (Eppendorf, Hamburg, Germany). The fold difference for mRNA expression was calculated as the difference between the average change in cycle threshold (ΔCT) of the experimental model (Atg16l1hm/hm) and the average ΔCT of the control model (Atg16l1+/+) divided by the control ΔCT.
Table 1.
Primer sequences
| Gene | Forward | Reverse |
|---|---|---|
| Scgb1a1 | ATGAAGATCGCCATCACAATCAC | GGATGCCACATAACCAGACTCT |
| Sftpa1 | GAGGAGCTTCAGACTGCACTC | AGACTTTATCCCCCACTGACAG |
| Sftpd | ACGTGGACTAAGTGGACCTCC | CCTTTTGCCCCTGTAGATCCTT |
| Cyp2f2 | CTCTGTCCCTGACCTTCAGC | CCCCTGGTGAAGTTGAAAAA |
| Nkx2-1 | AGGACACCATGCGGAACAG | CCATGCCGCTCATATTCATGC |
| Foxj1 | CCCTGACGACGTGGACTATG | GCCGACAGAGTGATCTTGGT |
| Spdef | GGACGGACGACTCTTCTGACAG | GCTCCTGATGCTGCCTTCTCC |
Scgb1a1, secretoglobulin family 1A member 1; Sftpa1 and Sftpd, surfactant proteins A and D; Cyp2f2, cytochrome P-450 family 2, subfamily f, polypeptide 2; Nkx2-1, NK2 homeobox 1; Foxj1, forkhead box J1; Spdef, SAM-pointed domain containing Ets transcription factor.
Electron microscopy.
For transmission electron microscopy, 1-cm2 sections were dissected from distal mouse lung tissue containing small bronchioles and fixed in 2% paraformaldehyde-2% glutaraldehyde. Sections were prepared for electron microscopy as previously described (34) and viewed with a JEOL model 100C microscope (JEOL USA, Peabody, MA).
Naphthalene treatment.
Mice were injected with intraperitoneal corn oil only or 0.2 mg/g naphthalene (Sigma-Aldrich) dissolved in corn oil. Lungs were fixed for processing on postinjection day 14.
Smoking exposure.
Four- to six-week-old mice were exposed to smoke from 4 cigarettes [Kentucky research cigarette 3R4F (with filters removed), University of Kentucky, Lexington, KY] per day for 5 days per week according to a previously described protocol (9, 17). Mice were exposed to smoke for 2 weeks then euthanized for tissue collection.
RESULTS
A club cell lineage knockout of Atg5 shows diminished expression of secretory proteins.
Autophagy is required for the proper function of several types of secretory cells, including airway goblet and intestinal Paneth cells (3, 10). Accordingly, we tested the role of autophagy in airway club cells, based on morphology and function similar to Paneth cells. We first examined the expression of LC3B [encoded by the microtubule-associated protein 1 LC3B (Map1lc3b) gene], a marker of autophagy that has been detected in tissue sections of human and mouse lung by immunofluorescence localization (1, 39, 50). The cytoplasmic LC3B-positive puncta observed in this assay indicate the presence of organelles that utilize autophagy proteins (i.e., LC3B-associated phagosomes, autophagosomes, and amphisomes) (34). In mouse intraparenchymal airways, we found that ~40% of Scgb1a-positive club cells (40) were LC3B-positive (Fig. 1, A–E). Ciliated cells in the same airways (marked by acetylated α-tubulin) showed a similar percentage (~50%) of LC3B-positive cells (Fig. 1, A–E). Based on recent single-cell sequencing studies (31, 36), it is likely that these autophagy proteins are expressed broadly in basal, club, and ciliated epithelial cell types. As a control, similarly processed sections of Map1lc3b−/− lungs showed no detectable LC3B immunoreactivity in either cell type (Fig. 1, B, D, and F). These data indicate that, similar to ciliated cells, autophagy proteins can be detected in club cells of mouse intraparenchymal airways.
Fig. 1.

Detection of autophagy in airway epithelial cells by endogenous light chain 3B (LC3B). A–D: images of small airway sections from microtubule-associated protein LC3B (Maplc3b) wild-type (Maplc3b+/+) and Maplc3b-deficient (Maplc3b−/−) mice. Sections were immunostained for LC3B (red) and acetylated α-tubulin (AAT, green, cilia) or LC3B (red) and secretoglobulin family 1A member 1 (Scgb1a1, green). Nuclei were stained with bis-benzamide (blue). Arrowheads indicate club cells; arrows indicate ciliated cells. Scale bars = 50 µm. E: percentage of LC3B-positive club and ciliated cells from immunostained sections of Maplc3b+/+ mice. F: percentage of LC3B-positive airway cells by genotype. Values are means ± SE; n = 3–4 mice/group from 2 independent experiments analyzed in cross sections of 4–6 small airways per mouse. ***P < 0.001 (by Mann-Whitney U-test). G and H: representative images of small airway sections from autophagy-related 5 (Atg5f/f) and Scgb1a1Cre/+;Atg5f/f mice. Scale bars = 100 µm. Sections were stained with polyclonal rabbit anti-LC3B (red) and AAT (green); nuclei were stained with bis-benzamide (blue). I: percentage of LC3B-positive airway cells. Values are means ± SE; n = 4–6 mice/group from 3 separate experiments. *P < 0.05 (by Mann-Whitney U-test).
We anticipated that autophagy-deficient club cells would have a defect in packaging secretory proteins, as do autophagy-deficient Paneth cells (3). Elongation of the autophagy vesicle membrane for packaging in the autophagosome requires activity of the Atg5-Atg12-Atg16 complex. We examined the role of autophagy specifically in club cells by targeted deletion of Atg5 (Scgb1a1Cre/+;Atg5f/f mice) (26). Atg5, a binding partner of Atg16l1, is well known to have similar functions (12). Club cells from adult Scgb1a1Cre/+;Atg5f/f mice showed diminished levels of LC3B-positive puncta (Fig. 1, G–I).
