

Abstract
Prostate cancer (PCa) is the second leading cause of death in men. Apart from androgen receptor, 5α-reductase has also been recognized as a potential target. In this study, a series of androst-17β-amide compounds have been designed and synthesized targeting both AR and 5α-reductase. Their anti-proliferation activities were evaluated in AR + cell line 22RV1 and AR − cell line PC-3. The results indicated that most of the synthesized compounds inhibited the testosterone-stimulated cell proliferation with good selectivity and safety. Among all the compounds, androst[3,2-c]pyrazole derivatives (9a–9d) displayed the best inhibition activity comparable with flutamide. Moreover, most of the synthesized compounds displayed good 5α-reductase inhibitory activities with IC50 lower than 1 μM. The docking result of 9d-AR indicated that AR was forced to expands its binding cavity and maintain an antagonistic conformation since the steric hindrance of 9d impeded H12 transposition. Overall, compound 9d can be identified as a potential dual 5α-reductase inhibitor and AR antagonist, which might be of therapeutic importance for PCa treatment.
Keywords: 5α-reductase Inhibitors, androgen receptor antagonists, prostate cancer
1. Introduction
Prostate cancer (PCa) is the second leading cause of death in men only less than lung cancer. It was estimated that around 220,800 cases were diagnosed in the United States in 2015 alone. In China, that number was 60,300 and has increased rapidly over the last 10 years1. Androgens, including testosterone (T) and dihydrotestosterone (DHT), and androgen receptor (AR) signalling pathway are essential for prostate development and homeostasis2. Huggins et al.3 introduced androgen deprivation therapy (ADT) for advanced and metastatic PCa in 1941. Thereafter, androgen ablation therapy has been shown to produce the most beneficial responses in multiple settings in PCA patients.
As the substantial clinical efficacy with AR blockade in PCa patients, AR has been recognized as an attractive target for the treatment of PCa. A number of small molecular AR antagonists, such as bicalutamide and flutamide (Figure 1) have been shown good therapeutic effects in clinic4. Newly approved enzalutamide (Figure 1) is a new generation AR antagonist which is effective for bicalutamide-resistant tumours and has been used in CRPC patients5. Although AR antagonists shown great benefits in treating PCa, drug resistances caused by AR mutation occur spontaneously in PCa6,7 as well as altered steroidogenesis8 underlies the emergence of castration-resistant prostate cancer (CRPC) within 2–3 years after starting ADT. Therefore, the exploration of developing new anti-PCa agents with increased activity and fewer side effects is still in urgent.
Figure 1.

The AR antagonists and 5α-reductase inhibitors in clinical.
Recently, it has been shown that the low levels of DHT in the prostate after ADT is a result of intratumoral androgen synthesis9. 5α-reductases, which are NADPH-dependent enzymes, are responsible for the reduction of T to DHT. The 5α-reductase family is composed of three isozymes, with the types 1 and 2 being the most known. Type 1 5α-reductase (5αR-1) is mainly expressed in prostate epithelial cells as well as in skin and liver at an optimal pH range of 6.0–8.5, while the type 2 5α-reductase (5αR-2) at an optimal pH 5.5, is mainly found in prostatestromal compartment and other genital tissues10. More recently, type 3 isozyme was identified in castration-resistant PCa cells as well as in other tissues such as the pancreas, brain, skin and adipose tissues11. The 5α-reductase levels, particularly type 1, appear to increase during the disease course of prostatic intraepithelial neoplasia and PCa, with greater expression occurring as the disease progresses12. Therefore, the inhibition of 5α-reductase could potentially reduce the risk of PCa development, prevent disease progression, and treat existing disease.
5α-reductase inhibitors, like finasteride and dutasteride (Figure 1), were used in the clinic for the treatment of BPH and were also proposed for chemoprevention and treatment of PCa. Dutasteride blocks both type 1and type 2 isoenzymes and has an inhibitory effect in PCa13,14. It is currently being studied as a chemopreventive agent and in combination with other androgen-reducing agents in PCa treatment. It was found that combination of dutasteride and enzalutamide synergistically inhibited tumour cell proliferation15. In our previous study, several androstene-17β-carboxamides has been reported as dual 5α-reductase inhibitors and antiandrogens showing good anti-proliferative activities in a human PCa cell line LNCaP cells and PC-3 cells16. These findings supported clinical studies with combinations of a 5α-reductase inhibitor and antiandrogens for the first line treatment of PCa and CRPC.
Considerable studies have been focused on the modifications of substitutions at C-17 position of steroid skeleton. Among them, aniline moiety has been widely used as functional groups in compounds targeting androgen axis16,17. This bulk in antagonist drugs, though small, is thought to be essential in pushing AR helix12 (H12) away from the binding pocket, disturbing the agonist conformation of the receptor18 (Figure 2).
Figure 2.

The rational design strategy of dual AR/5α-reductase inhibitors.
As the 5α-reductase isozymes have not been purified and crystallized due to their unstable nature, the design and optimisation of novel 5α-reductase inhibitor mainly relies on structure-based optimisation approach. 5α-reductase inhibitors are possible to postulate two different transition states (TS): the “substrate like” TS (S-TS) and the “product like” TS (P-TS) (Figure 2)19. For S-TS inhibitors, stabilisation of the proposed enolate intermediate is thus an obvious strategy for the design of potential inhibitors, and a number of compounds that satisfy this requirement have been produced. Thus it can be seen that introducing electron-withdrawing group in A ring and B ring is beneficial for inhibitory activity.
Thus, we set out to design and synthesis a number of novel dual 5α-reductase and AR antagonists by involving electron-withdrawing group at A ring of androst-17β-amides (Figure 2). The anti-proliferative activities of synthesized compounds were evaluated against 2 different PCa cell lines. Their inhibitory activity 5α-reductase isozyme types 1 and 2 were further assessed, respectively.
