

Abstract
Frankia strains induce the formation of nitrogen-fixing nodules on roots of actinorhizal plants. Phylogenetically, Frankia strains can be grouped in four clusters. The earliest divergent cluster, cluster-2, has a particularly wide host range. The analysis of cluster-2 strains has been hampered by the fact that with two exceptions, they could never be cultured. In this study, 12 Frankia-enriched metagenomes of Frankia cluster-2 strains or strain assemblages were sequenced based on seven inoculum sources. Sequences obtained via DNA isolated from whole nodules were compared with those of DNA isolated from fractionated preparations enhanced in the Frankia symbiotic structures. The results show that cluster-2 inocula represent groups of strains, and that strains not represented in symbiotic structures, that is, unable to perform symbiotic nitrogen fixation, may still be able to colonize nodules. Transposase gene abundance was compared in the different Frankia-enriched metagenomes with the result that North American strains contain more transposase genes than Eurasian strains. An analysis of the evolution and distribution of the host plants indicated that bursts of transposition may have coincided with niche competition with other cluster-2 Frankia strains. The first genome of an inoculum from the Southern Hemisphere, obtained from nodules of Coriaria papuana in Papua New Guinea, represents a novel species, postulated as Candidatus Frankia meridionalis. All Frankia-enriched metagenomes obtained in this study contained homologs of the canonical nod genes nodABC; the North American genomes also contained the sulfotransferase gene nodH, while the genome from the Southern Hemisphere only contained nodC and a truncated copy of nodB.
Keywords: Frankia; metagenomes; transposases; genome instability; Nod factors, nodU
Introduction
Actinorhizal plants, a diverse group of dicotyledonous plants from eight families within three different orders, can form nitrogen fixing root nodules that host actinobacteria from the genus Frankia. Phylogenetically, Frankia strains can be grouped in four clusters. Three clusters represent host specificity groups (fig. 1; Pawlowski and Demchenko 2012). Cluster-1 strains nodulate actinorhizal Fagales, that is, Betulaceae; members of the Casuarinaceae except for Gymnostoma sp.; and members of the Myricaceae except for Morella species. Cluster-3 strains nodulate most actinorhizal members of the Rosales, that is, Elaeagnaceae; Rhamnaceae except for Ceanothus sp.; and Gymnostoma and Morella, two outlier genera of the Fagales. Strains of cluster-2, the phylogenetically basal cluster of Frankia, nodulate all actinorhizal Cucurbitales, that is, taxa within the Datiscaceae and Coriariaceae; and some of the Rosales, that is, the actinorhizal Rosaceae and Ceanothus sp. (Rhamnaceae). The fourth cluster contains noninfective or noneffective strains (Normand et al. 1996; Pozzi et al. 2018).
Fig. 1.

—Core genome tree of sequenced Frankia strains from clusters-1, -3, and -4 and of all cluster-2 (meta-)genomes available thus far (status: March 2019). The tree was calculated by means of EDGAR, deduced from concatenated core gene alignments using the neighbor-joining algorithm as implemented in the PHYLIP package PHYLIP (Felsenstein 1989). Bootstrap values were 100 for every branch (Blom et al. 2009). The scale bar denotes 0.01 substitutions. The host plant orders (red for Cucurbitales, blue for Rosales) and geographic origins (black for Eurasia, green for North America, purple for the Southern hemisphere) of the original cluster-2 inocula are color-coded. Outgroups were two actinobacterial genomes, Acidothermus cellulolyticus 11B (Barabote et al. 2009) and Jatrophihabitans endophyticus DSM45617 (GenBank accession nr. FQVU00000000.1). References for the published Frankia genomes are Normand et al. (2007) for ACN14a, CcI3, and EAN1pec, Sen et al. (2013) for QA3, Swanson et al. (2015) for ACN1ag, Nouioui et al. (2019) for CpI1-S, Normand et al. (2018) for ARgP5, Mansour et al. (2014) for CcI6, Oshone et al. (2016) for Allo2, Ghodhbane-Gtari et al. (2014) for BMG5.23, Hurst et al. (2014) for Thr, Ngom et al. (2016) for CeD, Wall et al. (2013) for BCU110501, Nouioui et al. (2013) for BMG5.12, Ktari et al. (2017) for NRRLB-16219, Swanson et al. (2017) for Cc1.17, Pujic et al. (2015) for R43, Ghodhbane-Gtari et al. (2013) for CN3, Tisa et al. (2015) for DC12, Nouioui, Ghodhbane-Gtari, Del Carmen Montero-Calasanz, et al. (2017) for EU1c, Nouioui, Gueddou, et al. (2017) for NRRL B-16386, Gtari et al. (2015) for BMG5.1, Gueddou et al. (2019) for BMG5.30; Persson et al. (2011) for Dg1_Dg_vc and Nguyen et al. (2016) for Dg2_Dg_vc.
Frankia strains grow as a mycelium. In contrast with rhizobia, the nodule microsymbionts of legumes, Frankia strains can fix nitrogen ex planta under aerobic conditions. This is achieved by forming specialized cells, vesicles. The vesicle envelopes restrict oxygen access, thereby allowing nitrogenase function (Meesters et al. 1987; Parsons et al. 1987). In culture, vesicles are spherical to ovoid and septate, while in planta, their shape and subcellular localization is determined by the host (Newcomb and Wood 1987; Huss-Danell 1997). The fact that Frankia does not depend on the host in order to protect nitrogenase from oxygen is reflected in the diversity of oxygen protection systems present in different actinorhizal systems (Pawlowski and Demchenko 2012).
Since based on the latest phylogenetic studies, it represents the earliest branching cluster of Frankia strains, cluster-2 is of particular interest for the evolution of actinorhizal symbioses (Sen et al. 2014; Gtari et al. 2015; Persson et al. 2015; Nguyen et al. 2016; Pozzi et al. 2018). In this context, it was striking that the first cluster-2 strain to be sequenced—based on DNA isolated from symbiotic structures, namely vesicle clusters isolated from nodules—Candidatus Frankia datiscae Dg1, contained homologs of the canonical nod genes nodABC that encode the three enzymes responsible for synthesizing the common part of rhizobial signal factors, lipochitooligosaccharide (LCO) Nod factors, and these nod genes were expressed in nodules (Persson et al. 2015). Cluster-2 strains were considered unculturable until recently Gtari et al. (2015) published the isolation of an alkaliphilic strain from nodules of Coriaria myrtifolia, Frankia sp. BMG5.1, that fulfilled Koch’s postulate. The genome of this strain did not contain homologs of the canonical nod genes. However, in the next cluster-2 genome to be published, Dg2, a metagenome consisting of two major and one minor strain, both major strains contained the canonical nod genes and also a gene encoding the Nod factor sulfotransferase nodH, and also here, the nod genes were expressed in symbiosis (Nguyen et al. 2016). The configuration of the nod operons made clear that Dg1 and Dg2 had a common ancestor. It should be pointed out that both the Dg1 and the Dg2 genomes were isolated from nodules of the Californian species Datisca glomerata. However, Dg1 originated from Coriaria nepalensis growing in Pakistan and had been propagated in nodules of D. glomerata for more than a decade, while Dg2 originated from native Ceanothus velutinus nodules in California, and had undergone one round of propagation in nodules of greenhouse-grown plants.
