

ABSTRACT
Although best understood as a degradative pathway, recent evidence demonstrates pronounced involvement of the macroautophagic/autophagic molecular machinery in cellular secretion. With either overexpression or inhibition of autophagy mediators, dramatic alterations in the cellular secretory profile occur. This affects secretion of a plethora of factors ranging from cytokines, to granule contents, and even viral particles. Encompassing a wide range of secreted factors, autophagy-dependent secretion is implicated in diseases ranging from cancer to neurodegeneration. With a growing body of evidence shedding light onto the molecular mediators, this review delineates the molecular machinery involved in selective targeting of the autophagosome for either degradation or secretion. In addition, we summarize the current understanding of factors and cargo secreted through this unconventional route, and describe the implications of this pathway in both health and disease.
Abbreviations: BECN1, beclin 1; CAF, cancer associated fibroblast; CUPS, compartment for unconventional protein secretion; CXCL, C-X-C motif chemokine ligand; ER, endoplasmic reticulum; FGF2, fibroblast growth factor 2; HMGB1, high mobility group box 1; IDE, insulin degrading enzyme; IL, Interleukin; MAP1LC3/LC3, microtubule associated protein 1 light chain 3; MAPS, misfolding associated protein secretion; MEF, mouse embryonic fibroblast; MTORC1, MTOR complex I; PtdIns, phosphatidyl inositol; SEC22B, SEC22 homolog B, vesicle trafficking protein (gene/pseudogene); SFV, Semliki forest virus; SNCA, synuclein alpha; SQSTM1, sequestosome 1; STX, Syntaxin; TASCC, TOR-associated spatial coupling compartment; TGFB, transforming growth factor beta; TRIM16, tripartite motif containing 16; UPS, unconventional protein secretion; VWF, von Willebrand factor
KEYWORDS: Autophagy-dependent secretion, cancer, IL1B, infection, neurodegeneration, secretory autophagy
Introduction
Autophagy, since its discovery, is predominantly known as a degradative process. Increased autophagosomes in nutrient-deprived and/or stressed cells supports the concept that this pathway serves a pro-survival, metabolite-generating role. Mitochondrial structures identified within lysosomes led to the name ‘self-eating’ (autophagy), and prompted the degradative connotation of this pathway.
However, over the past 40 years, a subtle but growing body of evidence points to an interesting function of this pathway in cellular secretion. Even in the original report on autophagy in yeast, one outcome of increased autophagy under nutrient-starvation conditions was enhanced secretion of labeled leucine [1]. Others have demonstrated the fusion of autophagosomes with the plasma membrane for the expulsion of cellular cargo [2]. Additionally, cells with enhanced basal autophagy have a distinct secretome compared to low-autophagy cells [3]. With growing evidence, autophagy serves pleiotropic roles within the cell as both a degradative and secretory pathway.
Autophagy facilitates secretion in both normal physiology and pathology. Understanding autophagy-dependent secretion relies on an understanding of the autophagic molecular machinery. Enhancement or attenuation of these components leads to consequential alterations in secretion. Perturbation of this pathway has demonstrated that key signaling molecules and cytokines, such as IL1B (interleukin 1 beta) and IL6 (interleukin 6), alter with changes in autophagy [4]. Even entire viruses and bacteria are secreted through autophagic machinery-labeled vesicles [5]. As such, this pathway affects many diseases ranging from cancer to viral infections, asthma, and Crohn disease. With surmounting evidence regarding the implications of autophagy-dependent secretion, and the lack of drugs targeting this pathway, biologists across disciplines will see benefits from the current understanding of this process.
The autophagic machinery
The autophagic machinery is conserved across yeast and mammalian cells. In order to assess autophagy-dependent secretion, one needs a basic understanding of the molecular machinery involved. Broadly, the pathway comprises the initiation of a double-membrane structure, termed a phagophore, which sequesters cellular cargo; the phagophore matures into an autophagosome and traffics toward degradation in the lysosome (vacuole in yeast) (Figure 1) [6]. A catalytic cascade initiates the pathway, and nutrient sensor complexes AMPK and MTORC1 regulate this initiation. The phagophore incorporates the lipidated form of the LC3 protein. Cargo receptors bind and carry autophagic cargo destined for the phagophore, and facilitate the molecular tethering of cargo to LC3 on the phagophore membrane. The subsequent fusion of the autophagosome with the lysosome involves specialized SNARE complexes (Figure 2) [7]. This facilitates the delivery and degradation of autophagic cargo.
Figure 1.
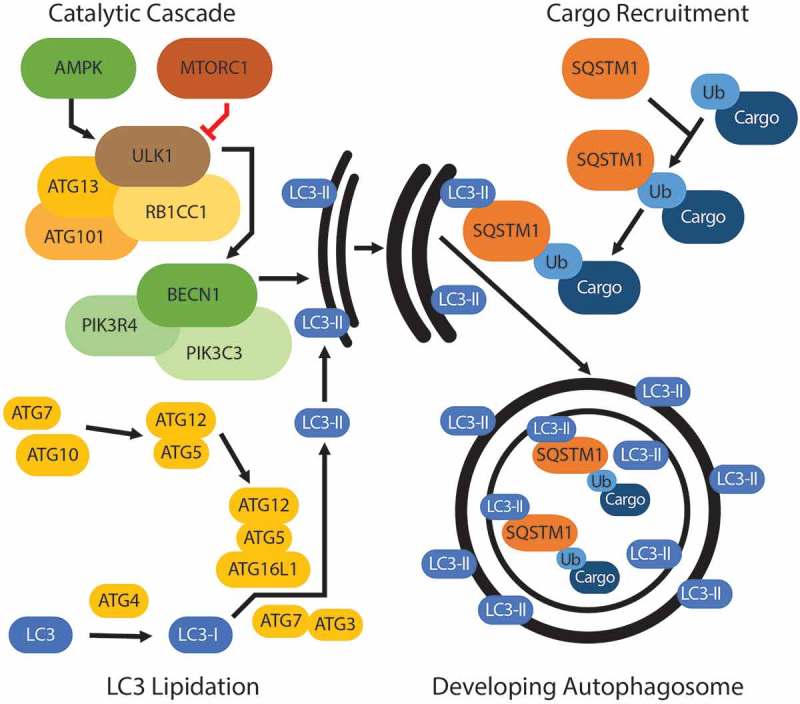
Initiating events in autophagy. The autophagic pathway centers around a convergence of 3 broad initiating events: 1) a catalytic cascade regulated by nutrient sensors, AMPK and MTORC1, leads to the phosphorylation of ULK1 and the subsequent activation of the BECN1 complex, which is essential for membrane nucleation and expansion; 2) the lipidation of LC3, which incorporates as LC3-II into the autophagic membrane; and 3) cargo recruitment by SQSTM1 or another autophagy cargo receptor, which bind primarily ubiquitinated (represented as ‘Ub’) cargo and traffic the cargo to the developing autophagic membrane.
Figure 2.