2. Materials and methods
2.1. Chemistry
Melting points of compounds were measured on a RY-1 melting point apparatus and were uncorrected. Nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker AV-300 (300 MHz) spectrometer as deuterochloroform (CDCl3) solutions using tetramethylsilane (TMS) as an internal standard (δ = 0) unless noted otherwise. Electron impact mass spectral (EI-MS) data were obtained on a SHIMADZU GCMS-QP2010 system. All chemicals were purchased from commercial sources and were used without further purification unless otherwise noted. The solvents (such as MeOH, EtOAc, EtOH, CH2Cl2, and others) were C.P. grade purchased from Nanjing Chemical Co., Ltd. and used without further purification. Column chromatography (CC) was carried out on silica gel (200–300 mesh, Qingdao Ocean Chemical Company, China). Thin-layer chromatography (TLC) analyses were carried out on silica gel GF254 (Qingdao Ocean Chemical Company, China) glass plates (2.5 × 10 cm with 250 µm layer). Concentration and evaporation of the solvent after reaction or extraction was carried out on a rotary evaporator operated at reduced pressure.
2.1.1. Synthesis of 3β-hydroxyl-5-pregnen-20-one-3-acetate(2)
To the solution of 3β-hydroxyl-5,16-Pregnadien-20-one-3-acetate (6 g, 16.8 mmol) in 180 ml EtOAc, 0.6 g Raney Ni was slowly added at 0 °C. The reaction mixture was stirred at 25 °C for 1 h. After filtration, the filtrate was evaporated to provide white solid 5.924 g, yield 98.18%. ESI-MS m/z: 381[M + Na]+.
2.1.2. Synthesis of 3β-hydroxyl-5-pregnen-20-one(3)
To the solution of 2 (5.92 g, 16.5 mmol) in 36 ml MeOH, 9 ml 20% K2CO3 aqueous solution was added. The reaction mixture was refluxed for 2 h. After cooled to room temperature, the resulting precipitate was filtered, washed with water, and dried to give white solid 4.85 g, yield 92.81%. ESI-MS m/z: 339[M + Na]+.
2.1.3. Synthesis of 3-oxo-4-pregnen-20-one(4)
To the solution of 3 (7 g, 22 mmol) in 112 ml totuene, cyclohexanone (21 ml, 0.2 mol) was added. After circumfluence to repel water for 2 h, aluminiumisopropoxide (1.05 g, 5.1 mmol) was added. After refluxed for another 1.5 g, the resulting mixture was poured into 10 ml cold 10% NaOH aqueous solution. The solvent was evaporated by steam distillation to provide yellow solid 6.3 g, yield 90.03%
2.1.4. Synthesis of 17β-carboxy-androst-4-ene-3-one(5)
To the solution of NaOH (5.9 g, 143 mmol) in 50 ml water, Br2 (1.9 ml, 37 mmol) was slowly added at 0 °C to provide NaOBr aqueous solution. Resolving 4 (3.5 g, 11 mmol) in 105 dioxane and 36 ml water, the NaOBr aqueous solution was added. After stirred at room temperature for 1.5 h, 19 ml 10% Na2SO3 was added and refluxed for 15 min. After cooled to room temperature, 2 N hydrochloric acid was added to adjust PH to 2 in ice bath. Dioxane was evaporated and the resulting precipitate was filtered, washed with water, and dried to give crude product. Recrystallized by acetone to provide white solid 2.6 g, yield 75.48%.
2.1.5. General procedure for the preparation of 2-hydroxymethylene-17β-formamido-androst- 4-ene-3-one (7a–7e)
To a solution of 5 (1 g, 3.16 mmol), anhydrous pyridine (0.32 ml, 4.12 mmol) in 15 ml anhydrous toluene, oxalyl chloride (0.34 ml, 3.56 mmol) in 7 ml anhydrous toluene was added drop wise at 0 °C. The resulting solution was stirred at room temperature 1.5 h then 7.9 mmol corresponding amine was added. The mixture was stirred at room temperature for another 8 h. 30 ml water was added and the reaction mixture was extracted three times with ethyl acetate. It was dried with sodium sulfate and the solvent was removed in vacuum. This compound was used in the next step without purification.
To the solution of 1.23 mmol corresponding amido product in 6 ml THF, NH (0.3 g, 7.4 mmol) was added. The mixture was stirred at room temperature for 0.5 h. 4 ml ethyl formate was added and the reaction mixture was refluxed for 2 h. After cooled to room temperature, the reaction mixture was poured into 20 ml water. Adjust PH to 2 by 2 N hydrochloric acid then extracted three times with ethyl acetate. It was dried with sodium sulfate and the solvent was removed in vacuum. The compound was purified by column chromatography (PE/Ethyl acetate, 2:1).
2.1.6. General procedure for the preparation of 2-hydroxymethylene-17β-(N-isobutyl-carboxamide)-androst-4-ene-3-one (7a)
Yield: 55% of pure product, m.p(0).152–158 °C. 1H NMR (CDCl3, 300 Hz) δ: 0.8 (s, 3H, 18-CH3), 1.08 (s, 3H, 19-CH3), 1.47 (s, 9H, t-Bu), 5.83 (s, 1H, 4-H), 7.30 (s, 1H, 2-ethylene), 13.82 (s, 1H, –OH) ppm. 13C NMR (CDCl3, 300 Hz) δ: 13.30, 17.58, 20.89, 24.22, 25.34, 26.84, 30.95, 31.96, 35.30, 37.01, 38.09, 42.7, 46.62, 50.16, 52.66, 55.35, 102.94, 109.85, 122.50, 123.96, 164.69, 169.55 ppm. HRMS (ESI): m/z [M + H]+. Calcd for C25H37NO3: 399.2773; Found: 399.2778.
