

Abstract
Recent epidemiological data indicate that the popularity of electronic cigarettes (e-cigarettes), and consequently nicotine use, is rising in both adolescent and adult populations. As nicotine is a known developmental neurotoxin, these products present a potential threat for those exposed during early life stages. Despite this, few studies have evaluated the toxicity of e-cigarettes on the developing central nervous system. The goal of this study was to assess neurotoxicity resulting from early-life exposure to electronic cigarette aerosols in an in vivo model. Specifically, studies here focused on neuro-parameters related to neuroinflammation and neurotrophins. To accomplish this, pregnant and neonatal C57BL/6 mice were exposed to aerosols produced from classic tobacco flavor e-cigarette cartridges (with [13 mg/ml] and without nicotine) during gestation (∼3 weeks) and lactation (∼3 weeks) via whole-body inhalation. Exposure to e-cigarette aerosols with and without nicotine caused significant reductions in hippocampal gene expression of Ngfr and Bdnf, as well as in serum levels of cytokines IL-1β, IL-2, and IL-6. Exposure to e-cigarette aerosols without nicotine enhanced expression of Iba-1, a specific marker of microglia, in the cornus ammonis 1 region of the hippocampus. Overall, our novel results indicate that exposure to e-cigarette aerosols, with and without nicotine, poses a considerable risk to the developing central nervous system. Consequently, e-cigarettes should be considered a potential public health threat, especially early in life, requiring further research and policy considerations.
Keywords: neurotoxicity, developmental neurotoxicology, neurotransmitter, neurotoxicology, astrocytes
Electronic cigarettes (e-cigarettes), nicotine delivery products whose potential health effects are poorly defined, have had a controversial reception from both the public and scientific communities around the world. Typically, e-cigarette liquid is a mixture of propylene glycol, glycerol, nicotine, flavorants, and other additives (Farsalinos et al., 2014). Although there is some consensus that e-cigarettes could reduce harm and dependency for conventional cigarette smokers, they also create a potential threat for those exposed during early life stages (Farsalinos et al., 2015; Gostin and Glasner, 2014). The consequences of consumption of these products by adolescents and young adults remain a public health question, partly due to nicotine’s previously identified negative impacts on the developing brain (Centers for Disease Control and Prevention, 2013; Dwyer et al., 2009; Schraufnagel, 2015). Moreover, early toxicological evaluations of e-cigarette devices have shown their capability of producing cellular damage and behavioral alterations (Bahl et al., 2012; Hom et al. 2016; Hwang et al., 2016; Martin et al., 2016; Ponzoni et al., 2015; Shen et al., 2016; Smith et al., 2015).
A limited number of studies exist pertaining to the usage and/or effects of e-cigarette use during pregnancy, or its impact on the mother and/or unborn child. A study by Wagner et al. (2017) reported that e-cigarette usage among pregnant women was approximately as prevalent as tobacco cigarette usage, but that e-cigarettes were considered to be safer, with that perceeption possibly resulting from the influence of advertising. A previous study (Kahr et al., 2015) examined attitudes among women of reproductive age surrounding e-cigarette and hookah use, given that use of both products is on the rise in that demographic. Although hookah use during pregnancy was viewed negatively, the study elicited mixed responses toward e-cigarette use during pregnancy. E-cigarettes were viewed positively when used as a smoking cessation device during pregnancy, but was viewed negatively overall. However, e-cigarette use during pregnancy was not met with the same stigma as tobacco cigarette use during pregnancy. Oncken et al. (2017) found that 53% of pregnant smokers had tried e-cigarettes in an effort to quit smoking (Oncken et al., 2017). However, Suter et al. (2015) considered the subject from a different standpoint, looking at existing data on nicotine delivery in animal models in the absence of cigarette smoke. This group noted numerous deleterious effects on the fetus resulting from nicotine exposure absent cigarette smoke, concluding that e-cigarettes with nicotine have the capacity to harm the developing fetus even in the absence of tobacco smoke (Suter et al., 2015). This does not, however, address the issue of potential harm from the e-cigarette vehicle solution, which will be addressed in this paper. This critical issue needs to be more thoroughly investigated, because cigarette smoke and thus, nicotine delivery during critical windows of brain development, is thought to lead to functionality deficits (Arrazola et al., 2015; England et al., 2015).
Cigarette smoke has been shown to directly and indirectly induce neuroinflammation, which can lead to neurodevelopmental damage and lasting effects. In a recent study, rats were exposed to cigarette smoke throughout gestation and after birth cerebral hemispheres were collected to develop astrocyte cultures. Cultures were incubated with hydrogen peroxide for 1 h and the astroglial cells from rats that were exposed prenatally to cigarette smoke had significantly reduced antioxidant levels and cell survival (Hamdi et al., 2016). As astrocytes are integral in maintaining central nervous system (CNS) homeostasis, astroglial cell loss could result in impairment of the antioxidant response leading to oxidative stress in the brain. In addition, cigarette smoke has been found to pose a direct risk for the development of Alzheimer’s disease (Durazzo et al., 2015; Giunta et al., 2012). This association is thought to involve decreases in microglial phagocytosis and/or an increased proinflammatory profile instigated by microglia (Giunta et al., 2012).
Both microglia and astrocytes play vital roles in neuroinflammation and dysfunction of either cell type is associated with neurodevelopmental disorders and neurodegenerative diseases (Floyd and Hensley, 2002; Guizzetti et al., 2014). Activation of astrocytes and microglia with concomitant morphological and signaling changes is an intrinsic part of neuroinflammation. The role of reactive glia, including both astrocytes and microglia, in neuroinflammation and neurodegenerative disease was reviewed by Ben Haim et al. (2015) with emphasis on glial fibrillary acidic protein (GFAP) as a marker for activated astrocytes (Ben Haim et al., 2015). Microglia are resident macrophage-like innate immune cells in the CNS that work to maintain homeostasis, and compose about 10% of the CNS population (DiSabato et al., 2016; Norden et al., 2015). Upon activation, microglia have morphological changes that can be detected using ionized calcium-binding adaptor molecule 1 (Iba-1); Iba-1 is a macrophage/microglia-specific calcium-binding protein that is involved in membrane ruffling and phagocytosis (Kanazawa et al., 2002; Ohsawa et al., 2000, 2004). Astrocytes, the most abundant cell type in the CNS, are a heterogeneous population involved in variety of processes including blood-brain barrier formation and maintenance, CNS metabolism, and neurotransmission (Chaboub and Deneen, 2012; Hu et al., 2016). Astrocyte dysfunction due to chronic stress or trauma can lead to the release of pro-inflammatory cells, loss of white matter, inhibition of axonal regeneration, formation of glial scarring, and inhibition of neurogenesis (Hu et al., 2016). Many studies have found that mature fibrous astrocytes and reactive astrocytes express the cytoskeletal component, GFAP, and up-regulation of GFAP occurs with aging, trauma, and disease (Chaboub and Deneen, 2012; Hu et al., 2016; Norden et al., 2015).
Micoglia and astrocytes are well known to be important in the homeostasis of the adult brain, and their activation is implicated in neurodegenerative disease. It is becoming increasingly clear that these cells also play an active role in the developing brain, thus, activation of glia during development as a result of e-cigarette exposure could have profound developmental consequences. Glia actively signal with neurons and disruption in this signaling may contribute to defects in synapse formation, pruning, and ultimately to cognitive impairment and neurodegenerative disease (Chung et al., 2015). Synaptic pruning is a part of the normal developmental process of the brain, occurring at high levels in the first weeks after birth. Microglia are constantly scanning neurons by extending and retracting processes, all the while signaling with neurons and actively maintaining the health of synapses.
Microglia are known to be active in brain remodeling at postnatal and perinatal stages; however, microglia are present in the brain at mid-embryogenesis and may play a role in earlier brain development. Microglia associate with dopaminergic axons and enter the cortical plate in a coordinated fashion, suggesting a role in patterning the neocortex (Squarzoni et al., 2014). Inhibition or absence of microglia in this region resulted in altered patterning of subsets of cortical interneurons, suggesting an active role for microglia in the wiring of the forebrain (Squarzoni et al., 2014).