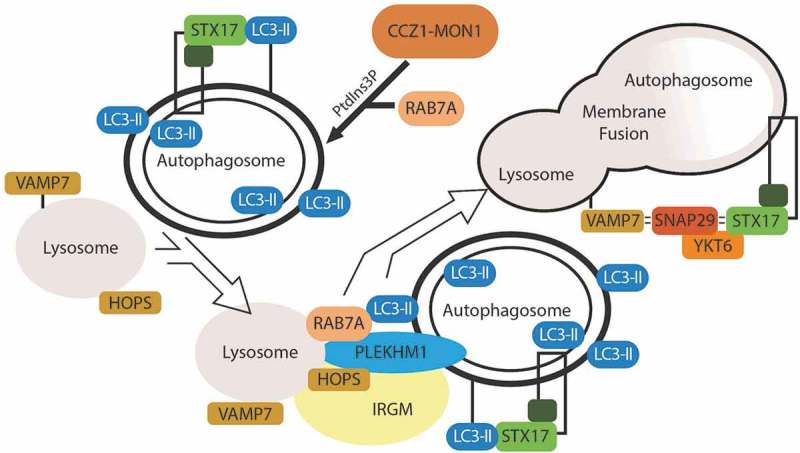
Degradative events in autophagy. Autophagosome degradation centers around the fusion of the lysosome with the autophagosome. The pool of PtdIns3P surrounding the autophagosome recruits RAB7A. RAB7A facilitates binding of the autophagosome to the HOPS complex on the lysosome, and PLEKHM1 mediates this binding. As the membranes converge, a SNARE-mediated fusion event occurs between VAMP7 on the lysosome and STX17 on the autophagosome, with SNAP29 being recruited and acting as a Qab SNARE. This allows for fusion of the autophagosome and lysosome membranes and the degradation of autophagosome content.
Secretory mechanism
Autophagy-dependent secretion stems from observations of the unconventionally secreted protein Acb1 (Acyl-CoA binding protein, dictyostelium homolog: AcbA). Functionally, Acb1 is released from pre-spore yeast cells to induce sporulation [8]. Under autophagy-enhancing conditions, such as low nitrogen concentrations, cells secrete increased levels of Acb1 [9]. Following individual knockdown of autophagy components, Atg5, Atg7, Atg8, and Atg12, Acb1 secretion significantly decreases, despite constant Acb1 concentrations within the cell. This foundational report suggested a role for the autophagosome in secretion of extracellular proteins.
Intriguingly, Acb1 is a leaderless protein, lacking a secretion signal sequence. Under conventional secretion, an N-terminal signal sequence directs proteins to the ER for folding, and then successive modification in the Golgi apparatus to facilitate secretion (Reviewed in [10]). This signal peptide consists of a positively charged amino terminus, a hydrophobic central region, and a cleavable carboxyl terminus, which allows for removal of the signal sequence in the ER. Bioinformatics estimates upwards of 30 percent of all proteins have a secretory signal sequence. However, many proteins do not have this cleavable, targeting sequence, even though they are readily detected extracellularly.
Proteins lacking a signal sequence are secreted through unconventional secretory routes, which bypass the conventional ER-Golgi route. There are 4 main Types of unconventional protein secretion (UPS), and types I-III involve the secretion of leaderless peptides [11]. Type I involves a pore-mediated translocation process across the plasma membrane. FGF2 (fibroblast growth factor 2) provides an example for this type of secretion. FGF2 binds the phosphatidylinositol 4,5-bisphosphate (PtdIns [4,5]P2) on the cytoplasmic leaflet of the plasma membrane [12]. Here, FGF2 self-oligomerizes, and forms a lipidic membrane pore, which facilitates the translocation of FGF2 to the extracellular plasma membrane [13]. The mechanism of type II UPS requires ABC transporters to translocate proteins across the plasma membrane. This is the least studied of UPS types, and appears to be dedicated to acetylated peptides or yeast pheromones [14–16]. Type III UPS relies on intracellular, membrane-bound intermediates to transport proteins for secretion. Type III secretion was first noted when IL1B was determined to be secreted by means of an intracellular vesicle [17]. Additionally, Acb1 was determined to be secreted by means of a membrane-bound intermediate [18]. Examples of membrane-bound intermediates proposed to be involved with type III UPS include late endosomes/exosomes, lysosomes, and autophagosomes (note, although lysosomes and autophagosomes are approximately 2 µm in diameter and larger than the size of a typical vesicle, they are sometimes referred to as a vesicle when described in the context of type III UPS). Exosomes are vesicles that form from late endosomal membranes, are packaged in multivesicular bodies and are then destined for secretion. Exosomes are small vesicles (40–90 nm), and a classic example of type III UPS via exosomes is MHC-II (major histocompatibility complex II) [19]. Secretory lysosomes are specialized lysosomes that are trafficked to the membrane following an intracellular influx of calcium. Secretory lysosomes contain specific cargo unique to particular cell types. For example, histamine in mast cells, or granzymes in cytotoxic lymphocytes, are secreted via secretory lysosomes in a type III UPS manner.
The connection of autophagic machinery with type III UPS arose from observations surrounding the secretion of Acb1. Acb1 secretion is dependent upon nutrient starvation and several autophagy-mediating proteins [9,20]. However, Acb1, and type III UPS as a whole, also has resemblances to endosome trafficking. Endosome sorting components, such as Stp22/Vps23, Grh1, and the endosome-specific t-SNARE Tlg2 are necessary for the secretion of Acb1 [9,21]. Grh1 is trafficked to a unique, cup-shaped membrane upon nutrient starvation. This membrane is termed the compartment for unconventional protein secretion (CUPS). The CUPS contains both endosomal sorting components, such as Stp22, as well as autophagic machinery, such as Atg8 and Atg9 [22]. CUPS associations with autophagy include that it is induced by nutrient starvation (specifically glucose starvation), and that Atg8, Atg9, and a pool of PtdIns3P are necessary for its development. However, CUPS do not lead to an LC3+ bilayered membrane-bound intermediate that is degraded in the lysosome. Indicating that although autophagic machinery and induction mechanisms play a role in CUPS development, CUPS and CUPS-dependent secretion are not involved in canonical autophagic flux. As a further example of a separation of CUPS from autophagic degradation, rapamycin, which induces autophagy, does not induce CUPS formation, nor does nitrogen starvation, which induces autophagy but not CUPS [20,22]. However, the secretion of Acb1 can be triggered by rapamycin and nitrogen starvation [20]. Thus, although Acb1 secretion resembles endosomal/exosomal-mediated secretion, autophagy and the autophagic machinery are necessary for Acb1’s secretion, though the mechanism differs from degradative autophagy. This also provides an example of pleiotropic roles for the autophagic machinery in cellular secretion.
It is peculiar that the 3 major membrane-bound intermediates (autophagosomes, exosomes, and lysosomes) mediating type III UPS arise from converging pathways. The late endosome and autophagosome both terminate with the lysosome. As such, a careful definition of autophagy-dependent secretion is necessary to distinguish from other type III UPS. Autophagy-dependent secretion refers to the secretion of cytoplasmic entities that depend on autophagic machinery for their secretion. With awareness that the autophagic machinery has pleiotropic roles, this review will focus on secreted entities whose secretion has been observed to be modulated by at least 2 separate components of the autophagic machinery, whether the modulation was genetic or pharmacological. This definition accounts for the non-autophagy and alternative roles of autophagic machinery; for example, the involvement of Atg8 and Atg9 in CUPS formation. Thus, the term autophagy-dependent secretion used in this review describes the secretion of a factor that depends upon functional autophagic machinery for its secretion, even if these machineries have roles outside of the canonical degradative mechanism of autophagic flux. With this definition, factors implicated in autophagy-dependent secretion most likely dynamically change with differences in autophagic flux [3]. One important caveat of this definition is that not all factors included by it are necessarily packaged and destined for secretion by a canonical, double-membrane, LC3+ autophagosome, but evidence does suggest that this occurs with some of the factors described, for instance IL1B or TGFB [4,23]. Table 1 provides the entities that meet this definition of autophagy-dependent secretion, where the entities’ secretion has been modulated by modifying at least 2 separate components of the autophagic machinery.
Table 1.
