

Abstract
Protein catalyzed capture (PCC) agents are synthetic antibody surrogates that can target a wide variety of biologically relevant proteins. As a step towards developing a high-throughput PCC pipeline we report on the preparation of a barcoded rapid assay platform for the analysis of hits from PCC agent library screens. The platform is constructed by first surface patterning a micrometer scale barcode composed of orthogonal ssDNA strands onto a glass slide. The slide is then partitioned into microwells, each of which contains multiple copies of the full barcode. Biotinylated candidate PCCs from a click screen are assembled onto the barcode stripes using a complementary ssDNA-encoded cysteine-modified streptavidin library. This platform was employed to evaluate candidate PCC ligands identified from an epitope targeted in situ click screen against the two conserved allosteric switch regions of the KRas protein. A single microchip was utilized for the simultaneous evaluation of fifteen PCC candidate fractions under more than a dozen different assay conditions. The platform also permitted more than a 10-fold savings in time and a more than 100-fold reduction in biological and chemical reagents relative to traditional multi-well plate assays. The best ligand was shown to exhibit an in vitro inhibition constant (IC50) of ~24 μM.
Graphical Abstract
Protein-catalyzed capture agents (PCCs) have been demonstrated to mimic the epitope targeting ability and high avidity of monoclonal antibodies for a number of protein targets.1 Further, PCCs can be engineered to have combined properties that are difficult to achieve for biologics, such as combinations of physical and biological stability, or, in one example, cell penetration.2 State-of-the-art PCCs are identified by carrying out an in situ click screen3 of a synthetic, strategically modified polypeptide fragment (the synthetic epitope, or SynEp) of the protein target against a synthetic one-bead-one compound (OBOC) library of macrocyclic peptides. The comprehensive OBOC library typically contains the roughly two million sequences that result from using all combinations of an 18–20 amino acid basis set to construct the variable 5-mer portion of the peptide.
PCC lead compounds are identified through a single generation screen that will typically yield on the order of five to ten hits. Once identified, those hit peptides are tested for binding to the full-length protein, often in various levels of serum background and under different blocking conditions. These assays, which are carried out on 96-well plates using a sandwich Enzyme-Linked Immunosorbent Assay (ELISA) format, represent a limiting factor in the production of high quality PCCs. Characterization of the ligands may involve running 50 to 100 multi-point assays in series. In addition to being laborious, the assays also consume significant amounts of chemical and biological reagents. Finding a more efficient solution to carrying out such assays should be useful for the production of other artificial antibody-type ligands, such as other classes of peptides or aptamers.4–7
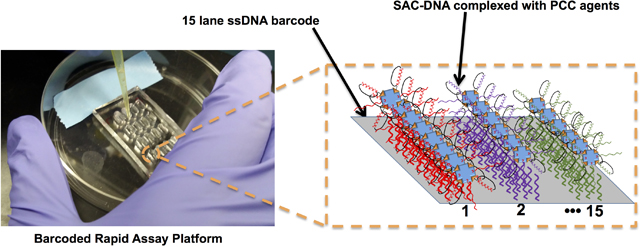
We report here on the barcoded rapid assay platform (B-RAP) (Figure 1), which is a microchip platform designed so that an entire set of candidate PCC ligands may be rapidly evaluated in parallel, using minimal quantities of reagents. Simultaneous testing of all PCCs under identical environments means that all assays are subject to the same uncertainties, which permits ready comparison of the EC50 values for the entire set of hit peptides. The B-RAP technology draws from the Nucleic Acid Cell Sorting (NACS)8 and DNA-Encoded Antibody Library (DEAL) methods.9–12 The B-RAP process starts with a microscope slide that is patterned, using microfluidic flow channels, with a distinct set of orthogonal ssDNA oligomers. The PCC candidates are prepared with a biotin label, and then assembled onto cysteine-modified streptavidin (SAC) scaffolds that have been labelled with complementary ssDNA oligomers.13–15 Once assembled, these reagents are combined into a cocktail, and assembled onto specific stripes of the barcode pattern using DNA hybridization.16 The microchip surface itself is partitioned into microliter volume wells, each of which contains multiple copies of the full barcode. The B-RAP technology can simultaneously assay a full panel of candidate PCCs over a range of target protein concentrations (or other conditions), such that the EC50 binding values for each candidate PCC are concurrently measured.
Figure 1. Overview of the B-RAP Technology.

