

Abstract
Purpose
Traumatic brain injury (TBI) is a risk factor for developing chronic neurodegenerative conditions including Alzheimer's disease (AD). The purpose of this study was to examine chronic effects of blast TBI on retinal ganglion cells (RGC), optic nerve, and brain amyloid load in a mouse model of AD amyloidosis.
Methods
Transgenic (TG) double-mutant APPswePSENd19e (APP/PS1) mice and nontransgenic (Non-TG) littermates were exposed to a single blast TBI (20 psi) at age 2 to 3 months. RGC cell structure and function was evaluated 2 months later (average age at endpoint = 4.5 months) using pattern electroretinogram (PERG), optical coherence tomography (OCT), and the chromatic pupil light reflex (cPLR), followed by histologic analysis of retina, optic nerve, and brain amyloid pathology.
Results
APP/PS1 mice exposed to blast TBI (TG-Blast) had significantly lower PERG and cPLR responses 2 months after injury compared to preblast values and compared to sham groups of APP/PS1 (TG-Sham) and nontransgenic (Non-TG-Sham) mice as well as nontransgenic blast-exposed mice (Non-TG-Blast). The TG-Blast group also had significantly thinner RGC complex and more optic nerve damage compared to all groups. No amyloid-β (Aβ) deposits were detected in retinas of APP/PS1 mice; however, increased amyloid precursor protein (APP)/Aβ-immunoreactivity was seen in TG-Blast compared to TG-Sham mice, particularly near blood vessels. TG-Blast and TG-Sham groups exhibited high variability in pathology severity, with a strong, but not statistically significant, trend for greater cerebral cortical Aβ plaque load in the TG-Blast compared to TG-Sham group.
Conclusions
When combined with a genetic susceptibility for developing amyloidosis of AD, blast TBI exposure leads to earlier RGC and optic nerve damage associated with modest but detectable increase in cerebral cortical Aβ pathology. These findings suggest that genetic risk factors for AD may increase the sensitivity of the retina to blast-mediated damage.
Keywords: amyloid, chronic, vision, brain injury, transgenic mice
Traumatic brain injury (TBI) is a risk factor for developing chronic neurodegenerative diseases including chronic traumatic encephalopathy (CTE), dementia pugilistica, pugilistic parkinsonism, and Alzheimer's disease (AD).1 Mild TBI (mTBI) due to low-level exposure to explosive blast waves (blast TBI) can initiate multiple pathological mechanisms leading to chronic dysfunction of the central nervous system (CNS) and increased risk for posttraumatic stress disorder (PTSD) in veterans returning from the battlefield.2,3 A history of exposure to one or more blast TBI is common among military personnel—nearly 75% of combat-related injuries in recent military conflicts were due to explosive devices,4 and approximately 50% of such blast injuries resulted in mTBI.5 Despite the high prevalence of blast exposure in veterans, the type, severity, and chronicity of functional deficits and neuropathological changes after blast TBI are not well understood.2
Individuals exposed to a TBI often have dysfunction of multiple sensory systems, including vision, olfaction, and auditory perception, which can persist chronically.6 Sensory system dysfunction is present after blast injury and can persist chronically following exposure,7 and the severity of dysfunction increases with cumulative blast exposure.8 Visual system pathology is particularly common after blast TBI in humans and experimental models.9 In line with this, visual deficits manifest immediately after blast TBI and tend to persist over time in preclinical blast TBI models. In rats exposed to blast overpressure by a shock tube device, immediate visual dysfunction was observed with delayed structural pathology in the cornea and lens,10 and significant visual and retinal changes persisted months after injury.11 These findings were consistent with other models of rodent single or multiple blast TBI that reported sustained visual system impairment.12–14 Preclinical studies at least partially replicate early and persistent visual dysfunction reported in veterans exposed to one or more blast TBI.15,16
In addition to injury, visual dysfunction can be caused by neurodegenerative disease. AD patients often report visual problems, including decreased contrast sensitivity, visual acuity, and perception of motion and color, which can impact complex visual–cognitive tasks.17–19 It was suggested that changes in visual structure and function may be useful as early biomarkers that precede the onset of cognitive decline in AD patients.20,21 The visual system dysfunction experienced by AD patients could be due to changes in retina22 or amyloid pathology in association visual cortex23 or both. The degree to which retinal dysfunction observed from blast TBI exposure resembles what is reported in AD is not known and requires further investigation because of the documented risk of developing neurologic disease after brain injury. This is particularly true of athletes in contact sports and veterans exposed to mTBI, including blast TBI, where an increasing number of studies have linked neuropsychological and pathological changes after injury to the characteristics of a wide spectrum of chronic neurodegenerative diseases.
Proteolysis of amyloid precursor protein (APP) into its metabolic product amyloid beta (Aβ), in addition to tau protein, gives rise to histopathological characteristics of AD, and these molecules are implicated in the pathological cascades linking TBI to AD and CTE (for reviews see Refs. 24 and 25). There are postmortem studies of humans and experimental animals that consistently show APP accumulation in damaged axons and dysregulated tau in the somatodendritic compartment of neurons and glia following TBI.26–30 Using anti-APP immunohistochemistry, marked axonopathy and APP accumulation was observed in veterans with blast TBI28 and rats exposed to blast.29 Phosphorylated tau and cleaved tau were also elevated after blast exposure in adult wild-type (WT) mice.31
Collectively, these data point to a role of Aβ and tau molecules in developing a polypathology characteristic of a chronic neurodegenerative amyloidosis and tauopathies. Evaluation of blast TBI-induced pathological cascades involving APP, Aβ, and tau in relation to those typically observed in chronic neurodegenerative diseases with dementia in an experimental setting is needed. Recently, we and others10–14 demonstrated long-term functional and structural visual deficits in rodents subjected to blast TBI by a shock tube device. In the current work we extend these studies to determine if the combination of genetic risk factors to develop AD and blast TBI modifies the progression of retinal damage and dysfunction as well as accelerated brain amyloidosis in the APPswePSENd19e mouse model of AD.
Methods
Animals
All animal procedures were conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Iowa City Department of Veterans Affairs Institutional Animal Care and Use Committee. The APPswePSENdE9 (B6C3-Tg(APPswe,PSEN1dE9)85Dbo/Mmjax, abbreviated APP/PS1 in this report) mouse model of AD-like amyloidosis32,33 was purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Male and female mice (8–12 weeks of age at time of injury; n = 65, females denoted where utilized) were exposed to a single blast injury or sham injury and divided into four groups: (1) APP/PS1 transgenic animals exposed to a sham blast TBI (TG-Sham), (2) APP/PS1 transgenic animals exposed to blast TBI (TG-Blast), (3) nontransgenic littermates exposed to sham blast (Non-TG-Sham), and (4) nontransgenic littermates exposed to blast (Non-TG-Blast). The APP/PS1 mouse model was chosen because the age of onset of amyloid pathology is between 5 and 6 months, and because progressive increases in amyloid pathology are observed in mice up to age 12 months and beyond. This time course of amyloid deposition allows for studies of effects of TBI in young adult mice prior to onset of amyloid pathology (as well as at the onset and after the onset) and evaluation of effects of TBI on amyloid pathology over extended survival intervals after injury. Mice in group 4 (nontransgenic littermates exposed to blast) underwent the two primary outcome measurements, pattern-evoked electroretinography, and spectral-domain optical coherence tomography only (see below). Analyses of Aβ immunoreactivity could not be performed on these mice because unlike their transgenic littermates, nontransgenic mice do not overproduce Aβ and consequently levels of endogenous mouse Aβ are at the limits of detection of the histologic procedures used in this study.