Secreted entities regulated by autophagy-dependent secretion.
| Entity | Association with Autophagy | Species or Cell Type | Reference |
|---|---|---|---|
| Acb1 | Atg1 mutant decreases secretion | P. pastoris | [20] |
| Atg5 mutant decreases secretion | S. cerevisiae | [9] | |
| Vps30/Atg6 mutant decreases secretion | P. pastoris | [20] | |
| Atg7 mutant decreases secretion | S. cerevisiae | [9] | |
| Atg8 mutant decreases secretion | S. cerevisiae | [9] | |
| Atg9 mutant decreases secretion | P. pastoris | [20] | |
| Atg11 mutant decreases secretion | P. pastoris | [20] | |
| Atg12 mutant decreases secretion | S. cerevisiae | [9] | |
| Atg17 mutant decreases secretion | P. pastoris | [20] | |
| Nitrogen deficiency enhances secretion |
S. cerevisiae; P. pastoris |
[9,20] | |
| Rapamycin induces secretion | P. pastoris | [20] | |
| IL1B | Starvation enhances secretion | Murine bone marrow-derived macrophages; human monocyte line, THP-1; human cervical cancer line, HeLa; human embryonic kidney cell line, HEK293T; MEFs |
[4,27,29] |
| ATG2 knockdown decreases secretion | HEK293T cells | [29] | |
| ATG4 mutant decreases secretion | HEK293T cells | [29] | |
| Atg5 knockout decreases secretion | Murine bone marrow-derived macrophages; MEFs | [27,29] | |
| ATG16L1 knockdown decreases secretion | THP-1 | [4] | |
| Cells with low autophagic flux have less secretion of this factor compared to cells with greater autophagic flux | Human breast cancer cells | [3] | |
| BECN1 overexpression enhances secretion | Human breast cancer cells | [3] | |
| Colocalizes with LC3 in the cytoplasm | Murine bone marrow-derived macrophages | [27] | |
| Cofractionates with LC3 vesicles in sucrose gradient | HEK293T cells | [29] | |
| Bafilomycin A1 decreases secretion | Murine bone marrow-derived macrophages | [27] | |
| 3-methyladenine and wortmannin decrease secretion | MEFs | [29] | |
| IL6 | ATG7 knockdown decreases secretion | Human pancreatic stellate cells; human breast cancer cells; human brain endothelial cells | [38,40,41] |
| Atg7 knockout decreases secretion | Mouse brain | [41] | |
| BECN1 knockdown decreases secretion | Human head and neck cancer-associated fibroblasts; human breast cancer cells | [34,40] | |
| Associates with TASCC, which is inhibited by ATG5 knockdown | Human fibroblast line, IMR90; human promyelocytic leukemia line, HL60 | [37] | |
| CXCL8 | BECN1 knockdown decreases secretion | Human head and neck cancer-associated fibroblasts | [34] |
| Produced near TASCC, which is inhibited by ATG5 knockdown | HL60 | [37] | |
| Cells with low autophagic flux have less secretion of this factor compared to cells with greater autophagic flux | Human breast cancer cells | [3] | |
| BECN1 overexpression enhances secretion | Human breast cancer cells | [3] | |
| IL18 | Bafilomycin A1 decreases secretion | Murine bone marrow-derived macrophages | [27] |
| Atg5 knockout decreases secretion | Murine bone marrow-derived macrophages | [27] | |
| HMGB1 | Atg5 knockout decreases secretion | Murine bone marrow-derived macrophages | [27] |
| ATG7 knockdown decreases secretion | Human glioblastoma cell line U87MG | [44] | |
| ATG12 knockdown decreases secretion | U87MG | [44] | |
| ATP | ATG5 knockdown decreases secretion | Murine colon carcinoma line CT26; MEFs; Human osteosarcoma line U20S | [45, 46] |
| Atg7 knockout and knockdown inhibits secretion | MEFs, U2OS | [46] | |
| ATG10 knockdown decreases secretion | U2OS | [46] | |
| BECN1 knockdown decreases secretion | U2OS | [46] | |
| TGFB1 | Ultrastructural colocalization with LC3 in double-membrane vesicles | Wi26 fibroblasts | [23] |
| BECN1 knockdown decreases secretion | MEFs | [23] | |
| Atg5 knockout decreases secretion | MEFs | [23] | |
| ATG7 knockdown decreases secretion | MEFs, THP-1 | [23] | |
| 3-MA decreases secretion | MEFs, THP-1, murine primary macrophages | [23] | |
| IDE (insulin degrading enzyme) | 3-methyladenine decreases secretion | Mouse primary astrocytes | [56] |
| Bafilomycin A1 decreases secretion | Mouse primary astrocytes | [56] | |
| Rapamycin induces secretion | Mouse primary astrocytes | [56] | |
| BECN1 knockdown decreases secretion | Mouse primary astrocytes | [55] | |
| ATG5 knockdown decreases secretion | Mouse primary astrocytes | [56] | |
| ATG7 mutant mice decrease secretion | Mouse brain | [56] | |
| Amyloid beta | Atg7 knockout reduces cellular secretion | Mouse forebrain | [75] |
| Rapamycin induces secretion | Mouse forebrain | [75] | |
| Spautin-1 inhibits secretion | Mouse forebrain | [75] | |
| SNCA | ATG5 knockdown decreases secretion | Rat pheochromocytoma cell line, PC12 | [77] |
| 3-MA inhibits secretion | PC12 | [77] | |
| Secretory Granules | |||
| Paneth cell contents (LYZ [lysozyme]) | Dysregulated and reduced LYZ exocytosis from Atg5 knockout | Mouse intestinal crypt | [48] |
| Dysregulated and reduced lysozyme exocytosis from ATG16L1 mutant | Mouse intestinal crypt | [48,49] | |
| Weibel-Palade bodies | ATG5 knockdown decreases secretion | Human primary endothelial cells | [50] |
| ATG7 knockdown decreases secretion | Human primary endothelial cells | [50] | |
| Secretory lysosomes | ATG4B or LC3 mutant decreases secretion | Murine osteoclasts | [51] |
| Atg5 knockout decreases secretion | Murine osteoclasts | [51] | |
| Atg7 knockout decreases secretion | Murine osteoclasts and bone marrow-derived mast cells | [51,53] | |
| Reflect Autophagy Dynamics | |||
| LIF, FAM3C, DKK3 | Cells with low autophagic flux have less secretion of these factors compared to cells with greater autophagic flux | Human breast cancer cells | [3] |
| BECN1 overexpression enhances secretion | Human breast cancer cells | [3] | |
| Viruses | |||
| Poliovirus | BECN1 knockdown decreases extracellular viral titer; whereas overexpression increases viral titer | HeLa cells with poliovirus infection | [64] |
| LC3 knockdown decreases extracellular viral titer | HeLa cells with poliovirus infection | [5,64] | |
| ATG12 knockdown decreases extracellular viral titer | HeLa cells with poliovirus infection | [5] | |
| Rapamycin increases extracellular titer | HeLa cells with poliovirus infection | [5] | |
| 3-MA treatment inhibits secretion | HeLa cells with poliovirus infection | [5] | |
| Colocalizes with LC3+ vesicle | HeLa cells with poliovirus infection | [64] | |
| Rhinovirus | LC3+ membrane associated | HeLa cells with rhinovirus infection | [5] |
| Coxsackie virus | LC3+ membrane associated | HeLa cells with coxsackie virus infection | [5] |
| Dengue virus | Spautin-1 decreases viral yield | Baby hamster kidney cell line BHK-21, and HeLa cells with dengue virus | [67] |
| Mycobacteria | Atg1 knockdown decreases bacterial ejection | Dictyostelium | [72] |
| Atg6A knockdown decreases bacterial ejection | Dictyostelium | [72] | |
| Atg7 knockdown decreases bacterial ejection | Dictyostelium | [72] | |
With a foundation in Acb1, most mechanistic work on autophagy-dependent secretion arises from studies analyzing IL1B secretion. The initial cloning of the gene encoding this protein indicated that IL1B lacks a secretion signal sequence [24]. The secretion mechanism of IL1B has had a plethora of proposed models; these include type I UPS, and other vesicles associated with type III UPS, such as exosomes [25]. Yet, membrane translocation of IL1B depends on a permeabilized membrane of a cell committed to cell death, and IL1B has not been observed to be localized within multivesicular bodies or exosomes [25,26]. A connection with autophagy arose with the observation that IL1B secretion is enhanced following starvation of bone marrow-derived macrophages, similar to the enhancement of Acb1 secretion following nutrient starvation [27]. Knockdown of ATG5 and colocalization with LC3 provides stronger evidence that autophagy mediates the secretion of IL1B [27]. Since this finding, the molecular mechanisms involved in autophagy-dependent secretion, including cargo recruitment, autophagosome trafficking, and membrane release, have been primarily worked out using IL1B as the released protein of interest.
