

Abstract
Sepsis has a well-studied inflammatory phase, with a less understood secondary immunosuppressive phase. Elevated blood lactate and slow lactate clearance are associated with mortality, however, regulatory roles are unknown. We hypothesized that lactic acid (LA) contributes to the late phase and is not solely a consequence of bacterial infection. No studies have examined LA effects in sepsis models in vivo or a mechanism by which it suppresses LPS-induced activation in vitro. Because mast cells can be activated systemically and contribute to sepsis, we examined LA effects on the mast cell response to LPS. LA significantly suppressed LPS-induced cytokine production and NFκB transcriptional activity in mouse bone marrow derived mast cells and cytokine production in peritoneal mast cells. Suppression was MCT-1-dependent and reproducible with sodium lactate or formic acid. Further, LA significantly suppressed cytokine induction following LPS-induced endotoxemia in mice. Because glycolysis is linked to inflammation and LA is a by-product of this process, we examined changes in glucose metabolism. LA treatment reduced glucose uptake and lactate export during LPS stimulation. LA effects were mimicked by glycolytic inhibitors and reversed by increasing ATP availability. These results indicate that glycolytic suppression and ATP production are necessary and sufficient for LA effects. Our work suggests that enhancing glycolysis and ATP production could improve immune function, counteracting LA suppressive effects in the immunosuppressive phase of sepsis.
Keywords: Lactic acid, LPS, mast cell, glycolysis, ATP, sepsis
Introduction
Sepsis is a common and deadly condition with life-threatening organ dysfunction and a dysregulated host response to infection. Sepsis can progress to septic shock, which is defined as a severe form of sepsis with circulatory, cellular, and metabolic dysfunction and has a higher risk of mortality (1). While exact estimates of sepsis-induced death vary due to the occurrence of co-morbidities such as cancer and aging-induced immunosuppression, conservative estimates suggest a fatality rate of at least 30% which contributes to the death of more than 150,000 Americans annually (2). Alarmingly, there are no clinically available targeted molecular treatments for sepsis, as we still do not understand the underlying disease progression and mechanisms.
Sepsis is traditionally characterized by high levels of pro-inflammatory cytokines leading to hypotension, vascular leak, tissue hypoxia, and organ failure (3). While the inflammatory response is beneficial to fight the infection, prolonged or exaggerated inflammation can damage host tissue and lead to death. There have been more than 100 clinical trials aimed to dampen the immune response following sepsis, which had no effect or worsened patient outcomes and mortality (4, 5). Importantly, recent studies suggest that fluid resuscitation and antibiotics allow most patients to survive the initial hyperinflammatory phase, at which time they enter an immunosuppressed state marked by impaired immune cell activation (4, 6, 7). Patient death in the immunosuppressive phase is often attributed to failure to clear the initial infection or acquisition of a secondary infection. Additionally, more than 100 distinct biochemical mediators are intricately involved in sepsis pathogenesis (5), but little is known about their role and the optimal response for survival and bacterial clearance. In order to devise more effective interventions for sepsis, a better understanding of the cellular mechanisms behind immunosuppression is required.
Lactate and associated H+ ions are produced following glycolytic energy production by hypoxic tissues or in tissues requiring a rapid supply of ATP (i.e. during cellular activation) (8–11). While normal blood lactate is approximately 0.5–2.5 mM (12), blood lactate concentrations ≥4 mM and impaired clearance have been independently associated with increased sepsis mortality (13–16). Due to these associations, lactate clearance has been added as a treatment guideline for sepsis to speed diagnosis and initial treatment for patients (1). This has reduced mortality in at least one study (17). While these data suggest lactate and mortality are related, it is not known whether elevated lactate is a cause or consequence of the infection.
In the tumor microenvironment, evidence suggests that lactic acid suppresses immune cell activation and promotes regulatory subsets (e.g. M2 macrophage lineage and myeloid derived suppressor cells) (18, 19). In the context of bacterial infection, lactic acid has been typically shown to suppress LPS-induced activation of monocytes, macrophages, and dendritic cells (20–23). Our lab recently found that lactic acid suppresses IL-33-induced mast cell activation in vitro and in vivo (24), however, no studies have examined lactic acid effects on mast cells in the context of bacterial activation. While mast cells are most often considered for their role in allergic disease, they contribute importantly to bacterial defense and septic inflammation. Specifically, local mast cell activation improves neutrophil recruitment and sepsis outcomes by the release of chemoattractants (25–31). Mast cell mMCP-4 enhances survival after cecal ligation and puncture (CLP)-induced sepsis by degrading TNF and limiting the detrimental effects of TNF on the host (32). Furthermore, mast cells have been shown to contribute to control of gram positive (TLR2-mediated) sepsis (33). Given the clinical correlations of lactate with sepsis outcomes and the role for mast cells in sepsis, understanding how lactic acid alters mast cell function is an important question.
The aim of this study was to understand lactic acid effects on LPS-mediated mast cell function and determine its mechanism of action. Our data suggest that lactic acid significantly suppresses LPS-induced mast cell responses by inhibiting glycolysis, effects that are reversed by increasing ATP availability. These results indicate that glycolytic suppression and ATP production are necessary and sufficient for lactic acid effects, and suggest a means of circumventing immunosuppression associated with sepsis.
Methods
Reagents
Recombinant mouse IL-33 was purchased from Shenandoah Biotechnology (Warwick, PA). L-lactic acid was purchased from MP Biosciences (Santa Ana, CA). Lipopolysaccharide (LPS) from Escherichia coli (E.Coli) 055:B5 (catalog L4524) for in vitro studies, LPS from E.coli 0111:B4 (catalog L3024) for in vivo studies, polyinosinic-polycytodylic acid (Poly (I:C; catalog P1530), L-sodium lactate, and formic acid were purchased from Sigma (St Louis, MO). Lipoteichoic acid (LTA) was purchased from AbD Serotec (BioRad, Hercules, CA). The MCT-½ inhibitor AR-C155858 was purchased from Tocris Bioscience (Minneapolis, MN). Sodium oxamate and 2-deoxyglucose (2DG) were purchased from Alfa Aesar (Tewksbury, MA). Etomoxir and rotenone were purchased from Cayman Chemical (Ann Arbor, MI). Antimycin A was purchased from Chem Cruz via Santa Cruz Biotechnology (Dallas, TX). ATP disodium salt was purchased from Tocris (Minneapolis, MN).
Mice
C57BL/6J and NFκB-luc transgenic mice, which have a luciferase transgene under the control of NFκB binding sites, were purchased from The Jackson Laboratory (Bar Harbor, ME). Colonies were maintained in a pathogen free facility with a standard chow diet. Bone marrow was extracted from mice at a minimum of 10 weeks old. Sepsis studies were conducted between ages 8–10 weeks with approval from the Virginia Commonwealth University Institutional Animal Care and Use Committee.
Mast cell culture
Mouse bone marrow was differentiated in IL-3-containing supernatant from WEHI-3 cells and SCF-containing supernatant from BHK-MKL cells as described to yield 90–99% FcεRI+ and cKit+ bone marrow derived mast cells (BMMC) at 21 days (24, 34). Additionally, mast cells from mouse peritoneal lavage were expanded in complete RPMI 1640 medium containing 10% FBS and IL-3+SCF (10 ng/mL) for 7–10 days to yield ~85% FcεRI+ and c-Kit+ peritoneal mast cells (PMC). For most experiments, BMMC or PMC were plated at 2×106 cells/mL with IL-3 and SCF (20 ng/mL). Media or 25 mM lactic acid was added 1:1 for a final concentration of: 1×106 cells/mL, 10 ng/mL IL-3 and SCF, and 12.5 mM lactic acid for all experiments unless otherwise noted. LPS was added at 1 μg/mL for 16 hours for ELISA and glucose/lactate analysis, or for 2 hours for qPCR and luciferase analyses.
ELISA
ELISA analysis was used to measure cytokine concentrations in the cell culture supernatant. IL-6, TNF, and MCP-1 (CCL2) murine ELISA kits were purchased from Biolegend; IL-13 and MIP-1α (CCL3) murine ELISA kits were purchased from Peprotech (Rocky Hill, NJ). Assays were performed in duplicate or triplicate according the manufacturers’ protocols.
RT-qPCR
TRIzol was purchased from Life Sciences (Grand Island, NY) and used to extract total RNA. Nucleic acid purity was measured using a Nanodrop 1000 UV-Vis Spectrophotometer (Thermo Scientific, Waltham, MA). cDNA was synthesized with the qScript cDNA synthesis kit (Quanta Biosciences, Gaithersburg, MD) following the manufacturer’s protocol. To determine IL-6 and GAPDH mRNA expression, real time quantitative PCR (RT-qPCR) was performed with the CFX96 Touch Real-Time PCR Detection System (Bio-Rad) and PerfeCTa SYBR Green SuperMix (Quanta Biosciences). Primers for IL-6 (forward: 5’-TCCAGTTGCCTTCTTGGGAC-3’, reverse: 5’-GTGTAATTAAGCTCCGACTTG-3’) and GAPDH (GAPDH forward, 5’-GATGACATCAAGAAGGTGGTG-3’, reverse, 5’-GCTGTAGCCAAATTCGTTGTC-3’) were purchased from Eurofins MWG Operon (Huntsville, AL). Amplification conditions were as follows: 95°C (2 min) followed by 40 cycles of 95°C (15 s), 55°C (30 s), and 60°C (1 min). A melting curve analysis was performed between 50°C and 95°C. Results were normalized to GAPDH and the H2O control by using the Relative Livak Method (ΔΔCt).
Endotoxemia Model
Age-matched groups of mice (~8–10 weeks old) were injected intraperitoneally (IP) with ketoprofen (1 mg/kg) for pain relief 30 minutes prior to lactic acid (80 mg/kg) or PBS. After 20 hours, a lethal dose of LPS (25 mg/kg) was injected IP to elicit septic shock. Core body temperature over 4 hours was measured using a rectal microprobe (Physitemp Instruments). Additionally, observational score was assessed for posture, grooming, stool, locomotion, respiration, and eye discharge, with 0 as normal and 4 as the worst score for each measure. Mice were sacrificed prior to endpoint if they reached a 3 on three or more categories or if they reached a score of 4 on two or more categories. After 4 hours of temperature and observational measures, mice were sacrificed. Blood was collected using cardiac puncture, and plasma cytokines were measured using ELISA.