A role for activated microglia has also been noted in white matter injury (WMI) of the brain associated with preterm birth. White matter injury is a leading cause of neurological morbidity due to cerebral palsy and other neurological damage (Back, 2017). White matter injury occurs during the maturation process of the oligodendrocyte lineage as a result of oxidative stress or other insult, and is accompanied by gliosis and disruption of the extracellular matrix, mediated by reactive astrocytes. It is unclear in the case of WMI to what degree glial cells are passive responders to injury, versus active participants in causing damage, but it is observed that microglia and activated astrocytes surround focal regions of necrosis in WMI.
This study aimed to assess the in vivo effects of e-cigarettes on neuroinflammation (ie, expression of Iba-1 and GFAP) in juvenile C57BL/6 offspring exposed during critical early life stages. The frontal cortex and hippocampus were selected as the major regions in the brain for investigating neuroinflammation. The frontal cortex, thought to be responsible for executive function, comprises many higher order cognitive processes (eg, impulse control, attention, working memory, planning, and decision making) and abilities that allow individuals to demonstrate goal-oriented behavior and self-control (Logue and Gould, 2014). The hippocampus was also selected for study as it plays a key role in spatial navigation, learning, and memory (Buzsáki and Moser, 2013; Eichenbaum and Cohen, 2014; Gruart et al., 2015). Studies using a rodent model have shown that the frontal cortex and hippocampus are susceptible to early-life damage, with prenatal stress leading to significant genomic alterations (Mychasiuk et al., 2011a,b).
The present study also assessed peripheral inflammatory markers (ie, IL-6, TNFα, IL-1β, IFNγ, IL-2, and MCP-1) in the serum of offspring to evaluate whether inflammation was localized in the CNS or if e-cigarettes induced a systemic inflammatory response. Although conventional cigarettes are known to cause low-grade systemic inflammation and have been linked to chronic inflammatory conditions (Rohleder and Kirschbaum, 2006), the inflammatory potential of e-cigarettes are not well known, though recent in vitro studies have shown that e-cigarette aerosols can elicit inflammation in lung epithelial cells (Hom et al., 2016; Scheffler et al., 2015; Schweitzer et al., 2015). In addition, this study assessed gene expression levels of neurotrophins (ie, Nerve Growth Factor Receptor [Ngfr], Brain Derived Neurotrophic Factor [Bdnf], and Glial Cell-Line Derived Neurotrophic Factor [Gdnf]) in the hippocampus of offspring (gene expression for these genes in the frontal cortex have been previously published [Lauterstein et al., 2016]). The CNS requires adequate levels of such trophic factors for proper development and function (Berry et al., 2012). Results from this investigation contribute to the limited toxicological database regarding developmental exposure to e-cigarettes, and emphasizes their potential to cause damage to the CNS during early life stages.
METERIALS AND METHODS
Animal care and exposures
C57BL/6 male and female mice (The Jackson Laboratory, Bar Harbor, ME) were maintained, paired, and exposed by inhalation to e-cigarette aerosols in polycarbonate cages as previously described in Lauterstein et al. (2016). Briefly, animals 8–9 week-of-age were paired for four nights (2 F/1 M per cage). Following pairing, males were removed and females (two/cage) were exposed to e-cigarette aerosols with or without nicotine or filtered air for 3 h/day, 5 days/week via whole-body inhalation in separate 1-m3 flow-through exposure chambers. Each day, mice were removed from their housing cages and placed in separate polycarbonate cages for exposures to minimize possible second- and/or third-hand exposures. No food or water was available in the exposure cages in order to prevent potential contamination by ingestion of the mice. Control mice exposed to filtered air underwent the same daily procedure (i.e., cage change, placement in 1-m3 flow-through exposure chamber, and food and water withdrawal for the same period of time) to those exposed to e-cigarette aerosols. At approximately day 15 of gestation, dams were separated and housed in individual cages, and exposures continued to parturition (∼3 weeks). Subsequently, lactating dams and their pups were exposed together via whole-body inhalation starting on postnatal days 4–6, and exposures continued throughout the lactation period (for a total of ∼3 weeks). Approximately 4 days following exposure cessation, mice were euthanized by intraperitoneal injection of 120 mg/kg pentobarbital sodium (Sleepaway; Fort Dodge Laboratories, Fort Dodge, IA) when the offspring were 1-month-old. All animal procedures were conducted under New York University Institutional Animal Care and Use Committee (IACUC) approval. Animals were housed and treated in the NYU Animal Exposure Facility of the NYU NIEHS Center.
E-cigarette exposures
E-cigarette aerosols were generated from blu classic tobacco flavor cartridges with (13 mg/ml) or without nicotine as previously described in Lauterstein et al. (2016). Briefly, an automated 3-port e-cigarette aerosol generator (e∼Aerosols, LLC, Central Valley, NY) was used to produce aerosols from the heating of e-cigarette cartridges. The puff regime consisted of 35 ml puff volumes of 4-s duration at 30-s intervals. Mean chamber levels of total suspended particulates for e-cigarette aerosols with and without nicotine were 25.6 and 30.7 mg/m3, respectively. Reported concentrations were measured using a portable DataRAM 4TM (Thermo Scientific, Waltham, MA).
Tissue processing, immuno-staining, and analysis for Iba-1 and GFAP
Following removal, whole brains were fixed for 48 h in 4% paraformaldehyde (Sigma, St. Louis, MO) and then placed in 30% sucrose/PBS solution (Sigma, St. Louis, MO) until they were no longer able to float. Samples (n = 6 [3 M and 3 F] offspring per treatment group) were brought (on ice) to Albert Einstein School of Medicine Histology Core for embedding, sectioning, staining and slide preparation. Brains were cut sagittally and processed routinely for paraffin embedding. Samples were sectioned to the area of hippocampus and frontal cortex, and five sections (10 µm thick) were collected at intervals separated by a distance of 40 µm. Each section was deparaffinized in xylene followed by graded alcohol treatment, and then samples were rehydrated in water. Antigen retrieval was performed in 10 mM sodium citrate buffer at pH 6.0 (Vector Labs, Burlingame, CA) at 96°C for 20 min. Slides were then placed into a Dako Cytomation Autostainer Plus (Dako North America, Inc., Carpinteria, CA) for staining. Blocking was performed by incubating tissue sections in 5% normal donkey serum with 2% BSA for 30 min.
The primary antibody to rabbit Iba-1 (#019-19741, Wako Chemicals USA, Inc., Richmond, VA) was used at a 1:500 dilution, and monoclonal antibody for GFAP (#835301, BioLegend, San Diego, CA) was also used at a 1:500 dilution; tissues and their respective primary antibodies were incubated for 1 h at room temperature. Tissue sections were then incubated for 30 min with biotinylated goat anti-rabbit IgG (#E0432, Dako North America, Inc., Carpinteria, CA), prepared at a 1:250 dilution. Following incubation with the secondary antibody, tissue sections were incubated for 1 h (at room temperature) with a 1:200 dilution of Streptavidin Alexa Fluor 555 and anti-mouse Alexa Fluor 488 (#S32355 and #A21202, Invitrogen, Carlsbad, CA). Slides were mounted with Prolong Gold Anti-fade reagent with DAPI (Invitrogen, Carlsbad, CA) and then stored in the dark at –4°C until microscopy analysis.
Whole sagittal slice images were captured at ×20 magnification using a PerkinElmer P250 High Capacity Slide Scanner (Fig. 1, example of image produced from slide scanner) and imported into Pannoramic Viewer imaging software. Using Pannoramic Viewer imaging software, images of the cornus ammonis 1 (CA1), cornus ammonis 3 (CA3), and dentate gyrus (DG) regions of the hippocampus, and frontal cortex region of each sagittal slice, were captured. Images for each brain region were imported into Volocity 6.3, the RGB channels split, and the three resultant images (channels) were combined into a three-channel image sequence for measurement. A standardized measurement protocol for each cell population (microglia, Iba-1 and astrocytes, GFAP) was created and used for each area of the brain, and the same measurements applied to all slides from that area. As fluorescence intensities for Iba-1 and GFAP were different in each brain area, a different measurement protocol was developed for each (with the exception of CA1 and CA3). To control for differences in background staining protocols were established in Velocity to filter the size of the object recognized for measurement. This has led to reduction in the overall brightness in slides with higher background staining. The goal of this procedure was to normalize background intensities and reduce variance related to differences in staining intensities. For each brain region sample imaged and analyzed in Volocity 6.3, summary data were exported and the mean intensity values for Iba-1 and GFAP were analyzed.