Unlike Paneth cells, club cells have a distinctive apical protrusion into the lumen of the airway. We found noticeable flattening of the apical extension of the club cell cytoplasm from Scgb1a1Cre/+;Atg5f/f mice compared with Atg5f/f littermate controls (Fig. 2, A and B). In contrast, sections of ciliated cells of Scgb1a1Cre/+;Atg5f/f mice showed no obvious alterations in morphology. The abnormal club cell morphology did not appear secondary to an inflammatory process, as hematoxylin-eosin- and periodic acid-Schiff-stained sections showed no obvious inflammation or goblet cell metaplasia, respectively (Fig. 2, C and D).
Fig. 2.

Scgb1a1Cre/+;Atg5 f/f mice have abnormal club cells. A–D: representative images of small airways stained with hematoxylin and eosin (A and B) and periodic acid-Schiff (C and D) for autophagy-related 5 (Atg5f/f) and secretoglobulin family 1A member 1 (Scgb1a1Cre+);Atg5f/f mice. Scale bars = 100 µm. Insets show regions within black boxes (scale bars = 10 µm). Images of similar magnification from parallel sections stained with Scgb1a1 and acetylated α-tubulin (AAT) are shown below hematoxylin-and-eosin insets. Arrows indicate ciliated cells; arrowheads indicate club cells. E–H: sections of lung from indicated genotypes immunostained for Scgb1a1 (green), surfactant protein A1 (Sftpa1) antibody (red), surfactant protein D (Sftpd, green), and anti-cytochrome P-450 family 2, subfamily f, polypeptide 2 (Cyp2f2, red); nuclei are stained with bis-benzamide (blue). Scale bars = 10 µm. I: percentage of high and sparse Scgb1a1-positive airway cells. Scgb1a1-positive cells were subdivided into those with high or sparse amounts of cytoplasmic puncta (see materials and methods). Values are means ± SE; n = 4–6 mice/group performed in 3 independent experiments; n = 4–6 well-orientated airways/mouse. **P < 0.01 (by one-way ANOVA with Tukey’s multiple comparison test). J: percentage of club cells that are positive for the indicated markers comparing Atg5f/f and Scgb1a1Cre+;Atg5 f/f mice. Values are means ± SE; n = 4–5 mice/group performed in 3 independent experiments; n = 4–6 well-orientated airways/mouse. **P < 0.01 (by Mann-Whitney U-test).
After observing that the volume of apical cytoplasm of Scgb1a1Cre/+;Atg5f/f club cells was reduced in tissue sections, we evaluated the expression of proteins known to be apically secreted, with a focus on those with important roles in host defense (40). Immunostained tissue sections of control mice for Scgb1a1, Sftpa1, Sftpd, and Cyp2f2 showed discrete cytoplasmic signals consistent with known localization of these proteins within vesicles of club cells (Fig. 2, E–H) (2, 19, 41). Scgb1a1 staining is diminished in Scgb1a1Cre/+;Atg5f/f mice, and the majority of positive club cells contain sparse granules (Fig. 2, E and I). In addition, the club cells from Scgb1a1Cre/+;Atg5f/f control mice showed markedly diminished staining for Sftpa1 and Sftpd (Fig. 2, F, G, and J). In contrast, Cyp2f2 staining of lungs from Scgb1a1Cre/+;Atg5f/f mice was similar to controls (Fig. 2, H and J).
We used transmission electron microscopy to investigate the ultrastructure of the Atg5-deficient club cells. Normally, club cells contain densely packed electron-dense granules within their apical cytoplasm (44) (Fig. 3, A and C). Our analysis showed no overall defect in cytoplasmic organization of Atg5-deficient club cells, as indicated by the presence of apically localized granules. However, Atg5-deficient club cells had a flattened luminal extension and significantly fewer granules than controls (Fig. 3, A–F). These findings confirm our initial conclusion that autophagy was required for the normal levels of secreted club cell proteins.
Fig. 3.

Autophagy-related 5 (Atg5)-deficient club cells have a diminished apical protrusion and numbers of secretory granules. A–D: representative transmission electron micrographs of club cells at 2 different magnifications [scale bars = 2 µm (A and C) and 500 nm (B and D)] in Atg5f/f and secretoglobulin family 1A member 1 (Scgb1a1Cre+);Atg5f/f mice. White dashed lines in A and B demarcate apical protrusions of club cells. In C and D, large black arrows mark high-density granules (G) and small black arrows mark mitochondria (M). E: height of apical club cell cytoplasmic protrusion (measured from apex of cell to dashed white line). F: number of club cell granules in the apical protrusion. Values are means ± SE; n = 3 mice/group performed in 2 separate experiments. ns, Not significant.
Atg5-deficient club cells show altered recovery after airway injury.
As the Atg5 mutation is constitutive in club cells during their development, we could not exclude the possibility that the morphological changes were a result of altered differentiation. Therefore, we used naphthalene to specifically ablate club cells, leading to regeneration of new club cells in the adult mouse (43, 46). Examination of the airway epithelial cells 14 days after naphthalene injury showed a reduction in Scgb1a1-positive club cells in Scgb1a1Cre/+;Atg5f/f mice compared with littermate controls (Fig. 4). The histological appearance of the club cell morphology was similar to that observed in the developmentally deficient Scgb1a1Cre/+;Atg5f/f mice. Together, these findings support a role for autophagy in the regeneration of club cells in response to airway injury and independent of the developmentally regulated differentiation of club cells.
Fig. 4.

Altered secretoglobulin family 1A member 1 (Scgb1a1) expression in lungs from Scgb1a1Cre/+;autophagy-related 5 (Atg5f/f) mice after naphthalene injury. A–D: representative images of lung sections from Atg5f/f and Scgb1a1Cre/+;Atg5f/f mice 14 days after naphthalene administration. Sections were stained with hematoxylin and eosin (A and B) or immunostained for Scgb1a1 (red) and acetylated-α-tubulin (AAT, green); nuclei were stained with bis-benzamide (blue) (C and D). Scale bars = 100 µm. E and F: percentage of Sgcb1a- and AAT-positive airway cells. Values are means ± SE; n = 5–8 mice/group performed in 2 separate experiments. **P < 0.01 (by one-way ANOVA and Tukey's multiple comparison test); ns, not significant.
Club cells in Atg16l1 hypomorphic mice have diminished expression of secreted proteins.