2.1.7. General procedure for the preparation of 2-hydroxymethylene-17β-(piperidine-1-carbonyl)-androst-4-ene-3-one(7b)
Yield: 42% of pure product, m.p. 105–110 °C. 1H NMR (CDCl3, 300 Hz) δ: 0.82 (s, 3H, 18-CH3), 1.04 (s, 3H, 19-CH3), 3.2–3.0 (m, 4H, CH2-N-CH2), 5.76 (s, 1H, 4-H), 13.80 (s, 1H, −OH) ppm. 13C NMR (CDCl3, 300 Hz) δ: 12.98, 13.36, 14.18, 17.51, 20.8, 24.21, 25.25, 29.16, 30.9, 31.92, 35.2, 36.99, 38.22, 39.36, 39.75, 41.39, 44.39, 50.38, 52.58, 55.24, 105.9, 122.41, 164.62, 169.65, 171.69 ppm. HRMS (ESI): m/z [M + H]+. Calcd for C26H37NO3: 411.2777; Found: 411.2773.
2.1.8. General procedure for the preparation of 2-hydroxymethylene-17β- (N,N-diethy-carboxamide)-androst-4-ene-3-one(7c)
Yield: 68% of pure product, m.p. 153–159 °C.1H NMR (CDCl3, 300 Hz) δ: 0.84 (s, 3H, 18-CH3), 1.03 (s, 3H, 19-CH3), 3.2 8–3.02 (m, 4H, CH2–N–CH2), 5.37 (s, 1H, –CONH–), 5.83 (s, 1H, 4-H), 7.30 (s, 1H, 2-ethylene), 13.78 (s, 1H, –OH) ppm. 13C NMR (CDCl3, 300 Hz) δ: 12.68, 19.72, 20.83, 23.15, 23.97, 28.12, 29.20, 30.83, 31.91, 35.22, 37.07, 37.92, 39.40, 43.14, 46.34, 52.60, 55.04, 56.69, 105.92, 122.53, 152.91, 156.49, 164.72, 173.14 ppm. EI-MS m/z: 387 (M+); HRMS (ESI): m/z [M + H]+. Calcd for C25H37NO3: 399.2779; Found: 399.2773.
2.1.9. General procedure for the preparation of 2-hydroxymethylene-17β-(N-(3,4-dimethoxyphenethyl)-carboxamide)-androst-4-ene-3-one(7d)
Yield: 65% of pure product, m.p. 121–129 °C. 1H NMR (CDCl3, 300 Hz) δ: 0.74 (s, 3H, 18-CH3), 1.01 (s, 3H, 19-CH3), 3.53 (m, 4H-, N–CH2–CH2–Ar), 3.92 (s, 6H, –OCH3), 5.35 (s, 1H, –CONH–), 5.89 (s, 1H, 4-H), 6.00 (s, 1H, Ar-2H), 6.79 (d, J = 6.70, 1H, Ar-5H), 6.88 (d, J = 6.87, 1H, Ar-6H) ppm. 13C NMR (101 MHz, CDCl3) δ: 13.07, 13.24, 18.04, 21.25, 23.57, 24.44, 31.33, 32.38, 35.44, 35.73, 37.57, 38.2, 39.88, 40.56, 43.57, 53.1, 55.5, 55.87, 55.97, 56.91, 57.03, 76.71, 77.03, 77.35, 106.38, 111.41, 111.94, 120.66, 123, 131.44, 147.75, 149.13, 165.39, 169.76, 172.47, 188.95 ppm. HRMS (ESI): m/z [M + H]+. Calcd for C31H41NO5: 507.2990; Found: 507.2985.
2.1.10. General procedure for the preparation of 2-hydroxymethylene-17β-(N-cyclopropylamino-carbonyl)-androst-4-ene-3-one (7e)
Yield: 73% of pure product, m.p. 125–130 °C. 1H NMR (CDCl3, 300 Hz) δ: 0.72 (s, 3H, 18-CH3), 1.09 (s, 3H, 19-CH3), 2.76 (m, 1H, –NH-CH–), 5.82 (s, 1H, 4-H), 7.41 (s, 1H, 2-ethylene), 13.79 (s, 1H, –OH) ppm. 13C NMR (CDCl3, 300 Hz) δ: 5.85, 6.30, 6.46, 12.66, 17.55, 20.87, 22.04, 22.49, 23.01, 23.95, 30.82, 31.89, 35.19, 37.04, 37.87, 39.39, 43.2, 52.58, 55.02, 55.70, 56.20, 105.9, 122.50, 164.75, 169.43, 179.44, 188.59 ppm. HRMS (ESI): m/z [M + H]+. Calcd for C24H33NO3: 383.2459; Found: 383.2460.
2.1.11. General procedure for the preparation of 2-methylene-17-formamido-androst-4-ene-3-one (8a–8e)
To the solution of 1 mmol corresponding 2-hydroxymethylene product in 8 ml acetone, 0.33 ml formaldehyde was added. The resulting mixture was stirred at room temperature for 2 h. Upon completion, the reaction mixture was poured into 30 ml water and extracted with ethyl acetate. The combined organic layers were dried and the solvent was evaporated in vacuo to provide yellow solid. The compound was purified by column chromatography (PE/ethyl acetate, 2:1).
