

Abstract
In Europe, Trichomonas gallinae recently emerged as a cause of epidemic disease in songbirds. A clonal strain of the parasite, first found in the United Kingdom, has become the predominant strain there and spread to continental Europe. Discriminating this epidemic strain of T. gallinae from other strains necessitated development of multilocus sequence typing (MLST). Development of the MLST was facilitated by the assembly and annotation of a 54.7 Mb draft genome of a cloned stabilate of the A1 European finch epidemic strain (isolated from Greenfinch, Chloris chloris, XT-1081/07 in 2007) containing 21,924 protein coding genes. This enabled construction of a robust 19 locus MLST based on existing typing loci for Trichomonas vaginalis and T. gallinae. Our MLST has the sensitivity to discriminate strains within existing genotypes confidently, and resolves the American finch A1 genotype from the European finch epidemic A1 genotype. Interestingly, one isolate we obtained from a captive black-naped fruit dove Ptilinopsus melanospilus, was not truly T. gallinae but a hybrid of T. gallinae with a distant trichomonad lineage. Phylogenetic analysis of the individual loci in this fruit dove provides evidence of gene flow between distant trichomonad lineages at 2 of the 19 loci examined and may provide precedence for the emergence of other hybrid trichomonad genomes including T. vaginalis.
Keywords: trichomonosis, trichomoniasis, genome, molecular epidemiology, emerging infectious disease, wild bird, MLST, hybrid
Introduction
A clonal strain of Trichomonas gallinae has recently emerged as a cause of epidemic disease in passerines in Europe: Whilst this strain also causes trichomonosis in columbids and birds of prey, it is a minority strain in healthy columbids (Lawson, Cunningham, et al. 2011; Chi et al. 2013). This epidemic was first reported in finches in the United Kingdom (Pennycott et al. 2005). Since when there has been unprecedented large-scale mortality of greenfinches, Chloris chloris, which has led to a 66% reduction in the British breeding population; from a peak of circa 4.3 million when the disease first emerged, to circa 1.5 million individuals in 2016 (Lawson et al. 2018). Lethal spillover to other British passerines has also been observed (Robinson et al. 2010; Lawson, Robinson, et al. 2011). Since some British raptors feed on passerine species (Cotgreave 1995), concern has been raised regarding the potential for an increase in raptor mortality due to trichomonosis as a result of the finch epidemic (Chi et al. 2013). Since 2008, finch trichomonosis has been reported in southern Fennoscandia and continental Europe (Peters et al. 2009; Forzan et al. 2010; Neimanis et al. 2010; Ganas et al. 2014; Gourlay et al. 2014), with chaffinch migration believed to be the primary vector of spread (Lawson, Robinson, et al. 2011).
Sequence data from the 5.8S ribosomal RNA (rRNA) and surrounding internal transcribed spacer regions 1 and 2 (ITS1, ITS2) have been increasingly used to detect T. gallinae infection (Gaspar da Silva et al. 2007) and to identify genetic heterogeneities in the parasite (Felleisen 1997; Gerhold et al. 2008; Anderson et al. 2009; Grabensteiner et al. 2010). Sequence analyses of the ITS1/5.8S/ITS2 region (hereafter called ITS region) have identified marked variation amongst isolates obtained from a wide geographical region and from different host taxa, with some 15 distinct ITS region sequences identified as discrete ITS ribotypes (Gerhold et al. 2008; Anderson et al. 2009; Sansano-Maestre et al. 2009; Grabensteiner et al. 2010).
ITS ribotypes of T. gallinae parasites obtained from 11 species of affected British passerines showed that they had 100% identity to each other and to ITS ribotype A isolates from the United States (Gerhold et al. 2008; Anderson et al. 2009), Brazil (Kleina et al. 2004), Spain (Sansano-Maestre et al. 2009), and Austria (Grabensteiner et al. 2010). Using the (hydrogenosomal) iron hydrogenase (Fe-hydrogenase) gene as a second genotyping marker (one which is particularly useful for a mitochondrial protists that lack widely used mitochondrial house-keeping protein encoding genes; Voncken et al. 2002), we detected finer-scale genetic variation between T. gallinae sequences (Lawson, Cunningham, et al. 2011). Although we found no variation amongst British passerine samples at this locus, when we compared columbid and bird of prey isolates collected from the United Kingdom and elsewhere, marked sequence diversity at the Fe-hydrogenase gene was observed which was not detected from the ITS region alone (Chi et al. 2013). Thus we proposed a simple alphanumeric genotype with the letter being drawn from the ITS ribotype and the number from the Fe-hydrogenase subtype and in which A1 was the genotype of the European finch epidemic strain (Chi et al. 2013).
Recently the A1 genotype has also been reported as an emerging cause of trichomonosis outbreaks in wild finches in the Canadian Maritimes (McBurney et al. 2015) and as an infection of wild columbids in the United States (Girard et al. 2014). It is possible, therefore, that there are multiple strains of T. gallinae within the A1 genotype worldwide. In order to determine the transmission pathways of this parasite between bird species and the spread of the recently emerged strains causing finch trichomonosis outbreaks in Europe and Canada, tools to further discriminate between parasites strains are required.
Multilocus sequence typing (MLST) is a nucleotide sequence based method that is used to characterize the genetic relationships between microbial species. It has been successfully applied to study populations of bacterial and eukaryotic organisms (Maiden et al. 1998; Hanage et al. 2005; Tibayrenc 2009). Selected loci are normally single copy housekeeping genes so that the variation within these genes is nearly neutral but less prone to homoplasy than using an alternative approach such as multilocus microsatellite typing, and thus they are better able to serve as robust markers of ancient and modern ancestry.