Blast Injury Induction
An enclosed blast chamber was used for the purpose of these studies, one-half of which was pressurized, with a 13-cm diameter opening between the chamber halves, as described previously.14,34 A Mylar membrane (Mylar A, 0.00142 gauge; Country Plastics, Ames, IA, USA) was placed over the opening on the pressurized side of the chamber. The unpressurized side of the tank contained a padded polyvinyl chloride (PVC) protective restraint for positioning of an anesthetized mouse 30 cm from the Mylar membrane (see Ref. 14 for diagram). To create the blast wave, air was pumped into the pressurized side of the tank to 20 psi, the pressure at which the membrane ruptures. Using this model, a blast wave is produced with the following characteristics: 137.8 ± 1.3 kPa at the point of exposure (head only) and with a 10- to 15-ms blast duration.35 The pressure was calculated by using a sensor 1 cm in diameter placed directly below the head of the mouse. Prior to blast wave induction, mice were anesthetized with a combination of ketamine (0.03 mg/g body weight, intraperitoneal, IP) and xylazine (0.005 mg/g body weight, IP) and positioned within the unpressurized half of the blast chamber with the left side of the head oriented toward the source of the blast wave. Only the head of the mouse was exposed to the blast wave, with the rest of the body shielded. The head of the mouse was unrestrained during blast wave exposure. After blast or sham-blast exposure, mice were placed on a heating pad to facilitate recovery from general anesthesia and to prevent hypothermia. Xylazine anesthesia was reversed with yohimbine chloride (0.001 mg/g, IP) to facilitate the recovery from anesthesia. Non–blast-exposed mice were anesthetized and placed in the blast chamber, but did not receive a blast exposure (referred to as sham blast). Blast TBI and sham groups of mice received analgesic via subcutaneous injection (0.1 mL/20 g body weight) of buprenorphine (0.003 mg/mL) immediately after recovery from the procedure.
Pattern-Evoked Electroretinography
Pattern-evoked electroretinography (PERG) was used to objectively measure RGC function 2 months following blast TBI or sham blast. Baseline measurements were obtained 1 week prior to blast exposure, and follow-up exams were performed 2 months following blast exposure. Anesthetized mice were positioned on a temperature-controlled platform with the eye of each mouse positioned 10 cm from the video monitor on which stimuli were displayed. All recordings were obtained from the eye facing the blast wave (i.e., the left eye). Recordings from the right eye were not reported as that eye was facing the padded portion of the holder, and interaction of the blast wave with this surface cannot be excluded. Pattern ERG responses were evoked using alternating, reversing, black and white vertical stimuli displayed on an LED monitor (Jorvec, Miami, FL, USA). A recording electrode was placed subcutaneously on the snout of the animal equidistant from each eye as previously described.36 A reference needle electrode was placed subcutaneously at the base of the head, and a ground electrode was placed subcutaneously at the base of the tail to complete the circuit. All animals were placed equivalently in a fixed position in front of the monitors to prevent recording variability due to variation in animal placement. Stimuli (18° radius visual angle subtended on full-field pattern, two reversals per second, 372 averaged signals with cutoff filter frequencies of 1 to 30 Hz, 98% contrast, 80 cd/m2 average monitor illumination intensity using luminance-matched pattern reversals to exclude outer retinal contributions) were delivered under mesopic conditions without dark adaptation. A diffuser placed over the pattern on the monitor also did not elicit a measurable evoked potential, further ensuring that the electrical responses were elicited from retinal ganglion cells (RGC). The PERG response was evaluated by measuring the amplitude (peak to trough) and implicit time of the waveform, as previously described.14,37
Chromatic Pupil Light Reflex
The anterior afferent cPLR was tested using a small animal pupilometer (NeurOptics, Irvine, CA, USA). Mice were dark adapted for 30 minutes, and lightly anesthetized using a combination of ketamine (0.046 mg/g, IP) and xylazine (0.0046 mg/g, IP) in the dark. Mice were placed on a platform and given sequential light stimulations. The light stimulation in scotopic room lighting was a 1-second blue 465- to 470-nm light 0.00 log (1 lux), followed by 15 seconds off. Averages of three stimulation profiles are presented. Baseline measurements were obtained 1 week prior to blast exposure, with follow-up analysis occurring 2 months post blast exposure.
Spectral-Domain Optical Coherence Tomography
Spectral-domain optical coherence tomography (SD-OCT) analysis was performed 2 months following blast TBI or sham procedure using a Spectralis SD-OCT (Heidelberg Engineering, Vista, CA, USA) imaging system coupled with a 25 D lens for mouse ocular imaging (Heidelberg Engineering). Anesthetized mice were placed on a temperature-controlled platform and pupils were dilated using a 1% tropicamide solution. The cornea was moisturized with a saline solution applied every 20 to 30 seconds during the procedure. Volume scans (49-line dense array) positioned directly over the optic nerve head were performed to quantify the RGC complex (inner plexiform layer + ganglion cell layer + retinal nerve fiber layer) thickness. Scans were analyzed by an individual masked to the treatment of the mouse in the superior and inferior central retina, approximately 150 μm from the peripapillary region. All scans were analyzed by excluding blood vessels from the thickness calculation. Baseline measurements were obtained 1 week prior to blast exposure, with follow-up analysis occurring 2 months post blast exposure.
Balance Beam
Vestibulomotor function was examined using the balance beam test 2 months post blast injury. A beam 1 cm in diameter and 40 cm long was used in these experiments. Mice were trained for 3 days to traverse the span of the beam to obtain a food reward. A probe trial was conducted on the fourth day. An experimenter naïve to the treatment and genotype of each mouse counted the number of hindlimb errors (slips or misplacements made by the mouse).
Histopathology of Brain and Retina
Mice were deeply anesthetized with carbon dioxide, perfused with normal saline (0.9% NaCl in distilled water), and killed by decapitation. For each animal, the left eye was dissected and immersion-fixed in 4% paraformaldehyde (4% PFA) made in 0.1 M sodium phosphate buffer (PB, pH 7.4) for 24 hours. The retinas of a subset of 4% PFA-fixed eyes were dissected and prepared as retina whole mounts. The remaining 4% PFA-fixed eyes were embedded in paraffin and cut into 8-μm sections. Brains were extracted from saline-perfused mice and bisected into left and right hemispheres. The left hemisphere was immersion-fixed in 4% PFA, cryoprotected in graded sucrose solutions (15% and 30% made in PB, pH 7.4), and cut coronally at 40 μm through the rostrocaudal extent of the cerebral cortex on a freezing sledge microtome. Every fifth section through the extent of the cerebral cortex was collected for analysis resulting in 10 to 12 sections per mouse, with all major cortical areas included in the analysis. Sections were processed using immunohistochemistry (IHC) and polyclonal antibodies specific for Aβ40 or Aβ42 peptides (AB5074P and AB5078P; Millipore, St. Louis, MO, USA),38 as well as the Aβ/APP antibodies clone 6E10 (SIG39320; Covance, Dedham, MA, USA) and 4G8 (SIG39220, Covance), which target epitopes within Aβ amino acids 4 to 13 and 17 to 24, respectively. Positive controls for each Aβ antibody were included in all immunohistochemical experiments and consisted of sections of frontal cortex from a case with severe AD (male, 68 years, Braak stage VI) and from a 12-month-old male APP/PS1 mouse. Retinal whole mounts, eye cross sections, and brain sections were also processed using the pan amyloid dye, X-34 (a highly fluorescent derivative of Congo red)39 and the antibodies listed above. Brain sections processed for quantitative Aβ IHC were analyzed by light microscopy. Grayscale digital images were acquired at ×10 magnification, and composites of the entire left hemisphere cerebral cortex were constructed. Percent area coverage of amyloid plaques (plaque load) was then calculated using a standardized region of interest grayscale thresholding analysis40 using ImageJ freeware,41 and modified algorithms to automate the procedure. In secondary analyses, plaque load values were calculated separately in male and female mice, and separately for primary/association visual cortex and remaining (“nonvisual”) cortex. Data were expressed as percent area coverage, corresponding to the total amount of area covered with amyloid deposits relative to area of cerebral cortex.42 Retinal whole mounts and cross sections were examined qualitatively.
Histology and Microscopy of Optic Nerve
Blast-exposed and sham-blast mice were deeply anesthetized with carbon dioxide, lightly perfused with normal saline, and euthanized by decapitation. Optic nerves were removed and placed at 4°C for 16 hours in half-strength Karnovsky's fixative (2% paraformaldehyde, 2.5% gluteraldehyde in 0.1 M sodium cacodylate). Nerves were rinsed in 0.1 M Na cacodylate buffer, postfixed with 1% osmium tetroxide for 1 hour, and dehydrated with a series of 40-minute incubations in graded acetone. Nerves were infiltrated overnight at 4°C in 33%, 66%, and 100% resin (Low Viscosity Spurr Epoxy Resin; Ted Pella, Redding, CA, USA) diluted in propylene oxide. Specimens were embedded in 100% resin and 1-μm cross sections were cut, transferred to glass slides, stained with 1% paraphenylenediamine (PPD), and mounted to glass slides (Permount; Fisher Scientific, Pittsburgh, PA, USA). Light micrograph images were obtained with an Olympus BX-52 microscope (Cedar Valley, PA, USA) at total magnifications of ×100 and ×1000.
