

Abstract
Objective:
Arrhythmogenic right ventricular dysplasia (ARVD) is a myocardial genetic disease that occurs primarily in the right ventricle. Patients with ARVD may present with severe ventricular arrhythmias, syncope, and cardiac arrest. The purpose of this study is to evaluate the clinical features and arrhythmic complications of patients with pediatric-onset ARVD.
Methods:
Patients diagnosed with ARVD between January 2010 and January 2019 were included in this study.
Results:
A total of 19 patients with ARVD were evaluated. Of them, 15 patients were male, and their mean age was 12±4 years. The most common symptoms were palpitations (n=6), syncope (n=4), and heart failure symptoms (n=2). Five patients were asymptomatic. Thirteen patients had an epsilon wave; all patients ≥14 years had a T wave inversion in V1–3. Premature ventricular contractions (PVCs) were observed in 15 patients, and ventricular tachycardia (VT) was observed in 9 patients. All patients underwent cardiac magnetic resonance imaging (MRI). Echocardiography and cardiac MRI of two patients were normal at the time of admission; patients were in the concealed phase, and the diagnosis was made by ECG, Holter monitoring, and genetic findings. We administered a beta-blocker in all patients. Two patients underwent an electrophysiological study and ablation because of PVC/VT. An implantable cardiac defibrillator was implanted in 8 patients. The mean follow-up period was 21.5±11 months. Two patients were deceased with incessant VT and heart failure, and one patient was deceased with multiorgan dysfunction after biventricular assist device implantation (n=3).
Conclusion:
Diagnosis of pediatric-onset ARVD might be much more difficult in children. Sudden cardiac death might be prevented in the early period by raising the awareness of physicians about the disorder. Prevention of sudden death with implantable cardiac defibrillators is crucial in the management of these patients. It should be kept in mind that children with structurally normal hearts may present with an earlier concealed phase and can be diagnosed with ARVD.
Keywords: arrhythmogenic right ventricular dysplasia, children, ventricular arrhythmia
Introduction
Arrhythmogenic right ventricular dysplasia (ARVD) is a progressive heritable cardiomyopathy that results from the fibrous or fibrofatty replacement of myocardium that predisposes to ventricular arrhythmias and sudden cardiac death (SCD) in young people (1, 2). The clinical presentation of ARVD in individuals either under 12 years or over 60 years of age is very rare (3). The disease affects either only right ventricle (RV) or both ventricles with right-sided predominance. Left-dominant arrhythmogenic cardiomyopathy is another form of ARVD that is characterized by a predominant and early left ventricle (LV) impairment (4). Diagnosis is based on structural manifestations, histopathologic evaluation of the myocardium, electrocardiographic (ECG) abnormalities, arrhythmias, and familial history (5). Four stages of ARVD that had been reported previously (4, 6) may be used to determine the presentation and clinical course of the disease. Patients may present with a broad clinical spectrum that includes ventricular arrhythmias, heart failure, or sudden death. An asymptomatic presentation is also possible (4). It is implicated that physical activity is a factor that speeds up the emergence and structural progression of ARVD (4, 7). In addition to the other treatment choices like antiarrhythmic drugs, an implantable cardiac defibrillator (ICD) may be a life-saving therapy that should be considered in appropriate and high-risk cases (8, 9).
In this study, we present our experience to demonstrate the clinical features, arrhythmic complications, and follow-up of patients with pediatric-onset ARVD, from a tertiary pediatric cardiac referral center.
Methods
Patients diagnosed with ARVD by clinical, morphological, and imaging methods between January 2010 and January 2019 were included in this study. The demographic data, symptoms at the time of admission, familial history, 12-lead electrocardiography (ECG), transthoracic echocardiography, 24-hour Holter monitoring (three channel and 12-lead if required), exercise testing, cardiac magnetic resonance imaging (MRI), genetic testing, and myocardial biopsy results of the patients were evaluated retrospectively.
Electrocardiograms and Holter-ECG monitoring
Twelve-lead ECGs of the patients were recorded at rest with 10 mm/mV and 25 mm/s paper speed. Electrocardiograms were evaluated to determine the ECG findings of ARVD and all Holter recordings were reviewed for evaluating arrhythmias. All available 12-lead Holter recordings were reviewed to determine the type of premature ventricular contractions (PVCs) whether they originated from the right ventricle or the left ventricle. It was determined whether the PVCs were monomorphic or polymorphic and whether there was ventricular tachycardia (VT).