Western Blot
Cell lysates were collected using Protease arrest (GBiosciences, Maryland Heights, MO) in cell lysis buffer (Cell Signaling Technology, Danvers, MA). Protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermofisher, Waltham, MA). 4–20% Mini-PROTEAN® TGX™ Precast Protein Gels (Bio–Rad, Hercules, CA) were loaded with 30 μg protein, electrophoresed and transferred to nitrocellulose (Pall Corporation, Ann Arbor, MI), and membranes were blocked for 60 minutes in Blocker casein in tris-buffered saline (TBS) (from Thermofisher, Waltham, MA). Blots were incubated with primary antibodies overnight in block buffer + Tween20 (1:1000) with rabbit anti-IκB and rabbit anti-actin (1:1000, antibodies all purchased from Cell Signaling, Danvers, MA). Blots were washed six times for 5 minutes each in TBS-Tween-20, followed by incubation with secondary antibody (1:10,000) for 60 minutes at room temperature (Cell Signaling, Danvers, MA). Size estimates for proteins were obtained using molecular weight standards from Bio–Rad (Hercules, CA). Blots were visualized and quantified using a LiCor Odyssey CLx Infrared imaging system (Lincoln, NE). After background subtraction, fluorescence intensity for the protein of interest was normalized to the signal intensity for the actin calculated relative to unactivated samples, using Image Studio 4.0 (LiCor).
Luciferase
BMMC were differentiated from NFκB-luc transgenic mouse bone marrow as above. Following lactic acid treatment and LPS activation, cells were lysed and luciferase activity was measured with the Promega Luciferase Assay Substrate and Glomax 20/20 Luminometer (Promega, Madison, WI). Luciferase expression is reported relative to protein concentration (Pierce BCA Protein Assay Kit, Thermofisher, Waltham, MA) and normalized to the unactivated control.
Flow Cytometry
To determine viability, BMMC were incubated for 1 min with propidium iodide (10 μg/mL) prior to flow cytometry analysis with a BD FACSCelesta (Franklin Lakes, NJ). The gating strategy used was doublet exclusion (FSC-A x FSC-H) and size and granularity (FSC x SSC). Percent positive and negative were recorded.
Cellular Metabolism
To determine glucose uptake and lactate export, supernatant concentrations were measured using the Glucose Assay Kit 1 and L-Lactate Assay Kit 1 from Eton Bioscience (San Diego, CA). Glucose uptake was calculated as [glucose in unactivated supernatant] – [glucose in activated supernatant]. Lactate export was calculated as [lactate in activated supernatant] – [lactate in unactivated supernatant].
Statistical analyses
For the glucose uptake, lactate export, and IκB data, a t-test was used. For the rest of the data, a one-way analysis of variance (ANOVA) was used to detect differences between groups. Post hoc testing using Tukey’s multiple comparisons was used to determine which conditions were significantly different from the media/vehicle control. GraphPad Prism software was used for all statistical analyses. Data are expressed as mean ± standard error of mean (SEM) with statistical significance: *p< .05, **p< .01, and ***p< .001.
Results
Lactic acid suppresses cytokine secretion and transcription in LPS-activated BMMC
We have previously shown suppressive effects of lactic acid on mast cell activation by IL-33 (24). Therefore, we hypothesized that these effects could extend to LPS-induced mast cell function and determined the kinetics of this response. BMMC were pretreated ± lactic acid (12.5 mM) for various times prior to LPS activation (1 μg/mL). Pre-treatment from 0–48 hours significantly suppressed LPS-induced IL-6 and TNF (Figure 1A). In sepsis, lactate increases following infection due to tissue hypoperfusion, impaired pyruvate dehydrogenase activity, elevated catecholamine secretion, and increased immune cell glycolysis (8, 35–37), factors that may emerge after infection has begun. Therefore, we also treated BMMC with lactic acid after LPS activation. Lactic acid significantly suppressed IL-6 and TNF when given up to 4 hours post-LPS activation for all cytokines measured (Figure 1B). To determine optimal lactic acid concentrations for suppression, BMMC were treated ± lactic acid for 24 hours prior to LPS. IL-6 was significantly reduced with ≥6 mM lactic acid, while TNF was significantly decreased with ≥12.5mM treatment (Figure 1C). Importantly, there was no significant change in cell viability at these concentration (Supplemental Figure 1A).
Figure 1. Lactic acid suppresses cytokine secretion and transcription in LPS-activated BMMC.
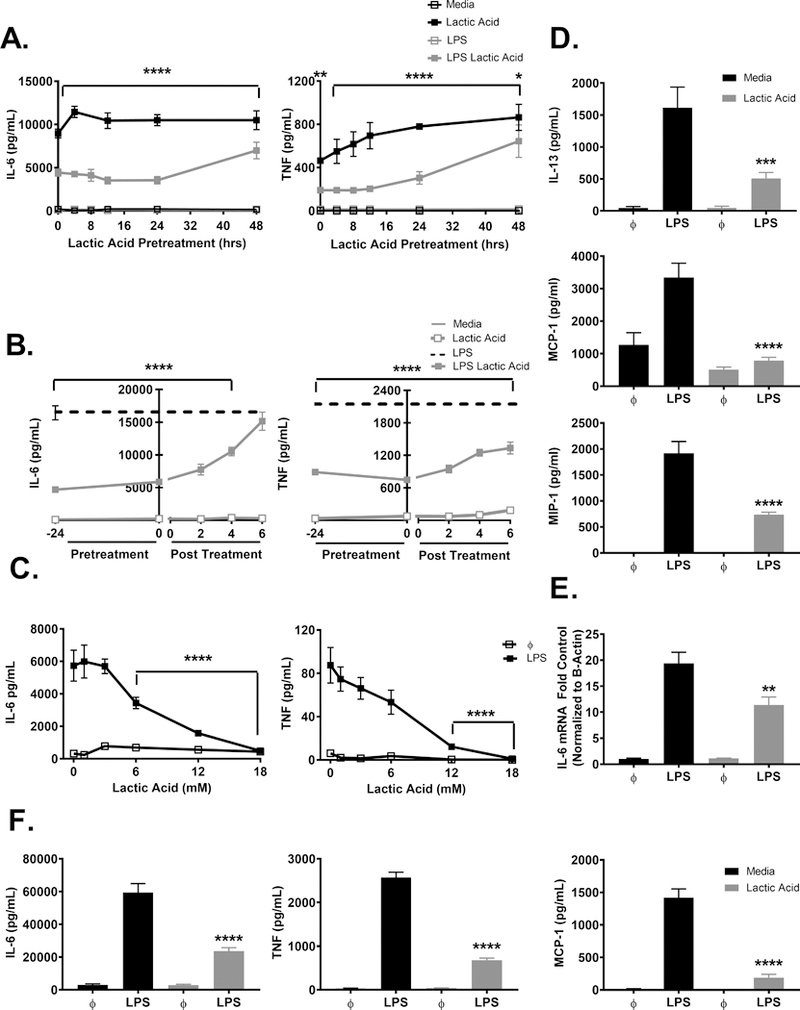
A) BMMC were pre-treated ± 12.5 mM lactic acid for the indicated time points and activated ± LPS (1 μg/mL) for 16 hours. B) BMMC were treated ± 12.5 mM lactic acid prior to, during, or following activation ± LPS (1 μg/mL) for 16 hours. C) BMMC were treated for 24 hours ± the indicated doses of lactic acid and activated ± LPS (1 μg/mL) for 16 hours. D) BMMC were treated for 24 hours ± 12.5 mM lactic acid and activated ± LPS (1 μg/mL) for 16 hours. ELISA analysis was used to determine supernatant cytokines. E) BMMC were treated for 24 hours ± 12.5 mM lactic acid and activated ± LPS (μg/mL) for 2 hours. RT-qPCR was used to determine IL-6 and actin mRNA expression. F) PMC were treated for 24 hours ± 12.5 mM lactic acid and activated ± LPS (1 μg/mL) for 16 hours. ELISA analysis was used to determine supernatant cytokine concentrations. Data are means ± SEM of 3 populations, representative of 3 independent experiments. **p < .01, *** p <0.001, ****p < .0001
For the remainder of the experiments, lactic acid treatment for 24 hours at 12.5 mM was used for optimal suppression unless otherwise stated. Additional cytokines and chemokines were examined under these conditions. IL-13, MCP-1, and MIP-1α concentrations were significantly reduced by lactic acid treatment (Figure 1D). Furthermore, lactic acid significantly suppressed IL-6 mRNA expression (Figure 1E), suggesting the effects were at or before the transcriptional level, not solely at the level of translation or secretion.
To ensure our results were not related to BMMC differentiation in vitro, PMC were used as a comparison population. PMC were extracted by peritoneal lavage and expanded in culture prior to treatment with lactic acid (12.5mM) for 24 hours prior to LPS activation. Lactic acid significantly suppressed LPS-induced IL-6, TNF, and MCP-1 in PMC, similar to the results observed with BMMC (Figure 1F). These results suggest that lactic acid consistently suppresses LPS-mediated mast cell function in vitro and ex vivo.
Lactic acid similarly suppresses TLR-2, −3, and −4 effects
Sepsis can occur in response to a multitude of bacterial, viral, or fungal infections (38). While LPS activation of toll-like receptor-4 (TLR4) is often used to mimic bacterial activation in vitro, thirteen TLRs have been classified with different bacterial and viral ligands. Extracellular signals from bacterial cell walls, like LPS, and intracellular signals from bacterial and viral replication, like dsRNA, have similar signaling pathways in mast cells (39). Therefore we extended this study to determine the effects of lactic acid on other common TLR signaling pathways. BMMC were treated with lactic acid prior to stimulation with 1 μg/mL LPS (TLR4 ligand), 5 μg/mL lipoteichoic acid (LTA; a TLR2 ligand), or 100 μg/mL polyinosinic:polycytidylic acid (PolyI:C; a TLR3 ligand). Lactic acid significantly suppressed cytokine production by all three ligands (Figure 2), suggesting the effects are not specific to TLR4, and that lactic acid may play a broader role in the context of bacterial and viral infections. Since results were similar, we used LPS as our model stimulus for the remainder of the study.
