

Abstract
Secondary coordination sphere interactions are critical in facilitating the formation, stabilization, and enhanced reactivity of high-valent oxidants required for essential biochemical processes. Herein, we compare the C–H bond oxidizing capabilities of spectroscopically characterized synthetic heme iron(IV) oxo complexes, F8Cmpd-II (F8 = tetrakis(2,6-difluorophenyl)porphyrinate), and a 2,6-lutidinium triflate (LutH+) Lewis acid adduct involving ferryl O-atom hydrogen-bonding, F8Cmpd-II(LutH+). Second-order rate constants utilizing C–H and C–D substrates were obtained by UV–vis spectroscopic monitoring, while products were characterized and quantified by EPR spectroscopy and gas chromatography (GC). With xanthene, F8Cmpd-II(LutH+) reacts 40 times faster (k2 = 14.2 M−1 s−1; −90 °C) than does F8Cmpd-II, giving bixanthene plus xanthone and the heme product [F8FeIIIOH2]+. For substrates with greater C–H bond dissociation energies (BDEs) F8Cmpd-II(LutH+) reacts with the second order rate constants k2(9,10-dihydroanthracene; DHA) = 0.485 M−1 s−1 and k2(fluorene) = 0.102 M−1 s−1 (−90 °C); by contrast, F8Cmpd-II is unreactive toward these substrates. For xanthene vs xanthene-(d2), large, nonclassical deuterium kinetic isotope effects are roughly estimated for both F8Cmpd-II and F8Cmpd-II(LutH+). The deuterated H-bonded analog, F8Cmpd-II(LutD+), was also prepared; for the reaction with DHA, an inverse KIE (compared to F8Cmpd-II(LutH+)) was observed. This work originates/inaugurates experimental investigation of the reactivity of authentic H-bonded heme-based FeIV═O compounds, critically establishing the importance of oxo H-bonding (or protonation) in heme complexes and enzyme active sites.
Graphical Abstract
INTRODUCTION
In heme metalloenzymes, specific amino acid residues are utilized in either the distal or the proximal pocket to provide favorable hydrogen-bonding interactions with iron-bound ligands, influencing the electronics (e.g., redox potential, acidity, geometry) of the iron metal center to facilitate the formation, stability, and reactivity of high-valent oxidants (e.g., iron(IV) oxo porphyrin π-cation radical species, Compound-I (Cmpd-I), or iron(IV) oxo porphyrin species, Compound-II (Cmpd-II)), the reactive intermediates commonly responsible for carrying out their essential biochemical functions (Figure 1).1–5 Secondary sphere coordination interactions can also be key for tuning the oxidative capabilities of the iron(IV) oxo moiety to enhance reactivity toward difficult substrates, allowing for efficient catalysis.5–9 The heme proteins catalase and cytochrome P450 highlight the importance of hydrogen-bonding with ferric hydroperoxo intermediates allowing heterolytic cleavage of the O–O bond to concomitantly release water and form the key intermediate, Cmpd-I, responsible for the necessary two-electron oxidation of the reactive oxygen species H2O2 in catalases or substrate oxygenation in P450.1,3,5,10 Initial substrate C–H hydrogen-atom abstraction from the cysteine-ligated Cmpd-I complex in cytochrome P450 yields protonated Cmpd-II (FeIV–OH, an example of such a species is depicted in Figure 1B), whose stability relies on hydrogen-bonding interactions with nearby amino acid residues to prevent deleterious enzymatic oxidative damage.8,9,11–13 Investigations are needed to fully understand and take advantage of H-bonding interactions and their implications.
Figure 1.

Hydrogen-bonding interactions in (A) cytochrome c peroxidase (CcP) Cmpd-I wherein the iron(IV) oxo group is coupled to a Trp residue radical (PDB 4CVJ)9 and (B) ascorbate peroxidase (APX) protonated Cmpd-II (PDB 5JPR).8 It is of note in A the hydrogen-bonding between the oxo ligand with distal residues Trp51 and Arg48 and with interstitial water molecules. In B hydrogen-bonding interactions are seen between Trp51 and the oxo ligand as well as between the hydroxide proton and an interstitial water and Arg38. Also, there are proximal hydrogen-bonding interactions in CcP and APX involving the histidine axial ligands in CcP (His175 with Asp235 and Trp191) and in APX (His163 with Asp208 and Trp179). Crystal structures were obtained from the Protein Data Bank (PDB) and generated in PyMOL.14
Due to the transient nature of strong heme-based biological oxidants, understanding the factors that govern their electronics can be difficult. Synthetically modeling ferryl species allows aspects of design and control that differ from or are not always feasible within enzymatic systems. Thus, systematic change of variables (e.g., substituents around the periphery of the porphyrin, axial ligation, environment around the oxo ligand, etc.) can be synthetically designed, incorporated and investigated. Such studies can allow us to gain fundamental chemical insights relevant to enzyme structure and reaction mechanisms. They could also potentially lead to the development of highly efficient and selective biomimetic heme catalysts15–19 to carry out similar oxidative transformations seen in the enzyme, such as hydroxylation and epoxidation,20–25 and, further, these systems could be promising for energy research.
Many studies have examined the addition of Brønsted acids to synthetic nonheme iron(IV) oxo complexes, which seemed to protonate (or act as a hydrogen-bonding source) to the oxo ligand.26–32 Interestingly, it has been observed that in the presence of a Lewis acid such as a proton source26–29 (or a nonredox active metal ion33–36), reactivity is enhanced toward such processes as oxygen-atom transfer,27,35 electron transfer,26–29,34,35 C–H bond cleavage,28,35 or epoxidation reactions29 (wherein the Lewis acid enhances the electron-acceptor capabilities of the iron(IV) oxo complexes leading to increased substrate oxidation rates).
For heme-based systems, limited studies have been reported on the effect of H-bonding on synthetic heme high-valent iron oxo species. Groves and co-workers explored the effect of various pH’s on the reactivity of synthetic heme Cmpd-I complexes, showing enhanced C–H cleavage at lower pH.37 Upon initial substrate hydrogen-atom abstraction (HAA) the authors suggested a protonated Cmpd-II species is formed under acidic conditions; however, the exact electronic state of this complex is a bis-aqua iron(III) porphyrin cation radical and as such is not a distinct iron(IV) hydroxide compound. There have been no studies that have examined the reactivity between Brønsted acids and synthetic heme iron(IV) oxo (Cmpd-II) complexes despite the importance of hydrogen-bonding interactions for high-valent intermediates in the catalytic cycles of heme proteins. However, the increase in the driving force for HAA reactions by synthetic high-valent iron(IV) oxo complexes by the addition of a proton (from the medium) during substrate oxidation as observed by Groves and co-workers37 suggests that addition of Brønsted acids to synthetic heme iron(IV) oxo (Cmpd-II) complexes will also enhance the reactive complexes’ oxidative capabilities as will be demonstrated herein (vide infra).
As described elsewhere, Solomon, Karlin, and co-workers reported the characterization (using UV–vis, 2H NMR, X-band EPR, 57Fe Mössbauer, Fe X-ray absorption (XAS), and 54/57Fe resonance Raman (rR) spectroscopies) of the synthetic high-valent heme iron(IV) oxo complexes: F8Cmpd-II (F8 = tetrakis(2,6-difluorophenyl)porphyrinate) and the resulting hydrogen-bonded ferryl species in the presence of the protic Lewis acid, 2,6-lutidinium triflate (LutH+), F8Cmpd-II-(LutH+).38,39 Mössbauer, rR, and XAS spectroscopic data suggest that addition of 1 equiv of LutH+ to F8Cmpd-II did not result in full proton transfer from the acid to the oxo ligand, but an H-bonding interaction does occur, yielding the first synthetic heme hydrogen-bonded iron(IV) oxo (Cmpd-II) adduct.40 This is characterized by an observed slight elongation of the iron–oxo bond (versus authentic FeIV–OH species in heme proteins such as chloroperoxidase, ascorbate peroxidase, peroxygenase, or cytochrome P450, which contain considerably longer Fe–O bond lengths and have more significant effects on the Mössbauer parameters).1,6,41–43 Resonance Raman spectroscopy revealed a shift in the Fe═O stretching frequency from 833 to 819 cm−1. Another change in properties coming about from H-bonding of this Lewis acid (LA) to the oxo atom, compared to the properties of F8Cmpd-II, is that the reduction potential of the iron(IV) oxo LA adduct significantly increased, making it a better oxidant by almost 1 V, and thereby experimentally supporting the as yet untested (until this present work) hypothesis that H-bonding interactions can enhance the substrate reactivity of heme Cmpd-II species.38
Herein, efforts are continued to investigate the effect of LutH+ on F8Cmpd-II by examining the complexes’ oxidative capabilities with C–H substrates containing moderate bond dissociation energies (BDEs). Reactivity studies were followed by UV–vis spectroscopy to determine if the hydrogen-bonding interaction from LutH+ with the oxo atom of the synthetic high-valent heme complex enhanced the rate of hydrogenatom abstraction from various C–H substrates (and their isotopically labeled C–D analogs). Further studies were also carried out with C–D substrates to quantify kinetic isotope effects (KIEs) that may suggest a tunneling mechanism. The deuterated analog, F8Cmpd-II(LutD+), was also prepared to examine its reactivity with C–H substrates (and obtain a KIE) versus the proteo analog to provide evidence for the role of H-bonding in these reactivity studies. This work experimentally highlights the importance of H-bonding on synthetic heme iron(IV) oxo complexes and the enhanced reactivity that results, thus also pointing out the benefits of synthetic models to probe the effects of secondary coordination effects or to gain mechanistic insights.
RESULTS AND DISCUSSION
Reactivity of F8Cmpd-II and F8Cmpd-II(LutH+) with Various C–H Substrates.