Transthoracic echocardiography
Transthoracic echocardiographic studies were performed using an HD11 XE ultrasound system (Philips Healthcare, Andover, Massachusetts), an iE33 ultrasound system (Philips Healthcare, Andover, Massachusetts), and an EPIQ 7 ultrasound system (Philips Healthcare, Andover, Massachusetts) using an appropriate transducer (8-3 or 5-1 MHz phased array transducer). Echocardiography was performed to determine the cardiac structure and also to evaluate the ventricular function. Two-dimensional imaging, M-mode sonography, color-flow Doppler, continuous-wave Doppler, and pulsed-wave Doppler echocardiography were used. Right ventricular outflow tract (RVOT) dimensions were measured in the parasternal long-axis and short-axis views; right ventricular volumes were measured using the Simpson method. Ejection fractions were estimated from the volumes (10). Right ventricular myocardium was assessed for hypokinetic or dyskinetic areas, thinning of the ventricular wall, diastolic bulgings, and segmental or global dilatation of the right ventricle.
Exercise testing
Exercise testing using the modified Bruce treadmill protocol was carried out in suitable patients (≥6 years). During the exercise, 12-lead ECG and blood pressure were monitored, and whether PVCs are augmented or suppressed by exercise was determined. The exercise test was suggested to have stopped when there was a failure of heart rate to increase with exercise, severe hypertension or progressive fall in systolic blood pressure, the presence of ≥2 mm ST-segment changing, increasing in ventricular ectopy with exercise or the presence of VT, and finally if complaints or signs of fatigue, dyspnea, or dizziness occurred in patients during exercise testing (11).
Cardiac magnetic resonance imaging
Cardiac MRI was performed to assess cardiac structures, ventricular motion abnormalities, ventricular volumes (RV, in particular) and functions, ventricular fibrofatty infiltrations, and RV or RVOT dilatations in all patients with suspected ARVD, on a 1.5 Tesla MRI scanner (MAGNETOM Aera; Siemens Healthcare, Erlangen, Germany). An RV end-diastolic volume of at least 110 mL/m2 (boys) or at least 100 mL/m2 (girls) or an RV ejection fraction of ≤40% was defined as the major criterion; an RV end-diastolic volume of at least 100 to less than 110 mL/m2 (boys) or at least 90 to less than 100 mL/m2 (girls) or an RV ejection fraction of >40% to ≤45% was defined as the minor criterion according to the revised Task Force criteria of ARVD (5).
Treatment
All patients were told to restrict exercise. All patients with/without symptoms were given treatment, primarily they were administered beta-blockers (metoprolol, propranolol, sotalol). Flecainide or amiodarone was added if necessary. An electrophysiological study and radiofrequency ablation were performed in selected severely symptomatic patients. An ICD was placed in the patients according to the indications in the International Task Force Consensus Statement about the treatment of ARVD (12).
Statistical analysis
Statistical analyses were performed using SPSS software version 21 (IBM Analytics, Armonk, New York, USA). All continuous data are presented as mean ± standard deviation (SD) and categorical variables as numbers and percentages of the total.
Results
Demographic and clinical characteristics
A total of 24 patients fulfilled Task Force criteria for ARVD (5) in our institution, and of them, 5 patients were excluded from the study due to insufficient data. The demographics and clinical characteristics of the study population are shown in Table 1. Out of 19 ARVD patients, 15 were male, and their mean age was 12±4 years; the youngest patient was a 1.5-year-old infant boy. The most common presenting symptoms were palpitations (n=6) and syncope (n=4). Two patients were presented with heart failure symptoms. One patient aged 14 years was presented with resuscitated/aborted cardiac arrest. Five patients had no symptoms at the time of admission. One of the asymptomatic patients was able to be found by the familial screening after the diagnosis of ARVD in his brother. The other four asymptomatic patients were referred to our clinic after being found to have arrhythmia incidentally during examinations. A review of familial history revealed that 2 patients had sudden deaths in their families at a young age, 2 patients were siblings, and 18 patients were probands.
Table 1.
Demographic and clinical characteristics of patients
| Number of patients (n) | 19 |
| Age (mean±standard deviation, range) | 12±4 (1.5-17 years) |
| Gender, male | 14 |
| Weight (kg) | 44±21 |
| BSA (m2) | 1.2±0.4 |
| Symptomatic patients (n) | 14 |
| Palpitation | 5 |
| Chest pain | 1 |
| Syncope | 4 |
| Fatigue | 1 |
| Heart failure | 2 |
| Cardiac arrest | 1 |
| Asymptomatic patients | 5 |
| Follow-up period (mean±standard deviation, range) | 21.5±11 (7-50 months) |
Electrocardiographic findings
Twelve-lead surface electrocardiogram showing abnormalities of depolarization and repolarization is a valuable diagnostic tool to investigate ARVD in patients (5). Thirteen patients had an epsilon wave. T wave inversion in the right precordial leads was detected in all patients, and all patients over the age of 14 had inverted T waves in V1-3 with the absence of complete right bundle-branch block. Thirteen patients had a low QRS voltage. According to arrhythmic properties, premature ventricular contractions (PVCs) more than 500 ectopies per day were observed in 15 patients, and PVCs were polymorphic in 7 of them. PVC with left bundle-branch block was seen in all patients with monomorphic ectopies, 6 of them had an inferior axis, and 2 had a superior axis. While the origin of the PVCs could not be determined in 3 of the patients with polymorphic ventricular ectopies, PVCs were left bundle-branch block with inferior and superior axis in the remaining patients. VT was detected in 9 patients on ECG and Holter monitoring. Table 2 presents the diagnostic characteristics of patients at the time of admission. Figure 1 shows examples of ECG and Holter recordings.