Since the draft genome of the closely related Trichomonas vaginalis was completed, two overlapping sets of MLST loci have been proposed (Conrad et al. 2011; Cornelius et al. 2012). We have sought to extend these analyses by applying them to T. gallinae and have therefore produced a draft annotated genome of the European finch epidemic strain of T. gallinae and used it initially to identify the loci involved. Primers were then produced to enable a parallel 19 locus MLST analysis from nine isolates of T. gallinae from captive and wild birds for comparison with a T. vaginalis reference strain.
Materials and Methods
Isolation and Cloning of the Genome Clonal Stabilate GF1c
For the genome stabilate, T. gallinae was isolated from a greenfinch found dead with esophageal thickening, consistent with trichomonosis, in Norfolk in October 2007 (isolate XT1081-07) (table 1). A sample was taken from the infected bird during postmortem examination at the time of initial presentation and inoculated into Trichomonas Medium No. 2 (Oxoid, United Kingdom), incubated at 30 °C and screened for motile trichomonads at days 1, 2, and 5 (Robinson et al. 2010). The initial stabilate for the isolate was obtained and cryopreserved in liquid nitrogen with 5% dimethyl sulfoxide (DMSO) at −196 °C until further processing. This stabilate was subsequently revived and cultured axenically in pH 7.2 Trypticase-yeast extract-maltose (TYM) supplemented with 10% heat inactivated horse serum at 37 °C (Diamond 1954), parasites were subcultured three times prior to cloning. Cultures were diluted to 50 parasites per milliliter, so that one parasite could be found on average in every 2 (10 μl) wells of Terasaki plates (Greiner Bio-One, Kremsmünster, Austria). The parasites were allowed to settle to the bottom of the wells, the plates were then scanned using an inverted microscope, the positions of wells containing only a single motile parasite were recorded. Wells containing no parasites or more than one were noted and ignored for cloning purposes. The plates were incubated for 24 h at 37 °C in an atmosphere containing 5% carbon dioxide (CO2) at 80% relative humidity before wells where the parasites had replicated were used to inoculate larger cultures. Clones were subcultured three times in TYM and in one case, for the genome clonal stabilate GF1c, the cloning procedure repeated. Finally, clones were recultured and adjusted to a final concentration of 5 × 106 living organisms per milliliter in TYM before cryopreservation. When in the logarithmic phase of growth, all cloned trichomonads exhibited normal morphology and more than 95% motility. GF1c was cryopreserved in liquid nitrogen with 5% DMSO at −196 °C. A clonal stabilate of the captive black-naped fruit dove isolate (BND1c) was subsequently produced by the same method as described for GF1c.
Table 1.
List of Case Isolate ID, Bird Species, Year Found, Location, Evidence of Upper Alimentary Tract Lesions Consistent With Trichomonosis, Genotype
| Case No. | Host Species | Year Found | Isolate Origin | Oropharyngeal Lesions | Genotype * |
|---|---|---|---|---|---|
| XT-1081/07 (GF1c) |
|
2007 |
|
Yes | A1 Europe |
| HF1 |
|
2006 |
|
Yes | A1 United States |
| R11 |
|
2004 |
|
No | A2 |
| R-1604/13 (BND1c) |
|
2013 |
|
No | M1 |
| 5 UEA |
|
2012 |
|
No | C2 |
| Fh49001 |
|
2014 |
|
Yes | C4 |
| Norfolk31/15 |
|
2015 |
|
No | C8 |
| Norfolk32/15 |
|
2015 |
|
No | C9 |
| R-138/14 |
|
2015 |
|
No | C10 |
Note.—Location refers to United Kingdom counties and United States state where the bird was found. *Genotyping scheme according to Chi et al. 2013.
Preparing DNA Extraction for Whole Genome (Illumina) Sequencing
Extraction of T. gallinae genomic DNA (gDNA) from parasite cultures was performed using DNAzol (Invitrogen, United Kingdom) essentially as described in the manufacturer’s instructions. A TruSeq PCR-Free kit was used with the gDNA to generate an Illumina shotgun library (insert size 200–300 bp). Paired-end sequencing (2 × 150 bp) was performed at the Centre for Genomic Research, University of Liverpool, on an Illumina HighSeq 2500 platform. The draft genome sequence was assembled de novo using Velvet (Zerbino and Birney 2008). The theoretical size of the genome was estimated from k-mer frequencies calculated in kAT (Mapleson et al. 2017) and shown in supplementary figure S1B, Supplementary Material online. Genome size was calculated as the total number of k-mers (area under the curve) divided by the coverage (mean coverage/curve peak), using the R statistical package. The peak was also compared with a Poisson distribution (supplementary fig. S1C, Supplementary Material online). Genome assembly quality was assessed using a k-mer spectra plot generated in kAT by comparing the k-mer content between the final assembly and the trimmed paired end reads using a k-mer size of 27 (supplementary fig. S1A, Supplementary Material online).