Cargo recruitment
A modified autophagosome routes IL1B for extracellular secretion. This begins with cargo recruitment to the developing autophagosome. Mature IL1B binds to TRIM16/ERBBP (tripartite motif containing 16) [28]. This IL1B-TRIM16 complex traffics to an autophagy sequestration membrane [4]. The sequestration membrane is not an autophagosome, which corresponds to a sealed terminal compartment, but an intermediate membrane necessary for lipidation of LC3-I to LC3-II [29]. Without TRIM16, IL1B cannot arrive at the sequestration membrane, or be found within the resulting autophagosome [4].
Secretory autophagosome trafficking
At the sequestration membrane, SEC22B (SEC22 homolog B, vesicle trafficking protein [gene/pseudogene]) binds the IL1B-TRIM16 complex. SEC22B consists of a longin domain (involved in protein transport to the plasma membrane) and a SNARE motif [30], with this SNARE motif critical to the vesicle fusion events involved in IL1B secretion. Originally identified as part of the vesicle fusion machinery involved in COP-II coated vesicle fusion in the ER-Golgi intermediate compartment [31], SEC22B is of particular importance to autophagy. Upon knockdown of SEC22B, LC3 lipidation is decreased [32]. Paradoxically, SEC22B depletion leads to an increase in LC3-II levels by immunoblot, and LC3 puncta by immunofluoresence [4,33], but no overall differences in autophagic flux. Reconciling this finding, SEC22B depletion blocks trafficking of lysosomal proteases to the lysosome, thereby rendering the lysosome ineffective [33]. With SEC22B depletion, IL1B secretion decreases [4]. Therefore, an autophagosome destined for secretion would have LC3-II, SEC22B, and TRIM16 on its cytosolic membrane.
Membrane fusion
To fuse with the plasma membrane, the secretory autophagosome undergoes a SNARE-mediated fusion event. The R-SNARE, SEC22B, on the secretory autophagosome binds to Qbc-SNAREs, SNAP23 and SNAP29 on the plasma membrane [4]. Together with STX3 (syntaxin 3) and STX4 (syntaxin 4) on the plasma membrane, these proteins mediate a SNARE complex allowing fusion of the secretory autophagosome with the plasma membrane [4]. The fusion of the secretory autophagosome with the plasma membrane facilitates secretion of IL1B.
The modified autophagosome involved in IL1B secretion has characteristics similar to a degradative autophagosome, but differs in a few key cytosolic membrane elements to facilitate the trafficking to the plasma membrane. Similar to the degradative autophagosome, a secretory autophagosome has a double membrane labeled with LC3-II. Cargo recruitment in both secretion and degradation appear to rely on trafficking of cellular cargo to LC3. However, the destinations of the LC3+ double-membrane intermediate differ based upon the SNARE machinery coating the cytosolic membrane (Figure 3). In a degradative autophagosome, STX17 allows for fusion with the lysosome. In a secretory autophagosome, SEC22B facilitates fusion with the plasma membrane. These subtle differences in the cytosolic membrane proteins determine whether the contents are degraded or expelled.
Figure 3.
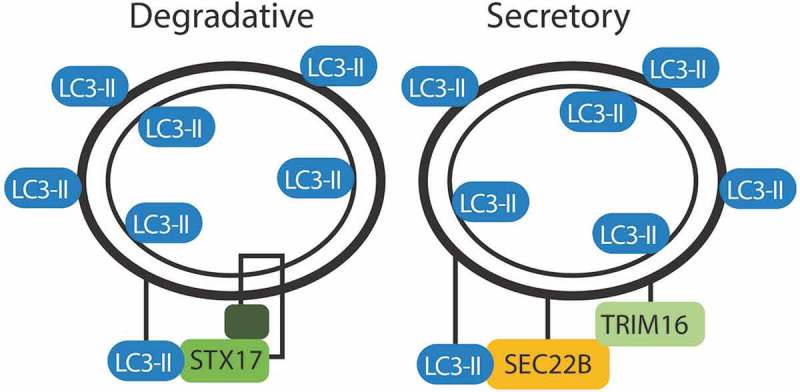
Markers of degradative and secretory autophagosomes. Trafficking of the autophagosome depends on the proteins decorating the outer membrane. Both degradative and secretory routes are labeled with LC3. STX17 directs fusion of the degradative autophagosome with the lysosome. SEC22B and TRIM16 direct an autophagosome for secretion.
Additionally, the role of the autolysosome in secretion remains unclear. Although the secretion of IL1B seems to bypass the lysosome, other secreted cargo depend upon lysosomal function. For instance, chloroquine inhibition of the lysosome alters the secretory profile of cancer-associated fibroblasts in a way that mimics BECN1 knockdown [34]. Even IL1B seems to rely on the lysosome, as the same group that delineated the mechanism of autophagic secretion of IL1B also demonstrated in an earlier report that bafilomycin A1, which inhibits the acidification of the lysosome, inhibits IL1B secretion [27]. Autophagy-dependent secreted cargo may all have the same route bypassing the lysosome, but an alternative pathway, which incorporates the lysosome, cannot be excluded based on the current data.
Although the best understanding of autophagy-dependent secretion comes from investigation of IL1B secretion, our understanding of the molecular mechanism is relatively nascent and could potentially differ with different cargo chaperoned by the autophagosome. For instance, there seems to be cross-communication between major vesicular mediators of type III UPS. For example, exosomal fragments derived from prostate cancer cells demonstrate the presence of autophagy markers LC3, SQSTM1, and others [35]. Within the cell, multivesicular bodies and exosomes are targeted to the autophagic intermediate when autophagy increases [36]. This may allow for the fusion of membranes and the directing of an autophagosome to the plasma membrane. The mechanisms regulating the cross-communication between exosomes, multivesicular bodies, and secreted autophagosomes are ill-defined.
The TOR-associated spatial coupling compartment (TASCC) provides an alternative mechanism for autophagy-dependent secretion, and enhances CXCL8 (C-X-C motif chemokine ligand 8) or IL8 (interleukin 8) secretion. At the Golgi apparatus, the TASCC brings together MTORC1, rough ER, autophagosomes and lysosomes [37]. This creates a dynamic metabolism center functioning to sequester material through degradative autophagy to reconstitute new proteins for secretion. CXCL8 and IL6 are both located in this compartment, and CXCL8 mRNA is detected at the marginal regions [37]. Under autophagy inhibition, the TASCC cannot form, preventing the translation of CXCL8 and IL6. This compartment identifies an alternative role of the autophagosome in secretion, as some secretory cargo are reliant upon a functioning degradative autophagy pathway, but not necessarily a secretory autophagosome.