Figure 1.
Example of whole sagittal slide image produced from PerkinElmer P250 high capacity slide scanner.
Quantitative real time RT-PCR analyses
After mice were euthanized with pentobarbital, brains were removed, the hippocampus section recovered and stored at –80 °C until RNA was extracted from the samples. A standard protocol for RNA isolation using TRIzol Reagent (Invitrogen, Carlsbad, CA) was used, and the RNA samples were subsequently purified using a RNeasy Mini Kit (Qiagen, Valencia, CA). RNA was quantified using a NanoDrop ND-1000 fluorospectrometer (Thermo Scientific, Waltham, MA). Gene expression levels for neurotrophins were assessed in offspring hippocampus samples using qRT-PCR. The genes selected for follow-up evaluation included: Ngfr, Bdnf, and Gdnf (Table 1). Commercially available TaqMan gene expression assays (Table 1) for mice (Life Technologies, Grand Island, NY) were used to prepare samples, which were processed using a QuantStudio 6K Flex Real Time PCR System (Applied Biosystems, Foster City, CA). Quantified mRNA expression levels were calculated using the ΔΔCT method and normalized to the reference gene, glyceraldehyde-3-phosphate dehydrogenase (Gapdh).
Table 1.
Taqman Gene Expression Assays Used in RT-qPCR
| Gene | Gene Symbol | Assay ID | Amplicon Length | Reference Sequence |
|---|---|---|---|---|
| Nerve growth factor receptor | Ngfr | Mm00446296_m1 | 77 | NM_033217.3 |
| Brain derived neurotrophic factor | Bdnf | Mm04230607_s1 | 92 | NM_007540.4 |
| Glial cell line derived neurotrophic factor | Gdnf | Mm00599849_m1 | 101 | NM_010275.2 |
| Glyceraldehyde-3-phosphate dehydrogenase | Gapdh | Mm99999915_g1 | 109 | NM_001289726.1 |
Serum cytokine/chemokine analysis
Blood from the torso was collected from mice at the time of sacrifice, and serum was stored at –20°C until analysis. Samples were sent on dry ice to the Immune Monitoring Core at NYU Langone Medical Center, where they were analyzed for levels of IL-6, TNFα, IL-1β, IFNγ, IL-2, and MCP-1. Serum analytes were measured using Milliplex mouse cytokine/chemokine magnetic bead panel MCYTOMAG-70K (Millipore, Billerica, MA) 96-well plate assay. The plate was scanned on a Luminex 200 instrument, where the Median Fluorescent Intensity data were analyzed using a five-parameter logistic curve-fitting method for calculating cytokine/chemokines concentrations in samples.
Ingenuity pathway analysis of genes associated with CNS inflammation using frontal cortex transcriptome data
The analysis of the frontal cortex transcriptome in offspring after early-life exposure to e-cigarette aerosols was completed in this laboratory and has been previously published (Lauterstein et al., 2016). The RNA-Seq data are available in the Gene Expression Omnibus, and can be retrieved through series accession number GSE75858 (Lauterstein et al., 2016). Previously published frontal cortex RNA-Seq data were reanalyzed here to identify genes related to CNS inflammation using Ingenuity Pathway Analysis (IPA). The process used to identify genes related to CNS inflammation in the frontal cortex from RNA-Seq data is summarized in Figure 2. Briefly, a comparison analysis between IPA Diseases and Functions molecule/gene list “Affects Inflammation of the CNS,” and molecules/genes found to be directly affected for each treatment group and sex was completed. Overlapping genes between “Affects Inflammation of the CNS” molecule list and individual treatment/sex groups affected molecules were identified.
Figure 2.

Process used to identify genes related to central nervous system inflammation.
Statistics
Mean intensity values for Iba-1 and GFAP and data from qRT-PCR were analyzed using a two-way ANOVA and Bonferroni post hoc testing. Cytokine/chemokine data were analyzed using a Student’s t-test. Statistical analyses were performed using GraphPad Prism 4 (GraphPad Software, Inc., San Diego, CA), and the differences were considered significant when p-values were ≤0.05. All data are shown as the mean ± SEM.
Differential statistical gene analysis was conducted using the DESeq2 R/Bioconductor package in the R statistical program. Alterations in gene expression were studied by comparing female and male frontal cortex samples from e-cigarette groups to their sex-matched filtered air control counterparts. The Benjamini-Hochberg procedure (used R function p.adjust) for multiple testing, which controls false discovery rate, was used to determine adjusted p-values. For each treatment, group/sex dataset which included: (1) female offspring exposed to e-cigarettes with nicotine; (2) female offspring exposed to e-cigarettes without nicotine; (3) male offspring exposed to e-cigarettes with nicotine; and (4) male offspring exposed to e-cigarettes without nicotine, fold changes of genes with an adjusted p-value of <0.01 were imported into IPA software to examine biological effects and disease pathway outcomes associated with the gene expression data.
RESULTS
Iba-1 and GFAP Expression in the Frontal Cortex and Hippocampus From Offspring
Fluorescent microscopy images for frontal cortex, CA1, CA3, and DG brain regions taken in Pannoramic Viewer software were imported into Volocity 6.3, where the mean intensity for slides stained with Iba-1 and GFAP antibodies were determined. No expression changes of GFAP were observed as a result of early-life exposure to e-cigarettes with or without nicotine (Figs. 3A–D). Alternatively, a significant increase in mean intensity for Iba-1 was observed in the CA1 region of male and female offspring exposed early in life to e-cigarette aerosols without nicotine; similar effects were not observed in male or female offspring exposed to e-cigarettes with nicotine (Figure 3A). Additionally, Iba-1 mean intensity tends to be increased in the frontal cortex region of offspring exposed to e-cigarettes without nicotine, but again not in those exposed to e-cigarettes with nicotine (Figure 3B). Iba-1 expression was not altered in the CA3 and DG regions of the hippocampus for either e-cigarette treatment group (Figs. 3C and 3D, respectively). Representative images for CA1 region from each treatment group are shown in Figure 4.
Figure 3.
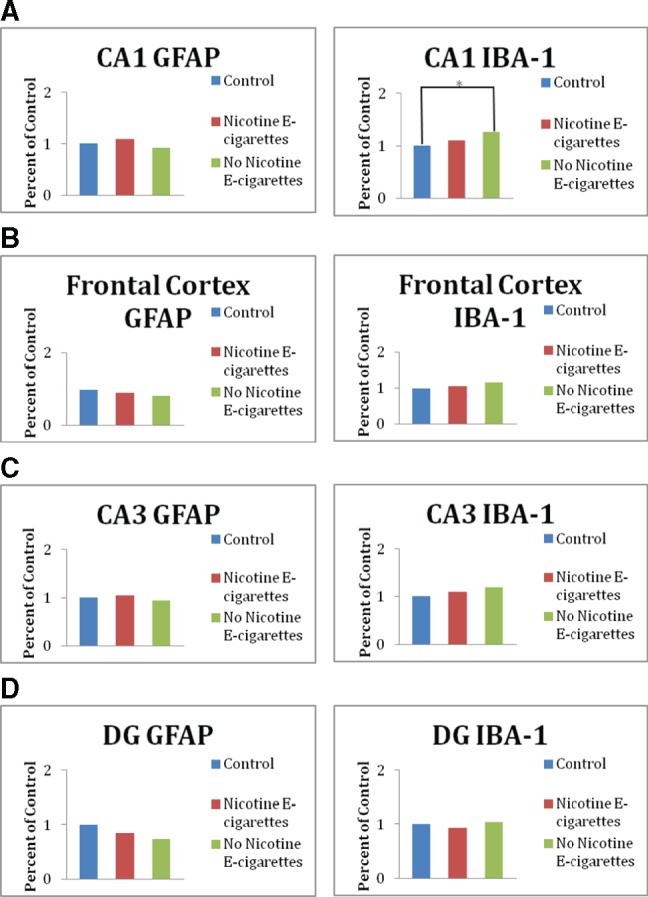
Expression of ionized calcium-binding adaptor molecule 1 (Iba-1) and glial fibrillary acidic protein (GFAP) in the hippocampus of offspring exposed to e-cigarette aerosols with and without nicotine early in life. A, Expression of Iba-1 and GFAP in the CA1 region of the hippocampus. B, Expression of Iba-1 and GFAP in the frontal cortex. C, Expression of Iba-1 and GFAP in the CA3 region of the hippocampus. D, Expression of Iba-1 and GFAP in the DG region of the hippocampus. All data are represented as mean values (n = 3 M and 3 F) ± SEM; for each individual sample three to six slide replicates were analyzed. *Significantly different from filtered air control p < .01.