We next examined the role of autophagy in whole body Atg16l1 hypomorphic (hm) mice, in which Atg16l1 expression is reduced throughout the airway (2). As anticipated, autophagy is diminished in club cells of Atg16l1hm/hm mice, as determined by LC3B immunostaining (data not shown). The club cells of Atg16l1hm/hm mice demonstrated markedly diminished apical cytoplasm (Fig. 5, A–D). In addition, protein levels (as determined by immunostaining) for Scgb1a1, Sftpa1, and Sftpd were reduced in Atg16l1hm/hm mice compared with controls (Fig. 5, E–G, I, and J). Staining for Cyp2f2 was not different between Atg16l1hm/hm mice and controls (Fig. 5, H and J). Together, these findings are similar to the effects of loss of Atg5 function in club cells (Fig. 2).
Fig. 5.

Autophagy-deficient [hypomorphic (hm)] autophagy-related 16-like 1-deficient (hm) Atg16L1hm/hm mice have abnormal club cells. A–D: representative images of small airways from Atg16l1+/+and Atg16l1hm/hm littermate mice stained with hematoxylin and eosin (A and B) and periodic acid-Schiff (C and D). Scale bars = 100 µm. Insets show regions within black boxes (scale bars = 10 µm). Images of similar magnification from parallel sections stained with secretoglobulin family 1A member 1 (Scgb1a1) and acetylated α-tubulin (AAT) are shown below hematoxylin-and-eosin insets. Arrows indicate ciliated cells; arrowheads indicate club cells. E–H: localization of club cell proteins in airways of Atg16l1+/+ and Atg16l1hm/hm mice. Lung sections were immunostained as indicated for Scgb1a1 (green), surfactant protein A1 (Sftpa1, red), surfactant protein D (Sftpd, green), and cytochrome P-450 family 2, subfamily f, polypeptide 2 (Cyp2f2, red), and nuclei were stained with bis-benzamide (blue). Scale bars = 10 µm. I: percentage of high and sparse Scgb1a1-positive airway cells. Scgb1a1-positive cells were subdivided into those with high or sparse amounts of cytoplasmic puncta (see materials and methods). Values are means ± SE; n = 4–6 mice/group performed in 3 independent experiments; n = 4–6 well-orientated airways/mouse. **P < 0.01 (by one-way ANOVA with Tukey’s multiple comparison test). J: percentage of club cells that are positive for the indicated markers in Atg16l1+/+ and Atg16l1hm/hm mice. Values are means ± SE; n = 4–6 mice/group performed in 3 independent experiments; n = 4–6 well-orientated airways/mouse. **P < 0.01 (by Mann-Whitney U-test); ns, not significant.
To further evaluate the basis for the autophagy-dependent club cell defect, we evaluated protein and mRNA expression for Scgb1a1, Sftpa1, Sftpd, and Cyp2f2 in whole nonperfused mouse lungs. Expression of Scgb1a1 and Cyp2f2 is specific to club cells in the lung, while both Sftpa1 and Sftpd are also expressed by type II pneumocytes (40). Immunoblot analysis showed significantly less Scgb1a1, Sftpa1, and Sftpd proteins in lungs from Atg16L1hm/hm mice than controls (Fig. 6, A–C). In contrast, Cyp2f2 protein levels were equivalent in the two mouse strains (Fig. 6, B and C). These findings suggest that secreted and intracellular levels of Scgb1a1, Sftpa1, and Sftpd are diminished in lungs of these mice. Interestingly, when we compared Atg16L1hm/hm with control mice, mRNA levels were similar for all four genes (Fig. 6D). We observed no differences in mRNA levels of transcription factors known to drive differentiation of lung epithelial cells [NK2 homeobox 1 (Nkx2-1)], airway multiciliated cells [forkhead box J1 (Foxj1)], or goblet cells [SAM-pointed domain containing Ets transcription factor (Spdef)] (data not shown). These results suggest that Atg16l1 expression affects protein levels of a subset of club cell-secreted proteins similar to loss of Atg5.
Fig. 6.
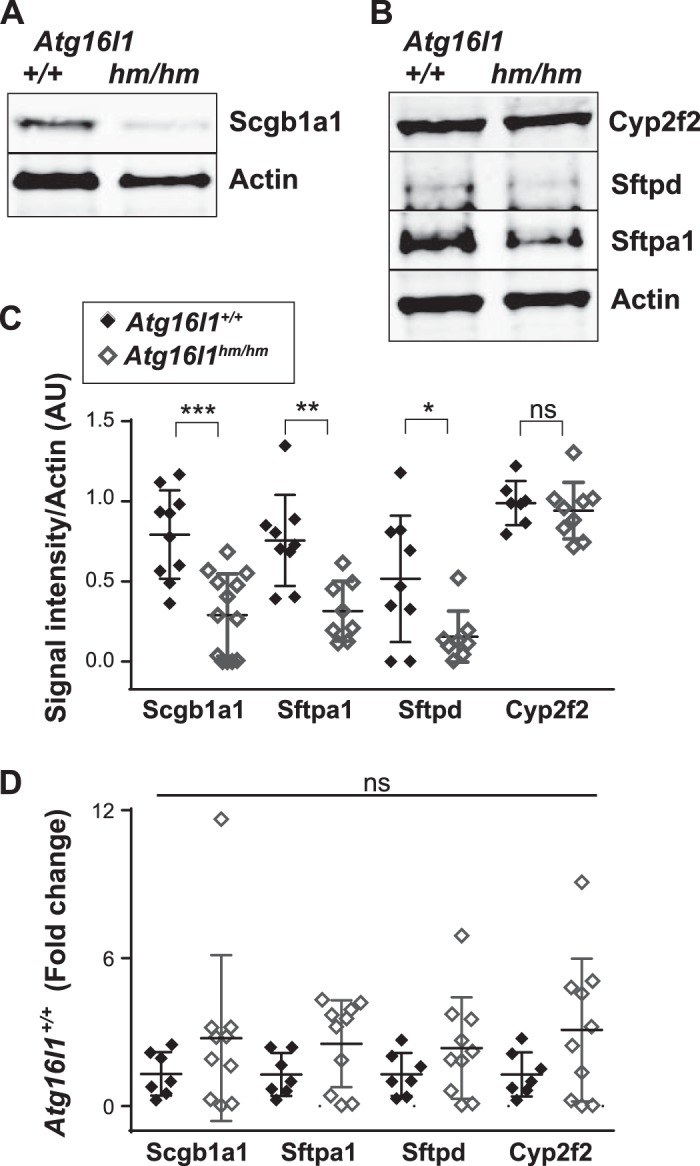
Diminished host defense protein expression in autophagy-related 16-like 1 hypomorphic (Atg16l1hm/hm) mouse lungs. A and B: representative immunoblots of secretoglobulin family 1A member 1 (Scgb1a1) and actin and cytochrome P-450 family 2, subfamily f, polypeptide 2 (Cyp2f2), surfactant proteins D and A1 (Sftpd and Sftpa1), and actin from nonperfused lungs of Atg16l1+/+and Atg16l1hm/hm littermate mice. C: band intensity compared with actin. AU, arbitrary units. Values are means ± SE; n = 5–9 samples/group performed in 3 separate experiments. *P < 0.05, **P < 0.01, ***P < 0.001 (by Mann-Whitney U-test); ns, not significant. D: fold change in mRNA of Scgb1a1, Sftpa1, Sftpd, and Cyp2f2 in lungs of Atg16l1+/+ and Atg16l1hm/hm mice compared with baseline expression in lungs of Atg16l1+/+ mice. Values are means ± SE; n = 7–10 samples/group performed in 3 separate experiments.