These findings raise several questions. First, the fact that Dg2 represents a metagenome while Dg1 represents a single Frankia strain provides further evidence that in the field, cluster-2 strains exist as assemblages and that different member strains dominate in nodules of different host plants, as indicated in Frankia strain marker analyses in host and nonhost rhizosphere soils (Battenberg et al. 2017). The Dg1 inoculum comes from an area where both C. nepalensis and Datisca cannabina are endemic (Mirza et al. 1994; Persson et al. 2015), so it is possible that only one of its member strains was well suited for the host species D. glomerata. On the other hand, cluster-2 strains might occur in the field as single strains or assemblages. Second, if an assemblage of cluster-2 strains infects a host species, is the identity and contribution of the dominant strains dependent on the assemblage, as suggested by Battenberg et al. (2017), or on the host species? Can all strains that can enter the host plant differentiate nitrogen-fixing vesicles, or is vesicle formation restricted to the strain(s) best suited for the host plant species? And since the infection mechanism giving rise to root nodules in the Cucurbitales (Datiscaceae, Coriariaceae) is unknown, the question has to be asked whether individual nodules contain (an) individual strains, or whether the assemblage is more or less equally distributed over all nodules.
The third question concerns the role of the canonical nod genes and nodH. Like rhizobial Nod factors, Frankia signals are transduced in the host via the common symbiotic signaling pathway (CSSP), which was recruited from arbuscular mycorrhizal symbioses (Gherbi et al. 2008; Markmann et al. 2008). This pathway is commonly initiated by the binding of rhizobial LCOs to LysM receptor kinase dimers; a LysM receptor then interacts with the first component of the CSSP, SymRK (Ried et al. 2014). In view of this fact, it seemed likely that the cluster-2 Frankia nod genes are responsible for production of a rhizobial LCO Nod factor equivalent in Frankia. This is supported by the fact that Frankia nod genes are expressed in nodules of D. glomerata and of Ceanothus thyrsiflorus (Persson et al. 2015; Nguyen et al. 2016). However, genomes of the cultured cluster-2 strains BMG5.1 and BMG5.30 and of cluster-1 or cluster-3 Frankia strains, which should, and in case of Frankia casuarinae CcI3 have been shown to signal via the CSSP (Gherbi et al. 2008), do not contain the canonical nod genes (Normand et al. 2007; Gtari et al. 2015; Gueddou et al. 2019), with one exception that is likely due to lateral gene transfer (Ktari et al. 2017). Furthermore, their signal factors do not share the chemical characteristics of LCOs (Cérémonie et al. 1999; Chabaud et al. 2016). Thus, it is clear that an LCO-independent signal transduction pathway exists in the host plants of cluster-1 and cluster-3 strains and for cluster-2 hosts, at least in the Coriaria species that can be nodulated by BMG5.1. This pathway could be identical with the CSSP and would just require signal factor receptors with a different substrate specificity, or the direct interaction of the bacterial signal factor with SymRK. Nevertheless, the finding of conserved nodABC genes in strains from Pakistan and California indicated a function that was maintained under selection pressure.
In order to answer these questions, we obtained cluster-2 inocula from different places all over the world to sequence the corresponding Frankia-enriched metagenomes (referred to as “(meta-)genomes” in the rest of this article for simplicity, since in the majority of cases 50–70% of the sequences came from one strain). These places were Japan, where Coriaria japonica is the only endemic Frankia cluster-2 host plant; Alaska, where this role is fulfilled by Dryas drummondii (Rosaceae); France, where currently C.myrtifolia is the only host plant species, but where in the 19th century D. cannabina was grown to provide a dye for silk (Stenhouse 1856); and Papua New Guinea, where Coriaria papuana is the only host plant species. Nodules of Cea. thyrsiflorus harvested in California were included in the analysis to provide a second (meta-)genome from California which represents an area, where several cluster-2 host plants (D. glomerata, Ceanothus sp., Purshia sp., Cercocarpus sp., Chamaebatia sp.) are endemic; and to see whether the new (meta-)genome would also belong to the species Candidatus Frankia californiensis (Normand et al. 2017). For two of these inocula, genomes were sequenced using nodules from two different host plant species each.
Materials and Methods
Obtaining Field Samples
The inoculum sources are summarized in supplementary table S1, Supplementary Material online. The Cv1 inoculum originates in Sagehen Experimental Forest (near Truckee, CA) from soil and nodules collected from Cea.velutinus plants. The Cj1 inoculum originates from nodules harvested from C.japonica plants growing under pine trees in a coastal area at Tokai (Ibaraki Prefecture, Japan). A voucher sample has been deposited in the herbarium of the Swedish Museum of Natural History (S), leg. K. Pawlowski s.n. (S; Reg. No. S18-27870 (S)). The Cm1 inoculum originated from nodules harvested from a C.myrtifolia plant in the outskirts of Montpellier (France). Nodules from C.papuana Warb. harvested at Pengar River at Mt. Wilhelm, Chimbu Province, Papua New Guinea, formed the original Cppng1 inoculum. A voucher sample of the plant has been deposited in the Botanical Collection of National Herbarium Papua New Guinea Forest Research Institute, number LAE 90743. The Dd1 inoculum originates from Dryas drummondii nodules in the Matanuska River floodplain in Alaska (a plant voucher sample, collection number MLC2015-005, has been deposited in the herbarium of the University of Alaska at Anchorage).
Propagation of Inocula
The isolation of vesicle clusters required 5–10 g of fresh nodules, while the isolation of total DNA from nodules required 150 mg of young nodule material (fresh or frozen). Therefore, the field samples had to be propagated before they could be used for DNA isolation. The Cv1 inoculum was propagated using Cea.thyrsiflorus plants that were purchased from a nursery, Corn Flower Farms (CA) in July 2012 as cuttings. For successful nodulation, the plants were repotted into new media (UC mix: perlite = 1:1) to remove any fertilizer added by the nursery. Nodulation status of each plant was checked at this point to ensure that no plants were nodulated prior to any further treatment. After 1 week, the plants were inoculated with the inoculum from Sagehen Experimental Forest. Since then the plants were maintained in a greenhouse with only deionized water and ¼-strength Hoagland solution without nitrogen (Hoagland and Arnon 1938). No exogenous nitrogen was given to the plants. The plants were kept under normal daylight except during winter when they were kept under extended artificial daylight. Nodules were harvested 8 months after inoculation. The nonlignified tips of lobes were cut off, frozen in liquid nitrogen, and kept at −80 °C until DNA isolation.