F8Cmpd-II and F8Cmpd-II-(LutH+) were prepared as reported in detail elsewhere.38 A solution of F8FeII in a mixture of 1:9 2-methyltetrahydrofuran (MeTHF):toluene was cooled to −90 °C and allowed to equilibrate for 10 min. Subsequently, 1 equiv of the oxidant m-chloroperoxybenzoic acid (mCPBA) was added to form F8Cmpd-II; further addition of 1 equiv of LutH+ generated F8Cmpd-II(LutH+) (Scheme 1). A spectral change via UV–vis spectroscopy was observed changing from absorption maxima at 422, 542 nm to 415, 544 nm upon addition of mCPBA to F8FeII (Figure S3, Scheme 1). A rather slight further change is seen when 1 equiv of LutH+ is added to F8Cmpd-II, from 415, 544 nm to 413, 546 nm (Figure S4, Scheme 1); however, as mentioned above, other physical properties of F8Cmpd-II(LutH+) are significantly changed from those observed for F8Cmpd-II. Various C–H substrates were added to the ferryl complexes, and their reactions were monitored via UV–vis spectroscopy to determine the oxidative capabilities of the iron(IV) oxo species.
Scheme 1.
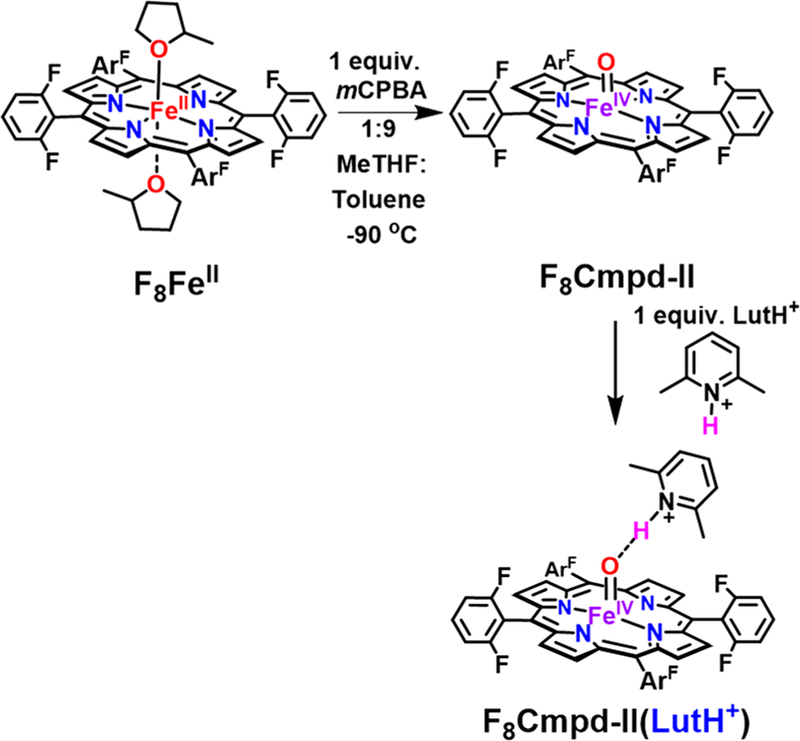
Formation of F8Cmpd-II and Protic Lewis Acid (LutH+-triflate) Adduct F8Cmpd-II(LutH+) in 1:9 MeTHF:toluene at −90 °C
Addition of excess xanthene, which has a C–H bond dissociation energy of 75 kcal/mol,44–47 to both F8Cmpd-II and F8Cmpd-II(LutH+) resulted in their decay over time to the ferric aqua complex, [F8FeIIIOH2]+, indicated by the appearance of characteristic UV–vis spectral ferric features such as the multiple peaks between 500 and 600 nm and the broad low-energy band from 700–1000 nm48 (Figures 2A, S5, and S8). X-Band EPR spectroscopy at 10 K was then utilized to support the ferric oxidation state of the final inorganic product of these substrate oxidation reactions. As previously reported, the complexes F8Cmpd-II and F8Cmpd-II(LutH+) are EPR silent, consistent with their S = 1 spin state.38 However, addition of 20 equiv of xanthene to a 2 mM solution of F8Cmpd-II or F8Cmpd-II(LutH+) resulted in an EPR-active spectrum with characteristic features at g = 6 and 2, which can be ascribed to a typical high-spin ferric species (Figures 2B, S6, and S9). The UV–vis and EPR spectroscopic properties of authentically generated [F8FeIIIOH2]+ match those described here and in Figure 2; see below and the SI for further details.
Figure 2.

UV–vis (A) and EPR (B) spectra monitoring the decay of F8Cmpd-II(LutH+) upon addition of 50 or 20 equiv of xanthene, respectively, at −90 °C in 1:9 MeTHF:toluene and the subsequent formation of [F8FeIIIOH2]+ over time (blue to green).
Substrates of higher bond dissociation energies were then added to the heme iron(IV) oxo complexes; the results are summarized in Figure 3. No reaction was observed upon addition of 9,10-dihydroanthracene (DHA, BDE = 78 kcal/mol49) to F8Cmpd-II, even after monitoring the reaction mixture for 10 h exclusively at −90 °C.50 However, addition of DHA and fluorene (BDE = 80 kcal/mol46,47) to F8Cmpd-II(LutH+) resulted in identical UV–vis spectral changes (Figures S11 and S14) as observed in its reaction with xanthene, forming the same ferric aqua final product as also determined by the ferric signal (g = 6 and 2) observed via EPR spectroscopy (Figures S12 and S15). No reaction occurred, as shown by the lack of UV–vis spectral changes and lack of ferric features via EPR spectroscopy upon addition of triphenylmethane (BDE = 81 kcal/mol) to F8Cmpd-II(LutH+). These results support the suggestion that the same mechanism occurs when xanthene, DHA, or fluorene is added to F8Cmpd-II(LutH+) in that initial HAA transforms F8Cmpd-II(LutH+) to F8FeIIIOH (H atom from the substrate), which then is protonated to form [F8FeIIIOH2]+ (vide infra). The source of the second proton in [F8FeIIIOH2]+ could be either the LutH+ which is present or 3-chlorobenzoic acid (mCBA), the byproduct after mCPBA transfers an oxygen atom to F8FeII, in the synthesis/generation of F8Cmpd-II. The resulting cation [F8FeIIIOH2]+ presumably exists in solution with m-chlorobenzoate as the counterion (which may possibly be coordinated).
Figure 3.
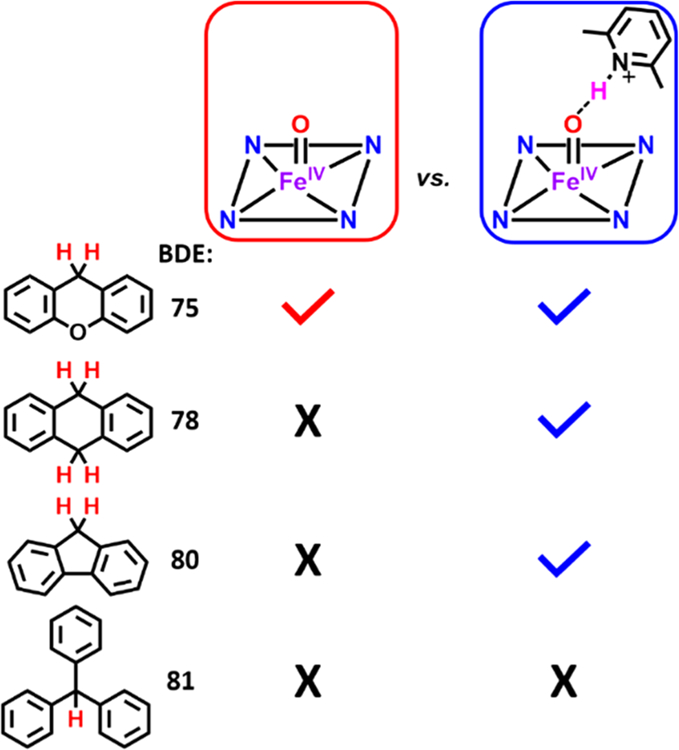
Reactivity between various C–H substrates (xanthene, dihydroanthracene (DHA), fluorene, and triphenylmethane) and F8Cmpd-II or F8Cmpd-II(LutH+). Also see the text for further details.
Identification and confirmation that [F8FeIIIOH2]+ is the inorganic product from F8Cmpd-II(LutH+) reactivity with the C–H substrates comes from its independent generation. Addition of 1 equiv of acid (mCBA) to the well-described F8FeIIIOH complex51 led to the formation of a UV–vis spectrum (Figure S16) identical to that observed from the reaction between xanthene and F8Cmpd-II (Figure S5). In the case of the substrate oxidation reactions performed by F8Cmpd-II(LutH+), however, the F8FeIIIOH complex formed from initial HAA from xanthene, DHA, or fluorene could easily be protonated by LutH+ (either concomitantly with HAA or as a fast second step, vide infra), generating [F8FeIIIOH2]+ (Figures 2A, S11, and S14); in fact, the ferric aqua complex can indeed also be independently generated by addition of 1 equiv of LutH+ to F8FeIIIOH (Figure S17).
This authentically generated product, [F8FeIIIOH2]+, also yielded an identical EPR spectrum (g = 6 and 2 signals typical of a high-spin ferric species) to the resulting reaction mixtures between the ferryl complexes and the various C–H substrates, further providing evidence of the identity (particularly as concerns the ferric oxidation state) of the resulting inorganic product (Figure S18). The independently formed product, [F8FeIIIOH2]+, was prepared at different concentrations (1.25–2.25 mM), and the ferric g = 6 signal was doubly integrated, generating a standard curve to quantify the resulting inorganic product (Figure S19, Table S1). It could then be determined that a yield of >96% (based on EPR spectroscopy) of the high-spin ferric complex [F8FeIIIOH2]+ was obtained at the end of the reactions between F8Cmpd-II with xanthene or between F8Cmpd-II(LutH+) with xanthene, DHA, or fluorene (Table S2).
It is worth emphasizing the presence of mCBA in the reaction mixture, derived from the original use of mCPBA to form F8Cmpd-II. The mCBA itself does not affect the reactivity of the iron complex, F8Cmpd-II, and we posit that they do not form a hydrogen-bonding complex. The data that supports this supposition is as follows: (1) chemical reduction of F8Cmpd-II results in a ferric product that is not [F8FeIIIOH2]+,38 (2) chemical reduction of F8 Cmpd-II(LutH+) gives [F8FeIIIOH2]+,38 and (3) F8FeIIIOH can be protonated by mCBA to make [F8FeIIIOH2]+. One would assume that if mCBA was forming an H-bonding complex, reduction of F8Cmpd-II would give [F8FeIIIOH2]+ (similar to the reactivity of F8Cmpd-II(LutH+)). However, this does not occur. It is also of note that in situ generation of F8Cmpd-II and F8Cmpd-II(LutH+) yields 1 equiv of mCBA, yet these two species have dramatically different bracketed reduction potentials38 and rates of reactivity. This then indicates that LutH+ (not mCBA) is activating the Fe═O moiety in F8Cmpd-II(LutH+).