Table 2.
Electrocardiographic, echocardiographic magnetic resonance imaging, and genetic screening findings of patients
| n | % | |
|---|---|---|
| Electrocardiographic findings | ||
| Epsilon wave | 13 | 68 |
| Low QRS voltage | 13 | 68 |
| ≥14 years T wave inversion in V1-3 | 10 | 100 |
| >500 PVC/ 24 h | 15 | 79 |
| PVC morphology | ||
| Monomorphic | 8 | 53 |
| Left branch block inferior axis | 6 | 40 |
| Left branch block superior axis | 2 | 13 |
| Polymorphic | 7 | 47 |
| Undetermine PVC origine | 4 | 26 |
| Ventricular tachycardia | 9 | 45 |
| Echocardiographic findings | ||
| Patients with echocardiography findings | 17/19 | 89 |
| Right ventricular dysfunction | 6 | 31.5 |
| Right ventricular aneurysm | 13 | 68 |
| Hypokinetic area in right ventricle | 11 | 57.8 |
| RVOT aneurysm | 17 | 89 |
| Left ventricular involvement | 4 | 21 |
| RVOT diameter PSAX (mean±SD) | 35.9±9.9 mm | 31±9.8 mm/m2 |
| RVOT diameter PLAX (mean±SD) | 32±8.5 mm | 28±10 mm/m2 |
| MRI findings | ||
| Patients underwent MRI | 19 | 100 |
| ARVD criterias in MRI | ||
| Fibro-fatty infiltration of the right ventricle | 12 | 63 |
| Right ventricle/RVOT aneurysm | 13 | 68 |
| Hypokinetic area in right ventricle | 13 | 6 |
| Left ventricular involvement | 7 | 36.8 |
| RV end-diastolic volume (mean±SD) | 157±64 mL/m2 | |
| RV EF % (mean±SD) | 33±16 | |
| Genetic screening | 11 | 57.8 |
| Resulted | 6 | 54.5 |
| Pathological mutation | 4 | 36 |
| PKP2 gene mutation | 1 | 9 |
| DSG2 gene mutation | 2 | 18 |
| RYR2 gene mutation | 1 | 9 |
| No pathological mutation | 2 | 18 |
| Expected results | 5 | 45.5 |
ARVD - arrhythmogenic right ventricular dysplasia, MRI - magnetic resonance imaging, RV - right ventricle, RVOT - right ventricle outflow tract, PVC - premature ventricular contraction
Figure 1.

Twelve-lead ECG and Twelve-lead Holter recording images of patients with ARVD. (a) Twelve-lead ECG demonstrated epsilon waves (blue arrows) in precordial leads. (b) Twelve-lead Holter recording demonstrated PVCs in a 15-year-old patient; LBBB with the inferior axis (red arrows) and RBBB with the superior axis (blue arrows). (c) Twelve-lead ECG demonstrated VT with large QRS with left branch block superior axis.
ARVD - arrhythmogenic right ventricular dysplasia, ECG - electrocardiography, LBBB - left bundle-branch block, PVCs - premature ventricular contractions, RBBB - right bundle-branch block
Echocardiographic findings
Structural abnormalities were detected by echocardiography in 17 patients. The hypokinetic area in the right ventricle, right ventricular aneurysm, and RVOT dilatation was detected in 11, 13, and 17 patients, respectively. The mean RVOT diameter at the parasternal short axis (PSAX) view was measured to be 35.9±9.9 mm and 31±9.8 mm/m2, and at the parasternal long axis (PLAX) view, it was 32±8.5 mm and 28±10 mm/m2. Right ventricular functions were preserved in 13 patients at the time of admission, and of them, 7 patients had worsened RV functions during follow-up. Left ventricular involvement was present in 4 patients at the time of admission (Table 2).
Cardiac magnetic resonance imaging findings
Medical records revealed that all patients had undergone cardiac MRI (12 patients at our center’s radiology department, remaining MRIs in radiology units of other institutions). Out of 19 patients, 12 patients had fibrofatty infiltration of the RV, 13 patients had hypokinetic areas, and 15 patients had aneurysms of RVOT and/or right ventricle. Different from echocardiography findings, left ventricular involvement was detected in 6 patients at the time of admission. The mean right ventricular end-diastolic volume was revealed to be 157±64 mL/m2 and mean RV EF was measured as 33±16% by MRI (Table 2). Figure 2 presents echocardiographic and cardiac MRI images of patients.