The Cj1, Cm1, and Dd1 inocula were propagated using D.glomerata plants; origin and conditions of cultivation and infection were as described by Nguyen et al. (2016). Cppng1 was propagated using Coriaria arborea plants. Seeds collected at Manganui o te Ao River near Raetihi central North Island NZ GR 39 19 04.41 S 175 13 34.95 E, elevation 305 m, were germinated for 3 weeks at 4 °C on sand wetted with tap water before transfer to the greenhouse. Further cultivation and infection conditions were the same as for D. glomerata. Nodules were collected 3 months after infection. For propagation of the inoculum in C. myrtifolia nodules, nodules of D. glomerata induced by the Cm1 inoculum were used to infect C. myrtifolia plants. Seeds of C. myrtifolia collected in Jijel (Algeria) were vernalized on wetted sand at 7 °C for 1 week before transfer to the greenhouse. Infection took place when the plants were ∼5 cm high. Further cultivation conditions were the same as for D. glomerata. Nodules were collected 8 months after infection.
A C.nepalensis plant was obtained from Crug Farm Plants (Caernarfon, United Kingdom). Cuttings were rooted in water, transferred to a soil/sand mixture, and infected with the Dg1 inoculum as described by Nguyen et al. (2016). Nodules were harvested 16 weeks after infection. Seeds of Coriaria terminalis var. xanthocarpa were obtained from www.plant-world-seeds.com, last accessed October 2, 2017 and germinated on a soil/sand mixture. Nodulation was performed as described for C. myrtifolia.
Isolation of Bacterial Genomic DNA
Whole nodule gDNA was isolated from 400 mg of fresh root nodules using the GenElute Bacterial Genomic DNA Kit (Sigma–Aldrich, Stockholm, Sweden). The bacterial hyphae and vesicles were broken using the ultrasonic homogenizer Sonoplus HD 2070 (Bandelin Electronic, Berlin, Germany) at 30% pulsing for three times with 25 s each time.
Purification of Frankia vesicle clusters, and isolation of gDNA from them, was performed as described in Nguyen et al. (2016).
Sequencing
Genomic sequencing libraries were constructed from 1 ng of gDNA with the Nextera XT DNA Sample Preparation Kit (Illumina) according to the manufacturer’s protocol. The libraries were quality controlled by analysis on an Agilent 2000 Bioanalyzer with Agilent High Sensitivity DNA Kit (Agilent Technologies) for fragment sizes of ∼200–500 bp. Sequencing on a MiSeq sequencer (Illumina; 2×250 bp paired-end sequencing, v3 chemistry) was performed in the Genomics Service Unit (LMU Biocenter, Martinsried, Germany). Raw reads were trimmed for quality (>Q20) and adapter sequences.
In order to avoid problems with GC-rich regions, for the genome sequences of Cppng1_Ca_nod and Dd1_Dg_vc whole-genome-shotgun PCR-free libraries (Nextera DNA Sample Prep Kit; Illumina, Munich, Germany) were generated based on the manufacturer’s protocol and sequenced on the MiSeq platform at the Center for Biotechnology (CeBiTec, Bielefeld University, Bielefeld, Germany).
Genome Reconstruction and Comparative Genome Analyses
After sequencing and processing of the Frankia data sets, de novo assemblies were performed using the gsAssembler 2.8 (Roche) with default settings. In a next step, all raw reads were aligned to the corresponding assembled (meta-)genome contigs using Bowtie 2 (v2.2.4; Langmead and Salzberg 2012). By means of SAMtools (v1.0; Li et al. 2009), the SAM file was converted to BAM, the alignment file was sorted, and read mapping statistics were calculated. To divide the (meta-)genome contigs into genome bins, MetaBAT (v0.21.3; Kang et al. 2015) was applied with default settings. Resulting bins that represented the Frankia genomes were used as reference to reconstruct the corresponding Frankia genome. Raw reads were exported by means of mapping to the bins and reassembled using again the gsAssembler 2.8 (Roche) with default settings. Completeness, contamination, and strain heterogeneity were estimated with BUSCO (v2.0; Simão et al. 2015), using the bacterial-specific single-copy marker genes database (odb9). Data were plotted by BUSCO plot (v2). For Cj1_Dg_nod and Cm1_Dg_nod, to estimate the relationship between the detected strains and the corresponding vc-strains, all binned contigs of were compared with the final draft genome of their vc-variants by applying BLASTN (threshold >1×10−20; Altschul et al. 1997).
Read mapping and SNP calling were performed as recently described (Rupp et al. 2015). Briefly, reads were mapped to the final draft genome sequences of Dg1_Cn_nod and Dg1_Dg_nod with Bowtie2 (Langmead and Salzberg 2012). The Genome Analysis Toolkit (GATK) IndelRealigner algorithm (McKenna et al. 2010) was applied for indel realignment, whereas SNPs were called using the GATK HaplotypeCaller algorithm (McKenna et al. 2010). Identified SNPs were exemplarily checked with ReadXplorer 2.2.3. (Hilker et al. 2014, 2016).
For the annotation of the genomes, Prokka (Seemann 2014) and GenDB (Meyer et al. 2003) were applied. Draft genome sequences were deposited at the EMBL/GenBank/DDBJ databases in BioProjects PRJEB19438–49 (for details, see supplementary table S2, Supplementary Material online).
Completeness of the reconstructed draft genomes and bins were estimated by calculating the content of bacterial BUSCOs (e-value: 0.001, data set v.3.0.2) (Waterhouse et al. 2018). The results are shown in supplementary table S3, Supplementary Material online.
The reconstructed and annotated Frankia (meta-)genomes were used for comparative genome analyses. Comparative analyses between the different available Frankia (meta-)genomes were accomplished using the comparative genomics program EDGAR 2.0 (Blom et al. 2009, 2016). Comparative analyses comprised identification of orthologous genes and classification of genes as core genes or singletons as well as the creation of phylogenetic tree based on the core genome.
Localization of DNA in Ceanothus thyrsiflorus Nodules
Nodules of Cea. thyrsiflorus were harvested 8 months after infection with Cv1. Several nonlignified tips of nodule lobes were cut off and fixed in 3% paraformaldehyde, 0.1% Tween-20, 0.1% Triton X-100 in 10 mM phosphate buffer pH 7.2 overnight before being washed and dehydrated in a graded EtOH series until 70% EtOH and stored at room temperature. Later, they were rehydrated in a graded EtOH series and embedded in 2% agarose (SeaKem LE agarose, Cambrex, Karlskoga, Sweden). Longitudinal sections (45 µm) were prepared on an vibrating blade microtome HM 650 V (Microm, Walldorf, Germany), stained with 0.001% 4′,6-diamidino-2-phenylindole (DAPI) for 30 min and analyzed under a confocal laser scanning microscope LSM 780 (Carl Zeiss, Jena, Germany).
