

Abstract
Background
Ivacaftor is a safe, effective treatment for cystic fibrosis (CF) in patients aged ≥6 years with a CFTR gating mutation. This open-label, multicenter, phase 3 study assessed ivacaftor in children aged 2–5 years.
Methods
Children aged 2–5 years with a CFTR gating mutation received ivacaftor (<14 kg: 50 mg every 12 hours; ≥14 kg: 75 mg every 12 hours) for 4 days (Part A; n=9) or 24 weeks (Part B; n=34). Primary outcomes were pharmacokinetics (PK) and safety. Other markers of disease were also evaluated.
Findings
Ivacaftor PK was similar to that reported in adults. Common adverse events in Part B included cough (n=19; 55·9%) and vomiting (n=10; 29·4%). Five patients (15%) experienced liver function test (LFT) elevations >8× ULN (study drug interrupted, n=4; study discontinuation, n=1). At week 24, sweat chloride improved on average by −46·9 mmol (SD 26·2, p<0·0001), BMI z-score by 0·4 (SD 0·4, p<0·0001), fecal elastase by 99·8 (138·4) μg/g (p=0·0009) and immunoreactive trypsinogen by −20·7 ng/mL (SD 24·0, p=0.0002).
Interpretation
Ivacaftor appears safe and beneficial in young CF children with a gating mutation followed for 24 weeks. The safety profile was similar to that seen in older CF patients, although the frequency of LFT elevations suggests consideration of more frequent monitoring in younger children, particularly those with a history of elevated LFTs. Improvements in sweat chloride and nutritional parameters, with a suggestion of enhanced pancreatic function were observed.
Funding
Vertex Pharmaceuticals Incorporated
www.clinicaltrials.gov identifier:
Keywords: children, clinical trial, phase 3, cystic fibrosis transmembrane conductance regulator, Kalydeco, nutritional status
INTRODUCTION
The consequences of cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction, pancreatic insufficiency, poor nutritional status, and structural lung damage, begin early in infancy.1–3 Computerized tomography scans in children with CF aged 1 month through 6 years4 reveal structural lung disease that precedes detection by spirometry. Nutritional deficits are typically the earliest clinical manifestations of CF and are not completely reversed by pancreatic enzyme replacement therapy.5,6 To improve the trajectory of long-term outcomes, early interventions should be implemented before structural lung disease is established. Few studies have explored chronic therapies for children <6 years, and there have been no studies of treatments targeting the basic defect in CFTR in this population.7,8
Ivacaftor, a CFTR potentiator, improves chloride transport through CFTR channels by increasing the channel gating activity of CFTR protein at the cell surface.9,10 The most common gating mutation is Gly551Asp-CFTR, present in approximately 4% of individuals with CF.11 Phase 3 studies showed ivacaftor to be well tolerated, safe, and effective in patients aged ≥6 years with CF and the Gly551Asp or other CFTR gating mutations, as evidenced by sustained reduction in sweat chloride concentrations and substantial, durable improvements in lung function, respiratory symptoms, and weight gain.12–14
This study evaluated the safety, pharmacokinetics (PK) and pharmacodynamics (PD, based on sweat chloride concentrations) of ivacaftor in children with CF aged 2 to 5 years with a CFTR gating mutation on ≥1 allele and explored the efficacy of ivacaftor in this population.
METHODS
This open-label, single-arm, phase 3 study (clinicaltrials.gov-identifier NCT01705145) was conducted from January 8, 2013 to March 28, 2014 at 15 sites in the United States, United Kingdom, and Canada. An independent ethics committee or institutional review board for each site approved the study protocol. Written informed consent was obtained from each child’s parent or legal guardian.
Patients
Children aged 2 to 5 years weighing ≥8 kg with a confirmed diagnosis of CF15 and a CFTR gating mutation on ≥1 allele (Gly551Asp, Gly178Arg, Ser549Asn, Ser549Arg, Gly551Ser, Gly970Arg, Gly1244Glu, Ser1251Asn, Ser1255Pro, or Gly1349Asp) were eligible for enrollment. Exclusion criteria were current or recent (within 4 weeks) pulmonary exacerbation, anemia, liver disease, or renal dysfunction; infection with organisms associated with a more rapid decline in pulmonary function (e.g., Burkholderia cenocepacia, Burkholderia dolosa, Mycobacterium abscessus); or use of moderate or strong cytochrome P450–3A inhibitors or inducers within 2 weeks of ivacaftor dosing.
Study Design
This study comprised two phases (Figure 1). Part A (4 days) assessed PK and safety of ivacaftor and its metabolites. Patients who completed Part A were eligible to enroll in Part B, which assessed longer-term (24-week) safety of ivacaftor. Patients completing Part B could enroll in the treatment arm of an ongoing open-label extension study; those who discontinued early or did not wish to continue ivacaftor therapy could enroll in an observational arm.
Figure 1.
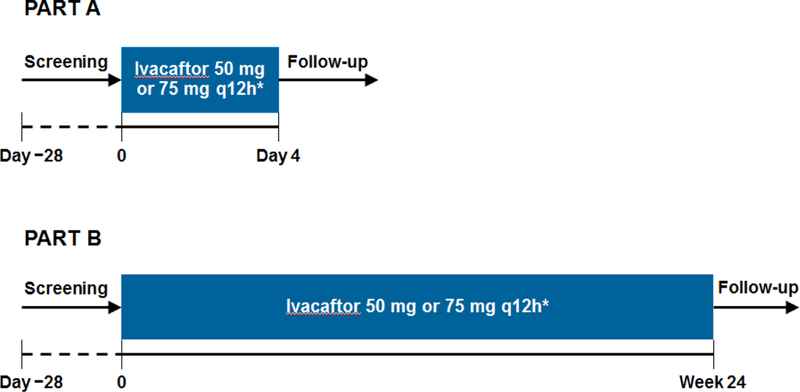
Study design.
*Weight-based dosing: 50 mg every 12 h for patients with body weight <14 kg; 75 mg every 12 h for patients ≥14 kg.