Figure 2. Lactic acid suppresses TLR-2, −3, and −4 effects on BMMC.
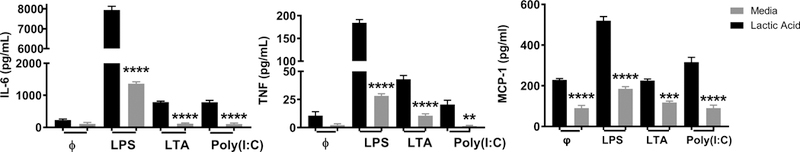
A) BMMC were treated ± 12.5 mM lactic acid for 24 hours and activated ± LPS (1 μg/mL), LTA (25 μg/mL), or Poly (I:C) (100 μg/mL) for 16 hours. Data are means ± SEM of 3 populations, representative of 3 independent experiments. **p < .01, ****p < .0001
Lactic acid suppresses cytokine production in a model of LPS-induced endotoxemia
Our results are consistent with lactic acid effects on other immune cell lineages (20–23). Therefore we expanded our questions to examine the effects of lactic acid on LPS-induced cytokine production in vivo. Because there is a correlative relationship between septic mortality and blood lactate in patients, we modulated lactic acid levels in a model of sepsis to provide causal data. An intraperitoneal (IP) injection of lactic acid (80 mg/kg) or PBS was given 20 hours prior to an IP injection of LPS (25 mg/kg) or PBS (Figure 3A). Lactic acid significantly suppressed LPS-induced plasma IL-6, TNF, and MCP-1 compared to the PBS control (Figure 3B). It is important to note that LPS and lactic acid act on many other immune cell lineages in this model, supporting the interpretation that lactic acid-mediated suppression is a consistent immune response. We also noted that lactic acid did not affect body temperature or observational score (Figure 3C and 3D), perhaps due to LPS effects on the vasculature. Overall, these data show that lactic acid can suppress LPS-induced cytokine production in vivo.
Figure 3. Lactic acid suppresses cytokine production in a model of LPS-induced septic shock.
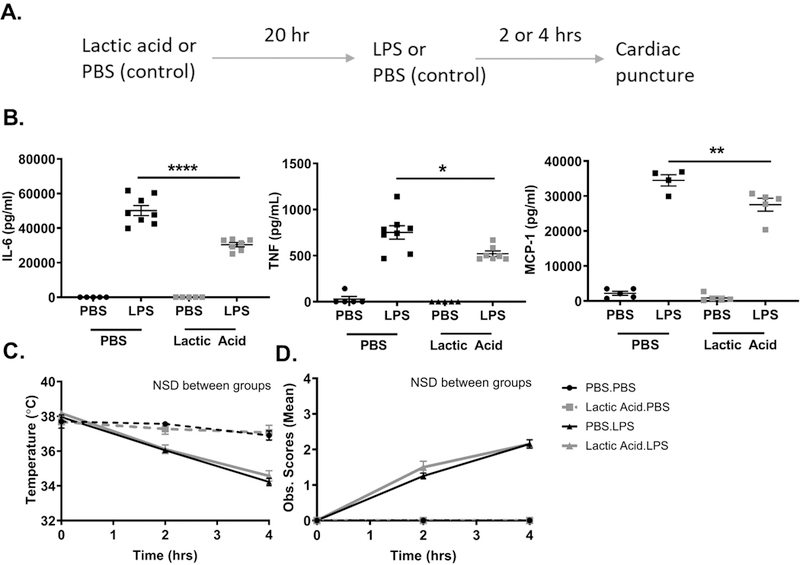
A) Schematic. Lactic acid (80 mg/kg) or PBS was injected IP 20 hours prior to LPS (25 mg/kg) or PBS. Mice were sacrificed at 2 or 4 hour. B) Plasma IL-6 was measured at 4 hours, TNF and MCP-1 were measured at 2 hours by ELISA. C) Rectal temperature and D) Observational scores were measured over 4 hours. Data are means of 5 mice/control group and 8 mice/experimental group, representative of 2 independent experiments. NSD= no significant difference. *p < .05, **p < .01, ****p < .0001, NSD= no significant difference
LPS effects involve the MCT-1 transporter, lactate, and H+ ions
Monocarboxylic transporters (MCTs) are a group of proton-linked transporters that shuttle single carboxylate molecules, including lactate, across the plasma membrane (40, 41). We previously found that MCT-1 is required for lactic acid to suppress IL-33-mediated mast cell activation (24), and therefore examined the role of MCT-1 in the LPS response. BMMC were cultured ± AR-C155858 (MCT-½ inhibitor) or DMSO for 1 hour prior to lactic acid treatment and LPS activation. While lactic acid significantly suppressed IL-6 and TNF in the DMSO control, it had no effect in the presence of AR-C155858 (Figure 4A). There was no effect on cell viability (Supplemental Figure 1B). Mast cells express MCT-1 but not MCT-2 (24), therefore, MCT-1 transport appears to be required for the suppressive effects of lactic acid on mast cells.
Figure 4. Lactic acid effects are dependent on MCT-1, lactate, and H+ ions.
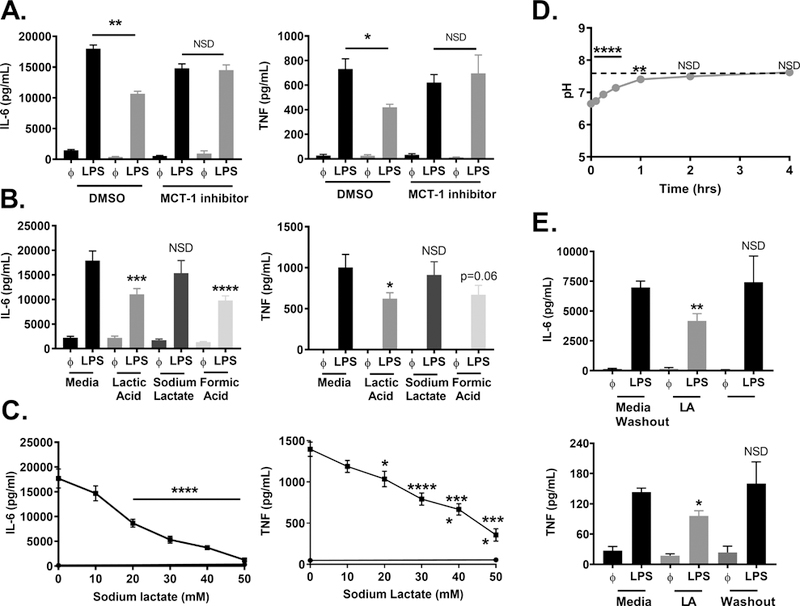
A) BMMC were treated ± AR-C155858 (100 nM) or DMSO (vehicle) for 1 hour, ± lactic acid (12.5) mM for 16 hours, and activated ± LPS (1 μg/mL) for 16 hours. B) BMMC were treated ± 12.5 mM lactic acid, sodium lactate, or formic acid for 24 hours, then activated ± LPS (1 μg/mL) for 16 hours. C) BMMC were treated with varying doses of sodium lactate for 24 hours and activated with LPS (1 μg/mL) for 16 hours. D) Lactic acid was added to cell culture media with BMMC plated at 1×106/mL. pH was measured over 4 hours. Dashed line indicates starting pH. E) BMMC were treated ± 12.5 mM lactic acid (LA) for 24 hours, then re-plated in new media ± 12.5 mM lactic acid and activated ± LPS (1 μg/mL) for 16 hours. ELISA was used to measure cytokine concentrations in the cell supernatant. Data are means ± SEM of 3 populations, representative of 3 independent experiments. *p < .05, **p < .01, ***p < .001, ****p < .0001, NSD= no significant difference
MCTs transport both lactate and H+ ions (40, 41). We previously published that lactic acid, but not lactate, suppresses IL-33-induced BMMC cytokine production (24). To determine if acidification is required for lactic acid effects on LPS activation, BMMC were cultured ± 12.5 mM lactic acid (pKa 3.86), sodium lactate, or formic acid (pKa 3.75) for 24 hours prior to LPS activation. Lactic acid and formic acid significantly suppressed IL-6 and TNF production (Figure 4B). There was no significant effect with sodium lactate at 12.5 mM. Additionally, there was no effect on cell viability with formic acid or sodium lactate (Supplemental Figure 1A). While these data support our previous findings that indicate a pH-dependent mechanism (24), other studies have shown suppression by lactate with effects often observed at higher doses (23, 42, 43). Therefore, a dose response was conducted with sodium lactate for 24 hours prior to LPS activation. At doses ≥20 mM, sodium lactate significantly suppressed cytokine production with no effects on viability (Figure 4C and Supplemental Figure 1A), indicating suppressive effects independent of pH. These data suggest that both acidity and the lactate molecule have similar effects on cytokine production.
Once lactate and H+ ions are released from the cell and into the bloodstream, buffering systems such as bicarbonate help to prevent acidosis (44, 45). Additionally, metabolic alkalosis sometimes occurs in patients with hyperlactemia due to saline resuscitation comorbidities (46, 47). Because our cultures are buffered by HEPES and FBS buffering systems, we monitored pH after lactic acid addition and examined kinetics of the buffering capacity. Upon addition to BMMC cultures, pH decreased to 6.7 (Figure 4D). Within 2 hours, the pH returned to 7.4 and was not different than our media control. These results suggest that lactic acid is buffered within our assays, similar to effects observed in blood systemically. Finally, because the pH returned to basal values within hours of addition, we investigated whether lactic acid effects persisted after plating treated cells in new media. BMMC were cultured ± lactic acid for 24 hours, washed, then activated in fresh media and compared to cells cultured ±lactic acid throughout the duration of the experiment. Continuous culture in lactic acid suppressed cytokine production, but there was no suppression of IL-6 and TNF secretion once lactic acid was washed out of the media (Figure 4E). Together, these data suggest that both acidity and lactate impact mast cell responses to LPS and that lactic acid effects are transient, requiring its presence at the time of activation.