A. The individual SAC-DNA conjugates B. The biotinylated PCC ligands are added to the SAC-DNAs and allowed to complex. C. The individual SAC-DNA-ligand solutions are pooled, and the cocktail is added to individual microwells on the DNA barcode. D. The SAC-DNA-ligand conjugates self-assemble with the DNA barcode to produce a fully assembled B-RAP assay.
We used the B-RAP technology to analyze the resulting hits from an epitope targeted in situ click screen against the Kirsten rat sarcoma (KRas) protein.17 Oncoprotein variants of KRas are implicated in driving ~20–25% of all human cancers including almost all pancreatic cancers.18 Oncogenic Ras proteins have largely evaded targeting by traditional therapeutic techniques,19–22 but recent work has shown that specific mutant isoforms may be targetable.23,24 We targeted conserved epitopes denoted Switch I (aa 25–40) and Switch II (aa 56–75), which are known to allosterically influence KRas activity.25 To our knowledge, these allosteric regions have not previously been targeted, perhaps because there is no obvious hydrophobic binding pocket. After screening, we tested the resultant hit compounds for their relative binding strengths. The strongest binders were then tested in a functional assay for in vitro KRas GTPase activity inhibition.
METHODS26
Preparation of the Barcode Rapid Assay Platform
DNA flow-patterned barcode chips, biotinylated peptides, and SAC-DNA were all used to assemble a miniaturized barcode of candidate PCCs for testing in a surface Immunofluorescent assay (IFA). Microfluidic flow patterning of 50 μm wide, 100 μm pitch ssDNA barcodes starts with adhering a polydimethylsiloxane (PDMS) microchannel mold onto a poly-L-lysine (PLL) coated glass microscope slide (Figure S1, Supporting Information). Reagents were flowed through the microchannels using a “pins-and-tubing-free” system that greatly simplified the preparation of barcoded microchips relative to the previous protocols (Figure S2 and Table S1, Supporting Information).27,28 The PDMS mold was patterned with microwells at each microchannel inlet (Figure S1A (i, ii), Supporting Information). Reagents (3–5 μL) are micropipetted into the wells, and two machined acrylic plates are clamped across the top and bottom of the inlet region. The top acrylic plate contains a cavity that encompasses all of the inlet microwells. This cavity is pressurized to fill the microchannels in about 20 minutes (Figure S2B, Supporting Information). The increased pressure tolerance of the design can enable the use of microchannels of widths as small as 10 μm. Initially 3 μL of poly-L-lysine (0.1% (w/w) in H2O) is flow patterned and dried overnight before flowing 5 μL of 300 μM of each ssDNA (Table S2) with 2 mM bis(sulfosuccinimidyl)suberate) (BS3) crosslinker. Approximately 20 to 25 DNA barcoded chips may be prepared in parallel (Figure S2C, Supporting Information). The bottom edge of the barcode is used to validate the coverage density and uniformity of the molecular patterns using fluorophore-labelled complementary ssDNA (Figures S2 and S3, Supporting Information). Once validated, the barcoded slides may be vacuum-sealed for up to six months storage before use (See Figure S4 and Table S3, Supporting Information).
The second component of the B-RAP technology, which is also independent of the specific identities of any PCC candidates to be tested, is the library of DNA-bound SAC (SAC-DNA) conjugates used to assemble individual biotinylated PCC candidates onto specific barcode lanes. The SAC protein (see Supplementary Methods) was conjugated with ssDNA strands complementary to the barcode DNA oligomers. This was done with N-succinimidly-4-formylbenzaldehyde (S-4FB) and maleimide 6-hydrazino-nicotinamide (MHPH), followed by fast protein liquid chromatography (FPLC) purification (Figure S5, Supporting Information).
The performance of the library of fifteen SAC-DNAs 16,29 was evaluated by hybridizing library elements onto the flow patterned ssDNA barcodes. The barcodes were then incubated with varying amounts of the fluorophore probe biotin-A20-Cy3 (Biotin*, 50–400 nM) (Figure S6, Supporting Information). The resulting surface fluorescence was measured and compared to the fluorescence signal from the bottom edge barcode validation region. The fluorescent output with 532 nm excitation (F532) of the captured biotinylated probe was lower than that of the validation region (45 to 65k fluorescence units (f.u.)), likely reflecting the size of the SAC protein relative to the Cy3 fluorophore.