Despite some remaining questions, this mechanism of secretion is enthralling. Machinery once thought to be only involved in degradation now appear to have pleiotropic roles. Alongside this, over the last decade, the variety of cargo secreted has expanded to include everything from metabolites to entire organelles.
Secreted factors
Autophagy-dependent secretion provides a wealth of extracellular factors. These secreted components range from inflammatory mediators to granule contents. By perturbing the canonical autophagic machinery, researchers identified several components that depend on autophagy for secretion. The list has expanded substantially from the identification of Acb1 secreted through an autophagic mechanism less than a decade ago.
Interleukins
The primary class of autophagy-dependent components are interleukins. As described above, IL1B has been extensively studied in relation to autophagic secretion, and provides the main understanding of the machinery involved in secretion. Similar to IL1B, the IL1 family member IL18 undergoes a similar secretory route, as inhibition of autophagosome fusion or formation by bafilomycin A1 or ATG5 knockdown attenuate IL18 secretion [27]. Beyond the IL1 family, other interleukins have been demonstrated to have significant involvement with autophagy, notably IL6 and CXCL8.
IL6 provides an interesting anecdote in the story of autophagy-dependent secretion. Multiple groups observe that the secretion of IL6 depends upon autophagy. With ATG7 knockdown, IL6 secretion reduces in pancreatic stellate cells [38], fibroblasts [39], breast cancer cells [40], and human brain endothelial cells [41]. Thus, IL6 is secreted in an autophagy-dependent mechanism. IL6 then feeds back, and further enhances autophagy. Observed in pancreatic cancer cells [42], and fibroblasts [34], IL6 provides a fascinating feed-forward loop accelerating autophagy-dependent secretion.
Damage response mediators
Although primarily studied in a healthy cell population, autophagy-dependent secretion proves important even in apoptotic and necrotic cells. Necrotic cells selectively release HMGB1 (high mobility group box 1), whereas apoptotic cells retain this immune stimulus within their nuclei [43]. Upon knockdown of key autophagy proteins, ATG5, ATG7, and ATG12, HMGB1 secretion during necrosis ceases [44]. Thus, autophagy-dependent secretion mediates regulation of microenvironment damage responses.
Additionally, extracellular ATP release serves as a chemokine to mediate an immune response towards a damaged region. For example, following irradiation, ATP release signals immune cells to the damaged region. However, knockdown of ATG5 inhibits ATP release, and a normal immune response cannot occur [45]. Chemotherapy-treated cells normally release ATP in a similar manner, and autophagy blockade attenuates ATP release from pharmacologically damaged cells [46]. Therefore, autophagy-dependent secretion mediates appropriate damage responses by regulating HMGB1 and ATP release from apoptotic or necrotic cells.
Secretory granule contents
In a variety of tissue types, secretory granules regulate tissue development and homeostasis. Autophagy-dependent secretion facilitates a variety of these secretory granules from widely different tissue types.
Intestinal paneth cells secrete many antimicrobial proteins necessary for both innate defense and regulation of the microbiome. Dysregulation of these cells is one feature of Crohn disease. ATG16L1 mutations predict susceptibility to Crohn disease [47]. Dysregulation of ATG16L1 leads to dysfunctional granule exocytosis from Paneth cells [48]. Disruption of normal granule exocytosis causes retention of key antimicrobials, such as lysozyme, resulting in an incompatible response to bacterial infection [49]. Without autophagy-dependent secretion, normal Paneth cell secretion could not occur.
In endothelial cells, autophagy-dependent secretion of secretory granules allows for homeostasis following vascular injury. VWF (von Willebrand factor) assembles long multimers, which, when tethered together, provide adhesion of circulating platelets and facilitates clotting. Weibel-Palade bodies contain VWF in endothelial cells. With impairment of autophagy, through knockdown of ATG5 or ATG7, Weibel-Palade bodies are retained intracellularly, VWF cannot be secreted, and there is impaired healing of the vessel wall [50].
Secretory lysosomes have physiological importance in tissue homeostasis and immune responses. For example, bone resorption relies on osteoclast-mediated secretion of lysosomal enzymes into an extracellular resorptive space. Once thought to have only a lysosome function, when autophagy proteins ATG5, ATG7, ATG4, and LC3 are knocked down or mutated, bone resorption dramatically decreases [51]. This provides evidence of the involvement of autophagy-dependent secretion in trafficking secretory lysosomes. This may occur in other myeloid-derived secretory cells, such as natural killer cells, the granular contents of which are contained within a secretory lysosome [52].
Furthermore, mast cells, components of the innate immune response, rely on autophagy-dependent secretion for degranulation of secretory lysosomes. Mast cells play a crucial role in maintenance of the allergic response. Degranulation of these cells releases histamine and other cytokines into the microenvironment to mount an immune response. LC3-II localizes with secretory granules within mast cells, and is secreted with colocalized CD63, a secretory lysosome marker. Knockout of ATG7 results in impaired degranulation of mast cells, and an impaired anaphylaxis reaction [53]. Autophagic machinery mediates the trafficking of granule components during the immune response.
Ranging from intestinal cells, vasculature, osteoclasts, and immune cells, autophagy-dependent secretion provides an essential homeostatic mechanism of granule release throughout an organism. Extensive characterization of the mechanisms of granule release compared to cytokine release remains to be carried out. However, the core autophagic machinery proves essential to this secretory process.
Extracellular matrix components
Of note, an association of autophagy-dependent secretion with extracellular matrix components has been observed. In pancreatic stellate cells, which synthesize the pancreatic stromal matrix, autophagy knockdown diminishes matrix synthesis. Upon knockdown of ATG7, key matrix components are significantly reduced in expression [38]. These include COL1A1/Collagen 1α1, FN1 (fibronectin 1), and POSTN (periostin) mRNA [38]. The role of autophagy-dependent secretion from fibroblasts of other tissues [34,39] provides support to the concept that extracellular matrix synthesis occurs through an autophagy-dependent secretory manner.
Further, IDE (insulin degrading enzyme) degrades a number of peptides, most notably insulin and amyloid beta, in the extracellular space [54]. IDE does not have a secretion signal sequence. Autophagy dynamics reflect IDE secretion, in that secretion of IDE increases with increases in autophagic flux [55]. In mouse primary astrocytes, BECN1 knockdown, ATG5 knockdown or ATG7 mutation decrease IDE secretion [55,56]. Additionally, pharmacological inhibition of autophagy with 3-methyladenine (3-MA, which inhibits PtdIns3K) or bafilomycin A1 (which inhibits the vacuolar-type H+-translocating ATPase on the lysosome) decreases secretion, whereas rapamycin induces secretion [56]. Although clearly an entity secreted in an autophagy-dependent manner, IDE has also been identified in exosomes; and both the exosome-secreted and non-exosome secreted levels of IDE increase with induction of autophagy [55].
Role in disease
With such a plethora of factors secreted, autophagy-dependent secretion affects both normal physiology and pathophysiology. The understanding of autophagy-dependent secretion in disease has grown primarily out of 3 disease classes: cancer, infection and neurodegeneration.
Cancer
Relative to normal tissue, cancer tissue of almost all organ sites upregulates autophagic flux. Increased degradative autophagy provides a mechanism for renewal of damaged organelles and proteins in a metabolically active microenvironment. Increased autophagy also promotes cancer cell survival by facilitating therapy resistance [57]. As discussed above, autophagy-dependent secretion facilitates secretion of cancer-promoting factors, such as IL1B and IL6. Within the microenvironment, both the cancer cells themselves and stromal supporting cells rely on autophagy-dependent secretion for progression of the disease as well as therapy resistance.
