

ABSTRACT
Several lines of evidence support the occurrence of cross-regulation between the endocytic pathway and autophagy, but the molecular mechanisms regulating this process are not well-understood. Here, we show that the calcium sensor UNC13D regulates the molecular mechanism of late endosomal trafficking and endosomal maturation, and defects in UNC13D lead to macroautophagy upregulation. unc13d-null cells showed impaired endosomal trafficking and defective endocytic flux. The defective phenotypes were rescued by the expression of UNC13D but not by its STX7-binding-deficient mutant. This defective endosomal function in UNC13D-deficient cells resulted in increased autophagic flux, increased long-lived protein degradation, decreased SQSTM1/p62 protein levels and increased autolysosome formation as determined by biochemical, microscopy and structural methods. The autophagic phenotype was not associated with increased recruitment of the UNC13D-binding proteins and autophagy regulators, RAB11 or VAMP8, but was caused, at least in part, by TFEB-mediated upregulation of a subset of autophagic and lysosomal genes, including Atg9b. Downregulation of TFEB decreased Atg9b levels and decreased macroautophagy in unc13d-null cells. UNC13D upregulation corrected the defects in endolysosomal trafficking and decreased the number of accumulated autophagosomes in a cellular model of the lysosomal-storage disorder cystinosis, under both fed and starvation conditions, identifying UNC13D as an important new regulatory molecule of autophagy regulation in cells with lysosomal disorders.
Abbreviations ACTB: actin, beta; CTSB: cathepsin B; EEA1: early endosome antigen 1; ESCRT: endosomal sorting complex required for transport; FHL3: familial hemophagocytic; lymphohistiocytosis type 3; HEX: hexosaminidase; HLH: hemophagocytic lymphohistiocytosis; LSD: lysosomal storage disorder; MEF: mouse embryonic fibroblast; SEM: standard errors of the mean; SNARE: soluble n-ethylmaleimide-sensitive-factor attachment receptor; STX: syntaxin; SYT7: synaptotagmin VII; TFE3: transcription factor E3; TFEB: transcription factor EB; TIRF: total internal reflection fluorescence ULK1: unc-51 like kinase 1; UNC13D: unc-13 homolog d; VAMP: vesicle-associate membrane protein; WT: wild-type
KEYWORDS: Autophagy, endosomal trafficking, FHL3, fusion, lysosomal signaling, secretion, TFEB
Introduction
Autophagy is an essential cellular process that consists of the digestion of cytoplasmic components through lysosomal degradation [1]. Autophagic pathways represent a major protective mechanism that, in addition to their role in providing cells with amino acids and nutrients, also allow for cell survival in response to multiple stressors, including starvation and oxidative stress, and help defend organisms against degenerative and neoplastic diseases. Several autophagic pathways have been described, including macroautophagy [2], microautophagy [3] and chaperone‐mediated autophagy (CMA) [4], which differ in their regulation and selectivity. Macroautophagy (hereon autophagy) is a degradative process by which bulky cytoplasmic components and organelles are sequestered in a structure known as the autophagosome and digested upon its fusion to the lysosome, a process highly regulated by nutrient availability and cell stress [2]. Macroautophagy is also dysregulated under stress conditions initiated by a variety of genetic human diseases including lysosomal storage disorders [5] and hyperinflammation [6]; however, very little information regarding a possible role of autophagy in hemophagocytic lymphohistiocytosis syndrome (HLH) is available [7] and therefore, further investigation on their association is critically needed [8].
Increasing evidence supports the cross-talk between the endocytic and the autophagic pathways and several non-ATG molecules that are both endosomal regulators and involved in autophagy have been described [9]. This includes small GTPases, including RAB7 [10] and RAB11 [11–14], ESCRT (endosomal sorting complexes required for transport) [15], and the SNAREs (soluble n-ethylmaleimide-sensitive-factor attachment receptors) STXs (syntaxins) [16] and VAMPs (vesicle-associated membrane proteins), which also control endosomal tethering and fusion [17]. Similar to autophagosomes, lysosomes have many important roles in the regulation of cellular homeostasis and specialized cellular functions. In addition to the function of lysosomes as the final degradation compartment of the endocytic and autophagic pathways, endo-lysosomes and subsets of lysosomes engage in the secretory pathway through regulated mechanisms [18,19], and control cellular processes as diverse as signaling [20], protein degradative pathways [21], receptor maturation [22], late endosomal maturation [23] and autophagy [24]. These processes facilitate the cellular elimination of degradation products and are used by eukaryotic cells to maintain normal cellular homeostasis [18]. Both endosomal and autophagosomal maturation are characterized by dynamic fusion processes with lysosomes to form a degradative compartment where lysosomal lumenal proteins, including cathepsins [25,26], digest macromolecules. Autophagosomes have also been demonstrated to fuse with endosomes to form chimeric vesicles named amphisomes, which are considered precursor structures [27,28] but can also play specialized roles in specific cells [29]. A sustained concept in autophagy research is the occurrence of cross-talk between the endocytic and the autophagic pathways. However, contradicting ideas have been proposed to explain how these mechanisms are mutually regulated. On one hand, based on the shared machinery utilized by these pathways, a functional endocytic pathway has been considered an essential requirement for efficient autophagic flux [9]. This view is supported by studies performed in neurons where the autophagic machinery is proposed to fuse with late endosomes to hijack their trafficking apparatus to engage with the retrograde transport system and then reach the lysosome-enriched soma [30]. On the other hand, based on studies performed using ESCRT mutants, which have defective endosomal maturation, it is proposed that a possible dysregulation of cellular homeostasis due to an endocytosis defect could trigger an enhanced autophagic response [15]. This is supported by the observation that autophagosomes are formed and are able to fuse with the lysosome in ESCRT mutants, thus supporting an induction of autophagic flux, as demonstrated, for example, for the ESCRT mutant vacuolar protein sorting 32 in C. elegans [15], although the effect is species-dependent [31]. In the same way, defective endosomal pathways are frequently observed in lysosomal storage disorders (LSDs) [32] and in some but not all LSDs autophagy is dysregulated. Altogether, whether defective endosomal function impairs or enhances autophagy is not completely elucidated and may depend on specific defective molecular pathways. Thus, the molecular mechanisms regulating the selectivity of endosome-lysosomal and autophagosome-lysosomal fusion and the possible cross-regulation of these mechanisms need further elucidation. To address these questions in mammalian cells, we utilize biochemical and microscopic approaches to study the autophagic function in cells deficient in the protein UNC13D/MUNC13-4 (unc-13 homolog D), a calcium sensor that regulates endosomal maturation through binding and regulation of the endocytic SNAREs STX7 and VAMP8 [33].
UNC13D was originally characterized as a regulator of exocytosis of secretory organelles [34,35] but was later characterized as an important regulator of endocytic maturation and endosomal function [33,36]. UNC13D function was originally demonstrated in hematopoietic cells but it is also expressed in many additional cellular systems and tissues, including lung, kidney, muscle and eye [37]. UNC13D deficiency causes familial hemophagocytic lymphohistiocytosis type 3 (FHL3) a life-threatening immunodeficiency. Although UNC13D is a RAB27A-effector [34] and together they coordinate lytic granule secretion in T-cells [34] and neutrophils [38,39], UNC13D also regulates trafficking mechanisms in a RAB27A-independent fashion [36]. For example, UNC13D was recently characterized as a calcium sensor [40], and as an important regulator of late endosomal maturation [33], a mechanism that is controlled through the calcium-dependent binding of UNC13D to the SNARE proteins STX7 and VAMP8 [33]. UNC13D also regulates recycling endosome function through its interaction with the small GTPase RAB11 [36]. Despite its close association with the endocytic pathway [33], a possible function for UNC13D in the cross-regulation of endosomal maturation and autophagy has not been explored. In this regard, a possible role for UNC13D in autophagy regulation has important connotations for the development of hemophagocytic lymphohistiocytosis (HLH), a syndrome in which a role for autophagy has been suggested [8].
TFEB (transcription factor EB) and the related molecule TFE3 play central roles in the control of lysosomal functions through the regulation of genes that encode lysosomal and autophagic proteins involved in several processes associated with macromolecule clearance [21,41]. In particular, TFEB regulates excretion of waste materials through the upregulation of lysosomal biogenesis, lysosomal exocytosis and autophagy. TFEB function is regulated by post-transcriptional modifications. Thus, MTOR-mediated phosphorylation of TFEB at Ser142 and Ser211 induces cytosolic localization and prevents the activation of the transcription factor [41], while mutations at Ser3 and Arg4 or deletion of 30 amino acids of the amino-terminal domain of TFEB prevent binding to RAGs, and induces TFEB nuclear translocation and activity [42]. The role of TFEB in promoting cellular clearance has been demonstrated in several lysosomal storage disorders (LSDs) including cystinosis [43] and Pompe disease [44], but a possible role for TFEB in the regulation of immunodeficiencies has not been demonstrated so far and the function of TFEB in the cross-regulation of late endosomal and autophagy functions has not been elucidated.
Here, we have uncovered molecular mechanisms of endosomal impairment in UNC13D-deficiency that trigger TFEB activation and autophagy upregulation in unc13d-null cells. Furthermore, we demonstrate that upregulation of UNC13D corrects endolysosomal functions and reduces autophagosome number in the lysosomal storage disorder cystinosis, thus identifying the involvement of UNC13D in lysosomal disease.
Results
UNC13D-deficient cells have impaired late endosomal trafficking and maturation
Although UNC13D is associated with exocytosis of lysosomal-related organelles in hematopoietic cells, it is also expressed in tissues without a major secretory function where its function is unknown. To study the role of UNC13D in the mechanisms of endosomal dynamics and function, we developed primary embryonic fibroblasts (MEFs) from the unc13d-null mouse model Jinx (Figure 1(a)). Jinx was generated by random germline mutagenesis using the alkylating agent N-ethyl-N-nitrosourea generating a premature termination [45]. The expected truncation of 859 aa is not expressed in unc13d Jinx/Jinx cells and therefore, Jinx mice have a null phenotype [38]. In this work, unc13d-null cells showed increased number of LAMP1-positive puncta and homogeneous distribution of LAMP1-positive organelles over the cells, as opposed to the less peripheral distribution observed in wild-type cells (Figure 1(b)). unc13d-null cells also showed increased LAMP1 expression (Figure 1(c)). Spread out distribution of RAB7-labeled late endosomes was also observed in UNC13D-deficient cells, an endosomal marker that largely colocalized with endogenous LAMP1 (Figure S1). In wild-type cells, UNC13D colocalized with LAMP1 (Figure 1(d)) but not with the lysosomal marker CTSB (cathepsin B), with the early endosome marker EEA1 (early endosome antigen 1) or with the recycling endosome marker RAB11 (Figure S2(a–d)), supporting its role in late endosomal function.
Figure 1.
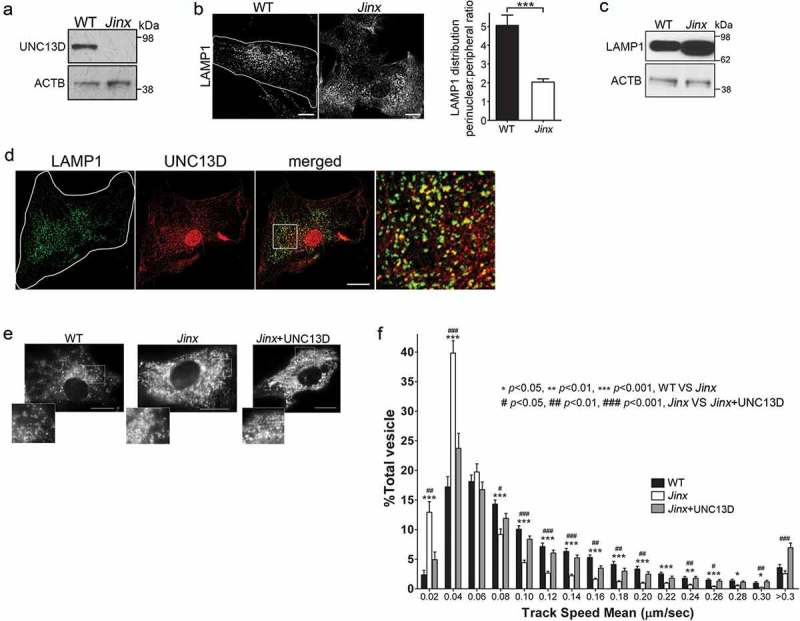
UNC13D regulates endo-lysosome trafficking in non-secretory cells. (a) Immunoblot analysis of the expression of UNC13D in wild-type (WT) and unc13d-null (Jinx) mouse embryonic fibroblasts (MEFs). (b) Left panel, Immunofluorescence and confocal microscopy analysis of the distribution of endogenous LAMP1 in WT and Jinx MEFs. Scale bar: 20 µm. Right panel, quantification of LAMP1 distribution. Results are represented as mean ± SEM. At least 25 cells from 3 separate experiments were analyzed. ***p < 0.001, Student’s t-test. (c) Immunoblot analysis of the expression of LAMP1 in WT and Jinx MEFs. (d) Immunofluorescence analysis of endogenous proteins. Quantification of UNC13D and LAMP1 colocalization is shown in Figure 7(d). (e,f) Pseudo-TIRF microscopy analysis of the trafficking of acidic organelles in WT and Jinx cells. (e) Representative images of WT, Jinx and rescued Jinx MEFs labeled with LysoTracker. Scale bar: 20 µm. (f) Quantification of vesicular dynamics, analyzed by pseudo-TIRFM. The histograms represent the track speeds of LysoTracker-labeled vesicles in WT (black bars), Jinx (white bars) and rescued Jinx MEFs (gray bars). The speeds for the independent vesicles were binned in 0.02 µm/s increments and plotted as a percentage of total vesicles for a given cell. Results are represented as mean ± SEM from at least 20 cells. The statistically significant differences between the two groups are indicated in the figure. Student’s t-test.