Following screening, patients received weight-based ivacaftor every 12 hours: 50 mg if <14 kg, and 75 mg if ≥14 kg. Study medication was supplied as granules in a capsule, with instructions to break open the capsule and mix the contents in approximately 5 mL of applesauce (or other suitable food) at the time of a high-calorie, high-fat meal.
In Part A, patients received ivacaftor every 12 hours for 4 days. Analysis of PK data from Part A confirmed that these doses were appropriate to use in Part B, in which patients received ivacaftor every 12 hours for 24 weeks. Details regarding study site visits and blood sample collection for PK analysis are provided in the supplemental online appendix.
Outcome Measures
The primary outcomes were PK and safety of ivacaftor. Safety evaluations included adverse events (AEs), clinical laboratory parameters, vital signs, 12-lead electrocardiogram (ECG), coagulation studies, and physical and ophthalmologic examinations. Adverse events were monitored throughout the study, including the screening period and up to 4 weeks ± 7 days after last dose in Part B. Ophthalmologic examinations were performed at screening before each study part, at 12 weeks after last ivacaftor dose in Part A, and at weeks 12 and 24 in Part B.
PK parameters included area under the plasma drug concentration-vs-time curve (AUC) and minimum plasma drug concentration (Cmin). Secondary outcome measures in Part B included absolute change from baseline in sweat chloride concentration (as a measure of PD) and weight, body mass index (BMI), and height z-scores. Exploratory post-hoc analyses evaluated changes in percent predicted FEV1 in children ≥3 years of age,16 fecal elastase-1, and immunoreactive trypsinogen (IRT).
Additional details regarding study procedures, including palatability assessment and outcome measures, are provided in the supplemental online appendix.
Statistical Analysis
Plasma concentrations of ivacaftor from Parts A and B were evaluated using nonlinear mixed-effects modeling software, version 7 (ICON Development Solutions; Hanover, MD). The PK samples were analyzed using a previously developed population PK model for ivacaftor to describe ivacaftor disposition in patients aged 2 to 5 years.
Because the primary goal was confirmation of ivacaftor PK and safety, the study was not designed to detect statistically significant treatment effects. For Part B, a post-hoc analysis using a one-sample two-sided t test was performed to assess mean absolute change from baseline at week 24 for secondary and exploratory endpoints. In addition, for fecal elastase-1, a Fisher’s exact test was performed to compare the proportion of patients with a value >200 μg/g between baseline and the on-treatment period.
Role of funding source
The sponsor designed the protocol in collaboration with the authors, analyzed data, participated in data interpretation, and provided editorial/writing assistance. All authors had full access to the study data and made the final decision to submit for publication.
RESULTS
Study Population
Part A.
Nine patients were enrolled (six male, three female); aged 2 years (n=3), 3 years (n=4), and 4 to 5 years (n=2). All had a Gly551Asp-CFTR mutation on ≥1 allele; the most common genotype was Gly551Asp/Phe508del (n=7). All patients received ivacaftor 50 mg every 12 h (n=4) or 75 mg every 12 h (n=5) for the 4-day treatment period and were included in the safety analysis. Eight of these patients were enrolled in Part B. The other patient turned 6 years of age between study periods and switched to commercially available ivacaftor.
Part B.
Of 34 patients enrolled, 32 had Gly551Asp and two had Ser549Asn mutations. Ten received ivacaftor 50 mg every 12 h and 24 ivacaftor 75 mg every 12 h. Thirty-three patients (97%) completed the 24-week treatment period; one receiving ivacaftor 50 mg discontinued study drug treatment because of an AE (elevated liver transaminase). Demographic and baseline characteristics are summarized in Table 1.
Table 1.
Patient Demographics and Baseline Characteristics (Part B)
| Characteristic | Overall (N=34) |
|---|---|
| Sex, n (%) | |
| Male | 28 (82·4) |
| Female | 6 (17·6) |
| Race, n (%) | |
| White | 34 (100·0) |
| Ethnicity, n (%) | |
| Hispanic | 1 (2·9) |
| Genotype, n (%) | |
| Gly551Asp/Phe508del | 26 (76·5) |
| Gly551Asp/Lys447ArgfsX2 | 1 (2·9) |
| Gly551Asp/c.1585–1G>A | 1 (2·9) |
| Gly551Asp/Leu1258PhefsX7 | 1 (2·9) |
| Gly551Asp/Leu88IlefsX22 | 1 (2·9) |
| Gly551Asp/Gly551Asp | 1 (2·9) |
| Gly551Asp/Arg117His | 1 (2·9) |
| Ser549Asn/Phe508del | 1 (2·9) |
| Ser549Asn/Arg553X | 1 (2·9) |
| Age category, years, n (%) | |
| 2 | 9 (26·5) |
| 3 | 11 (32·4) |
| 4 to 5 | 14 (41·2) |
| Weight z-score, mean (SD) | −0·2 (0·8) |
| Height z-score, mean (SD) | −0·3 (0·8) |
| Fecal elastase-1 <200 μg/g, n (%)* | 26 (96.3) |
| Immunoreactive trypsinogen (ng/mL), mean (SD) | 33·6 (29·8) |
| Sweat chloride (mmol/L), mean (SD) | 97·9 (14·0) |
Of patients with fecal elastase-1 data available (n=27); FE-1 <200 mcg/g is diagnostic if exocrine pancreatic insufficiency15; Gly551Asp was formerly G551D
Primary Outcomes:
Pharmacokinetics
Population PK results (Parts A and B) for ivacaftor indicated that exposures to ivacaftor (Figure 2) for both doses in patients aged 2 to 5 years were similar to each other and to that reported in adults in phase 3 studies. Body weight was the most important predictor of ivacaftor disposition, which was consistent with the previous population PK model for ivacaftor (data not shown). Age and sex did not contribute to variability in ivacaftor PK in a meaningful manner after accounting for body weight.
Figure 2.