Identifying Transposases and Inverted Repeats
To identify transposases in the Frankia genomes, the draft genomes (see table 1) were subjected to multiple BLASTX searches (Ye et al. 2006) against a database of transposase amino acid sequences. Each (meta-)genome was searched against 5,180 transposase ORF amino acid sequences, mostly from the ISfinder web site (Siguier et al. 2006) as of May 2015.
Table 1.
List of (Meta-)Genomes from Nodules Induced by Frankia Cluster-2 Inocula
| Inoculum | Plant Species of Origin | Plant Species for Propagation of Inoculum | Isolated from Whole Nodules (nod) or Vesicle Clusters (vc) | Metagenome Name | Number of Strains | Number of Major Strains | Ratio of Strain Contributions | Genome Size (Mb) | % GC | Sequence Similarity between Major Strains (16S) | nod Genes |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cj1 | Coriaria japonica | Datisca glomerata a | nod | Cj1_Dg_nod | 2 | 2 | 59/41 | 8.526 | 68.24 | 86–92% | nodABCnltIJ |
| Datisca glomerata | vc | Cj1_Dg_vc | 1 | 1 | n.a. | 5.044 | 69.94 | n.a. | nodABCnltIJ | ||
| Cm1 | Coriaria myrtifolia | Coriaria myrtifolia | nod | Cm1_Cm_nod | 1 | 1 | n.a. | 4.953 | 70.22 | n.a. | nodABCnltIJ |
| Datisca glomerata a | nod | Cm1_Dg_nod | 2 | 2 | 72/28 | 9.779 | 69.28 | 86–92% | nodABCnltIJ | ||
| Datisca glomerata | vc | Cm1_Dg_vc | 1 | 1 | n.a. | 5.069 | 69.82 | n.a. | nodABCnltIJ | ||
| Cv1 | Ceanothus velutinus | Ceanothus thyrsiflorus b | nod | Cv1_Ct_nod | 2 | 1 | 93/7 | 5.499 | 68.07 | n.a. | nodABCHnltIJ |
| Cppng1 | Coriaria papuana | Coriaria arborea | nod | Crpng1_Ca_nod | 1 | 1 | n.a. | 5.046 | 67.50 | n.a. | nodB2'CnltIJU |
| Dd1 | Dryas drummondii | Datisca glomerata | nod | Dd1_Dg_nod | 1 | 1 | n.a. | 5.435 | 67.80 | n.a. | nodABCHnltIJ |
| Datisca glomerata | vc | Dd1_Dg_vc | 1 | 1 | n.a. | 5.573 | 67.97 | n.a. | nodABCHnltIJ | ||
| Dg1 | Coriaria nepalensis | Datisca glomerata c | vc | Dg1_Dg_vc | 1 | 1 | n.a. | 5.323 | 70.04 | n.a. | nodABCnltIJ |
| Datisca glomerata | nod | Dg1_Dg_nod1 | 1 | 1 | n.a. | 4.888 | 70.14 | n.a. | nodABCnltIJ | ||
| Datisca glomerata d | nod | Dg1_Dg_nod2 | >1 | n.d. | n.d. | 5.548 | 69.32 | n.d. | nodABCnltIJ | ||
| Coriaria nepalensis d | nod | Dg1_Cn_nod | >1 | n.d. | n.d. | 5.191 | 69.55 | n.d. | nodABCnltIJ | ||
| Dg2 | Datisca glomerata | Datisca glomerata e | vc | Dg2_Dg_vc | 3 | 2 | 60/40/1 | 5.929 | 67.90 | 99% | nodABCHnltIJ |
For numbers and relatedness of strains in Cj1_Dg_nod and Cm1_Dg_nod, see supplementary figure S2, Supplementary Material online.
Relatedness of strains in Cv1_Ct_nod was not quantified.
Maintained in D. glomerata over a period of 10 years before isolation of vesicle clusters for sequencing; Persson et al. (2015).
Dg1_Dg_nod2 and Dg1_Cn_nod contain several very similar strains (supplementary fig. S3, Supplementary Material online) the exact relatedness of which could not be quantified.
Nguyen et al. (2016).
n.a., not applicable; n.d., not determined.
To parse and analyze search results with accuracy and repeatability, a multistep process was employed: First, all areas of a (meta-)genome that contained one or more transposase hits (e-value cutoff 10−4) were designated as “footprints.” Then, the footprints were subjected to BLASTX searches (e-value cutoff 10−4) to identify the transposase occupying it. In the case of several BLASTX hits, the transposase hit with the highest score was chosen. If this hit did not cover the entire footprint sequence, the remainders were searched until the footprint had transposases designated to its entire sequence. Then, since transposases were occasionally identified as several fragments, an in-house script was used to join these fragments into single units. The criteria for joining two fragments were 1) that the fragments should be within 1 kb of each other; 2) that the fragments should be hits to transposases on the same strand; and 3) that the fragments should be hits to consecutive parts (±50 amino acids) of transposases.
A fraction of transposase value was computed for each transposase hit to evaluate its completeness. This value was calculated as the number of nucleotides covered by the transposase hit divided by the full nucleotide length of that transposase. Joined hits were considered as one transposase for these purposes, and the fraction of transposase value was computed as the sum of their coverages divided by the shortest of the originating transposases. For statistical analysis, Wilcoxson rank sum test was performed with the wilcox.test function of the R program for statistical computing (R Core Team 2019).
Scripts and data used, as well as GenBank files with annotated transposase hits for the investigated Frankia (meta-)genomes, are available in supplementary file S1, Supplementary Material online.
Inverted repeats were identified with the “Find repeats” function in Unipro UGENE v. 1.18.0 using the following settings: Window size = 12, Minimum identity per window = 92%, min and max distance between repeats = 0, 11,000 bp and the “Search for inverted repeats” option checked (Okonechnikov et al. 2012).
Nucleotide Alignments
Nucleotide alignments were performed in Geneious 7.1.7 (Kearse et al. 2012) using the Geneious algorithm, “global alignment with free end gaps,” 65% similarity cost matrix, open penalty 12, extension penalty 3, automatic sequence direction.
Protein Phylogeny
To investigate the evolutionary history of Frankia sp. Cppng1 putative nodU gene (cmcH), a phylogenetic analysis was conducted in combination with three sets of sequences. First, known NodU sequences of 11 rhizobial genera were collected from GenBank. Up to three sequences were collected from each genus from different species or strains. Species with their genome sequences available in Integrated Microbial Genomes (IMG) (Markowitz et al. 2014) were preferred over species without. Next, for each species with their genome available in IMG, by using its NodU sequence as a query for BLASTP (Altschul et al. 1990) against its genome, all NodU-like (but not NodU) carbamoyl transferases with similarity score better than 1e−20 were collected. Finally, by using the putative NodU sequence from Frankia sp. Cppng1 as a query for BLASTP against NCBI nonredundant database, carbamoyl transferases highly similar to Frankia sp. Cppng1 putative NodU (1e−150 or better, 45% amino acid identity or better) were collected from a phylogenetically diverse group of Actinobacteria (Sen et al. 2014).