Figure 4.

Representative images of the cornus ammonis 1 (CA1) hippocampal region from each treatment group. Immunofluorescence staining for Iba-1 (red), GFAP (green), and DAPI (blue) was identified using a PerkinElmer P250 high capacity slide scanner. A, CA1 region from a mouse treated with e-cigarette aerosols with no nicotine. B, CA1 region from a mouse treated with e-cigarette aerosols with nicotine. C, CA1 region from control mouse treated with filtered-air.
qRT-PCR Analysis of Neurotrophins in the Hippocampus of Offspring
Significant (p < .001) decreases in gene expression levels of Ngfr and Bdnf were observed in sex-pooled hippocampus samples from offspring exposed early in life to both e-cigarette aerosols with and without nicotine when compared with sex-pooled filtered air controls (Figure 5). No significant differences were observed for Gdnf (data not shown).
Figure 5.
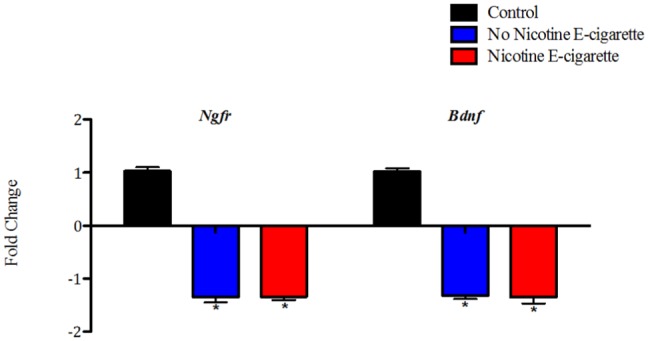
Decreases in hippocampal expression levels for Ngfr and Bdnf after early-life exposure to e-cigarette aerosols with and without nicotine. All data are represented as mean values (n = 6–10) ± SEM. *Significantly different from filtered air control p < .001. Results were calculated using the 2^(–ΔΔCT) method.
Serum Cytokine/Chemokine Analysis in Offspring
A Milliplex mouse cytokine/chemokine magnetic bead panel was used to measure serum levels of IL-6, TNFα, IL-1β, IFNγ, IL-2, MCP-1 in 1-month-old offspring. Significant decreases in serum levels of IL-1β were observed for both sexes of offspring exposed to e-cigarettes with and without nicotine when compared with sex-pooled filtered air controls (Figure 6A). Moreover, a significant decrease in serum levels of IL-2 was observed for offspring exposed to e-cigarette aerosols with nicotine; offspring exposed to e-cigarette aerosols without nicotine were modestly (albeit, not significantly) reduced compared with sex-pooled filtered air controls (Figure 6B). Serum levels of IL-6 tend to be decreased in offspring exposed to e-cigarette aerosols with and without nicotine compared with sex-pooled filtered air controls (Figure 6C). No effects on serum levels of TNFα, MCP-1, or IFNγ were observed compared with controls (data not shown).
Figure 6.
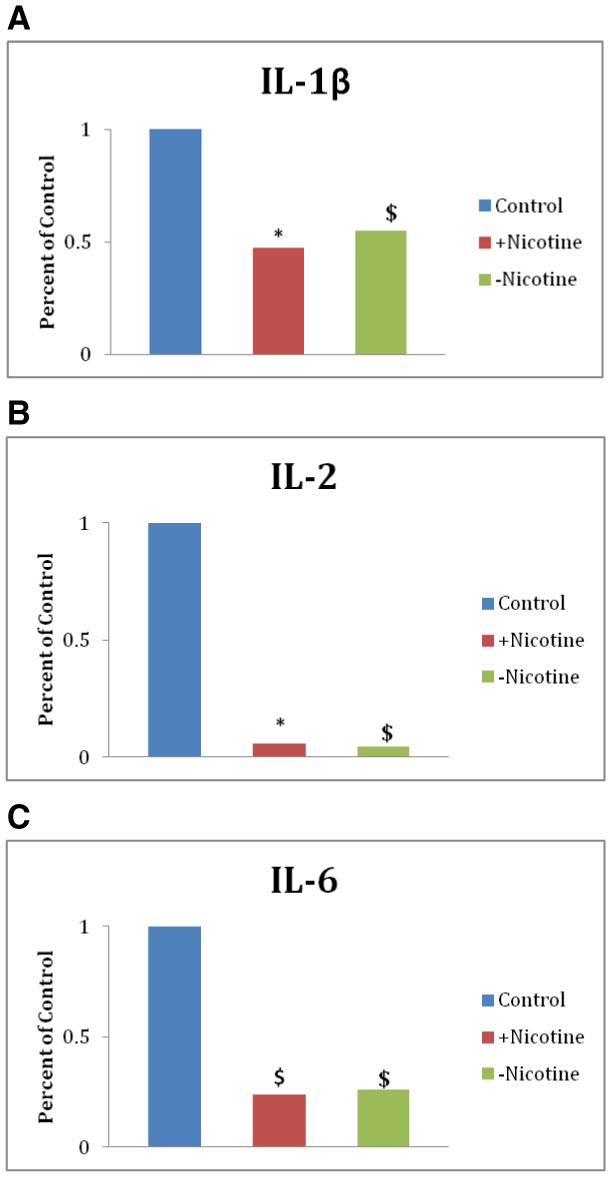
Early-life exposure to e-cigarette aerosols with nicotine decreases serum cytokine levels. A, Serum levels of IL-1β. B, Serum levels of IL-2. C, Serum levels of IL-6. All data are represented as mean values (n = 3–9) ± SEM. *Significantly different from filtered air control p < .05. $Difference from filtered air control (p < .1).
Analysis of Genes Associated With CNS Inflammation in Offspring Using Frontal Cortex Transcriptome Data
Results from these analyses revealed that male offspring exposed early in life to e-cigarette aerosols without nicotine had 27 altered genes that overlapped (genes in common) with the IPA “affects inflammation of the CNS” molecule list (Table 2A). Female offspring exposed early in life to e-cigarette aerosols without nicotine had 24 altered genes that overlapped with IPA “affects inflammation of the CNS” molecule list (Table 2B). Whereas, male offspring exposed early in life to e-cigarette aerosols containing nicotine only had six altered genes that overlapped with IPA “affects inflammation of the CNS” molecule list (Table 2C); and, female offspring exposed early in life to e-cigarette aerosols with nicotine had 13 altered genes that overlapped with IPA “affects inflammation of the CNS” molecule list (Table 2D).
Table 2.