Figure 2.
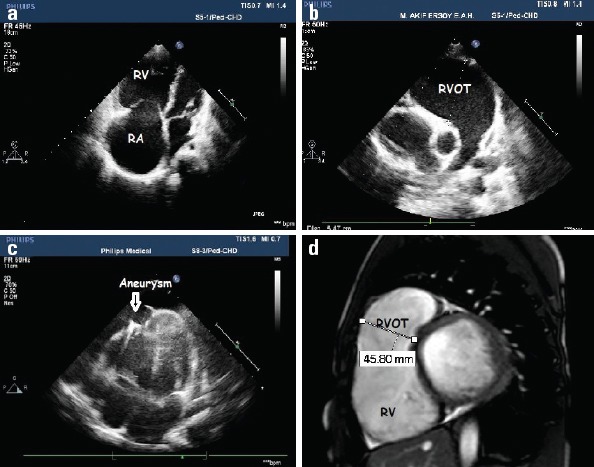
Two-dimensional echocardiography and cardiac magnetic resonance images of patients with ARVD. (a) Two-dimensional echocardiographic image demonstrated dilatation of RV and RA. (b) Two-dimensional echocardiographic image demonstrated significant dilatation of RVOT. (c) Two-dimensional echocardiographic image demonstrated a localized right ventricular apical aneurysm in a patient with ARVD. (d) Cardiac MRI image demonstrated significant dilatation of RVOT and thinning of the walls in the RV myocardium in the sagittal plane
ARVD - arrhythmogenic right ventricular dysplasia, MRI - magnetic resonance imaging, RA - right atrium, RV - right ventricle, RVOT - right ventricle outflow tract
Exercise testing findings
Exercise testing was carried out for 14 suitable patients, and all patients had PVCs with exercise. PVCs increased at the maximum heart rates and induced VT in 2 patients, and PVCs were decreased with exercise in 12 patients.
Genetic testing
All patients diagnosed with ARVD were requested to carry out genetic analysis, but genetic screening could be performed in 11 patients. Pathological mutations were detected in 4 of the 6 patients (Table 2). Genetic screening results of the remaining 5 patients are yet to be obtained.
With the findings mentioned above, patients were diagnosed based on revised Task Force criteria (5). Table 3 outlines the diagnostic criteria of the patients in our study population.
Table 3.
Diagnostic criteria of patients based on revised Task Force criteria (5)
| Patient number | Age at presentation (years) | Global or regional dysfunction and structural alterations | Repolarization abnormalities | Depolarization/conduction abnormalities | Arrhythmias | Family history | Tissue characterization of Wall | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2D echo | MRI | ||||||||||||||
| Major | Minor | Major | Minor | Major | Minor | Major | Minor | Major | Minor | Major | Minor | Major | Minor | ||
| P01 | 12 | x | x | + | x | x | x | ||||||||
| P02 | 15 | x | x | * | x | x | x | x | |||||||
| P03 | 10 | x | x | + | x | x | |||||||||
| P04 | 14 | * | x | x | |||||||||||
| P05 | 8 | x | x | x | x | ||||||||||
| P06 | 16 | x | x | * | x | x | x | ||||||||
| P07 | 11 | x | x | + | x | x | |||||||||
| P08 | 16 | x | * | x | x | ||||||||||
| P09 | 14 | x | x | * | x | x | |||||||||
| P10 | 15 | x | x | * | x | x | |||||||||
| P11 | 10 | x | x | + | x | x | x | ||||||||
| P12 | 9 | x | x | + | x | x | x | ||||||||
| P13 | 6 | x | x | x | |||||||||||
| P14 | 15 | x | x | * | x | x | |||||||||
| P15 | 10 | x | x | + | x | ||||||||||
| P16 | 14 | x | x | * | |||||||||||
| P17 | 17 | x | x | * | x | x | |||||||||
| P18 | 15 | * | x | x | x | ||||||||||
| P19 | 1.5 | x | x | + | x | x | |||||||||
x: Diagnostic criterias of patients,
Patients ≥ 14 years with T wave invertion at V1–3, +: Patients <14 years with T wave invertion at V1–3. MRI - magnetic resonance imaging
Management of patients
First, exercise restriction was recommended to all diagnosed patients. Also, beta-blocker therapy was started in all patients. Eleven patients were taking beta-blockers alone and eight patients were taking either flecainide or amiodarone as a second drug. Two patients underwent electrophysiological study and radiofrequency ablation due to VT. One patient underwent RVOT-induced monomorphic VT ablation, and the other underwent right ventricle free wall-induced polymorphic VT ablation. ICDs were placed in a total of 8 patients, and 4 patients received the device for primary prevention. ICDs were implanted epicardially in 2 patients and transvenously in others. Although the decision to implant ICDs was given for 3 additional patients, they could not be placed in because familial consent could not be obtained. Assist device was inserted in one patient who was in the terminal phase of the heart failure. A six-year-old patient had right ventricle outflow tract aneurysm at presentation, and surgical aneurysm resection was performed. Histopathological evaluation revealed dense fibrosis, fatty tissue, and myocyte loss in the aneurysm specimen. Also, myocardial fibrofatty infiltration was detected in the right ventricular free wall.