Grading of Optic Nerve Damage
Assessment of levels of damage in optic nerves was performed using a three-level grading scale (1, mild; 2, moderate; 3, severe) as previously described.43,44 Light micrographs of optic nerves were assessed by two independent investigators who were blinded to both the identity of the nerves and the scores given by the other investigator. Investigators assigned the same score for 77% of the nerves and different scores for 23% of nerves. In these cases of damage grade disagreement, a third investigator, blinded to both the identity of nerves and scores given by the first two investigators, assigned a third damage grade, and the most common grade (among all three investigators) was used in the final grading for each nerve specimen.
Statistical Analysis
Results are expressed as mean ± SEM unless otherwise indicated. Statistical comparisons were performed using Graphpad Prism (version 4.0; Graphpad Software, San Diego, CA, USA) using both paired t-tests and ANOVA testing. Aβ plaque load values for sham and blast-exposed groups were compared using the Mann-Whitney U test since the data were not normally distributed (D'Agostino and Pearson normality test). Optic nerve analysis was performed using Kruskal-Wallis and post hoc Dunn's test for multiple comparisons.
Results
Balance Beam Test
Mice were trained on the balance beam prior to exposure to blast TBI or sham surgery. Performance on the balance beam was tested 2 months after the sham or blast procedure (Fig. 1): Non-TG-Sham mice made 4.23 ± 0.35 errors, TG-Sham mice made 6.57 ± 0.65 errors, and TG-Blast mice made 9.67 ± 1.18 errors. Statistical analysis revealed no differences between Non-TG-Sham mice and TG-Sham mice (P = 0.1037, q = 2.94, degrees of freedom [df] = 49, 1-way ANOVA with Tukey's multiple comparison test). TG-Blast mice made significantly more errors compared to both Non-TG-Sham mice (P < 0.0001, q = 6.87, df = 49, 1-way ANOVA with Tukey's multiple comparison test) and TG-Sham mice (P = 0.0231, q = 3.86, df = 49, respectively, 1-way ANOVA with Tukey's multiple comparison test).
Figure 1.
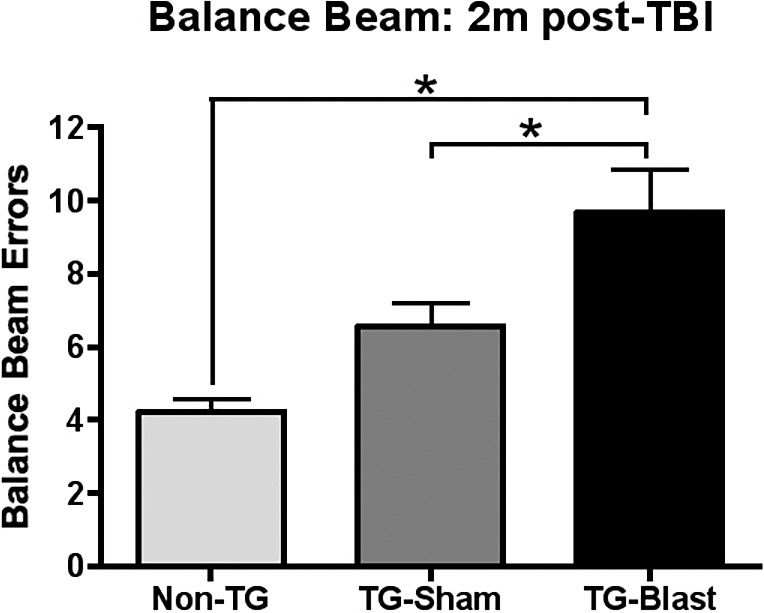
Blast exposure results in loss of coordinated motor function in APP/PS1 transgenic (TG) mice. Balance beam analysis showed increased foot placement errors in TG-Blast mice compared to Non-TG-Sham (P < 0.0001, 1-way ANOVA with Tukey's multiple comparison test) and TG-Sham mice (P = 0.0231, 1-way ANOVA with Tukey's multiple comparison test) 60 days after injury. There was no significant difference between TG-Sham and Non-TG mice (P = 0.1037, 1-way ANOVA with Tukey's multiple comparison test).
Pattern-Evoked Electroretinography
Baseline PERG measurements were obtained for all mice 1 week prior to injury (Fig. 2A). The baseline PERG amplitudes prior to sham or blast injury were 21.91 ± 1.98 μV (Non-TG-Sham, n = 7), 22.71 ± 2.49 μV (Non-TG-Blast, n = 11), 17.60 ± 0.78 μV (TG-Sham, n = 10), and 18.12 ± 1.55 μV (TG-Blast, n = 12). There were no significant differences in baseline PERG responses between groups (1-way ANOVA, P = 0.1268, F = 2.03, df = 3.36). A significant difference was observed between PERG responses 2 months following sham or blast (1-way ANOVA, P = 0.028, F = 3.403, df = 3.35). PERG amplitudes were 20.14 ± 1.17 μV (Non-TG-Sham), 18.08 ± 2.95 μV (Non-TG-Blast), 20.04 ± 2.8 μV (TG-Sham), and 11.52 ± 1.45 μV (TG-Blast), with the TG-Blast group having significantly lower PERG responses compared to the TG-Sham (1-way ANOVA with Tukey's multiple comparison test, P = 0.044, q = 3.89, df = 35). There were no significant differences between baseline and 2-month follow-up PERG values for TG-Sham (paired t-test, P = 0.4287, t = 0.8288, df = 9), Non-TG-Sham (paired t-test, P = 0.4688, t = 0.7731, df = 6) or Non-TG-Blast (paired t-test, P = 0.3751, t = 0.9331, df = 9) groups. There was, however a significant decrease in PERG responses from baseline to 2-month follow-up in the TG-Blast group (paired t-test, P = 0.0329, t = 2.44, df = 11).
Figure 2.

Blast-induced visual functional loss in APP/PS1 transgenic (TG) mice 2 months following blast exposure. (A) Analysis of preblast and postblast PERG responses shows no significant difference in the amplitude of Non-TG-Sham mice (P = 0.4688, paired t-test), Non-TG-Blast mice (P = 0.3751, paired t-test), and TG-Sham mice (0.4287, paired t-test). There was, however, a significant decrease in the PERG amplitude in APP/PS1 mice exposed to blast (TG-Blast, P = 0.0329, paired t-test). (B) Analysis of the pupil light reflex showed no difference in comparison of the percentage of pupil constriction baselines and subsequent analysis after 2 months for Non-TG-Sham mice (P > 0.99, paired t-test) or TG-Sham mice (P = 0.27, paired t-test). There was a significant reduction in the baseline measurements of the percentage of pupil constriction and subsequent analysis 2 months later in the TG-Blast group (P = 0.0185, paired t-test).
Chromatic Pupil Light Reflex
Baseline percent pupil constrictions obtained 1 week prior to injury after stimulation with light were 36.57 ± 1.88% (Non-TG-Sham, n = 7), 31.6 ± 2.82% (TG-Sham, n = 10), and 36.8 ± 1.32% (TG-Blast, n = 10, Fig. 2B) with no significant differences among the groups (1-way ANOVA, P = 0.168, F = 1.92, df = 2.24). Two months following sham or blast procedure, the pupil constriction percentages after stimulation were 36.57 ± 2.16% (Non-TG-Sham), 28.1 ± 2.63% (TG-Sham), and 30.70 ± 2.09% (TG-Blast). There was not a significant difference between baseline and 2-month follow-up values for the TG-Sham (paired t-test, P = 0.27, t = 1.15, df = 9) or the Non-TG-Sham (paired t-test, P > 0.99, t = 0, df = 6) groups. However, the TG-Blast group had a significant decrease from baseline to 2-month follow-up (paired t-test, P = 0.0185, t = 2.87, df = 9).
Optical Coherence Tomography
The RGC complex thickness from each group was quantified 2 months after blast or sham exposure in the superior (Fig. 3A) and inferior retina (Fig. 3B). Analysis of the superior retina revealed thicknesses of 74.36 ± 1.19 μm (Non-TG-Sham, n = 7, Fig. 3C), 73.5 ± 1.02 (Non-TG-Blast, n = 12, Fig. 3D), 76.71 ± 2.50 μm (TG-Sham, n = 6, Fig. 3E), and 64.65 ± 3.31 μm (TG-Blast, n = 5, Fig. 3F) groups. A significant decrease was detected in the TG-Blast group compared to the TG-Sham (P = 0.0018, q = 5.84, df = 26), Non-TG-Sham (P = 0.0101, q = 4.86, df = 26), and the Non-TG-Blast (P = 0.0099, q = 4.87, df = 26) (all 1-way ANOVA with Tukey's multiple comparison test) groups. Inferior retina thickness was 74.38 ± 0.93 μm for Non-TG-Sham, 73.95 ± 2.08 μm for TG-Sham, and 67.98 ± 4.37 μm for TG-Blast group, with no significant differences among the groups (P > 0.05 for all groups, 1-way ANOVA with Tukey's multiple comparison test).