The increased peripheral distribution of LAMP1-positive organelles in UNC13D-deficient cells, together with the lack of colocalization of UNC13D with lysosomal markers, suggested that late endosomes utilize UNC13D to mediate trafficking in non-secretory cells. To directly analyze whether UNC13D regulates the transport of endosomal acidic organelles, we labeled the cells with LysoTracker and quantitatively analyzed organelle dynamics using pseudo-TIRFM (oblique illumination), a technique that allows highly specific analysis of fluorescent proteins and probes localization and dynamics by cancelling intracellular background fluorescence from deeper layers in the cytosol and facilitates the analysis of organelles that may not necessarily be in areas adjacent to the plasma membrane [46]. Representative images and time-lapse of wild-type and unc13d-null cells labeled with LysoTracker are shown in Figure 1(e) and in Movies 1 to 3, respectively. Kinetic analyses showed that acidic organelles from UNC13D-deficient fibroblasts are characterized by impaired trafficking, compared to wild-type cells (Figure 1(f)). Thus, unc13d-null cells showed a significant decrease in the number of acidic organelles moving at high speed and a concomitant increase in the number of acidic organelles with restricted movement. The defective phenotype was rescued by exogenous reconstitution of UNC13D expression (Figure 1(f)), further confirming that UNC13D is important for endo-lysosomal trafficking.
In a previous study, we showed that UNC13D is a calcium sensor for heterotypic fusion of late endosomes and lysosomes in hematopoietic cells [33]. To directly analyze whether late endosomes in UNC13D-deficient cells have a defective processing of endosomal substrates, we utilized a pulse-chase approach to label the late endosomal lumen with the trackable cargo FITC-dextran, an indigestible substrate largely utilized to measure endosome and lysosome function and localization both in vitro and in vivo [47]. Using a pulse period of 3 hours, followed by a chase period in complete medium for 3 hours, we show that the cargo accumulated entirely in vesicles that are positive for the endosomal marker RAB7 (Figure 2(a)). Dextran-positive vesicles were also positive for LAMP1 (Figure S2(e)) and UNC13D (Figure S2(f)), but negative for EEA1 (early endosomes) (Figure S2(g)) or SYT7/synaptotagmin VII (lysosomes) (Figure S2(h)). This supports the localization of dextran at late endosomal compartments [48] and differs from its localization at SYT7-positive lysosomes at longer chase periods [49]. Poor colocalization between endogenous SYT7 and LAMP1 was also confirmed by immunofluorescence (Figure S2(i)) supporting that LAMP1 mainly localized at SYT7-negative endolysosomal structures in our assay conditions. Altogether, the observations that endogenous UNC13D largely colocalizes with LAMP1-positive structures (Figure 1(d)), that colocalization of UNC13D with the lysosomal enzyme CTSB or with the early and recycling endosomes EEA1 and RAB11 was not observed in fibroblasts (Figures S2(a–d)), and that UNC13D colocalized with dextran puncta when expressed in unc13d-null cells (Figure S2(f)) support that UNC13D localizes at dextran-loaded late endosomal compartments.
Figure 2.
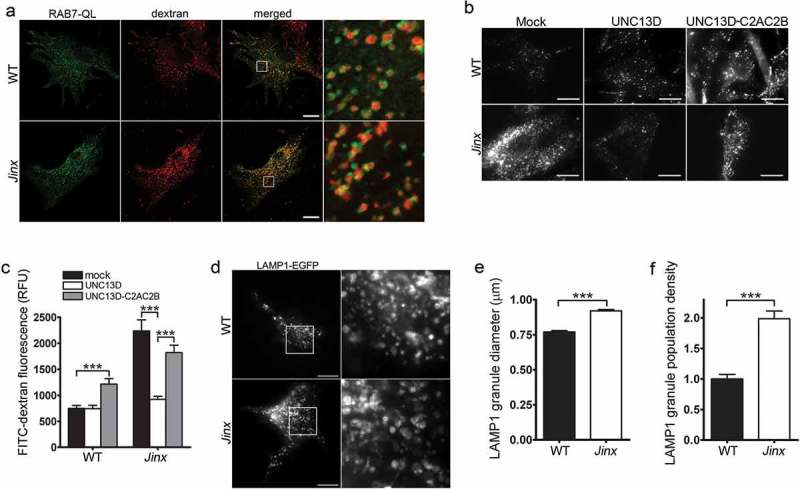
Defective endosomal cargo processing in UNC13D-deficiency is rescued by UNC13D but not by a calcium- and STX7-binding-deficient mutant. (a) Confocal microscopy analysis of the distribution of GFP-RAB7QL and dextran in wild-type (WT) and unc13d-null (Jinx) cells. Scale bar: 20 µm. After 3-hour loading and 3-hour chase dextran was observed exclusively inside RAB7-positive vesicles. (b) Analysis of endosomal cargo processing. WT and Jinx MEFs were mock transfected (mock) or transfected with vectors for the expression of mCherry-UNC13D or the calcium- and STX7-binding-deficient mutant mCherry-UNC13D-C2AC2B, with point mutations D127A, D133A, D941A and D947A to knockout the Ca2+-binding sites in the C2A and C2B domains. After 48 h, the cells were loaded with FITC-dextran at 37°C for 3 h, then washed and chased in complete medium for 3 h. Representative images of accumulated FITC-dextran in cells are shown. Scale bar: 20 µm. (c) Quantification of the results shown in (b). Results are represented as mean ± SEM. At least 60 cells from 3 separate experiments were analyzed. ***p < 0.001, Student’s t-test. RFU, Relative Fluorescence Units. (d) Representative images of pseudo-TIRFM analysis of LAMP1-EGFP-positive organelles in WT and Jinx cells. Scale bar: 20 µm. (e) Quantitative analysis of late endosomal size. Quantification was performed by measuring the diameter of LAMP1-EGFP-positive late endosomes in each cell using ImageJ. Results are represented as mean ± SEM from at least 20 cells. ***p < 0.001, Student’s t-test. (f) Quantitative analysis of late endosome population density was performed by the establishing the ratio of EGFP-LAMP1-positive late endosomes numbers in each cell per cell area. Results are represented as mean ± SEM from at least 20 cells. ***p < 0.001, Student’s t-test.
In dextran-loading assays, unc13d-null cells showed significantly increased levels of fluorescent dextran compared to wild-type cells (Figure 2(b,c)). Because dextran is indigestible but fluorescein is pH-sensitive (pKa of 6.4, decreased fluorescence at lower pH), the phenotype is most likely explained by the aforementioned defective endosomal-lysosomal fusion that characterizes UNC13D-deficient cells [33], which would lead to the accumulation of FITC-dextran in the endosomal compartment which is less acidic than endolysosomes as the lumenal pH decreases along the endosomal-lysosomal pathway [50]. Furthermore, the increased accumulation of FITC-dextran in the sub-population of endocytic vesicles distributed at the periphery could also contribute to the increased-fluorescence phenotype, as it was previously described that peripheral endocytic vesicles are characterized by slightly increased alkalization [51]. Of note, in a previous study, we show no overt defects in acidification per-se in UNC13D-deficiency [33] and the number of acidic organelles (LysoTracker) is increased in UNC13D-deficiency (Figure S3), thus ruling out acidification defects in UNC13D deficiency. Expression of wild-type UNC13D in unc13d-null cells rescued the phenotype and the rescued cells showed reduced FITC-dextran fluorescence signal (Figure 2(b,c)), suggesting that increased trafficking and/or endosomal-lysosomal fusion is upregulated by UNC13D gain-of-function. Contrarily, expression of a calcium- and STX7-binding-deficient mutant of UNC13D [33], not only failed to rescue the phenotype in UNC13D-deficient cells, but also induced a defective phenotype in wild-type cells (Figure 2(b,c)), further supporting the idea that UNC13D plays an important role in endosomal trafficking and endolysosomal fusion in non-hematopoietic cells, in a STX7-dependent manner.
Next, to directly demonstrate that UNC13D regulates endocytic flux in non-hemophagocytic cells, we transfected wild-type and unc13d-null fibroblasts with the endosomal marker GFP-LAMP1 which, as mentioned above, colocalized with RAB7 (Figure S1), and analyzed endosomal flux in real time. To this end, we quantitatively analyzed the size and number of late endosomes and found that UNC13D-deficient cells are characterized by enlarged LAMP1-positive vesicles (Figure 2(d,e)). Furthermore, consistent with a role for UNC13D in late endosome-lysosome fusion rather than in the up-regulation of homotypic or heterotypic late endosomal fusion, which may also lead to enlarged endosomes albeit in reduced numbers, quantification of the number of LAMP1-positive organelles revealed that UNC13D deficiency leads to an increased in the number of enlarged vesicles (Figure 2(f)). This is in agreement with our previous study showing that UNC13D regulates RAB7-positive endosomal function in neutrophils [33] and supports a defective mechanism of late endosomal fusion with lysosomes also in non-hematopoietic cells lacking UNC13D.
Next, since vesicular trafficking towards the plasma membrane is a prerequisite for late endosomes and lysosomes to undergo exocytosis, and UNC13D is known to regulate secretion [38], we next sought to investigate the role of UNC13D in the regulation of lysosome exocytosis. To this end, wild-type and UNC13D-deficient fibroblasts were stimulated with the calcium ionophore ionomycin, and the secretion of HEX/hexosaminidase was analyzed immunologically. UNC13D deficiency only moderately decreased endo/lysosome exocytosis while over-expression of UNC13D had a slight effect on HEX secretion (Figure S4). Further confirming a minor role for UNC13D in lysosomal exocytosis, expression of the fusion-deficient mutant of UNC13D induced a mild defective exocytosis phenotype in wild-type cells (Figure S4). These results, together with the observation that UNC13D colocalized with endosomal markers and with dextran-loaded endosomes but that the endosomal markers did not show strong colocalization with the secretory lysosomal marker SYT7, suggest that UNC13D may regulate exocytosis of a small subpopulation of endo/lysosomes and that endosomal substrate accumulation is not explained by defective exocytosis.
UNC13D-deficient cells are characterized by increased autophagic flux and macroautophagy
To analyze a possible cross-talk between endocytic defects and autophagic pathways in unc13d-null cells, we next studied whether UNC13D-deficiency causes impairment in autophagy and proteostasis regulation. To this end, we first measured degradation of long‐lived proteins using metabolic labeling, a protein degradation assay that represents a well-established methodology for measuring autophagic flux quantitatively [52]. The analysis of long‐lived protein degradation showed significantly increased levels of proteolysis in unc13d-null cells compared to wild-type cells under both fed and starved conditions (Figure 3(a)), suggesting increased autophagic flux in UNC13D-deficiency. Next, in order to analyze autophagic flux in unc13d-null cells under stress conditions, we first measured the degradation rate of the autophagic marker SQSTM1/p62, an ubiquitin‐binding scaffold protein that binds to LC3 and GABARAP proteins, is degraded by autophagy and guides ubiquitinated proteins to the autophagic machinery for degradation in the lysosome [53]. Decreased levels of SQSTM1 protein expression were observed in unc13d-null cells under fed conditions and its expression was further decreased by induction of autophagy by serum starvation (Figures 3(b) and S5), suggesting that in the absence of UNC13D expression macroautophagy is active, inducible, and mediates SQSTM1 degradation. Quantitative analyses of SQSTM1 protein expression confirmed that SQSTM1 levels are significantly decreased in UNC13D-deficient cells under both fed and starvation conditions (Figure 3(c)). SQSTM1 protein expression was recovered by the inhibition of autophagic flux using bafilomycin A, further supporting that UNC13D is dispensable for autophagosome-lysosome fusion and macroautophagy (Figure 3(b,c)). Treatment with the proteasome inhibitor clasto-Lactacystin β-lactone did not significantly affect SQSTM1 levels or the differences in SQSTM1 protein expression between wild-type and unc13d-null cells. SQSTM1 degradation was confirmed using two independent antibodies, both detecting a single band at mw ~50 under the assay conditions (Figure S5) and [54]. The band is specific as it is not detected in sqstm1−/- cells as extensively shown in Ref. [54]. In agreement with a phenotype of macroautophagy upregulation in UNC13D-deficiency, we observed significantly higher levels of LC3B-II expression in unc13d-null cells as compared to wild-type cells at both fed and starvation conditions (Figure 3(b,d)). LC3B-II levels in unc13d-null cells were further increased by bafilomycin A treatment and decreased by recovery of the fed condition (Figure 3(b,d) and data not shown), suggesting that autophagosome formation and degradation are functional and the autophagic flux is upregulated in UNC13D-deficiency.