2.1.12. General procedure for the preparation of 2-methylene-17β-(N-isobutyl-carboxamide)-androst-4-ene-3-one (8a)
Yield: 55% of pure product, m.p. 168–175 °C. 1H NMR (CDCl3, 300 Hz) δ: 0.75 (s, 3H, 18-CH3), 1.07 (s, 3H, 19-CH3), 1.47 (d, 6H, CH-(CH3)2), 3.52 (m, 2H, –NH–CH2–), 5.20 (s, 1H, 2-ethylene), 5.81 (s, 1H, 4-H), 5.91 (s, 1H, 2-ethylene) ppm. 13C NMR (75 MHz, CDCl3) δ: 13.00, 13.40, 14.22, 18.04, 20.69, 24.19, 25.30, 31.34, 32.20, 35.12, 38.22, 39.75, 40.28, 41.39, 44.39, 44.52, 50.41, 52.61, 55.32, 76.17, 76.59, 77.02, 119.75, 123.95, 140.79, 171.45, 171.65, 188.33 ppm. HRMS (ESI): m/z [M + H]+. Calcd for C25H37NO2: 383.2824; Found: 383.2897.
2.1.13. General procedure for the preparation of 2-methylene-17β-(piperidine-1-carbonyl)-androst-4-ene-3-one (8 b)
Yield: 42% of pure product, m.p. 150–155 °C. 1H NMR (CDCl3, 300 Hz) δ: 0.67 (s, 3H, 18-CH3), 1.00 (s, 3H, 19-CH3), 2.9 6–3.55 (m, 4H, –NH–(CH2)2–), 5.09 (s, 1H, 2-ethylene), 5.71 (s, 1H, 4-H), 5.81 (s, 1H, 2-ethylene) ppm. 13C NMR (75 MHz, CDCl3) δ: 13.49, 13.9, 14.71, 18.54, 21.22, 24.7, 25.84, 31.88, 32.7, 35.66, 38.77, 40.27, 40.79, 41.91, 44.9, 45.06, 50.97, 53.17, 55.88, 76.61, 77.03, 77.46, 120.21, 124.48, 141.31, 171.86, 172.18, 188.81 ppm. HRMS (ESI): m/z [M + H]+. Calcd for C26H37NO2: 395.2832; Found: 395.2824.
2.1.14. 2-methylene-17β-(N,N-diethy-carboxamide)-androst-4-ene-3-one (8c)
Yield: 68% of pure product, m.p. 98–105 °C. 1H NMR (CDCl3, 300 Hz) δ: 0.61 (s, 3H, 18-CH3), 1.12 (s, 3H, 19-CH3), 2.8 7–3.09 (m, 4H, –NH– (CH2)2–), 5.09 (s, 1H, –CONH–), 5.30 (s, 1H, 2-ethylene), 5.70 (s, 1H, 4-H),5.80 (s, 1H, 2-ethylene) ppm. 13C NMR (75 MHz, CDCl3) δ: 13.49, 13.90, 14.71, 18.54, 21.22, 24.70, 25.84, 31.88, 32.7, 35.66, 38.77, 40.27, 40.79, 41.91, 44.9, 45.06, 50.97, 53.17, 55.88, 76.61, 77.03, 77.46, 120.21, 124.48, 141.31, 171.86, 172.18, 188.81 ppm. HRMS (ESI): m/z [M + H]+. Calcd for C25H37NO2: 283.2826; Found: 283.2824.
2.1.15. General procedure for the preparation of 2-methylene-17β-(N-(3,4-dimethoxyphenethyl)-carboxamide)-androst-4-ene-3-one (8d)
Yield: 65% of pure product, m.p. 74–78 °C. 1H NMR (CDCl3, 300 Hz) δ: 0.74 (s, 3H, 18-CH3), 1.01 (s, 3H, 19-CH3), 3.53 (m, 4H, N-CH2-CH2-Ar), 5.29 (s, 1H, 2-ethylene), 5.35 (s, 1H, –CONH–), 3.92 (s, 6H, -OCH3), 5.89 (s, 1H, 4-H), 6.00 (s, 1H, 2-ethylene), 6.85 (m, 3H, Ar-H) ppm. 13C NMR (75 MHz, CDCl3) δ: 12.59, 17.54, 20.72, 23.04, 23.93, 30.81, 31.88, 34.94, 35.18, 37.03, 37.63, 39.36, 40.09, 43.07, 52.55, 54.95, 55.34, 55.43, 56.47, 76.18, 76.6, 77.03, 110.77, 111.33, 120.14, 122.49, 130.91, 147.14, 148.52, 164.75, 169.43, 172.02, 188.58 ppm. HRMS (ESI): m/z [M + H]+. Calcd for C31H41NO4: 491.3036; Found: 491.3107.
2.1.16. General procedure for the preparation of 2-methylene-17β-(N-cyclopropylamino-carbonyl)-androst-4-ene-3-one (8e)
Yield: 73% of pure product, m.p. 135–142 °C. 1H NMR (CDCl3, 300 Hz) δ: 0.53 (m, 4H, –CH2–CH2–), 0.69 (s, 3H, 18-CH3), 1.06 (s, 3H, 19-CH3), 2.75 (m, 4H, –NH–CH–), 5.45 (s, 1H, 4-H), 6.23 (s, 2H, 2-ethylene) ppm. 13C NMR (101 MHz, CDCl3) δ: 13.18, 18.56, 20.20, 21.20, 23.68, 24.42, 28.61, 29.69, 31.75, 32.65, 35.63, 38.39, 40.77, 43.64, 45.07, 46.86, 53.11, 55.59, 57.19, 76.70, 77.01, 77.33, 120.25, 124.54, 141.28, 171.70, 172.39, 188.77 ppm. HRMS (ESI): m/z [M + H]+. Calcd for C24H33NO2: 367.2520; Found: 367.2511.
2.1.17. General procedure for the preparation of General procedure for the preparation of 4-oxa-17-formamido-5α-androst-3-one (9a–9e)
To the solution of 1 mmol, corresponding 2-hydroxymethylene product in 8 ml hydrazine hydrate. The resulting mixture was refluxed for 2 h. Upon completion, the reaction mixture was poured into 30 ml water. The resulting precipitate was filtered, washed with water, and dried to give white solid. The compound was purified by column chromatography (PE/Ethyl acetate, 1:1).