Addition of 100 equiv of DHA to 0.01 mM F8Cmpd-II(LutH+) exhibited a Soret band for [F8FeIIIOH2]+ at 411 nm but also the characteristic five sharp-lined UV–vis signature of anthracene between 300 and 380 nm that grew in over time (Figure S11). Thus, as expected, addition of DHA to F8Cmpd-II(LutH+) resulted in the abstraction of two hydrogen atoms from DHA, yielding the dehydrogenated product, anthracene (Scheme 2).
Scheme 2.
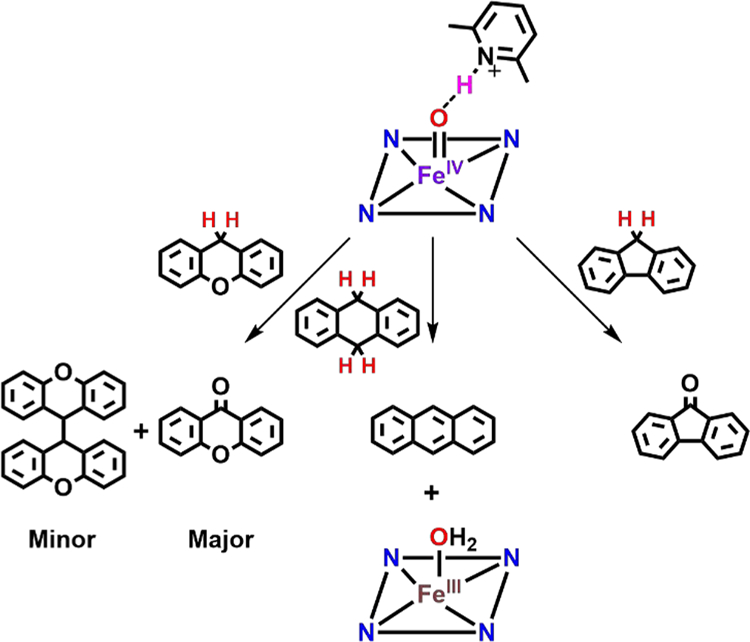
Reactivity of F8Cmpd-II(LutH+) with Xanthene and Fluorene Yields Oxygenated Major Products (with xanthene also affording a coupled minor product), While DHA Is Oxidized to Anthracenea
aThe final inorganic product in all cases is the ferric aqua complex, [F8FeIIIOH2]+.52
However, C–H cleavage reactions with xanthene and fluorene most often result in a more complex mixture of products, as the hydroxylated, oxidized, and radical coupled products (i.e., xanthydrol, xanthone, and 9,9′-bixanthene, respectively) are known to form (Scheme 2). Thus, the resulting reaction mixtures with xanthene and fluorene were analyzed by gas chromatography equipped with a flame ionization detector (GC/FID); see the SI for additional experimental details. Addition of xanthene to F8Cmpd-II resulted in the formation of primarily xanthone in 74–85% yield by GC/FID (using authentic xanthone as an integration standard) along with a small amount of 9,9′-bixanthene (13–14% yield by GC/FID) (similar to Mayer and co-workers,53 9,9′-bifluorenyl was used as an integration standard), giving an overall reaction yield of 88–98% (Figure S38 and Table S14).
Oxidation/oxygenation of C–H substrates with F8Cmpd-II(LutH+) occurs with significantly differing kinetic behavior, as described below. F8Cmpd-II(LutH+) plus xanthene resulted in the formation of primarily xanthone in 58–64% yield along with the minor product 9,9′-bixanthene yield of 6–12%, giving an overall reaction yield of ~70% (Figure S39 and Table S14). Addition of fluorene to F8Cmpd-II(LutH+) resulted in only the formation of fluorenone in 83% yield by GC/FID (using authentic fluorenone as an integration standard); no 9,9′-bifluorenyl was observed (Figure S42 and Table S16). Quantification of products was based on preparing calibration curves of authentic standards (Figures S36, S37, S40 and S41 and Tables S13 and S15). The present findings are analogous to previous reports54–57 in which xanthene and fluorene react with high-valent metal oxo compounds similarly; typically, xanthone and fluorenone or 9,9′-bixanthene and 9,9′-bifluorene are detected as the major products.
A mechanistic pathway for the oxidation/oxygenation of xanthene or fluorene by F8Cmpd-II(LutH+) can be proposed based on the products determined by UV–vis and EPR spectroscopy and GC, thus taking into account the reaction stoichiometry observed (Schemes S1 and S2). In the initial step, 1 equiv of F8Cmpd-II(LutH+) abstracts a hydrogen atom from xanthene, releasing the ferric aqua complex and xanthyl radical. This can either dimerize (via interaction with a second equivalent of xanthyl radical) to form 9,9′-bixanthene or further react with a second equivalent of F8Cmpd-II(LutH+) via “rebound”, forming xanthydrol bound to an iron(III) ion. This xanthydrol can then be oxidized by two more equivalents of F8Cmpd-II(LutH+) in order to give xanthone as the major product of the reaction (Scheme S2). The significant product yields reported herein take into consideration a 4:1 Cmpd-II:product stoichiometry for the oxygenated products (xanthone and fluorenone) and a 2:1 Cmpd-II:product stoichiometry for the coupled 9,9′-bixanthene product. The observed yields (by GC) are 14.5–18.5% for xanthone and 21% for fluorenone. However, the maximum yield based on the 4:1 stoichiometry is 25%, therefore, we can conclude that high yields of these oxidation products were obtained. Similarly, the yields reported for 9,9′-bixanthene are based on the maximum yield of 50%.
Kinetic Analysis of F8Cmpd-II and F8Cmpd-II(LutH+) with Various C–H Substrates.
Second-order rate constants (k2) were determined for the reactions of F8Cmpd-II and F8Cmpd-II(LutH+) with C–H substrates using the method of initial rates (due to the slow rate of some of the reactions, vide infra) under pseudo-first order conditions (Figures S20, S22, S24, S26, S28, S30, and S32). We found that F8Cmpd-II(LutH+) reacted 40 times faster with xanthene than F8Cmpd-II (Figures 4, S21, and S23 and Tables 1, S3, and S4; k2 = 14.2 M−1 s−1 vs k2 = 0.369 M−1 s−1).58 Second-order rate constants were also obtained for the reactions with F8Cmpd-II(LutH+) (k2(DHA) = 0.485 M−1 s−1 and k2(fluorene) = 0.102 M−1 s−1, Figures S25 and S27 and Tables 1, S5, and S6), showing decreased rates for these stronger C–H substrates (vide infra). Recall, DHA and fluorene do not react with F8Cmpd-II (vide supra).
Figure 4.
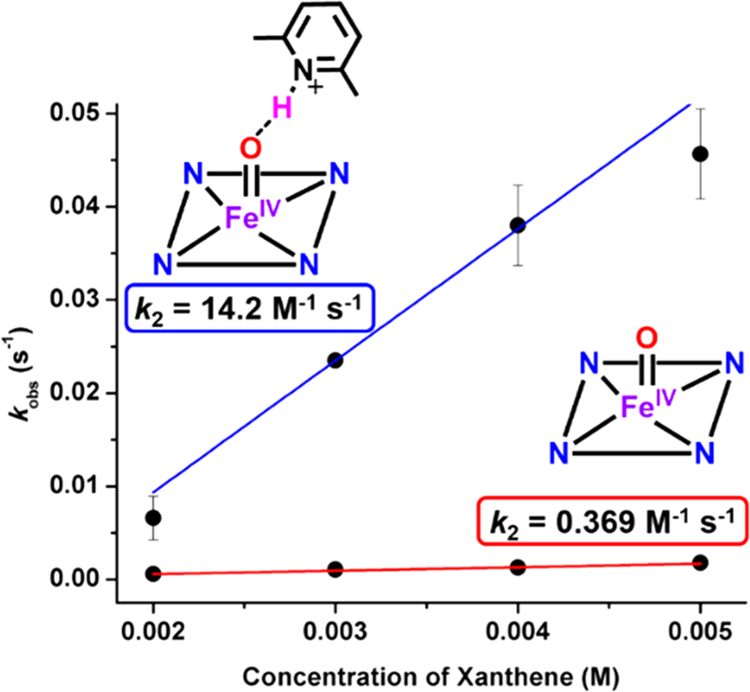
Rates of reactivity as determined via UV–vis spectroscopy of 0.1 mM F8Cmpd-II (red) or F8Cmpd-II(LutH+) (blue) with various concentrations of xanthene (2–5 mM) at −90 °C in 1:9 MeTHF:toluene. Each data point represents three trials with error bars denoting the standard deviation between the trials. F8Cmpd-II(LutH+) reacts 40 times faster than F8Cmpd-II with xanthene under pseudo-first-order conditions. See also Figures S21 and S23.
Table 1.
Second-Order Rate Constants (k2, M−1 s−1) Determined via UV–vis Spectroscopy Monitoring the Addition of Various C–H Substrates under Pseudo-First-Order Conditions to 0.1 mM F8Cmpd-II or F8Cmpd-II(LutH+) at −90 °C in 1:9 MeTHF:toluene
| C–H substrate (BDE in kcal/mol) |
F8Cmpd-II (M−1 s−1) |
F8Cmpd-II(LutH+) (M−1 s−1) |
|---|---|---|
| xanthene (75) | 0.369 | 14.2 |
| DHA (78) | 0.485 | |
| fluorene (80) | 0.102 |
As expected, the rate of C–H cleavage by F8Cmpd-II(LutH+)was shown to be dependent on the substrate bond dissociation energy (Table 1, Figure 5). There was a 30-fold decrease in the reaction rate from xanthene to DHA (Δ(C–HBDE) = +3 kcal/mol) and a 140-fold decrease from xanthene to fluorene (Δ(C–HBDE) = +5 kcal/mol). A linear correlation was observed between the log of the rate constants and the BDE of the C–H substrates (Figure 5), providing supporting evidence that hydrogen-atom abstraction is occurring by F8Cmpd-II(LutH+). Thus, hydrogen-bonding from LutH+ to F8Cmpd-II resulted in an enhancement of C–H cleavage reactivity and the ability to react with C–H substrates containing stronger BDEs. These kinetic studies thereby emphasize the importance of H-bonding to tune the active site of metalloproteins to allow potentially energetically unfavorable reactions to occur in biology. For example, Cmpd-II species in peroxidases such as chloroperoxidase and horseradish peroxidase (HRP) are unreactive toward C–H bonds; however, Groves and co-workers recently reported on the P-450 like APO-II enzyme, an aromatic peroxygenase, in which the Cmpd-II species is basic and is protonated (Fe(IV)–OH), exhibiting enhanced reactivity toward C–H substrates.59 A parallel can be drawn with the synthetic systems reported herein, where the parent F8Cmpd-II complex is not very reactive but the H-bonded adduct F8Cmpd-II(LutH+) shows greatly enhanced reactivity toward C–H substrates.