Overlapping Molecules (Genes in Common) That Affect CNS Inflammation in Juvenile Offspring Exposed Early in Life to E-Cigarette Aerosols
| Symbol | Entrez Gene Name | Type(s) | Entrez Gene ID for Mouse |
|---|---|---|---|
| (A) Males exposed to e-cigarette aerosols without nicotine | |||
| CST3 | Cystatin C | Other | 13010 |
| CYP51A1 | Cytochrome P450 family 51 subfamily A member 1 | Enzyme | 13121 |
| DHFR | Dihydrofolate reductase | Enzyme | 13361 |
| ESR2 | Estrogen receptor 2 (ER beta) | Ligand-dependent nuclear receptor | 13983 |
| GRIA3 | Glutamate receptor, ionotropic, ampa 3 | Ion channel | 53623 |
| HIF1A | Hypoxia inducible factor 1, alpha subunit (basic helix-loop-helix transcription factor) | Transcription regulator | 15251 |
| Igh (family) | — | Group | |
| ITGA4 | Integrin subunit alpha 4 | Transmembrane receptor | 16401 |
| LGALS1 | Lectin, galactoside-binding, soluble, 1 | Other | 16852 |
| LGI1 | Leucine-rich, glioma inactivated 1 | Other | 56839 |
| MAPK11 | Mitogen-activated protein kinase 11 | Kinase | 19094 |
| MAPK14 | Mitogen-activated protein kinase 14 | Kinase | 26416 |
| MBP | Myelin basic protein | Other | 17196 |
| MOG | Myelin oligodendrocyte glycoprotein | Other | 17441 |
| NFATC2 | Nuclear factor of activated t-cells, cytoplasmic, calcineurin-dependent 2 | Transcription regulator | 18019 |
| NGFR | Nerve growth factor receptor | Transmembrane receptor | 18053 |
| NR3C1 | Nuclear receptor subfamily 3 group c member 1 | Ligand-dependent nuclear receptor | 14815 |
| PLAT | Plasminogen activator, tissue type | Peptidase | 18791 |
| PLP1 | Proteolipid protein 1 | Other | 18823 |
| PPP3CB | Protein phosphatase 3, catalytic subunit, beta isozyme | Phosphatase | 19056 |
| PTGS2 | Prostaglandin-endoperoxide synthase 2 (prostaglandin g/h synthase and cyclooxygenase) | Enzyme | 19225 |
| RORA | Rar-related orphan receptor a | Ligand-dependent nuclear receptor | 19883 |
| SCN1B | Sodium channel, voltage gated, type i beta subunit | Ion channel | 20266 |
| SCN5A | Sodium channel, voltage gated, type v alpha subunit | Ion channel | 20271 |
| SERPINE1 | Serpin peptidase inhibitor, clade e (nexin, plasminogen activator inhibitor type 1), member 1 | Other | 18787 |
| SOD1 | Superoxide dismutase 1, soluble | Enzyme | 20655 |
| Thymidine kinase | — | Group | |
| (B) Females exposed to e-cigarette aerosols without nicotine | |||
| APP | Amyloid beta precursor protein | Other | 11820 |
| C4A/C4B | Complement component 4B (Chido blood group) | Other | 625018|12268 |
| ESR2 | Estrogen receptor 2 (ER beta) | Ligand-dependent nuclear receptor | 13983 |
| GRIA3 | Glutamate receptor, ionotropic, AMPA 3 | Ion channel | 53623 |
| ITGA4 | Integrin subunit alpha 4 | Transmembrane receptor | 16401 |
| LGI1 | Leucine-rich, glioma inactivated 1 | Other | 56839 |
| LRRK2 | Leucine-rich repeat kinase 2 | Kinase | 66725 |
| MAPK11 | Mitogen-activated protein kinase 11 | Kinase | 19094 |
| MAPK14 | Mitogen-activated protein kinase 14 | Kinase | 26416 |
| MBP | Myelin basic protein | Other | 17196 |
| MOG | Myelin oligodendrocyte glycoprotein | Other | 17441 |
| NFATC2 | Nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent 2 | Transcription regulator | 18019 |
| NGFR | Nerve growth factor receptor | Transmembrane receptor | 18053 |
| NR3C1 | Nuclear receptor subfamily 3 group C member 1 | Ligand-dependent nuclear receptor | 14815 |
| PLP1 | Proteolipid protein 1 | Other | 18823 |
| PPP3CA | Protein phosphatase 3, catalytic subunit, alpha isozyme | Phosphatase | 19055 |
| PPP3CB | Protein phosphatase 3, catalytic subunit, beta isozyme | Phosphatase | 19056 |
| PPP3CC | Protein phosphatase 3, catalytic subunit, gamma isozyme | Phosphatase | 19057 |
| PTGS2 | Prostaglandin-endoperoxide synthase 2 (prostaglandin G/H synthase and cyclooxygenase) | Enzyme | 19225 |
| RORA | RAR-related orphan receptor A | Ligand-dependent nuclear receptor | 19883 |
| SCN1B | Sodium channel, voltage gated, type I beta subunit | Ion channel | 20266 |
| SCN5A | Sodium channel, voltage gated, type V alpha subunit | Ion channel | 20271 |
| SOD1 | Superoxide dismutase 1, soluble | Enzyme | 20655 |
| TH2 Cytokine | — | group | |
| (C) Males exposed to e-cigarette aerosols with nicotine | |||
| APP | Amyloid beta precursor protein | Other | 11820 |
| MAPK11 | Mitogen-activated protein kinase 11 | Kinase | 19094 |
| NGFR | Nerve growth factor receptor | Transmembrane receptor | 18053 |
| NOS2 | Nitric oxide synthase 2 | Enzyme | 18126 |
| NR3C1 | Nuclear receptor subfamily 3 group C member 1 | Ligand-dependent nuclear receptor | 14815 |
| XBP1 | X-box binding protein 1 | Transcription regulator | 22433 |
| (D) Females exposed to e-cigarette aerosols with nicotine | |||
| ESR2 | Estrogen receptor 2 (ER beta) | Ligand-dependent nuclear receptor | 13983 |
| ITGA4 | Integrin subunit alpha 4 | Transmembrane receptor | 16401 |
| KIT | KIT proto-oncogene receptor tyrosine kinase | Transmembrane receptor | 16590 |
| LGI1 | Leucine-rich, glioma inactivated 1 | Other | 56839 |
| MBP | Myelin basic protein | Other | 17196 |
| MOG | Myelin oligodendrocyte glycoprotein | Other | 17441 |
| NFATC2 | Nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent 2 | Transcription regulator | 18019 |
| NGFR | Nerve growth factor receptor | Transmembrane receptor | 18053 |
| NR3C1 | Nuclear receptor subfamily 3 group C member 1 | Ligand-dependent nuclear receptor | 14815 |
| PLP1 | Proteolipid protein 1 | Other | 18823 |
| PPP3CB | protein phosphatase 3, catalytic subunit, beta isozyme | Phosphatase | 19056 |
| RORA | RAR-related orphan receptor A | Ligand-dependent nuclear receptor | 19883 |
| Thymidine kinase | — | Group | |
DISCUSSION
In this study, a significant increase in Iba-1 expression was observed in the CA1 hippocampal region of 1-month-old offspring exposed to e-cigarette aerosols without nicotine (compared with controls). This result suggests that e-cigarette components other than nicotine are capable of causing localized inflammation in the hippocampus, an area of the brain that plays an essential role in long-term memory and learning (Lazarov and Hollands, 2016). In support of this finding, the IPA analysis of genes associated with CNS inflammation in offspring using frontal cortex transcriptome data revealed a greater number of overlapping molecules that affect CNS inflammation for offspring exposed to e-cigarettes without nicotine, compared with the number of overlapping genes in offspring exposed to e-cigarettes with nicotine. Prolonged activation of proinflammatory microglia can lead to neurotoxicity, neurological deficits and neurodegeneration, indicating that e-cigarette components other than nicotine pose a sizable threat to the developing CNS. Based on the results from this study (i.e., that Iba-1 expression was elevated in offspring exposed to e-cigarette aerosols without nicotine, but was not altered in offspring exposed to e-cigarette aerosols with nicotine), and published literature indicating that nicotine has the potential to be neuroprotective (Huang et al., 2007; Opanashuk et al., 2001; Wielgus et al., 2004), it is possible that nicotine exerts some degree of neuroprotection by preventing/mitigating the activation of microglia, as reviewed later.
In support of the proposed hypothesis that nicotine exposure during early life resulted in some level of neuroprotection, Huang et al. (2007) demonstrated that treatment from PND 1-7 with nicotine (via oral gastric intubation, total daily dose of 6 mg/kg/day) decreased the number of dying cells in hippocampal CA3 areas (i.e., in the CA3 strata oriens and radiatu areas). In another study, Wielgus et al. (2004) showed that embryonic chick yolks exposed (embryonic days 1–7) to low doses of nicotine (daily doses of 100 ng nicotine) produced nicotine-induced inhibition of motor neuron apoptosis and enhanced neuronal growth (Wielgus et al., 2004). In addition, Opanashuk et al. (2001) found that nicotine (1 μM treatment for 7 days) increased proliferation and survival of cerebellar neuronal precursors purified from 5-day-old mice (Opanashuk et al., 2001). Taken together, these studies suggest that exposure to low doses of nicotine during early life may have a neuroprotective effect by restricting developmental cell death, and promoting neurogenesis in certain neuronal populations (Huang et al., 2007; Opanashuk et al., 2001; Wielgus et al., 2004).