A New Draft Trichomonad Genome
A total of 32,936,526 paired-end reads were generated from sequencing, which were adapted and quality trimmed using cutadapt (version 1.2.1). The draft genome sequence was assembled de novo using a version of SPAdes. We used a homology-based approach with the aim of annotating the draft genome of T. gallinae using the OrthoMCL software (version 1.4) (Li et al. 2003) via shared gene cluster membership after including genomic data from closely related organisms. Initially we downloaded 541 coding sequences (CDS) of T. vaginalis genes from RefSeq to prepare a model for use with the GlimmerHMM software (Majoros et al. 2004). Using this model, we predicted 22,348 open reading frames (ORFs) in the contigs of the T. gallinae genomic sequence. Finally, we performed an OrthoMCL analysis of these ORFs against the T. vaginalis proteome downloaded from RefSeq (Carlton et al. 2007). Annotations for 16,651 ORFs (74.51%) were transferred from T. vaginalis, with 10,444 genes (46.73%) being annotated as hypothetical genes. It should be noted, that while the T. vaginalis draft genome contains 59,681 predicted genes, only about 26,000 are supported by experimental evidence (which is corroborated by the number of genes found in our study); the same study suggesting that the T. vaginalis genome contains large gene families and many repetitions (Carlton et al. 2007). This situation would make overcompression of the draft genome, by grouping repeated reads in the same contig, more likely, hence decreasing the apparent size of the genome.
To assess the completeness of our draft genome and annotation BUSCO analysis was used to assess the number of Benchmarking Universal Single-Copy Orthologs. The results shown in table 2 show that the draft genome and annotation are high quality and comparable. Overall the T. gallinae annotation has fewer fragmented BUSCOs than the T. vaginalis annotation despite the large difference in predicted proteome sizes. The difference between the genome and annotation scores stems from the protein coding prediction method employed by BUSCO, which is not trained for specific genomes. The fact that not all of the BUSCOs are present may be attributed to the BUSCOs that were searched for being generic eukaryote BUSCOs rather than specific for Trichomonads. Collinearity between T. vaginalis and T. gallinae was determined for contigs and predicted CDS’s using the MUMmer programs NUCmer and PROmer, respectively. Results were visualized and plotted using mummerplot (Delcher et al. 2003).
Table 2.
BUSCO Reports for T. gallinae and T. vaginalis Giving a Measure of the Completeness of the Genome and Showing that the T. gallinae Genome and Annotation is Comparable to the T. vaginalis Genome
| BUSCO Content | T. gallinae Genome | T. gallinae Proteome | T. vaginalis Genome | T. vaginalis Proteome |
|---|---|---|---|---|
| Complete | 186 | 207 | 186 | 209 |
| Single copy | 163 | 158 | 154 | 160 |
| Duplicated | 23 | 49 | 32 | 49 |
| Fragmented | 13 | 9 | 15 | 16 |
| Missing | 104 | 87 | 102 | 78 |
| Completeness (%) | 65.67 | 71.28 | 66.33 | 74.25 |
Parasite Strains and DNA Extraction for MLST
Eight additional reference strains (table 1) of T. gallinae from captive and wild birds from the United Kingdom and United States, collected between 2007 and 2015, were analyzed using MLST in addition to the GF1c isolate. Most of these strains have been widely used in previous studies (Gerhold et al. 2008; Lawson, Cunningham, et al. 2011; Chi et al. 2013). Cryopreserved isolates were revived and cultured in modified TYM medium at 37 °C and gDNA was extracted as per GF1c (above).
Choice of Loci for MLST Typing
Using the whole genome sequence of GF1c, genes were verified as single copy by performing an all-against-all, basic local alignment search tool (BLAST) comparison of nucleotide sequences. Based on their diversity and their ability to be readily amplified and sequenced on both DNA strands, 19 housekeeping loci were identified for the final MLST scheme. Each of the 19 loci was on a different contig of the sequenced genome. Primers were designed for each of the 19 loci using Primer-BLAST at the GenBank website (http://www.ncbi.nlm.nih.gov/tools/primer-blast/; last Accessed August 1, 2019). All primers were designed from common sequence for the T. gallinae genes and T. vaginalis orthologs, using our new draft genome and the published draft genome sequence of T. vaginalis (version 1.2, http://trichdb.org; last Accessed August 1, 2019).
PCR Amplification for MLST
Primers for the sequencing of MLST loci were designed to amplify gene fragments of 655–966 base pairs in length. Details of primers and reaction conditions are provided in supplementary table S1, Supplementary Material online. All genes were amplified as follows: 30 μl PCR reactions containing 20 μl of HotStarTaq Master Mix Kit (Qiagen, Inc., United Kingdom), 2 μl of distilled water, 3 μl of forward primer, 3 μl of reverse primer, and 2 μl of template of DNA. We used the same thermocycler program parameters for all loci as follows: Initial denaturation of template DNA at 95 °C for 3 min; 40 cycles of 95 °C for 45 s, 55–57 °C for 1 min, 72 °C for 2 min with a 5-s extension after each cycle, followed by the final extension at 72 °C for 7 min. Samples were visualized on 1% agarose gels to verify amplification. Bidirectional Sanger sequencing was conducted by Source BioScience (Nottingham, United Kingdom). Sequence alignments were carried out using MEGA software version 6.06.
Sequence Analysis of MLST and Phylogenetic Trees
The nucleotide sequence of each of the selected housekeeping genes from T. gallinae was aligned with its respective gene sequence from the nine T. gallinae isolates using maximum likelihood (ML) trees generated using Molecular Evolutionary Genetics Analysis (MEGA) software version 6.06 (Tamura et al. 2011), both for individual genes and for the concatenated gene sequences. Felsenstein’s bootstrap test was used to evaluate the support for tree topologies and clustering of taxa (2,000 times) (Felsenstein 1985).