Figure 5.

Linear correlation of the logarithmic rate constants and substrate C–H BDEs suggests a Bell–Evans–Polanyi relationship,60,61 supporting a rate-limiting hydrogen-atom abstraction step.
Kinetic Isotope Effects (KIEs) of F8Cmpd-II and F8Cmpd-II(LutH+): Tunneling.
To evaluate the influence of hydrogen-bonding on the rate of reactivity and to also provide additional evidence that hydrogen-atom abstraction was occurring as the rate-determining step, various C–D isotopically labeled substrates were reacted with the ferryl complexes. The rate of C–D homolytic cleavage by the ferryl complexes was determined by following the decay of the ferryl species at 544 or 546 nm (for F8Cmpd-II and F8Cmpd-II(LutH+), respectively) via UV–vis spectroscopy. Second-order rate constants were determined by addition of C–D substrates under pseudo-first-order conditions (20–300 equiv of substrate) utilizing the method of initial rates (due to the greatly increased reaction time scales, Figures S28, S30, and S32). Addition of the C–D substrates to F8Cmpd-II and F8Cmpd-II(LutH+) resulted in similar reactions as those for the C–H substrates, in which the high-valent UV–vis signature decayed and the appearance of the same ferric complex belonging to [F8FeIIIOH2]+ grew in over time (Figures S7, S10, and S13). As expected, the rate of C–D cleavage was significantly slower (versus C–H cleavage) by the iron(IV) oxo complexes (Tables 1 and S10). As determined directly from the data obtained, for example, oxidation of xanthene-d2 by F8Cmpd-II and F8Cmpd-II(LutH+) was 176 and 215 times slower than their respective reactions with xanthene (Figures S29 amd S31 and Tables S7, S8, and S10). The kinetic isotope effect study was extended for F8Cmpd-II(LutH+) reacting with DHA-d4, giving a KIE of 81 (Figure 6 and Tables S9 and S10).
Figure 6.

Kinetic isotope effect (kH/kD) determined upon monitoring the rate of reactivity via UV–vis spectroscopy by F8Cmpd-II(LutH+) toward various concentrations of DHA and DHA-d4 under pseudo-first-order conditions. Each data point is an average of three trials, and error bars represent the standard deviation. Note: The scale of concentrations (the abscissa range) differs for plots A and B. Error bars in A, for 50 equiv of DHA, are smaller than the plot marker.
However, it is important to note that the deuterium KIE values reported here utilizing xanthene-d2 and 9,10-dihydroan-thracene-d4 are uncorrected for residual protium in these samples, less than 1–2% as determined from the 1H NMR spectra following synthesis (see Experimental Section; Figures S1 and S2). This follows the approach carried out in a good number of other published studies where KIE’s were determined utilizing C–D substrates with similar percent deuterium incorporation as here with iron28,35,55,62–64 or manganese56 containing oxidants. However, in those cases such a methodology is/was warranted. In the present situation, the 1–2% protium within the deuterated substrates will induce very fast apparent kinetics, and thus, exceptionally high KIE values cannot be determined with accuracy; the existing situation suggests that only KIE values of ~50 or less can be accurately measured; therefore, we can only say that KIE values for the reactions described here are ~50.65
The large KIE values exhibited by these complexes indicate a nonclassical primary kinetic isotope effect, suggesting the rate-determining step is hydrogen-atom abstraction from the C–H(D) substrates with tunneling effects.1,66 In a reaction accompanied by tunneling, the hydrogen atom does not need to go over the transition state barrier but instead “tunnels” through it, increasing the rate of hydrogen-atom transfer beyond the predicted value (Figure 7).1 Many previous reports67–71 of large apparent KIEs have been observed from reactions of synthetic iron(IV) oxo complexes (mainly nonheme) with C–H substrates, and it was suggested that a hydrogen-atom tunneling mechanism was occurring (some-times accompanied by two-state reactivity); such a finding is also a common occurrence found with HAA processes by various nonheme and heme metalloenzymes.69,72–77 Thus, hydrogen-atom tunneling may be occurring here, giving rise to the large nonclassical KIE values for F8Cmpd-II and F8Cmpd-II(LutH+).
Figure 7.
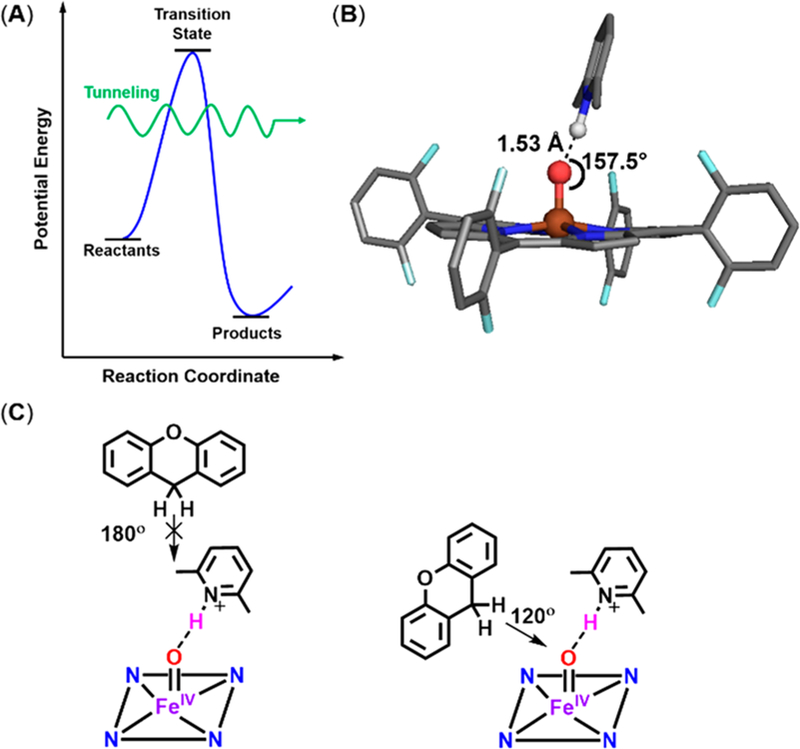
(A) Representative scheme of tunneling (green arrow), resulting in a lower activation energy than needed to reach the transition state.1 (B) DFT-calculated structure of F8Cmpd-II-(LutH+)38 showing the steric encumbrance by the lutidine molecule. (C) For HAA by iron(IV) oxo complexes, the substrate can approach at either a 180° or a 120° angle with respect to the Fe=O bond (as previously suggested by Shaik and co-workers);66 these approaches are consistent with the sp2-hybridized oxo ligand.78 However, in the present system with the lutidinium present, substrates can only approach at a 120° angle.
While the KIE values reported herein are larger than those previously reported for heme and nonheme iron(IV) oxo systems,1,54,66,67 this may be due to the lower cryogenic temperatures at which the reactions are carried out, −90 °C (and possibly due to the unique solvent system of 1:9 MeTHF:toluene). In fact, many examples of temperature-dependent KIE values have been reported, with higher KIEs being observed at lower temperatures.79 For example, Que, Shaik, and co-workers reported67 on the temperature-dependent KIE for hydrogen-atom abstraction from ethylbenzene by a nonheme iron(IV) oxo complex in CH3CN with a 10-fold KIE increase at −40 °C compared to 40 °C (KIE of 400 vs 40, respectively). The authors suggested that their temperature-dependent KIE was due to hydrogen-atom tunneling, as the difference in activation energies for the C–H and C–D reactions were found to be less than the zero-point energy differences of the substrate C–H and C–D bonds (i.e., the reactions proceed with less activation energy needed than the theoretical value based solely on zero-point energies).67 Thus, the high KIE values observed for F8Cmpd-II and F8Cmpd-II(LutH+), respectively, are most likely due to hydrogen-atom tunneling (with the larger than normal values coming from the especially low temperatures used herein, −90 °C). Unfortunately, we are unable to obtain variable-temperature kinetics for these reactions as the ferryl compounds are not stable above −90 °C and the solvent mixture used freezes at −95 °C, thus preventing us from obtaining activation parameters. Without these parameters, we cannot estimate the KIE of these reactions at room temperature, which would allow for a better comparison with literature values previously reported.
The iron(IV) oxo complex’s spin state and the geometrical approach of the substrate can also affect the magnitude of KIE values, as shown by Shaik and co-workers.66 However, there have not been any reports on two-state reactivity occurring in synthetic heme Cmpd-II systems, so we suspect it is unlikely that it is occurring in our system. The substrate can approach in two different ways, either directly from above the iron(IV) oxo (with a transition state having an O···H–C angle of 180°) or from a side angle (with a transition state having an O···H–C angle of 120°) (Figure 7C). Previously, DFT calculations on F8Cmpd-II(LutH+) showed that the lutidinium molecule interacts with the iron(IV) oxo moiety with an Fe=O–H(N) angle of 157.5° (Figure 7B).38 However, since there is not full proton transfer, lutidine is still present, blocking approach of substrate from a 180° angle to the iron(IV) oxo. While neither trajectory is blocked for F8Cmpd-II, the similarities in the large KIE values suggest that both reactions occur via the same substrate approach trajectory. Since only one such trajectory is available for F8Cmpd-II(LutH+), it is assumed that the same trajectory may occur for F8Cmpd-II. Thus, the most likely pathway for C–H abstraction is with substrate approach from a 120° angle, denoted the π pathway by Shaik and co-workers.66 Their calculated KIE values for heme iron(IV) oxo complexes reacting with C–H substrates via this pathway are much higher than those for a 180° substrate approach.66 Thus, another factor in the large KIE values experimentally observed herein may be due to this mechanism and nature of substrate approach.