KRas Protein Expression and Purification
The KRas protein isoform 4B was expressed from transformed BL21(D3) E. coli cells as a His6-tagged protein30 and purified by FPLC using a Ni-NTA resin (Figure S7, Supporting Information). The fractions with pure KRas protein were dialyzed into tris-buffered saline (TBS, pH = 7.4), aliquotted, and stored at −80.0 °C until needed.
Preparation of Switch I and Switch II SynEps and Scrambled SynEps
The synthetic epitopes (SynEp1 and SynEp2) were 11–12 amino acid polypeptides with sequences extracted from the allosteric switch regions of KRas (Figure 2 and Table S4, Supporting Information). The SynEp1 differs from the wild-type sequence as it is missing a valine residue. An azido click handle was added by substituting residue-similar azido-amino acids, as shown in Figure 2A. Rearranged version of the SynEps were also prepared and used in a pre-screen step to remove promiscuous binders. All epitopes were synthesized on biotin Novatag resin and purified using semi-preparative high-performance liquid chromatography (semi-prep HPLC). The appropriate fractions were identified using matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) (Figures S8–S12, Supporting Information). Each SynEp was dissolved in dimethyl sulfoxide (DMSO), quantified using a Nanodrop 2000 spectrophotometer, and stored at 4 °C until use.
Figure 2. Identification of SynEps for the dual epitope in-situ click screen, and the resulting PCC candidate hit sequences.
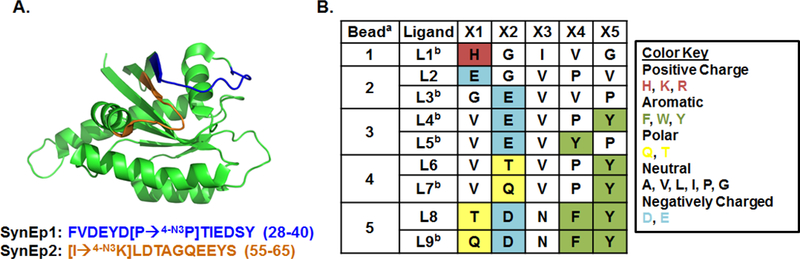
A. The allosteric KRas switch epitopes (pdb: 4dst) from which the SynEps were designed are highlighted in orange and dark blue, with SynEp sequences given below the protein structure. B. The hit sequences of the PCC candidates. Positions with high homology exhibit color pooling. a Similar sequences can arise from a single hit bead due to uncertainty in the Edman degradation peptide sequencing. b These ligands had two correct mass fractions following HPLC purification, arising from either epimerization or differential protonation. Both fractions were tested
Library Preparation and In-situ Library Click Screen
A comprehensive OBOC library of 5-mer variable peptide macrocycles, using an 18 amino acid basis set, was prepared as previously reported.1 The macrocyclic peptides were closed with a 1,4-triazole using Cu(I)-catalyzed click chemistry. These macrocycles were designed to present a propargylglycine click handle. The in situ library click screen was a dual SynEp version of a previously reported protocol.1 After removing the beads that bound to the scrambled SynEps during a pre-clear screen the remaining library was incubated with both SynEp1 and SynEp2 (Experimental Methods, Supporting Information). After incubating with an anti-biotin capture antibody and an alkaline-phosphatase conjugated secondary antibody, the hit beads were identified by their deep purple color. The isolated hit beads were stored at RT in 0.1 M hydrochloric acid. Just prior to sequencing by Edman degradation, the beads were decolorized in N-methyl 2-pyrrolidone (NMP). The hit compounds (Table S4, Supporting Information) were then scaled up on biotin Novatag resin following previously established protocols,1 purified, lyophilized, reconstituted in DMSO, quantified, and then stored at 4 °C until ready for use.
Surface Immunofluorescent Assays on the Barcoded Rapid Assay Platform
The barcode patterned microchip surface was partitioned into 16 individual microwells using a pre-fabricated PDMS slab. Individual biotinylated PCC candidates were complexed to specific SAC-DNA conjugates, combined into a cocktail, and then self-assembled, via DNA hybridization, onto designated barcode stripes (Figure 1). Incubation with a specific concentration of the target protein preceded incubation with a primary capture antibody and then a fluorophore-conjugated secondary detection antibody. During assay execution, each well represents a different target concentration or assay condition. Once developed, the fluorescence of the barcodes is digitized using a GenePix 4400A array scanner, with an excitation laser power optimized to a power level of 40% (60 W), which maximizes detection sensitivity while also minimizing signal saturation. Data extraction occurs using 10 μm radius circles, taken along the length of a barcode stripe. A fluorescence signal representing the average of all the pixels within a given circle is collected. A total of ten circles (data-blocks) are measured along a 180 μm span of the middle portion for each individual barcode lane in a given well. After extraction the data is background corrected. The background signal arises from (a) non-specific binding of the primary and secondary antibodies (independent of [KRas]), but can vary across different barcode stripes), and (b) non-specific binding of KRas protein ([KRas] dependent). Background (a) was assessed by measuring the average signal in the null protein well for each stripe. Background (b) was assessed by measuring the average fluorescence for the dummy ligand (Biotin* probe) that was in each well. The background subtracted data was then graphed in Graphpad Prism 7 and fitted to a sigmoidal curve (Hill coefficient = 1).