Lactic acid suppresses NFκB function and miR-155 expression
TLR4 signaling via MyD88 induces phosphorylation of IRAK, TRAF, TAK1, MAP kinases, and NFκB (39). Previous publications have observed reduced p-NFκB and nuclear translocation in monocytes with lactate and lactic acid treatment (22, 43). To determine the effects on NFκB activity in mast cells, we first examined canonical NFκB signaling by measuring IκB degradation. BMMC were treated ± lactic acid for 24 hours prior to LPS activation for 30 minutes. While LPS activation decreased IκB expression relative to unactivated controls, this change was attenuated with lactic acid treatment (Figure 5A). Because these results suggested that lactic acid reduces NFκB activation, we further examined BMMC from NFκB-luc transgenic mice, which have a luciferase transgene under the control of NFκB binding sites. BMMC were treated ± lactic acid for 24 hours prior to LPS activation for 2 hours. Lactic acid significantly suppressed LPS-induced NFκB transcriptional activity as determined by luciferase expression (Figure 5B).
Figure 5. Lactic acid suppresses NFκB function and miR-155 expression.
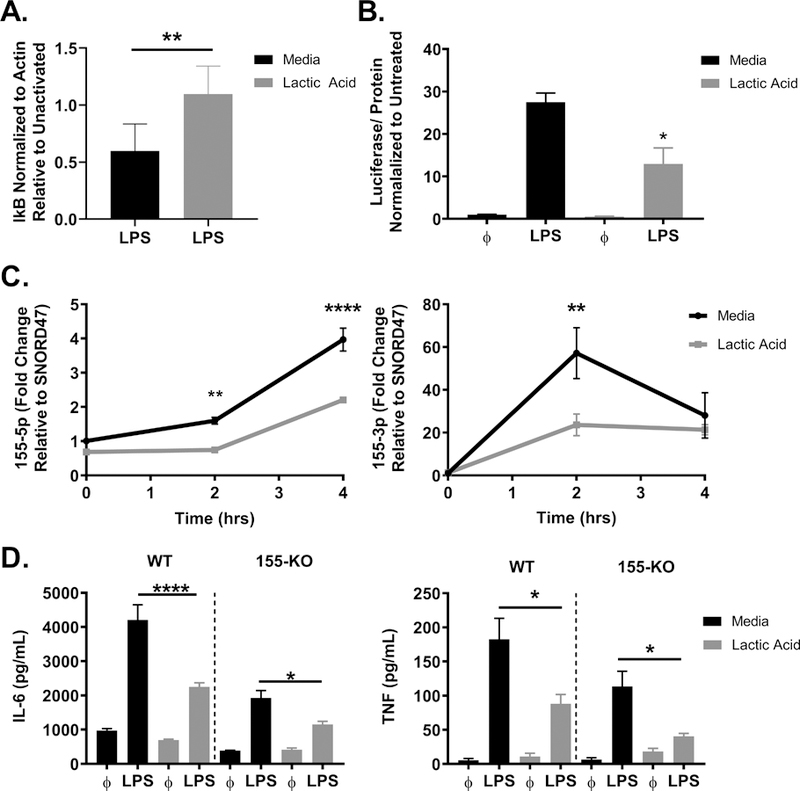
A) BMMC were treated ± lactic acid (12.5 mM) for 24 hours, then activated ± LPS (1 μg/mL) for 30 minutes. IκB degradation was determined by LPS-activated expression normalized to β-actin and relative to unstimulated controls. B) NFκB-luc transgenic BMMC were treated ± lactic acid (12.5 mM) for 24 hours, then activated ± LPS (1 μg/mL) for 2 hours. Luciferase activity was measured with the Promega Luciferase Assay Substrate and Glomax Luminometer. C) BMMC were cultured ± lactic acid (12.5 mM) for 24 hours, then activated ± LPS (1 μg/mL) for 2–4 hours. miRs were measured via qPCR and normalized relative to SNORD47. D) C57Bl/6 or miR-155 KO BMMC were treated ± lactic acid (12.5 mM) for 24 hours and activated ± LPS (1 μg/mL) for 16 hours. ELISA was used to measure cytokine concentrations in the cell supernatant. Data are means ± SEM of 3 populations, representative of 2 (A) 3 (B-D) independent experiments. *p < .05, **p < .01, ****p < .0001
miR-155 is an NFκB-induced micro-RNA that enhances inflammation (48). We previously found that lactic acid inhibited IL-33-induced miR-155 expression, an effect that was required for lactic acid mediated-suppression (24). To determine if LPS-mediated miR-155 expression was similarly inhibited, BMMC were cultured ± lactic acid for 24 hours prior to activation. Lactic acid did not affect baseline miR-155 levels, but antagonized LPS-induced miR-155–5p and miR-155–3p expression (Figure 5C). To determine if changes in miR-155 expression are required for lactic acid effects, we compared wild type (WT) and miR-155 KO BMMC. As anticipated by the pro-inflammatory role of miR-155, LPS-stimulated KO cells had lower LPS-induced cytokine production than WT cells. Lactic acid similarly inhibited LPS-induced IL-6 (54% vs. 60%) and TNF suppression (48% vs. 35%) in WT compared to the miR-155 KO mast cells (Figure 5D). Together, these data suggest that lactic acid suppresses NFκB activation and downstream miR-155 induction, but while suppressing miR-155 (a pro-inflammatory mediator) inhibits LPS effects, miR-155 reduction is not the sole mechanism explaining suppression.
Lactic acid effects on glucose metabolism suppress cytokine production in BMMC
Lactate and associated H+ ions are the byproduct of glycolysis, an important cellular pathway for the production of energy and intermediates for additional chemical reactions. Glycolysis produces ATP from glucose, ending with the conversion of pyruvate to lactate in order to recycle NADH and continue rapid ATP production during cellular activation. Importantly, other studies have shown that lactic acid and lactate suppress intracellular ATP, glucose uptake, and extracellular acidification in monocytes and CD4+ T cells (21, 23, 49). Therefore, we examined how lactic acid modulates glucose metabolism in our system. BMMC were cultured ± 12.5 mM lactic acid for 24 hours prior to LPS activation for 16 hours. LPS increased glucose uptake and lactate export, effects that were significantly reduced by lactic acid (Figure 6A), and suggest that lactic acid suppresses glycolysis in our assays. These results support data from other systems, and indicate that lactic acid acts as a negative regulator of glycolysis during LPS-mediated mast cell activation.
Figure 6. Lactic acid suppresses glycolysis, a necessary component of the LPS response.
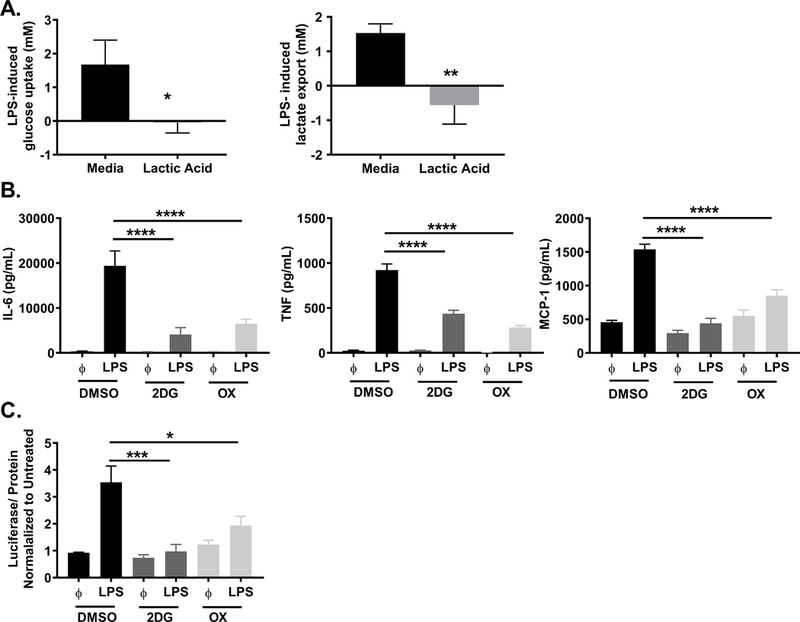
A) BMMC were treated ± 12.5 mM lactic acid for 24 hours, then activated ± LPS (1 μg/mL) for 16 hours. Glucose and lactate were measured in the cell supernatant by colorimetric assay. Uptake and export were calculated from the difference between controls and activated samples. B) BMMC were treated ± 2DG (1mM) or OX (20 mM) for 1 hour prior to LPS activation (1 μg/mL). After 16 hours, cytokines were measured in supernatant by ELISA. C) BMMC were treated with 2DG (1mM) or OX (20 mM) for 1 hour prior to LPS activation (1 μg/mL). After 2 hours, luciferase activity was measured from the NFκB-luc transgenic BMMC on the Glomax Luminometer. Data are means ± SEM of 3 populations analyzed in triplicate, representative of 3 independent experiments. *p < .05, **p < .01, ***p < .001, ****p < .0001.
In the past 10 years, ample research has shown that cellular metabolism is closely linked to immune cell phenotype and effector function (50). To examine if glycolysis has a functional role in LPS-induced cytokine production, 2-deoxyglucose (2-DG) was used to block hexokinase activity, directly reducing glycolysis, and sodium oxamate (OX) was used to block lactate dehydrogenase activity, slowing glycolysis by preventing the recycling of NADH needed for glycolytic enzyme activity. BMMC were cultured ± 2-DG (1 mM) or OX (40 mM) for 1 hour prior to activation with LPS for 16 hours. 2DG and OX significantly suppressed LPS-induced cytokine production (Figure 6B). Importantly, there was no effect on cell viability (Supplemental Figure 1C). Additionally, 1 hour treatment with 2-DG and OX reduced NFκB transcriptional activity, as determined by luciferase expression in NFκB-luc BMMC (Figure 6C). Because oxidative phosphorylation (OX PHOS) is also used to produce energy, we examined if inhibiting OX PHOS reduced cytokine production. BMMC were treated with etomoxir (200 μM) or rotenone and antimycin A (1 mM each) to inhibit OX PHOS 1 hour prior to activation with LPS. There was no significant change in cytokine production following OX PHOS inhibition (Supplemental Figure 2). Together, these data show that inhibiting glycolysis mimics lactic acid effects and is sufficient to suppress both cytokine production and cell signaling.