Figure 3.
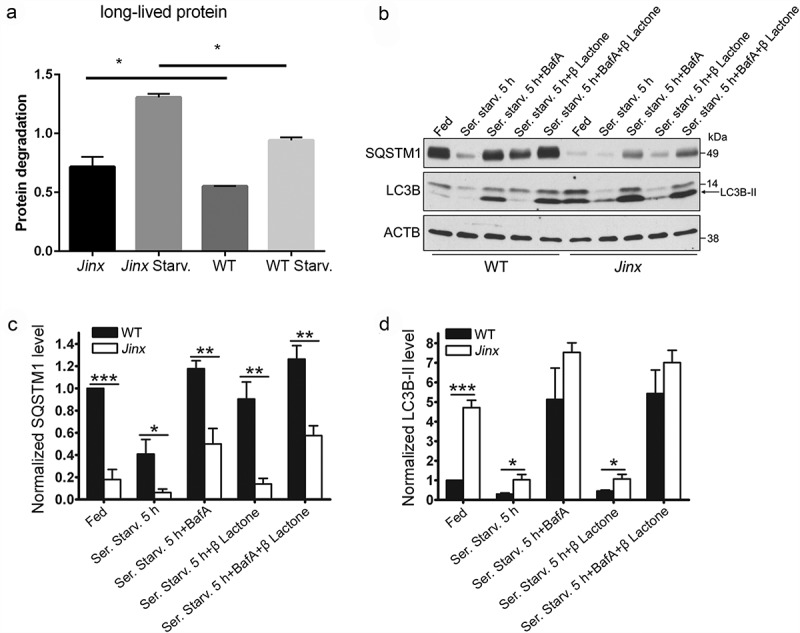
Increased autophagic flux in UNC13D-deficiency. (a) Long-lived protein degradation assays were performed in 35S-methionine labeled wild-type (WT) and unc13d-null cells (Jinx) under fed or starvation (Starv.) conditions as described in Materials and Methods. The data represent the mean ± SEM of 3 biological replicates. *p < 0.05, Student’s t-test. (b) SQSTM1 and LC3B-II protein expression in WT and Jinx MEFs were analyzed by immunoblotting under fed and serum starvation (starv) conditions, in the presence or absence of 100 nM bafilomycin A (BafA) or 1 µM of the proteasome inhibitor Clasto-Lactacystin β-lactone, for 5 h. Quantitation of SQSTM1 (c) and LC3B-II (d) protein expression levels. The data represent mean ± SEM from 3 independent experiments. *p < 0.05, **p < 0.01 and ***p < 0.001, Student’s t-test.
Ultrastructural analysis using transmission electron microscopy further demonstrated increased double-membrane autophagosome numbers in UNC13D-deficient cells under fed conditions (Figure 4(a)). Next, to study autophagosome formation and maturation in UNC13D-deficient cells, we quantified autophagosomes and autolysosomes using the tandem fluorescently-tagged macroautophagy sensor mCherry‐GFP‐LC3B. Using this biosensor, we showed increased macroautophagy flux in UNC13D-deficiency (Figure 4(b,c)). Thus, we observed increased basal levels of fully fused autolysosomes in fed unc13d-null cells, a condition that did not increase further under starvation due to maximal formation-reformation activity. The data showing that treatment with chloroquine dramatically reduces the number of autolysosomes (red-only vesicles) further confirmed active autophagosome-lysosome fusion activity in unc13d-null cells. Finally, we analyzed the level of autophagosomes in UNC13D-deficiency in vivo using a model of inflammation and showed increased autophagosome formation in UNC13D-deficient mice skeletal muscle (Figure 4(d,e)), a tissue that is affected in FHL3 [55] and is characterized by active autophagy [56]. Altogether, our data indicate that, in the absence of UNC13D, both autophagic flux and autophagy-dependent protein degradation are upregulated. Our results also suggest that although UNC13D is dispensable for autophagosome formation and degradation, and for the degradation of long-lived proteins in the autophagosome, its deficiency induces autophagy.
Figure 4.
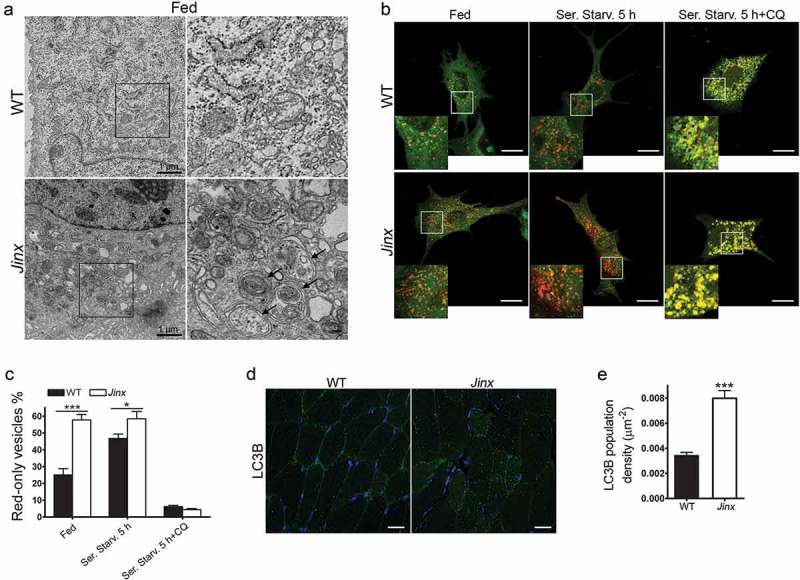
Increased number of autophagosomes in UNC13D-deficiency. (a) Electron microscopy analysis in wild-type (WT) and unc13d-null (Jinx) MEFs, respectively, under fed conditions. Black arrows, autophagosomes. (b) Representative images of WT and Jinx cells transfected with the mCherry-GFP-LC3B macroautophagy biosensor under resting conditions (Fed), or after serum starvation (Ser. Starv.) in the presence or absence of the alkalinizing drug chloroquine (CQ). GFP and mCherry florescence was analyzed by confocal microscopy. Yellow vesicles are indicative of autophagosomes. Red-only vesicles are indicative of autolysosomes as GFP fluorescence decreases in the acidic environment of autolysosomes. Scale bar: 20 µm. (c) The percentage of mature autolysosomes (red-only vesicles) was calculated based on the ratio between the number of red-only puncta and the total number of autophagosomes. Quantitative results are represented as mean ± SEM from at least 20 cells. *p < 0.05 and ***p < 0.001, Student’s t-test. (d) Immunofluorescence analysis of mouse skeletal muscle tissue for the expression of endogenous LC3B (green) shows that Jinx tissue has increased LC3B puncta as compared to WT control tissue. Nuclei are shown in blue (DAPI). Scale bar: 20 µm. (e) Quantitative analysis of the LC3B population density was performed using Image Pro. Data are represented as mean ± SEM. At least 35 cells were analyzed. ***p < 0.001, Student’s t-test.
Increased macroautophagy in UNC13D-deficiency is not induced by increased availability of RAB11-positive membranes or enhanced fusion of autophagosomes with VAMP8-positive vesicles
Previous studies from our laboratory characterized UNC13D as a recycling endosome regulator through specific binding to the small GTPase RAB11 in hematopoietic cells [36]. Fader et al. showed that GFP-RAB11 localizes at LC3B puncta in autophagosomes and that functional RAB11 is involved in the interaction between multivesicular bodies and the autophagic pathway [12]. In addition, Longatti and collaborators showed that RAB11-dependent vesicular transport from recycling endosomes regulates autophagy [14,57]. Here, to determine whether UNC13D may interfere with the autophagic flux by sequestering RAB11 and preventing RAB11-positive membranes delivery to the autophagosome, we analyzed the distribution of RAB11 in relationship to that of LC3B in wild-type and UNC13D-deficient cells. We observed poor colocalization of RAB11 with LC3B under both fed and starvation conditions (Figures S6(a,b)). Furthermore, treatment with chloroquine, a basic agent that interferes with lysosomal pH and fusion, and induces autophagosome accumulation, did not have a significant effect on RAB11-LC3B colocalization either in wild-type or unc13d-null cells. Different to that observed in neutrophils [36], the colocalization between UNC13D and RAB11 in MEFs was negligible (Figure S2(c)). Altogether these data suggest that the RAB11-dependent autophagosome formation pathway may not be highly active in MEFs and thus ruling out a role for UNC13D in RAB11 sequestration to downregulate autophagy in these cells.
UNC13D regulates late endosomal maturation by controlling VAMP8-positive vesicle fusion with late endosomes [33]. Furthermore, VAMP8 has been demonstrated to participate in autophagosome-lysosome fusion through the autophagosomal SNARE STX17 (syntaxin 17) [58], therefore, to rule out that increased availability of VAMP8+ vesicles explains the increased autophagic flux in UNC13D-deficient cells, we studied the distribution of VAMP8 related to LC3B. Here, we show that in the absence of UNC13D, the colocalization of VAMP8 with LC3B-positive autophagosomes was not significantly increased under fed or starvation conditions (Figure S6(c,d)), thus suggesting that increased autophagy in unc13d-null cells is not explained by enhanced fusion between VAMP8+ and LC3B-positive structures.
UNC13D-deficient cells are characterized by TFEB activation
The increased autophagic flux in UNC13D deficiency suggested that this mechanism could be caused by a compensatory upregulation of autophagic pathways induced by either defective endocytic flux and/or by impaired endosomal maturation. The transcription factors TFEB and TFE3 regulate lysosomal function and autophagy to maintain cellular homeostasis [20,59]. Here, we next analyzed whether TFEB activation was exacerbated in UNC13D-deficiency. First, we quantitatively analyzed the nuclear localization of wild-type TFEB in wild-type and UNC13D-deficient cells using GFP-TFEB to emulate the distribution of the endogenous transcription factor, as described before [43,44]. We found that while TFEB localized at the cytosol in wild-type cells, the transcription factor showed nuclear localization in around 50% of unc13d-null cells (Figure 5(a,b)). This differs from that observed for two different constitutively active mutants of TFEB (TFEB-delta30 and TFEBS3AR4A), which, as expected [42], showed nuclear localization in both wild-type and unc13d-null cells (Figure 5(c,d)). As discussed above, unc13d-null cells showed defective endosomal trafficking characterized by homogeneous distribution of LAMP1-positive organelles throughout the cell, rather than the less peripheral distribution observed in wild-type cells. Interestingly, unc13d-null cells with nuclear localization of overexpressed TFEB mutants, which have increased transcriptional activity [42], showed a marked change in the distribution of LAMP1-positive puncta with a clear re-localization towards the perinuclear area (Figure 5(c,d), arrows), suggesting that the upregulation of transcriptional activation by TFEB increases endosomal trafficking in UNC13D-deficient cells.
Figure 5.