Figure 3.
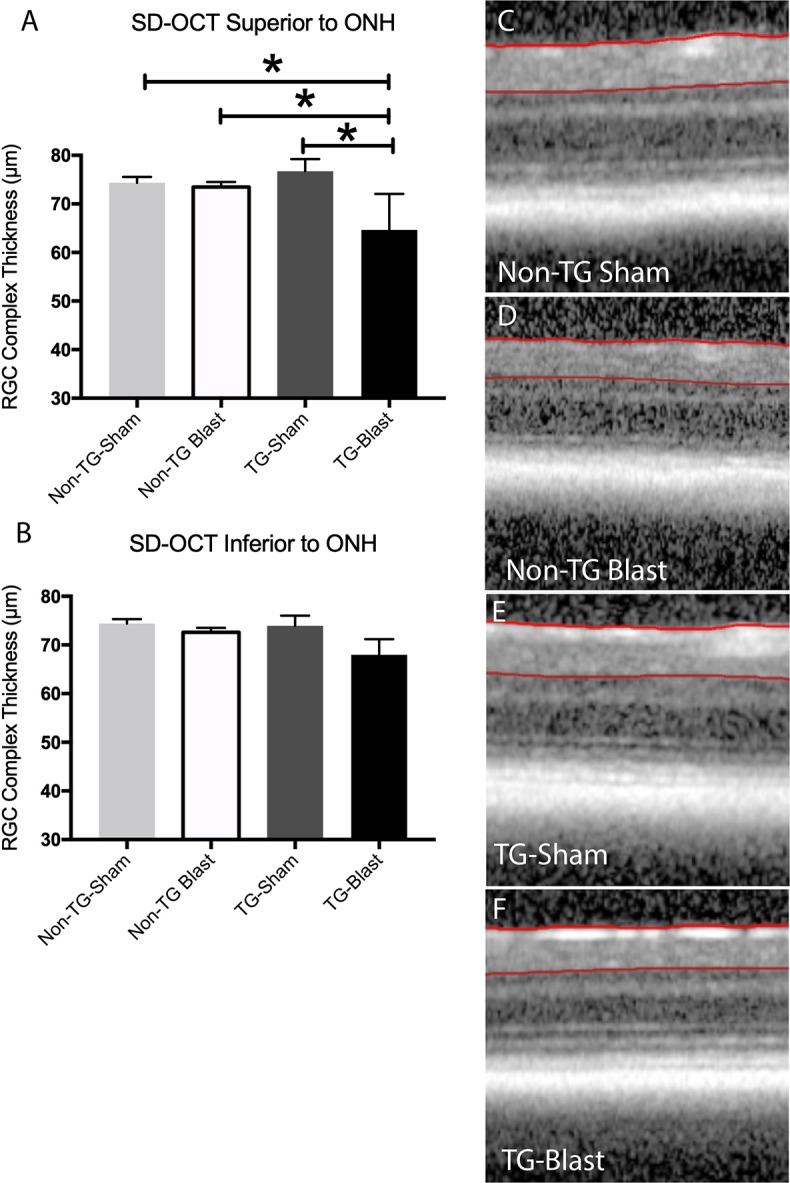
Blast exposure results in the loss of superior retinal thickness in APP/PS1 transgenic (TG) mice. Analysis of retinal OCT scans in the superior (A) retina demonstrated there was a significant decrease in the thickness of the RGC complex in TG-Blast mice compared to Non-TG-Sham mice (P = 0.0101, 1-way ANOVA with Tukey's multiple comparison test), Non-TG-Blast (P = 0.0099, 1-way ANOVA with Tukey's multiple comparison test), or TG-Sham (P = 0.0018, 1-way ANOVA with Tukey's multiple comparison test). There was not a significant difference in TG-Sham mice compared to Non-TG-Sham mice (P > 0.05, 1-way ANOVA with Bonferroni's multiple comparison test). No significant difference was observed in the inferior retina (B), although there was a trend toward reduced RGC complex thickness in TG-Blast mice. Representative SD-OCT images of the superior retina for each group are shown (C–F).
Analysis of the Optic Nerve
Optic nerve (ON) integrity in each group was quantified 2 months after blast or sham blast using a rating procedure outlined in Methods. The average ON scores were 1.00 ± 0.00 (Non-TG-Sham, n = 6), 1.67 ± 0.82 (TG-Sham, n = 6), and 2.80 ± 0.45 (TG-Blast, n = 5; Fig. 4). Compared to TG-Sham mice, TG-Blast mice had significantly more ON damage (P = 0.048, nonparametric Mann-Whitney test) as determined by naïve evaluators. There was no significant difference in ON damage between Non-TG-Sham and TG-Sham mice.
Figure 4.
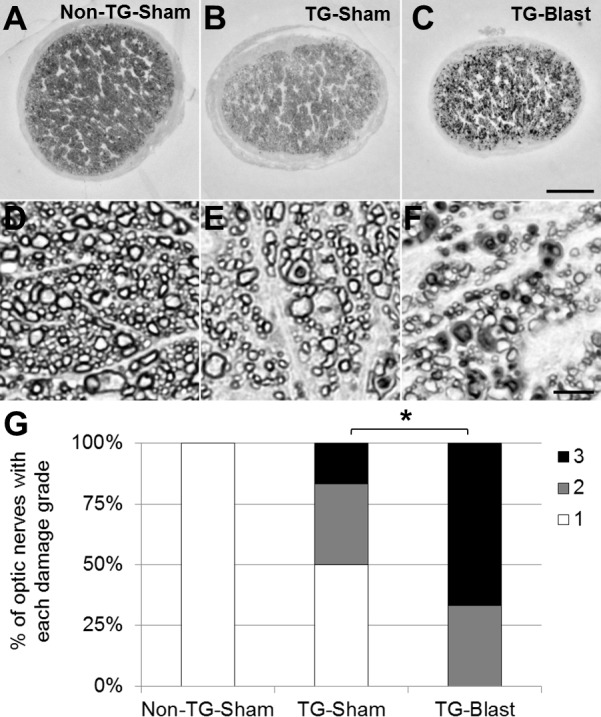
Variable optic nerve damage in APP/PS1 transgenic (TG) mice is exacerbated by exposure to blast. Qualitative analyses of nerve damage in paraphenylenediamine (PPD)-stained histologic cross sections of optic nerve from TG-Sham and TG-Blast mice. Representative light microscopy images of optic nerve at (A–C) low (10× objective, 100× total magnification) and (D–F) high magnification (100× objective, 1000× total magnification). There is a decrease in the density of myelinated axons in nerves from TG-Sham compared to Non-TG-Sham mice (E versus D) and TG-Blast compared to TG-Sham mice (F versus E). Also note the increased prevalence of damaged axons (PPD stains the myelin sheath of intact axons and the axoplasm of damaged axons) in TG-Blast compared to TG-Sham mice (F versus E) and TG-Blast compared to Non-TG-Sham mice (F versus D). Scale bars: 100 μm (A–C) and 5 μm (D–F). (G) Stacked bar graph showing the percentage of optic nerves with the indicated damage grade for each cohort of mice using an established qualitative damage grading scale, whereby increasing values represent increasing damage severity (1 = no to mild damage [white], 2 = moderate [gray], 3 = severe [black]). Each bar represents the percentage of nerves with each damage grade for each cohort of mice: Non-TG-Sham (n = 6 mice), TG-Sham (n = 6), and TG-Blast (n = 5). Statistical significance was assessed using a nonparametric Mann-Whitney test. Asterisk represents a P < 0.05.