ATP reverses lactic acid suppression
Glycolysis rapidly generates ATP, which is required for kinase activity and signaling, tRNA synthetase function, ion transport, and chromatin remodeling. These processes all play a role in cell signaling and cytokine production. In addition, utilizing glycolysis allows immune cells to generate metabolites for many anabolic processes such as cell growth, differentiation, and inflammatory function (50). Because LPS induced glycolysis and lactic acid suppressed glycolysis, we wanted to determine if reduced glycolytic ATP production was a critical mechanism of lactic acid-mediated suppression. BMMC were cultured ± lactic acid and ± ATP (10 μM) at the time of LPS activation. ATP reversed lactic acid effects on both cytokine secretion and NFκB transcriptional activity (Figure 7A,B). In a previous manuscript, we showed that μM quantities of labeled ATP diffuse into BMMC within 20 minutes (51), which can increase the ATP available in the cell for energy and bypass the need for ATP produced by glycolysis. This data, together with the experiments above, suggest that lactic acid suppresses LPS-induced glycolytic ATP production, effectively attenuating energy available for cell signaling and cytokine production.
Figure 7. ATP reverses lactic acid effects.
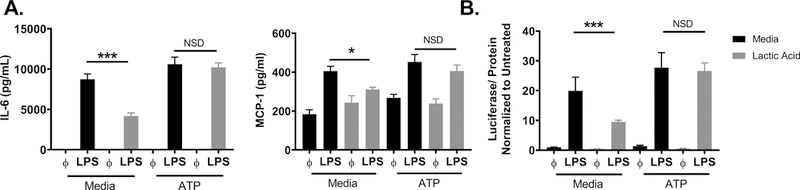
A) BMMC were treated ± 12.5 mM lactic acid, ± ATP (10 μM) and ± LPS (1 μg/mL) for 16 hours, and ELISA analysis was used to determine supernatant cytokine concentrations. B) BMMC were treated ± 12.5 mM lactic acid, ± ATP (10 μM) and ± LPS (1 μg/mL). After 2 hours, luciferase activity was measured from the NFκB-luc transgenic BMMC on the Glomax Luminometer. Data are means ± SEM of 3 populations, representative of 3 independent experiments. *p < .05, ***p < .001
Discussion
Elevated blood lactate consistently correlates with increased mortality from sepsis (2, 13, 52–55), however there is little understanding of a cause-and-effect relationship. Previous studies showed that lactic acid suppresses macrophages, DCs, and T cell activation in vitro (18, 20–24, 43), but there has been no clear mechanism of action and no in vivo studies to determine the direct effects of lactic acid in a sepsis model. Because mast cell function reduces sepsis-induced death in mouse models (25–33), we used mast cells as a model system to study lactic acid. We demonstrate that lactic acid suppresses LPS-mediated activation of BMMC and PMC. Additionally, these are the first data to demonstrate that lactic acid suppresses cytokine production in vivo, using a model of LPS-induced endotoxemia. Lactic acid has been shown to suppress glycolysis and intracellular ATP (21, 23, 49), and our data demonstrate glycolytic/ATP suppression is necessary and sufficient to inhibit LPS signaling and cytokine production (Figure 8). These findings suggest that lactic acid, the end product of glycolysis, can act as a feedback regulator of mast cell activation to reduce the inflammatory response, specifically in sepsis. Additionally, these data support the hypothesis that targeting metabolic pathways may be an effective mechanism to modulate lactic acid effects in disease.
Figure 8. Schematic diagram.
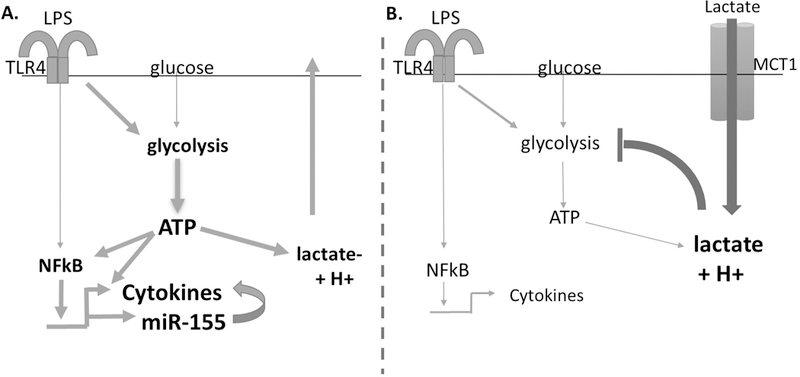
A) LPS activates TLR4, signaling through NFκB to induce cytokines and miR-155. LPS activation also increases glycolysis, providing ATP to augment NFκB transcription and cytokine production. B) Exogenous lactic acid imported through MCT-1 suppresses glycolysis, effectively suppressing NFκB transcriptional activity.
In BMMC and PMC, physiologically relevant lactic acid concentrations suppressed the LPS-induced production of several cytokines and chemokines. We observed significant effects between 6–12 mM, which are relevant to wound tissue lactate concentrations (5–20mM; maximum 80 mM) (56, 57) and blood lactate concentrations (2–10mM; maximum 20 mM) (15, 17, 54, 58). Importantly, many studies define high lactate as blood concentrations greater than 4 mM (13, 14, 59). However, changes in tissue lactate concentration and pH can be more dynamic than blood. For example, one study showed that septic patients had no difference in blood pH between low and high lactate groups, but there was a significant difference in the gastric intramucosal pH (60). Additionally, another study demonstrated that muscle lactate concentrations were ~1.5 mM higher than arterial lactate concentrations in septic shock patients (61). These data suggest that our results are particularly relevant to tissue resident immune cells, such as mast cells, and immune cells recruited into the tissue.
Importantly, lactic acid suppressed cytokine production in a model of endotoxemia in vivo. We chose the LPS-induced model and the associated time points to directly extend our in vitro studies. Lactic acid significantly reduced LPS-induced IL-6, MCP-1, and TNF, supporting the effects we observed in vitro. It is important to note that LPS-induced endoxemia only mimics the initial inflammatory stage of sepsis, which limits our ability to understand the role of lactic acid on the secondary immunosuppressive phase. Additionally, while we show that pretreatment with lactic acid reduces the production of inflammatory cytokines, our model does not directly show that lactic acid following the onset of sepsis has the same effect. Future experiments should modulate lactic acid after shock initiation and include study of the secondary immunosuppressive phase with re-infection models. In vitro, we observed that lactic acid suppressed activation by TLR-2, −3, and −4 agonists, suggesting that lactic acid may antagonize immune responses more broadly in vivo. We hypothesize that after the onset of sepsis, lactate levels rise in the blood and could suppress inflammatory cytokine production, contributing to pathological immunosuppression.
Interestingly, there were no effects of lactic acid on hypothermia or observational score in the LPS endotoxemia model. We postulate that these clinical phenotypes are at least partly immune-independent. LPS and lactic acid have been shown to have overlapping effects on endothelial cells, increasing nitric oxide production, vascular permeability, and vasodilation (62–70). In mice, low dose infusions of HCl have been reported to drop blood pH to 7.13, increase endothelial NO, reduce blood pressure, and reduce temperature (66). Furthermore, these authors mentioned that lactic acid also reduced pH and blood pressure and increased endothelial NO. Thus, we hypothesize that lactic acid contributes to vasodilation and vascular leak in sepsis, while reducing inflammatory cytokine production from immune cells. Furthermore, lactate and/or the associated changes in pH may contribute to pain within incisions or wounds (71), influencing locomotion, grooming, and overall observational scores. For ethical considerations and animal care, ketoprofen was injected prior to lactic acid. This limits our model, in that arachidonic acid metabolite production and pain responses are reduced. Future work should further examine the effects of lactic acid on different cell types and organ systems within a sepsis model to better study sepsis as a systemic disease.
Lactic acid effects on LPS-induced cytokine production were MCT-1-dependent, similar to our findings with lactic acid effects on IL-33-induced activation (24). MCT-1 is the primary transporter of lactic acid, known to co-transport lactate and H+ (40), and in our experiments, there was no effect of lactic acid with MCT-1 inhibition, which suggests that lactate and/or H+ must be transported into the cell for suppressive effects. We also observed that both acidification with formic acid and elevated concentrations of sodium lactate could mimic the suppressive effects of lactic acid. These results support many studies that have reported immunosuppressive effects of acidification (21, 72, 73), high concentrations of lactate (10–100 mM) (20–22), and slightly lower concentrations of lactic acid (5–20 mM) (23, 42, 43). These results also support our data showing that lactic acid suppresses glycolysis because H+ inhibits phosphofructokinase, the rate limiting enzyme in the glycolytic pathway, and lactate can inhibit different isoforms of lactate dehydrogenase, an example of product inhibition which can further reduce glycolysis (74, 75). Our data have been corroborated in DCs, in which the inhibitory effects of 10mM lactic acid were reverted after normalizing the pH (20). Together, these results suggest that without a change in pH, a much higher concentration of lactate is required for transport into the cell by MCT-1. We also observed a pH reduction from 7.4 to 6.7 upon lactic acid addition to media, rebounding within 2 hours. This suggests that our in vitro media buffering systems at least partly mimic blood, where deprotonated lactic acid and H+ ions are buffered by the bicarbonate system.
When seeking a molecular mechanism, we noted that lactic acid reduced IκB degradation and the activity of NFκB, a major transcription factor in LPS signaling that is important for cytokine expression (76–78). Previous studies reported reduced NFκB phosphorylation and nuclear translocation in monocytes after lactate or lactic acid treatment (22, 43), data supported by our study. Our results suggest that lactic acid inhibits NFκB activation and transcriptional activity, which may contribute to reduced cytokine production. We also examined miR-155, which functions to suppress the negative regulators SHIP1 and SOCS1, with an overall pro-inflammatory role in immune cell signaling (34, 79). We have previously published that lactic acid suppresses the IL-33-induced, NFκB-dependent induction of miR-155 (24). In this study, lactic acid similarly suppressed LPS-mediated miR-155–5p and miR-155–3p expression. While miR-155 KO BMMC were hyporesponsive to LPS, lactic acid had similar effects on LPS-stimulated cytokine secretion in these cultures. miR-155 suppression may be one anti-inflammatory effect of lactic acid, however miR-155 suppression could not fully explain lactic acid effects, supporting additional mechanisms.