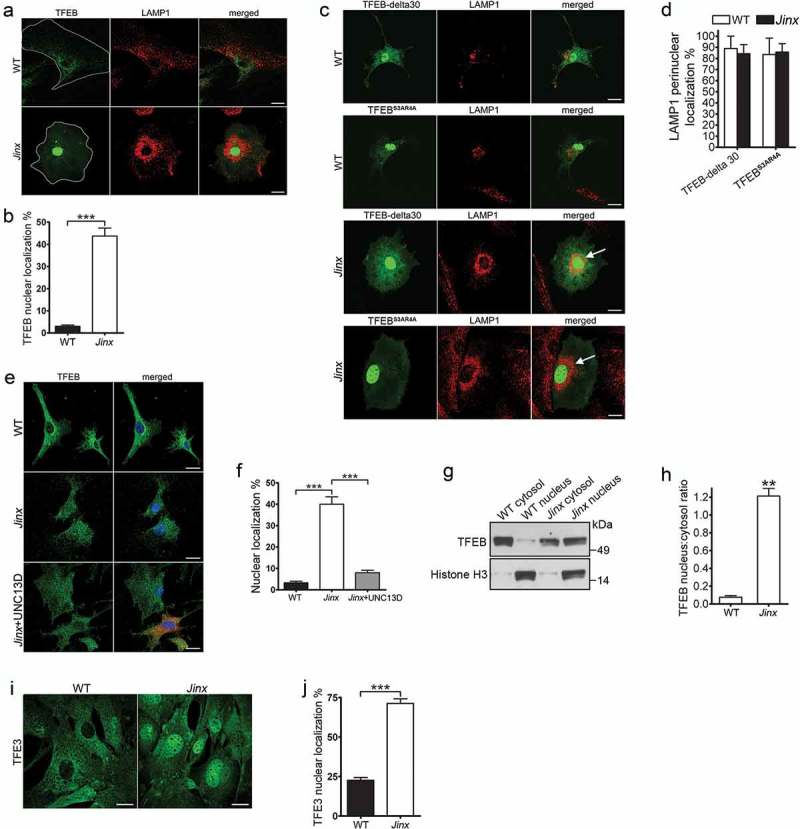
TFEB is activated in UNC13D-deficient cells. (a) Confocal microscopy analysis of the localization of TFEB and LAMP1. Wild-type (WT) and unc13d-null (Jinx) cells were transfected with a TFEB-EGFP expression vector and immuno-stained with anti-LAMP1 antibody. Representative images are shown. Scale bar: 20 µm. (b) Quantification of the distribution of TFEB-GFP in WT and Jinx cells. The data is represented as mean ± SEM. At least 20 cells for each group were analyzed. ***p < 0.001, Student’s t-test. (c) Confocal microscopy analysis of the localization of constitutively active TFEB and LAMP1. WT and Jinx MEFs were transfected with EGFP-TFEB-delta30 or EGFP-TFEBS3AR4A expression vectors and immuno-stained with anti-LAMP1 antibody. Representative images are shown. Scale bar: 20 µm. The arrows indicate the change in the distribution of LAMP1-positive puncta showing re-localization towards the perinuclear area. (d) Quantification of the data show in (c). The data is represented as mean ± SEM. At least 20 cells were analyzed. (e) Confocal microscopy analysis of the distribution of endogenous TFEB (green) and nuclei (blue) in WT, Jinx and Jinx MEFs rescued by the expressing mCherry-UNC13D (red). Scale bar: 20 µm. (f) Quantification of the data shown in E. The percentage of MEFs with nuclear localized TFEB is presented as mean ± SEM from at least 50 cells. ***p < 0.001, Student’s t-test. (g) Cytosol and nuclear fractions from WT and Jinx MEFs were analyzed by Western blot. Histone H3 was used as a marker of the nuclear fraction. (h) Quantification of the nucleus:cytosol ratio of TFEB from 3 independent experiments. The results are presented as mean ± SEM. **p < 0.01, Student’s t-test. (i) Confocal microscopy analysis of the distribution of endogenous TFE3 in WT and Jinx MEFs. Scale bar: 20 µm. (j) The percentage of cells with nuclear localized TFE3 are presented as mean ± SEM from the analysis of at least 50 cells. ***p < 0.001, Student’s t-test.
The analysis of the subcellular localization of endogenous TFEB by immunofluorescence has been challenging as most available antibodies underperform for this application. To study endogenous TFEB activation, we next used an anti-TFEB antibody that has been previously used for this application [60]. In these assays, we detected weak, but significantly increased TFEB staining in the nuclei of UNC13D-deficient cells. Quantitative analysis showed that around 40% of unc13d-null cells have nuclear localization of TFEB under basal conditions, while only less than 5% wild-type cells showed this phenotype (Figure 5(e,f)). Furthermore, the nuclear distribution of TFEB decreased to basal levels by exogenously expressing UNC13D in co-cultured cells, serving as internal control (Figure 5(e,f)).
To confirm that the TFEB pathway is activated in UNC13D-deficient cell, we next took two independent approaches: first, we performed cell fractionation studies to analyze the distribution of endogenous TFEB by immunoblotting using validated antibodies. In Figure 5(g,h), we show that TFEB was significantly increased in the nuclear fraction of unc13d-null cells. Second, we studied the activation of TFE3, a member of the MiTF/TFE family of transcription factors that together with TFEB serves as a major regulator of lysosomal homeostasis and autophagy and that usually follows the activation pattern of TFEB [61]. Using a largely validated anti-TFE3 antibody [61], we showed significantly increased activation (nuclear translocation) of TFE3 in UNC13D-deficiency (Figure 5(i,j)). These results confirm the increased nuclear localization of transcription factors of the TFE family in UNC13D-deficiency.
The increased autophagic flux observed in UNC13D-null cells is associated with the induction of a selective cohort of autophagy genes
To better understand the state of the autophagic pathway at the transcriptional level, we used a quantitative RT-PCR array of autophagy and lysosomal genes, and identified several autophagic and lysosomal transcriptional targets that were induced in UNC13D-deficiency (Figure 6(a) and Table S1). This includes the autophagic regulatory genes Atg9b, Ulk1 (Unc-51 like kinase 1) and Death associated protein kinase 1; the lysosomal genes, Ctsb (cathepsin B), Ctsd and Cln3 (ceroid lipofuscinosis, neuronal 3, juvenile [Batten, Spielmeyer-Vogt disease]), and the regulatory genes Pik3c/phosphoinositide 3-kinase and Pten. Many of these genes, including Atg9b, Ctsb, Ctsd and Cln3, are well known transcriptional targets of the regulator of lysosomal biogenesis and autophagy, TFEB [21,41]. The upregulation of several autophagic and lysosomal genes in UNC13D-deficiency, including Atg9b, Ulk1, Ctsb and Ctsd were further confirmed by quantitative RT-PCR (Figure 6(b)). Other autophagy-related genes that are not targeted by TFEB, including Atg9a [41], where not induced in UNC13D-deficiency. Notably, not all known autophagy genes where upregulated in UNC13D-deficient cells; however, this is not surprising as even under TFEB overexpression conditions, this transcription factor induces both upregulation and downregulation of autophagy-associated genes (Table S1 in Reference [61]) and only few autophagic genes show upregulation by greater than 2-fold under TFEB overexpression conditions in that work. Notably, Atg9b, whose expression was dramatically upregulated in Jinx cells (Figure 6(a–c)), is one of the genes that shows marked upregulation upon TFEB overexpression [61].
Figure 6.
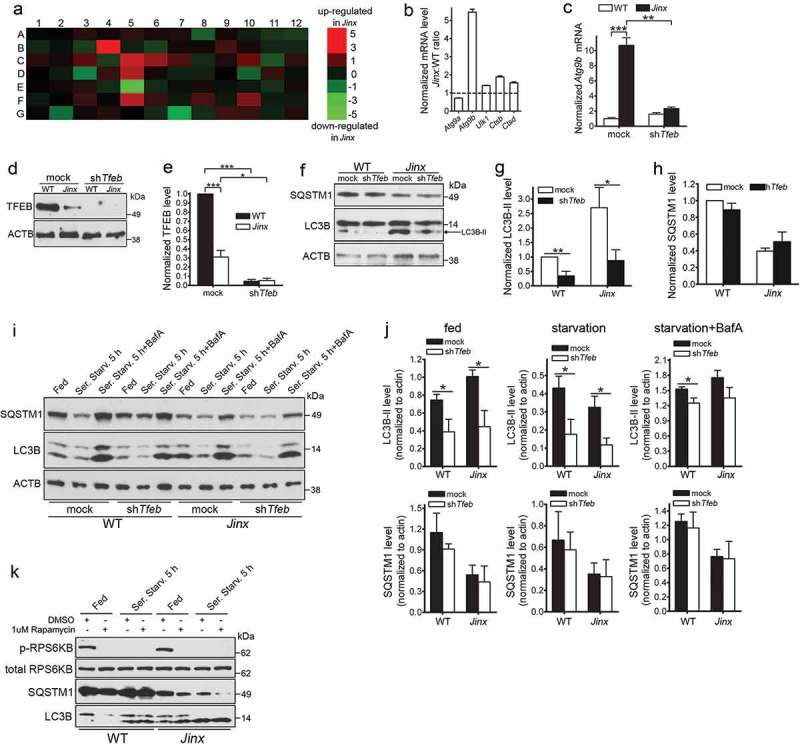
Activation of TFEB induces autophagy upregulation in UNC13D-deficient cells. (a) qPCR array was performed using the RT2 Profiler™ PCR Array System and 84 autophagy and lysosomal genes were analyzed in wild-type (WT) and unc13d-null (Jinx) MEFs. The amount of mRNA in the samples, standardized to 5 housekeeping genes, was expressed as ΔΔCT as described in Material and Methods. See Table S1 for gene information. (b) The upregulation of autophagic and lysosomal genes in UNC13D-deficient cells was validated by quantitative RT-PCR. The target gene expression level was calculated using the 2-ΔΔCT method, which was normalized to actin. The expression levels were relative to the fold change of the corresponding control (WT), which were defined as 1. (c) Atg9b mRNA level was analyzed by qPCR in mock and Tfeb-shRNA (shTfeb) treated WT and Jinx MEFs. (d) WT and Jinx MEFs were infected with lentiviral mouse shRNA against Tfeb (TRCN0000085548, Dharmacon) for 96 h. Expression levels of TFEB were analyzed by Western blot. (e) Quantitative analysis of (d), presented as mean ± SEM from 3 independent experiments. *p < 0.05, ***p < 0.001, Student’s t-test. (f) The indicated macroautophagy markers in mock transfected and Tfeb-knockdown cells were analyzed by Western blot and quantified using ImageJ. (g) Quantitative analysis of LC3B-II expression. Results are represented as mean ± SEM from 3 independent experiments. *p < 0.05, **p < 0.01, Student’s t-test. (h) Quantitative analysis of SQSTM1 protein expression. The results are presented as mean ± SEM from 3 independent experiments. (i) SQSTM1 and LC3B-II protein expression levels were analyzed by Western blot in mock and Tfeb-shRNA transfected WT and Jinx cells under fed, serum starvation (starv) and serum starvation with BafA conditions. (j) Quantitative results of (i) are represented as mean ± SEM from 3 independent experiments. *p < 0.05, Student’s t-test. (k) Phospho-RPS6KB, total RPS6KB, SQSTM1 and LC3B levels in wild-type (WT) and unc13d-null (Jinx) MEFs were analyzed by western blot under fed and serum starvation (Starv.) conditions in the presence or absence of 1 µM rapamycin.
Next, to determine whether the regulation of genes upregulated in UNC13D-deficiency was dependent on TFEB activation, we downregulated TFEB in wild-type and unc13d-null cells using lentiviral shRNA and subsequently analyzed gene expression by quantitative RT-PCR. We show that the upregulation of Atg9b in unc13d-null cells was dependent on TFEB expression as Tfeb downregulation prevented the upregulation of Atg9b in unc13d-null cells (Figure 6(c)). Thus, our data suggest that loss of UNC13D function leads to the expression of a cohort of genes, which are regulated by TFEB.
To further test the hypothesis that that the increased autophagic flux observed in unc13d-null cells could be explained, at least in part, by an elevated induction of transcription mediated by TFEB, we used lentiviral shRNA Tfeb-knockdown cells for biochemical analysis of the autophagic pathway (Figure 6(d,e)). We observed that Tfeb-knockdown decreased autophagosome formation bringing LC3B-II expression in unc13d-null cells to those levels observed in wild-type cells (Figure 6(f,g)). LC3B-II levels were also reduced in Tfeb-knockdown wild-type cells (Figure 6(f,g)). In contrast, no significant differences were observed in the protein expression levels of SQSTM1 after Tfeb-knockdown (Figure 6(f,h)), most likely reflecting a delay in re-synthesis of SQSTM1 in the time frame of this study. Tfeb-knockdown reduced LC3B-II levels not only under fed but also under conditions of autophagy induction by starvation (Figure 6(i,j)). Decreased levels were also observed under blocking conditions (Figure 6(i,j)), although based on the relative increase in LC3B-II levels under blocking conditions, it was evident that other mechanisms still operate to induce autophagy, likely TFE3-dependent. Similar results were previously reported for TFEB-downregulation under blocking conditions by Settembre et al. [41]. Of note, we observed decreased TFEB expression in UNC13D-deficiency (Figure 6(d,e)) but did not observe a shift in the TFEB electrophoretic migration pattern, usually caused by phosphorylation of the transcription factor. This is most likely explained by increased degradation of the phosphorylated form of TFEB in UNC13D-deficient cells. Similar observations were recently reported, and a major regulatory role in phospho-TFEB degradation has been attributed to a proteasome-dependent degradative mechanism mediated by STUB1 [62]. Although the expression levels of STUB1 were not affected in unc13d-null cells (not shown), it is likely that similar mechanisms may mediate degradation of inactive (phosphorylated) TFEB in these cells.