Analysis of RGC Density and Aβ/APP Immunoreactivity in Brain and Eyes
No amyloid deposits or plaques of fibrillar Aβ (assessed with X-34 or Aβx-40 and Aβx-42 immunoreactivity) were seen in retina whole mounts from TG-Sham and TG-Blast mice. Aβ/APP (6E10 or 4G8) immunoreactivity was detected in retinal blood vessels and perivascular cell-like profiles in the ganglion cell layer as well as inner and outer plexiform layers in retina cross sections of TG-Blast mice (Figs. 5A, 5B). Retinal ganglion cell densities in each group were quantified in retinal cross sections 2 months after blast or sham exposure. We detected lower RGC density in TG-Blast mice (11.63 ± 1.32) compared to Non-TG-Sham mice (22.67 ± 1.74) (P < 0.05, 1-way ANOVA with Bonferroni's multiple comparison test) (Fig. 5C). There was not a significant difference in TG-Sham (16.38 ± 3.78) mice compared to Non-TG-Sham mice or TG-Blast mice (P > 0.05, 1-way ANOVA with Bonferroni's multiple comparison test). Aβ-immunoreactive plaques were detected in all transgenic (TG-Sham and TG-Blast) mice, while no plaques were detected in Non-TG mice. Two months after blast or sham procedure, there was a trend for greater plaque load in the TG-Blast group compared to the TG-Sham group when the Aβ/APP 6E10 antibody was used (Mann-Whitney U = 56, P = 0.08), and similar but weaker trends when C-terminal neoepitope antibodies were used (Fig. 5D). Similar results were obtained using histology with the pan-amyloid marker X-34 (not shown). No difference in plaque load was detected when primary and association visual cortex and remaining neocortex were analyzed separately in both TG-Sham (visual versus nonvisual: 0.295 ± 0.14 vs. 0.230 ± 0.17) and TG-Blast mice (visual versus nonvisual: 0.781 ± 0.23 vs. 0.812 ± 0.21). When separating mice by sex, overall plaque load values in female mice were higher than in male mice in TG-Sham (0.469 ± 0.12 and 0.319 ± 0.24) and TG-Blast (0.898 ± 0.40 and 0.860 ± 0.33, respectively) groups; neither difference was significant by Mann-Whitney U test.
Figure 5.
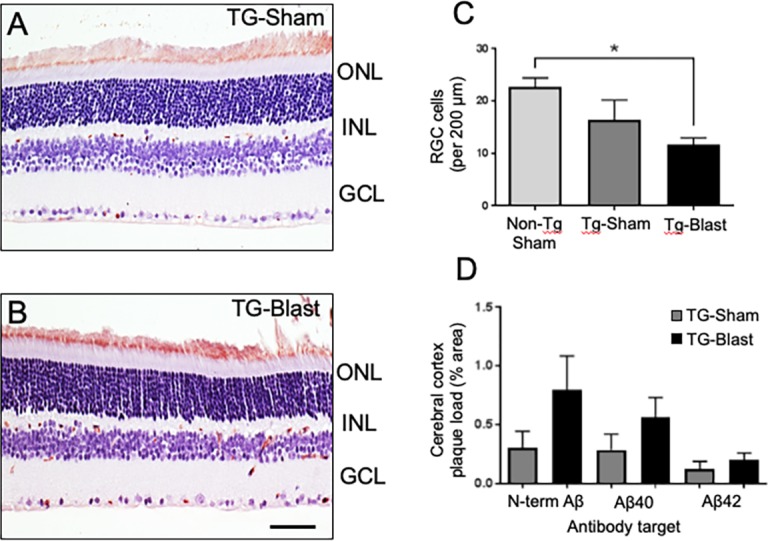
Retinal ganglion cell loss in APP/PS1 transgenic (TG) mice. Analyses of retinal cross sections show Aβ/APP immunoreactivity (brown color) in different layers of retina and more pronounced in TG-Blast compared to TG-Sham mice (A, B). RGC density is lower in TG-Blast mice compared to Non-TG-Sham mice (C, P < 0.05, 1-way ANOVA with Bonferroni's multiple comparison test). There was not a significant difference in TG-Sham mice compared to Non-TG-Sham mice or TG-Blast mice (P > 0.05, 1-way ANOVA with Bonferroni's multiple comparison test). Amyloid plaque load in the cerebral cortex of TG-Sham and TG-Blast mice, as percent area immunoreactive with an Aβ/APP antibody 6E10 and two Aβ antibodies directed toward neoepitopes at C-terminal amino acid 40 (valine, Aβx-40) or C-terminal amino acid 42 (alanine, Aβx-42). Plaque load values in each group were not normally distributed (D'Agostino and Pearson normality test, P < 0.01); consequently, for each antibody type, TG-Sham and TG-Blast groups were compared using the Mann-Whitney t-test. The greater 6E10 plaque load in TG-Blast mice was not statistically significantly different from the TG-Sham group (Mann-Whitney U test = 56, P = 0.08). Similar though weaker trends were observed when Aβx-40 (P = 0.0956, Mann-Whitney t-test) and Aβx-42 (P = 0.1444, Mann-Whitney t-test) were used to mark plaques (D). Scale bar: 35 μm.
Discussion
The purpose of this study was to explore the interaction between genetic predisposition for AD-like brain amyloidosis and blast wave–induced TBI on structure and function of the visual sensory system as well as CNS (cerebral cortex and retina) amyloidosis in adult transgenic APP/PS1 mice. These mice are genetically modified to overexpress the APP and overproduce Aβ peptide and exhibit age-related cerebral amyloidosis between ages 5 and 6 months,33 with amyloid deposits reported in the retina of old mice (12–19 months).45 Mice used in this study were exposed to blast TBI at 2 to 3 months of age and evaluated 2 months later (at age 4 to 5 months) and were at the earliest stage of amyloidosis at the time of evaluation. This age was chosen to determine if blast TBI in early life accelerates amyloid pathology, a process that might be masked in older mice with existing severe amyloid burden. Active service members exposed to blast TBI are typically young adults; thus use of young adult mice in this study is applicable to individuals with a history of blast TBI in their young adult years.
The experiments reported here begin to provide information about how the interaction between genetic predisposition for amyloidosis (excess APP expression and autosomal dominant mutations in the presenilin gene, resulting in early-onset AD) and blast exposure influences visual system structure and function and brain amyloid pathology. Our results demonstrate chronic impairment of visual and coordinated motor function as well as moderate enhancement of brain amyloidosis 2 months after blast TBI in young adult APP/PS1 mice. PERG decrements, ON degeneration, decrease in the RGC complex thickness, and decreases in coordinated motor function were seen in APP/PS1 mice exposed to blast TBI compared to the sham-exposed transgenic littermates. A common result of blast injury is combat ocular trauma (up to 43%)46 and there is a strong association between cranial and ocular injuries in military personnel,47 yet closed-eye injuries associated with TBI are often overlooked. In addition to widespread axonal damage in the brain,48 blast injury induces pathological changes and functional deficits in the retina, ON, and brain areas subserving sensorimotor and behavioral/cognitive processes. The mechanism of blast injury damage on the visual system is not clearly understood.46 It is also unknown if retinal and ON functional responses may serve as biomarkers for brain dysfunction and degeneration, in addition to visual dysfunction, in veterans with a history of blast TBI.
The retina has been explored recently as a “window” to brain amyloidosis in AD,49 and direct imaging of amyloid plaques is currently being explored in living patients.50,51 Structural and functional changes in the retina/ON (independent of presence of amyloid) have also been considered as biomarkers for AD. Recent studies in a mouse AD model reported that deficits in retinal physiology and visual evoked potentials precede other brain and cognitive deficits, which suggests that this measure is particularly sensitive to dysregulation in AD.52 Analysis of patients with AD suggests that the retina undergoes significant remodeling during the course of the disease, with both increases53 and decreases54,55 in retinal thickness observed using OCT. AD patients also have profound changes in oxygen metabolism,56 and an overall decrease in the retinal vasculature density,57 which may contribute to or result from abnormal functional and structural retinal outcomes in this disease. It is possible that the RGC functional loss on the PERG response analysis was related to the observed reductions in density of ganglion cells (see Fig. 5).
The primary outcomes used in this study were PERG and inner retinal layer thickness using OCT. These outcomes were chosen because they are direct measures of function and structure of an individual class of neurons, the RGCs. A notable result from our study was the reduced effect of blast exposure on APP/PS1 WT littermates compared to our previously published studies using C57BL6/J mice with respect to PERG and SD-OCT outcomes.14,34,58 In our previous study using WT C57Bl6/J mice, PERG responses decreased an average of 23%, while the RGC complex thickness decreased an average of 6%. In this study the PERG of APP/PS1 WT littermates decreased an average of 20% with a 1.2% decrease in OCT thickness. The baseline OCT thickness was 74 μm, compared to 68-μm baseline for C57Bl6/J mice. These differences may help to explain the relative lack of effect of blast injury on Non-TG-Blast mice, which may be useful to explore in future studies. APP/PS1 transgenic mice had an overall larger response to injury compared to WT littermates or previously studied WT C57Bl6/J mice, with an average group decline of 27% for PERG responses and 16% for RGC complex thickness. These declines may be due to a greater sensitivity of neurons in the APP/PS1 mice to be damaged by blast, an exacerbation of molecular processes causing neuronal degeneration, or both.