The dynamics of autophagy-dependent secretion in cancer cells is best exemplified by differing secretomes between cells of the same cancer site with differing basal rates of autophagic flux [3]. Paired melanoma cells from the same patient with differing basal levels of autophagy provide a unique material to study autophagy-dependent secretomes [3]. Low-autophagy cells have markedly reduced secreted levels of CXCL8, IL1B, LIF, FAM3C, and DKK3 compared to cells with high basal levels of autophagy [3]. BECN1 overexpression provides confirmation in these low-autophagy cells that the factors secreted are indeed dependent upon autophagy. These factors support tumor progression, and allow for useful biomarkers in response to autophagy-modulating cancer therapy.
In the microenvironment, cancer cells induce autophagy in the surrounding fibroblasts. These cancer-associated fibroblasts undergo autophagy-dependent secretion to enhance tumor-promoting factors. Pancreatic stellate cells activate in an autophagy-dependent mechanism [38]. With autophagy inhibition by knockdown of ATG7, expression levels of several secreted tumor-promoting factors are reduced, including IL1B, and IL6 [38]. Additionally, we have observed head and neck cancer cells to induce stromal fibroblast autophagy [34]. Autophagy inhibition in head and neck cancer-associated fibroblasts, through both pharmacological use of chloroquine or knockdown of the autophagy genes BECN1 or ATG7, suppresses fibroblast-secreted contributors to disease progression [34]. Notably, we observe reductions in IL6, CXCL8, CXCL1/GROa (C-X-C motif chemokine ligand 1), and LIF/leukemia inhibitory factor (LIF interleukin 6 family cytokine) following BECN1 knockdown in cancer-associated fibroblasts (CAFs) [34]. In breast cancer, CAFs have increased autophagy relative to normal fibroblasts [58]. Conditioned media collected from cancer-associated fibroblasts promotes cancer migration, whereas autophagy inhibition in cancer-associated fibroblasts using 3-MA diminishes cancer migration [58]. Additionally, HMGB1 secreted by autophagy-dependent secretion in CAFs promotes the maintenance of breast cancer stem cells [59]. Across organ sites, stromal autophagy-dependent secretion facilitates tumor progression and resistance to therapy.
Infection
Viruses hijack normal cell mechanisms to reproduce, and the autophagic machinery provide a fascinating example of this. Early on, picornaviruses, such as polio, were observed in double-membrane vesicles, with an appearance similar to the autophagosome [60,61]. Intriguingly, poliovirus components 2BC and 3A induce the formation of these double-membrane structures [62]. 2BC directly modifies MAP1LC3/LC3, allowing its incorporation into the membrane [63]. Poliovirus then uses the autophagosome-like membrane as a lipid source during viral replication. Once formed, the LC3-positive double-membrane vesicle traffics the enclosed viral particles to the plasma membrane, where LC3-positive vesicles are observed to bleb off the host cell surface [5]. This creates a secreted autophagosome, coated with the host’s own cellular components, but packed with poliovirus cargo (much like a Trojan horse). This method of non-lytic viral production significantly enhances infection and reproduction of the viruses.
Poliovirus transmission tightly intertwines with autophagy-dependent secretion. With BECN1 overexpression, poliovirus transmission increases [64]. Accordingly, BECN1 knockdown significantly reduces poliovirus yield [64]. Pharmacological inducers of autophagy, such as rapamacyin, enhance poliovirus yield by more than 3 fold [5]. Thus, autophagy dynamics correlate with poliovirus secretion.
Beyond poliovirus, other viruses undergo such autophagy-dependent secretion. Rhinovirus and coxsackievirus also exit host cells in vesicles harboring LC3-II [65]. Varicella zoster virus exits the host in a single-membrane vesicle harboring LC3, but also RAB11A, an endocytic marker, which may indicate the convergence of the autophagy and endocytic pathways in varicella zoster expulsion [66]. Dengue virus induces LC3 puncta, in a manner that relies on BECN1 [67]. During dengue virus infection, BECN1 inhibition significantly reduces extracellular virus [67]. Within the last decade, this method of non-lytic viral transmission has just been uncovered, and much work is still to be done with the exact mechanisms in the transmission of each viral type.
Additionally, outside of viral secretion, unconventional dynamics of autophagy-related proteins play an important role in viral replication. Out of 44 ATG proteins knocked down in an siRNA screen of U2OS and HeLa cells, the knockdown of 16 ATG proteins resulted in significant changes in viral replication [68]. However, no single gene was identified that regulates viral replication consistently across the 6 viruses tested in the 2 cell lines. For example, whereas ATG7 knockdown decreased herpes simplex virus-1 replication in both cell types, this increased Semliki Forest virus (SFV) replication in both cell types. Yet, ATG5 knockdown decreased SFV replication in U2OS cells, but increased SFV replication in HeLa cells [68]. All the while, atg5 knockout mouse embryonic fibroblasts (MEFs) have an unchanged SFV viral titer [69]. These viral replication studies point towards pleiotropic roles of the autophagic machinery that are cell-type specific, and draw caution on the interpretation of results if only one ATG gene product is targeted [70].
Ejected bacteria also rely on autophagy-dependent secretion. Mycobacteria tuberculosis and M. marinum expel themselves from the host cell in an ejectosome [71]. The autophagic machinery is recruited to the distal pole of the ejectosome, and facilitates bacterial expulsion [72]. When Dictyostelium atg1, atg5, atg7, and atg8 are knocked down, non-lytic cell-to-cell transmission of mycobacteria is significantly reduced [72]. Thus, autophagy-dependent secretion facilitates viral and bacterial transmission.
Neurodegeneration
Two neurodegenerative conditions, Alzheimer and Parkinson disease, have long been associated with defective autophagy. In both diseases, an intracellular inclusion body forms: amyloid beta aggregates in Alzheimer disease, and SNCA/α-synuclein (synuclein alpha) inclusions in Parkinson disease. The dysregulation of autophagy in these diseases was thought to prevent the degradation of these aggregates, which led to their abundance within the cell. However, recent evidence points to defective autophagy limiting their secretion, which results in intracellular accumulation.
In Alzheimer disease, dystrophic neurites have an accumulation of autophagosomes [73]. These autophagosomes contain the bulk intracellular reservoir of amyloid beta [74]. Although this finding led to the initial conclusion that the accumulation of amyloid beta and autophagosomes resulted from a dysfunctional autolysosome, studies of Atg7 knockout transgenic mice have connected amyloid beta accumulation with a decreased secretory autophagy pathway. ATG7 knockout neurons have diminished amyloid beta secretion, and reconstitution of ATG7 restores the secretion of amyloid beta [75]. Further, pharmacological induction of autophagy with rapamycin enhances amyloid beta secretion, whereas inhibition of autophagy with spautin-1 diminishes secretion [75]. Thus, autophagy influences secretion of amyloid beta in Alzheimer disease.
In Parkinson disease, SNCA aggregates accumulate within dopaminergic neurons [76]. Both autophagy and the proteasome degrade SNCA. In neurons, TPPP/brain specific protein p25α (tubulin polymerization promoting protein), traffics SNCA to autophagic structures, while also preventing autophagosome-lysosome fusion [77]. This promotes secretion of an SNCA-containing autophagosome [77]. Upstream autophagy inhibitors, such as 3-MA, attenuate SNCA release [77]. Thus, autophagy-dependent secretion facilitates the secretion of SNCA in neurons.