Meanwhile, another e-cigarette constituent propylene glycol has been shown to have pro-apoptotic effects. Lau et al. (2012) reported that a single intraperitoneal injection of propylene glycol (doses as low as 2 ml/kg)-induced widespread apoptosis in the developing mouse brain (Lau et al., 2012). The observed degeneration was similar to that which followed injection with ethanol. Although Lau et al. (2012) did not investigate the role of glial cell activation in propylene glycol-induced apoptosis observed in their study, aberrant microglia activation has been associated with neuronal cell death (Cotman et al., 1996; Floyd and Hensley, 2002). It is possible that exposure to propylene glycol in this study brought about increases in Iba-1 expression in offspring exposed to e-cigarette aerosols without nicotine. Although these deductions are currently speculative in nature, and suffer from a lack of literature evaluating the toxicity of propylene glycol in the brain, they outline the importance of further studies needed to evaluate the potential of propylene glycol to bring about such effects.
The aforementioned studies have shown potential neuroprotective effects from early-life exposure to nicotine, and nicotine is known to have neuroprotective effects in adults. However, reports indicating that early-life exposure to nicotine can inhibit or drive damaging neurogenesis and induce neuroinflammation are contradictory. Chang et al. (2013) reported that rats exposed to nicotine via osmotic pumps (1.5 mg/kg/day) during gestation exhibited increased neurogenesis of orexigenic peptide-expressing neurons in the hypothalamus and amygdala (Chang et al., 2013). The aforementioned changes were associated with increased vulnerability to consummatory behavior (ie, increased consumption of nicotine, ethanol, and a fat-rich diet in rats prenatally exposed to nicotine) later in life. However, no changes were observed in the gliogenesis markers, GFAP, and Iba-1 (Chang et al., 2013). A study by Orellana et al. (2014) demonstrated that prenatal nicotine exposure via osmotic pumps (60 mg/kg/day) combined with a postnatal high-fat/cholesterol-rich diet enhanced release of gliotransmitters in mice (Orellana et al., 2014). Another study using a rat model, found that gestational exposure to nicotine via osmotic pumps (3.3 mg/kg/day) resulted in decreases in surviving neurons, and increases in GFAP in the cerebellum and CA1 region of the hippocampus at PND 30 and 60 (Abdel-Rahman et al., 2005).
Thus, the literature concerning the effects of nicotine exposure during early life stages are divergent, with findings demonstrating that nicotine is both a developmental neuroprotectant and neurotoxin. Conflicts between the studies are likely due, at least in part, to differences between exposure route, nicotine dose, and experimental design used in each study.
Although enhanced expression of Iba-1 was only observed in offspring exposed to e-cigarette aerosols without nicotine, early-life exposure to both e-cigarettes with and without nicotine-induced alterations in the other biological parameters (ie, gene expression of neurotrophins in the hippocampus, and serum levels of cytokines) examined in this study. During early postnatal development, the CNS is sensitive to external stimuli and neurotrophins, and nerve growth factor (NGF) and Brain-Derived Neurotrophic factor (BDNF) modulate brain plasticity to adapt to the environment (Berry et al., 2012). Several studies suggest that reduced NGF and BDNF are associated with the pathophysiology of psychiatric disorders (Altar, 1999; Castrén, 2004; Cirulli and Alleva, 2009; Sen et al., 2008). In this study, the reductions seen in Ngfr and Bdnf in the hippocampus could indicate decreased synaptic plasticity, and/or impair development of coping responses to new or stressful situations in offspring exposed to e-cigarettes (with and without nicotine) early in life. Moreover, reductions in these two neurotrophins could increase the susceptibility of the CNS to damage arising from oxidative stress and inflammation.
A significant decrease in serum levels of IL-1β and IL-2 was observed following exposure to e-cigarette aerosols containing nicotine in both sexes; serum levels of IL-6 were modestly reduced in both treatment groups compared with pooled controls. Serum levels of IL-1β and IL-2 were modestly reduced in mice following exposure to e-cigarette aerosols without nicotine, but levels failed to reach statistical significance compared with pooled control values. In support of these toxicological studies, prospective birth cohorts have been reported to exhibit decreased cytokine levels in cord blood from infants whose mothers smoked cigarettes. In prospective studies, Noakes et al. (2006) reported significantly decreased cord blood serum levels of IL-6, IL-10, and TNFα, and Macaubas et al. (2003) found significantly decreased cord blood serum levels of IL-4 and IFNγ in infants whose mothers smoked compared with cord blood samples from infants whose mothers did not smoke (Macaubas et al., 2003; Noakes et al., 2006).
In vivo studies have demonstrated varying inflammatory outcomes from gestational nicotine exposure. Mohsenzadeh et al. (2014) reported that prenatal exposure to various doses of nicotine leads to significant dose-dependent increases in serum levels of acute phase reactant hs-CRP, and proinflammatory cytokines IL-6 and TNFα in newborn rats (Mohsenzadeh et al., 2014). In contrast to these findings, Yang et al. (2014) demonstrated that pregnant rats first injected with lipopolysaccharide (LPS) and subsequently treated with nicotine exhibited reductions in LPS-induced inflammatory responses (ie, serum levels of IL-6 and TNFα), as well as increased viability of the fetus (Yang et al., 2014).
Although the observed decreases in peripheral cytokines that were seen herein are consistent with published literature for cigarette smoke and nicotine exposures, the effects on cytokine levels seen in our studies without nicotine are likely due to other e-cigarette constituent(s). The e-cigarette-induced decreases in peripheral cytokines observed here suggest that early-life exposure to e-cigarette aerosols may modulate immunity compromising the ability of offspring to mount an effective immune response. In support of our findings presented here, other recent studies have observed immune-suppression resulting from e-cigarette exposure in humans and animals. For example, a recent study by Martin et al. (2016) examined immune-related gene changes in the nasal mucosa of e-cigarette (various brands, containing nicotine) and conventional cigarette users. Results from that study revealed that e-cigarette users had a higher quantity of gene expression alterations, as well as greater levels of immune-suppression compared with conventional cigarette users (Martin et al., 2016). In addition, animal studies have shown that inhalation of e-cigarette aerosols that contained nicotine (aerosols that did not contain nicotine were not examined) compromised host resistance to viral and bacterial infections in mice (Hwang et al., 2016; Sussan et al., 2015).
CONCLUSIONS
To our knowledge, this is the first study to examine e-cigarette aerosol-induced effects on activation of astrocytes and microglia in the developing brain. Results from this study revealed that only offspring exposed early in life to e-cigarette aerosols without nicotine-induced microglia activation. This effect could arise as a result of the dual function of nicotine as both a developmental neurotoxicant and neuroprotectant. On the other hand, offspring exposed during early life to both e-cigarette aerosols with and without nicotine exhibited reductions in gene expression levels of neurotrophins and serum levels of cytokines. Findings from this study indicate that e-cigarette constituents other than nicotine, such as flavorings, propylene glycol or glycerin, could induce biological disruptions in the developing CNS. Prior studies investigating propylene glycol and glycerin have demonstrated that these compounds are relatively harmless when ingested, and thus have been designated as “generally regarded as safe.” However, studies concerning the potential toxicity of propylene glycol and glycerin when inhaled, rather than ingested, are scarce (Farsalinos et al., 2015; Tayyarah and Long, 2014).
The present study highlights the need for increased investigations on e-cigarette constituents, including nicotine, using a relevant exposure route. In conclusion, although e-cigarettes are often viewed as “safe,” our data reveal a substantial threat to the developing CNS. Thus, considering their public health risk potential, particularly during susceptible life stages, e-cigarettes require additional study and appropriate policy considerations.
ACKNOWLEDGMENTS
The authors thank the Immune Monitoring Core at NYU Langone Medical Center and the Albert Einstein School of Medicine Histology and Analytical Imaging core facilities for all their help with experimental design and analysis.
FUNDING
National Institutes of Health (R01 ES 07731, R01 ES10563, R03 ES024849, R21 ES025415) to M.A. and NYU NIEHS P30ES000260-50 pilot grant (JTZ).