Our qualitative analyses of ON phenotypes indicate that adult APP/PS1 mice develop mild nerve damage by 5 months of age that can be exacerbated by blast TBI. In addition to grading the severity of damage in ONs using an ordinal scale,43,44 visual inspection of PPD-stained ON cross sections revealed overt features of damage from blast TBI compared to sham controls. These features included increases in the relative proportion of damaged axons (i.e., PPD stains the axoplasm of damaged axons), gliosis (i.e., increased prevalence and hypertrophy of glial cells), and areas of nerve devoid of myelinated axons. In comparing ONs from sham mice differing only by genotype, we observed mild to moderate degrees of axon damage and gliosis in nerves from TG-Sham mice compared to those from Non-TG-Sham controls. Among nerves from TG mice, we observed a striking increase in the prevalence of damaged axons, an increased proportion of nerve area (cross-sectional) devoid of myelinated axons, and more severe gliosis in TG-Blast mice compared to TG-Sham controls. These results demonstrate that APP/PS1 mice develop mild phenotypes of ON damage that are exacerbated by blast TBI.
In addition to our primary functional and structural outcomes that analyze one type of retinal neuron, we also utilized two outcomes that rely on neuronal circuits and networks. The pupil light reflex (PLR) was used as an indicator of overall retinal and ON function, although the decrease observed in the current study could have resulted from damage to axons in cranial nerves II or III, or neuronal synaptic partners in the pretectal nucleus, Edinger-Westphal nucleus, or ciliary ganglion. Based on the overall level of ON/myelin damage observed in the TG-Blast group it is not surprising that a deficit was observed. It is worth pointing out that WT C57Bl6/J mice analyzed in our previous study had PLR deficits for only 1 day, although ongoing ON degeneration occurred for up to 10 months. This suggests that the interaction of genetic predisposition for amyloidosis may cause more severe functional visual impairment than in WT mice. The second outcome that we chose to examine was the balance beam test. This test of coordinated motor function relies on the integration of multiple sensory and motor neuronal networks. Declines in motor function have been shown to occur in patients before cognitive symptoms of AD manifest clinically.59 In our study we observed significant decreases in coordinated motor function in TG mice receiving blast injury. The worsening of motor function observed in TG-Blast compared to TG-Sham was not seen in our study of WT mice.14 TG mice also had decrements in the PLR response, PERG response, and a decrease in the superior SD-OCT RGC complex, all before a significant increase in Aβ plaques was observed. Taken together, these data suggest that these responses alone or in combination may have greater sensitivity to detect patients with amyloidosis when compared to currently used methods.
The effects of blast TBI on amyloid pathology in adult APP/PS1 mice were modest at 2 months after injury. At this time point, amyloid load in cortical brain regions of TG-Blast mice was modestly higher relative to TG-Sham counterparts, suggesting that in autosomal dominant familial AD mutation carriers (i.e., PSEN1, PSEN2) or APP mutation carriers and subjects with Down syndrome, blast injury (and possibly other forms of head trauma) could exacerbate or accelerate the disease process. However, aside from infrequent intracellular and vessel-associated APP/Aβ immunoreactive profiles, we did not detect Aβ-immunoreactive or X-34–labeled amyloid plaque deposits in retinas from TG-Blast or TG-Sham mice, possibly due to the early age at which mice were analyzed. This makes it unlikely that observed changes in structure and function of the retina after blast TBI were due to local amyloid deposition. Instead, soluble oligomeric Aβ or gliosis independent of human Aβ could underlie retinal dysfunction. Reports of retinal amyloid pathology have been inconsistent in the literature. Several studies used thioflavin-S histology or Aβ/APP IHC and reported only diffuse extracellular or intracellular signal, primarily in the RGC layer, but no evidence of retinal amyloid plaques in APP/PS1 mice up to 27 months of age.60–62 However, small (<5 μm) Aβ-immunoreactive plaque-like deposits were reported in retinas of >10-month-old APP/PS1 mice,45,63,64 and curcumin-labeled radial glia-like cells in retina were also Aβ/APP immunoreactive (antibody clone 4G8—labels both APP and Aβ) in 2.5- to 5-month-old transgenic APP mice. Compared to studies reporting retinal Aβ-immunoreactive accumulates, the APP/PS1 mice we analyzed were younger (5–6 months old versus >10 months). However, our observation of vessel-associated profiles immunoreactive for APP/Aβ in retina of young APP/PS1 mice, together with the report of curcumin uptake and Aβ/APP immunolabeling in retina of 2.5-month-old transgenic APP-overexpressing mice,63 supports the idea that intracellular Aβ precedes amyloid plaques in the APP/PS1 mouse.65
In naïve (no injury) APP/PS1 mice, memory impairment begins around age 7 to 8 months,66,67 later than the age at completion of our current study (4–5 months of age). Thus, the APP/PS1 mice we examined in this study could be considered a model of preclinical AD, similar to amyloid positivity preceding the onset of clinical symptoms in humans at risk for AD. It will be important to determine in future studies if blast TBI accelerates the onset of memory impairment in APP/PS1 mice, and if retinal degeneration continues to progress as amyloid burden increases. The contribution of RGC loss and ON pathology to amyloid pathology in contralateral visual cortex was not assessed in the current study, as our analyses of retina and cortex ipsilateral to blast exposure was aiming to determine the effects of blast injury on cerebral cortical amyloidosis in general, independent from potential degeneration of multiple crossed sensory pathways. Future studies will focus more specifically on the interactions of sensory system trauma due to blast exposure on brain amyloidosis in all components of the visual pathway including visual cortex.
In conclusion, our study indicates that blast TBI facilitates progression of amyloid pathology and supports the potential utility of retinal functional (PERG, cPLR) and structural (SD-OCT) analyses as tools to evaluate the visual system impairment associated with this condition, even before the development of significant plaque pathology in the brain. Because we demonstrate chronic effects of blast injury on both retinal physiology and structure as well as amyloid load, new translational biomarker development studies could focus on combined assessments of retinal function and brain amyloidosis by positron emission tomography imaging in aging veterans with past exposure to blast TBI.
Acknowledgments
The authors thank Lan Shao and Elena Hernandez-Merino for their technical expertise. The authors alone are responsible for the content and writing of this paper.
Supported by Department of Veterans Affairs Grant RX000952 (MDI, MMH), Department of Defense Grant W81XWH-14-1-0583 (MMH), a T32 training grant (T32DK112751-01) (AHB), and the Iowa City Center for the Prevention and Treatment of Visual Loss (MMH).