Follow-up results
The mean follow-up period was 21.5±11 months (7-50 months). One year after the diagnosis, the right ventricular function deteriorated in one patient, and a significant left ventricular dysfunction was developed. Another patient who did not comply with both drug recommendation and exercise restriction was presented with significant biventricular heart failure 1.5 years after the first diagnosis. He has been in intensive care unit for 5 months and is awaiting transplantation. During follow-up, 2 patients were deceased with incessant VT and heart failure, and one patient was deceased with multiorgan dysfunction after biventricular assist device implantation (n=3). Figure 3 presents the follow-up data of our study population.
Figure 3.
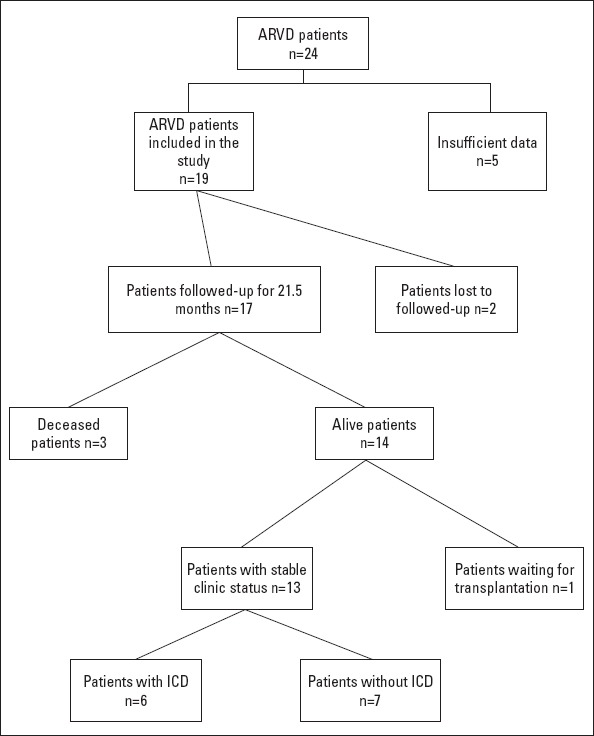
Follow-up data of the study population
Discussion
The results of this study describe the clinical characteristics, arrhythmic complications, and follow-up of 19 patients with pediatric-onset ARVD from a single center in Turkey. Although there are many case reports (13-16), to our knowledge, this is the first reported study about pediatric ARVD from our country. The most remarkable findings in this study are:
ARVD is usually common at adolescence in the pediatric age group, but it can also be detected in infancy.
Patients with ARVD may be symptomatic; however, it is not uncommon for ARVD patients to be asymptomatic.
In patients without any structural abnormalities, ARVD can be diagnosed by clinical, arrhythmic, and genetic features.
ICD is a life-saving treatment in patients diagnosed with ARVD.
Numerous studies on ARVD include adult patients, but there is not enough information about the incidence/prevalence, clinical characteristics, and natural course of the disease in infants and children. Although ARVD has been reported as a major cause of sudden cardiac death in the young patients (1), the prevalence of ARVD in the pediatric population might be higher than reported (17). In previous studies, affected patients usually present during the second to fifth decades of life (3, 18). In our study, the mean age of the patients was 12 years, and the youngest patient was an infant boy aged 1.5 years. These results are similar to those of Deshpande et al. in which the mean age of patients was 12.6 years and the youngest patient’s age was 3 (17). Te Riele et al. (19) defined ARVD as a disease of adolescence at pediatric age groups, and they found the mean age of patients to be 15 years in their study. Nucifora et al. (20) reported the case of a 4-month-old male infant who died suddenly in his sleep. Postmortem examination revealed the presence of ARVD.
The clinical spectrum of this disease varies from asymptomatic presentation to admission with sudden cardiac death or aborted cardiac arrest similar to our study (4, 21). Four patients presented with symptoms of heart failure at the time of admission. An infant with right ventricular dilatation and dysfunction had no complaints at the time of diagnosis. A 14-year-old patient presented with resuscitated cardiac arrest, and a 6-year-old patient with an aneurysm at the RV apex had complaint of fatigue. In a report of 16 pediatric cases of ARVD (17), 5 patients were presented with cardiac arrest, 6 were found to have VT, and 2 patients were presented with heart failure.