Our data suggest a primary mechanism of action is glycolytic inhibition, which is necessary and sufficient for lactic acid to suppress LPS-induced cytokine production. In BMMC, we observed a significant reduction in LPS-mediated glucose uptake and lactate export with lactic acid treatment. This is consistent with results in human monocytes and mouse macrophages stimulated with LPS (21, 23), as well as the known ability of H+ ions to inhibit phosphofructokinase activity (80, 81), which reduces glycolysis and lactate production (82). Elevated lactate also suppresses pyruvate-to-lactate conversion by lactate dehydrogenase, an equilibrium enzyme. This effectively reduces NADH recycling required for glycolysis (83, 84). Interestingly, another study reported that blocking endogenous lactate export by MCT-4 in macrophages following LPS activation suppresses cytokine production and glycolysis (11). Together, these results suggest that elevated lactic acid, whether endogenous or exogenous, suppresses cytokine production and immune cell glycolysis needed for inflammatory responses.
Notably, inhibiting glycolysis mimicked lactic acid effects on cytokine production. Treatment with the glycolytic inhibitors 2DG and OX significantly suppressed cytokine production and NFκB transcription, similar to lactic acid. While 2DG has been shown to suppress LPS-induced cytokine production (21, 23, 79), this is the first report to show an effect on NFκB-mediated transcription. Collectively, our studies suggest that suppressing glycolysis is sufficient to limit inflammatory cytokine responses. ATP, the main product of glycolysis, is required for many cellular processes including enzyme activity involved in cell signaling and cytokine production. Increasing ATP availability reversed the effects of lactic acid, restoring both NFκB function and cytokine production. Thus by acting on glycolysis, lactic acid can blunt LPS-induced ATP production that is broadly required for the inflammatory response.
Together these data support the theory that high lactate levels following the initiation of sepsis may suppress immune cell glycolysis and function, potentially contributing to the immunosuppression observed in the secondary phase of sepsis. In immunosuppression, immune cells have reduced glucose metabolism, cytokine production, antigen presentation, and cytolytic function (6), mirroring the effects of lactate treatment (19–23, 49, 85). We hypothesize that reducing lactate accumulation, improving lactate clearance, or reversing lactic acid effects may improve immune function in the secondary phase of sepsis. However, no studies have directly examined the effects of these interventions on immune cell function. Some studies conflict with this hypothesis, reporting no significant improvement in patient outcomes after reducing blood lactate, however, study designs may limit the interpretation of these results. In one study, a randomized clinical trial was conducted to determine if targeting lactate clearance provided lower patient mortality than targeting central venous oxygen saturation (SCVO2) (86). While management to normalize blood lactate clearance did not improve survival compared with interventions normalizing SCVO2, both patient groups received the same interventions, including isotonic crystalloid, vasopressors, packed red blood cells, and dobutamine. The interventions were administered due to different lactate or SCVO2 indicators, however the protocol was nearly identical and the lactate and SCVO2 levels were not significantly different between the groups at either time point measured. These results suggest that protocols to normalize lactate are similarly effective to protocols to normalize SCVO2, however there was no control group to adequately compare the overall effectiveness of these interventions. Another study administered dichloroacetate (DCA) to reduce the production of blood lactate in sepsis patients (87). DCA treatment worsened patient survival, leading researchers to conclude that modulating blood lactate does not improve survival in sepsis. However, while DCA reduces lactate secretion by increasing pyruvate conversion to acetyl co-A in the mitochondria, it also inhibits glycolysis, effectively augmenting lactic acid effects (88–90). Furthermore, despite lowering blood lactate levels, intravenous DCA administration has been shown to reduce tissue ATP (91), mimicking lactic acid effects. Future studies should examine the effects of early lactate clearance on immune cell metabolism and function in septic patients during the immunosuppressive phase of the disease. Additionally, future studies should aim to increase glycolysis to reverse lactic acid suppression, potentially by targeting known glycolytic regulators such as mTORC, Akt2, Myc, or HIF-1 (92, 93).
In summary, lactic acid suppresses LPS-induced mast cell activation and LPS-induced cytokine production in vivo. These effects are dependent upon MCT-1 transport into the cell, and both lactate and H+ ions independently reduce activation. Moreover, lactic acid inhibits LPS-induced glycolysis, suppressing cytokine production and NFκB function, partly by reducing ATP availability. Future studies should directly examine lactic acid effects in a model of immunosuppression and target enhancing glycolysis and ATP production to improve immune function and counteract lactic acid effects in sepsis.
Supplementary Material
Key Points:
LA suppresses LPS-mediated mast cell function.
Suppression required the MCT-1 transporter and reduced NFkB function.
LA suppresses LPS-induced glycolysis, effects linked to suppression.
Acknowledgements
We would like to thank Dr. Frank Fang at VCU for providing us access to the Glomax 20/20 Luminometer.
This work was supported by U.S. National Institutes of Health, National Institute of Allergy and Infectious Diseases Grants 1R01AI101153 and 2R01AI059638 (to J.J.R.) and from National Institutes of Health via HL125353 (CEC), HD087198 (CEC), RR031535 (CEC). This work was also supported by research grants from the Veteran’s Administration (VA Merit Review, I BX001792 (CEC) and a Research Career Scientist Award, 13F-RCS-002 (CEC)). The contents of this manuscript do not represent the views of the Department of Veterans Affairs or the United States Government.
References
- 1.Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, Kumar A, Sevransky JE, Sprung CL, Nunnally ME, Rochwerg B, Rubenfeld GD, Angus DC, Annane D, Beale RJ, Bellinghan GJ, Bernard GR, Chiche J-D, Coopersmith C, De Backer DP, French CJ, Fujishima S, Gerlach H, Hidalgo JL, Hollenberg SM, Jones AE, Karnad DR, Kleinpell RM, Koh Y, Lisboa TC, Machado FR, Marini JJ, Marshall JC, Mazuski JE, McIntyre LA, McLean AS, Mehta S, Moreno RP, Myburgh J, Navalesi P, Nishida O, Osborn TM, Perner A, Plunkett CM, Ranieri M, Schorr CA, Seckel MA, Seymour CW, Shieh L, Shukri KA, Simpson SQ, Singer M, Thompson BT, Townsend SR, Van der Poll T, Vincent J-L, Wiersinga WJ, Zimmerman JL, and Dellinger RP. 2017. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock. Critical Care Medicine 1. [DOI] [PubMed] [Google Scholar]
- 2.Epstein L 2016. Varying Estimates of Sepsis Mortality Using Death Certificates and Administrative Codes — United States, 1999–2014. MMWR Morb Mortal Wkly Rep 65. [DOI] [PubMed] [Google Scholar]
- 3.Lewis DH, Chan DL, Pinheiro D, Armitage-Chan E, and Garden OA. 2012. The Immunopathology of Sepsis: Pathogen Recognition, Systemic Inflammation, the Compensatory Anti-Inflammatory Response, and Regulatory T Cells. Journal of Veterinary Internal Medicine 26: 457–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delano MJ, and Ward PA. 2016. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? Journal of Clinical Investigation 126: 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marshall JC 2014. Why have clinical trials in sepsis failed? Trends in Molecular Medicine 20: 195–203. [DOI] [PubMed] [Google Scholar]
- 6.Hotchkiss RS, Monneret G, and Payen D. 2013. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nature Reviews Immunology 13: 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD, Kreisel D, Krupnick AS, and others 2011. Immunosuppression in patients who die of sepsis and multiple organ failure. Jama 306: 2594–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allen SE, and Holm JL. 2008. Lactate: physiology and clinical utility. Journal of Veterinary Emergency and Critical Care 18: 123–132. [Google Scholar]
- 9.Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, and Pearce EJ. 2010. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115: 4742–4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Everts B, Amiel E, van der Windt GJW, Freitas TC, Chott R, Yarasheski KE, Pearce EL, and Pearce EJ. 2012. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 120: 1422–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan Z, Xie N, Banerjee S, Cui H, Fu M, Thannickal VJ, and Liu G. 2015. The Monocarboxylate Transporter 4 Is Required for Glycolytic Reprogramming and Inflammatory Response in Macrophages. Journal of Biological Chemistry 290: 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wacharasint P, Nakada T, Boyd JH, Russell JA, and Walley KR. 2012. Normal-Range Blood Lactate Concentration in Septic Shock Is Prognostic and Predictive: Shock 38: 4–10. [DOI] [PubMed] [Google Scholar]
- 13.Trzeciak S, Dellinger RP, Chansky ME, Arnold RC, Schorr C, Milcarek B, Hollenberg SM, and Parrillo JE. 2007. Serum lactate as a predictor of mortality in patients with infection. Intensive Care Medicine 33: 970–977. [DOI] [PubMed] [Google Scholar]