Cigarette smoking is not required to trigger diminished Scgb1a1 in Atg16l1T300A/T300A mice.
Previous studies have shown that expression of Scgb1a1 can be reduced in wild-type mice exposed to cigarette smoke (24, 51). Here, we tested if cigarette smoke could further diminish Scgb1a1 expression in Atg16l1hm/hm mice or evoke this phenotype in Atg16l1T300A/T300A mice. The latter strain of mice contains a single nucleotide polymorphism of Atg16l1 known in humans to alter expression of this protein in response to cellular stress (22). In addition, when exposed to cigarette smoke, these mice develop abnormal Paneth cells (27). Contrary to our expectation, we found a decrease Scgb1a1 in the Atg16l1hm/hm and Atg16l1T300A/T300A strains in sham-treated conditions. As anticipated, expression of Scgb1a1 was reduced after 2 wk of smoke exposure in wild-type mice (littermates of Atg16L1hm/hm mice) compared with sham-treated wild-type mice. However, exposure to cigarette smoke did not further reduce levels of Scgb1a1 expression in Atg16l1hm/hm and Atg16l1T300A/T300A mice (Fig. 7). One possible interpretation of these results is that cigarette smoke can reduce autophagy in club cells and an additional environmental perturbation (cigarette smoking) is not required to reduce expression of Scgb1a1 in club cells from Atg16l1T300A/T300A mice. However, these experiments are limited by a 2-wk cigarette smoke-exposure model. A longer-term trial may be important to further test this idea.
Fig. 7.
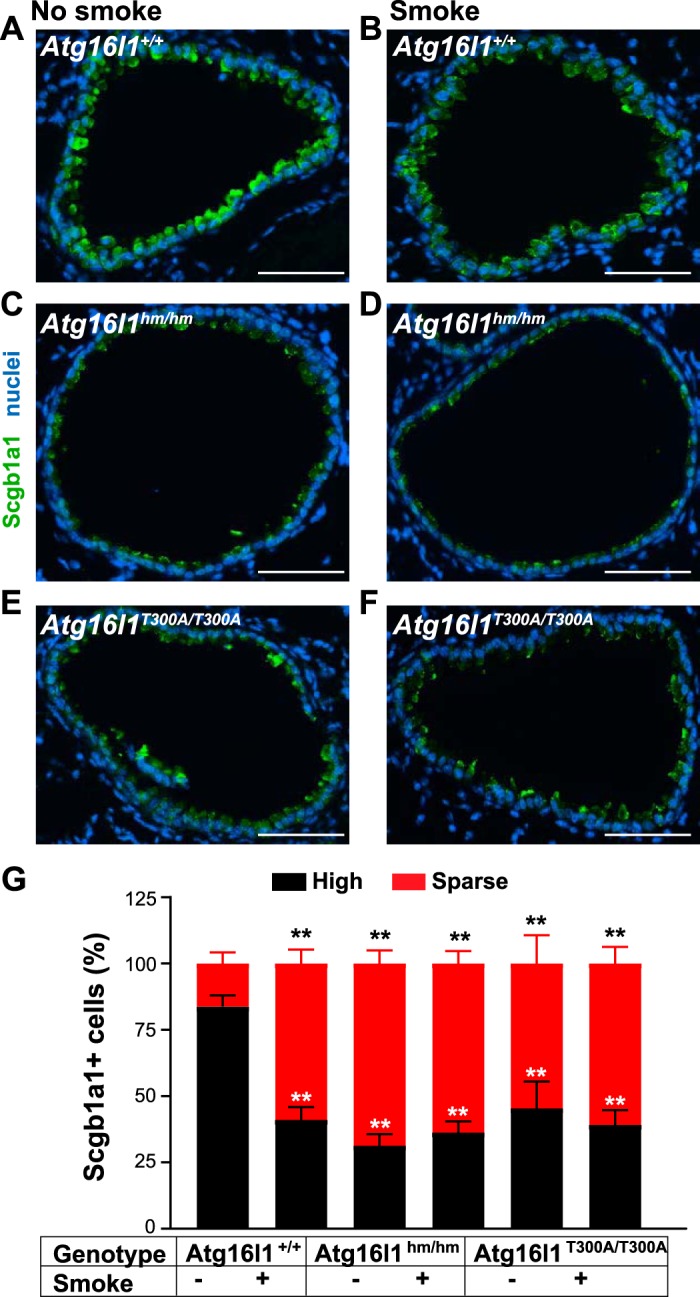
Exposure to cigarette smoke decreases secretoglobulin family 1A member 1 (SCGB1A1) expression in club cells in lungs of wild-type [hypomorphic (hm)] autophagy-related 5 (Atg16L1hm/hm) and Atg16L1T300A/T300A mice. A–F: lung sections from cigarette smoke-exposed and non-smoke-exposed Atg16L1+/+, Atg16L1hm/hm, and Atg16l1T300A/T300A mice stained for Scgb1a1 (green) (see materials and methods); nuclei were stained with bis-benzamide (blue). Scale bars = 100 µm. G: percentage of high and sparse Scgb1a1-positive airway cells. Values are means ± SE; n = 4–8 mice/group performed in 3 independent experiments; n = 4–6 well-orientated airways/mouse. **P < 0.01 (by one-way ANOVA with Tukey’s multiple comparison test).