Next, to rule out possible alterations in autophagy-related signaling, we analyzed the integrity of the MTOR pathway. To determine whether the increased autophagosome numbers in UNC13D-deficient cells was due to aberrant MTOR activity, we analyzed the phosphorylation levels of the MTOR substrate RPS6KB/p70‐S6 kinase, both in fed and starved WT and unc13d-null fibroblasts. This assay showed that the levels of p-RPS6KB were comparable in non‐starved WT and UNC13D-deficient cells (Figure 6(k)). Furthermore, both cell starvation and treatment with the autophagy inducer rapamycin induced loss of MTOR activity in both WT and unc13d-null cells (Figure 6(k)). Furthermore, MTOR activity was also not impaired under starvation recovery conditions (not shown), suggesting that MTOR-dependent phosphorylation of RPS6KB is functional in UNC13D-deficiency. This is in agreement with several works showing that TFEB activity does not necessarily correlate with the phosphorylation of ‘canonical’ MTORC1 substrates [63–65]. Also, enhanced autophagic flux with normal MTOR activity has previously been shown in other systems (for example, Ref. [66].). The indicated previous works, provide correlative evidence that alternative pathways may operate under our experimental conditions to increase TFEB activation under normal MTOR activity.
UNC13D upregulation decreases endolysosomal overload in the lysosomal storage disease cystinosis
In the LSD cystinosis, cells are characterized by increased lysosomal overload caused by the accumulation of the amino acid cystine in lysosomes [67,68]. We showed that cystinotic cells have decreased expression of the UNC13D-binding partner, the small GTPase RAB27A, decreased endolysosomal trafficking [32], and increased autophagy [5] but a possible defect in UNC13D function in this LSD is currently unknown. Here, we show that despite normal expression, UNC13D distribution was defective in cystinosis. We show that cystinotic fibroblasts have normal expression levels of UNC13D (Figure 7(a)). UNC13D expression was also observed in wild-type and cystinotic mice, in colocalization with LAMP1, at proximal tubule cells (PTCs) (Figure 7(b)), the most affected cell type in cystinosis [69]. Different from wild-type cells, LAMP1-positive structures showed a favored apical distribution in cystinotic PTCs (Figure 7(b)). Furthermore, very few LAMP1-positive organelles showed defined colocalization with endogenous UNC13D in ctns−/- fibroblasts, which showed decrease presence of the calcium sensor at endo-lysosomes (Figure 7(c,d)). This phenotype is possibly explained by decreased expression levels of the UNC13D-binding protein RAB27A in these organelles in cystinosis [32]. However, the correct localization of RAB27A at LAMP1-positive vesicles described in cystinotic cells and tissues [32] would be sufficient to recruit UNC13D to some endo-lysosomes upon over expression, as indicated by our immunofluorescence analysis (Figure 7(c), lower panel, arrows and Figure 7(d), grey column). Of note, although several cells showed nuclear localization of UNC13D, and previous studies have shown a similar distribution not only for UNC13D but also for its binding partner DOC2α [70], the significance of this finding is currently unclear.
Figure 7.
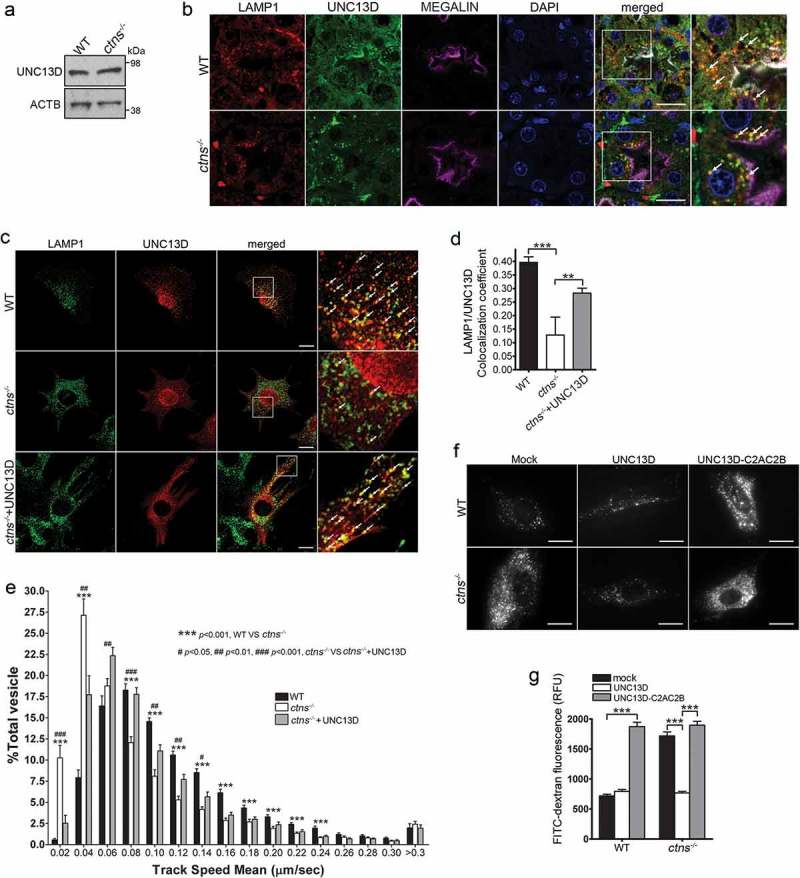
UNC13D but not the STX7-binding-deficient mutant UNC13D-C2AC2B rescues the endosomal defective phenotype in cystinosin-deficient (ctns−/-) cells. (a) UNC13D expression in ctns−/- cells. The expression level of UNC13D in WT and ctns−/- MEFs were analyzed by Western blot. (b) Immunofluorescence analysis of WT and ctns−/- mouse kidney proximal tubule cells, identified by the expression of the apical receptor LRP2/megalin, and showing the distribution of endogenous LAMP1 and UNC13D. Scale bar: 20 µm. (c) Localization of UNC13D in WT and ctns−/- cells. Confocal microscopy analysis of the distribution of endogenous LAMP1 and UNC13D in WT, ctns−/- and ctns−/- MEFs expressing mCherry-UNC13D was performed as described in Material and Methods. Scale bar: 20 µm. The arrows indicate that UNC13D localizes at LAMP1-positive structures. (d) Quantitative analysis of the colocalization of LAMP1 and UNC13D in WT, ctns−/- and ctns−/-+UNC13D MEFs. Data are presented as mean ± SEM, n = 15. **p < 0.01 and ***p < 0.001. (e) Vesicular dynamics analysis of acidic endolysosomes in WT and ctns−/- cells was performed using pseudo-TIRFM. The histograms represent the track speeds of LysoTracker-labeled vesicles in WT (black bars), ctns−/- (white bars) and ctns−/- MEFs expressing mCherry-UNC13D (gray bars). The speeds of the independent vesicles were binned in 0.02 µm/s increments and plotted as a percentage of total vesicles for a given cell. Results are represented as mean ± SEM from at least 20 cells. The statistical significant differences between the groups are indicated in the figure. Student’s t-test. (f) Analysis of endosomal cargo processing in WT and ctns−/- cells. WT and ctns−/- MEFs were mock-transfected or transfected with vectors for the expression of mCherry-UNC13D or the calcium/STX7-binding-defective mutant mCherry-UNC13D-C2AC2B (described in Figure 2). The cells were used in dextran processing assays as described under Materials and Methods. Representative images of accumulated FITC-dextran in the cells are shown. Scale bar: 20 µm. (g) Quantitative analysis of fluorescent FITC-dextran represented as Mean ± SEM. At least 60 cells from 3 independent experiments were analyzed. ***p < 0.001, Student’s t-test. RFU, Relative Fluorescence Units.
To determine whether UNC13D function is important for endolysosomal transport in cystinosis, we labeled endolysosomes with LysoTracker and measured vesicular trafficking. In Figure 7(e) we show that cystinotic cells have defective endolysosomal transport and that over-expression of UNC13D rescued the defective trafficking phenotype. In order to test whether the UNC13D effect on endolysosomal transport may have a significant effect on decreasing endosomal cargo processing in cystinotic cells, we next analyzed the effect of UNC13D upregulation on the accumulation of endocytic substrates in cystinotic cells. To this end, wild-type mCherry-UNC13D or the dominant negative calcium- and STX7-binding-deficient UNC13D mutant mCherry-UNC13D-C2A*-C2B* [33] were transfected into wild-type and ctns−/- fibroblasts and endolysosomal cargo accumulation was analyzed by the FITC-dextran loading assay. We show that cystinotic cells have significantly increased endolysosomal accumulation of FITC-Dextran fluorescence compared to wild-type cells, and that this returned to basal, wild-type levels when UNC13D was expressed in ctns−/- cells (Figure 7(f,g)). Further supporting a role for UNC13D in increasing endolysosomal trafficking and function in this LSD, expression of the dominant negative UNC13D mutant UNC13D-C2A*-C2B* failed to restore normal endosomal cargo processing in cystinotic cells and induced a defective phenotype in wild-type cells (Figure 7(f,g)).
Similar to UNC13D-deficient cells, ctns−/- fibroblasts are characterized by impaired late endosomal trafficking (Ref. [32]. and Figure 7(e)), and accumulation of autophagosomes (Ref. [5]. and Figure 8(a)). Because UNC13D expression rescued endosomal localization and trafficking in ctns−/- cells, we raised the question as whether the increased number of autophagosomes accumulated in ctns−/- cells also decreases upon UNC13D expression. To investigate whether UNC13D expression rescues autophagy in cystinosis, we first quantified the number of autophagosomes by analysis of endogenous LC3 puncta, by immunofluorescence, under fed conditions. Here, we show that expression of wild-type UNC13D (Figure 8(b,d)), but not its STX7-binding-deficient mutant UNC13D-C2A*C2B* (Figure 8(c,d)), decreased the number of autophagosomes in cystinotic cells to the levels observed in wild-type cells. To further understand the effect of UNC13D restoration on the autophagic pathways in cystinosis, we analyzed LC3B lipidation under fed, starved and blocking conditions. UNC13D expression reduced the number of autophagosomes in cystinosis, an effect that was especially manifested under bafilomycin treatment conditions (Figure 8(e,f)). Furthermore, SQSTM1 levels, which were decreased in cystinotic cells, were increased in ctns−/- cells that express UNC13D (Figure 8(e,g)). Of note, the apparent differences observed in the rescue of LC3 puncta vs LC3B lipidation by UNC13D under basal conditions is most likely explained by the fact that lipidation assays were performed using lysates of cells transiently expressing UNC13D (~70% transfection efficiency) and thus containing a subpopulation of cells not expressing UNC13D. Increased (dysregulated) autophagic flux in cystinosis goes hand-in-hand with our previous observations using GFP-LC3B ctns−/- mice and is also confirmed by previous LC3B lipidation and long-live protein degradation analyses in cystinotic MEFs. To further study the effect of UNC13D in cystinosis, we analyzed the effect of UNC13D expression on TFE3 nuclear localization in ctns−/- cells. In Figure 8(h,i), we show that TFE3 nuclear localization was increased in ctns−/- cells and that the phenotype was rescued by UNC13D expression. Altogether, our data suggest that UNC13D deficiency triggers an endosomal defective phenotype and that re-establishing or increasing UNC13D function might be a plausible mechanism to improve endosomal trafficking and potentially improve cell function in lysosomal diseases.
Figure 8.