For the phylogenetic tree based on the housekeeping genes, ML tree evolutionary distances were computed using the Tamura-Nei method in units of the number of base substitutions per site (Tamura and Nei 1993). Bootstrap values of the ML tree with the highest log likelihood were obtained. For all trees, all positions containing gaps and missing data were eliminated. Constrained and unconstrained ML trees were used to test topological significance (supplementary fig. S3 and table S3, Supplementary Material online). Trees were built using GTR model and Gamma correction in iqtree (Nguyen et al. 2015). Evolutionary analyses were conducted using MEGA 6.06 (Tamura et al. 2011) and network analyses were performed using Splitstree (Huson and Bryant 2006) to construct a Neighbor-Net analysis from catenated MLST fragments after which a final consensus network, obtained from the combination of the individual ML tree for each gene, was constructed. These networks were also used to test for evidence of recombination using the Pairwise Homoplasy Index, which is included in the Splitstree package.
Results
Isolates Used
For this study, we investigated the potential for increased resolution offered by MLST. Nine isolates of T. gallinae of known genotype were selected for the study; seven of the isolates were obtained in the United Kingdom (five wild birds and two captive), and two from wilds birds in the United States (table 1). Three isolates were ribotype A, one subtype A2, two subtype A1; one A1 was isolated from a UK greenfinch and the other from a US house finch (Haemorhous mexicanus). Five of the isolates, from captive and wild columbids in the United Kingdom, were ribotype C. One isolate was a highly divergent ribotype M, which was isolated in the United Kingdom from a captive-bred black-naped fruit dove.
Genome Sequence Data
MLST genes were selected based on the previously validated MLST of T. vaginalis proposed by Conrad et al. (2011). Initially, we attempted amplification and sequencing using the published T. vaginalis primers, however, few of the loci amplified using template DNA from T. gallinae isolates (data not shown). To engineer our own primers for T. gallinae orthologs, we produced a draft genome sequence of T. gallinae which we present here, the draft genome sequence and annotation of a clone from an isolate of the European finch epidemic strain of T. gallinae (A1) GF1c (GenBank accession number MRSU00000000).
The final assembly of the T. gallinae draft genome sequence was determined to consist of 11,704 contigs and an N50 contig size of 20,741. The assembly size was 54,799,485 bp with a coverage of 90× and a G + C content of 33.77%. The BUSCO analysis suggested a completeness comparable to the published T. vaginalis draft genome (table 2). The annotation results revealed 22,251 predicted genes, and 21,924 protein-coding sequences, including three pseudogenes and four rRNAs. There were also 319 transfer RNAs (tRNAs) including predicted tRNAs for all amino acids (table 3).
Table 3.
Summary for the Trichomonas gallinae GF1c genome assembly (GenBank Accession Number MRSU00000000)
| Feature | Value |
|---|---|
| Genome | |
| Size of assembly (bp) | 54,799,485 |
| Gene G + C content (%) | 38.77 |
| No. of scaffolds | 11,704 |
| N50 scaffold size (bp) | 20,741 |
| Protein-coding genes | |
| No. of predicted genes | 21,924 |
| Mean gene length (bp) | 1,392 |
| Gene G + C content (%) | 38.25 |
| Gene density (genes/Mb) | 400.08 |
| Nonprotein-coding genes | |
| No. of nonprotein-coding genes | 327 |
| Predicted tRNA genes | 319 |
| Ribosomal RNA (rRNA) | 4 |
| Predicted snRNAs | 4 |
To test for collinearity between the T. vaginalis and T. gallinae assemblies, the contigs were plotted against each other in MUMmer (fig. 1A). This analysis revealed collinearity between 667 unique T. vaginalis contigs and 1,487 unique T. gallinae contigs representing 1% and 12% of the total number of contigs, respectively. Given this low proportion of collinearity between contigs, the predicted coding sequences of T. vaginalis and T. gallinae were tested as well (fig. 1B). This showed 1,585 coding sequences in T. vaginalis are colinear with 13,551 coding sequences in T. gallinae representing 2.7% and 61% of the total number of coding sequences, respectively.
Fig. 1.
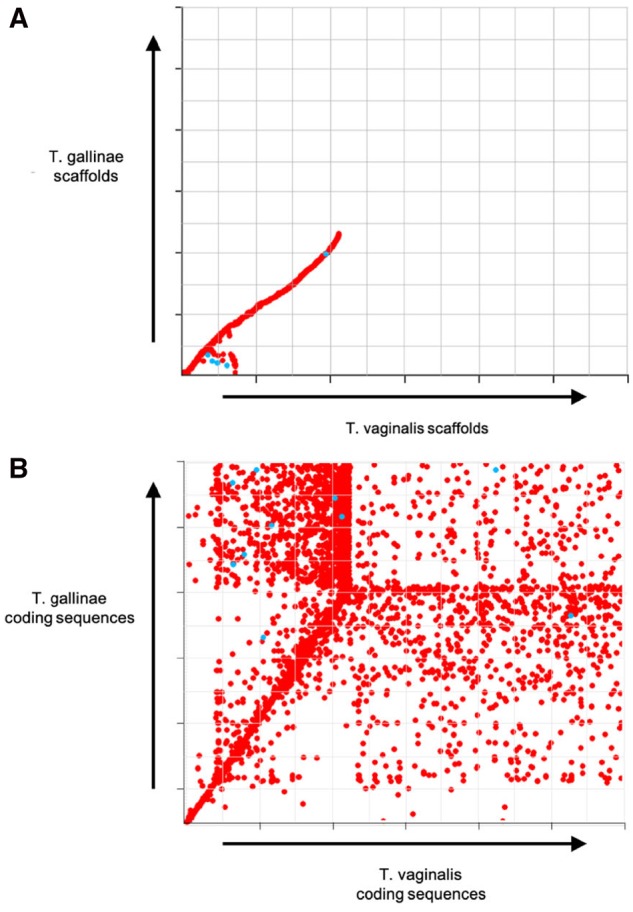
—MUMmer plot showing collinearity between Trichomonas gallinae and Trichomonas vaginalis. Red points denote collinearity matches >75% sequence identity and blue matches are inverted for both the contigs (A) and the coding sequences (B) for T. gallinae and T. vaginalis.