2.1.18. General procedure for the preparation of 17β-(N-isobutyl-carboxamide)-androst[3,2-c]pyrazole-4-ene (9a)
Yield: 20.01% of pure product, m.p.:189–195 °C.1H NMR(CDCl3, 300 MHz) δ: 0.51 (m, 2H, −CHCH2CH2−), 0.75 (s, 3H, 18-CH3), 0.87 (m, 2H, −CHCH2CH2−), 1.11 (s, 3H, 19-CH3), 2.76 (s, 1H, −N(CH)), 5.47 (s, 1H, −CONH−), 5.82 (s, 1H, 4-H), 7.41 (s, 1H, 5-pyrazole) ppm. 13C NMR (75 MHz, CDCl3) δ: 13.02, 13.46, 14.21, 17.94, 21.11, 24.38, 25.35, 31.14, 31.79, 33.6, 36.04, 38.63, 39.77, 41.42, 44.59, 50.6, 53.65, 55.55, 76.15, 76.57, 77, 111.71, 111.84, 127.16, 148.76, 171.95 ppm. HRMS (ESI): m/z [M + H]+. Calcd for C24H33N3O: 380.2628; Found: 380.2951.
2.1.19. General procedure for the preparation of 17β-(Piperidine-1-carbonyl)- androst[3,2-c]pyrazole -4-ene (9 b)
Yield: 45.20% of pure product, m.p.:154–162 °C;1H NMR(CDCl3, 300 MHz) δ: 0.77 (s, 3H, 18-CH3), 0.95 (s, 3H, 19-CH3), 3.56 (m, 4H,-N(CH2)2), 6.18 (s, 1H, 4-H), 6.54 (s, 1H, –CONH–), 7.22 (s, 1H, 5-pyrazole) ppm. 13C NMR (75 MHz, CDCl3) δ: 13.01, 13.42, 14.2, 17.95, 22.12, 24.39, 25.36, 31.15, 31.79, 33.58, 36.06, 38.62, 38.65, 39.73, 41.37, 44.55, 50.64, 53.67, 55.60, 92.74, 111.72, 112.07, 166.15 ppm. HRMS (ESI): m/z [M + H]+. Calcd for C26H37N3O2: 408.2940; Found: 408.3009.
2.1.20. General procedure for the preparation of 17β-(N,N-diethy-carboxamide)-androst[3,2-c]pyrazole-4-ene (9c)
Yield: 47.91% of pure product, m.p.:111–118 °C; 1H NMR(CDCl3, 300 MHz) δ: 0.77 (s, 3H, 18-CH3), 3.01 (m, 4H, –NCH2), 3.76 (m, 4H, –NCH2), 6.18 (s, 1H, 4-H), 0.95 (s, 3H, 19-CH3), 7.21 (s, 1H, 5-pyrazole) ppm. 13C NMR (101 MHz, CDCl3) δ: 13.07, 18.03, 21.25, 23.57, 24.44, 31.33, 32.38, 35.44, 35.73, 37.57, 38.2, 39.88, 40.56, 43.57, 53.1, 55.5, 55.87, 55.97, 57.03, 76.71, 77.03, 77.34, 106.37, 111.41, 111.94, 120.66, 123, 131.44, 147.75, 149.13, 165.39, 169.76, 172.47, 188.95 ppm. HRMS (ESI): m/z [M + H]+. Calcd for C29H37N3O: 396.2941; Found: 396.3013.
2.1.21. General procedure for the preparation of 17β-(N-(3,4-dimethoxyphenethyl)-carboxamide)-androst[3,2-c]pyrazole-4-ene(9d)
yield: 37.40%of pure product, m.p.:137–145 °C; 1H NMR(CDCl3, 300 MHz) δ: 0.65 (s, 3H, 18-CH3), 0.87 (s, 3H, 19-CH3), 3.74 (s, 6H, −OCH3), 6.08 (s, 1H, 4-H), 6.79(m, 2H, Ar-H), 7.32(s, 1H, -CONH), 7.45 (s, 1H, 5-pyrazole), 12.15(s, 1H, 1-pyrazole)ppm. 13C NMR (CDCl3, 75 MHz) δ: 5.85, 6.46, 12.66, 17.55, 22.04, 22.49, 23.91, 30.82, 31.89, 35.9, 37.04, 37.87, 39.39, 43.2, 52.58, 55.2, 55.7, 56.2, 105.9, 122.5, 164.75, 169.43, 173.44, 188.59 ppm. HRMS (ESI): m/z [M + H]+.Calcd for C31H41N3O3: 504.3156; Found: 504.3225.
2.1.22. General procedure for the preparation of 17β-(N-cyclopropylamino-carbonyl)-androst[3,2-c]pyrazole-4-ene (9e)
Yield: 73% of pure product, m.p.:91–97 °C; 1H NMR(CDCl3, 300 MHz) δ: 0.66 (s, 3H, 18-CH3), 0.86 (s, 3H, 19-CH3), 1.67 (m, 6H, −CH(CH3)2), 6.08 (s, 1H, 4-H), 7.31(s, 1H, −CONH),7.50 (s, 1H, 5-pyrazole), 12.22 (s, 1H, 1-pyrazole) ppm. 13C NMR (CDCl3, 75 MHz) δ: 5.85, 6.31, 6.47, 12.68, 18.05, 20.63, 22.43, 23.04, 23.9, 32.12, 32.14, 35.08, 37.84, 40.26, 41.37, 43.19, 44.53, 44.59, 44.9, 44.9, 45.07, 52.55, 52.55, 52.58, 53.1, 55.06, 56.2, 119.83, 124.2, 140.74, 171.28, 173.43 ppm. HRMS (ESI): m/z [M + H]+. Calcd for C29H37N3O: 396.2941; Found: 396.3013.