- 14.Arnold RC, Shapiro NI, Jones AE, Schorr C, Pope J, Casner E, Parrillo JE, Dellinger RP, and Trzeciak S. 2009. MULTICENTER STUDY OF EARLY LACTATE CLEARANCE AS A DETERMINANT OF SURVIVAL IN PATIENTS WITH PRESUMED SEPSIS: Shock 32: 35–39. [DOI] [PubMed] [Google Scholar]
- 15.Marty P, Roquilly A, Vallée F, Luzi A, Ferré F, Fourcade O, Asehnoune K, and Minville V. 2013. Lactate clearance for death prediction in severe sepsis or septic shock patients during the first 24 hours in Intensive Care Unit: an observational study. Annals of intensive care 3: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nguyen HB, Rivers EP, Knoblich BP, Jacobsen G, Muzzin A, Ressler JA, and Tomlanovich MC. 2004. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock*: Critical Care Medicine 32: 1637–1642. [DOI] [PubMed] [Google Scholar]
- 17.Kuttab HI, Sterk E, Rech MA, Nghiem T, Bahar B, and Kahn S. 2018. Early Recognition and Treatment of Sepsis After the Addition of Lactate to the Laboratory’s Critical Result Call List. Journal of Intensive Care Medicine 33: 111–115. [DOI] [PubMed] [Google Scholar]
- 18.Colegio OR, Chu N-Q, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, Cline GW, Phillips AJ, and Medzhitov R. 2014. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513: 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Husain Z, Huang Y, Seth P, and Sukhatme VP. 2013. Tumor-Derived Lactate Modifies Antitumor Immune Response: Effect on Myeloid-Derived Suppressor Cells and NK Cells. The Journal of Immunology 191: 1486–1495. [DOI] [PubMed] [Google Scholar]
- 20.Gottfried E 2006. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 107: 2013–2021. [DOI] [PubMed] [Google Scholar]
- 21.Dietl K, Renner K, Dettmer K, Timischl B, Eberhart K, Dorn C, Hellerbrand C, Kastenberger M, Kunz-Schughart LA, Oefner PJ, Andreesen R, Gottfried E, and Kreutz MP. 2010. Lactic Acid and Acidification Inhibit TNF Secretion and Glycolysis of Human Monocytes. The Journal of Immunology 184: 1200–1209. [DOI] [PubMed] [Google Scholar]
- 22.Peter K, Rehli M, Singer K, Renner-Sattler K, and Kreutz M. 2015. Lactic acid delays the inflammatory response of human monocytes. Biochemical and Biophysical Research Communications 457: 412–418. [DOI] [PubMed] [Google Scholar]
- 23.Errea A, Cayet D, Marchetti P, Tang C, Kluza J, Offermanns S, Sirard J-C, and Rumbo M. 2016. Lactate Inhibits the Pro-Inflammatory Response and Metabolic Reprogramming in Murine Macrophages in a GPR81-Independent Manner. PLOS ONE 11: e0163694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abebayehu D, Spence AJ, Qayum AA, Taruselli MT, McLeod JJA, Caslin HL, Chumanevich AP, Kolawole EM, Paranjape A, Baker B, Ndaw VS, Barnstein BO, Oskeritzian CA, Sell SA, and Ryan JJ. 2016. Lactic Acid Suppresses IL-33–Mediated Mast Cell Inflammatory Responses via Hypoxia-Inducible Factor-1α–Dependent miR-155 Suppression. The Journal of Immunology 197: 2909–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seeley EJ, Sutherland RE, Kim SS, and Wolters PJ. 2011. Systemic mast cell degranulation increases mortality during polymicrobial septic peritonitis in mice. Journal of Leukocyte Biology 90: 591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramos B, Zhanfg Y, Quereshi R, and Jackshik B. 1991. Mast cells are critical for the production of leukotrienes responsible for neutrophil recruitment in immune complex-induced peritonitis in mice. Journal of Immunology 147: 1636–1641. [PubMed] [Google Scholar]
- 27.Malaviya R, and Abraham SN. 2000. Role of mast cell leukotrienes in neutrophil recruitment and bacterial clearance in infectious peritonitis. Journal of leukocyte biology 67: 841–846. [DOI] [PubMed] [Google Scholar]
- 28.Sutherland R, Olsen JS, McKinstry A, Villalta SA, and Wolters PJ. 2008. Mast cell IL-6 improves survival from Klebsiella pneumonia and sepsis by enhancing neutrophil killing. Journal of Immunology 181: 5598–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bone-Larson CL, Hogaboam CM, Steinhauser ML, Oliveira SH, Lukacs NW, Strieter RM, and Kunkel SL. 2000. Novel protective effects of stem cell factor in a murine model of acute septic peritonitis: Dependence on MCP-1. The American journal of pathology 157: 1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Echtenacher B, Mannel DN, and Hultner L. 1996. Critical Role of Mast Cells in a Model of Acute Septic Peritonitis. Nature 381: 75–77. [DOI] [PubMed] [Google Scholar]
- 31.Malaviya R, Ikeda T, Ross E, and Abraham SN. 1996. Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-α. Nature 381: 77–80. [DOI] [PubMed] [Google Scholar]
- 32.Piliponsky AM, Chen C-C, Rios EJ, Treuting PM, Lahiri A, Abrink M, Pejler G, Tsai M, and Galli SJ. 2012. The Chymase Mouse Mast Cell Protease 4 Degrades TNF, Limits Inflammation, and Promotes Survival in a Model of Sepsis. The American Journal of Pathology 181: 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carlos D, Frantz FG, Souza-Júnior DA, Jamur MC, Oliver C, Ramos SG, Quesniaux VF, Ryffel B, Silva CL, Bozza MT, and Faccioli LH. 2009. TLR2-dependent mast cell activation contributes to the control of Mycobacterium tuberculosis infection. Microbes and Infection 11: 770–778. [DOI] [PubMed] [Google Scholar]
- 34.Qayum AA, Paranjape A, Abebayehu D, Kolawole EM, Haque TT, McLeod JJA, Spence AJ, Caslin HL, Taruselli MT, Chumanevich AP, Baker B, Oskeritzian CA, and Ryan JJ. 2016. IL-10–Induced miR-155 Targets SOCS1 To Enhance IgE-Mediated Mast Cell Function. The Journal of Immunology 196: 4457–4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haji-Michael PG, Ladrière L, Sener A, Vincent J-L, and Malaisse WJ. 1999. Leukocyte glycolysis and lactate output in animal sepsis and ex vivo human blood. Metabolism 48: 779–785. [DOI] [PubMed] [Google Scholar]
- 36.Vary TC. 1996. Sepsis induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock 6: 89–94. [DOI] [PubMed] [Google Scholar]
- 37.McCarter FD, James JH, Luchette FA, Wang L, Friend LA, King J-K, Evans JM, George MA, and Fischer JE. 2001. Adrenergic Blockade Reduces Skeletal Muscle Glycolysis and Na+, K+-ATPase Activity during Hemorrhage. Journal of Surgical Research 99: 235–244. [DOI] [PubMed] [Google Scholar]
- 38.Martin GS 2012. Sepsis, severe sepsis and septic shock: changes in incidence, pathogens and outcomes. Expert Review of Anti-infective Therapy 10: 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sandig H, and Bulfone-Paus S. 2012. TLR signaling in mast cells: common and unique features. Frontiers in Immunology 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Halestrap AP 2012. The monocarboxylate transporter family-Structure and functional characterization. IUBMB Life 64: 1–9. [DOI] [PubMed] [Google Scholar]
- 41.Halestrap AP 2013. The SLC16 gene family – Structure, role and regulation in health and disease. Molecular Aspects of Medicine 34: 337–349. [DOI] [PubMed] [Google Scholar]
- 42.Goetze K. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. International Journal of Oncology. 2011 doi: 10.3892/ijo.2011.1055. [DOI] [PubMed] [Google Scholar]
- 43.Hoque R, Farooq A, Ghani A, Gorelick F, and Mehal WZ. 2014. Lactate Reduces Organ Injury in Toll-like Receptor- and Inflammasome-Mediated Inflammation via GPR81-mediated Suppression of Innate Immunity. Gastroenterology 146: 1763–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beaver WL, Wasserman K, and Whipp BJ. 1986. Bicarbonate buffering of lactic acid generated during exercise. Journal of Applied Physiology 60: 472–478. [DOI] [PubMed] [Google Scholar]
- 45.Zhang YY, Sietsema KE, Sullivan CS, and Wasserman K. 1994. A method for estimating bicarbonate buffering of lactic acid during constant work rate exercise. European journal of applied physiology and occupational physiology 69: 309–315. [DOI] [PubMed] [Google Scholar]
- 46.Kellum JA 2004. Metabolic acidosis in patients with sepsis: epiphenomenon or part of the pathophysiology? Crit Care Resusc 6: 197–203. [PubMed] [Google Scholar]
- 47.Tuhay G, Pein MC, Masevicius FD, Kutscherauer DO, and Dubin A. 2008. Severe hyperlactatemia with normal base excess: a quantitative analysis using conventional and Stewart approaches. Crit Care 12: R66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kluiver J, van den Berg A, de Jong D, Blokzijl T, Harms G, Bouwman E, Jacobs S, Poppema S, and Kroesen B-J. 2007. Regulation of pri-microRNA BIC transcription and processing in Burkitt lymphoma. Oncogene 26: 3769–3776. [DOI] [PubMed] [Google Scholar]
- 49.Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D’Acquisto F, Bland EJ, Bombardieri M, Pitzalis C, Perretti M, Marelli-Berg FM, and Mauro C. 2015. Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLOS Biology 13: e1002202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O’Neill LAJ, Kishton RJ, and Rathmell J. 2016. A guide to immunometabolism for immunologists. Nature Reviews Immunology 16: 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Caslin HL, Taruselli MT, Haque T, Pondicherry N, Baldwin EA, Barnstein BO, and Ryan JJ. 2018. Inhibiting Glycolysis and ATP Production Attenuates IL-33-Mediated Mast Cell Function and Peritonitis. Frontiers in Immunology 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shapiro NI, Howell MD, Talmor D, Nathanson LA, Lisbon A, Wolfe RE, and Weiss JW. 2005. Serum Lactate as a Predictor of Mortality in Emergency Department Patients with Infection. Annals of Emergency Medicine 45: 524–528. [DOI] [PubMed] [Google Scholar]