Disclosure: M.M. Harper, None; A. Hedberg-Buenz, None; J. Herlein, None; E.E. Abrahamson, None; M.G. Anderson, None; M.H. Kuehn, None; R.H. Kardon, None; P. Poolman, None; M.D. Ikonomovic, None
References
- 1.DeKosky ST, Ikonomovic MD, Gandy S. Traumatic brain injury—football, warfare, and long-term effects. N Engl J Med. 2010;363:1293–1296. doi: 10.1056/NEJMp1007051. [DOI] [PubMed] [Google Scholar]
- 2.Elder GA, Mitsis EM, Ahlers ST, Cristian A. Blast-induced mild traumatic brain injury. Psychiatr Clin North Am. 2010;33:757–781. doi: 10.1016/j.psc.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Kontos AP, Kotwal RS, Elbin RJ, et al. Residual effects of combat-related mild traumatic brain injury. J Neurotrauma. 2013;30:680–686. doi: 10.1089/neu.2012.2506. [DOI] [PubMed] [Google Scholar]
- 4.Owens BD, Kragh JF, Jr, Wenke JC, Macaitis J, Wade CE, Holcomb JB. Combat wounds in operation Iraqi Freedom and operation Enduring Freedom. J Trauma. 2008;64:295–299. doi: 10.1097/TA.0b013e318163b875. [DOI] [PubMed] [Google Scholar]
- 5.Murray CK, Reynolds JC, Schroeder JM, Harrison MB, Evans OM, Hospenthal DR. Spectrum of care provided at an echelon II Medical Unit during Operation Iraqi Freedom. Mil Med. 2005;170:516–520. doi: 10.7205/milmed.170.6.516. [DOI] [PubMed] [Google Scholar]
- 6.Swan AA, Nelson JT, Pogoda TK, Amuan ME, Akin FW, Pugh MJ. Sensory dysfunction and traumatic brain injury severity among deployed post-9/11 veterans: a chronic effects of neurotrauma consortium study. Brain Inj. 2018;32:1197–1207. doi: 10.1080/02699052.2018.1495340. [DOI] [PubMed] [Google Scholar]
- 7.Belanger HG, Proctor-Weber Z, Kretzmer T, Kim M, French LM, Vanderploeg RD. Symptom complaints following reports of blast versus non-blast mild TBI: does mechanism of injury matter? Clin Neuropsychol. 2011;25:702–715. doi: 10.1080/13854046.2011.566892. [DOI] [PubMed] [Google Scholar]
- 8.Reid MW, Miller KJ, Lange RT, et al. A multisite study of the relationships between blast exposures and symptom reporting in a post-deployment active duty military population with mild traumatic brain injury. J Neurotrauma. 2014;31:1899–1906. doi: 10.1089/neu.2014.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeWalt GJ, Eldred WD. Visual system pathology in humans and animal models of blast injury. J Comp Neurol. 2017;525:2955–2967. doi: 10.1002/cne.24252. [DOI] [PubMed] [Google Scholar]
- 10.Shedd DF, Benko NA, Jones J, Zaugg BE, Peiffer RL, Coats B. Long term temporal changes in structure and function of rat visual system after blast exposure. Invest Ophthalmol Vis Sci. 2018;59:349–361. doi: 10.1167/iovs.17-21530. [DOI] [PubMed] [Google Scholar]
- 11.Allen RS, Motz CT, Feola A, et al. Long-term functional and structural consequences of primary blast overpressure to the eye. J Neurotrauma. 2018;35:2104–2116. doi: 10.1089/neu.2017.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi JH, Greene WA, Johnson AJ, et al. Pathophysiology of blast-induced ocular trauma in rats after repeated exposure to low-level blast overpressure. Clin Exp Ophthalmol. 2015;43:239–246. doi: 10.1111/ceo.12407. [DOI] [PubMed] [Google Scholar]
- 13.Hines-Beard J, Marchetta J, Gordon S, Chaum E, Geisert EE, Rex TS. A mouse model of ocular blast injury that induces closed globe anterior and posterior pole damage. Exp Eye Res. 2012;99:63–70. doi: 10.1016/j.exer.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mohan K, Kecova H, Hernandez-Merino E, Kardon RH, Harper MM. Retinal ganglion cell damage in an experimental rodent model of blast-mediated traumatic brain injury. Invest Ophthalmol Vis Sci. 2013;54:3440–3450. doi: 10.1167/iovs.12-11522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Capo-Aponte JE, Urosevich TG, Temme LA, Tarbett AK, Sanghera NK. Visual dysfunctions and symptoms during the subacute stage of blast-induced mild traumatic brain injury. Mil Med. 2012;177:804–813. doi: 10.7205/milmed-d-12-00061. [DOI] [PubMed] [Google Scholar]
- 16.Magone MT, Kwon E, Shin SY. Chronic visual dysfunction after blast-induced mild traumatic brain injury. J Rehabil Res Dev. 2014;51:71–80. doi: 10.1682/JRRD.2013.01.0008. [DOI] [PubMed] [Google Scholar]
- 17.Kirby E, Bandelow S, Hogervorst E. Visual impairment in Alzheimer's disease: a critical review. J Alzheimers Dis. 2010;21:15–34. doi: 10.3233/JAD-2010-080785. [DOI] [PubMed] [Google Scholar]
- 18.Risacher SL, Wudunn D, Pepin SM, et al. Visual contrast sensitivity in Alzheimer's disease, mild cognitive impairment, and older adults with cognitive complaints. Neurobiol Aging. 2013;34:1133–1144. doi: 10.1016/j.neurobiolaging.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polo V, Rodrigo MJ, Garcia-Martin E, et al. Visual dysfunction and its correlation with retinal changes in patients with Alzheimer's disease. Eye (Lond) 2017;31:1034–1041. doi: 10.1038/eye.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O'Bryhim BE, Apte RS, Kung N, Coble D, Van Stavern GP. Association of preclinical Alzheimer disease with optical coherence tomographic angiography findings. JAMA Ophthalmol. 2018;136:1242–1248. doi: 10.1001/jamaophthalmol.2018.3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shariflou S, Georgevsky D, Mansour H, et al. Diagnostic and prognostic potential of retinal biomarkers in early on-set Alzheimer's disease. Curr Alzheimer Res. 2017;14:1000–1007. doi: 10.2174/1567205014666170329114445. [DOI] [PubMed] [Google Scholar]
- 22.Golzan SM, Goozee K, Georgevsky D, et al. Retinal vascular and structural changes are associated with amyloid burden in the elderly: ophthalmic biomarkers of preclinical Alzheimer's disease. Alzheimers Res Ther. 2017;9:13. doi: 10.1186/s13195-017-0239-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mendez MF, Mendez MA, Martin R, Smyth KA, Whitehouse PJ. Complex visual disturbances in Alzheimer's disease. Neurology. 1990;40:439–443. doi: 10.1212/wnl.40.3_part_1.439. [DOI] [PubMed] [Google Scholar]
- 24.Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer's disease? Nat Rev Neurosci. 2010;11:361–370. doi: 10.1038/nrn2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ikonomovic MD, Mi Z, Abrahamson EE. Disordered APP metabolism and neurovasculature in trauma and aging: combined risks for chronic neurodegenerative disorders. Ageing Res Rev. 2017;34:51–63. doi: 10.1016/j.arr.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cernak I, Wang Z, Jiang J, Bian X, Savic J. Ultrastructural and functional characteristics of blast injury-induced neurotrauma. J Trauma. 2001;50:695–706. doi: 10.1097/00005373-200104000-00017. [DOI] [PubMed] [Google Scholar]
- 27.Goldstein LE, Fisher AM, Tagge CA, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. 2012;4:134ra60. doi: 10.1126/scitranslmed.3003716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryu J, Horkayne-Szakaly I, Xu L, et al. The problem of axonal injury in the brains of veterans with histories of blast exposure. Acta Neuropathol Commun. 2014;2:153. doi: 10.1186/s40478-014-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verma SK, Kan EM, Lu J, et al. Multi-echo susceptibility-weighted imaging and histology of open-field blast-induced traumatic brain injury in a rat model. NMR Biomed. 2015;28:1069–1077. doi: 10.1002/nbm.3351. [DOI] [PubMed] [Google Scholar]
- 30.Kuehn R, Simard PF, Driscoll I, et al. Rodent model of direct cranial blast injury. J Neurotrauma. 2011;28:2155–2169. doi: 10.1089/neu.2010.1532. [DOI] [PubMed] [Google Scholar]
- 31.Huber BR, Meabon JS, Martin TJ, et al. Blast exposure causes early and persistent aberrant phospho- and cleaved-tau expression in a murine model of mild blast-induced traumatic brain injury. J Alzheimers Dis. 2013;37:309–323. doi: 10.3233/JAD-130182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee MK, Borchelt DR, Kim G, et al. Hyperaccumulation of FAD-linked presenilin 1 variants in vivo. Nat Med. 1997;3:756–760. doi: 10.1038/nm0797-756. [DOI] [PubMed] [Google Scholar]
- 33.Jankowsky JL, Fadale DJ, Anderson J, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- 34.Dutca LM, Stasheff SF, Hedberg-Buenz A, et al. Early detection of subclinical visual damage after blast-mediated TBI enables prevention of chronic visual deficit by treatment with P7C3-S243. Invest Ophthalmol Vis Sci. 2014;55:8330–8341. doi: 10.1167/iovs.14-15468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harper MM. Author response: pressure wave dosimetry for “retinal ganglion cell damage in an experimental rodent model of blast-mediated traumatic brain injury.”. Invest Ophthalmol Vis Sci. 2014;55:1350–1351. doi: 10.1167/iovs.13-13692. [DOI] [PubMed] [Google Scholar]