2.2. Biological activity
2.2.1. Cell lines and culture conditions
Human prostate carcinoma 22RV1 (CRL-2505, ATCC) and PC-3 (CRL-1435, ATCC) cells were cultured in RPMI-1640 medium (Invitrogen), supplemented with 10% (v/v) foetal calf serum (FCS) (Invitrogen), 1% (v/v), penicillin and streptomycin (Invitrogen) at 37 °C with 5% CO2 in a humidified atmosphere. Fresh medium was given every second day and on the day before the experiments were done. Cells were passaged at preconfluent densities, using a solution containing 0.05% trypsin.
2.2.2. Cell viability assay (MTT)
The anti-cancer activity in vitro was measured using the MTT assay. Exponentially growing cells were harvested and plated in 96-well plates at a concentration of 1 × 104 cells/well. After 24 h incubation at 37 °C under a humidified 5% CO2 to allow cell attachment, the cells in the wells were respectively treated with target compounds at various concentrations for 48 h. The concentration of DMSO was always kept below 0.5%, which was found to be nontoxic to the cells. A solution of 3–(4,5-dimethylthizao1-2-y1)-2,5-diphenyltetrazolium bromide (MTT), was prepared at 5 mg/ml in phosphate buffered saline (PBS: 1.5 mM KH2PO4, 6.5 mM Na2HPO4, 137 mM NaCl, 2.7 mM KCl; pH 7.4). Of this solution 20 µl was added to each well. After incubation for 4 h at 37 °C in a humidified incubator with 5% CO2, the medium/MTT mixtures were removed, and the formazan crystals formed by the mitochondrial dehydrogenase activity of vital cells were dissolved in 100 µl of DMSO per well. The absorbance of the wells was read with a microplate reader at 570 nm. Effects of the drug cell viability were calculated using cell treated with DMSO as control.
2.2.3. Enzyme inhibition test
Female rats were sacrificed and liver were taken within 5 min. All the following steps were performed at 0–4 °C. 1 g liver was cut into 3 mm pieces and added to 3 times volume of PBS. The tissue was homogenized and centrifuged at 10,000 g for 30 min. The supernatant was collected and centrifuged repeatedly at 100,000 for 1 h. The pellet obtained was resuspended in phosphate buffer (contained 30% glycerol, 1:3 v/v) and stored at −70 °C. The incubation mixture contained 50 µL enzyme solution, 6 µL NADPH (80 µM), 6 µL T (20 µM), and 108 µL buffer with or without test compound. The negative control contained 50 µL enzyme solution, 6 µL NADPH (80 µM) and 114 µL buffer. The incubation was carried out for 30 min at 37 °C. The fluorescence decay rate was tested in 10 min at 37 °C.
2.3. Molecular modelling
The molecular modelling was performed with Discovery Studio.3.0/CDOCK protocol (Accelrys Software Inc.). The crystal structures of AR (PDB code: 2PNU) were downloaded from Protein Data Bank. Compound 9d was drowned and optimize using Hyperchem v7.0. The protein and ligands were optimized and charged with CHARMm force field to perform docking. Up to 10 conformations were retained and binding modes presented graphically are representative of the highest-scored conformations.
3. Results and discussion
3.1. Chemistry
Taking commercial available compound 16-dehydropregnenolone acetate (1) as starting material, the targeting compounds were synthesized through 8 steps (Scheme 1). Compound 2 was afforded by the selective reduction of 1 with Raney Ni in EtOH. After filtration, the hydrolysis of the acetate group at C-3 was produced by directly adding KOH to resulting filtrate to obtain compound 3. The resulting secondary alcohol at C-3 was oxidized to the α,β-unsaturated ketone compound 4 by Oppenauer oxidation using aluminium isopropoxide and cyclohexanone in refluxing toluene. The17-amide derivatives 6a–6e were then obtained by treating 5 with bromine and sodium hydroxide followed by acylation with different amide, respectively. The 2-hydroxymethylene derivatives 7a–7e were prepared by the formulation of 6a–6e with NaH and HCOOEt in toluene. Compound 8a–8e were then obtained by the reduction of HCHO. The cyclisation of compound 8a–8e under hydrazinium hydroxide in EtOH eventually afford compound 9a–9e. The representative 1H NMR, 13C NMR and MS spectra are depicted in the supplementary material.
Scheme 1.

The synthesis of compound 7a–9e.
3.2. Anti-proliferative activity
To investigate their anti-cancer effects, all the synthesized compounds were tested for their anti-proliferative activities in 2 PCa cell lines by taking finasteride and flutaminde as positive control.
The anti-proliferative activities were firstly evaluated against 22RV1 stimulated by 0.1 µM T or not. As shown in Table 1, most compounds presented better growth inhibition activity in T-stimulated group than the non-stimulated group, suggesting that the anti-proliferative effects were attributed to androgen-related signalling pathway due to their competitive binding to AR with T. For T-stimulated group, most compounds displayed good activity with the IC50 less than 30 µM. It was obvious that androst[3,2-c]pyrazoleis derivatives (9a∼9e) were more potent than others with their IC50 lower than 20 µM. Among them, compound 9d was the most potent with IC50 of 8.85 µM, much better than the positive control. Although androst[3,2-c]pyrazoleis common in anabolic steroid, luckily these derivatives shown no androgen agonistic activity. The phenethylamino substituted compounds (7d, 8d, and 9d) and piperidine substituted compounds (7b, 8b, and 9b) were more efficient than others. The small sterically hindered derivatives (7a, 8a, and 9a) showed lower growth inhibition activity, which proved that increasing of the steric hindrance of amide substitution led to better inhibitory activity. As presented in Figure 3, 0.1 µm of T was selectedas the most adequate to induce proliferation of 22RV1 cells. Results shown in Figure 3(A,B) indicate that compound 9d and flutamide inhibited cell proliferation in a dose-dependent manner. Compound 9d caused a much more significant decrease in cell viability for all concentrations than flutamide.