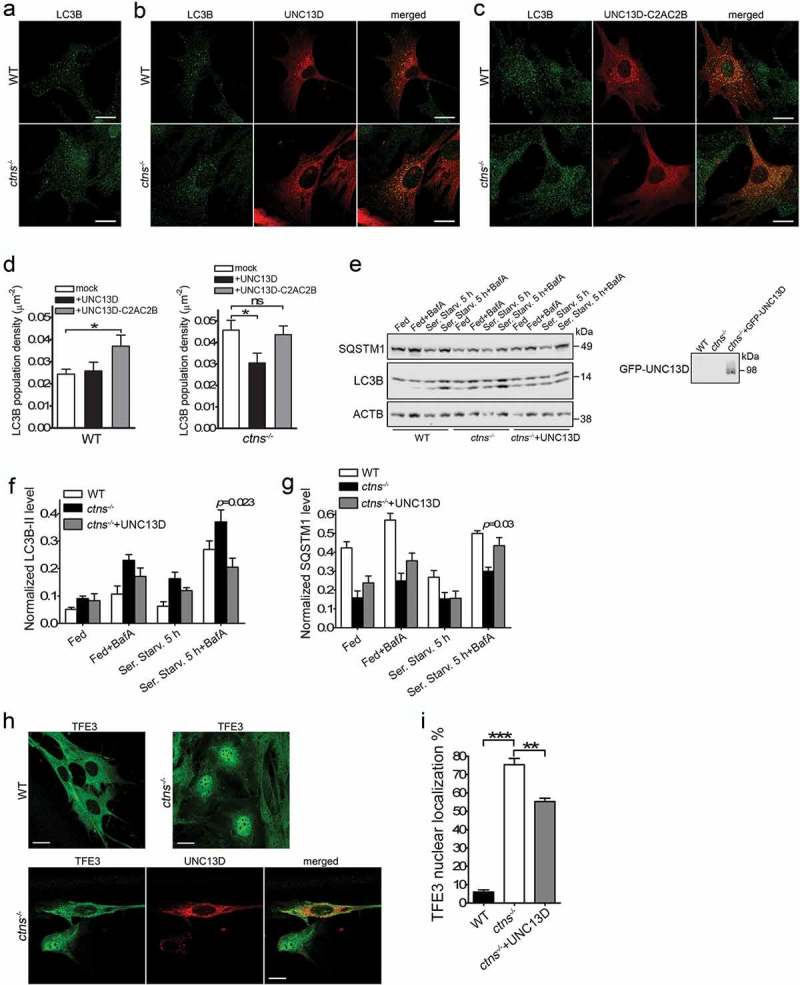
UNC13D expression improves autophagy in a model of the lysosomal-storage disease cystinosis. (a–c) WT and ctns−/- mouse embryonic fibroblasts were mock transfected (a), transfected with mCherry-UNC13D (b) or with mCherry-UNC13D-C2AC2B (C) expression vectors and immuno-stained with an anti-LC3B antibody. Representative confocal images are shown. Scale bar: 20 µm. (d) LC3B puncta density was calculated using Image Pro. Data are presented as mean ± SEM. At least 20 cells were analyzed. *p < 0.05. ANOVA followed by Fisher’s post-hoc test. (e–g) Analysis of the effect of UNC13D exogenous expression on autophagic flux in ctns−/- cells under fed, starved and blocked conditions. ctns−/- cells and ctns−/- cells expressing GFP-UNC13D were fed, starved and/or treated with bafilomycin and analyzed for the expression of LC3B and SQSTM1 levels by Western blot. (e) Representative of 4 independent experiments. (f) and (g) The data are presented as Mean ± SEM. (h) and (i), Immunofluorescence analysis and quantification of the nuclear localization of TFE3 in wild-type, ctns−/- and ctns−/- cells expressing mCherry-UNC13D. Scale bar: 20 µm. n = 3; Mean ± SEM. At least 50 cells were analyzed. **p < 0.01 and ***p < 0.001.
Discussion
Endosomal trafficking and function regulate autophagic pathways but the mechanisms mediating this regulation are not fully elucidated. Here, using a model of defective endosomal trafficking and maturation caused by the downregulation of the calcium-binding fusion-sensor UNC13D, we uncovered a mechanism of endosomal trafficking impairment associated with TFEB activation and autophagy induction. We show that UNC13D deficiency is associated with defective endosomal trafficking and endocytic substrate clearance. Exogenous expression of wild-type UNC13D but not a calcium and STX7-binding-deficient mutant of UNC13D rescued the phenotypes and the mutant emulated the defective phenotype when expressed in wild-type cells. Furthermore, despite its role in endosomal-lysosome fusion [33], our data indicate that UNC13D is dispensable for autophagosome-lysosome fusion during autophagy. Furthermore, we show that UNC13D deficiency is associated with the upregulation of macroautophagy, a process reverted by TFEB downregulation. Concordantly, a subset of autophagy-related genes was upregulated in UNC13D deficiency with Atg9b showing the most pronounced upregulation, a process reverted by TFEB silencing. The expression of UNC13D increased late endosomal trafficking and normalized autophagosome balance under fed and starved conditions in cystinosis, a lysosomal-storage disorder (LSDs) with an endosomal function-defective phenotype. The data suggest that UNC13D upregulation may help correct LSDs cellular defects associated with endosomal malfunction.
Endolysosome trafficking, and autophagosome fusion with endosomes and lysosomes are essential mechanisms that regulate macroautophagy [58]. We found that UNC13D regulates endosomal trafficking, but autophagosome-lysosome fusion is independent of UNC13D expression or function. This is supported by our data showing that normal autolysosome maturation takes place in both UNC13D-deficient cells and in cells that overexpress a calcium-binding-deficient mutant of UNC13D that is unable to bind to SNAREs. The lack of positive regulation by UNC13D in the process of fusion of lysosomes with autophagosomes correlates with previous studies from our laboratory showing that UNC13D binds to STX7 [33], which regulates endo-lysosomal fusion [17], but not to STX17 [33], a SNARE that localizes to the outer membrane of autophagosomes and regulates autophagosome-lysosome fusion [58,71]. In a recent work, the cross-regulation between the endocytic and autophagic pathways has been suggested to be dependent on the formation of amphisomes, a product of late endosomes and autophagosomes fusion [31]. Here, we show that in a setting of decreased late endosomal trafficking and fusion induced by UNC13D-deficiency, TFEB was activated and autophagic flux was upregulated, thus connecting defective endosomal pathways with increased autophagy through transcriptional regulation mechanisms.
Here we show that the absence of UNC13D induced a significant increase in autophagic flux, characterized by increased degradation of long-lived proteins, increased protein degradation of the autophagic substrate SQSTM1 and increased numbers of autophagosomes, and this increment was further manifested when autophagosome-lysosome fusion was pharmacologically blocked. This suggest that UNC13D, either directly or indirectly, acts as a negative regulator of autophagy. UNC13D is a RAB11 and VAMP8 binding protein [33,36] and both RAB11 and VAMP8 regulate autophagy [12,72] by the delivery of recycling endosome membranes for autophagosome formation [12] and by recruiting STX17 for autophagosome-lysosome fusion [58], respectively. However, we show that the level of fusion of RAB11- and VAMP8-positive organelles to autophagosomes in UNC13D-deficient cells was not significantly different from that observed in wild-type cells, suggesting that increased autophagic flux in cells lacking UNC13D is not explained by increased RAB11 or VAMP8 availability. We cannot rule out at this time that other mechanisms may operate through UNC13D sequestration of important autophagic regulators that mediate membrane fusion since multiple small GTPases and SNARE proteins both interact with UNC13D and are well known regulators of macroautophagy, and we are currently searching for these mechanisms in our laboratory.
In this work, we found that UNC13D-deficient cells showed increased nuclear localization of the transcription factor TFEB, known to drive the expression of autophagy and lysosomal genes [20] and whose overexpression significantly increases the LC3B levels and the number of autophagosomes [41]. Our observations that UNC13D deficiency is accompanied by increased nuclear translocation of TFEB, increased LC3B-II levels, long-lived protein degradation and increased autophagic flux, goes hand-in-hand with the idea that the upregulation of autophagy in these cells is a mechanism induced, at least in part, by TFEB activation. This is supported by the observation that TFEB target genes including Atg9b and Ulk1 were upregulated in unc13d-null cells, and that Tfeb-knockdown decreased LC3B-II levels and Atg9b upregulation. However, similar to that shown in previous reports that utilized TFEB overexpression conditions, not all autophagy genes showed upregulation under TFEB activation. Of note, genes like Sqstm1, which in our array shows downregulation, have been shown to have little or no significant upregulation under more physiological or pathological conditions of TFEB activation and, different from Atg9b, Sqstm1 is only partially downregulated in siTFEB cells (Figure S5(b) in Ref. [41].) suggesting that Sqstm1 transcriptional regulation is complex and not just TFEB-dependent, as also supported by other works [73]. Finally, increased nuclear distribution of TFEB in unc13d-null cells was not sufficient to rescue all defective phenotypes in these cells. In particular, unc13d-null cells showed increased peripheral distribution of endolysosomes despite their increased TFEB nuclear localization, a phenotype that was only rescued when constitutively active, nuclear-localized TFEB was exogenously expressed. Our data suggest that additional compensatory mechanisms are necessary to repair the late endosomal trafficking system in unc13d-null cells. Of note, the UNC13D-binding partner RAB27A not only regulates late endosomal trafficking but it also contains three possible TFEB-mediated regulatory elements in the promoter regions, both in the mouse and human genes [43]. Altogether, our data suggest that the activation of endogenous TFEB and the consequent increase in autophagic flux are cellular responses in an attempt to overcome the defective endosomal trafficking observed in UNC13D deficiency. In principle, exogenous activation of TFEB could potentially improve the phenotype even further by increasing late endosomal trafficking and upregulating autophagy, as demonstrated by our data showing that active TFEB improved late endosomal distribution in UNC13D-deficient cells, and by recent data showing that prolonged expression of TFEB rescues certain abnormalities of the lysosomal compartment in cells with lysosomal disorders [43].
UNC13D deficiency is characterized by an immunodeficiency phenotype caused by defective cytotoxicity and consequent impaired killing of infected cells, a process initiated by defective exocytosis of cytotoxic T lymphocytes, natural killer cells and neutrophils. UNC13D deficiency is associated with hemophagocytic lymphohistiocytosis (HLH) and although a link between HLH and macroautophagy has been suggested [8], no direct evidence connecting UNC13D-deficiency and macroautophagy has been demonstrated so far. Our data presented here strongly suggest that autophagy dysregulation constitute an important component of familial hemophagocytic syndrome. Cytokine storm constitute a clinical feature of both Familial Hemophagocytic Lymphohistiocytosis (FHL) and Macrophage Activating Syndrome (MAS) [74], and both syndromes have been associated with UNC13D genetic defects [35,75]. Cytokine storm has been suggested to promote ATP-induced autophagy and impair the degradation of phagocytosed bacteria [76] further supporting an association between the clinical features of FHL and autophagic pathways. Our results introduce a mechanistic explanation for the exacerbated autophagy present in UNC13D-deficiency linking the defective endosomal trafficking phenotype to TFEB-mediated autophagy upregulation, a mechanism that may also help explain the increased autophagic flux induced by other endosomal defects, for example those induced by ESCRT mutants.
Lysosomal storage disorders (LSDs) are characterized by defects in lysosomal function, lysosomal overload and by several cellular defects including increased endoplasmic reticulum (ER) stress [32,77], defective autophagy and increased cell death [5,78,79]. Defects in the regulation of late endosomal trafficking is a mechanism associated with lysosomal storage disorders, and the upregulation of this trafficking mechanism improves cellular function in some LSDs [32,80]. Thus, upregulation of endo-lysosomal trafficking not only decreases the accumulated substrate in lysosomes of cells with LSDs but also decreases endoplasmic reticulum stress [32], a characteristic of cells with lysosomal overload [77]. Here, we show that UNC13D-deficiency, the cause of human immunodeficiency FHL3 [35], phenocopies many of the lysosomal defects observed in other LSDs, thus identifying previously unnoticed, hematopoietic cell-independent defects affecting the FHL3 phenotype. As proof-of-concept of the importance of UNC13D in endosomal function, we demonstrated that in the LSD cystinosis, UNC13D upregulation increases late endosomal trafficking, decreases endolysosomal substrate accumulation and normalizes the autophagic pathways by reducing the number of autophagosomes. Our data support previous studies that upregulation of lysosomal trafficking by the expression of small Rab GTPases decreases overload and may help improve cellular function in cystinosis [32].
Altogether, we have identified a mechanism of cross-regulation between the endocytic and autophagic pathways in which defective late endosomal trafficking and function leads to a compensatory upregulation of macroautophagy. We have also identified UNC13D-deficiency as a regulator of autophagy, a process that requires TFEB activation and has implications for the immunodeficiency FHL3. Finally, our data suggest that upregulation of the late endosomal pathway is a possible target to improve cell function in lysosomal diseases with defective endosomal transport.
Materials and methods
Animals
C57BL/6 unc13djinx/jinx mice (here referred to as Jinx) are UNC13D-deficient mouse generated by random germline mutagenesis using the alkylating agent N-ethyl-N-nitrosourea [45]. Using an antibody directed to the amino terminal domain of UNC13D, we have previously demonstrated that the expected truncation of 859 aa is not expressed in Jinx and therefore, Jinx is a null phenotype [38]. The C57BL/6 ctns−/− mice [81] (ctns−/- or ctns KO) and their parental strains, C57BL/6 (wild-type) were used in this work. Mice (6–12weeks old) were maintained in a pathogen-free environment and had access to food and water ad libitum. All animal studies were performed in compliance with the United States Department of Health and Human Services Guide for the Care and Use of Laboratory Animals. All studies were conducted according to National Institutes of Health and institutional guidelines and with approval from the animal review board at The Scripps Research Institute.
Constructs and transfections
The mCherry-GFP-LC3B tandem reporter was a kind gift from Dr. Fernando Macian (Albert Einstein School of Medicine). The TFEB-EGFP, TFEB-EGFP-delta30 and TFEB-EGFP-S3AR4A plasmids were purchased from Addgene (38119, 44445, 44446, deposited by Shawn Ferguson). LAMP1-EGFP, mCherry-UNC13D, mCherry-UNC13D-C2AC2B, GFP-RAB11, GFP-RAB7QL and EGFP-VAMP8 were previously described [32,33,36]. Previously validated mouse-specific Tfeb shRNA [82] lenti-vector TRCN0000085548 was obtained from Dharmacon, GE. MEFs were transfected using the Neon transfection system (Thermo Fisher Scientific, MPK5000) or using GenJetTM transfection reagent for MEFs (SignaGen Laboratories, SL100489-MEF), and 293T cells were transfected using Lipofectamine® 2000 (Thermo Fisher Scientific, 11668027) following the manufacturer’s instructions. Where indicated wild-type and unc13d-null cells were infected using shRNA lenti-vectors as described [5].