Inverse Kinetic Isotope Effect of F8Cmpd-II(LutH+/D+).
To further investigate the role of H-bonding on the oxidative reactivity of F8Cmpd-II(LutH+), the ferryl deuterated lutidinium analog, F8Cmpd-II(LutD+), was synthesized by addition of 1 equiv of LutD+ triflate to F8Cmpd-II at −90 °C in 1:9 MeTHF:toluene. F8Cmpd-II(LutD+) had the same UV–vis and Mössbauer spectroscopic parameters (Figure S33 and Table S11) as F8Cmpd-II(LutH+), suggesting that the incorporation of deuterium into the H-bonding complex does not have a significant effect on the electronic properties of the complex (i.e., the proteo and deutero analogs behave similarly in terms of the H-bonding interaction with the iron(IV) oxo moiety). However, when kinetic studies were explored for the oxidation of DHA by F8Cmpd-II(LutD+), it was found that this complex reacts slightly faster (1.5 times) than its proteo analog (k2 = 0.713 ± 0.050 M−1 s−1) (Figures 8, S34, and S35 and Table S12). This gives an inverse KIE (kH/kD) of 0.68, providing further evidence that H-bonding plays a direct role in substrate oxidation since the identity of the H-bond donor (H+ vs D+) affects the rate of reaction. A similar inverse KIE has been observed by Fukuzumi, Nam, and co-workers for proton-coupled electron transfer (PCET) reactions of a nonheme iron(IV) oxo complex in the presence of triflic acid.35,80
Figure 8.

Kinetic isotope effect (kH/kD) determined upon monitoring the rate of reactivity via UV–vis spectroscopy of F8Cmpd-II(LutH+) and F8Cmpd-II(LutD+) toward various concentrations of DHA under pseudo-first-order conditions. Each data point is an average of three trials, and error bars represent the standard deviation. F8Cmpd-II(LutD+) reacted 1.5× faster, indicating both HAA and subsequent protonation are part of the rate-determining step.81
The observation of an inverse KIE may arise because the N–H(D) bond of LutH(D)+ is also broken in the rate-determining step of the reaction (along with the C–H bond of the substrate), causing a difference in rates between the H-bonding adducts with LutH+ vs LutD+. Thus, even though F8Cmpd-II(LutH+) is a hydrogen-bonding adduct, without full proton transfer, hydrogen-atom abstraction from the substrate and deprotonation of LutH(D)+ may occur simultaneously during the reaction; the fact that [F8FeIIIOH2]+ is the final reaction product, with no observable intermediates, also supports this. Alternatively, the inverse KIE value could be due to a secondary KIE, with the H-bonding properties of LutH+ vs LutD+ acting differently during the course of the reaction. The inverse KIE, whether primary or secondary, arises from the change in hybridization of the oxo ligand during the reaction. In the starting complex, the oxo ligand is sp2 hybridized, while in the final product, the resulting aqua ligand is sp3 hybridized; thus, there is a larger activation energy for the O–H bond over the O–D bond.82 This allows for the deuterated Lewis acid to react faster than the proteo analog. These results highlight the importance of H-bonding to synthetic heme iron(IV) oxo complexes and support the direct interaction of LutH+ with the oxo ligand.
CONCLUSION
Secondary coordination sphere effects such as hydrogen-bonding greatly influence the active site in heme proteins.1–5 Formation of reactive intermediates or the properties of the iron metal center depend on extensive distal and proximal hydrogen-bonding networks found in heme metalloenzymes, which tune the heme environment appropriately for the desired catalysis to occur.5–9 This work highlights the dramatic effects H-bonding can induce on reactivity, especially considering crystal or neutron diffraction structures6,8,9 depicting significant hydrogen-bonding interactions with high-valent intermediates in heme metalloenzymes. The implications of hydrogen-bonding were systematically investigated on the mechanism of substrate oxygenation with a hydrogen-bonded synthetic heme iron(IV) oxo (Cmpd II) complex. In the presence of hydrogen-bonding a synthetic heme ferryl complex (vs the parent complex) the following were observed:
Enhanced rate of reactivity with C–H substrates. F8Cmpd-II(LutH+) reacted 40× faster with xanthene than F8Cmpd-II.
Enhanced oxidative capabilities with substrates of higher C–H bond dissociation energies. F8Cmpd-II(LutH+) was shown to react with 9,10-dihydroanthracene and fluorene (BDEs = 78 and 80 kcal/mol, respectively); however, F8Cmpd-II could only react with xanthene (BDE = 75 kcal/mol).
A linear correlation was observed between the logarithm of the rates of reactivity with C–H substrates and their BDEs for F8Cmpd-II(LutH+), wherein the higher the BDE, the slower the rate of reactivity, supporting a hydrogen-atom abstraction mechanism.
Apparent large nonclassical KIEs were observed upon reacting various C–H and C–D substrates with F8Cmpd-II and F8Cmpd-II(LutH+), possibly or likely due to tunneling.
An inverse KIE was observed for F8Cmpd-II(LutD+) vs F8Cmpd-II(LutH+), wherein the deuterated analog reacted 1.5× faster than the proteo analog, supporting the influence of hydrogen-bonding in the reaction mechanism.
Experimentally, for the first time, reactivity studies have been carried out with a hydrogen-bonded synthetic heme iron(IV) oxo complex (a heme protein Cmpd II model), and results support that hydrogen-bonding may be a key factor in the catalysis of heme metalloenzymes by aiding in the formation or stability of various intermediates or tuning the reactivity of the active site. Potentially, addition of proton sources may lead to the development of more promising biomimetic catalysts for energy research or for synthetic applications. The present work should be followed by exploration employing other Lewis or Brønsted acids, as has been carried out in the case of many high-valent nonheme iron or manganese oxo complexes.4,36,83 It would also be of interest to generate the 18O-labeled Cmpd-II species to establish the origin of the incorporated O atom in xanthone (or fluorenone).
EXPERIMENTAL SECTION
General.
All reagents and solvents purchased and used were of commercially available quality except as noted. Inhibitor-free 2-methyltetrahydrofuran (MeTHF) was distilled over Na/benzophe-none under Ar and deoxygenated with Ar before use. Toluene was distilled over calcium hydride and deoxygenated with Ar before use. Butyronitrile was distilled over sodium carbonate and potassium permanganate and deoxygenated with Ar before use. Solvent deoxygenation was achieved by bubbling Ar through the desired solvent for ≥45 min via an addition funnel connected to a receiving Schlenk flask. All solvents were stored in amber bottles under 4 Å sieves. Air-free manipulations were performed in a Vac atmosphere OMNI-LAB drybox or under argon atmosphere using standard Schlenk techniques. Low-temperature UV–vis spectroscopy experiments were carried out using a Cary-50 Bio spectrophotometer equipped with an Unisoku USP-203A cryostat using a modified Schlenk cuvette with a 1 cm path quartz cell. The spectrometer was equipped with Cary WinUV Scanning Kinetics software. All NMR spectra were recorded in 9 in., 5 mm o.d. NMR tubes on a Bruker 300 MHz NMR instrument. The 1H chemical shifts were calibrated to residual proton solvent peaks. EPR spectra were collected with an ER 073 magnet equipped with a Bruker ER041 X-Band microwave bridge and a Bruker EMX 081 power supply: microwave frequency = 9.42 GHz, microwave power = 0.201 mW, attenuation = 30 dB, modulation amplitude = 10 G, modulation frequency = 100 kHz, temperature = 10 K. Mössbauer spectra were recorded on a spectrometer from SEE Co. (Edina, MN) operating in the constant acceleration mode in a transmission geometry. The sample was kept in an SVT-400 cryostat from Janis (Wilmington, MA) using liquid N2 as a cryogen for 80 K measurements. Isomer shifts were determined relative to the centroid of the spectrum of a metallic foil of α-Fe collected at room temperature. Data analysis was performed using version F of the program WMOSS (www.wmoss.org), and quadrupole doublets were fit to Lorentzian line shape. Elemental analyses were performed by Midwest Microlab, Indianapolis, IN. Gas chromatography (GC) was performed on an Agilent 6890N gas chromatograph using a DB-5 5% phenylmethyl siloxane capillary column and equipped with a flame-ionization detector (FID).
High-resolution EI mass spectra (EI-MS) were obtained using a VG70S double-focusing magnetic sector mass spectrometer (VG Analytical, Manchester, UK, now Micromass/Waters) equipped with an MSS data acquisition system (MasCom, Bremen, Germany). The resolution of the instrument was set at 10 000 (100 ppm peak width). Samples were introduced into the source (block temperature = 200 °C) using a heated direct insertion probe fitted with a deep quartz cup with a heating rate of 1 C/s. Spectra were acquired under control of the computer data system. The electron energy was 70 eV. Nominal mass scan spectra were acquired for a scan mass range of 10–1000 amu using a magnet scan rate of 20 s/dec. Accurate mass measurements were obtained across a narrower mass range using high-boiling Perfluorokerosene (PFK) as the reference.
The oxidant 3-chloroperbenzoic acid (mCPBA) was purified according to published procedures.84 The starting materials F8FeII complex,85–87 2,6-lutidinium triflate,88 and C–D substrates55,64,89 xanthene-d2 (XAN-d2) and 9,10-dihydroanthracene-d4 (DHA-d4) were synthesized as previously described. C–H substrates utilized for reactivity studies were purified according to literature methods, affording white crystalline solids.55,64,89 9,10-Dihydroanthracene was sublimed and recrystallized twice from ethanol. Fluorene and xanthene were recrystallized twice from ethanol.
Xanthene-d2.
In the glovebox, purified xanthene (0.500 g, mmol) and 5 equiv of sodium hydride (0.330 g, mmol) were dissolved in 3 C–mL of dimethyl sulfoxide-d6 and allowed to react for 4 h, resulting in a deep red reaction mixture. Subsequently, 5 mL of water-d2 was added to the reaction to quench the excess sodium hydride and stirred for 1 h, yielding a white solid suspended in a pale yellow solution. The crude product was filtered and washed with 100 mL of water, affording a white solid. The white solid was recrystallized twice from ethanol. 1H NMR (400 MHz, CDCl3): δ 7.17 (4H, m), 7.02 (4H, m), 4.05 (0.037H). NMR data indicated >99% deuteration. The percent incorporation was similarly determined by Groves and co-workers.55 See Figure S1 for 1H NMR spectra. EI-MS: 184.0856 (M+).