Epitope Binding Study
The biotinylated ligand fractions L1a, L2, and L8 and blank (biotin-PEG5-NHAc) were immobilized to a black Neutravidin coated 96-well plate for two hours before blocking overnight at 4 °C. The immobilized ligands were then incubated with 25 μM of the Cy3-labeled polypeptide corresponding to either Switch I or Switch II. The fluorescence was read after filling the wells with 200 μL PBS (excitation λ= 532 nm, emission λ= 568 nm, filter cut-off λ= 550 nm, 6 reads/well), and doubly background corrected by first subtracting the average RFU from wells without any SynEp followed by subtracting the average RFU from the wells containing blank ligand. The data was then plotted in Graphpad Prism 7.
Measuring the Effect of the Allosteric Ligands on the Intrinsic KRas GTPase Activity
KRas inhibition assays were carried out using a GTPase Glo Assay kit from Promega (Figure S19, Supporting Information). Each candidate inhibitor PCC was initially tested by combining a concentration series of the ligand with 10 μM KRas protein in an opaque white 96-well plate and incubated with 5 μM 5’-guanosine triphosphate (GTP) for two hours The remaining GTP was converted to 5’-adenosine triphosphate (ATP) over 30 minutes using the reconstituted GTPase Glo reagent before a ten-minute incubation with the detection reagent. Chemiluminescence was measured using a Flexstation 3 plate reader (All wavelengths mode, 500 ms integration), and plotted using Graphpad Prism 7. A full inhibition curve of the most potent inhibitor was then generated using a four-hour incubation with GTP and a 2.5 μM to 100 μM concentration range. All measurements were done in triplicate.
RESULTS AND DISCUSSION
Optimizing B-RAP Assay conditions
The in situ click screen against the Switch I and II KRas protein SynEps (Figure 2A) yielded five beads from which nine candidate sequences were determined (Figure 2B). Biotinylated candidate ligands were then tested using a single-point IFA with the B-RAP technology (Figure S13, Supporting Information) to identify appropriate blocking conditions. Modification of the protein incubation solution to include the nonionic surfactant Polysorbate 20 (Tween20) was found to minimize non-specific binding between the KRas protein and the unmodified PLL surface.
Validation of the B-RAP Technology
Following optimization of the assay conditions the B-RAP technology was subjected to statistical tests to assess the variance in assay results measured within an individual microwell, between microwells on the same chip, and between different microchips. The average percent coefficient of variation (%CV) seen along an individual barcode stripe in the wells above background (500 nM to 400 μM KRas) using the values from the data-block extraction method was ~15%. Each microwell contains between two to three full copies of the DNA barcode. For the same barcode lane in the different full barcode sets in the same microwell, the fluorescence output was measured to have an average %CV of ~14% (Figure S14A, Supporting Information). The %CV between wells on the same microchip run under identical conditions was ~9%. The average %CV for identical barcode lanes between two separate platforms run in parallel by different users was ~18% with an average %CV of ~15% for the 1 μM to 400 μM range of KRas protein (Figure S14B, Supporting Information). Additionally, to validate that our data-block extraction method of a portion of the barcode lane captured the F635 for the entire barcode lane the average F635 from a full-line line scan of the barcode lane was compared to the average F635 resulting from our data-block extraction method. The values from the full-line scan were contained within two standard deviations of the data-block extraction’s average F635 (Figure S15, Supporting Information). This was compared to taking the measurement of individual pixels along the entirety of the barcode lanes in one full set of the 10 μM well for one plate (Figure S16A, Supporting Information) then graphing to find the centroid region (Figure S16B, Supporting Information), which is the region that is roughly stable in fluorescent intensity. The average F635, the standard deviation, and the %CV for each lane was calculated for the full lane, the centroid region of the lane, and the different parts of the centroid region (Table S5, Supporting Information). The full lane %CVs were in the 20–30% range, while the % CVs of the centroid regions were 10–20%. This arises from edge effects near the microwell walls. Assays of individual PCC candidates (different barcode stripes) collected within a single microwell, and so representing a single point of a binding curve, could be readily distinguished (Table S7, Supporting Information). These results indicated that the centroid region of a barcode stripe yielded the most reliable data, but also that assay results from different microwells, or different B-RAP chips, could be readily compared.