MLST Improves Strain Resolution and Discriminates the American A1 Strains from the European Finch A1 Epidemic Strain
The whole genome data were successfully mined for the T. gallinae MLST orthologous genes. Primers were then designed to amplify a long internal fragment of each gene. All 19 housekeeping gene fragments were successfully amplified using PCR and were sequenced for each of the T. gallinae isolates (table 5; supplementary table S1, Supplementary Material online) affording comparison of 14,770 bp, and yielding 2,043 discriminating characters of 19 concatenated sequences for all 9 isolates of T. gallinae and the single T. vaginalis isolate.
Table 5.
List of GenBank Accession Numbers for Each Locus of the MLST
The size of the fragments analyzed for the selected housekeeping genes ranged between 655 bp (TGA-000730800) and 966 bp (TGA-000731500) (cf. table 4). For each T. gallinae isolate, the sequence obtained for each of the 19 loci was compared with that of every other isolate, and single nucleotide polymorphisms (SNPs) were numbered consecutively. In total 773 T. gallinae SNPs were detected across these loci from 14,770 nucleotides analyzed, corresponding to an average of 1 SNP every 19 bp. The number of SNPs varied for each gene, ranging from 1 SNP every 9 bp for TGA-000478600 to 1 SNP every 202 bp for TGA-000149300 (table 4).
Table 4.
Multilocus Sequence Typing (MLST) Loci Employed
| T. gallinaeGene ID | T. vaginalisGene ID | Locus (supplementary fig. S2, Supplementary Material online) label | Gene Name | Length | SNPs | Nucleotides per SNP |
|---|---|---|---|---|---|---|
| TGA-000149300 | TVAG-087140 | A | Arp2/3, putative | 808 | 4 | 202 |
| TGA-001385000 | TVAG-400860 | B | Clan MA, family M8, leishmanolysin-like metallopeptidase (GP63a) | 682 | 16 | 43 |
| TGA-00112400 | TVAG-005070 | C | Mismatch repair MutL homolog (MIh1A) | 708 | 59 | 12 |
| TGA-000731500 | TVAG-302400 | D | Mismatch repair MutL homolog (MIh1A) | 966 | 32 | 30 |
| TGA-000149500 | TVAG-021420 | E | Coronin (CRN) | 904 | 18 | 50 |
| TGA-000080800 | TVAG-364940 | F | Antigenic protein P1, putative (VSA) | 741 | 71 | 10 |
| TGA-002154000 | TVAG-216430 | G | Clan MA, family M8, leishmanolysin-like metallopeptidase (GP63b) | 771 | 53 | 15 |
| TGA-001611300 | TVAG-303420 | H | Vesicular mannose-binding lectin, putative (LLF4) | 725 | 36 | 20 |
| TGA-000024800 | TVAG-291830 | I | Vesicular mannose-binding lectin, putative, PS(LLF1) | 736 | 32 | 23 |
| TGA-000367600 | TVAG-228710 | J | Clan CA, family C1, cathepsin L-like cysteine peptidase | 817 | 53 | 15 |
| TGA-000478600 | TVAG-485880 | K | Clan CA, family C1, cathepsin L-like cysteine peptidase | 721 | 80 | 9 |
| TGA-001175900 | TVAG-171780 | L | HIV-1 rev binding protein, putative | 759 | 53 | 14 |
| TGA-001325800 | TVAG-086190 | M | Vesicular mannose-binding lectin, putative | 910 | 40 | 28 |
| TGA-002155200 | TVAG-291970 | N | Multidrug resistance pump, putative | 743 | 50 | 15 |
| TGA-000818700 | TVAG-459080 | O | Aspartic peptidase | 900 | 29 | 31 |
| TGA-000730800 | TVAG-414100 | P | Tropomyosin isoforms1/2, putative | 655 | 33 | 20 |
| TGA-000739900 | TVAG-192620 | Q | Actin depolymerizing factor, putative | 657 | 31 | 21 |
| TGA-001849400 | TVAG-094560 | R | Clan CE, family C48, cysteine peptidase | 867 | 52 | 17 |
| TGA-001506800 | TVAG-309150 | S | Conserved hypothetical protein (with PF03388 Domain) | 700 | 31 | 23 |
We used our panel of nine isolates and T. vaginalis as the outgroup to demonstrate that strains of T. gallinae are only partially resolved into three groups by ribotyping (fig. 2A) or seven groups using genotyping with Fe-hydrogenase (fig. 2B). However, our MLST sequences yield a tree with enough resolution to discriminate all nine of the isolates while providing for reduced variance in distance estimates. Our ML tree (fig. 2C) was based on 14,770 bp of concatenated sequence comprising all 19 loci. The tree was rooted with T. vaginalis as the outgroup. On this tree two groups of T. gallinae are evident. All of the ribotype C isolates cluster together, although it is notable the C4 subtype is significantly divergent from the other ribotype C isolates. The ribotype A isolates cluster with the ribotype M isolate. Notably the UK and US A1 subtype isolates are resolved by the MLST and, importantly, the US A1 genotype appears to be more closely related to the US A2 genotype in the panel than to the European finch epidemic strain A1 genotype, supporting different origins for these strains.
Fig. 2.