These sequences were first aligned using MAFFT v7.272 (Katoh and Standley 2013) with accuracy-oriented alignment parameters (–localpair –retree 2 –maxiterate 1,000). Then the best substitution model for this multiple sequence alignment was calculated using ProtTest3 (Darriba et al. 2011). PROTTEST3 predicted the best substitution model to be LG with invariable sites, with gamma distribution, and with empirical base frequencies (LG+I + G+F) based on Bayesian information criterion (BIC), Akaike information criterion (AIC), and corrected Akaike information criterion (cAIC). These model parameters were transferred to RAxML v8.2.8 which was used to reconstruct the phylogeny based on maximum likelihood (Stamatakis 2014). Four parallel runs were conducted and only the best one was kept. 100 bootstrap replicates were conducted.
RNA Isolation, DNase Digestion, and RT-qPCR Analyses
Nodules were ground in liquid nitrogen with mortar and pestle. The Frankia vesicles were broken using the ultrasonic homogenizer Sonoplus HD 2070 (Bandelin Electronic, Berlin, Germany) at 90% amplitude and 30% pulsing three times for 25 s each. RNA samples were isolated according to the protocol of the Spectrum Plant Total RNA kit from Sigma–Aldrich (Stockholm, Sweden) with on-column gDNA digestion by the RNase-Free DNase Set (Qiagen, Minden, Germany).
Three biological samples of 100 mg each were analyzed for each type of nodule (100 mg were represented by one to three nodules). The integrity of RNA samples were analyzed by the Agilent 2100 Bioanalyzer system (Agilent Technologies). All RNA samples that were chosen for further analyses had RNA Integrity Number (RIN) values >8.5. Reverse transcription were performed using the SuperScript IV First Strand Synthesis System (Thermo Fisher Scientific). For each gene, primers were designed based on the conserved regions of sequences from the Dg1 genome and the metagenome Dg2, except for nodU, which was based on the Cppng1 metagenome sequence, using Gemi (Sobhy and Colson 2012). Each qPCR reaction contained 1× Maxima SYBR Green qPCR Master Mix (ThermoFisher Scientific), 300 nM of each primer, and 4 ng of cDNA in a reaction volume of 10 µl. The conditions of qPCR was as followed: after 10 min at 95 °C were 40 cycles of 15 s at 95 °C, 30 s at 60 °C, and 30 s at 72 °C, followed by melt curve program (15 s at 95 °C, 15 s at 60 °C, and 15 s at 95 °C). Gene expression values were normalized against infC, the gene encoding translation initiation factor IF3. Data preprocessing and normalization were performed using GenEX (MultiD Analyses, Sweden). Primer sequences are given in supplementary table S4, Supplementary Material online.
Results and Discussion
Sequencing of 12 Different (Meta-)Genomes Based on Five Different Inocula
The inocula from nodules of C.japonica, Dryas drummondii, and C.myrtifolia were used to nodulate D.glomerata and (meta-)genomes were sequenced from these D. glomerata nodules. They were sequenced in two approaches: first, from vesicle clusters isolated from nodules and second, by direct sequencing of total DNA isolated from nodules and bioinformatic removal of non-Frankia sequences. Some of the D. glomerata nodules were used to infect other host plant species, and the resulting nodules were used for direct genome sequencing. All (meta-)genome sequences are described in table 1. To distinguish between the different (meta-)genome sequences derived from one inoculum, the following nomenclature was developed: name of inoculum, followed by the initials of the host plant species from which the metagenome was isolated, followed by either “vc” for (meta-)genomes obtained from DNA from isolated vesicle clusters or “nod” for (meta-)genomes obtained by direct sequencing of DNA isolated from whole nodules. Thus, for example, the inoculum Cm1 from C. myrtifolia nodules collected in Montpellier (France) gave rise to three different (meta-)genomes, Cm1_Dg_vc, Cm1_Dg_nod, and Cm1_Cm_nod. The previously published genome sequence Dg1 (Persson et al. 2015) becomes Dg1_Dg_vc while Dg2 (Nguyen et al. 2016) becomes Dg2_Dg_vc. Cv1_Ct_nod was directly sequenced from Cea.thyrsiflorus nodules harvested in California. Three _nod versions were sequenced of the previously published Candidatus Frankia datisca Dg1 inoculum (Persson et al. 2011, 2015), Dg1_Dg_nod1 and Dg1_Dg_nod2 from nodules of D. glomerata and Dg1_Cn_nod from nodules of C.nepalensis induced by Dg1.
Datisca glomerata was used for propagation of the different inocula because for this species, the procedure for isolating vesicle clusters from nodules was well established (Persson et al. 2015; Nguyen et al. 2016). Attempts to obtain Frankia genomic DNA from nodules of Cea. thyrsiflorus had been successful when nodules were used as starting material. However, it was never possible to obtain significant amounts of DNA isolated from vesicle clusters isolated from Cea. thyrsiflorus nodules. This could not be due to the absence of DNA from vesicles in Cea. thyrsiflorus nodules since they showed strong staining with DAPI (supplementary fig. S1, Supplementary Material online). For the only inoculum from the Southern Hemisphere, Cppng1, C.arborea had to be used for propagation since the inoculum did not nodulate either D. glomerata or D. cannabina. The features of all (meta-)genomes obtained in the course of this study, together with the already published (meta-)genomes of Frankia cluster-2 strains or strain assemblages, are summarized in table 1.
Thus, in this study, altogether 14 (meta-)genomes were compared that originated in seven different Frankia cluster-2 inocula from four continents (table 1 and supplementary table S1, Supplementary Material online). Five of these (meta-)genomes were sequenced from DNA isolated from symbiotic structures, vesicle clusters (Dg1_Dg_vc, Dg2_Dg_vc, Dd1_Dg_vc, Cm1_Dg_vc, Cj1_Dg_vc; Persson et al. 2015; Nguyen et al. 2016; this study) while the other nine were sequenced based on DNA isolated from whole nodules and thus could include strains that were not able to form vesicles. Two different DNA isolations from Dg1-induced nodules were used for sequencing; one of them (Dg1_Dg_nod1) represented one strain and the other one (Dg1_Dg_nod2) represented three strains (table 1). In short, a single inoculum can lead to considerable variety in nodule occupancy.