Measuring the EC50 of the Allosteric Binding PCC Ligands
After characterizing the B-RAP technology, we used the platform to generate complete binding curves for 15 PCC ligand fractions simultaneously, (Figure 3B, for the binding curves without dummy ligand correction see Figure S17, Supporting Information) and determined the EC50 values for each (Figure 3C) (for goodness of fit measurements for the curves see Table S8, Supporting Information). These measurements were comprised of a 13-point concentration series, with each point collected in decaplicate. The EC50 values enabled the ranking of the ligands, and the best binders were identified to be L1, L2, and L8. The true amino acid sequences for each hit peptide were also distinguished from the artifact sequences that arose from sequencing uncertainties. The true on-bead sequences for the hit beads are identified as L1, L2, L5, L7, and L8.
Figure 3. Full binding curves for L1-L9 and the corresponding EC50 values.

A. The raw scan of the barcode after running the KRas protein binding curves. B. The worked up graphs for the allosteric PCC ligands. C. The EC50 values derived from the B-RAP technology and the multi-well ELISA technology. aNot calculated due to non-saturation of graph. bSelect ligands that had the uncertainty for their EC50 values greater than twice their EC50 value and thus their binding curves were considered poorly resolved by the multi-well ELISA.
We also provide a comparison of the EC50 values from multi-well ELISA assays (triplicate measurements). While both assays identify L1a and L1b as the strongest binders, the ELISA assays are significantly noisier, with binding saturation not achieved for several ligands (for the ELISA curves see Figure S18, Supporting Information). The poor relative performance of the ELISAs arises from a few factors. First, the ligands tested are relatively weak (μM-level) binders, and this exacerbates certain issues associated with the ELISAs. ELISAs are absorbance measurements, and thus have a significantly smaller dynamic range than the B-RAP fluorescence assays. Second, ELISA signal arises from enzymatic amplification, while the B-RAP assays are not amplified. For weak binders, amplified assays tend to be noisy, as both signal and noise are amplified. The improved relative sensitivity and statistics afforded by the B-RAP technology readily enables the comparative evaluation of these relatively weak KRas binders.
Further Characterization of the Allosteric Ligand Leads
The allosteric lead PCCs L1a, L2, and L8 were then subjected to further characterization of their binding to the switch regions, and for their selectivity for Ras proteins.
The ligands were identified through a dual-SynEp in-situ library click screen and could bind to either switch 1, corresponding to SynEp1, and switch 2, corresponding to SynEp2. The polypeptide sequences for both SynEp1 and SynEp2 without the azido-click handle substitution, called 1 and 2, respectively, were prepared and coupled to Cy3 dye through a short PEG2-linker. An IFA epitope binding study (Figure S19, Supporting Information) revealed that the allosteric PCC leads all bind to switch 1, which is the more conformationally dynamic switch.
A protein selectivity assay using the B-RAP technology was carried out with the recombinant human proteins WT KRas, WT HRas, and WT MEK1 (Figure S20, Supporting Information) and allosteric leads L1a and L2. The Ras proteins are 100% conserved at the allosteric switch regions, and as expected the ligands bound both Ras proteins with comparable selectivity. The binding for MEK1, however, was sharply reduced. MEK1 is a non-homologous kinase that is downstream of KRas in the MAPK signaling pathway.