Table 1.
The anti-proliferation activities of synthesized compounds against 22RV1.
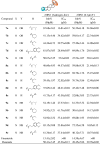 |
Figure 3.

Effects of compounds 9d (A) and flutamide (B) in cell viability of T-treated 22RV1 cells. 22RV1 cells were cultured with different concentrations of tested compounds and 0.1 μM T. Values are mean ± SD (n = 3). ***p < 0.001 versus T group.
To investigate the selectivity and safety of the synthesized compounds, the anti-proliferative activity was further assessed against PC-3 cell line. Since PC-3 is AR-independent cell, the anti-proliferative effect was mainly attributed to the non-androgen signal pathway mechanism or cytotoxicity. As shown in Table 2, the majority of compounds presented much weaker anti-proliferation activity toward PC-3 with IC50 twice higher than the IC50 toward 22RV1, which proves their good androgen antagonist activity and safety. The highest IC50 was observed among androst[3,2-c]pyrazoleis (9a∼9e), in which compound 9d was the most efficient with IC50 9.74 times higher than its IC50 toward 22RV1. The low toxicity and good selectivity warrant its further development for identifying more potent anticancer agents.
Table 2.
The anti-proliferation activities of synthesized compounds against PC-3.
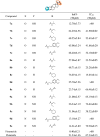 |
3.3. Enzyme inhibitory activity
The inhibitory activities of the synthesized compounds were then assessed towards the 5α-reductase 1 and 2 isozymes, respectively. Finasteride was used as positive control. As shown in Table 3, many compounds exhibited good inhibitory activities with IC50 lower than 1 µM, comparable to the positive control finasteride. For most compounds, the inhibitory activities towards the type 1 isozyme were slightly higher than type 2. Among them, the androst[3,2-c]pyrazole derivatives (9a–9e) were much more potent with the inhibition rate higher than 50%. The 2-methylene derivatives (8a–8e) presented a much inferior inhibitory activity to the others, which suggested that the electron-withdrawing group at 2-C was essential for the inhibitory activity. Consistent with the anti-proliferative result, it was also found that the increasing of the steric hindrance of amide substitution led to better inhibitory activity. The compounds with phenethylamino substitution (7d, 8d, and 9d) showed better inhibitory activities than others, in which, the most potential compounds 9d, with IC50 of 0.09 and 0.08 µM for 1 and 2 isozymes, respectively, was better than the positive control finasteride.
Table 3.
The inhibitory activities of synthesized compounds towards two 5α-reductaseiozymes.
| Compound | Type 1 (pH 6.6) |
Type 2 (pH 5.5) |
||
|---|---|---|---|---|
| Inh% (1 μM) | IC50 (μM) | Inh% (1μM) | IC50 (μM) | |
| 7a | 24.96 ± 9.68 | – | 24.16 ± 1.13 | – |
| 7b | 52.20 ± 12.33 | – | 49.91 ± 3.08 | – |
| 7c | 27.92 ± 4.01 | – | 24.52 ± 8.47 | – |
| 7d | 66.42 ± 4.37 | 0.58 ± 0.06 | 69.03 ± 9.30 | 0.41 ± 0.03 |
| 7e | 54.70 ± 5.94 | – | 45.75 ± 9.86 | – |
| 8a | 31.14 ± 6.49 | – | 19.88 ± 12.91 | – |
| 8b | 35.76 ± 9.58 | – | 21.17 ± 1.31 | – |
| 8c | 25.82 ± 4.22 | – | 18.29 ± 4.61 | – |
| 8d | 24.82 ± 9.02 | – | 26.06 ± 10.34 | – |
| 8e | 34.09 ± 10.05 | – | 20.94 ± 10.06 | – |
| 9a | 53.15 ± 9.71 | – | 48.69 ± 3.08 | – |
| 9b | 59.25 ± 2.28 | 0.63 ± 0.14 | 49.62 ± 5.75 | 1.01 ± 0.06 |
| 9c | 68.02 ± 11.96 | 0.52 ± 0.06 | 63.56 ± 12.47 | 0.41 ± 0.14 |
| 9d | 80.14 ± 6.73 | 0.09 ± 0.02 | 71.92 ± 3.40 | 0.08 ± 0.07 |
| 9e | 70.77 ± 9.74 | 0.23 ± 0.09 | 79.41 ± 2.47 | 0.21 ± 0.09 |
| Finasteride | 73.69 ± 4.22 | 1.04 ± 0.08 | 70.04 ± 4.12 | 0.04 ± 0.02 |
3.4. Molecular docking studies
To further rationalize the prospective activities of designed compound against AR, molecular docking studies were performed using the Discovery Studio 3.0/CDOCKER protocol. The docking orientation and interactions of 9d and DHT within the ligand binding domain (LBD) of AR (2PNU, PDB) are shown in Figure 4. As shown in Figure 4(B), AR was forced to expands its binding cavity and maintain an antagonistic conformation because of the steric hindrance that impede the transposition of H12. As an antagonist conformation of AR, there is only one hydrogen bond between DHT and AR forming with Arg752, but lacking the “latch” effect of the two hydrogen bond with Thr877 and Asn705 (Figure 4(A)). For 9d, its steroidal-core was favorably positioned similar to DHT while the bulky substitution at 17-amide pointing to H12 (Figure 4(A)). The detailed docking analysis in 2D diagram (Figure 4(C)) showed that the aromatic group at the substitution of C-17 established a Pi-Pi interaction with Trp741 when the amide group picked up hydrogen bonds with Thr877.
Figure 4.

(A) The detailed docking result of compound 9d (yellow) and DHT (green) with AR. The hydrogen bonds were indicated by green-dotted lines. The Pi–Pi interactions were indicated by orange lines. (B) The expended binding cavity (grey) of AR. (C) The 2D diagram of 9d-AR binding mode.