REFERENCES
- Abdel-Rahman A., Dechkovskaia A. M., Sutton J. M., Chen W. C., Guan X., Khan W. A., Abou-Donia M. B. (2005). Maternal exposure of rats to nicotine via infusion during gestation produces neurobehavioral deficits and elevated expression of glial fibrillary acidic protein in the cerebellum and CA1 subfield in the offspring at puberty. Toxicology 209, 245–261. [DOI] [PubMed] [Google Scholar]
- Altar C. A. (1999). Neurotrophins and depression. Trends Pharmacol. Sci. 20, 59–61. 10.1016/S0165-6147(99)01309-7 [DOI] [PubMed] [Google Scholar]
- Arrazola R. A., Singh T., Corey C. G., Husten C. G., Neff L. J., Apelberg B. J., Bunnell R. E., Choiniere C. J., King B. A., Cox S. (2015). Tobacco use among middle and high school students—United States, 2011–2014. MMWR Morb. Mortal Wkly Rep. 14, 381–385. [PMC free article] [PubMed] [Google Scholar]
- Back S. A. (2017). White matter injury in the preterm infant: pathology and mechanisms. Acta Neuropathol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahl V., Lin S., Xu N., Davis B., Wang Y. H., Talbot P. (2012). Comparison of electronic cigarette refill fluid cytotoxicity using embryonic and adult models. Reprod. Toxicol. 34, 529–537. 10.1016/j.reprotox.2012.08.001 [DOI] [PubMed] [Google Scholar]
- Ben Haim L., Carrillo-de Sauvage M.-A., Ceyzériat K., Escartin C. (2015). Elusive roles for reactive astrocytes in neurodegenerative disease. Fron. Cell. Neurosci. 9, 278.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry A., Bindocci E., Alleva E. (2012). NGF, brain and behavioral plasticity. Neural Plast. 2012, 784040.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G., Moser E. I. (2013). Memory, navigation and theta rhythm in the hippocampal-entorhinal system. Nat. Neurosci. 16, 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrén E. (2004). Neurotrophins as mediators of drug effects on mood, addiction, and neuroprotection. Mol. Neurobiol. 29, 289–302. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (2013). Notes from the field: electronic cigarette use among middle and high school students—United States, 2011–2012. MMWR Morb. Mortal. Wkly Rep. 35, 729–730. [PMC free article] [PubMed] [Google Scholar]
- Chaboub L. S., Deneen B. (2012). Developmental origins of astrocyte heterogeneity: the final frontier of CNS development. Dev. Neurosci. 5, 379–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang G. Q., Karatayev O., Leibowitz S. F. (2013). Prenatal exposure to nicotine stimulates neurogenesis of orexigenic peptide-expressing neurons in hypothalamus and amygdala. J. Neurosci. 33, 13600–13611. 10.1523/JNEUROSCI.5835-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W. S., Welsh C. A., Barres B. A., Stevens B. (2015). Do glia drive synaptic and cognitive impairment in disease? Nat. Neurosci. 18, 1539–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli F., Alleva E. (2009). The NGF saga: from animal models of psychosocial stress to stress-related psychopathology. Front. Neuroendocrinol. 30, 379–395. 10.1016/j.yfrne.2009.05.002 [DOI] [PubMed] [Google Scholar]
- Cotman C. W., Tenner A. J., Cummings B. J. (1996). beta-Amyloid converts an acute phase injury response to chronic injury responses. Neurobiol. Aging 17, 723–731. 10.1016/0197-4580(96)00117-0 [DOI] [PubMed] [Google Scholar]
- DiSabato D., Quan N., Godbout J. P. (2016). Neuroinflammation: the Devil is in the Details. J. Neurochem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durazzo T. C., Meyerhoff D. J., Murray D. E. (2015). Comparison of regional brain perfusion levels in chronically smoking and non-smoking adults. Int. J. Environ. Res. Public Health 12, 8198–8213. 10.3390/ijerph120708198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer J. B., McQuown S. C., Leslie F. M. (2009). The dynamic effects of nicotine on the developing brain. Pharmacol. Ther. 2, 125–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichenbaum H., Cohen N. J. (2014). Can we reconcile the declarative memory and spatial navigation views on hippocampal function? Neuron 83, 764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England L. J., Bunnell R. E., Pechacek T. F., Tong V. T., McAfee T. A. (2015). Nicotine and the developing human: a neglected element in the electronic cigarette debate. Am. J. Prev. Med. 2, 286–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farsalinos K. E., Romagna G., Tsiapras D., Kyrzopoulos S., Voudris V. (2014). Characteristics, perceived side effects and benefits of electronic cigarette use: a worldwide survey of more than 19,000 consumers. Int. J. Environ. Res. Public Health 4, 4356–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farsalinos K. E., Le Houezec J. (2015). Regulation in the face of uncertainty: the evidence on electronic nicotine delivery systems (e-cigarettes). Risk Manag. Healthc. Policy 8, 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farsalinos K. E., Kistler K. A., Gillman G., Voudris V. (2015). Evaluation of electronic cigarette liquids and aerosol for the presence of selected inhalation toxins. Nicotine Tob. Res. 17, 168–174. 10.1093/ntr/ntu176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd R. A., Hensley K. (2002). Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol. Aging 5: 795–807. [DOI] [PubMed] [Google Scholar]
- Giunta B., Deng J., Jin J., Sadic E., Rum S., Zhou H., Sanberg P., Tan J. (2012). Evaluation of how cigarette smoke is a direct risk for Alzheimer’s disease. Technol. Innov. 14, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gostin L. O., Glasner A. Y. (2014). E-cigarettes, vaping, and youth. JAMA 6, 595–596. [DOI] [PubMed] [Google Scholar]
- Gruart A., Leal-Campanario R., López-Ramos J. C., Delgado-García J. M. (2015). Functional basis of associative learning and their relationships with long-term potentiation evoked in the involved neural circuits: lessons from studies in behaving mammals. Neurobiol. Learn. Mem. 124, 3–18. [DOI] [PubMed] [Google Scholar]
- Guizzetti M., Zhang X., Goeke C., Gavin D. P. (2014). Glia and neurodevelopment: focus on fetal alcohol spectrum disorders. Front. Pediatr. 2, 123.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdi Y., Madfai H., Belhareth R., Mokni M., Masmoudi-Kouki O., Amri M. (2016). Prenatal exposure to cigarette smoke enhances oxidative stress in astrocytes of neonatal rat. Toxicol. Mech. Methods 26, 231–237. [DOI] [PubMed] [Google Scholar]
- Hom S., Chen L., Wang T., Ghebrehiwet B., Yin W., Rubenstein D. A. (2016). Platelet activation, adhesion, inflammation, and aggregation potential are altered in the presence of electronic cigarette extracts of variable nicotine concentrations. Platelets 1–9. [DOI] [PubMed] [Google Scholar]
- Hu X., Yuan Y., Wang D., Su Z. (2016). Heterogeneous astrocytes: active players in CNS. Brain Res. Bull. 125, 1–18. 10.1016/j.brainresbull.2016.03.017 [DOI] [PubMed] [Google Scholar]
- Huang L. Z., Abbott L. C., Winzer-Serhan U. H. (2007). Effects of chronic neonatal nicotine exposure on nicotinic acetylcholine receptor binding, cell death and morphology in hippocampus and cerebellum. Neuroscience 146, 1854–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J. H., Lyes M., Sladewski K., Enany S., McEachern E., Mathew D. P., Das S., Moshensky A., Bapat S., Pride D. T., et al. (2016). Electronic cigarette inhalation alters innate immunity and airway cytokines while increasing the virulence of colonizing bacteria. J. Mol. Med. (Berl.) 94, 667–679. [DOI] [PubMed] [Google Scholar]
- Kahr M. K., Padgett S., Shope C. D., Griffin E. N., Xie S. S., Gonzalez P. J., Levison J., Mastrobattista J., Abramovici A. R., Northrup T. F., et al. (2015). Quantitative assessment of the perceived risks of electronic cigarette and hookah use in pregnancy. BMC Public Health 15, 1273.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanazawa H., Ohsawa K., Sasaki Y., Kohsaka S., Imai Y. (2002). Macrophage/microglia-specific protein Iba1 enhances membrane ruffling and Rac activation via phospholipase C-gamma-dependent pathway. J. Biol. Chem. 277, 20026–22032. [DOI] [PubMed] [Google Scholar]