Gel electrophoresis and immunoblotting
Cells were lysed in RIPA lysis buffer in the presence of protease-inhibitors (Sigma-Aldrich, 11697498001) and phosphatase-inhibitors (Millipore, Sigma, 524625) cocktails. Following electrophoresis using NuPAGE 4–12% gels (Thermo Fisher Scientific, NP0322BOX), proteins were transferred onto 0.45-μm nitrocellulose membranes and the membranes were incubated overnight at 4°C in the indicated primary antibodies, followed by incubation with HRP-conjugated secondary antibodies. The following antibodies were used in this study: rat anti-LAMP1 (Santa Cruz Biotechnology, sc-19992), rabbit anti-actin (Sigma-Aldrich, A2066), rabbit anti-UNC13D (Santa Cruz Biotechnology, sc-50465), mouse anti-EEA1 (BD Medical Technology, 610456), mouse anti-RAB11 (BD Medical Technology, 610656), rabbit anti-LC3B (Cell Signaling Technology, 2775), rabbit anti-RPS6KB/p70 S6 Kinase (Cell Signaling Technology, 9202), mouse anti-phospho-RPS6KB/p70 S6 Kinase (Thr389; Cell Signaling Technology, 9206), goat anti-CTSB (Santa Cruz Biotechnology, sc-6490) and rabbit anti-SYT7 (SySy, 105 173) (gift from Dr. Maximov, The Scripps Research Institute). For SQSTM1 immunoblot detection, we used rabbit anti-SQSTM1 (Cell Signaling Technology, 5114) and guinea pig anti-SQSTM1 (Progen Biotechnik, GP62‐C). Of note, both antibodies detect a single band at mw ~50 when run in NuPAGE gels (Figure S5) and [54]. The band is specific for SQSTM1 as it is not detected in sqstm1−/- cells as extensively shown in [54].
Immunofluorescence and confocal microscopy analysis
Cells were seeded on 4-chamber 35-mm glass-bottom dish (In Vitro Scientific, D35C4-20-1.5-N). Where indicated, cells were treated with the indicated treatments, then fixed with 2% paraformaldehyde for 15 min and blocked with 10% BSA (Rockland Immunochemicals Inc., BSA-50) in phosphate-buffered saline (PBS; Corning Inc., 21-031-CV) for 1 h. Samples were labeled with the indicated primary antibodies overnight at 4°C in the presence of 0.01% saponin (Calbiochem, 558255) and 1% BSA. Samples were washed 3 times and subsequently incubated with the appropriate combinations of Alexa Fluor (488 or 594)-conjugated anti-rabbit, anti-rat, or anti-mouse secondary antibodies (Thermo Fisher Scientific, A-21206; A-21207; A-21208; A-21209; A-21202; A-21203, respectively). Samples were analyzed with a Zeiss LSM 710 or 880 laser scanning confocal microscope (LSCM) attached to a Zeiss Observer Z1 microscope at 21°C, using a 63× oil Plan Apo, 1.4 NA objective. Images were collected using ZEN-LSM software and processed using ImageJ. The exposure time and gain were maintained throughout the experiment to comparatively analyze wild-type and unc13d-null cells. Analysis of colocalization was performed using plugin Coloc 2 in ImageJ, which implements and performs the pixel intensity correlation over space using the methods of Pearson. For LAMP1 distribution analysis, fluorescence intensity in perinuclear and peripheral areas were measured using ImageJ. Anti-TFEB was from BETHYL Laboratories (A303-673A). For TFEB distribution analysis, the nuclear to cytosol signal threshold has been specified at 1 and thus, those cells with nuclear signal equal or larger than the signal in cytosol were considered to have TFEB nuclear localization.
Total internal reflection fluorescence (TIRF) microscopy
Pseudo-TIRF microscopy experiments were performed using a 100 × 1.45 numerical aperture TIRF objective (Nikon) on a Nikon TE2000U microscope custom modified with a TIRF illumination module as described [32]. Images were acquired on a 16-bit, cooled charge-coupled device camera (Hamamatsu) controlled through NIS-Elements software. For live experiments, the images were recorded using 300–500 ms exposures depending on the fluorescence intensity of the sample. The exposure time and gain were maintained throughout the experiment to comparatively analyze wild-type and unc13d-null cells. Late endosome diameters were analyzed using ImageJ software as described before [33]. For late endosomal size quantification, the longest diameter of the LAMP1-positive large vesicles was drawn manually using straight line tool, and the length was measured using the ‘Measure’ tool [33].
Late endosomal cargo processing
For late endosomal cargo processing quantitative assays, WT, Jinx or ctns−/- MEFs were mock transfected, or transfected with expression vectors for mCherry-UNC13D or mCherry-UNC13D-C2AC2B mutant with the point mutations D127A, D133A, D941A and D947A to knock out the Ca2+-binding sites in the C2A and C2B domains [33]. After 48 h, the cells were incubated in cell culture medium containing 5 mg/ml FITC-dextran (Sigma-Aldrich, FD10S) at 37°C for 3 hours, then washed and chased in complete medium for 3 hours. The subcellular localization of dextran was tested by immunofluorescence analysis using EEA1, LAMP1 and LysoTracker (Thermo Fisher Scientific, L7528) as markers. To comparatively analyze endolysosomal overload the fluorescence intensity of accumulated FITC-dextran in WT, Jinx and ctns−/- cells was analyzed by TIRFM while maintaining the exposure time and gain throughout the experiment and samples. The images were then analyzed using ImageJ software. To this end, the outline of the cells was selected using the ‘manual’ tool and the ratio of the fluorescence intensity/cell area was then calculated using the ‘measure’ tool.
Starvation protocols
For macroautophagy studies, cells were briefly washed in serum-free DMEM (containing 1x amino acids), media aspirated, and fresh serum-free DMEM was added followed by 5-hour incubation at 37°C, in the presence or absence 100 nM of the lysosomal inhibitor bafilomycin A1 (LC laboratories, B-1080) or 1 µM proteasome inhibitor Clasto-Lactacystin β-lactone (Cayman Chemical, 154226-60-5).
Hexb/β-hexosaminidase assay
HEXB activity was measured using the membrane impermeant fluorogenic substrate 4-methylumbelliferyl-N-acetyl-β-glucosaminide dehydrate (Sigma-Aldrich, M2133) as previously described [32]. Using 12-well plates, cells were incubated with 250 µl PBS with 1.5 mM Ca2+ (with or without 10 µM ionomycin Sigma-Aldrich, I3909) for 10 min at 37°C. Supernatant and cells were harvested separately. For each sample, 200 µl sample was incubated with 29 µl substrate (6 mM 4-methylumbelliferyl N-acetyl-β-D-glucosaminide in 0.1 M Sodium citrate + 0.1 M Sodium phosphate, pH 4.5) for 15 min at 37°C. The reaction was stopped by adding 57 µl stop solution, and the fluorescence was measured by fluorometry (SpectraMax GeminiEM, Molecular Devises, 365/450 nm). Cell extracts were obtained by incubation with 250 µl of PBS + 1% NP-40 (Calbiochem, 492015), followed by a 5-min centrifugation of the extract at 11,000 g.
qPCR array and RT-PCR analysis
Total RNA was extracted from WT and Jinx MEFs using RNeasy Plus Mini Kit (QIAGEN, 74134) according to the manufacturer’s instruction. Reverse transcription and qPCR were performed with RT2 Profiler™ PCR Array Mouse Autophagy (QIAGEN, PAMM-084ZD-2) based on the manufacturer’s instructions. The target gene expression levels were calculated using the 2-ΔΔCT method, which was normalized to 5 housekeeping genes (ΔΔCT = (CTGOI-CTAVG HKG)WT-(CTGOI-CTAVG HKG)Jinx). Abbreviations: GOI, gene of interest; HKG, housekeeping gene; AVG, average. The expression levels were relative to the fold change of the corresponding controls, which were defined as 1. See Table S1 for gene information. For quantitative RT-PCR analysis, RNA was isolated from wild-type or Jinx mouse fibroblasts using the RNeasy mini-kit for RNA purification. A total of 100 ng of RNA for each cell line was reverse-transcribed (RT) using iScript cDNA synthesis kit (Bio-Rad, 1708890). Quantitative RT-PCR was performed using QuantiTect SYBR Green PCR mix (QIAGEB, 204141), with the primers specified in Table S1.
Long-lived protein degradation assay
Degradation of long-lived proteins using radiolabeled amino acids. Degradation of long-lived proteins was measured essentially as described previously with modifications [5]. WT or unc13djinx/jinx cells were labeled with 11.7 µl of Easy-tag Methionine L35S (PerkinElmer, NEG709A) in growth medium containing 10% FCS. In these experiments, 11.7 µl of Easy tag Methionine L35S (10.25 mCi/ml) were added to cells to reach a of 0.010 mCi/ml. Cells were labeled for 48 h, washed with normal growth medium or starvation medium (normal growth medium without serum) and medium was replaced with the appropriate fresh media and cells were cultured for additional 15 h. The acid-soluble radioactivity (AcsoR) in the medium was determined by precipitation with trichloroacetic acid (10% v/v). Total radioactivity incorporated into cellular proteins (CellR) was determined as the amount of acid-precipitable radioactivity in labeled cells immediately after washing. Proteolysis was measured as the rate AcsoRx100/(AcsoR+CellR).
Transmission electron microscopy
Samples were immersed in modified Karnovsky’s fixative (2.5% glutaraldehyde and 2% paraformaldehyde in 0.15 M sodium cacodylate buffer, pH 7.4) for at least 4 hours, post-fixed in 1% osmium tetroxide in 0.15 M cacodylate buffer for 1 hour and stained in bloc in 2% uranyl acetate for 1 hour. Samples were dehydrated in ethanol, embedded in Durcupan epoxy resin (Sigma-Aldrich, 44611), sectioned at 50 to 60 nm on a Leica UCT ultramicrotome, and picked up on Formvar and carbon-coated copper grids. Sections were stained with 2% uranyl acetate for 5 minutes and Sato’s lead stain for 1 minute. Grids were viewed using FEI Tecnai™ Spirit transmission electron microscope.
Cell fractionation
Fully confluent 10-cm dishes of WT and Jinx MEFs were harvested and washed twice with cold PBS. The cells were resuspended in 200 μl of cold PBS contained protease inhibitor cocktail, and subsequently lysed by nitrogen cavitation. The cell particulate fraction containing the nuclear fraction was spun down at 500 × g for 10 min. The supernatant was saved as the cytosolic fraction. The pellets were resuspended in RIPA buffer and used for protein expression analysis by immunoblotting.
Statistical analysis
Data are presented as mean, and error bars correspond to standard errors of the means (SEMs) unless otherwise indicated. Statistical significance was determined using the unpaired Student’s t-test or the ANOVA test using GraphPad InStat (version 3) or Excel software, and graphs were made using GraphPad Prism (version 4) software.
Funding Statement
This work was supported by the Cystinosis Research Foundation [Fellowship 3]; Cystinosis Research Foundation [Fellowship 1]; Cystinosis Research Foundation [Fellowship 2]; National Heart, Lung, and Blood Institute [HL088256]; National Institute of Arthritis and Musculoskeletal and Skin Diseases [AR070837]; National Institute of Diabetes and Digestive and Kidney Diseases [R01DK110162].
Acknowledgments
This work was supported by U.S. Public Health Service grants HL088256, AR070837 and R01DK110162 to S. D. Catz and by Cystinosis Research Foundation fellowships to JZ, JH and MR. We thank Drs. Stephanie Cherqui, Anton Maximov and Corinne Antignac for the contribution of cells and reagents and Dr. Fernando Macian for contributing the GFP-mCherry-LC3B expression vector.