Late-stage COPD lungs have diminished expression of LC3B in club cells.
Given the limitations of cigarette smoke exposure to model chronic lung disease in mice (48), we evaluated lung tissue from patients with COPD. SCGB1A1 is reported to be diminished in airway epithelial cells in patients with COPD and related to cigarette smoke (23, 33). Furthermore, many studies show that autophagy is active in lung tissues from patients with COPD (7, 8). However, these studies focus on autophagy in the alveolar compartment and immune cells, sites where autophagy appears most active in reports to date. We analyzed lung tissue sections from patients with very severe COPD (former smokers whose lungs were resected during transplantation) and lungs donated for transplantation but not used (non-COPD donors). Tissues were assayed for the expression of autophagy markers LC3B and P62 (SQSTM1) and the relationship to SCGB1A1 in airway bronchioles (terminal airways and those lacking cartilage). Bronchiolar airway epithelial cells from patients with COPD had significantly less LC3B staining than controls (Fig. 8, A, B, and G). The number of SCGB1A1-LC3B dual-positive cells was also reduced (Fig. 8H). There was no difference in the total number of SCGB1A1-positive cells between the tissues from patients with COPD and controls. Similar to LC3B, expression of P62 was reduced in terminal airways (Fig. 8, C, D, and G), as was the number of SCGB1A1-P62 dual-positive cells (Fig. 8H). There was P62 expression in nonairway cells from donors and patients with COPD (Fig. 8, E and F). These findings suggest a parallel role for autophagy and SCGB1A1 protein expression in mouse and human club cells.
Fig. 8.

Club cells from lungs of patients with chronic obstructive pulmonary disease (COPD) have decreased secretoglobulin family 1A member 1 (SCGB1A1), light chain 3B (LC3B), and P62. A–D: representative lung tissue sections immunostained for SCGB1A1 (red), LC3B (green), and acetylated α-tubulin (AAT, purple) or SCGB1A1 (green) and p62 (red) from donor (non-COPD) subjects and COPD patients; nuclei were stained with bis-benzamide (blue). Scale bars = 100 µm. Images at right are higher magnifications (scale bars = 10 µm) of regions in white boxes. Arrows indicate ciliated cells; arrowheads indicate club cells. White dashed lines outline individual cells in the insets. E and F: magnified images of P62 (red)-stained nonairway cells; nuclei were stained with bis-benzamide (blue). Scale bars = 10 µm. G and H: percentage of single (G) and dual (H) positive airway cells for the indicated markers on the x-axis. Values are means ± SE; n = 4–14 subjects/group. *P < 0.05 (by Mann-Whitney U-test).
DISCUSSION
Using multiple mouse genetic developmental and injury models, we have shown a role for late-stage autophagy proteins in the function of lung secretory club cells. LC3B expression was reduced in club cells from mice with loss of function of Atg5. These autophagy-reduced club cells featured abnormal morphology with diminished apical cytoplasm. Atg5-deficient club cells also showed reduced protein (but not mRNA) expression for a subset of highly expressed secreted molecules with broad roles in host defense, including Scgb1a1, Sftpa1, and Sftpd. This phenotype was recapitulated in club cells that were regenerated postinjury, indicating that this finding is independent of developmental programs of differentiation. Mice with diminished function of Atg16l1 (a binding partner of Atg5) showed a phenotype in these cells similar to that in Atg5-deficient mice. Diminished expression of Scgb1a1 was also observed in club cells of Atg16l1T300A/T300A mice, as well club cells of patients with COPD, who also showed evidence of reduced autophagy.
Autophagy proteins have been shown to play broad roles in secretion and membrane trafficking (11, 42). Autophagy-associated protein secretion pathways address the overlap between autophagy and conventional protein secretion (38). The major cellular defect in Atg5-deficient club cells is that they contain fewer granules, as shown by electron microscopy. We could postulate that this is a failure of unconventional protein secretion; however, those mechanisms are typically invoked for proteins that lack signal peptides (leader sequence), which is not the case for Scgb1a1 (30). Additional unconventional protein secretion pathways have been described for proteins with leader sequences, including cystic fibrosis protein CFTR; however, molecular routes have been shown to vary with the specific protein (13, 38).
The link between autophagy proteins and vesicle number in club cells is not understood. The possibilities are broad. A leading hypothesis is that secretion and autophagy may be linked by shared structural protein requirements for vesicle packaging. An alternative hypothesis is that autophagy proteins direct control of protein translation to maintain cellular quality control. Loss of function of Atg5/Atg16l1 in secretory cells and other cell types is not instructive, since the role of autophagy also appears to be cell type-specific. For example, loss of function of Atg5 in goblet cells in the lung and gut leads to enhanced numbers of vesicles due to lack of secretion during inflammatory conditions (10). In Paneth cells, antimicrobial proteins are mislocalized to the cytoplasm and lead to enhanced apoptosis (3). Current concepts of how secretoglobulin and the collectins are produced and packaged in lung epithelial cells are lacking, perhaps because methods for their sustained culture are not well established. The future development of systems for the culture and manipulation of club cell populations may make it possible to begin to address questions related to Scgb1a1, Sftpa1, and Sftpd packaging, trafficking, and secretion.