- Lau K., Swiney B. S., Reeves N., Noguchi K. K., Farber N. B. (2012). Propylene glycol produces excessive apoptosis in the developing mouse brain, alone and in combination with phenobarbital. Pediatr. Res. 71, 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterstein D. E., Tijerina P. B., Corbett K., Akgol Oksuz B., Shen S. S., Gordon T., Klein C. B., Zelikoff J. T. (2016). Frontal cortex transcriptome analysis of mice exposed to electronic cigarettes during early life stages. Int. J. Environ. Res. Public Health 13, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov O., Hollands C. (2016). Hippocampal neurogenesis: learning to remember. Prog. Neurobiol. 138-140, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue S. F., Gould T. J. (2014). The neural and genetic basis of executive function: attention, cognitive flexibility, and response inhibition. Pharmacol. Biochem. Behav. 123, 45–54. 10.1016/j.pbb.2013.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaubas C., de Klerk N. H., Holt B. J., Wee C., Kendall G., Firth M., Sly P. D., Holt P. G. (2003). Association between antenatal cytokine production and the development of atopy and asthma at age 6 years. Lancet 362, 1192–1197. [DOI] [PubMed] [Google Scholar]
- Martin E. M., Clapp P. W., Rebuli M. E., Pawlak E. A., Glista-Baker E., Benowitz N. L., Fry R. C., Jaspers I. (2016). E-cigarette use results in suppression of immune and inflammatory-response genes in nasal epithelial cells similar to cigarette smoke. Am. J. Physiol. Lung Cell Mol. Physiol. 311, L135–L144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohsenzadeh Y., Rahmani A., Cheraghi J., Pyrani M., Asadollahi K. (2014). Prenatal exposure to nicotine in pregnant rat increased inflammatory marker in newborn rat. Mediators Inflamm. 2014, 274048.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mychasiuk R., Ilnytskyy S., Kovalchuk O., Kolb B., Gibb R. (2011b). Intensity matters: brain, behaviour and the epigenome of prenatally stressed rats. Neuroscience 180, 105–110. [DOI] [PubMed] [Google Scholar]
- Mychasiuk R., Gibb R., Kolb B. (2011a). Prenatal stress produces sexually dimorphic and regionally specific changes in gene expression in hippocampus and frontal cortex of developing rat offspring. Dev. Neurosci. 33, 531–538. [DOI] [PubMed] [Google Scholar]
- Noakes P. S., Hale J., Thomas R., Lane C., Devadason S. G., Prescott S. L. (2006). Maternal smoking is associated with impaired neonatal toll-like-receptor-mediated immune responses. Eur. Respir. J. 28, 721–729. [DOI] [PubMed] [Google Scholar]
- Norden D. M., Muccigrosso M. M., Godbout J. P. (2015). Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology 96, 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsawa K., Imai Y., Kanazawa H., Sasaki Y., Kohsaka S. (2000). Involvement of Iba1 in membrane ruffling and phagocytosis of macrophages/microglia. J. Cell Sci. 113(Pt 17), 3073–3084. [DOI] [PubMed] [Google Scholar]
- Ohsawa K., Imai Y., Sasaki Y., Kohsaka S. (2004). Microglia/macrophage-specific protein Iba1 binds to fimbrin and enhances its actin-bundling activity. J. Neurochem. 88, 844–856. 10.1046/j.1471-4159.2003.02213.x [DOI] [PubMed] [Google Scholar]
- Oncken C., Ricci K. A., Kuo C. L., Dornelas E., Kranzler H. R., Sankey H. Z. (2017). Correlates of Electronic Cigarettes Use Before and During Pregnancy. Nicotine Tobacco Res. 19, 585–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opanashuk L. A., Pauly J. R., Hauser K. F. (2001). Effect of nicotine on cerebellar granule neuron development. Eur. J. Neurosci. 1, 48–56. [PMC free article] [PubMed] [Google Scholar]
- Orellana J. A., Busso D., Ramírez G., Campos M., Rigotti A., Eugenín J., von Bernhardi R. (2014). Prenatal nicotine exposure enhances Cx43 and Panx1 unopposed channel activity in brain cells of adult offspring mice fed a high-fat/cholesterol diet. Front. Cell. Neurosci. 8, 403.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponzoni L., Moretti M., Sala M., Fasoli F., Mucchietto V., Lucini V., Cannazza G., Gallesi G., Castellana C. N., Clementi F., et al. (2015). Different physiological and behavioural effects of e-cigarette vapour and cigarette smoke in mice. Eur. Neuropsychopharmacol. 25, 1775–1786. 10.1016/j.euroneuro.2015.06.010 [DOI] [PubMed] [Google Scholar]
- Rohleder N., Kirschbaum C. (2006). The hypothalamic-pituitary-adrenal (HPA) axis in habitual smokers. Int. J. Psychophysiol. 59, 236–243. 10.1016/j.ijpsycho.2005.10.012 [DOI] [PubMed] [Google Scholar]
- Scheffler S., Dieken H., Krischenowski O., Aufderheide M. (2015). Cytotoxic Evaluation of e-liquid aerosol using different lung-derived cell models. Int. J. Environ. Res. Public Health 12, 12466–12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraufnagel D. E. (2015). Electronic cigarettes: vulnerability of youth. Pediatr. Allergy Immunol. Pulmonol. 28, 2–6. 10.1089/ped.2015.0490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer K. S., Chen S. X., Law S., Van Demark M., Poirier C., Justice M. J., Hubbard W. C., Kim E. S., Lai X., Wang M., et al. (2015). Endothelial disruptive proinflammatory effects of nicotine and e-cigarette vapor exposures. Am. J. Physiol. Lung Cell Mol. Physiol. 309, L175–L187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S., Duman R., Sanacora G. (2008). Serum brain-derived neurotrophic factor, depression, and antidepressant medications: meta-analyses and implications. Biol. Psychiatry 64, 527–532. 10.1016/j.biopsych.2008.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y., Wolkowicz M. J., Kotova T., Fan L., Timko M. P. (2016). Transcriptome sequencing reveals e-cigarette vapor and mainstream-smoke from tobacco cigarettes activate different gene expression profiles in human bronchial epithelial cells. Sci. Rep. 6, 23984.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D., Aherrera A., Lopez A., Neptune E., Winickoff J. P., Klein J. D., Chen G., Lazarus P., Collaco J. M., McGrath-Morrow S. A., et al. (2015). Adult behavior in male mice exposed to e-cigarette nicotine vapors during late prenatal and early postnatal life. PLoS One 10, e0137953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squarzoni P., Oller G., Hoeffel G., Pont-Lezica L., Rostaing P., Low D., Bessis A., Ginhoux F., Garel S. (2014). Microglia modulate wiring of the embryonic forebrain. Cell Rep. 8, 1271–1279. [DOI] [PubMed] [Google Scholar]
- Sussan T. E., Gajghate S., Thimmulappa R. K., Ma J., Kim J. H., Sudini K., Consolini N., Cormier S. A., Lomnicki S., Hasan F., et al. (2015). Exposure to electronic cigarettes impairs pulmonary anti-bacterial and anti-viral defenses in a mouse model. PLoS One 10, e0116861.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter M. A., Mastrobattista J., Sachs M., Aagaard K. (2015). Is there evidence for potential harm of electronic cigarette use in pregnancy? Birth Defects Res. Part A, Clin. Mol. Teratol. 103, 186–195. 10.1002/bdra.23333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayyarah R., Long G. A. (2014). Comparison of select analytes in aerosol from e-cigarettes with smoke from conventional cigarettes and with ambient air. Regul. Toxicol. Pharmacol. 70, 704–710. 10.1016/j.yrtph.2014.10.010 [DOI] [PubMed] [Google Scholar]
- Wagner N. J., Camerota M., Propper C. (2017). Prevalence and perceptions of electronic cigarette use during pregnancy. Matern. Child Health J. 21, 1655–1661. 10.1007/s10995-016-2257-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielgus J. J., Corbin Downey L., Ewald K. W., Hatley M. E., Wilson K. C., Yeilding R. H. (2004). Exposure to low concentrations of nicotine during cranial nerve development inhibits apoptosis and causes cellular hypertrophy in the ventral oculomotor nuclei of the chick embryo. Brain Res. 1000, 123–133. [DOI] [PubMed] [Google Scholar]
- Yang J., Shi S. Q., Shi L., Fang D., Liu H., Garfield R. E. (2014). Nicotine, an α7 nAChR agonist, reduces lipopolysaccharide-induced inflammatory response rotects fetuses in pregnant rats. Am. J. Obstet. Gynecol. 211, 538.e1–537. [DOI] [PubMed] [Google Scholar]