Based on the fully assembled chromosome of Dg1 (Persson et al. 2015) and the genomes of Frankia coriariae BMG5.1 (Gtari et al. 2015; Nouioui, Ghodhbane-Gtari, Rohde, et al. 2017) and the (meta-)genomes containing one dominant strain (table 1), the average genome size of a cluster-2 strain is 5.2 ± 0.3 Mb. The metagenomes Cm1_Dg_nod and Cj1_Dg_nod consist of two strains each, and the genomes of these two strains are rather diverse (86–92% DNA sequence identity; supplementary fig. S2, Supplementary Material online). This was also reflected by the apparent genome size. If the major strains forming a metagenome were rather similar (99% identity of 16S rDNAs for the two dominant members of Dg2_Dg_vc; Nguyen et al. 2016), most sequence differences would appear as SNPs in the assembled metagenome, and the apparent genome size (5.9 Mb for Dg2_Dg_vc) would not be much larger than the average of metagenomes with only one dominant strain. If, however, the major strains showed strong sequence differences as in the cases of Cm1_Dg_nod and Cj1_Dg_nod, most genome regions would be assembled independently for both strains, leading to an apparent genome size of 8.5–9.8 Mb, that is, nearly double the size of metagenomes with only one dominant strain. However, even in those cases it was not possible to separate the genomes via binning, because only few contigs were strain-specific; in most cases, the divergent regions were interspersed with conserved ones.
A Phylogenetic Tree Based on the Entire Core Genome Shows That (Meta-)Genomes Coming from the Same Inoculum Can Display Strong Differences
All Frankia cluster-2 (meta-)genomes listed in table 1 and several published genomes from Frankia clusters-1, -3, and -4 were used to reconstruct a phylogenetic tree, build out of a core of 348 genes per genome, using EDGAR 2.0 (Blom et al. 2009, 2016) which is depicted in figure 1. In this tree, Cppng1_Ca_nod is sister to all other Frankia cluster-2 genomes. This placement is consistent with the fact that in previous phylogenies that contained sequences from strains from the Southern Hemisphere, these strains always occupied the basal position in cluster-2 (Benson et al. 1996; Clawson et al. 2004; Nouioui et al. 2014; Nguyen et al. 2016). Furthermore, a clear separation can be seen between the Eurasian and the North American cluster-2 (meta-)genomes.
At least one each of the (meta-)genomes from the inocula Dg1, Dg2 (Nguyen et al. 2016), Cj1, Cm1, and Cv1 represented more than one cluster-2 Frankia strain. The only exception in this regard was the Alaskan inoculum Dd1, and this might simply be due to the fact that the other strain(s) were underrepresented compared with the major strains as was the case for Dg1 (compare Dg1_Dg_vc; Persson et al. 2015, with Dg1_Dg_nod1 and Dg1_Dg_nod2 from this study). Thus, the data obtained in this study imply that cluster-2 Frankia strains often appear in groups, a fact that in combination with our inability to culture most of them may be responsible for their wide apparent host range in that 1) an inoculum can be made up from strains with different host specificity and 2) strains that do not nodulate the host plant used to prepare the inoculum might be carried over because they might grow on the nodule surface. (Meta-)genomes sequenced by Nguyen et al. (2016) and in this study contained maximally three different strains; however Frankia cluster-2 inocula are very likely to contain more than two to three strains since the (meta-)genomes shown here can only encompass strains that actually colonize nodules of a particular host plant. It is important to note that the fact that cluster-2 inocula represent assemblages makes cross-inoculation studies hard to interprete. Not only can different strains be responsible for the nodulation of different host plants but also negative results can be open to doubt as well as it is not clear whether the composition of an assemblage is stable during the propagation in one host plant. Strains able to nodulate other host plants might get lost.
When comparing the different (meta-)genomes derived from the same inoculum, two factors have to be taken into account. First, a _vc genome is based mostly on symbiotic structures, vesicle clusters. Since in planta, vesicle clusters are embedded in a pectin-rich matrix (Liu and Berry 1991), sequences from (a) Frankia strain(s) that do(es) not form vesicles in these nodules could still turn up, but only as minor contaminations. Second, the isolation of vesicle clusters that leads to _vc (meta-)genomes requires up to 10 g of nodule material, while the isolation of total DNA from nodules that leads to _nod (meta-)genomes requires only 400 mg, that is, two to five nodules. Thus, _nod (meta-)genomes will not necessarily be dominated by the strains that can form vesicles—that is, provide the plant with fixed nitrogen—and since they represent only a few nodules, they might be dominated by strains that represent minor members of the inoculum. This is underscored by the two different _nod (meta-)genomes from Dg1_Dg: Dg1_Dg_nod1 represents a single strain while Dg1_Dg_nod2 represents several closely related strains (supplementary fig. S3, Supplementary Material online). Furthermore, a comparison of Cm1_Dg_vc versus Cm1_Dg_nod and of Cj1_Dg_vc versus Cj1_Dg_nod shows that inocula can contain strains that differ dramatically with only 86–92% DNA sequence identity (table 1 and supplementary fig. S2, Supplementary Material online).
The phylogenetic tree (fig. 1) made clear that the strains represented by DNA isolated from vesicle clusters of all Eurasian inocula (Cj1_Dg_vc, Cm1_Dg_vc, Dg1_Dg_vc), as well as the dominant strains in the Eurasian _nod metagenomes (Cm1_Dg_nod and Cj1_Dg_nod; 60–70% of total Frankia DNA from nodules; table 1), showed striking sequence conservation (99–100% sequence identity; supplementary fig. S2, Supplementary Material online). This high similarity among the _vc versions of Eurasian cluster-2 genomes might be ascribed to the fact that they all were propagated in nodules of the same plant species, D.glomerata. It remains to be examined whether the _vc versions of the same inocula would change, when other host plants are used for propagation. At any rate, this result showed that strain assemblages from France, Pakistan, and Japan contained one strain the genome of which was very highly conserved.
A comparison of the different versions of (meta-)genomes from a single inoculum showed that the _nod metagenomes Dd1_Dg_nod, Dg1_Dg_nod1, Dg1_Dg_nod2, and Dg1_Cn_nod represented the strain that dominated the corresponding vesicle cluster-based genomes (Dd1_Dg_vc and Dg1_Dg_vc, respectively), or (a) very similar strain(s). In case of the Dg1 inoculum, this may be due to the fact that the inoculum goes back to C. nepalensis in Pakistan and was propagated in nodules of the Californian species D. glomerata in greenhouses for two decades (Persson et al. 2015) which may have led to the loss of strains from the assemblage.
However, two _nod versions, Cm1_Dg_nod and Cj1_Dg_nod, also contained strains of their respective assemblages that were not strongly represented in the corresponding _vc versions. These two outlier strains present in Cj1_Dg_nod (from Japan) and Cm1_Dg_nod (from France) were quite different from the strain in the respective vesicle clusters (86–92% sequence identity overall; supplementary fig. S2, Supplementary Material online); they were more similar to each other than to the genomes of the strains that dominated the _vc version of their inocula (fig. 1 and supplementary fig. S2, Supplementary Material online).