Although the release of both SNCA and amyloid beta depend on autophagy, other routes may coexist. Misfolding-associated protein secretion (MAPS) is a recently uncovered UPS mechanism [78]. Here, the ER-associated deubiquitinase USP19 (ubiquitin specific peptidase 19) acts as a chaperone to enrich misfolded proteins at the ER surface [78]. Two other chaperones, HSPA8/HSC70 (heat shock protein family A [Hsp70] member 8) and DNAJC5 (DnaJ heat shock protein family [Hsp40] member C5) function with USP19 to triage proteins in MAPS [79]. Both SNCA and amyloid beta have been linked to a MAPS pathway of secretion [78,80]. Further studies may provide a connection between MAPS and autophagy-dependent secretion.
Conclusions
A secretory pathway conserved across yeast to mammals, autophagy-dependent secretion supplies a wide variety of cargo in both normal physiology and pathophysiology. Despite such broad implications, our understanding of this pathway is in its infancy, with the molecular mediators only partially worked out with IL1B secretion. As such, few targeted inhibitors exist.
For example, hydroxychloroquine provides the lone clinically available autophagy inhibitor [81]. This, and other lysomotropic inhibitors, destabilize the lysosome, and prevents autophagosome degradation. Although this may destabilize the TASCC and prevent autophagy-dependent secretion of CXCL8 and IL6, it also inhibits autophagic degradation that is useful for homeostatic maintenance of the cell. Alternatively, inhibitors such as SAR405 and spautin-1 target autophagy upstream at the BECN1-PtdIns3K complex [82,83]. Effective at diminishing both secretory and degradative autophagy, these inhibitors cannot differentiate between the 2 routes for the autophagosome.
Developing a selective route inhibitor, to better delineate the secretory and degradative routes, would provide details of the pathways and vesicle traffickers involved. The primary difference in the final fate of the autophagosome relies on the differential expression of STX17 or SEC22B on the cytosolic membrane. However, these 2 proteins function in multiple cellular pathways, and an inhibitor solely targeting either of these would be of little use. Perhaps proteomic approaches assessing carriers like TRIM16, unified cargo carriers that bind both LC3 and SEC22B, would be of use. By understanding the binding of cargo carriers to LC3 and the trafficking intermediate SEC22B, an appreciation of the exact cargo secreted through a secretory autophagosome could develop.
Overall, autophagy-dependent secretion proves essential in a wide variety of cellular processes with a plethora of factors secreted. Despite clear importance in both normal physiology and disease, there is much more to understand about the molecular mediators of this process. A clearer understanding of the molecular mechanisms will facilitate therapeutic development to counteract disease and augment normal physiology.
Funding Statement
The University of Kansas Cancer Center under CCSG P30CA168524, NIH grant CA227838, philanthropic donations and a NIH Clinical and Translational Science Award grant (UL1TR000001, formerly UL1RR033179) awarded to the University of Kansas Medical Center and an internal Lied Basic Science Grant Program of the KUMC Research Institute (to S. M. Thomas) provided financial support for this review. The KUMC Biomedical Research Training Program supported Jacob New.
Acknowledgments
We thank Phil Shafer and Christopher Neal for assistance in figure design.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Takeshige K, Baba M, Tsuboi S, et al. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol. 1992;119:301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Schweers RL, Zhang J, Randall MS, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104:19500–19505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kraya AA, Piao S, Xu X, et al. Identification of secreted proteins that reflect autophagy dynamics within tumor cells. Autophagy. 2015;11:60–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kimura T, Jia J, Kumar S, et al. Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J. 2017;36:42–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jackson WT, Giddings TH Jr., Taylor MP, et al. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3:e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. 2014;24:24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wang Y, Li L, Hou C, et al. SNARE-mediated membrane fusion in autophagy. Semin Cell Dev Biol. 2016;60:97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Anjard C, Loomis WF.. Peptide signaling during terminal differentiation of Dictyostelium. Proc Natl Acad Sci U S A. 2005;102:7607–7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Duran JM, Anjard C, Stefan C, et al. Unconventional secretion of Acb1 is mediated by autophagosomes. J Cell Biol. 2010;188:527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rapoport TA. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature. 2007;450:663–669. [DOI] [PubMed] [Google Scholar]
- [11].Rabouille C. Pathways of unconventional protein secretion. Trends Cell Biol. 2017;27:230–240. [DOI] [PubMed] [Google Scholar]
- [12].La Venuta G, Zeitler M, Steringer JP, et al. The startling properties of fibroblast growth factor 2: how to exit mammalian cells without a signal peptide at hand. J Biol Chem. 2015;290:27015–27020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Steringer JP, Bleicken S, Andreas H, et al. Phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2)-dependent oligomerization of fibroblast growth factor 2 (FGF2) triggers the formation of a lipidic membrane pore implicated in unconventional secretion. J Biol Chem. 2012;287:27659–27669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ricardo S, Lehmann R. An ABC transporter controls export of a drosophila germ cell attractant. Science (New York, NY). 2009;323:943–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Maclean LM, O’Toole PJ, Stark M, et al. Trafficking and release of Leishmania metacyclic HASPB on macrophage invasion. Cell Microbiol. 2012;14:740–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rabouille C, Malhotra V, Nickel W. Diversity in unconventional protein secretion. J Cell Sci. 2012;125:5251–5255. [DOI] [PubMed] [Google Scholar]
- [17].Rubartelli A, Cozzolino F, Talio M, et al. A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence. EMBO J. 1990;9:1503–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cabral M, Anjard C, Malhotra V, et al. Unconventional secretion of AcbA in dictyostelium discoideum through a vesicular intermediate. Eukaryot Cell. 2010;9:1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Fevrier B, Raposo G. Exosomes: endosomal-derived vesicles shipping extracellular messages. Curr Opin Cell Biol. 2004;16:415–421. [DOI] [PubMed] [Google Scholar]
- [20].Manjithaya R, Anjard C, Loomis WF, et al. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J Cell Biol. 2010;188:537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kinseth MA, Anjard C, Fuller D, et al. The golgi-associated protein GRASP is required for unconventional protein secretion during development. Cell. 2007;130:524–534. [DOI] [PubMed] [Google Scholar]
- [22].Bruns C, McCaffery JM, Curwin AJ, et al. Biogenesis of a novel compartment for autophagosome-mediated unconventional protein secretion. J Cell Biol. 2011;195:979–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nüchel J, Ghatak S, Zuk AV, et al. TGFB1 is secreted through an unconventional pathway dependent on the autophagic machinery and cytoskeletal regulators. Autophagy. 2018;14:465–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Auron PE, Webb AC, Rosenwasser LJ, et al. Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proc Natl Acad Sci U S A. 1984;81:7907–7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Monteleone M, Stow JL, Schroder K. Mechanisms of unconventional secretion of IL-1 family cytokines. Cytokine. 2015;74:213–218. [DOI] [PubMed] [Google Scholar]
- [26].Martín-Sánchez F, Diamond C, Zeitler M, et al. Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. Cell Death Differ. 2016;23:1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dupont N, Jiang S, Pilli M, et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011;30:4701–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Munding C, Keller M, Niklaus G, et al. The estrogen-responsive B box protein: a novel enhancer of interleukin-1β secretion. Cell Death Differ. 2006;13:1938. [DOI] [PubMed] [Google Scholar]
- [29].Zhang M, Kenny SJ, Ge L, et al. Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion. eLife. 2015;4:e11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liu Y, Flanagan JJ, Barlowe C. Sec22p export from the endoplasmic reticulum is independent of SNARE pairing. J Biol Chem. 2004;279:27225–27232. [DOI] [PubMed] [Google Scholar]
- [31].Mancias JD, Goldberg J. The transport signal on Sec22 for packaging into COPII-coated vesicles is a conformational epitope. Mol Cell. 2007;26:403–414. [DOI] [PubMed] [Google Scholar]