—A concatenated phylogeny of Trichomonas gallinae based on 19 housekeeping genes of MLST created by using maximum likelihood provides resolution for all strains tested. Trichomonas vaginalis is included as an outgroup. Numbers on branches indicate bootstrap values based on 2,000 replicates. (A) ITS ribotypes alone resolve only 3 groups of avian trichomonads A, C, and M. (B) Fe-hydrogenase is able to resolve 8 genotypes from the 10 avian trichomonads strains tested. (C) MLST based on 19 loci (listed in table 3) provides confident resolution of all the avian trichomonad strains tested. A1 (epidemic) encompasses isolates of the European finch epidemic A1 strain.
Investigation of the M Isolate
The M isolate was cloned to ensure that the results obtained were not the result of a mixed infection. Both isolate and clone were found to be identical to the European finch epidemic strain at several loci, including the Fe-hydrogenase subtyping locus suggesting recent divergence from a common ancestor. The isolate is resolved in the concatenated tree because of the considerable divergence of a few loci. This incongruence is indicative of a hybrid origin and caused us to consider each of the MLST loci for evidence of introgressed genes.
Phylogenetic trees of individual genes were constructed using the ML and neighbor joining (NJ) methods. The tree topologies generated by these two methods were found to be entirely congruent. Individual phylogenetic trees for each of the 19 genes are presented in supplementary figure S2A–S, Supplementary Material online. Most of the coding genes (16/19) produced trees which were congruent with each other and with the Fe-hydrogenase subtyping gene, however, four loci (A, D, I, and the ITS region) produced incongruent trees. These four loci fell into two congruent groups: First, the D and I alleles were congruent with each other, showing greater similarity to alleles from ribotype C isolates than to other ribotype A isolates. Second, the trees produced from the ITS region and from A were congruent with each other but the M isolate gene sequences diverged substantially from those of the other isolates, suggesting that they had a non-T. gallinae origin.
We used trees and networks to further investigate the incongruent phylogenetic signals. Across all of our T. gallinae isolates, we compared 1) concatenated MLST, ITS region, and Fe-hydrogenase sequences (fig. 2); 2) with those from the congruent group of 16 loci (fig. 3), from D and I (fig. 4) and from A and the ITS region (fig. 5). We plotted the phylogenetic relationships using Neighbor-Net split network with the EqualAngle algorithm, which takes recombination (as evidenced for 6 of the MLST loci in supplementary table S2, Supplementary Material online) into account in formulating the network depictions. As evidenced in figure 3, the M1 isolate possesses alleles that segregate it with ribotype A isolates. Indeed, at most loci, it is indistinguishable from the A1 European finch epidemic strain. In contrast, the ITS ribotype and Arp2/3 gene locus are so divergent that they do not group the isolate with any of the other isolates examined. Notably though, at two loci the alleles group most closely with C ribotype isolates C8 and C9 on the Neighbor-Net, but show some association with isolates of ribotype A—suggestive of gene flow between these groups.
Fig. 3.

—Most markers group the M1 isolate with the A1 (European finch epidemic A1 strain). Phylogenetic trees and split network tree based on concatenated sequence analysis of Trichomonas gallinae strains, with 16 loci of MLST (all except loci A, D, and I) and the Fe-hydrogenase gene.
Fig. 4.

—Introgression of Trichomonas gallinae genes. The phylogenetic (A) and split network (B) trees based on concatenated sequence of Loci D and I are incongruous with other loci grouping the M1 isolate with C8 and C9 strains.
Fig. 5.

—Introgression of non-Trichomonas gallinae Trichomonad genes. The phylogenetic (A) and split network (B) trees based on concatenated sequence of Locus A and the ITS region are incongruous with other loci placing the M1 isolate outside of the T. gallinae group and thus suggesting a divergent trichomonad as the origin or these two loci.
Discussion
Improved Genotyping and Distinct Origins for UK and US Lineages of A1 Subtype
The 19 locus test described in this study demonstrated excellent consistency and discriminatory ability for assessing phylogenetic relationships for the T. gallinae isolates examined. These findings suggest that this MLST will be a valuable alternative to established genotyping and subtyping targets for T. gallinae. However, the test’s stability should be further assessed against a larger collection of isolates from different subtype strains and geographical locations to validate their discriminatory power.
Finch trichomonosis in the United States has been previously reported (Anderson et al. 2010) and identification of genotype A1 has also been reported from the United States in band-tailed doves—Patagioenas fasciata (Girard et al. 2014). Here, we are able to differentiate the American A1 strain from a house finch, from the A1 strain causing the European finch epidemic. It is of interest that the American A1 strain actually appears to be more closely related to the American A2 strain, than the European A1 strain, suggesting some modest homoplasy may occur in Fe-hydrogenase alleles.
Evidence for New Strain Emergence from Trans-Species, Introgressed Hybrids
Our phylogeny based on the ITS region (fig. 2A) shows that the isolate from a black-naped fruit dove possessed a distinct M ribotype which was highly divergent from the T. gallinae complex. This suggested that the black-naped fruit dove parasite is a novel trichomonad species. Surprisingly, when subtyped with the Fe-hydrogenase gene the result was incongruent and the sequences obtained were identical to the A1 genotype of the European finch epidemic strain.
An obvious possibility to reconcile the results was that DNA had been extracted from a mixed infection, hence the black-naped fruit dove isolate was cloned. Despite being derived from a single parasite, the initial sequencing results were reproduced, namely a novel and highly divergent M ribotype with an A1 genotype sequence for the Fe-hydrogenase locus.