We observed that autophagy deficiency disrupted Scgb1a1, Sftpa1, and Sftpd production independent of the cellular stress of cigarette smoke exposure. This is not the case for the intestinal secretory Paneth cells, as abnormal secretion in Paneth cells requires environmental triggers in conjunction with genetic susceptibility in autophagy genes. We previously reported that Paneth cell dysfunction in mice with the Atg16l1T300A/T300A genetic mutation requires cigarette smoke exposure as the environmental trigger (27). A cellular stress such as smoke exposure is required to make the Atg16l1T300A protein susceptible to cleavage and diminished function (22, 32). Thus it was surprising that, in mice bearing the human variant Atg16l1T300A/T300A, exposure to cigarette smoke is not required to elicit abnormal club cells. It is possible that lesser environmental stimuli are required to elicit abnormal club cells in Atg16l1T300A/T300A mice. An alternative explanation is that the anatomic consequence of normal lung exposure is sufficient stress. Thus the sham procedure or our mouse housing environment may be sufficient to act as a trigger for this cell type. It has been reported that bronchitis, bronchiolitis, and interstitial lung disease may be increased in patients with Crohn’s disease (29), the population at risk for the T300A variant. Certainly, examination of club cells in individuals with this single nucleotide polymorphism would be of interest.
We observed that cigarette smoke exposure in autophagy-deficient mice did not further diminish the low baseline levels of Scgb1a1. This outcome may be related to a detection issue or an autophagy pathway block, so that turnover of Scgb1a1 is interrupted. We found that the result in human tissue was somewhat similar. We also evaluated SCGB1A1 and autophagy in lung tissue from individuals who had smoked in the past but were abstinent as a requirement of transplantation, and we identified a correlation between decreased SCGB1A1 expression and diminished LC3B expression in the club cells. This finding matches the correlation of autophagy and Scgb1a1 in our mouse models. The low levels of p62 in these mice may indicate that not only is the level of autophagy low, so is likely the need for protein turnover. The finding of low SCGB1A1 staining (CC16) in patients with very severe COPD is also consistent with previous reports (23, 51). The implication is that there is interplay between the role of autophagy in chronic inflammation and production of club cell host defense proteins. Low levels of CC16 have been measured in sputum of patients who have cystic fibrosis and chronic lung infection, suggesting that similar events may occur in other chronic lung diseases (21). The requirement for autophagy to generate Scgb1a1 granules in the human tissue was consistent with the requirement in mice that were genetically deficient in autophagy proteins. However, it is likely that the high-inflammatory environment in the lung of individuals with COPD somehow depresses autophagy in the club cell. There is controversy in the literature about whether autophagy is impaired or elevated in emphysema and COPD. Our observations suggest that this may be cell-specific.
In summary, we show a novel effect of the autophagy pathway on the regulation of major secretory proteins of the airway epithelium. This effect appears to be distinct from the effects of autophagy deficiency in airway and intestinal goblet cells or intestinal Paneth cells, and dissimilar from the effects of global knockouts of the secretory proteins associated with these cells. Other pathways known to affect the secretion of club cell proteins do not appear to be able to overcome the effect of autophagy deficiency. This autophagy deficiency appears to be relevant to human disease states and may tie into some of the therapies that are being explored to combat COPD and other airway diseases.
GRANTS
This work was supported, in part, by National Heart, Lung, and Blood Institute Grant R01 HL-122582 (S. L. Brody).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.-C.L., S.L.B., and T.S. conceived and designed research; N.P.M., J.T.K., and T.-C.L. performed experiments; N.P.M., S.L.B., and T.S. analyzed data; N.P.M., S.L.B., and T.S. interpreted results of experiments; N.P.M. and S.L.B. prepared figures; N.P.M., S.L.B., and T.S. drafted manuscript; N.P.M., S.L.B., and T.S. edited and revised manuscript; N.P.M., J.T.K., T.-C.L., S.L.B., and T.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Barry Stripp for antibodies to Scgb1a1 and Derek Byers for assistance with human tissue samples.
REFERENCES
- 1.Araya J, Kojima J, Takasaka N, Ito S, Fujii S, Hara H, Yanagisawa H, Kobayashi K, Tsurushige C, Kawaishi M, Kamiya N, Hirano J, Odaka M, Morikawa T, Nishimura SL, Kawabata Y, Hano H, Nakayama K, Kuwano K. Insufficient autophagy in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 304: L56–L69, 2013. doi: 10.1152/ajplung.00213.2012. [DOI] [PubMed] [Google Scholar]
- 2.Bedetti CD, Singh J, Singh G, Katyal SL, Wong-Chong ML. Ultrastructural localization of rat Clara cell 10 KD secretory protein by the immunogold technique using polyclonal and monoclonal antibodies. J Histochem Cytochem 35: 789–794, 1987. doi: 10.1177/35.7.2438324. [DOI] [PubMed] [Google Scholar]
- 3.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, Stone CD, Brunt EM, Xavier RJ, Sleckman BP, Li E, Mizushima N, Stappenbeck TS, Virgin HW IV. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456: 259–263, 2008. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cadwell K, Patel KK, Komatsu M, Virgin HW IV, Stappenbeck TS. A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy 5: 250–252, 2009. doi: 10.4161/auto.5.2.7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cann GM, Guignabert C, Ying L, Deshpande N, Bekker JM, Wang L, Zhou B, Rabinovitch M. Developmental expression of LC3α and -β: absence of fibronectin or autophagy phenotype in LC3β knockout mice. Dev Dyn 237: 187–195, 2008. doi: 10.1002/dvdy.21392. [DOI] [PubMed] [Google Scholar]