Testing the Allosteric Ligands as Inhibitors of KRas GTPase Activity
The ligands identified here were screened for binding to epitopes that exhibit structural fluctuations as the KRas protein switches between its inactive 5’-guanosine diphosphate (GDP)-bound form and its active GTP-bound form.25 Consequently, the best three ligand fractions L1a, L2, and L8 were probed in a functional, solution phase assay for their ability to disrupt the intrinsic GTPase enzymatic activity of KRas protein (Figure 4A). This assay measures the enzymatic conversion of GTP to GDP by KRas – a process that can potentially be inhibited. After incubation, an added GTPase Glo™ reagent converts the remaining GTP to ATP, and the ATP is converted into a chemiluminescent signal. Thus, higher chemiluminescence translates to lower KRas enzymatic activity. For the measurements, a fixed [KRas protein] is incubated with varying ligand concentrations. A concentration of 10 μM KRas protein was selected after generating a standard curve for the intrinsic KRas GTPase activity (Figure S21A, Supporting Information). KRas is a slow acting enzyme, so a KRas/PCC incubation time of two hours was used for the initial survey scans. However, four-hour incubation times were used for the higher resolution data (Figure 4B). The survey assays indicated that all three ligands exhibited an inhibitory effect on the KRas protein’s GTPase activity, but L2 was the most potent (Figure S21, Supporting Information). Thus, the modulation of KRas activity by L2 was recorded with an expanded concentration range (Figure 4B). We found that L2 switches from weakly activating to strongly inhibiting above 20 μM. Less than 5% of the rGTP was hydrolyzed in the L2-only (no KRas) wells, and ~61% was hydrolyzed in the KRas-only wells. This result confirms that L2 lacks any innate GTPase enzymatic activity (Figure S22, Supporting Information). The sharp transition in the titration curve fits to a Hill coefficient of ~10, which suggests that upon full occupancy of the allosteric switch region, KRas flips into an inactive conformation. An IC50 value was 24.0 ± 1.2 μM for L2. The sharp L2 concentration-dependent switch between activating and inhibiting may involve dimers of L2. We did, in fact, identify that L2 does form dimers in solution, at least at concentrations above 10 μM (Figure S23, Supplementary Information), although we were unable to quantify the relative abundance of the dimers Regardless, the measured IC50 value indicates that L2 provides an excellent starting point for a first-generation allosteric inhibitor against this challenging target. We are currently pursuing chemical optimizations of L2 to develop it into a more potent inhibitor.
Figure 4. Measuring the inhibition of the GTPase activity of KRas by L2.
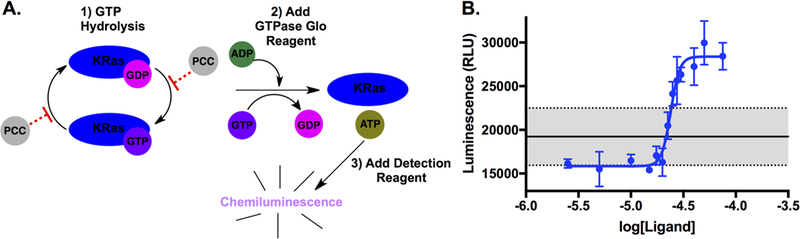
A. The KRas protein GTPase assay involves incubation with rGTP and hydrolysis by the KRas protein followed by conversion of the remaining GTP into ATP. A detection reagent uses the ATP to generate a chemiluminescent output. The PCC agents bind to allosteric regions of KRas and could either enhance or hinder its intrinsic GTPase activity. B. The inhibition curve generated with L2 when incubated with 10 μM KRas protein. The solid line represents the intrinsic GTPase activity of 10 μM KRas protein in the absence of ligand with one standard deviation shaded between the two dotted lines.
CONCLUSIONS
We report on the development and use of a barcoded rapid assay microchip, which allows for the simultaneous evaluation of fifteen PCC candidate ligands in up to sixteen unique assay conditions, with significant associated savings in terms of both time and reagent use (Table 1). In a single day the B-RAP technology was applied to identify the best allosteric KRas binders from a pool of 15 ligands identified from a dual SynEp PCC library in situ click screen. The B-RAP technology is designed to yield an equilibrium-based EC50 value for assessing relative binding strengths. For a number of PCCs, the EC50 value provides an upper limit for the dissociation constant (KD).1 Importantly, relative binding affinities can provide guidance for selecting ligands for further quantitative characterizations, such as the solution phase KRas activity assay explored here. To this end, the B-RAP technology works well. A comparison of the B-RAP assay metrics relative to standard 96-well plate ELISAs is presented (Table 1). Extending this platform to evaluating PCC binders, or other ligand classes, against new protein targets should work well, requiring only an optimization of both concentration ranges (determined by the candidate ligands) and blocking conditions (typically determined by the protein target).31
Table 1.