Four stages of ARVD were reported previously by Sen-Chowdhry et al. (4) and Marcus et al. (5): an early concealed phase (I), overt electrical disorder (II), isolated right ventricular failure (III), and biventricular cardiac failure (IV). Patients may present without any structural abnormalities, as in two of our patients. This early phase of the disease is called as the “concealed phase,” and the ventricular morphology with functions is preserved. However, the risk of sudden cardiac death due to ventricular arrhythmias exists in this stage, and it might be the first and only manifestation of the disorder (2, 4, 6, 18). We examined previous echocardiography and cardiac MRI findings of 2 patients who presented with cardiac arrest and the other with chest pain at the time of admission. Patients were eventually diagnosed by ECG, Holter monitoring, and genetic screening findings, and we considered them to be in the concealed phase.
Similarly, Novotny et al. (8) described a case of two brothers suffering from arrhythmogenic cardiomyopathy, in which the younger was detected at the concealed phase. The first patient was a 48-year-old man who presented with cardiac arrest that occurred during the sports activity, and then the younger brother (35-year-old) of the proband was examined for the history of the recurrent presyncopes and syncope during the sports. Patient’s echocardiography and cardiac MRI revealed normal cardiac functions and non-dilated ventricles, resting ECG was normal, but Holter monitoring exhibited frequent isolated monomorphic PVCs. In our study, we found that 4 of the patients were in the biventricular cardiac failure phase, 2 were in the concealed phase, and the others were in either overt electrical disorder or isolated right ventricular failure phases.
Intraventricular conduction delay occurs because of the localized fibrofatty infiltration of the right ventricular myocardium and causes ventricular arrhythmias that induce symptoms in patients (22). In the majority of our patients (n=12), we determined PVCs with mostly left bundle-branch block morphology at different frequencies, and most of these patients (n=9) had VT in ambulatory ECG monitoring, which was the only form of arrhythmia in our patients. Patients who previously had VT or fibrillation are at a high risk for life-threatening arrhythmic events (12). Mazzanti et al. (9) evaluated the predictors of arrhythmic risk of patients with ARVD, and atrial fibrillation, syncope, participation in heavy exercise after the diagnosis of disease, hemodynamically tolerated sustained monomorphic VT, and male sex were found to be predictive factors of lethal arrhythmias during follow-up. They stated that the high risk of life-threatening arrhythmias in patients with ARVD spans from adolescence to advanced age, reaching its peak between ages 21 and 40 years. In their study, no differences have been observed in the occurrence of life-threatening arrhythmic events before and after treatment with medications (amiodarone, beta-blockers, sotalol), or ablation (9).
Signal averaged ECG (SAECG) is one of the important diagnostic tools; however, we did not use it in our patients (5). SAECG shows delayed RV depolarization by detecting late potentials. Three separate parameters are recorded in SAECGs: filtered QRS duration (fQRS), duration of terminal QRS (low-amplitude signal duration-LAS) <40 µV, and root-mean-square voltage of terminal 40 ms (RMS40). Pathological values are considered to be fQRS ≥ 114 ms, LAS ≥ 38 ms, and RMS40 ≤ 20 µV. Terminal activation duration of QRS ≥ 55 ms measured from the nadir of the S wave to the end of the QRS, including R´, in V1, V2, or V3, in the absence of complete right bundle-branch block is also accepted as abnormal. Late potentials by SAECG in ≥1 of 3 parameters in the absence of a QRS duration of ≥110 ms on the standard ECG were noted as abnormal in Task Force criteria for ARVD (5). Liao et al. (23) stated that the 3+ SAECG could therefore be a novel and useful tool in the risk stratification process and may guide the physician in the early course of management of ARVD patients. In their study, they specified that the presence of 3+ SAECG had been predicted to be malignant in all patients with definite and non-definite ARVD (p<0.01, OR=30.5, 95% CI=2.5-373.7). Liao et al. (23) suggested consideration of an ICD implantation in the definite ARVD patients with 3+ SAECG, who demonstrated a cumulative adverse event rate of 60%. In conclusion of their study, they stated that the SAECG fulfilling all 3 Task Force criteria was an independent risk predictor of malignant events in ARVC patients (23).
Lifestyle changes, especially avoiding physical activities, have a crucial role in patients diagnosed with ARVD. Physical activity is a factor that accelerates the structural progression of the disease (4, 6). In our study population, one of our patients who did not comply with exercise restriction developed significant biventricular heart failure 1.5 years after the diagnosis.