Using all MLST markers together, a close relationship between the M ribotype and the European finch epidemic A1 genotype is clear. Considered individually, however, the markers used fell into three groups, each giving a discrete tree topology. Within each group for each single marker the tree topologies were congruent. The majority of loci (17 of 21) were identical or nearly identical to the sequence to the European finch epidemic A1 genotype and hence produced a tree which suggested that the majority of the genome could be considered to be A1. Two markers were clearly still T. gallinae alleles, but appeared to be more akin to isolates with C ribotypes, suggesting a recent cross between the European finch epidemic A1 strain with an as yet uncharacterized C type followed by introgression. Most intriguingly, two alleles appeared to be from a non-T. gallinae trichomonad: The ITS 5.8S ribosomal locus, as previously noted, and the locus encoding an actin related protein—two well conserved house-keeping genes. The implication of this result is that the parasite was not an entirely newly discovered diverged species, but rather a recent chimaera or hybrid between T. gallinae and a hitherto uncharacterized trichomonad species arising either by fusion of two distant lineages or by some manner of gene transfer. The black-naped fruit dove originates from Indonesia, Malaysia and the Philippines: It is possible that the loci from a divergent trichomonad species originated from the species’ native range. Indeed, it is noteworthy that other authors have identified evidence of a divergent trichomonad species in another frugivorous columbid from Australia, the Pied Imperial pigeon (Ducula bicolor) (Peters, 2013). The black-naped fruit dove from which the clonal isolate was derived was a captive bird from a UK zoological collection, whose parents were captive bred in a zoological collection in mainland Europe. It is therefore plausible that coinfection with multiple trichomonad parasite lineages, perhaps from different regions, occurred and led to hybrid formation.
Trichomonas vaginalis is morphologically indistinguishable from T. gallinae and it has been observed that some avian Trichomonas at least show high levels of sequence identity at some loci to T. vaginalis (Gerhold et al. 2008) prompting speculation as to the relationship of human and avian trichomonads (Maritz et al. 2014). It is therefore somewhat surprising to discover such little collinearity between the two draft genomes of these trichomonads. However, this does inform the results from the MLST analysis, not least by suggesting that the T. gallinae genome appears to be congruent with T. vaginalis over only a part of the total genome. We also show that there are small-scale repeats in the T. vaginalis draft genome that appear only once in the T. gallinae draft genome, coupled with the difference between the contig collinearity and the coding sequences this suggests that the majority of the genome where there is no collinearity is noncoding.
Our results indicate that the black-naped fruit dove isolate of the M strain shows evidence of introgression from two crosses one of which was likely to have been recent. On this basis, we speculate that occasional hybrid formation and introgression may be an important factor in allowing rapid colonization of new hosts as may have happened in the natural history of T. vaginalis. Future work utilizing high quality genomes and bespoke bioinformatic methods should help discriminate introgression from the possibility of lateral gene transfer, identify how and when and between what lineages genomic exchanges have occurred and elucidate the frequency of type of genetic exchange, particularly between more closely related lineages.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Shaheed Macgregor and other staff from the Zoological Society of London’s Veterinary Department for assistance with sample provision. A.A. was supported by King Saud University, Deanship of Scientific Research, College of Science Research Center. Wild bird T. gallinae isolates were provided by the Garden Wildlife Health project which receives financial support from the Department for Environment, Food and Rural Affairs (Defra) and the Welsh Government through the Animal & Plant Health Agency’s Diseases of Wildlife Scheme Scanning Surveillance Programme; the Esmée Fairbairn Foundation and the Universities Federation for Animal Welfare.
Data deposition: This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession MRSU00000000. The version described in this paper is version MRSU01000000.
Literature Cited
- Anderson NL, Grahn RA, Van Hoosear K, BonDurant RH.. 2009. Studies of trichomonad protozoa in free ranging songbirds: prevalence of Trichomonas gallinae in house finches (Carpodacus mexicanus) and corvids and a novel trichomonad in mockingbirds (Mimus polyglottos). Vet Parasitol. 161(3–4):178–186. [DOI] [PubMed] [Google Scholar]
- Anderson NL, et al. 2010. Clinical signs and histopathologic findings associated with a newly recognized protozoal disease (Trichomonas gallinae) in free-ranging house finches (Carpodacus mexicanus). J Zoo Wildl Med. 41(2):249–254. [DOI] [PubMed] [Google Scholar]
- Carlton JM, et al. 2007. Draft genome sequence of the sexually transmitted pathogen Trichomonas vaginalis. Science 315(5809):207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi JF, et al. 2013. The finch epidemic strain of Trichomonas gallinae is predominant in British non-passerines. Parasitology 140(10):1234–1245. [DOI] [PubMed] [Google Scholar]
- Conrad M, et al. 2011. Microsatellite polymorphism in the sexually transmitted human pathogen Trichomonas vaginalis indicates a genetically diverse parasite. Mol Biochem Parasitol. 175(1):30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelius DC, et al. 2012. Genetic characterization of Trichomonas vaginalis isolates by use of multilocus sequence typing. J Clin Microbiol. 50(10):3293–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotgreave P. 1995. Relative importance of avian groups in the diets of British and Irish predators. Bird Study 42(3):246–252. [Google Scholar]