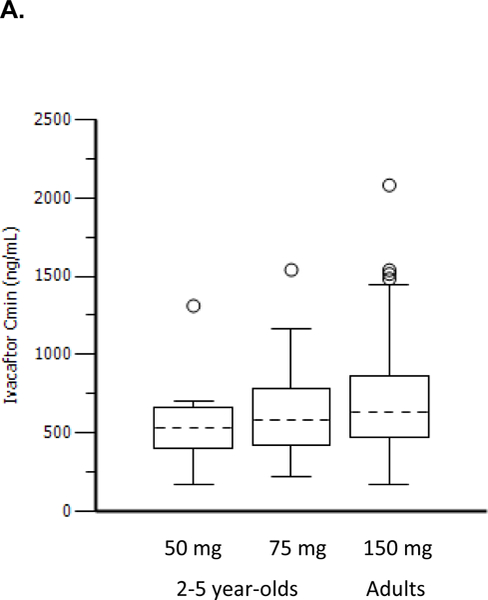
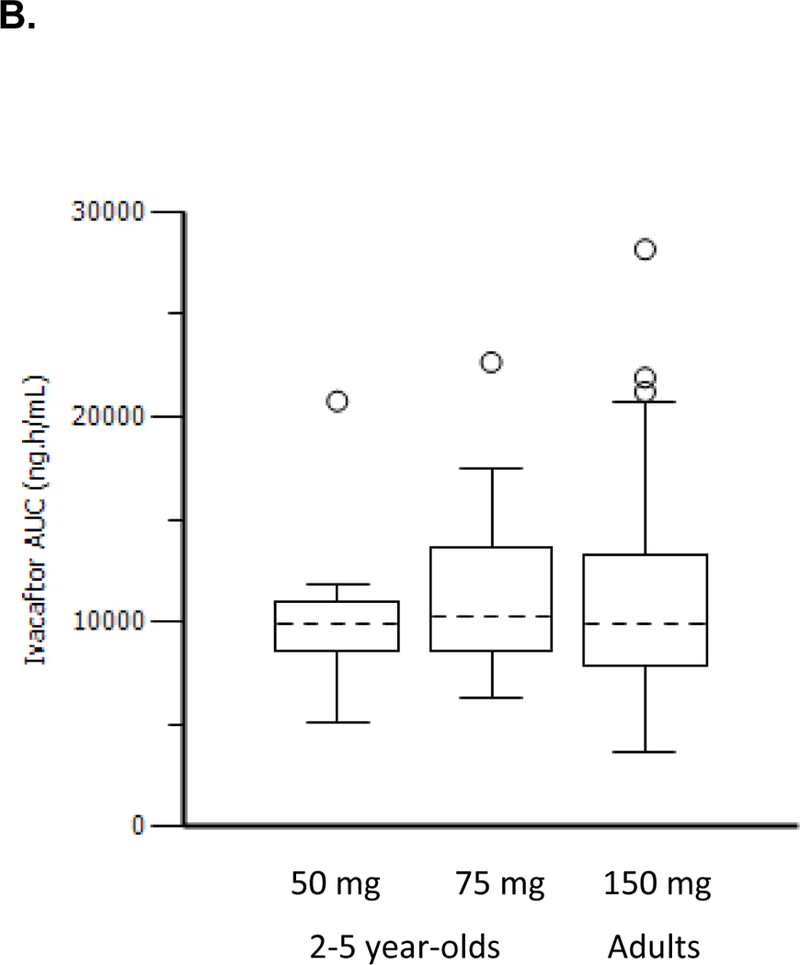
Ivacaftor Cmin distribution (A) and ivacaftor AUC distribution (B) in children aged 2–5 years compared with adults from previous Phase 3 studies.
Dashed black lines in the center of the boxes are medians, boxes are the interquartile range (IQR), whiskers are 1·5•IQR, and open circles are outliers.
AUC, area under the plasma drug concentration-vs-time curve; Cmin, minimum plasma drug concentration.
Safety and Tolerability
Part A.
Eight patients (88·9%) experienced AEs. The only AEs reported by ≥2 patients were pyrexia (n=4; 44·4%); vomiting, ecchymosis, and rhinorrhea (all n=2; 22·2%).
Part B.
In total, 33 patients (97·1%) experienced AEs (Table 2); the most common were respiratory or gastrointestinal in nature. Most AEs were mild or moderate in severity. In total, two patients (5·9%), both receiving ivacaftor 50 mg every 12 h, experienced severe AEs (transaminase increased and device-related sepsis); they were included in the serious AE (SAE) group (Table 2).
Table 2.
Serious Adverse Events and Adverse Events Occurring in ≥3 Patients
| Adverse Event, n (%) | Ivacaftor 50 mg Every 12 h (n=10) |
Ivacaftor 75 mg Every 12 h (n=24) |
Overall (N=34) |
|---|---|---|---|
| Serious Adverse Events (Part B) | |||
| Infective pulmonary exacerbation of CF | 1 (10·0) | 1 (4·1) | 2 (5·9) |
| Device related sepsis | 1 (10·0) | 0 | 1 (2·9) |
| Pseudomonas test positive | 1 (10·0) | 0 | 1 (2·9) |
| Transaminases (hepatic enzymes) increased | 1 (10·0) | 0 | 1 (2·9) |
| Vomiting | 0 | 1 (10·0) | 1 (2·9) |
| Convulsion | 0 | 1 (10·0) | 1 (2·9) |
| AEs Occurring in >3 Patients (Part B) | |||
| ≥1 AE | 10 (100·0) | 23 (95·8) | 33 (97·1) |
| Cough | 4 (40·0) | 15 (62·5) | 19 (55·9) |
| Vomiting | 3 (30·0) | 7 (29·2) | 10 (29·4) |
| Nasal congestion | 4 (40·0) | 5 (20·8) | 9 (26·5) |
| Upper respiratory tract infection | 1 (10·0) | 7 (29·2) | 8 (23·5) |
| Rhinorrhea | 2 (20·0) | 5 (20·8) | 7 (20·6) |
| Positive respiratory culture* | 0 | 6 (25·0) | 6 (17·6) |
| Pyrexia | 4 (40·0) | 2 (8·3) | 6 (17·6) |
| Hepatic enzyme increased | 3 (30·0) | 2 (8·3) | 5 (14·7) |
| Infective pulmonary exacerbation of CF | 1 (10·0) | 4 (16·7) | 5 (14·7) |
| Constipation | 0 | 4 (16·7) | 4 (11·8) |
| Rash | 2 (20·0) | 2 (8·3) | 4 (11·8) |
| Croup, infectious | 2 (20·0) | 1 (4·2) | 3 (8·8) |
| Otitis media | 2 (20·0) | 1 (4·2) | 3 (8·8) |
| Productive cough | 0 | 3 (12·5) | 3 (8·8) |
| Sinusitis | 2 (20·0) | 1 (4·2) | 3 (8·8) |
Adverse events were coded by system organ class and preferred terms using the Medical Dictionary for Regulatory Activities v. 15·1 and were summarized descriptively.