One of our patients with ICD, who presented with severe RV failure, had sustained VT and deceased 11 months after the referral to our hospital, and the second one presented with advanced biventricular involvement and deceased after an assist device was inserted. A 10-year-old girl who had no ICD was admitted to an external center with ventricular fibrillation 1.5 years after the admission.
Preventing sudden cardiac death is the primary aim of clinical management of this life-threatening disease. In addition to exercise restriction, medical therapy, electrophysiologic study, and ablation (if required) and ICD therapy are life-saving treatment modalities. Of our 3 deceased patients, 2 patients who had ICD were presented at the end-stage of disease and had appropriate ICD shocks, but unfortunately, they were deceased as a result of severe heart failure. The third patient, who had no ICD, died due to the incessant VT. Our alive and routinely followed-up patients who had ICD have not yet been shocked; however, our follow-up period is currently quite short. Even if ICD is implanted, patients with ventricular failure may decease due to congestive heart failure as in the case of our patients. Assist device and heart transplantation can be the curative treatment in these patients. Unfortunately, heart transplantations cannot be performed in our center.
Study limitations
Our study evaluated patients with ARVD through retrospective reviews with a small group of patients. Over the years, our awareness of the disease was heightened, and 80% of the patients were diagnosed after 2015. Moreover, genetic screening could not be performed in 100% of the patients.
Conclusion
Diagnosis of ARVD might be much more difficult in children. It is critical to remember that patients may present asymptomatically. Avoidance of physical exercise is vitally important to prevent or decelerate the progression of the disease. ICD is a life-saving treatment that prevents patients diagnosed with ARVD from fatal arrhythmic events. Arrhythmic complications and sudden cardiac death might be prevented in the early period by raising the awareness of physicians about the disease. It should be kept in mind that ARVD patients can present with an earlier concealed phase without any cardiac structural involvement.
Acknowledgments
We would like to thank our patients and their families for making this study possible.
Footnotes
Ethical approval: Study is reviewed by the Local Ethics Committee.
Informed consent: For this type of study formal consent is not required.
Conflict of interest: None declared.
Peer-review: Externally peer-reviewed.
Authorship contributions: Concept – A.G., Y.E.; Design – A.G., Y.E.; Supervision – F.S.Ş., Y.E.; Fundings – None; Materials – O.A.; Data collection &/or processing – F.S.Ş., G.T.Ş.; Analysis &/or interpretation – S.Ö.; Literature search – F.S.Ş., H.C.K., Y.E.; Writing – F.S.Ş., Y.E.; Critical review – A.G., Y.E.
References
- 1.Tabib A, Loire R, Chalabreysse L, Meyronnet D, Miras A, Malicier D, et al. Circumstances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmogenic right ventricular cardiomyopathy and/or dysplasia. Circulation. 2003;108:3000–5. doi: 10.1161/01.CIR.0000108396.65446.21. [DOI] [PubMed] [Google Scholar]
- 2.Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. Heart Rhythm Society (HRS);European Heart Rhythm Association (EHRA) HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. Europace. 2011;13:1077–109. doi: 10.1093/europace/eur245. [DOI] [PubMed] [Google Scholar]
- 3.Calkins H, Corrado D, Marcus F. Risk stratification in arrhythmogenic right ventricular cardiomyopathy. Circulation. 2017;136:2068–82. doi: 10.1161/CIRCULATIONAHA.117.030792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sen-Chowdhry S, Morgan RD, Chambers JC, McKenna WJ. Arrhythmogenic cardiomyopathy:etiology, diagnosis, and treatment. Annu Rev Med. 2010;61:233–53. doi: 10.1146/annurev.med.052208.130419. [DOI] [PubMed] [Google Scholar]
- 5.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia:Proposed modification of the task force criteria. Circulation. 2010;121:1533–41. doi: 10.1161/CIRCULATIONAHA.108.840827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Albakri A. Arrhythmogenic right ventricular dysplasia:A review of literature on clinical status and meta-analysis of diagnosis and clinical management methods. Clin Med Invest. 2018;3:1–17. [Google Scholar]
- 7.Kirchhof P, Fabritz L, Zwiener M, Witt H, Schäfers M, Zellerhoff S, et al. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation. 2006;114:1799–806. doi: 10.1161/CIRCULATIONAHA.106.624502. [DOI] [PubMed] [Google Scholar]