Comparison of the capacity, reagent quantities used, and assay times for multi-well ELISA plates relative to the barcoded rapid assay platform.
| Criteria | ELISA Platform |
B-RAP Chip |
|---|---|---|
| Full binding curves per Assay | 1 | 15 |
| Relative amount PCC per binding curve (nmol) | 7 | 0.15 |
| Relative amount protein per binding curve (nmol) | 300 | 2.7 |
| Protein concentration points per assay | 12 | 16 |
| Assay run time (h) | 10–36 | 8–10 |
| # Data Points per platform | 96 | 2400 |
Using the B-RAP platform coupled with the epitope-targeted in situ click screening approach, we identified a PCC ligand lead (L2) that serves as an allosteric inhibitor of the intrinsic GTPase enzymatic activity of KRas, with an IC50 value of around 20 μM. L2 is a first-generation ligand, and, as such, can surely be optimized, via medicinal chemistry methods, for increased potency and selectivity. Thus, given the well-known challenging nature of KRas as a drug target, L2 provides an excellent starting point for developing a more potent inhibitor.
Supplementary Material
ACKNOWLEDGMENTS
Dr. Songming Peng is acknowledged for providing transformed E. coli cells used in the preparation of SAC protein and helpful discussions. We acknowledge Anvita Mishra and Rachel Ng for help in the preparation of some peptides and SynEps. We gratefully acknowledge the following agencies and foundations: The B-RAP technology was largely developed with support from the Institute for Collaborative Biotechnologies 6.2 program (J.R.H., # W911NF-09-D-0001). The KRas protein work was supported by the National Cancer Institute (J.R.H., 1U54 CA199090–01) and the Jean Perkins Foundation. The NSF is acknowledged for fellowship support (A.M.M., Grant DGE-1144469). The results in this paper were generated through the use of the Caltech Center for the Chemistry of Cellular Signaling, the Caltech Center for Catalysis and Chemical Synthesis, the Caltech Protein Expression and Purification Center, and the Caltech CCE Multi-User Mass Spectrometry Laboratory, and the University of Washington mass spectrometry facility.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Material sources, experimental protocols, and supplemental figures (PDF). The Supporting Information is available free of charge on the ACS Publications website.
The authors declare the following competing financial interest(s): J.R.H. is a board member of InDi Molecular, which is a company seeking to commercialize the PCC technology.
REFERENCES
- (1).Das S; Nag A; Liang J; Bunck DN; Umeda A; Farrow B; Coppock MB; Sarkes DA; Finch AS; Agnew HD; Pitram S; Lai B; Yu MB; Museth AK; Deyle KM; Lepe B; Rodriguez-Rivera FP; McCarthy A; Alvarez-Villalonga B; Chen A; Heath J; Stratis-Cullum DN; Heath JR Angew. Chemie Int. Ed. 2015, 54 (45), 13219–13224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Farrow B; Wong M; Malette J; Lai B; Deyle KM; Das S; Nag A; Agnew HD; Heath JR Angew. Chemie Int. Ed. 2015, 54 (24), 7114–7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Agnew HD; Rohde RD; Millward SW; Nag A; Yeo WS; Hein JE; Pitram SM; Abdul Ahad Tariq V; Burns AM; Krom RJ; Fokin VV; Barry Sharpless K; Heath JR Angew. Chemie Int. Ed. 2009, 48 (27), 4944–4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Yüce M; Ullah N; Budak H Analyst 2015, 140 (16), 5379–5399. [DOI] [PubMed] [Google Scholar]
- (5).Jost C; Plückthun A Current Opinion in Structural Biology. 2014, pp 102–112. [DOI] [PubMed] [Google Scholar]
- (6).Mascini M; Palchetti I; Tombelli S Angew. Chemie - Int. Ed. 2012, 51 (6), 1316–1332. [DOI] [PubMed] [Google Scholar]
- (7).Csordas AT; Jørgensen A; Wang J; Gruber E; Gong Q; Bagley ER; Nakamoto MA; Eisenstein M; Soh HT Anal. Chem. 2016, 88 (22), 10842–10847. [DOI] [PubMed] [Google Scholar]