Test was positive for Haemophilus in three patients, for methicillin-resistant Staphylococcus aureus in two patients, and for Moraxella in one patient. AE, adverse event; CF, cystic fibrosis. A serious adverse event (SAE) was defined as any adverse event that met any of the following criteria: (1) Fatal (death, regardless of cause occurring during participation in the study or occurred after participation in the study and was suspected of being a delayed toxicity due to administration of the study drug), (2) Life-threatening, such that the subject was at immediate risk of death from the reaction as it occurred, (3) Inpatient hospitalization or prolongation of hospitalization, with the exception of planned or elective hospitalization, (4) Persistent or significant disability/incapacity, (5) Congenital anomaly or birth defect, (6) Important medical event, that, based upon appropriate medical judgment, jeopardized the subject or required medical or surgical intervention to prevent 1 of the outcomes
In total, 6 patients experienced 7 serious AEs (SAEs). Of patients receiving ivacaftor 50 mg every 12 hours, 4 SAEs occurred in 3 patients (30%): infective pulmonary exacerbation of CF, device-related sepsis, positive Pseudomonas aeruginosa culture, and increased transaminases. In the ivacaftor 75 mg every 12 hours group, 3 SAEs occurred in 3 patients (12·5%): infective pulmonary exacerbation of CF, vomiting, and convulsion. Increased transaminases was the only SAE considered related to ivacaftor and the only AE that resulted in study discontinuation.
Five patients (14·7%) experienced elevations in alanine transaminase (ALT) or aspartate transaminase (AST) >8× the upper limit of normal (ULN) (n=3, ivacaftor 50 mg; n=2, ivacaftor 75 mg). All had a history of elevated transaminases before enrollment and had elevated LFTs (>2× but <3x ULN) at baseline (screening exclusion criteria: ≥3× ULN). In four of these patients, ivacaftor treatment was interrupted, in accordance with the protocol. In all four patients, LFTs returned to baseline levels following interruption of ivacaftor. The remaining patient discontinued the study due to an SAE of severe transaminases after 18 days of treatment. No patients had notable bilirubin, alkaline phosphatase, or gamma-glutamyl transferase elevations or clinical symptoms related to the LFT elevations (Additional details in the supplemental online appendix).
Eight additional patients experienced AEs resulting in study drug interruption in Part B: In the 50 mg group, croup (n=1), and vomiting and device-related sepsis (n=1); in the 75 mg group, vomiting (n=2), gastroenteritis (n=1), retching (n=1), cough (n=1), and rash (n=1). These AEs were not deemed study-drug related by the site investigators.
No new cataracts or lens opacities or other clinically relevant findings for chemistry, hematology, or ECG were reported. The mean adherence to study treatment in Part B was 96·1%; 31 patients had an adherence of 80% or higher. No patients were discontinued because of non-adherence.
Secondary endpoints:
Pharmacodynamics (Sweat Chloride As a Measure of CFTR Function)
By week 2, a mean (SD) decrease of 44·0 (20·3) mmol/L in sweat chloride concentrations was observed (p<0·0001). This effect was sustained through week 24, with a mean (SD) absolute change from baseline of −46·9 (26·2) mmol/L (p<0·0001 vs baseline; Figure 3). Similar changes from baseline to week 24 in sweat chloride were observed in both groups (50 mg, −47·1 [24·3] mmol/L; p=0·002; 75 mg, −46·8 [27·6] mmol/L; p<0·0001).
Figure 3.
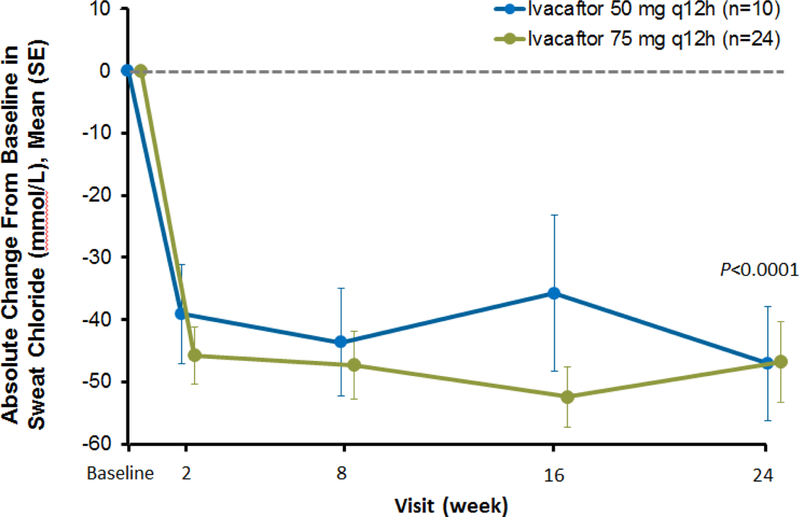
Absolute change from baseline in sweat chloride concentrations during the 24-week ivacaftor treatment period, presented as means and SD for each dose group at each time point
Palatability
Palatability assessments indicated that the ivacaftor granules were acceptable when administered in soft food. Overall, all 9 patients in Part A and 32 (94·1%) in Part B fully consumed the first dose of study drug.
Nutritional measures
Improvements in weight and BMI z-scores were similar in both dose groups. Mean weight z-scores increased by week 2 and were sustained throughout the treatment period to week 24 (0·2 [0·3]; p<0·0001; Figure 4A). Similarly, BMI z-scores increased during the 24-week treatment period (0·4 [0·4]; p<0·0001; Figure 4B). Height z-scores were similar to baseline at week 24 (−0·01 [0·3]; p=0·84; Figure 4C).
Figure 4.