4. Conclusion
In this study, a series of substituted androst-17β-amide derivatives were designed and synthesized. Biological evaluation was performed on their anti-proliferative activities against 2 PCa cell lines. The result anti-proliferative effects against 22RV1 showed that most compounds exhibited better anti-proliferative activity in T-stimulate group, which was attributed to androgen-related signalling pathway due to their competitive binding to AR. Among them, compound 9d showed the best anti-proliferative activity with the IC50 of 8.85 µM, more potent than the positive control. Moreover, the investigation of anti-proliferative activity against PC-3 showed that the majority of compounds exhibited good selectivity and low toxicity against androgen-independent cells, suggesting their acceptable safety profiles as anti-tumour agents. In the further study of the inhibitory activity of the 5α-reductase 1 and 2 isozymes, androst[3,2-c]pyrazole derivatives (9a–9d) were observed as more potent inhibitors, which was consisted with their anti-proliferative activities. Among all the compounds, 9d was found to be the most potential inhibitor with the IC50 of 0.09 and 0.08 µM, respectively. Overall, based on biological activities data, compound 9d can be identified as potential dual 5α-reductase inhibitors and AR antagonists lead molecule which might be of therapeutic importance for PCa treatment.
Supplementary Material
Funding Statement
This work was supported by grants from the Research Fund of Shaanxi Provincial Education Department [grant number 18JS104] and Natural Science Basic Research Plan in Shaanxi Province of China [grant number 2019JQ-202]. We thank the support of The Youth Innovation Team of Shaanxi Universities.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- 2.Zhou Y, Bolton EC, Jones JO. Androgens and androgen receptor signaling in prostate tumorigenesis. J Mol Endocrinol 2015;54:R15–29. [DOI] [PubMed] [Google Scholar]
- 3.Huggins C, Stevens RE Jr, Hodges CV. Studies on prostatic cancer: Ii. The effects of castration on advanced carcinoma of the prostate gland. JAMA Surg 1941;43:209–23. [Google Scholar]
- 4.Helsen C, Van den Broeck T, Voet A, et al. Androgen receptor antagonists for prostate cancer therapy. Endocr Relat Cancer 2014;21:T105–118. [DOI] [PubMed] [Google Scholar]
- 5.Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. New Engl J Med 2012;367:1187–97. [DOI] [PubMed] [Google Scholar]
- 6.Armstrong CM, Gao AC. Drug resistance in castration resistant prostate cancer: Resistance mechanisms and emerging treatment strategies. Am J Clin Exp Urol 2015;3:64–76. [PMC free article] [PubMed] [Google Scholar]
- 7.Culig Z. Molecular mechanisms of enzalutamide resistance in prostate cancer. Curr Mol Biol Rep 2017;3:230–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang KH, Ercole CE, Sharifi N. Androgen metabolism in prostate cancer: From molecular mechanisms to clinical consequences. Br J Cancer 2014;111:1249–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamid AR, Pfeiffer MJ, Verhaegh GW, et al. Aldo-keto reductase family 1 member c3 (akr1c3) is a biomarker and therapeutic target for castration-resistant prostate cancer. Mol Med 2013;18:1449–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aggarwal S, Thareja S, Verma A, et al. An overview on 5alpha-reductase inhibitors. Steroids 2010;75:109–53. [DOI] [PubMed] [Google Scholar]
- 11.Uemura M, Tamura K, Chung S, et al. Novel 5 alpha-steroid reductase (srd5a3, type-3) is overexpressed in hormone-refractory prostate cancer. Cancer Sci 2008;99:81–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J, Ding Z, Wang Z, et al. Androgen regulation of 5α-reductase isoenzymes in prostate cancer: Implications for prostate cancer prevention. PloS One 2011;6:e28840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore CM, Robertson NL, Jichi F, et al. The effect of dutasteride on magnetic resonance imaging defined prostate cancer: mapped-a randomized, placebo controlled, double-blind clinical trial. J Urol 2017;197:1006–13. [DOI] [PubMed] [Google Scholar]
- 14.Andriole GL, Humphrey P, Ray P, et al. Effect of the dual 5alpha-reductase inhibitor dutasteride on markers of tumor regression in prostate cancer. J Urol 2004;172:915–9. [DOI] [PubMed] [Google Scholar]
- 15.Hamid AR, Verhaegh GW, Smit FP, et al. Dutasteride and enzalutamide synergistically suppress prostate tumor cell proliferation. J Urol 2015;193:1023–9. [DOI] [PubMed] [Google Scholar]
- 16.Lao K, Sun J, Wang C, et al. Design, synthesis and biological evaluation of novel 3-oxo-4-oxa-5α-androst-17β-amide derivatives as dual 5α-reductase inhibitors and androgen receptor antagonists. Bioorg Med Chem Lett 2017;27:4212–7. [DOI] [PubMed] [Google Scholar]
- 17.Lao K, Sun J, Wang C, et al. Design, synthesis and biological evaluation of novel androst-3,5-diene-3-carboxylic acid derivatives as inhibitors of 5α-reductase type 1 and 2. Steroids 2017;124:29–34. [DOI] [PubMed] [Google Scholar]
- 18.Zhou J, Liu B, Geng G, Wu JH. Study of the impact of the t877a mutation on ligand-induced helix-12 positioning of the androgen receptor resulted in design and synthesis of novel antiandrogens. Proteins 2010;78:623–37. [DOI] [PubMed] [Google Scholar]
- 19.Salvador JA, Pinto RM, Silvestre SM. Steroidal 5α-reductase and 17α-hydroxylase/17,20-lyase (cyp17) inhibitors useful in the treatment of prostatic diseases. J Steroid Biochem Mol Biol 2013;137:199–222. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.