9,10-Dihydroanthracene-d4.
The C–D substrate was prepared in a similar fashion to xanthene-d2 (and the same color changes occurred). 1H NMR: δ 7.34 (4H, m), 7.26 (4H, m), 4.00 (0.039H). NMR data indicated >98% deuteration. Percent incorporation was similarly determined by Groves and co-workers.55 See Figure S2 for 1H NMR spectra. EI-MS: 184.1190 (M+).
Deuterium KIE values determined in this report utilizing xanthene-d2 and 9,10-dihydroanthracene-d4 are uncorrected for residual protium, as has also been the case for previously published kinetic studies29,36,56,57,63–65 on substrate oxidations by iron or manganese oxidants.
2,6-Lutidinium Triflate.
1H NMR (CD2Cl2): δ 14.78 (1H), 8.17 (1H, t), 7.50 (2H, d), 2.84 (6H, s). Anal. Calcd for C8H10NF3O3S: C, 37.36; H, 3.92; N, 5.45. Found: C, 37.14; H, 3.87; N, 5.34.
Deuterated 2,6-Lutidinium Triflate.
Prepared similarly to 2,6-lutidinium triflate.88 2,6-Lutidine (1.55 mL, 13.31 mmol) was dissolved in 20 mL of pentane in the open air and cooled to 0 °C. Trifluoromethanesulfonic acid-d (2 mL, 22.61 mmol, 1.70 equiv) was added rapidly, upon which a white solid immediately formed. The reaction was allowed to stir for 20 min, and this was followed by filtration. The crude solid was then washed with ether and dried overnight. 1H NMR (CD2Cl2): δ 14.61 (0.038), 8.22 (1H, t), 7.56 (2H, d), 2.83 (6H, s). NMR data indicated >96% deuteration. Anal. Calcd for C8H10NF3O3S: C, 37.36; H, 3.92; N, 5.45. Found: C, 37.15; H, 3.88; N, 5.39. Elemental analysis cannot distinguish D from H.
Spectroscopic Sample Preparations.
UV–vis. F8Cmpd-II and F8Cmpd-II(LutH+) were synthesized as previously described.38 High-valent iron(IV) oxo complexes were generated by preparing 3.0 mL of a 1:9 MeTHF:toluene solution (0.01 or 0.1 mM) of F8FeII in a 10 mm path length quartz Schlenk cuvette, which was then sealed with a rubber septum in the glovebox. The cuvette was then cooled in the cryostat chamber to −90 °C, and the solution was then allowed to thermally equilibrate (~10 min) before various reagents were added. The reduced iron(II) precursor was subjected to 1 equiv of mCPBA solution in toluene at −90 °C, forming F8Cmpd-II. Subsequently, 1 equiv of 2,6-lutidinium triflate was added in butyronitrile to F8Cmpd-II, generating F8Cmpd-II(LutH+). Various C–H or C–D substrates (xanthene, xanthene-d2, DHA, DHA-d4, or fluorene) were added to F8Cmpd-II or F8Cmpd-II(LutH+), wherein the reactions were monitored by UV–vis or EPR spectroscopy. The rate of decay of the high-valent species in the presence of the various substrates under pseudo-first-order rate conditions was ascertained by UV–vis spectroscopy allowing for second-order rate constants k2 and kinetic isotope effects (KIE) to be determined. See Supporting Information for further details.
X-Band EPR.
F8Cmpd-II or F8Cmpd-II(LutH+) complexes were prepared by adding 1 equiv of mCPBA and, depending on the derivative, 1 equiv of 2,6-lutidinium triflate in butyronitrile to a 1:9 MeTHF:toluene solution containing 2 mM F8FeII (total volume = 0.4 mL) at −90 °C (acetone/liquid nitrogen cold bath). Upon addition of reagent(s), the solution was bubbled with Ar for 20 s to ensure thorough mixing. In each case, upon adding 20 equiv of the various C–H substrates to the high-valent species the solution was allowed to react for 1 h. After the complexes were generated, tubes were frozen in N2(liq), and all spectra were recorded at 10 K. The resulting complex, [F8FeIIIOH2]+, was independently generated, and a standard curve was prepared to quantify the amount of inorganic product at the end of each reaction.
Product Analysis by GC.
The reaction mixtures of F8Cmpd-II or F8Cmpd-II(LutH+) with C–H substrates were carried out on a large scale in a 10 mL Schlenk flask under argon under the same reaction conditions as the UV–vis experiments. Upon completion of the reaction at −90 °C, dodecane, the internal standard (as a solution from the glovebox dissolved in DCM, an air-free solution), was added. Aliquots of the reaction mixture (still being kept under argon) were taken up in a purged argon syringe and immediately injected into the GC-FID for analysis. Very stringent precautions were taken to ensure the absence of O2 during product analysis. There is the need for the absence of O2 in the system to ensure proper product quantification/characterization. Xanthene is converted within minutes to the major species xanthone and the minor species 9,9′-bixanthene (using 9,9′-bifluorenyl as an integration standard as previously reported by Mayer and co-workers53). Fluorene is converted within minutes to the major species fluorenone. See SI for additional details.
Supplementary Material
ACKNOWLEDGMENTS
The research support of the U.S. National Institutes of Health (GM60353 to K.D.K.) is gratefully acknowledged.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b01253.
Synthetic and analytical details (methodologies and UV–vis, rR, Mossbauer, EXAFS, EPR, and NMR spectra) (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Huang X; Groves JT Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev 2018, 118, 2491–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Baglia RA; Zaragoza JPT; Goldberg DP Biomimetic Reactivity of Oxygen-Derived Manganese and Iron Porphyrinoid Complexes. Chem. Rev 2017, 117, 13320–13352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Adam SM; Wijeratne GB; Rogler PJ; Diaz DE; Quist DA; Liu JJ; Karlin KD Synthetic Fe/Cu Complexes: Toward Understanding Heme-Copper Oxidase Structure and Function. Chem. Rev 2018, 118, 10840–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Guo M; Corona T; Ray K; Nam W Heme and Nonheme High-Valent Iron and Manganese Oxo Cores in Biological and Abiological Oxidation Reactions. ACS Cent. Sci 2019, 5, 13–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Poulos TL Heme Enzyme Structure and Function. Chem. Rev 2014, 114, 3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Moody PCE; Raven EL The Nature and Reactivity of Ferryl Heme in Compounds I and II. Acc. Chem. Res 2018, 51, 427–435. [DOI] [PubMed] [Google Scholar]
- (7).Poulos TL Thirty Years of Heme Peroxidase Structural Biology. Arch. Biochem. Biophys 2010, 500, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kwon H; Basran J; Casadei CM; Fielding AJ; Schrader TE; Ostermann A; Devos JM; Aller P; Blakeley MP; Moody PCE; Raven EL Direct Visualization of a Fe(IV)–OH Intermediate in a Heme Enzyme. Nat. Commun 2016, 7, 13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Casadei CM; Gumiero A; Metcalfe CL; Murphy EJ; Basran J; Concilio MG; Teixeira SCM; Schrader TE; Fielding AJ; Ostermann A; Blakeley MP; Raven EL; Moody PC Neutron Cryo-Crystallography Captures the Protonation State of Ferryl Heme in a Peroxidase. Science 2014, 345, 193–197. [DOI] [PubMed] [Google Scholar]
- (10).de Montellano PRO Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th ed.; de Montellano PRO, Ed.; Springer International Publishing: New York, 2015. [Google Scholar]
- (11).Chreifi G; Baxter EL; Doukov T; Cohen AE; McPhillips SE; Song J; Meharenna YT; Soltis SM; Poulos TL Crystal Structure of the Pristine Peroxidase Ferryl Center and Its Relevance to Proton-Coupled Electron Transfer. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 1226–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Yosca TH; Rittle J; Krest CM; Onderko EL; Silakov A; Calixto JC; Behan RK; Green MT Iron(IV)Hydroxide pKa and the Role of Thiolate Ligation in C-H Bond Activation by Cytochrome P450. Science 2013, 342, 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Yosca TH; Ledray AP; Ngo J; Green MT A New Look at the Role of Thiolate Ligation in Cytochrome P450. JBIC, J. Biol. Inorg. Chem 2017, 22, 209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).The PyMOL Molecular Graphics System, Version 1.8; Schrodinger LLC, 2015. [Google Scholar]
- (15).Meunier B; de Visser SP; Shaik S Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem. Rev 2004, 104, 3947–3980. [DOI] [PubMed] [Google Scholar]
- (16).Denisov IG; Makris TM; Sligar SG; Schlichting I Structure and Chemistry of Cytochrome P450. Chem. Rev 2005, 105, 2253–2278. [DOI] [PubMed] [Google Scholar]
- (17).Shaik S; Cohen S; Wang Y; Chen H; Kumar D; Thiel W P450 Enzymes: Their Structure, Reactivity, and Selectivity Modeled by QM/MM Calculations. Chem. Rev 2010, 110, 949–1017. [DOI] [PubMed] [Google Scholar]
- (18).Ortiz de Montellano PR Hydrocarbon Hydroxylation by Cytochrome P 450 Enzymes. Chem. Rev 2010, 110, 932–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Groves JT High-Valent Iron in Chemical and Biological Oxidations. J. Inorg. Biochem 2006, 100, 434–447. [DOI] [PubMed] [Google Scholar]
- (20).Meunier B Metalloporphyrins as Versatile Catalysts for Oxidation Reactions and Oxidative DNA Cleavage. Chem. Rev 1992, 92, 1411–1456. [Google Scholar]