- 36.Chou TH, Bohorquez J, Toft-Nielsen J, Ozdamar O, Porciatti V. Robust mouse pattern electroretinograms derived simultaneously from each eye using a common snout electrode. Invest Ophthalmol Vis Sci. 2014;55:2469–2475. doi: 10.1167/iovs.14-13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohan K, Harper MM, Kecova H, et al. Characterization of structure and function of the mouse retina using pattern electroretinography, pupil light reflex, and optical coherence tomography. Vet Ophthalmol. 2012;15:94–104. doi: 10.1111/j.1463-5224.2012.01034.x. [DOI] [PubMed] [Google Scholar]
- 38.Wegiel J, Kuchna I, Nowicki K, et al. Intraneuronal αβ immunoreactivity is not a predictor of brain amyloidosis-beta or neurofibrillary degeneration. Acta Neuropathol. 2007;113:389–402. doi: 10.1007/s00401-006-0191-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ikonomovic MD, Abrahamson EE, Isanski BA, et al. X-34 labeling of abnormal protein aggregates during the progression of Alzheimer's disease. Methods Enzymol. 2006;412:123–144. doi: 10.1016/S0076-6879(06)12009-1. [DOI] [PubMed] [Google Scholar]
- 40.Fishman CE, Cummins DJ, Bales KR, et al. Statistical aspects of quantitative image analysis of beta-amyloid in the APP(V717F) transgenic mouse model of Alzheimer's disease. J Neurosci Methods. 2001;108:145–152. doi: 10.1016/s0165-0270(01)00381-8. [DOI] [PubMed] [Google Scholar]
- 41.Schneider CA, Rasband WS, Eliceiri KW. NIH. Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cohen AD, Ikonomovic MD, Abrahamson EE, et al. Anti-amyloid effects of small molecule αβ-binding agents in PS1/APP mice. Lett Drug Des Discov. 2009;6:437. doi: 10.2174/157018009789057526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Libby RT, Anderson MG, Pang IH, et al. Inherited glaucoma in DBA/2J mice: pertinent disease features for studying the neurodegeneration. Vis Neurosci. 2005;22:637–648. doi: 10.1017/S0952523805225130. [DOI] [PubMed] [Google Scholar]
- 44.Anderson MG, Libby RT, Gould DB, Smith RS, John SW. High-dose radiation with bone marrow transfer prevents neurodegeneration in an inherited glaucoma. Proc Natl Acad Sci U S A. 2005;102:4566–4571. doi: 10.1073/pnas.0407357102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perez SE, Lumayag S, Kovacs B, Mufson EJ, Xu S. Beta-amyloid deposition and functional impairment in the retina of the APPswe/PS1DeltaE9 transgenic mouse model of Alzheimer's disease. Invest Ophthalmol Vis Sci. 2009;50:793–800. doi: 10.1167/iovs.08-2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cockerham GC, Goodrich GL, Weichel ED, et al. Eye and visual function in traumatic brain injury. J Rehabil Res Dev. 2009;46:811–818. doi: 10.1682/jrrd.2008.08.0109. [DOI] [PubMed] [Google Scholar]
- 47.Cho RI, Bakken HE, Reynolds ME, Schlifka BA, Powers DB. Concomitant cranial and ocular combat injuries during Operation Iraqi Freedom. J Trauma. 2009;67:516–520. doi: 10.1097/TA.0b013e3181a5f08d. discussion 9–20. [DOI] [PubMed] [Google Scholar]
- 48.Mac Donald CL, Johnson AM, Cooper D, et al. Detection of blast-related traumatic brain injury in U.S. military personnel. N Engl J Med. 2011;364:2091–2100. doi: 10.1056/NEJMoa1008069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krantic S, Torriglia A. Retina: source of the earliest biomarkers for Alzheimer's disease? J Alzheimers Dis. 2014;40:237–243. doi: 10.3233/JAD-132105. [DOI] [PubMed] [Google Scholar]
- 50.Koronyo Y, Biggs D, Barron E, et al. Retinal amyloid pathology and proof-of-concept imaging trial in Alzheimer's disease. JCI Insight. 2017;2:93621. doi: 10.1172/jci.insight.93621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koronyo Y, Salumbides BC, Black KL, Koronyo-Hamaoui M. Alzheimer's disease in the retina: imaging retinal abeta plaques for early diagnosis and therapy assessment. Neurodegener Dis. 2012;10:285–293. doi: 10.1159/000335154. [DOI] [PubMed] [Google Scholar]
- 52.Criscuolo C, Cerri E, Fabiani C, Capsoni S, Cattaneo A, Domenici L. The retina as a window to early dysfunctions of Alzheimer's disease following studies with a 5xFAD mouse model. Neurobiol Aging. 2018;67:181–188. doi: 10.1016/j.neurobiolaging.2018.03.017. [DOI] [PubMed] [Google Scholar]
- 53.Lad EM, Mukherjee D, Stinnett SS, et al. Evaluation of inner retinal layers as biomarkers in mild cognitive impairment to moderate Alzheimer's disease. PLoS One. 2018;13:e0192646. doi: 10.1371/journal.pone.0192646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.den Haan J, Janssen SF, van de Kreeke JA, Scheltens P, Verbraak FD, Bouwman FH. Retinal thickness correlates with parietal cortical atrophy in early-onset Alzheimer's disease and controls. Alzheimers Dement (Amst) 2018;10:49–55. doi: 10.1016/j.dadm.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cunha JP, Proenca R, Dias-Santos A, et al. OCT in Alzheimer's disease: thinning of the RNFL and superior hemiretina. Graefes Arch Clin Exp Ophthalmol. 2017;255:1827–1835. doi: 10.1007/s00417-017-3715-9. [DOI] [PubMed] [Google Scholar]
- 56.Olafsdottir OB, Saevarsdottir HS, Hardarson SH, et al. Retinal oxygen metabolism in patients with mild cognitive impairment. Alzheimers Dement (Amst) 2018;10:340–345. doi: 10.1016/j.dadm.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bulut M, Kurtulus F, Gozkaya O, et al. Evaluation of optical coherence tomography angiographic findings in Alzheimer's type dementia. Br J Ophthalmol. 2018;102:233–237. doi: 10.1136/bjophthalmol-2017-310476. [DOI] [PubMed] [Google Scholar]
- 58.Yin TC, Voorhees JR, Genova RM, et al. Acute axonal degeneration drives development of cognitive, motor, and visual deficits after blast-mediated traumatic brain injury in mice. eNeuro. 2016;3:ENEURO.0220-16.2016. doi: 10.1523/ENEURO.0220-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Buchman AS, Bennett DA. Loss of motor function in preclinical Alzheimer's disease. Expert Rev Neurother. 2011;11:665–676. doi: 10.1586/ern.11.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ning A, Cui J, To E, Ashe KH, Matsubara J. Amyloid-beta deposits lead to retinal degeneration in a mouse model of Alzheimer disease. Invest Ophthalmol Vis Sci. 2008;49:5136–5143. doi: 10.1167/iovs.08-1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shimazawa M, Inokuchi Y, Okuno T, et al. Reduced retinal function in amyloid precursor protein-over-expressing transgenic mice via attenuating glutamate-N-methyl-d-aspartate receptor signaling. J Neurochem. 2008;107:279–290. doi: 10.1111/j.1471-4159.2008.05606.x. [DOI] [PubMed] [Google Scholar]
- 62.Dutescu RM, Li QX, Crowston J, Masters CL, Baird PN, Culvenor JG. Amyloid precursor protein processing and retinal pathology in mouse models of Alzheimer's disease. Graefes Arch Clin Exp Ophthalmol. 2009;247:1213–1221. doi: 10.1007/s00417-009-1060-3. [DOI] [PubMed] [Google Scholar]
- 63.Koronyo-Hamaoui M, Koronyo Y, Ljubimov AV, et al. Identification of amyloid plaques in retinas from Alzheimer's patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage. 2011;54(suppl 1):S204–S217. doi: 10.1016/j.neuroimage.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gupta VK, Chitranshi N, Gupta VB, et al. Amyloid beta accumulation and inner retinal degenerative changes in Alzheimer's disease transgenic mouse. Neurosci Lett. 2016;623:52–56. doi: 10.1016/j.neulet.2016.04.059. [DOI] [PubMed] [Google Scholar]
- 65.Wirths O, Multhaup G, Czech C, et al. Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett. 2001;306:116–120. doi: 10.1016/s0304-3940(01)01876-6. [DOI] [PubMed] [Google Scholar]
- 66.Serneels L, Van Biervliet J, Craessaerts K, et al. gamma-Secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer's disease. Science. 2009;324:639–642. doi: 10.1126/science.1171176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Radde R, Bolmont T, Kaeser SA, et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006;7:940–946. doi: 10.1038/sj.embor.7400784. [DOI] [PMC free article] [PubMed] [Google Scholar]