- 8.Novotny P, Panovsky R, Feitova V, Balcarkova P, Grochova I, Kincl V. Atypical form of arrhythmogenic cardiomyopathy. Cor et Vasa. 2014;56:e396–e402. [Google Scholar]
- 9.Mazzanti A, Ng K, Faragli A, Maragna R, Chiodaroli E, Orphanou N, et al. Arrhythmogenic Right Ventricular Cardiomyopathy:Clinical Course and Predictors of Arrhythmic Risk. J Am Coll Cardiol. 2016;68:2540–50. doi: 10.1016/j.jacc.2016.09.951. [DOI] [PubMed] [Google Scholar]
- 10.Foale R, Nihoyannopoulos P, McKenna W, Klienebenne A, Nadazdin A, Rowland E, et al. Echocardiographic measurement of the normal adult right ventricle. Br Heart J. 1986;56:33–44. doi: 10.1136/hrt.56.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paridon SM, Alpert BS, Boas SR, Cabrera ME, Caldarera LL, Daniels SR, et al. American Heart Association Council on Cardiovascular Disease in the Young, Committee on Atherosclerosis, Hypertension, and Obesity in Youth. Clinical stress testing in the pediatric age group:a statement from the American Heart Association Council on Cardiovascular Disease in the Young, Committee on Atherosclerosis, Hypertension, and Obesity in Youth. Circulation. 2006;113:1905–20. doi: 10.1161/CIRCULATIONAHA.106.174375. [DOI] [PubMed] [Google Scholar]
- 12.Corrado D, Wichter T, Link MS, Hauer RN, Marchlinski FE, Anastasakis A, et al. Treatment of arrhythmogenic right ventricular cardiomyopathy/ dysplasia- an International Task Force Consensus Statement. Circulation. 2015;132:441–53. doi: 10.1161/CIRCULATIONAHA.115.017944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atalay S, Imamoglu A, Gümüs H, Gürdal M, Ozenci M. Value of the echocardiographic findings of arrhythmogenic right ventricular dysplasia with left ventricular involvement in a child. Pediatr Cardiol. 1996;17:40–2. doi: 10.1007/BF02505810. [DOI] [PubMed] [Google Scholar]
- 14.Aykan HH, Gülgün M, Ertuğrul İ, Karagöz T. Electrical storm in an adolescent with arrhythmogenic right ventricle cardiomyopathy treated with cardiac transplantation. Anatol J Cardiol. 2015;15:513. doi: 10.5152/akd.2015.5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gülgün M, Fidancı MK, Genç AF. Electrical storm might be the initial presentation of arrhythmogenic right ventricular cardiomyopathy. Anatol J Cardiol. 2016;16:218–9. doi: 10.14744/AnatolJCardiol.2016.6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gorgulu S, Nurkalem Z, Celebi A, Bilal MS, Yalcin Y, Cine N, et al. Unusual presentation of a patient with arrhythmogenic right ventricular dysplasia treated with a Glenn shunt. Int J Cardiol. 2006;113:410–3. doi: 10.1016/j.ijcard.2005.09.048. [DOI] [PubMed] [Google Scholar]
- 17.Deshpande SR, Herman HK, Quigley PC, Shinnick JK, Cundiff CA, Caltharp S, et al. Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia (ARVC/D):Review of 16 Pediatric Cases and a Proposal of Modified Pediatric Criteria. Pediatr Cardiol. 2016;37:646–55. doi: 10.1007/s00246-015-1327-x. [DOI] [PubMed] [Google Scholar]
- 18.Orgeron GM, Crosson JE. Arrhythmogenic right ventricular dysplasia/cardiomyopathy. Cardiol Young. 2017;27:S57–S61. doi: 10.1017/S1047951116002249. [DOI] [PubMed] [Google Scholar]
- 19.Te Riele ASJM, James CA, Sawant AC, Bhonsale A, Groeneweg JA, Mast TP, et al. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy in the Pediatric Population:Clinical Characterization and Comparison With Adult-Onset Disease. JACC Clin Electrophysiol. 2015;1:551–60. doi: 10.1016/j.jacep.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 20.Nucifora G, Benettoni A, Allocca G, Bussani R, Silvestri F. Arrhythmogenic right ventricular dysplasia/cardiomyopathy as a cause of sudden infant death. J Cardiovasc Med (Hagerstown) 2008;9:430–1. doi: 10.2459/JCM.0b013e3282eee772. [DOI] [PubMed] [Google Scholar]
- 21.Yoo SJ, Grosse-Wortmann L, Hamilton RM. Magnetic resonance imaging assessment of arrhythmogenic right ventricular cardiomyopathy/dysplasia in children. Korean Circ J. 2010;40:357–67. doi: 10.4070/kcj.2010.40.8.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fontaine G. Arrhythmogenic right ventricular dysplasia. Curr Opin Cardiol. 1995;10:16–20. doi: 10.1097/00001573-199501000-00004. [DOI] [PubMed] [Google Scholar]
- 23.Liao YC, Lin YJ, Chung FP, Chang SL, Lo LW, Hu YF, et al. Risk stratification of arrhythmogenic right ventricular cardiomyopathy based on signal averaged electrocardiograms. Int J Cardiol. 2014;174:628–33. doi: 10.1016/j.ijcard.2014.04.169. [DOI] [PubMed] [Google Scholar]