- (21).Costas M Selective C–H Oxidation Catalyzed by Metalloporphyrins. Coord. Chem. Rev 2011, 255, 2912–2932. [Google Scholar]
- (22).Comba P; Rajaraman G Epoxidation and 1,2-Dihydroxylation of Alkenes by a Nonheme Iron Model System – DFT Supports the Mechanism Proposed by Experiment. Inorg. Chem 2008, 47, 78–93. [DOI] [PubMed] [Google Scholar]
- (23).Nam W; Ryu YO; Song WJ Oxidizing Intermediates in Cytochrome P450 Model Reactions. JBIC, J. Biol. Inorg. Chem 2004, 9, 654–660. [DOI] [PubMed] [Google Scholar]
- (24).Kang M-J; Song WJ; Han A-R; Choi YS; Jang HG; Nam W Mechanistic Insight into the Aromatic Hydroxylation by High-Valent Iron(IV)-Oxo Porphyrin π-Cation Radical Complexes. J. Org. Chem 2007, 72, 6301–6304. [DOI] [PubMed] [Google Scholar]
- (25).Que L The Road to Non-Heme Oxoferryls and Beyond. Acc. Chem. Res 2007, 40, 493–500. [DOI] [PubMed] [Google Scholar]
- (26).Fukuzumi S Electron-Transfer Properties of High-Valent Metal-Oxo Complexes. Coord. Chem. Rev 2013, 257, 1564–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Park J; Morimoto Y; Lee Y-M; Nam W; Fukuzumi S Proton-Promoted Oxygen Atom Transfer vs Proton-Coupled Electron Transfer of a Non-Heme Iron(IV)-Oxo Complex. J. Am. Chem. Soc 2012, 134, 3903–3911. [DOI] [PubMed] [Google Scholar]
- (28).Park J; Lee Y-M; Nam W; Fukuzumi S Brønsted Acid-Promoted C–H Bond Cleavage via Electron Transfer from Toluene Derivatives to a Protonated Nonheme Iron(IV)-Oxo Complex with No Kinetic Isotope Effect. J. Am. Chem. Soc 2013, 135, 5052–5061. [DOI] [PubMed] [Google Scholar]
- (29).Park J; Lee Y-M; Ohkubo K; Nam W; Fukuzumi S Efficient Epoxidation of Styrene Derivatives by a Nonheme Iron(IV)-Oxo Complex via Proton-Coupled Electron Transfer with Triflic Acid. Inorg. Chem 2015, 54, 5806–5812. [DOI] [PubMed] [Google Scholar]
- (30).Hill EA; Weitz AC; Onderko E; Romero-Rivera A; Guo Y; Swart M; Bominaar EL; Green MT; Hendrich MP; Lacy DC; Borovik AS Reactivity of an FeIV-Oxo Complex with Protons and Oxidants. J. Am. Chem. Soc 2016, 138, 13143–13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Weitz AC; Mills MR; Ryabov AD; Collins TJ; Guo Y; Bominaar EL; Hendrich MP A Synthetically Generated LFeIVOHn Complex. Inorg. Chem 2019, 58, 2099–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Zaragoza JPT; Yosca TH; Siegler MA; Moënne-Loccoz P; Green MT; Goldberg DP Direct Observation of Oxygen Rebound with an Iron-Hydroxide Complex. J. Am. Chem. Soc 2017, 139, 13640–13643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Park J; Morimoto Y; Lee Y-M; You Y; Nam W; Fukuzumi S Scandium Ion-Enhanced Oxidative Dimerization and N-Demethylation of N, N-Dimethylanilines by a Non-Heme Iron(IV)-Oxo Complex. Inorg. Chem 2011, 50, 11612–11622. [DOI] [PubMed] [Google Scholar]
- (34).Morimoto Y; Kotani H; Park J; Lee Y-M; Nam W; Fukuzumi S Metal Ion-Coupled Electron Transfer of a Nonheme Oxoiron(IV) Complex: Remarkable Enhancement of Electron-Transfer Rates by Sc3+. J. Am. Chem. Soc 2011, 133, 403–405. [DOI] [PubMed] [Google Scholar]
- (35).Park J; Morimoto Y; Lee YM; Nam W; Fukuzumi S Unified View of Oxidative C-H Bond Cleavage and Sulfoxidation by a Nonheme Iron(IV)-Oxo Complex via Lewis Acid-Promoted Electron Transfer. Inorg. Chem 2014, 53, 3618–3628. [DOI] [PubMed] [Google Scholar]
- (36).Nam W; Lee Y-M; Fukuzumi S Hydrogen Atom Transfer Reactions of Mononuclear Nonheme Metal–Oxygen Intermediates. Acc. Chem. Res 2018, 51, 2014–2022. [DOI] [PubMed] [Google Scholar]
- (37).Boaz NC; Bell SR; Groves JT Ferryl Protonation in Oxoiron(IV) Porphyrins and Its Role in Oxygen Transfer. J. Am. Chem. Soc 2015, 137, 2875–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ehudin MA; Gee LB; Sabuncu S; Braun A; Moënne-Loccoz P; Hedman B; Hodgson KO; Solomon EI; Karlin KD Tuning the Geometric and Electronic Structure of Synthetic High-Valent Heme Iron(IV)-Oxo Models in the Presence of a Lewis Acid and Various Axial Ligands. J. Am. Chem. Soc 2019, 141, 5942–5960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).The term Lewis acid adduct is used for F8Cmpd-II(LutH+) since the proton (an electron acceptor and thus a Lewis acid) on the lutidinium ion interacts with the oxo moiety (Lewis base) without full proton transfer. See ref 38.
- (40).Due to the fact that the Mössbauer parameters (e.g., isomer shift, quadrupole splitting) for F8Cmpd-II and F8Cmpd-II(LutH+) complexes are almost identical, virtually indistinguishable, determining the purity of F8Cmpd-II(LutH+) via Mössbauer spectroscopy was complicated and the data for the hydrogen-bonded complex could be fit either as being one species or two (in an ~50:50 mixture).
- (41).Green MT Oxoiron(IV) in Chloroperoxidase Compound II Is Basic: Implications for P450 Chemistry. Science 2004, 304, 1653–1656. [DOI] [PubMed] [Google Scholar]
- (42).Behan RK; Green MT On the Status of Ferryl Protonation. J. Inorg. Biochem 2006, 100, 448–459. [DOI] [PubMed] [Google Scholar]
- (43).Hohenberger J; Ray K; Meyer K The Biology and Chemistry of High-Valent Iron-Oxo and Iron-Nitrido Complexes. Nat. Commun 2012, 3, 720. [DOI] [PubMed] [Google Scholar]
- (44).Stein SE; Brown RL Prediction of Carbon-Hydrogen Bond Dissociation Energies for Polycyclic Aromatic Hydrocarbons of Arbitrary Size. J. Am. Chem. Soc 1991, 113, 787–793. [Google Scholar]
- (45).Burkey TJ; Majewski M; Griller D Heats of Formation of Radicals and Molecules by a Photoacoustic Technique. J. Am. Chem. Soc 1986, 108, 2218–2221. [DOI] [PubMed] [Google Scholar]
- (46).Mayer JM Biomimetic Oxidations Catalyzed by Transition Metal Complexes; Meunier B, Ed.; Imperial College Press: London, 2000; pp 1–43. [Google Scholar]
- (47).Roth JP; Mayer JM Hydrogen Transfer Reactivity of a Ferric Bi-Imidazoline Complex That Models the Activity of Lipoxygenase Enzymes. Inorg. Chem 1999, 38, 2760–2761. [DOI] [PubMed] [Google Scholar]
- (48).The origin of the 700–1000 absorption envelope likely arises as ligand-to-metal charge transfer transition(s) from the porphyrinate to the iron dxy/dyz orbitals. Time-dependendent density functional theory (TD-DFT) calculations, in the future, will be required to determine this, but these are beyond the scope of this current article.
- (49).Bordwell FG; Cheng J; Ji GZ; Satish AV; Zhang X Bond Dissociation Energies in DMSO Related to the Gas Phase Values. J. Am. Chem. Soc 1991, 113, 9790–9795. [Google Scholar]
- (50).Due to the thermal instability of F8Cmpd-II and F8Cmpd-II(LutH+), reactions were carried out and held at −90 °C and aliquots were taken directly from the −90 °C mixture and analyzed (see Experimental Section) to ensure reactivity with substrates was indeed due do these Cmpd-II species. Warming the reaction mixtures would result in thermal decay to other species, which might in fact be capable of effecting substrate oxidations (monitoring reactivity at −90 °C ensures we are following the reactivity of only the stable high-valent complexes).
- (51).Karlin KD; Nanthakumar A; Fox S; Murthy NN; Ravi N; Huynh BH; Orosz RD; Day EP X-Ray Structure and Physical Properties of the Oxo-Bridged Complex [(F8-TPP)Fe-O-Cu(TMPA)]+, F8-TPP = Tetrakis(2,6-Difluorophenyl)Porphyrinate-(2-), TMPA = Tris(2-Pyridylmethyl)Amine: Modeling the Cyto-chrome c Oxidase Fe-Cu Heterodinuclear Active Site. J. Am. Chem. Soc 1994, 116, 4753–4763. [Google Scholar]
- (52).Another possible (minor) product from the reaction of F8Cmpd-II(LutH+) with fluorene is 9,9′-bifluorenyl; however, this product was not observed by GC analysis.
- (53).Bryant JR; Taves JE; Mayer JM Oxidations of Hydrocarbons by Manganese(III) Tris(Hexafluoroacetylacetonate). Inorg. Chem 2002, 41, 2769–2776. [DOI] [PubMed] [Google Scholar]
- (54).Jeong YJ; Kang Y; Han AR; Lee YM; Kotani H; Fukuzumi S; Nam W Hydrogen Atom Abstraction and Hydride Transfer Reactions by Iron(IV)-Oxo Porphyrins. Angew. Chem., Int. Ed 2008, 47, 7321–7324. [DOI] [PubMed] [Google Scholar]
- (55).Gao H; Groves JT Fast Hydrogen Atom Abstraction by a Hydroxo Iron(III) Porphyrazine. J. Am. Chem. Soc 2017, 139, 3938–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Arunkumar C; Lee Y-M; Lee JY; Fukuzumi S; Nam W Hydrogen-Atom Abstraction Reactions by Manganese(V)– and Manganese(IV)–Oxo Porphyrin Complexes in Aqueous Solution. Chem. - Eur. J 2009, 15, 11482–11489. [DOI] [PubMed] [Google Scholar]
- (57).Bryant JR; Mayer JM Oxidation of C–H Bonds by [(Bpy)2(Py)RuIVO]2+ Occurs by Hydrogen Atom Abstraction. J. Am. Chem. Soc 2003, 125, 10351–10361. [DOI] [PubMed] [Google Scholar]
- (58).Since the oxidative substrate reactivity is greatly enhanced with LutH+ present in solution, any F8Cmpd-II present in solution is most likely rapidly transformed to F8Cmpd-II(LutH+) prior to reacting with substrate. Thus, the possibility of two species being present in solution should not have any influence on the kinetic analyses.