- Delcher AL, Salzberg SL, Phillippy AM.. 2003. Using MUMmer to identify similar regions in large sequence sets. Curr Protoc Bioinformatics Chapter 10:Unit 10.13. [DOI] [PubMed] [Google Scholar]
- Diamond LS. 1954. A comparative study of 28 culture media for Trichomonas gallinae. Exp Parasitol. 3(3):251–258. [DOI] [PubMed] [Google Scholar]
- Felleisen RSJ. 1997. Comparative sequence analysis of 5.8S rRNA genes and internal transcribed spacer (ITS) regions of Trichomonadid protozoa. Parasitology 115(2):111–119. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39(4):783–791. [DOI] [PubMed] [Google Scholar]
- Forzan M, Vanderstichel R, Melekhovets Y, McBurney S.. 2010. Trichomoniasis in finches from the Canadian Maritime provinces - an emerging disease. Can Vet J. 51(4):391–396. [PMC free article] [PubMed] [Google Scholar]
- Ganas P, et al. 2014. Multi-locus sequence typing confirms the clonality of Trichomonas gallinae isolates circulating in European finches. Parasitology 141(5):652–661. [DOI] [PubMed] [Google Scholar]
- Gaspar da Silva D, et al. 2007. Molecular identity and heterogeneity of trichomonad parasites in a closed avian population. Infect Genet Evol. 7(4):433–440. [DOI] [PubMed] [Google Scholar]
- Gerhold RW, et al. 2008. Molecular characterization of the Trichomonas gallinae morphologic complex in the United States. J Parasitol. 94(6):1335–1341. [DOI] [PubMed] [Google Scholar]
- Girard YA, et al. 2014. Trichomonas stableri n. sp., an agent of trichomonosis in Pacific Coast band-tailed pigeons (Patagioenas fasciata monilis). Int J Parasitol Parasites Wildl. 3(1):32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourlay P, et al. 2014. The potential capacity of French wildlife rescue centres for wild bird disease surveillance. Eur J Wildl Res. 60(6):865–873. [Google Scholar]
- Grabensteiner E, Bilic I, Kolbe T, Hess M.. 2010. Molecular analysis of clonal trichomonad isolates indicate the existence of heterogenic species present in different birds and within the same host. Vet Parasitol. 172(1–2):53–64. [DOI] [PubMed] [Google Scholar]
- Hanage WP, et al. 2005. Using multilocus sequence data to define the pneumococcus. J Bacteriol. 187(17):6223–6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Bryant D.. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 23(2):254–267. [DOI] [PubMed] [Google Scholar]
- Kleina P, Bettim-Bandinelli J, Bonatto SL, Benchimol M, Bogo MR.. 2004. Molecular phylogeny of Trichomonadidae family inferred from ITS-1, 5.8S rRNA and ITS-2 sequences. Int J Parasitol. 34(8):963–970. [DOI] [PubMed] [Google Scholar]
- Lawson B, Cunningham AA, et al. 2011. A clonal strain of Trichomonas gallinae is the aetiologic agent of an emerging avian epidemic disease. Infect Genet Evol. 11(7):1638–1645. [DOI] [PubMed] [Google Scholar]
- Lawson B, Robinson RA, et al. 2011. Evidence of spread of the emerging infectious disease, finch trichomonosis, by migrating birds. EcoHealth 8(2):143–153. [DOI] [PubMed] [Google Scholar]
- Lawson B, et al. 2018. Health hazards to wild birds and risk factors associated with anthropogenic food provisioning. Philos Trans R Soc Lond B Biol Sci. 373: 20170091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ Jr, Roos DS.. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13(9):2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiden MC, et al. 1998. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci. 95(6):3140–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoros WH, Pertea M, Salzberg SL.. 2004. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20(16):2878–2879. [DOI] [PubMed] [Google Scholar]
- Mapleson D, Accinelli GG, Kettleborough G, Wright J, Clavijo BJ.. 2017. KAT: a K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 33:574–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maritz JM, Land KM, Carlton JM, Hirt RP.. 2014. What is the importance of zoonotic trichomonads for human health? Trends Parasitol. 30(7):333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBurney S, et al. 2015. Molecular characterization of Trichomonas gallinae isolates recovered from the Canadian Maritime provinces' wild avifauna reveals the presence of the genotype responsible for the European finch trichomonosis epidemic and additional strains. Parasitology. 142(8):1053–1062. [DOI] [PubMed] [Google Scholar]
- Neimanis AS, et al. 2010. First report of epizootic trichomoniasis in wild finches (Family Fringillidae) in Southern Fennoscandia. Avian Dis. 54(1):136–141. [DOI] [PubMed] [Google Scholar]
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ.. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennycott T, Lawson B, Cunningham A, Simpson V, Chantrey J.. 2005. Necrotic ingluvitis in wild finches. Vet Rec. 157(12):360. [DOI] [PubMed] [Google Scholar]
- Peters, A. 2013. People, Pigeons and a Parasite, Studies on Columbiformes and Trichomonas. Australia: Charles Sturt University. [Google Scholar]
- Peters M, Kilwinski J, Reckling D, Henning K.. 2009. Epidemic mortality in greenfinches at feeder stations caused by Trichomonas gallinae - a recent problem in Northern Germany. Kleintierpraxis 54:433–438. [Google Scholar]
- Robinson RA, et al. 2010. Emerging infectious disease leads to rapid population declines of common British birds. PLoS One 5(8):e12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansano-Maestre J, Garijo-Toledo MM, Gomez MM.. 2009. Prevalence and genotyping of Trichomonas gallinae in pigeons and birds of prey. Avian Pathol. 38(3):201–207. [DOI] [PubMed] [Google Scholar]
- Tamura K, Nei M.. 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 10(3):512–526. [DOI] [PubMed] [Google Scholar]
- Tamura K, et al. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 28(10):2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibayrenc M. 2009. Multilocus enzyme electrophoresis for parasites and other pathogens. Methods Mol Biol. 551:13–25. [DOI] [PubMed] [Google Scholar]
- Voncken F, et al. 2002. Multiple origins of hydrogenosomes: functional and phylogenetic evidence from the ADP/ATP carrier of the anaerobic chytrid Neocallimastix sp. Mol Microbiol. 44(6):1441–1454. [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Birney E.. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18(5):821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.