The fact that a group of a few individual nodules can contain a strain (at 30–40% of total Frankia DNA from nodules) that does not seem to be competitive in forming vesicle clusters in nodules, that is, does not fix nitrogen in nodules, and that two such strains were identified in two attempts out of eight, suggests that not all strains that can colonize D. glomerata nodules can form vesicles in infected cells. In other words, D. glomerata is not very efficient in discriminating against symbiotically inefficient Frankia strains.
Average Nucleotide Identity Comparisons Show That Cppng1_Ca_nod Represents a Novel Species
In order to find out whether the new (meta-)genomes represented novel species of Frankia cluster-2, Average Nucleotide Identity (ANI) comparisons were performed for the 13 genomes of the cluster. The mean ANI values shown in figure 2 show that if the newly calculated ANI threshold range of 98.65% as equivalent of 70% DNA–DNA hybridization were applied (Kim et al. 2014), the inoculum from Alaska (Dd1_Dg_vc and Dd1_Dg_nod) would represent a new species. However, based on the usually applied ANI threshold range of 95–96% for species demarcation set by Goris et al. (2007) and Richter and Rosselló-Móra (2009), all North American inocula available thus far represent members of Candidatus Frankia californiensis (Normand et al. 2017). For the Eurasian strains, strong sequence conservation is found between Candidatus Frankia datiscae Dg1 (Persson et al. 2011) represented by Dg1_Dg_v, Dg1_Dg_nod1, Dg1_Dg_nod2, and Dg1_Cn_nod, as well as Cv1_Dg_vc, Cm1_Dg_vc, and Cm1_Cm_nod. As already seen in the core genome phylogeny (fig. 1), Frankia coriariae BMG5.1 (Gtari et al. 2015; Nouioui, Ghodhbane-Gtari, Rohde, et al. 2017) shows higher similarity with Candidatus Frankia datiscae than the two metagenomes Cm1_Dg_nod and Cv1_Dg_nod.
Fig. 2.

—Mean ANI values between (meta-)genomes of Frankia cluster-2. Comparisons between Eurasian/Mediterranean (meta-)genomes are labeled in green, comparisons between North American metagenomes are labeled in blue. The Southern hemisphere metagenome (Cppng1_Ca_nod) is labeled in pink. SDs are given in brackets.
The genome sequenced from the Southern hemisphere inoculum, Cppng1_Ca_nod, has <86% ANI with all other Frankia cluster-2 (meta-)genomes available. Thus, based on ANI values, the Southern hemisphere strain Cppng1_Ca_nod represents a novel species of cluster-2 Frankia. Therefore, we propose this strain as type strain of a new species, Candidatus Frankia meridionalis (me.riː.di.oːˈnaː.lis. N.L. masc./fem. adj. meridionalis, southern). The species nodulates and fixes nitrogen in the root nodules of Coriaria species growing in Papua New Guinea and New Zealand and is genetically different from all other Cluster-2 Frankia strains sequenced so far. It also could nodulate the Northern hemisphere species C.terminalis (supplementary fig. S4, Supplementary Material online).
Altogether, while the differences between strains of the same inoculum observed in this study were still within the species level, there was significant strain diversity within an inoculum. Nevertheless, the composition of the _vc versions, which represent ∼10 g of nodules each, implies that only (a) particular strain(s) can form vesicle clusters in a particular host plant species. Furthermore, the striking sequence conservation among the _vc version of Eurasian (meta-)genomes, that is, between Cm1_Dg_vc from France, Cj1_Dg_vc from Japan and Dg1_Dg_vc from Pakistan cannot, in contrast with the genetic conservation of Casuarina-infective strains (fig. 1), be ascribed to geographic isolation. This phenomenon might instead be ascribed to the low saprotrophic potential of cluster-2 Frankia. While there is evidence that cluster-2 strains can occasionally occur in the soil in the absence of their host plant species, this seems to be rare (Battenberg et al. 2017).
Frankia Cluster-2 Genome Instability: Transposase Abundance in the Different Species
Although genome stability is vital for the survival of any organism, a certain flexibility is required in order to adapt to a changing environment. Bacterial genomes achieve this flexibility using mobile DNA elements, horizontal gene transfer, and genome rearrangement (Juhas et al. 2009). In particular, insertion sequence (IS) elements confer genomic plasticity (Mugnier et al. 2009; Raeside et al. 2014). Previous analyses had shown that the genome of the cluster-2 strain Dg1 contained more full size IS elements—that is, IS elements capable of transposition—than genomes of representatives of the other Frankia clusters (Persson et al. 2015). Therefore, transposase abundance was examined in all cluster-2 (meta-)genomes. The results are depicted in figure 3.
Fig. 3.
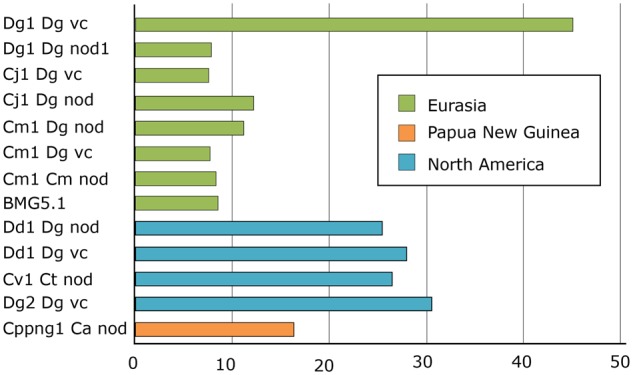
—Transposase abundance. Numbers of transposase ORFs per Mbp (meta-)genome in the cluster-2 Frankia (meta-)genomes are given. The genome of Candidatus Frankia Datiscae Dg1 sequenced from DNA isolated from vesicle clusters (Dg1_Dg_vc) contains at least four times more transposases in its genome than all other Eurasian genomes; this discrepancy is explained by the fact that this genome is the only one in the group which is not in draft stage, but fully assembled. Among the draft genomes, the Eurasian strains contain significantly less transposase ORFs than the North American strains (Wilcoxon’s P = 0.006).
The genome of Candidatus Frankia Datiscae Dg1 sequenced from DNA isolated from vesicle clusters (Dg1_Dg_vc) contains at least four times more transposases in its genome than all other Eurasian genomes; this discrepancy is explained by the fact that this genome is the only one in the group which is not in draft stage, but fully assembled. This result underlines the fact that transposase abundance can be compared between draft genomes or between fully assembled genomes, but not between a mixture of both. Since genome assembly usually condenses all copies of an insertion element into a single contig, fully assembled genomes will contain more repetitive elements than draft genomes. The results for the draft (meta-)genomes obtained in this study, including Dg2 (Nguyen et al. 2016), clearly show that the North American strains, that is, representatives of the species Candidatus Frankia californiensis (Normand et al. 2017), contain significantly more IS elements than the Eurasian strains, that is, the representatives of the species Candidatus Frankia datiscae (Persson et al. 2015) and Frankia coriariae BMG5.1 (Nouioui, Ghodhbane-Gtari, Rohde, et al. 2017). The number of IS elements in the Southern hemisphere metagenome Cppng1_Ca_nod seems to be closer to those of the Eurasian than of the North-American (meta-)genomes, but since so far the Southern hemisphere species is represented by a single strain, conclusions might be premature.