- (8).Kwong GA; Radu CG; Hwang K; Shu CJ; Chao M; Koya RC; Comin-Anduix B; Hadrup SR; Bailey RC; Witte ON; Schumacher TN; Ribas A; Heath JR J. Am. Chem. Soc. 2009, 131 (28), 9695–9703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bailey RC; Kwong GA; Radu CG; Witte ON; Heath JR J. Am. Chem. Soc. 2007, 129 (7), 1959–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Boozer C; Ladd J; Chen S; Yu Q; Homola J; Jiang S Anal. Chem. 2004, 76 (23), 6967–6972. [DOI] [PubMed] [Google Scholar]
- (11).Kozlov IA; Melnyk PC; Stromsborg KE; Chee MS; Barker DL; Zhao C Biopolymers 2004, 73 (5), 621–630. [DOI] [PubMed] [Google Scholar]
- (12).Adler M; Wacker R; Bootltink E; Manz B; Niemeyer CM Nat. Methods 2005, 2 (2), 147–149. [Google Scholar]
- (13).Sano T; Cantor CR Proc. Natl. Acad. Sci. 1990, 87 (1), 142–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Reznik GO; Vajda S; Cantor CR; Sano T Bioconjug. Chem. 2001, 12 (6), 1000–1004. [DOI] [PubMed] [Google Scholar]
- (15).Ramachandiran V; Grigoriev V; Lan L; Ravkov E; Mertens SA; Altman JD J. Immunol. Methods 2007, 319 (1–2), 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shin YS; Ahmad H; Shi Q; Kim H; Pascal TA; Fan R; Goddard WA; Heath JR ChemPhysChem 2010, 11 (14), 3063–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Cooper GM Science (80-. ). 1982, 217 (4562), 801–806. [DOI] [PubMed] [Google Scholar]
- (18).Cox AD; Fesik SW; Kimmelman AC; Luo J; Der CJ Nature Reviews Drug Discovery. 2014, pp 828–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Whitehead RP; Mccoy S; Macdonald JS; Rivkin SE; Neubauer MA; Dakhil SR; Lenz H-J; Tanaka MS; Abbruzzese JL Invest. New Drugs 2006, 24, 335–341. [DOI] [PubMed] [Google Scholar]
- (20).Macdonald JS; Mccoy S; Whitehead RP; Iqbal S; Wade Iii JL; Giguere JK; Abbruzzese JL Invest. New Drugs 2005, 23, 485–487. [DOI] [PubMed] [Google Scholar]
- (21).Winquist E; Moore MJ; Chi KN; Ernst DS; Hirte H; North S; Powers J; Walsh W; Boucher T; Patton R; Seymour L Urol. Oncol. 2003, 23 (3), 143–149. [DOI] [PubMed] [Google Scholar]
- (22).Sharma S; Kemeny N; Kelsen DP; Ilson D; O ‘reilly E; Zaknoen S; Baum C; Statkevich P; Hollywood E; Zhu Y; Saltz LB Ann. Oncol. 2002, 13, 1067–1071. [DOI] [PubMed] [Google Scholar]
- (23).Ostrem JM; Peters U; Sos ML; Wells JA; Shokat KM Nature 2013, 503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Sakamoto K; Kamada Y; Sameshima T; Yaguchi M; Niida A; Sasaki S; Miwa M; Ohkubo S; Sakamoto J ichi; Kamaura M; Cho N; Tani A Biochem. Biophys. Res. Commun. 2017, 484 (3), 605–611. [DOI] [PubMed] [Google Scholar]
- (25).Hall BE; Yang SS; Boriack-Sjodin PA; Kuriyan J; Bar-Sagi DJ Biol. Chem. 2001, 276 (29), 27629–27637. [DOI] [PubMed] [Google Scholar]
- (26).For detailed protocols of all experiments see the supporting information file.
- (27).Yu J; Zhou J; Sutherland A; Wei W; Shin YS; Xue M; Heath JR Annu. Rev. Anal. Chem. 2014, 7 (1), 275–295. [DOI] [PubMed] [Google Scholar]
- (28).Wei W; Shin YS; Xue M; Matsutani T; Masui K; Yang H; Ikegami S; Gu Y; Herrmann K; Johnson D; Ding X; Hwang K; Kim J; Zhou J; Su Y; Li X; Bonetti B; Chopra R; James CD; Cavenee WK; Cloughesy TF; Mischel PS; Heath JR; Gini B Cancer Cell 2016, 29 (4), 563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Xue M; Wei W; Su Y; Kim J; Shin YS; Mai WX; Nathanson DA; Heath JR J. Am. Chem. Soc. 2015, 137 (12), 4066–4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Boriack-Sjodin PA; Margarit SM; Bar-Sagi D; Kuriyan J Nature 1998, 394 (6691), 337–343. [DOI] [PubMed] [Google Scholar]
- (31).Gibbs J ELISA Tech. Bull. Corning Inc. Life Sci. Kennebunk, ME 2001, No. 3, 1–6. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.