- 6.Chen LC, Zhang Z, Myers AC, Huang SK. Cutting edge: altered pulmonary eosinophilic inflammation in mice deficient for Clara cell secretory 10-kDa protein. J Immunol 167: 3025–3028, 2001. doi: 10.4049/jimmunol.167.6.3025. [DOI] [PubMed] [Google Scholar]
- 7.Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, Dhir R, Landreneau RJ, Schuchert MJ, Yousem SA, Nakahira K, Pilewski JM, Lee JS, Zhang Y, Ryter SW, Choi AM. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One 3: e3316, 2008. doi: 10.1371/journal.pone.0003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen ZH, Lam HC, Jin Y, Kim HP, Cao J, Lee SJ, Ifedigbo E, Parameswaran H, Ryter SW, Choi AM. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci USA 107: 18880–18885, 2010. doi: 10.1073/pnas.1005574107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deslee G, Adair-Kirk TL, Betsuyaku T, Woods JC, Moore CH, Gierada DS, Conradi SH, Atkinson JJ, Toennies HM, Battaile JT, Kobayashi DK, Patterson GA, Holtzman MJ, Pierce RA. Cigarette smoke induces nucleic-acid oxidation in lung fibroblasts. Am J Respir Cell Mol Biol 43: 576–584, 2010. doi: 10.1165/rcmb.2009-0221OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dickinson JD, Alevy Y, Malvin NP, Patel KK, Gunsten SP, Holtzman MJ, Stappenbeck TS, Brody SL. IL13 activates autophagy to regulate secretion in airway epithelial cells. Autophagy 12: 397–409, 2016. doi: 10.1080/15548627.2015.1056967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farhan H, Kundu M, Ferro-Novick S. The link between autophagy and secretion: a story of multitasking proteins. Mol Biol Cell 28: 1161–1164, 2017. doi: 10.1091/mbc.e16-11-0762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 19: 2092–2100, 2008. doi: 10.1091/mbc.e07-12-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gee HY, Noh SH, Tang BL, Kim KH, Lee MG. Rescue of ΔF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell 146: 746–760, 2011. doi: 10.1016/j.cell.2011.07.021. [DOI] [PubMed] [Google Scholar]
- 14.Guerra S, Halonen M, Vasquez MM, Spangenberg A, Stern DA, Morgan WJ, Wright AL, Lavi I, Tarès L, Carsin AE, Dobaño C, Barreiro E, Zock JP, Martínez-Moratalla J, Urrutia I, Sunyer J, Keidel D, Imboden M, Probst-Hensch N, Hallberg J, Melén E, Wickman M, Bousquet J, Belgrave DC, Simpson A, Custovic A, Antó JM, Martinez FD. Relation between circulating CC16 concentrations, lung function, and development of chronic obstructive pulmonary disease across the lifespan: a prospective study. Lancet Respir Med 3: 613–620, 2015. doi: 10.1016/S2213-2600(15)00196-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han S, Mallampalli RK. The role of surfactant in lung disease and host defense against pulmonary infections. Ann Am Thorac Soc 12: 765–774, 2015. doi: 10.1513/AnnalsATS.201411-507FR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441: 885–889, 2006. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 17.Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 277: 2002–2004, 1997. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- 18.Hiemstra PS, Bourdin A. Club cells, CC10 and self-control at the epithelial surface. Eur Respir J 44: 831–832, 2014. doi: 10.1183/09031936.00089214. [DOI] [PubMed] [Google Scholar]
- 19.Karnati S, Graulich T, Oruqaj G, Pfreimer S, Seimetz M, Stamme C, Mariani TJ, Weissmann N, Mühlfeld C, Baumgart-Vogt E. Postnatal development of the bronchiolar club cells of distal airways in the mouse lung: stereological and molecular biological studies. Cell Tissue Res 364: 543–557, 2016. doi: 10.1007/s00441-015-2354-x. [DOI] [PubMed] [Google Scholar]
- 20.King BA, Kingma PS. Surfactant protein D deficiency increases lung injury during endotoxemia. Am J Respir Cell Mol Biol 44: 709–715, 2011. doi: 10.1165/rcmb.2009-0436OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laguna TA, Williams CB, Brandy KR, Welchlin-Bradford C, Moen CE, Reilly CS, Wendt CH. Sputum club cell protein concentration is associated with pulmonary exacerbation in cystic fibrosis. J Cyst Fibros 14: 334–340, 2015. doi: 10.1016/j.jcf.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lassen KG, Kuballa P, Conway KL, Patel KK, Becker CE, Peloquin JM, Villablanca EJ, Norman JM, Liu TC, Heath RJ, Becker ML, Fagbami L, Horn H, Mercer J, Yilmaz OH, Jaffe JD, Shamji AF, Bhan AK, Carr SA, Daly MJ, Virgin HW, Schreiber SL, Stappenbeck TS, Xavier RJ. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci USA 111: 7741–7746, 2014. doi: 10.1073/pnas.1407001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laucho-Contreras ME, Polverino F, Gupta K, Taylor KL, Kelly E, Pinto-Plata V, Divo M, Ashfaq N, Petersen H, Stripp B, Pilon AL, Tesfaigzi Y, Celli BR, Owen CA. Protective role for club cell secretory protein-16 (CC16) in the development of COPD. Eur Respir J 45: 1544–1556, 2015. doi: 10.1183/09031936.00134214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laucho-Contreras ME, Polverino F, Tesfaigzi Y, Pilon A, Celli BR, Owen CA. Club cell protein 16 (CC16) augmentation: a potential disease-modifying approach for chronic obstructive pulmonary disease (COPD). Expert Opin Ther Targets 20: 869–883, 2016. doi: 10.1517/14728222.2016.1139084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ledford JG, Pastva AM, Wright JR. Collectins link innate and adaptive immunity in allergic airway disease. Innate Immun 16: 183–190, 2010. doi: 10.1177/1753425910368446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Cho SN, Evans CM, Dickey BF, Jeong JW, DeMayo FJ. Cre-mediated recombination in mouse Clara cells. Genesis 46: 300–307, 2008. doi: 10.1002/dvg.20396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu TC, Kern JT, VanDussen KL, Xiong S, Kaiko GE, Wilen CB, Rajala MW, Caruso R, Holtzman MJ, Gao F, McGovern DP, Nunez G, Head RD, Stappenbeck TS. Interaction between smoking and ATG16L1T300A triggers Paneth cell defects in Crohn’s disease. J Clin Invest 128: 5110–5122, 2018. doi: 10.1172/JCI120453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mackay RM, Grainge CL, Lau LC, Barber C, Clark HW, Howarth PH. Airway surfactant protein D deficiency in adults with severe asthma. Chest 149: 1165–1172, 2016. doi: 10.1016/j.chest.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Majewski S, Piotrowski W. Pulmonary manifestations of inflammatory bowel disease. Arch Med Sci 11: 1179–1188, 2015. doi: 10.5114/aoms.2015.56343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malsky ML, Bullock DW, Willard JJ, Ward DN. Progesterone-induced secretory protein. NH2-terminal sequence of pre-uteroglobin. J Biol Chem 254: 1580–1585, 1979. [PubMed] [Google Scholar]