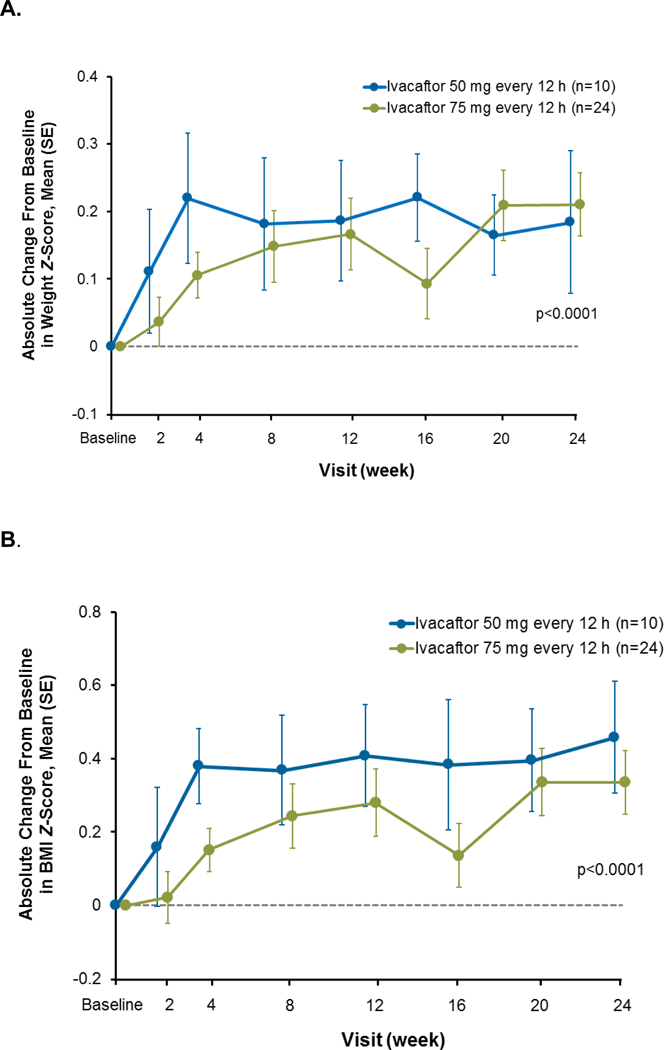
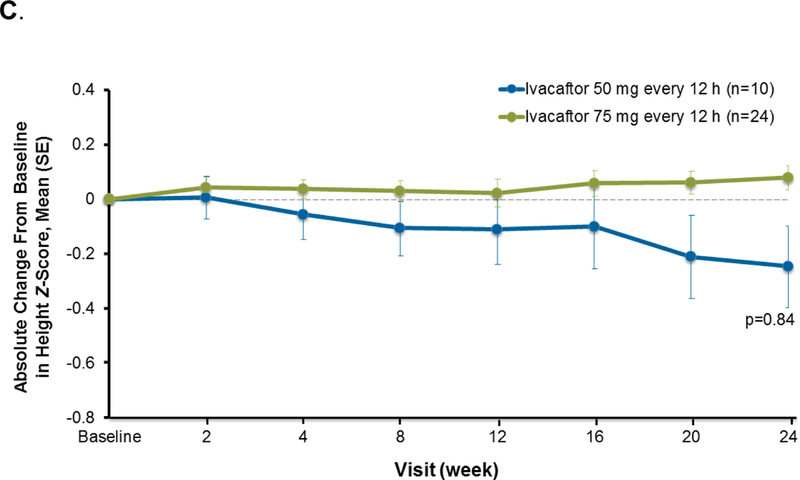
Absolute change from baseline in weight z-score (A), BMI z-score (B), and height z-score (C) during the 24-week ivacaftor treatment period, presented as means and SD for each dose group at each time point
Exploratory endpoints:
Pancreatic function
In total, 27 patients contributed samples for fecal elastase-1 measurements. At baseline, measurements were: <50 μg/g [n=25], 53 μg/g [n=1], and >500 μg/g [n=1]). Overall, fecal elastase-1 increased on average by 99·8 (138·4) μg/g after 24 weeks of treatment (p=0·0009); improvements were observed in both dose groups (50 mg: 127·9 μg/g [191·8]; 75 mg, 93·5 [128·3] μg/g; Figure 5). Overall, 26 (76·5%) patients at baseline and 24 (70·6%) patients at week 24 had fecal elastase-1 measurements <200 μg/g, and 1 (2·9%) patient at baseline and 7 (20·6%) patients at week 24 had fecal elastase-1 measurements ≥200 μg/g (Fisher’s exact test, P=0·0504). Eleven (42·3%) patients had ≥1 on-treatment value >100 μg/g and 10 patients (38·5%) demonstrated no improvement.
Figure 5.
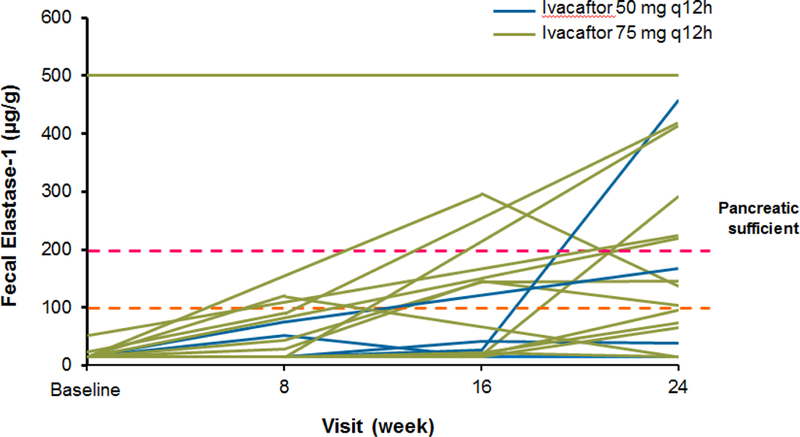
Fecal elastase-1 for individual patients during 24-week ivacaftor treatment period among the 27 patients with fecal elastase data available. The one patient with baseline fecal elastase-1 >500 μg/g (upper limit of quantitation) had the Gly551Asp/Arg117His mutation and was not receiving pancreatic enzyme supplementation. A fecal elastase <200 mcg/g is associated with exocrine pancreatic insufficiency.15
Overall, IRT (a marker of pancreatic stress) decreased from baseline by an average of 20·7 (24·0) ng/mL at week 24 (p=0·0002); similar improvements were noted in both ivacaftor doses (mean [SD] changes: 50 mg, −24·4 [21·7] ng/ml; 75 mg, −19·5 [25·1] ng/mL). Decreases in IRT were seen as early as week 2 (Figure 6). Patients with substantial improvements in fecal elastase-1 tended to be those with higher baseline IRT levels.