- 53.Chebl R. Bou, El Khuri C, Shami A, Rajha E, Faris N, Bachir R, and Dagher G. Abou. 2017. Serum lactate is an independent predictor of hospital mortality in critically ill patients in the emergency department: a retrospective study. Scandinavian Journal of Trauma, Resuscitation and Emergency Medicine 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Filho RR, Rocha LL, Corrêa TD, Pessoa CMS, Colombo G, and Assuncao MSC. 2016. Blood Lactate Levels Cutoff and Mortality Prediction in Sepsis—Time for a Reappraisal? a Retrospective Cohort Study: SHOCK 46: 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kruse O, Grunnet N, and Barfod C. 2011. Blood lactate as a predictor for in-hospital mortality in patients admitted acutely to hospital: a systematic review. Scandinavian journal of trauma, resuscitation and emergency medicine 19: 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Britland S, Ross-Smith O, Jamil H, Smith AG, Vowden K, and Vowden P. 2012. The lactate conundrum in wound healing: Clinical and experimental findings indicate the requirement for a rapid point-of-care diagnostic. Biotechnology Progress 28: 917–924. [DOI] [PubMed] [Google Scholar]
- 57.Löffler M, Zieker D, Weinreich J, Löb S, Königsrainer I, Symons S, Bühler S, Königsrainer A, Northoff H, and Beckert S. 2011. Wound fluid lactate concentration: a helpful marker for diagnosing soft-tissue infection in diabetic foot ulcers? Preliminary findings: Marker for soft-tissue infection. Diabetic Medicine 28: 175–178. [DOI] [PubMed] [Google Scholar]
- 58.Theerawit P, Petvicharn C. Na, Tangsujaritvijit V, and Sutherasan Y. 2018. The Correlation Between Arterial Lactate and Venous Lactate in Patients With Sepsis and Septic Shock. Journal of Intensive Care Medicine 33: 116–120. [DOI] [PubMed] [Google Scholar]
- 59.Mikkelsen ME, Miltiades AN, Gaieski DF, Goyal M, Fuchs BD, Shah CV, Bellamy SL, and Christie JD. 2009. Serum lactate is associated with mortality in severe sepsis independent of organ failure and shock*: Critical Care Medicine 37: 1670–1677. [DOI] [PubMed] [Google Scholar]
- 60.Friedman G, Berlot G, Kahn RJ, and Vincent JL. 1995. Combined measurements of blood lactate concentrations and gastric intramucosal pH in patients with severe sepsis. Crit. Care Med 23: 1184–1193. [DOI] [PubMed] [Google Scholar]
- 61.Levy B, Gibot S, Franck P, Cravoisy A, and Bollaert P-E. 2005. Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study. Lancet 365: 871–875. [DOI] [PubMed] [Google Scholar]
- 62.Bannerman DD, and Goldblum SE. 1999. Direct effects of endotoxin on the endothelium: barrier function and injury. Lab. Invest 79: 1181–1199. [PubMed] [Google Scholar]
- 63.Farias NC, Borelli-Montigny GL, Fauaz G, Feres T, Borges ACR, and Paiva TB. 2002. Different mechanism of LPS-induced vasodilation in resistance and conductance arteries from SHR and normotensive rats. Br J Pharmacol 137: 213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sayk F, Vietheer A, Schaaf B, Wellhoener P, Weitz G, Lehnert H, and Dodt C. 2008. Endotoxemia causes central downregulation of sympathetic vasomotor tone in healthy humans. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 295: R891–R898. [DOI] [PubMed] [Google Scholar]
- 65.Singh AK, Jiang Y, and Gupta S. 2007. Effects of bacterial toxins on endothelial tight junction in vitro: a mechanism-based investigation. Toxicol. Mech. Methods 17: 331–347. [DOI] [PubMed] [Google Scholar]
- 66.Pedoto A, Caruso JE, Nandi J, Oler A, Hoffmann SP, Tassiopoulos AK, McGRAW DJ, Camporesi EM, and Hakim TS. 1999. Acidosis stimulates nitric oxide production and lung damage in rats. American journal of respiratory and critical care medicine 159: 397–402. [DOI] [PubMed] [Google Scholar]
- 67.Beckert S, Farrahi F, Aslam RS, Scheuenstuhl H, Königsrainer A, Hussain MZ, and Hunt TK. 2006. Lactate stimulates endothelial cell migration. Wound Repair and Regeneration 14: 321–324. [DOI] [PubMed] [Google Scholar]
- 68.Ruan G-X, and Kazlauskas A. 2013. Lactate Engages Receptor Tyrosine Kinases Axl, Tie2, and Vascular Endothelial Growth Factor Receptor 2 to Activate Phosphoinositide 3-Kinase/Akt and Promote Angiogenesis. Journal of Biological Chemistry 288: 21161–21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hattori Y, Hattori S, and Kasai K. 2003. Lipopolysaccharide activates Akt in vascular smooth muscle cells resulting in induction of inducible nitric oxide synthase through nuclear factor-kappa B activation. European Journal of Pharmacology 481: 153–158. [DOI] [PubMed] [Google Scholar]
- 70.Sukriti S, Tauseef M, Yazbeck P, and Mehta D. 2014. Mechanisms Regulating Endothelial Permeability. Pulmonary Circulation 4: 535–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim TJ, Freml L, Park SS, and Brennan TJ. 2007. Lactate concentrations in incisions indicate ischemic-like conditions may contribute to postoperative pain. J Pain 8: 59–66. [DOI] [PubMed] [Google Scholar]
- 72.Bidani A, Wang CZ, Saggi SJ, and Heming TA. 1998. Evidence for pH sensitivity of tumor necrosis factor-alpha release by alveolar macrophages. Lung 176: 111–121. [DOI] [PubMed] [Google Scholar]
- 73.Fernandez SF, Fung C, Helinski JD, Alluri R, Davidson BA, and Knight PR. 2013. Low pH Environmental Stress Inhibits LPS and LTA-Stimulated Proinflammatory Cytokine Production in Rat Alveolar Macrophages. BioMed Research International 2013: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stambaugh R, and Post D. 1966. Substrate and Product Inhibition of Rabbit Muscle Lactic Dehydrogenase Heart (H4) and Muscle (M4) Isozymes. J. Biol. Chem 241: 1462–1467. [PubMed] [Google Scholar]
- 75.Spriet LL, Howlett R, and Heigenhauser G. 2000. An enzymatic approach to lactate production in human skeletal muscle during exercise. Medicine & Science in Sports & Exercise 32: 756–763. [DOI] [PubMed] [Google Scholar]
- 76.Sharif O, Bolshakov VN, Raines S, Newham P, and Perkins ND. 2007. Transcriptional profiling of the LPS induced NF-κB response in macrophages. BMC immunology 8: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kawai T, and Akira S. 2007. Signaling to NF-κB by Toll-like receptors. Trends in Molecular Medicine 13: 460–469. [DOI] [PubMed] [Google Scholar]
- 78.Blackwell TS, and Christman JW. 1997. The Role of Nuclear Factor Kappa B in Cytokine Gene Regulation. American Journal of Respiratory Cell and Molecular Biology 17: 3–9. [DOI] [PubMed] [Google Scholar]
- 79.Doxaki C, Kampranis SC, Eliopoulos AG, Spilianakis C, and Tsatsanis C. 2015. Coordinated Regulation of miR-155 and miR-146a Genes during Induction of Endotoxin Tolerance in Macrophages. The Journal of Immunology 195: 5750–5761. [DOI] [PubMed] [Google Scholar]
- 80.Dobson GP, Yamamoto E, and Hochachka PW. 1986. Phosphofructokinase control in muscle: nature and reversal of pH-dependent ATP inhibition. Am. J. Physiol 250: R71–76. [DOI] [PubMed] [Google Scholar]
- 81.Leite TC, Coelho RG, Silva DD, Coelho WS, Marinho-Carvalho MM, and Sola-Penna M. 2011. Lactate downregulates the glycolytic enzymes hexokinase and phosphofructokinase in diverse tissues from mice. FEBS Letters 585: 92–98. [DOI] [PubMed] [Google Scholar]
- 82.Halperin ML, Connors HP, Relman AS, and Karnovsky ML. 1969. Factors that control the effect of pH on glycolysis in leukocytes. Journal of Biological Chemistry 244: 384–390. [PubMed] [Google Scholar]
- 83.Gray LR, Tompkins SC, and Taylor EB. 2014. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci 71: 2577–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, Wang Z, Quinn WJ, Kopinski PK, Wang L, Akimova T, Liu Y, Bhatti TR, Han R, Laskin BL, Baur JA, Blair IA, Wallace DC, Hancock WW, and Beier UH. 2017. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metabolism 25: 1282–1293.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, and others 2007. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood 109: 3812–3819. [DOI] [PubMed] [Google Scholar]
- 86.Jones AE 2010. Lactate Clearance vs Central Venous Oxygen Saturation as Goals of Early Sepsis TherapyA Randomized Clinical Trial. JAMA 303: 739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stacpoole PW, Wright EC, Baumgartner TG, Bersin RM, Buchalter S, Curry SH, Duncan CA, Harman EM, Henderson GN, Jenkinson S, Lachin JM, Lorenz A, Schneider SH, Siegel JH, Summer WR, Thompson D, Wolfe CL, Zorovich B, and the D.-L. A. S. Group*. 1992. A Controlled Clinical Trial of Dichloroacetate for Treatment of Lactic Acidosis in Adults. New England Journal of Medicine 327: 1564–1569. [DOI] [PubMed] [Google Scholar]
- 88.Suzuki H, Hisamatsu T, Chiba S, Mori K, Kitazume MT, Shimamura K, Nakamoto N, Matsuoka K, Ebinuma H, Naganuma M, and Kanai T. 2016. Glycolytic pathway affects differentiation of human monocytes to regulatory macrophages. Immunology Letters 176: 18–27. [DOI] [PubMed] [Google Scholar]
- 89.Sanchez WY, McGee SL, Connor T, Mottram B, Wilkinson A, Whitehead JP, Vuckovic S, and Catley L. 2013. Dichloroacetate inhibits aerobic glycolysis in multiple myeloma cells and increases sensitivity to bortezomib. Br J Cancer 108: 1624–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Michelakis ED, Webster L, and Mackey JR. 2008. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 99: 989–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Levy B, Mansart A, Montemont C, Gibot S, Mallie J-P, Regnault V, Lecompte T, and Lacolley P. 2007. Myocardial lactate deprivation is associated with decreased cardiovascular performance, decreased myocardial energetics, and early death in endotoxic shock. Intensive Care Medicine 33: 495–502. [DOI] [PubMed] [Google Scholar]
- 92.Covarrubias AJ, Aksoylar HI, and Horng T. 2015. Control of macrophage metabolism and activation by mTOR and Akt signaling. Seminars in Immunology 27: 286–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Buck MD, O’Sullivan D, and Pearce EL. 2015. T cell metabolism drives immunity. The Journal of Cell Biology 210: 2104OIA169. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.