- (59).Wang X; Ullrich R; Hofrichter M; Groves JT Heme-Thiolate Ferryl of Aromatic Peroxygenase Is Basic and Reactive. Proc. Natl. Acad. Sci. U.S.A 2015, 112, 3686–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Mayer JM Acc. Chem. Res 1998, 31, 441–450. [Google Scholar]
- (61).Shaik S; Kumar D; de Visser SP A Valence Bond Modeling of Trends in Hydrogen Abstraction Barriers and Transition States of Hydroxylation Reactions Catalyzed by Cytochrome P450 Enzymes. J. Am. Chem. Soc 2008, 130, 10128–10140. [DOI] [PubMed] [Google Scholar]
- (62).Cong Z; Kinemuchi H; Kurahashi T; Fujii H Factors Affecting Hydrogen-Tunneling Contribution in Hydroxylation Reactions Promoted by Oxoiron(IV) Porphyrin π-Cation Radical Complexes. Inorg. Chem 2014, 53, 10632–10641. [DOI] [PubMed] [Google Scholar]
- (63).Boaz NC; Bell SR; Groves JT Ferryl Protonation in Oxoiron(IV) Porphyrins and Its Role in Oxygen Transfer. J. Am. Chem. Soc 2015, 137, 2875–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Goldsmith CR; Jonas RT; Stack TDP C-H Bond Activation by a Ferric Methoxide Complex: Modeling the Rate-Determining Step in the Mechanism of Lipoxygenase. J. Am. Chem. Soc 2002, 124, 83–96. [DOI] [PubMed] [Google Scholar]
- (65).We acknowledge and thank a reviewer who explained and corrected our statement of large KIE values (i.e., >100; Table S10) which come from a direct analysis of the experimentally observed kinetics and which we initially thought to report. Accurate KIE values cannot be obtained with the level (even though small) of protium contamination in our substrates. For example, the small amount of proteo-xanthene present in the reaction mixture reacts as fast as (or faster than) the much larger amount of deutero-xanthene, suggesting that the KIE values observed are only rough estimates and should be considered as qualitative minimum values. KIE values greater than ~50 cannot be accurately determined, given the current experimental conditions and methodologies.
- (66).Mandal D; Mallick D; Shaik S Kinetic Isotope Effect Determination Probes the Spin of the Transition State, Its Stereochemistry, and Its Ligand Sphere in Hydrogen Abstraction Reactions of Oxoiron(IV) Complexes. Acc. Chem. Res 2018, 51, 107–117. [DOI] [PubMed] [Google Scholar]
- (67).Klinker EJ; Shaik S; Hirao H; Que L A Two-State Reactivity Model Explains Unusual Kinetic Isotope Effect Patterns in C–H Bond Cleavage by Nonheme Oxoiron(IV) Complexes. Angew. Chem., Int. Ed 2009, 48, 1291–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Kwon YH; Mai BK; Lee Y-M; Dhuri SN; Mandal D; Cho K-B; Kim Y; Shaik S; Nam W Determination of Spin Inversion Probability, H-Tunneling Correction, and Regioselectivity in the Two-State Reactivity of Nonheme Iron(IV)-Oxo Complexes. J. Phys. Chem. Lett 2015, 6, 1472–1476. [DOI] [PubMed] [Google Scholar]
- (69).Layfield JP; Hammes-Schiffer S Hydrogen Tunneling in Enzymes and Biomimetic Models. Chem. Rev 2014, 114, 3466–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Mandal D; Ramanan R; Usharani D; Janardanan D; Wang B; Shaik S How Does Tunneling Contribute to Counterintuitive H-Abstraction Reactivity of Nonheme Fe(IV)O Oxidants with Alkanes? J. Am. Chem. Soc 2015, 137, 722–733. [DOI] [PubMed] [Google Scholar]
- (71).Mandal D; Shaik S Interplay of Tunneling, Two-State Reactivity, and Bell–Evans–Polanyi Effects in C–H Activation by Nonheme Fe(IV)O Oxidants. J. Am. Chem. Soc 2016, 138, 2094–2097. [DOI] [PubMed] [Google Scholar]
- (72).Klinman JP The Role of Tunneling in Enzyme Catalysis of C–H Activation. Biochim. Biophys. Acta, Bioenerg 2006, 1757, 981–987. [DOI] [PubMed] [Google Scholar]
- (73).Zhang Y; Lin H Quantum Tunneling in Testosterone 6β-Hydroxylation by Cytochrome P450: Reaction Dynamics Calculations Employing Multiconfiguration Molecular–Mechanical Potential Energy Surfaces. J. Phys. Chem. A 2009, 113, 11501–11508. [DOI] [PubMed] [Google Scholar]
- (74).Krauser JA; Guengerich FP Cytochrome P450 3A4-Catalyzed Testosterone 6β-Hydroxylation Stereochemistry, Kinetic Deuterium Isotope Effects, and Rate-Limiting Steps. J. Biol. Chem 2005, 280, 19496–19506. [DOI] [PubMed] [Google Scholar]
- (75).Price JC; Barr EW; Tirupati B; Bollinger JM; Krebs C The First Direct Characterization of a High-Valent Iron Intermediate in the Reaction of an α-Ketoglutarate-Dependent Dioxygenase: A High-Spin Fe(IV) Complex in Taurine/α-Ketoglutarate Dioxygenase (TauD) from Escherichia coli. Biochemistry 2003, 42, 7497–7508. [DOI] [PubMed] [Google Scholar]
- (76).Price JC; Barr EW; Glass TE; Krebs C; Bollinger JM Evidence for Hydrogen Abstraction from C1 of Taurine by the High-Spin Fe(IV) Intermediate Detected during Oxygen Activation by Taurine:α-Ketoglutarate Dioxygenase (TauD). J. Am. Chem. Soc 2003, 125, 13008–13009. [DOI] [PubMed] [Google Scholar]
- (77).Price JC; Barr EW; Hoffart LM; Krebs C; Bollinger JM Kinetic Dissection of the Catalytic Mechanism of Taurine:α-Ketoglutarate Dioxygenase (TauD) from Escherichia coli. Biochemistry 2005, 44, 8138–8147. [DOI] [PubMed] [Google Scholar]
- (78).A reviewer pointed out the possibility that π–π stacking interactions between the substrate and the lutidine molecule may facilitate substrate approach to the iron(IV) oxo moiety.
- (79).Pu J; Gao J; Truhlar DG Multidimensional Tunneling, Recrossing, and the Transmission Coefficient for Enzymatic Reactions. Chem. Rev 2006, 106, 3140–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Fukuzumi S; Ohkubo K; Lee Y-M; Nam W Lewis Acid Coupled Electron Transfer of Metal-Oxygen Intermediates. Chem. - Eur. J 2015, 21, 17548–17559. [DOI] [PubMed] [Google Scholar]
- (81).Extrapolation of the kinetic plots in Figure 8 suggest that at lower concentrations of DHA (i.e., 10 equiv or less) the rate of reaction of F8Cmpd-II(LutD+) becomes less than that of F8Cmpd-II(LutH+). One possibile explanation for this is that as the rate of the reaction between F8Cmpd-II(LutD+) and DHA decreases, there is the chance for exchange between LutD+ and mCBA, which would reduce the rate. It is also possible that at such low concentrations the reaction would no longer be under pseudo-first-order conditions, and thus, results in that region are not reliable.
- (82).Anslyn EV; Dougherty DA Modern Physical Organic Chemistry; University Science Books, 2006. [Google Scholar]
- (83).Ray K; Pfaff FF; Wang B; Nam W Status of Reactive Non-Heme Metal–Oxygen Intermediates in Chemical and Enzymatic Reactions. J. Am. Chem. Soc 2014, 136, 13942–13958. [DOI] [PubMed] [Google Scholar]
- (84).Chai C; Armarego WLF Purification of Laboratory Chemicals, 6th ed.; Elsevier Inc: Amsterdam, 2009. [Google Scholar]
- (85).Ghiladi RA; Chufán EE; del Río D; Solomon EI; Krebs C; Huynh BH; Huang H; Moënne-Loccoz P; Kaderli S; Honecker M; Zuberbühler AD; Marzilli L; Cotter RJ; Karlin KD Further Insights into the Spectroscopic Properties, Electronic Structure, and Kinetics of Formation of the Heme–Peroxo–Copper Complex [(F8TPP)FeIII–(O22−)–CuII(TMPA)]+. Inorg. Chem 2007, 46, 3889–3902. [DOI] [PubMed] [Google Scholar]
- (86).Garcia-Bosch I; Sharma SK; Karlin KD A Selective Stepwise Heme Oxygenase Model System: An Iron(IV)-Oxo Porphyrin π-Cation Radical Leads to a Verdoheme-Type Compound via an Isoporphyrin Intermediate. J. Am. Chem. Soc 2013, 135, 16248–16251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Ghiladi RA; Hatwell KR; Karlin KD; Huang H; Moënne-Loccoz P; Krebs C; Huynh BH; Marzilli LA; Cotter RJ; Kaderli S; Zuberbü AD Dioxygen Reactivity of Mononuclear Heme and Copper Components Yielding A High-Spin Heme–Peroxo–Cu Complex. J. Am. Chem. Soc 2001, 123, 6183–6184. [DOI] [PubMed] [Google Scholar]
- (88).Curley JJ; Bergman RG; Tilley TD Preparation and Physical Properties of Early-Late Heterobimetallic Compounds Featuring Ir-M Bonds (M = Ti, Zr, Hf). Dalton Trans 2012, 41, 192–200. [DOI] [PubMed] [Google Scholar]
- (89).Sastri CV; Lee JJ; Oh K; Lee YJ; Lee JJ; Jackson TA; Ray K; Hirao H; Shin W; Halfen JA; Kim J; Que L; Shaik S; Nam W Axial Ligand Tuning of a Nonheme Iron(IV)–Oxo Unit for Hydrogen Atom Abstraction. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 19181–19186 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.