Figure 6.
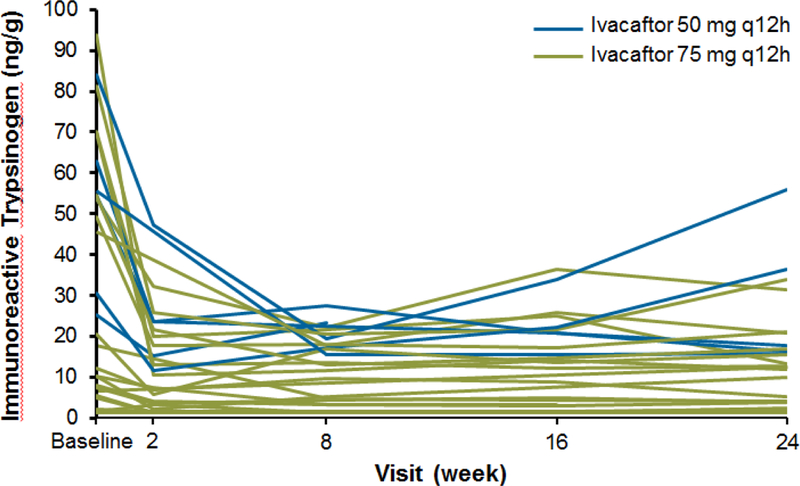
Immunoreactive trypsinogen concentrations for individual patients during the 24-week ivacaftor treatment period.
Lung function
Baseline spirometry measurements were submitted for 23 patients, of whom 21 had on-treatment measurements. Independent over-reading determined that only three participants (14·3%) had acceptable measurements at baseline and one time point on treatment, precluding any analysis of these data.
DISCUSSION
This report describes the first clinical study of a CFTR potentiator, ivacaftor, in children aged 2 to 5 years with CF and a CFTR gating mutation. Based on ivacaftor exposure similar to that observed in adults, ivacaftor 50 mg every 12 hours or 75 mg every 12 hours appear to be appropriate doses for children aged 2 to 5 years. The formulation was also determined to be palatable, with high treatment adherence (96% overall over 24 weeks). Over the 24-week open-label treatment period, improvements in sweat chloride and nutritional parameters, as well as a suggestion of improvement in markers of pancreatic function, were observed.
We selected an open-label, single-arm study design due to small group size, the Gly551Asp-CFTR mutation being present in only about 4% of CF patients worldwide.11 As the primary objective was to assess safety, the number of children exposed to active drug could be maximized with this design. Additionally, PK evidence of appropriate dose, along with evidence of CFTR functional correction via the biomarker sweat chloride, was sufficient to allow extrapolation of presumed clinical efficacy from the older groups.
Both ivacaftor doses were generally well tolerated; most AEs were consistent with those expected for patients with CF and largely considered unrelated to study drug. However, the frequency of elevated LFTs exceeded that in studies of ivacaftor in older patients.12,13 Five of 34 patients (15%) experienced significant (>8× ULN) increases in transaminases; four required dose interruption and one discontinued. All five had a history of elevated LFTs, with levels above the ULN, but within the protocol-accepted range. Although reduction in LFTs after interruption of ivacaftor suggests the drug played a role in these AEs, patients also had other potentially contributing factors (concomitant antibiotic use or intercurrent viral infection). Further, as all LFT elevations were asymptomatic, they were likely to have been undetected in standard clinical practice, in which monitoring is less frequent. The prevalence of transient LFT abnormalities in this age group is unknown and absence of a control group makes interpretation more difficult. Nonetheless, these data suggest that additional monitoring should be considered in children 2 to 5 years old receiving ivacaftor, particularly in the presence of any additional risk factors such as prior LFT elevation or whilst being treated with hepatotoxic drugs. An on-going long-term extension study, KLIMB, is collecting additional safety data.
The improvement observed in sweat chloride was similar in magnitude to that in studies in older age groups that showed associated improvements in pulmonary function and weight.12,13,17 In our view, this confirms functional efficacy at the cellular level, but is no surrogate for clinical improvement. However, improvement in nutritional status and a suggestion of improvement in markers of exocrine pancreatic function indicate a possible clinical effect. Children entered this study with a mean weight z-score of −0·2, which significantly (p<0·0001) increased by a mean of 0·2 after 24 weeks. Previous trials of ivacaftor in older populations similarly demonstrated a significant increase in weight. As previous studies have suggested the CF pancreas to be irreversibly damaged from early life,2,3 restoration of pancreatic exocrine function has been considered an unlikely explanation for nutritional improvements observed with ivacaftor.12–14 Studies based on intraluminal monitoring provide compelling evidence in support of improvement in intestinal pH related to restoration of CFTR-mediated bicarbonate secretion18; thus, improved absorption has been the leading hypothesis. Our study is the first to explore the effects of ivacaftor on pancreatic exocrine function. Fecal elastase-1, which was in the insufficient range in 26/27 (96·3%) patients at baseline, rose above the clinical cutoff for exocrine pancreatic insufficiency of 200 μg/g at least once in >25% of children, with more modest increases in others. There were also overall reductions in the serum pancreatic stress marker (IRT), which may support this observation; however, because this test is not widely used outside the neonatal screening period, results should be interpreted cautiously. These preliminary data suggest, for the first time, a window in early life wherein at least partial restoration of pancreatic exocrine function may be possible, and warrant further exploration of the effect of CFTR modulators on exocrine pancreatic function in infants with CF. The effect of ivacaftor on endocrine pancreatic function and risk of development of CF-related diabetes is not well understood and was not explored in this study.