Disclosure statement
The authors declare that they have no conflicts of interest with the contents of this article.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Klionsky D. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8(11):931–937. [DOI] [PubMed] [Google Scholar]
- [2].Boya P, Gonzalez-Polo RA, Casares N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25(3):1025–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mijaljica D, Prescott M, Devenish RJ.. Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy. 2011;7(7):673–682. [DOI] [PubMed] [Google Scholar]
- [4].Kaushik S, Cuervo A.. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 2012;22(8):407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Napolitano G, Johnson JL, He J, et al. Impairment of chaperone-mediated autophagy leads to selective lysosomal degradation defects in the lysosomal storage disease cystinosis. EMBO Mol Med. 2015;7(2):158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].de Luca A, Smeekens SP, Casagrande A, et al. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci U S A. 2014;111(9):3526–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Schneider EM, Lorenz M, Walther P.. Autophagy as a hallmark of hemophagocytic disease In: Gorbunov N, editor. Autophagy: principles, regulation and roles in disease. Hauppauge (NY): Nova Science Publisher; 2012. p. 45–60. [Google Scholar]
- [8].Bode SF, Ammann S, Al-Herz W, et al. The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis. Haematologica. 2015;100(7):978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lamb CA, Dooley HC, Tooze SA.. Endocytosis and autophagy: shared machinery for degradation. Bioessays. 2013;35(1):34–45. [DOI] [PubMed] [Google Scholar]
- [10].Jager S, Bucci C, Tanida I, et al. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117(Pt 20):4837–4848. [DOI] [PubMed] [Google Scholar]
- [11].Zhang J, Johnson JL, He J, et al. Cystinosin, the small GTPase Rab11, and the Rab7 effector RILP regulate intracellular trafficking of the chaperone-mediated autophagy receptor LAMP2A. J Biol Chem. 2017;292(25):10328–10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fader CM, Sanchez D, Furlan M, et al. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic. 2008;9(2):230–250. [DOI] [PubMed] [Google Scholar]
- [13].Szatmari Z, Kis V, Lippai M, et al. Rab11 facilitates cross-talk between autophagy and endosomal pathway through regulation of Hook localization. Mol Biol Cell. 2014;25(4):522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Longatti A, Lamb CA, Razi M, et al. TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J Cell Biol. 2012;197(5):659–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Michelet X, Legouis R. Autophagy in endosomal mutants: desperately seeking to survive. Worm. 2012;1(4):216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Itakura E, Mizushima N. Syntaxin 17: the autophagosomal SNARE. Autophagy. 2013;9(6):917–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mullock BM, Smith CW, Ihrke G, et al. Syntaxin 7 is localized to late endosome compartments, associates with Vamp 8, and Is required for late endosome-lysosome fusion. Mol Biol Cell. 2000;11(9):3137–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Andrews NW. Regulated secretion of conventional lysosomes. Trends Cell Biol. 2000;10(8):316–321. [DOI] [PubMed] [Google Scholar]
- [19].Monteiro P, Rosse C, Castro-Castro A, et al. Endosomal WASH and exocyst complexes control exocytosis of MT1-MMP at invadopodia. J Cell Biol. 2013;203(6):1063–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Settembre C, Fraldi A, Medina D, et al. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14(5):283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sardiello M, Palmieri M, Di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325(5939):473–477. [DOI] [PubMed] [Google Scholar]
- [22].Cuervo A, Mann L, Bonten E, et al. Cathepsin A regulates chaperone-mediated autophagy through cleavage of the lysosomal receptor. Embo J. 2003;22(1):47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Luzio JP, Hackmann Y, Dieckmann NM, et al. The biogenesis of lysosomes and lysosome-related organelles. Cold Spring Harb Perspect Biol. 2014;6(9):a016840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].He C, Klionsky D. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bhoopathi P, Chetty C, Gujrati M, et al. Cathepsin B facilitates autophagy-mediated apoptosis in SPARC overexpressed primitive neuroectodermal tumor cells. Cell Death Differ. 2010;17(10):1529–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Man SM, Kanneganti TD. Regulation of lysosomal dynamics and autophagy by CTSB/cathepsin B. Autophagy. 2016;12(12):2504–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Seglen PO, Gordon PB, Holen I, et al. Hepatocytic autophagy. Biomed Biochim Acta. 1991;50(4–6):373–381. [PubMed] [Google Scholar]
- [28].Sanchez-Wandelmer J, Reggiori F. Amphisomes: out of the autophagosome shadow? Embo J. 2013;32(24):3116–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Patel KK, Miyoshi H, Beatty WL, et al. Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. Embo J. 2013;32(24):3130–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cheng XT, Zhou B, Lin MY, et al. Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J Cell Biol. 2015;209(3):377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Manil-Segalen M, Lefebvre C, Culetto E, et al. Need an ESCRT for autophagosomal maturation? Commun Integr Biol. 2012;5(6):566–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Johnson J, Napolitano G, Monfregola J, et al. Upregulation of the Rab27a-dependent trafficking and secretory mechanisms improves lysosomal transport, alleviates endoplasmic reticulum stress, and reduces lysosome overload in cystinosis. Mol Cell Biol. 2013;33(15):2950–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].He J, Johnson JL, Monfregola J, et al. Munc13-4 interacts with syntaxin 7 and regulates late endosomal maturation, endosomal signaling, and TLR9-initiated cellular responses. Mol Biol Cell. 2016;27(3):572–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Neeft M, Wieffer M, de Jong AS, et al. Munc13-4 is an effector of rab27a and controls secretion of lysosomes in hematopoietic cells. MolBiolCell. 2005;16(2):731–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Feldmann J, Callebaut I, Raposo G, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell. 2003;115(4):461–473. [DOI] [PubMed] [Google Scholar]
- [36].Johnson JL, He J, Ramadass M, et al. Munc13-4 Is a Rab11-binding Protein That Regulates Rab11-positive Vesicle Trafficking and Docking at the Plasma Membrane. J Biol Chem. 2016;291(7):3423–3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Koch H, Hofmann K, Brose N. Definition of Munc13-homology-domains and characterization of a novel ubiquitously expressed Munc13 isoform. Biochem J. 2000;349(Pt 1):247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Brzezinska AA, Johnson JL, Munafo DB, et al. The Rab27a effectors JFC1/Slp1 and Munc13-4 regulate exocytosis of neutrophil granules. Traffic. 2008;9(12):2151–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Johnson JL, Hong H, Monfregola J, et al. MUNC13-4 restricts motility of RAB27A-expressing vesicles to facilitate lipopolysaccharide-induced priming of exocytosis in neutrophils. J Biol Chem. 2010;286:5647–5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Boswell KL, James DJ, Esquibel JM, et al. Munc13-4 reconstitutes calcium-dependent SNARE-mediated membrane fusion. J Cell Biol. 2012;197(2):301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Settembre C, Di Malta C, Va P, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Martina JA, Puertollano R. Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J Cell Biol. 2013;200(4):475–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rega LR, Polishchuk E, Montefusco S, et al. Activation of the transcription factor EB rescues lysosomal abnormalities in cystinotic kidney cells. Kidney Int. 2016;89(4):862–873. [DOI] [PubMed] [Google Scholar]
- [44].Spampanato C, Feeney E, Li L, et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol Med. 2013;5(5):691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Crozat K, Hoebe K, Ugolini S, et al. Jinx, an MCMV susceptibility phenotype caused by disruption of Unc13d: a mouse model of type 3 familial hemophagocytic lymphohistiocytosis. J Exp Med. 2007;204(4):853–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mudrakola HV, Zhang K, Cui B. Optically resolving individual microtubules in live axons. Structure. 2009;17(11):1433–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lencer WI, Weyer P, Verkman AS, et al. FITC-dextran as a probe for endosome function and localization in kidney. Am J Physiol. 1990;258(2 Pt 1):C309–C317. [DOI] [PubMed] [Google Scholar]
- [48].Nakamura N, Lill JR, Phung Q, et al. Endosomes are specialized platforms for bacterial sensing and NOD2 signalling. Nature. 2014;509(7499):240–244. [DOI] [PubMed] [Google Scholar]
- [49].Jaiswal JK, Andrews NW, Simon SM. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J Cell Biol. 2002;159(4):625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Pillay CS, Elliott E, Dennison C. Endolysosomal proteolysis and its regulation. Biochem J. 2002;363(Pt 3):417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Johnson DE, Ostrowski P, Jaumouille V, et al. The position of lysosomes within the cell determines their luminal pH. J Cell Biol. 2016;212(6):677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bjorkoy G, Lamark T, Pankiv S, et al. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–197. [DOI] [PubMed] [Google Scholar]
- [54].Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12(3):213–223. [DOI] [PubMed] [Google Scholar]
- [55].Garrett JP, Fung I, Rupon J, et al. Presentation of hemophagocytic lymphohistiocytosis due to a novel MUNC 13-4 mutation masked by partial therapeutic immunosuppression. Pediatr Rheumatol Online J. 2012;10(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sandri M. Autophagy in skeletal muscle. FEBS Lett. 2010;584(7):1411–1416. [DOI] [PubMed] [Google Scholar]
- [57].Longatti A, Tooze SA. Recycling endosomes contribute to autophagosome formation. Autophagy. 2012;8(11):1682–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Diao J, Liu R, Rong Y, et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature. 2015;520(7548):563–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Chauhan S, Ahmed Z, Bradfute SB, et al. Pharmaceutical screen identifies novel target processes for activation of autophagy with a broad translational potential. Nat Commun. 2015;6:8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Fan Y, Lu H, Liang W, et al. Endothelial TFEB (transcription factor EB) positively regulates postischemic angiogenesis. Circ Res. 2018;122(7):945–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Martina JA, Diab HI, Lishu L, et al. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal. 2014;7(309):ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sha Y, Rao L, Settembre C, et al. STUB1 regulates TFEB-induced autophagy-lysosome pathway. Embo J. 2017;36(17):2544–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Wada S, Neinast M, Jang C, et al. The tumor suppressor FLCN mediates an alternate mTOR pathway to regulate browning of adipose tissue. Genes Dev. 2016;30(22):2551–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Bartolomeo R, Cinque L, De Leonibus C, et al. mTORC1 hyperactivation arrests bone growth in lysosomal storage disorders by suppressing autophagy. J Clin Invest. 2017;127(10):3717–3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Palmieri M, Pal R, Nelvagal HR, et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat Commun. 2017;8:14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Liu P, Liu K, Gu H, et al. High autophagic flux guards ESC identity through coordinating autophagy machinery gene program by FOXO1. Cell Death Differ. 2017;24(10):1672–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kalatzis V, Cherqui S, Antignac C, et al. Cystinosin, the protein defective in cystinosis, is a H(+)-driven lysosomal cystine transporter. Embo J. 2001;20(21):5940–5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kalatzis V, Nevo N, Cherqui S, et al. Molecular pathogenesis of cystinosis: effect of CTNS mutations on the transport activity and subcellular localization of cystinosin. Hum Mol Genet. 2004;13(13):1361–1371. [DOI] [PubMed] [Google Scholar]
- [69].Cherqui S. Cysteamine therapy: a treatment for cystinosis, not a cure. Kidney Int. 2012;81(2):127–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Higashio H, Nishimura N, Ishizaki H, et al. Doc2 alpha and Munc13-4 regulate Ca(2+) -dependent secretory lysosome exocytosis in mast cells. J Immunol. 2008;180(7):4774–4784. [DOI] [PubMed] [Google Scholar]
- [71].Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151(6):1256–1269. [DOI] [PubMed] [Google Scholar]
- [72].Furuta N, Fujita N, Noda T, et al. Combinational soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins VAMP8 and Vti1b mediate fusion of antimicrobial and canonical autophagosomes with lysosomes. Mol Biol Cell. 2010;21(6):1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Jain A, Lamark T, Sjottem E, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285(29):22576–22591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Weaver LK, Behrens EM. Hyperinflammation, rather than hemophagocytosis, is the common link between macrophage activation syndrome and hemophagocytic lymphohistiocytosis. Curr Opin Rheumatol. 2014;26(5):562–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zhang K, Biroschak J, Glass DN, et al. Macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis is associated with MUNC13-4 polymorphisms. Arthritis Rheum. 2008;58(9):2892–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Schneider EM, Flacke S, Liu F, et al. Autophagy and ATP-induced anti-apoptosis in antigen presenting cells (APC) follows the cytokine storm in patients after major trauma. J Cell Commun Signal. 2011;5(2):145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Wei H, Kim SJ, Zhang Z, et al. ER and oxidative stresses are common mediators of apoptosis in both neurodegenerative and non-neurodegenerative lysosomal storage disorders and are alleviated by chemical chaperones. Hum Mol Genet. 2008;17(4):469–477. [DOI] [PubMed] [Google Scholar]
- [78].Settembre C, Fraldi A, Jahreiss L, et al. A block of autophagy in lysosomal storage disorders. Hum Mol Genet. 2008;17(1):119–129. [DOI] [PubMed] [Google Scholar]
- [79].Venugopal B, Mesires NT, Kennedy JC, et al. Chaperone-mediated autophagy is defective in mucolipidosis type IV. J Cell Physiol. 2009;219(2):344–353. [DOI] [PubMed] [Google Scholar]
- [80].Medina DL, Fraldi A, Bouche V, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell. 2011;21(3):421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Nevo N, Chol M, Bailleux A, et al. Renal phenotype of the cystinosis mouse model is dependent upon genetic background. Nephrol Dial Transplant. 2010;25(4):1059–1066. [DOI] [PubMed] [Google Scholar]
- [82].Pena-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, et al. Regulation of TFEB and V-ATPases by mTORC1. Embo J. 2011;30(16):3242–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.