- 31.Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, Yuan F, Chen S, Leung HM, Villoria J, Rogel N, Burgin G, Tsankov AM, Waghray A, Slyper M, Waldman J, Nguyen L, Dionne D, Rozenblatt-Rosen O, Tata PR, Mou H, Shivaraju M, Bihler H, Mense M, Tearney GJ, Rowe SM, Engelhardt JF, Regev A, Rajagopal J. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560: 319–324, 2018. doi: 10.1038/s41586-018-0393-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murthy A, Li Y, Peng I, Reichelt M, Katakam AK, Noubade R, Roose-Girma M, DeVoss J, Diehl L, Graham RR, van Lookeren Campagne M. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 506: 456–462, 2014. doi: 10.1038/nature13044. [DOI] [PubMed] [Google Scholar]
- 33.Park HY, Churg A, Wright JL, Li Y, Tam S, Man SF, Tashkin D, Wise RA, Connett JE, Sin DD. Club cell protein 16 and disease progression in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 188: 1413–1419, 2013. doi: 10.1164/rccm.201305-0892OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel KK, Miyoshi H, Beatty WL, Head RD, Malvin NP, Cadwell K, Guan JL, Saitoh T, Akira S, Seglen PO, Dinauer MC, Virgin HW, Stappenbeck TS. Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. EMBO J 32: 3130–3144, 2013. doi: 10.1038/emboj.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Plasschaert LW, Žilionis R, Choo-Wing R, Savova V, Knehr J, Roma G, Klein AM, Jaffe AB. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560: 377–381, 2018. doi: 10.1038/s41586-018-0394-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ponpuak M, Mandell MA, Kimura T, Chauhan S, Cleyrat C, Deretic V. Secretory autophagy. Curr Opin Cell Biol 35: 106–116, 2015. doi: 10.1016/j.ceb.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rabouille C. Pathways of unconventional protein secretion. Trends Cell Biol 27: 230–240, 2017. doi: 10.1016/j.tcb.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 39.Schläfli AM, Berezowska S, Adams O, Langer R, Tschan MP. Reliable LC3 and p62 autophagy marker detection in formalin fixed paraffin embedded human tissue by immunohistochemistry. Eur J Histochem 59: 2481, 2015. doi: 10.4081/ejh.2015.2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh G, Katyal SL. Clara cell proteins. Ann NY Acad Sci 923: 43–58, 2000. doi: 10.1111/j.1749-6632.2000.tb05518.x. [DOI] [PubMed] [Google Scholar]
- 41.Singh G, Katyal SL. Clara cells and Clara cell 10 kD protein (CC10). Am J Respir Cell Mol Biol 17: 141–143, 1997. doi: 10.1165/ajrcmb.17.2.f138. [DOI] [PubMed] [Google Scholar]
- 42.Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 16: 495–501, 2014. doi: 10.1038/ncb2979. [DOI] [PubMed] [Google Scholar]
- 43.Stripp BR, Maxson K, Mera R, Singh G. Plasticity of airway cell proliferation and gene expression after acute naphthalene injury. Am J Physiol Lung Cell Mol Physiol 269: L791–L799, 1995. doi: 10.1152/ajplung.1995.269.6.L791. [DOI] [PubMed] [Google Scholar]
- 44.Stripp BR, Reynolds SD, Boe IM, Lund J, Power JH, Coppens JT, Wong V, Reynolds PR, Plopper CG. Clara cell secretory protein deficiency alters Clara cell secretory apparatus and the protein composition of airway lining fluid. Am J Respir Cell Mol Biol 27: 170–178, 2002. doi: 10.1165/ajrcmb.27.2.200200270c. [DOI] [PubMed] [Google Scholar]
- 45.Torisu T, Torisu K, Lee IH, Liu J, Malide D, Combs CA, Wu XS, Rovira II, Fergusson MM, Weigert R, Connelly PS, Daniels MP, Komatsu M, Cao L, Finkel T. Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat Med 19: 1281–1287, 2013. doi: 10.1038/nm.3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Winkle LS, Buckpitt AR, Nishio SJ, Isaac JM, Plopper CG. Cellular response in naphthalene-induced Clara cell injury and bronchiolar epithelial repair in mice. Am J Physiol Lung Cell Mol Physiol 269: L800–L818, 1995. doi: 10.1152/ajplung.1995.269.6.L800. [DOI] [PubMed] [Google Scholar]
- 47.Wan H, Kaestner KH, Ang SL, Ikegami M, Finkelman FD, Stahlman MT, Fulkerson PC, Rothenberg ME, Whitsett JA. Foxa2 regulates alveolarization and goblet cell hyperplasia. Development 131: 953–964, 2004. doi: 10.1242/dev.00966. [DOI] [PubMed] [Google Scholar]
- 48.Wright JL, Cosio M, Churg A. Animal models of chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol 295: L1–L15, 2008. doi: 10.1152/ajplung.90200.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiang X, Yuan F, Zhao J, Li Z, Wang X, Guan Y, Tang C, Sun G, Li Y, Zhang W. Deficiency in pulmonary surfactant proteins in mice with fatty acid binding protein 4-Cre-mediated knockout of the tuberous sclerosis complex 1 gene. Exp Physiol 98: 830–841, 2013. doi: 10.1113/expphysiol.2012.069674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang HZ, Wang JP, Mi S, Liu HZ, Cui B, Yan HM, Yan J, Li Z, Liu H, Hua F, Lu W, Hu ZW. TLR4 activity is required in the resolution of pulmonary inflammation and fibrosis after acute and chronic lung injury. Am J Pathol 180: 275–292, 2012. doi: 10.1016/j.ajpath.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 51.Zhu L, Di PY, Wu R, Pinkerton KE, Chen Y. Repression of CC16 by cigarette smoke (CS) exposure. PLoS One 10: e0116159, 2015. doi: 10.1371/journal.pone.0116159. [DOI] [PMC free article] [PubMed] [Google Scholar]