Unfortunately, this study could not generate meaningful data on pulmonary function because only 3 patients produced research-quality measurements and we did not provide specific training or quality control in preschool lung function testing.19 Although some have reported meaningful spirometry in this age group,20,21 future studies in this group should consider lung clearance index (LCl) from multiple-breath washout tests as an alternative.22 LCI, is a non–effort-dependent test with increased sensitivity to detect early disease than spirometry.21,23–25
In conclusion, this study confirmed a safe and tolerable dose of an acceptable formulation of ivacaftor in children 2 to 5 years old with a CFTR gating mutation, although liver function appears to require closer monitoring in this age range, particularly among those with a history of elevated LFTs. Ivacaftor administered for 6 months led to significant declines in sweat chloride concentrations, nutritional benefits, and a suggestion of improvement in markers of pancreatic exocrine function. The cohort is undergoing long-term follow-up in an extension study, during which durability of these effects and longer-term safety will be assessed.
Supplementary Material
Research in context.
Evidence before this study
Ivacaftor, a CFTR potentiator that corrects a gating defect, is the first CFTR modulator to show a clinical benefit in adults. As CFTR-directed therapy is expected to be most beneficial before secondary sequelae of CFTR dysfunction take hold (e.g., infection and inflammation), it is imperative to define the safety of ivacaftor in younger populations. A search of PubMed was conducted on May 1, 2015, using the terms “ivacaftor” or “Kalydeco” or “VX770” AND “G551D” or “gating” and “pediatric,” with no restrictions on publication date or language.
Added value of this study
This study establishes the safety, dose, and therapeutic benefit of ivacaftor in young children (age 2 to 5 years) with CF. Ivacaftor at 50 mg (<14 kg) or 75 mg (≥14 kg) every 12 hours is well tolerated by CF toddlers, with improvements in sweat chloride and nutritional measures, and a suggestion of benefit to exocrine pancreatic function. Due to age limitations, impact on lung function was not assessed. Reported side effects are similar to those in the general CF population, although children with previous CF liver disease may experience transient elevation in transaminases.
Implications of all the available evidence
The findings of this study confirm the safety and potential therapeutic benefit of ivacaftor for CF children with a CFTR gating mutation. Based on the results of this study, ivacaftor should be considered (and has been approved by the FDA) for treatment of CF patients aged 2 years and older with a CFTR gating mutation (Gly551Asp, Gly1244Glu, Gly1349Asp, Gly178Arg, Gly551Ser, Ser1251Asn, Ser1255Pro, Ser549Asn, Ser549Arg).
ACKNOWLEDGMENTS
The authors thank Clare Saunders, BSc, for independent analysis of spirometry results. Medical writing and editorial support were provided by Dhrupad Patel, PharmD, and Bina J. Patel, PharmD. DP is an employee of Vertex Pharmaceuticals Incorporated and may own stock or stock options in that company. BJP is an employee of Peloton Advantage, Parsippany, NJ, which received funding from Vertex Pharmaceuticals Incorporated.
This project was supported by the NIHR Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London. The views expressed in this publication are those of the authors and not necessarily those of the NHS, The National Institute for Health Research, or the Department of Health.
The authors acknowledge the following investigators and study coordinators, who also participated as part of the KIWI Study Group: Margaret Rosenfeld, Janine Bufi, and Sharon McNamara, Seattle Children’s Hospital, Seattle, WA, USA; Ginger Reeves, Heather Hathorne, Katie Brand, and William Harris, University of Alabama at Birmingham, Birmingham, AL, USA; Jonathan Spahr and Elizabeth Hartigan, Children’s Hospital of Pittsburgh of UPMC, Pittsburgh, PA, USA; Brooke Noren, Patricia Grover, and Warren Regelmann, University of Minnesota, Minneapolis, MN, USA; Eric Hunter and Seth Walker, Emory University, Atlanta, GA, USA; Barbara Chatfield and Jane Vroom, University of Utah Hospitals & Clinics, Salt Lake City, UT, USA; April Williams, Candy Schmoll, and Philip Black, Children’s Mercy Hospitals & Clinics, Kansas City, MS, USA; Raquel Telfer and John Colombo, University of Nebraska Medical Center, Omaha, NE, USA; Lisa Bendy and Gregory Montgomery, Riley Hospital for Children, Indianapolis, IN, USA; Allen Lapey and Caitlin Doolittle, Massachusetts General Hospital, Boston, MA, USA; Thomas Symington, Cynthia Gile, and Susan Millard, Helen Devos Children’s Hospital, Grand Rapids, MI, USA; Jonathan Greenberg, Catherine Correia, and Gregory Sawicki, Children’s Hospital Boston, Boston, MA, USA; Matthew Ward and Howard Schmidt, Virginia Commonwealth University Medical Center, Richmond, VA, USA; Frank Accurso and Sheryl Faut, Children’s Hospital Colorado, Aurora, CO, USA; Mark Chilvers, Maggie McIlwaine, and Melissa Richmond, British Columbia’s Children’s Hospital, Vancouver, BC, Canada; Peter Murphy, Sandra Scott, and Jane Davies, Royal Brompton Hospital, London, UK; Steven Cunningham, Andrew Fall, and Debbie Miller, Royal Hospital for Sick Children, Edinburgh, Lothian Region, Edinburgh, UK; Kevin Southern and Kim Doolan, Alder Hey Children’s Hospital, Liverpool, Merseyside, UK.
Funding disclosure: This study was sponsored by Vertex Pharmaceuticals Incorporated. No author received an honorarium or other form of financial support related to the development of this manuscript.
This study was sponsored by Vertex Pharmaceuticals Incorporated. JCD has served on advisory boards and has undertaken educational activities for Vertex Pharmaceuticals Incorporated, for which her institution, Imperial College, has received payment. S Cunningham and WE Regelmann have no conflicts to declare. WT Harris has received research grants from Vertex Pharmaceuticals Incorporated, for which his institution, the University of Alabama at Birmingham, received payment.