- [32].Ge L, Zhang M, Schekman R. Phosphatidylinositol 3-kinase and COPII generate LC3 lipidation vesicles from the ER-Golgi intermediate compartment. eLife. 2014;3:e04135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Renna M, Schaffner C, Winslow AR, et al. Autophagic substrate clearance requires activity of the syntaxin-5 SNARE complex. J Cell Sci. 2011;124:469–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].New J, Arnold L, Ananth M, et al. Secretory autophagy in cancer-associated fibroblasts promotes head and neck cancer progression and offers a novel therapeutic target. Cancer Res. 2017;77:6679–6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hessvik NP, Øverbye A, Brech A, et al. PIKfyve inhibition increases exosome release and induces secretory autophagy. Cell Mol Life Sci. 2016;73:4717–4737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fader CM, Sanchez D, Furlan M, et al. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic. 2008;9:230–250. [DOI] [PubMed] [Google Scholar]
- [37].Narita M, Young ARJ, Arakawa S, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science (New York, NY). 2011;332:966–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Endo S, Nakata K, Ohuchida K, et al. Autophagy is required for activation of pancreatic stellate cells, associated with pancreatic cancer progression and promotes growth of pancreatic tumors in mice. Gastroenterology. 2017;152:1492–506.e24. [DOI] [PubMed] [Google Scholar]
- [39].Young ARJ, Narita M, Ferreira M, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Maycotte P, Jones KL, Goodall ML, et al. Autophagy supports breast cancer stem cell maintenance by regulating IL6 secretion. Mol Cancer Res. 2015;13:651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhuang S-F, Liu D-X, Wang H-J, et al. Atg7 regulates brain angiogenesis via NF-κB-dependent IL-6 production. Int J Mol Sci. 2017;18:968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kang R, Loux T, Tang D, et al. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc Natl Acad Sci U S A. 2012;109:7031–7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. [DOI] [PubMed] [Google Scholar]
- [44].Thorburn J, Horita H, Redzic J, et al. Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 2008;16:175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ko A, Kanehisa A, Martins I, et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ. 2014;21:92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Michaud M, Martins I, Sukkurwala AQ, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science (New York, NY). 2011;334:1573–1577. [DOI] [PubMed] [Google Scholar]
- [47].Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2006;39:207. [DOI] [PubMed] [Google Scholar]
- [48].Cadwell K, Liu JY, Brown SL, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bel S, Pendse M, Wang Y, et al. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science (New York, NY). 2017;357:1047–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Torisu T, Torisu K, Lee IH, et al. Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat Med. 2013;19:1281–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].DeSelm Carl J, Miller Brian C, Zou W, et al. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell. 2011;21:966–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lopez-Soto A, Bravo-San Pedro JM, Kroemer G, et al. Involvement of autophagy in NK cell development and function. Autophagy. 2017;13:633–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ushio H, Ueno T, Kojima Y, et al. Crucial role for autophagy in degranulation of mast cells. J Allergy Clin Immunol. 2011;127:1267–76.e6. [DOI] [PubMed] [Google Scholar]
- [54].Fernandez-Gamba A, Leal MC, Morelli L, et al. Insulin-degrading enzyme: structure-function relationship and its possible roles in health and disease. Curr Pharm Des. 2009;15:3644–3655. [DOI] [PubMed] [Google Scholar]
- [55].Son SM, Kang S, Choi H, et al. Statins induce insulin-degrading enzyme secretion from astrocytes via an autophagy-based unconventional secretory pathway. Mol Neurodegener. 2015;10:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Son SM, Cha M-Y, Choi H, et al. Insulin-degrading enzyme secretion from astrocytes is mediated by an autophagy-based unconventional secretory pathway in Alzheimer disease. Autophagy. 2016;12:784–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ma XH, Piao S, Wang D, et al. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin Cancer Res off J Am Assoc Cancer Res. 2011;17:3478–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wang M, Zhang J, Huang Y, et al. Cancer-associated fibroblasts autophagy enhances progression of triple-negative breast cancer cells. Med Sci Monit. 2017;23:3904–3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhao X-L, Lin Y, Jiang J, et al. High-mobility group box 1 released by autophagic cancer-associated fibroblasts maintains the stemness of luminal breast cancer cells. J Pathol. 2017;243:376–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Dales S, Eggers HJ, Tamm I, et al. Electron microscopic study of the formation of poliovirus. Virology. 1965;26:379–389. [DOI] [PubMed] [Google Scholar]
- [61].Schlegel A, Giddings TH Jr., Ladinsky MS, et al. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J Virol. 1996;70:6576–6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Suhy DA, Giddings TH Jr., Kirkegaard K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J Virol. 2000;74:8953–8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Taylor MP, Kirkegaard K. Modification of cellular autophagy protein LC3 by poliovirus. J Virol. 2007;81:12543–12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Chen Y, Du W, Hagemeijer M, et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell. 2015;160:619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Robinson SM, Tsueng G, Sin J, et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014;10:e1004045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Buckingham EM, Jarosinski KW, Jackson W, et al. Exocytosis of varicella-zoster virus virions involves a convergence of endosomal and autophagy pathways. J Virol. 2016;90:8673–8685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Mateo R, Nagamine CM, Spagnolo J, et al. Inhibition of cellular autophagy deranges dengue virion maturation. J Virol. 2013;87:1312–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Mauthe M, Langereis M, Jung J, et al. An siRNA screen for ATG protein depletion reveals the extent of the unconventional functions of the autophagy proteome in virus replication. J Cell Biol. 2016;214:619–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Eng KE, Panas MD, Murphy D, et al. Accumulation of autophagosomes in Semliki Forest virus-infected cells is dependent on expression of the viral glycoproteins. J Virol. 2012;86:5674–5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Subramani S, Malhotra V. Non-autophagic roles of autophagy-related proteins. EMBO Rep. 2013;14:143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Hagedorn M, Rohde KH, Russell DG, et al. Infection by tubercular mycobacteria is spread by nonlytic ejection from their amoeba hosts. Science (New York, NY). 2009;323:1729–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Gerstenmaier L, Pilla R, Herrmann L, et al. The autophagic machinery ensures nonlytic transmission of mycobacteria. Proc Nat Acad Sci. 2015;112:E687–E92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–4091. [DOI] [PubMed] [Google Scholar]
- [74].Yu WH, Cuervo AM, Kumar A, et al. Macroautophagy–a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol. 2005;171:87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Nilsson P, Loganathan K, Sekiguchi M, et al. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013;5:61–69. [DOI] [PubMed] [Google Scholar]
- [76].Luk KC, Kehm VM, Zhang B, et al. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J Exp Med. 2012;209:975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ejlerskov P, Rasmussen I, Nielsen TT, et al. Tubulin polymerization-promoting protein (TPPP/p25alpha) promotes unconventional secretion of alpha-synuclein through exophagy by impairing autophagosome-lysosome fusion. J Biol Chem. 2013;288:17313–17335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Lee JG, Takahama S, Zhang G, et al. Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat Cell Biol. 2016;18:765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Xu Y, Cui L, Dibello A, et al. DNAJC5 facilitates USP19-dependent unconventional secretion of misfolded cytosolic proteins. Cell Discov. 2018;4:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Fontaine SN, Zheng D, Sabbagh JJ, et al. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 2016;35:1537–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Pasquier B. Autophagy inhibitors. Cell Mol Life Sci. 2016;73:985–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Ronan B, Flamand O, Vescovi L, et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol. 2014;10:1013–1019. [DOI] [PubMed] [Google Scholar]
- [83].Liu J, Xia H, Kim M, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147:223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]