KW Southern:
S Robertson, J Cooke, and Y Green are employees of Vertex Pharmaceuticals Incorporated and may own stock or stock options in that company. M Rosenfeld has received research grants and served as a consultant for Vertex Pharmaceuticals Incorporated, for which her institution received payment.
Footnotes
These study results were presented in part at the European Cystic Fibrosis Society Conference, June 10–13, 2015, in Brussels, Belgium, and at the 28th Annual North American Conference of the Cystic Fibrosis Foundation, October 9–11, 2014, in Atlanta, GA, USA.
Declaration of interests
A Lapey:
GS Sawicki has served on an advisory board for Vertex Pharmaceuticals Incorporated.
References
- 1.Moss R Pathophysiology and therapeutic targets. Johns Hopkins Adv Studies Med 2010; 10: 9–13. [Google Scholar]
- 2.Couper RT, Corey M, Durie PR, Forstner GG, Moore DJ. Longitudinal evaluation of serum trypsinogen measurement in pancreatic-insufficient and pancreatic-sufficient patients with cystic fibrosis. J Pediatr 1995; 127: 408–13. [DOI] [PubMed] [Google Scholar]
- 3.Imrie JR, Fagan DG, Sturgess JM. Quantitative evaluation of the development of the exocrine pancreas in cystic fibrosis and control infants. Am J Pathol 1979; 95: 697–708. [PMC free article] [PubMed] [Google Scholar]
- 4.Stick SM, Brennan S, Murray C, et al. Bronchiectasis in infants and preschool children diagnosed with cystic fibrosis after newborn screening. J Pediatr 2009; 155: 623–8. [DOI] [PubMed] [Google Scholar]
- 5.Li L, Somerset S. Digestive system dysfunction in cystic fibrosis: challenges for nutrition therapy. Dig Liver Dis 2014; 46: 865–74. [DOI] [PubMed] [Google Scholar]
- 6.Sinaasappel M, Stern M, Littlewood J, et al. Nutrition in patients with cystic fibrosis: a European Consensus. J Cyst Fibros 2002; 1: 51–75. [DOI] [PubMed] [Google Scholar]
- 7.Nguyen TT, Thia LP, Hoo AF, et al. Evolution of lung function during the first year of life in newborn screened cystic fibrosis infants. Thorax 2014; 69: 910–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenfeld M, Ratjen F, Brumback L, et al. Inhaled hypertonic saline in infants and children younger than 6 years with cystic fibrosis: the ISIS randomized controlled trial. JAMA 2012; 307: 2269–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A 2009; 106: 18825–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu H, Burton B, Huang CJ, et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J Cyst Fibros 2012; 11: 237–45. [DOI] [PubMed] [Google Scholar]
- 11.Cystic Fibrosis Foundation Patient Registry 2012 Annual Data Report. Bethesda, MD: Cystic Fibrosis Foundation; 2013. [Google Scholar]
- 12.Davies JC, Wainwright CE, Canny GJ, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med 2013; 187: 1219–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011; 365: 1663–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Boeck K, Munck A, Walker S, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros 2014; 13: 674–80. [DOI] [PubMed] [Google Scholar]
- 15.Farrell PM, Rosenstein BJ, White TB, et al. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J Pediatr 2008; 153: S4–S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beydon N, Davis SD, Lombardi E, et al. An official American Thoracic Society/European Respiratory Society statement: pulmonary function testing in preschool children. Am J Respir Crit Care Med 2007; 175: 1304–45. [DOI] [PubMed] [Google Scholar]
- 17.De Boeck K, Munck A, Walker S, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros 2014; 13: 674–80. [DOI] [PubMed] [Google Scholar]
- 18.Pallagi P, Venglovecz V, Rakonczay Z Jr., et al. Trypsin reduces pancreatic ductal bicarbonate secretion by inhibiting CFTR Cl(−) channels and luminal anion exchangers. Gastroenterology 2011; 141: 2228–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arets HG, Brackel HJ, van der Ent CK. Forced expiratory manoeuvres in children: do they meet ATS and ERS criteria for spirometry? Eur Respir J 2001; 18: 655–60. [DOI] [PubMed] [Google Scholar]
- 20.Kerby GS, Rosenfeld M, Ren CL, et al. Lung function distinguishes preschool children with CF from healthy controls in a multi-center setting. Pediatr Pulmonol 2012; 47: 597–605. [DOI] [PubMed] [Google Scholar]
- 21.Aurora P, Bush A, Gustafsson P, et al. Multiple-breath washout as a marker of lung disease in preschool children with cystic fibrosis. Am J Respir Crit Care Med 2005; 171: 249–56. [DOI] [PubMed] [Google Scholar]
- 22.Horsley A Lung clearance index in the assessment of airways disease. Respir Med 2009; 103: 793–9. [DOI] [PubMed] [Google Scholar]
- 23.Horsley AR, Gustafsson PM, Macleod KA, et al. Lung clearance index is a sensitive, repeatable and practical measure of airways disease in adults with cystic fibrosis. Thorax 2008; 63: 135–40. [DOI] [PubMed] [Google Scholar]
- 24.Gustafsson PM, Aurora P, Lindblad A. Evaluation of ventilation maldistribution as an early indicator of lung disease in children with cystic fibrosis. Eur Respir J 2003; 22: 972–9. [DOI] [PubMed] [Google Scholar]
- 25.Aurora P, Gustafsson P, Bush A, et al. Multiple breath inert gas washout as a measure of ventilation distribution in children with cystic fibrosis. Thorax 2004; 59: 1068–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kent L, Reix P, Innes JA, et al. Lung clearance index: evidence for use in clinical trials in cystic fibrosis. J Cyst Fibros 2014; 13: 123–38. [DOI] [PubMed] [Google Scholar]
- 27.De Boeck K, Munck A, Walker S, et al. The effect of ivacaftor, a CFTR potentiator, in patients with cystic fibrosis and a non-G551D-CFTR gating mutation: the KONNECTION study [abstract WS1.1]. J Cyst Fibros 2014; 13(suppl 2): S1. [DOI] [PubMed] [Google Scholar]
- 28.Clinical Growth Charts. Centers for Disease Control and Prevention. Available at: www.cdc.gov/growthcharts/clinical_charts.htm. Accessed: 9 February 2015.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.