All Sequenced Eurasian Frankia Cluster-2 (Meta-)Genomes except for BMG5.1 Contain the Canonical Nod Genes nodABC, All (Meta-)Genomes from North America Also Contain nodH
The (meta-)genomes of Candidatus Frankia datiscae Dg1 and Candidatus Frankia californiensis Dg2 contained the canonical nod genes nodABC that are responsible for the synthesis of the common backbone of rhizobial symbiotic signal factors, LCOs (Persson et al. 2015; Nguyen et al. 2016). Dg2 additionally contained the sulfotransferase gene nodH. However, no nod genes were found in the genome of the only cluster-2 strain cultured thus far, BMG5.1 (Gtari et al. 2015). As listed in table 1, all but one of the (meta-)genomes sequenced in this study contain the canonical nod genes nodABC. All North American metagenomes also contained nodH. The exception is the Southern hemisphere metagenome, Cppng1_Ca_nod, which contained nodC, a truncated copy of nodB2, and no nodA.
The nodA’B1A Operon (nod1 Region) Is Part of a Transposable Unit That Also Contains a betAdegT Operon
In Dg1, the canonical nod genes were present as two operons in different locations on the chromosome, nodA’B1A (nod1 region) and nodB2CnltIJ (nod2 region; also contained homologs of the nodulation-related ABC transporter genes nodI and nodJ). Synteny analysis showed that in all Eurasian Frankia strains with the exception of BMG5.1 and BMG5.30, the nodA’B1A operon is part of a consensus region (shaded in pink in fig. 4). This region includes transposases on both sites of the betAdegT nodA’B1A operons. Nucleotide identity in this region is 100% for Eurasian (meta-)genomes with three exceptions (fig. 4). In the North American metagenomes, the nod1 region synteny has been affected by more transposition events leading, for example, to the transposition of nodA from the nodA’B1A operon in Dd1_Dg_vc while a truncated copy of nodA was retained in the operon, or simply to the duplication of nodA in Dd1_Dg_nod.
Fig. 4.

—NodA’B1A operons (nod1 region) in different (meta-)genomes. Illustration of the nod1 region of the Frankia cluster-2 (meta-)genomes available. This region contains the nodAB1A'degTbetA genes, enclosed by transposases. In all Eurasian strains, this region shows >99% sequence conservation (shaded in pink). The exceptions are two small insertions involving repeated CTAG tetramers, and a transposition: in Cm1_Dg_nod, a downstream transposase is replaced by a gene of unknown function, referred to as encoding hypothetical protein A. This gene is also found in Cj1_Dg_nod and BMG5.1, always adjacent to the gene padR. In Cppng_Ca_nod and BMG5.1, the nodAB1A'degTbetA region is missing; instead, suggested regions for its excision are shown. The sequence conservation as well as the surrounding transposases and inverted repeats suggests that the nodAB1A'degTbetA region is mobile. Inverted repeats of interest are indicated as double-pointed arrows. A version of this figure with gene names above the ORFs is available in the supplementary fig. S5, Supplementary Material online.
The nodB2CnltIJ Operon (nod2 Region) Is Enclosed by Inverted Repeats and Might Represent a Transposable Unit
The nod2 region sequences are also highly conserved in the Eurasian (meta-)genomes (shaded in pink in fig. 5), except for 18 bp between the nodB2C and nltIJ operons where the sequences differ due to insertions or deletions of the inverted repeat CTAGCTAGCTAG. Again, the sequences of the nod2 regions of the North American strains are less conserved and appear to have acquired multiple point mutations. In Dg2_Dg_vc (Nguyen et al. 2016) as well as in Dd1_Dg_vc and Dd1_Dg_nod, the nod1 and nod2 regions are linked. In these three metagenomes, there is at least one IS elements between the two linked regions.
Fig. 5.

—NodB2CnltIJ operon (nod2 region) in different (meta-)genomes. Illustration of the nod2 region of all cluster-2 Frankia (meta-)genomes available (BMG5.1 is not included here since it does not contain the nod2 region). This part of the genome contains the nodB2CnltIJ genes (shaded in pink). Like the nod1 region, the nod2 region is identical in the Eurasian (meta-)genomes, except for 18 bp between the nod and nlt genes involving repeated CTAG tetramers. In Dg2_Dg_vc, the region is located between two transposases and adjacent to an area rich in inverted repeats which may have been involved in the creation of the nodB1H-transposase-nodA’B region. Inverted repeats of interest are indicated as double-pointed arrows. A version of this figure with gene names above the ORFs is available in the supplementary fig. S5, Supplementary Material online.
The nod2 region is enclosed by inverted repeats (CTAGCTAGCTAG; fig. 5). No obvious mobile genetic element can be found upstream of the nodB2CnltIJ region of the Eurasian (meta-)genomes, but several strains have transposases immediately downstream of the region.
The Cppng1_Ca_nod Metagenome Contains an Operon with the nodB2’CnltIJ Genes and a Homolog of the Carbamoyl Transferase Encoding Gene nodU
The metagenome originating from C. papuana nodules collected in Papua New Guinea did not contain an nodA’B1A operon (nod1 region), but it did contain an operon containing a truncated copy nodB2 and complete copies of nodC and nltIJ (nod2 region; fig. 5). Interestingly, two ORFs were present between nodB2 and nodC, CPPNG1CANOD_5034 and CPPNG1CANOD_5033. The proteins encoded by these two ORFs had no homologs with >5e−26 with proteins encoded by any other of the cluster-2 (meta-)genomes known thus far, or with proteins encoded by any other publicly available Frankia genome. Nevertheless, they represent members of actinobacterial protein families with close homologs present in several other actinobacterial genomes (closest homolog with 5e−178 for CPPNG1CANOD_5034 and with 1e−66 for CPPNG1CANOD_5033). The closest homolog of the 259 amino acid transmembrane protein Cppng1_Ca_nod_5034 was PBC69544 from Streptomyces sp. CF124 (JGI) which was isolated from the Populus root microbiome. The closest homolog of the 636 amino acid cytosolic protein Cppng1_Ca_nod_5033 was WP_020524966 from Catelliglobosispora koreensis (JGI) which was isolated from a gold mine cave (Ara et al. 2008). NodC is followed by nltIJ which is followed by a gene predicted to encode a carbamoyl transferase.
This carbamoyl transferase has the highest homology (starting with E = 0.0) with carbamoyl transferases from the secondary metabolism of actinobacteria, hydroxymethyl cephem carbamoyltransferases (cmcH; Aharonowitz et al. 1992). Among its homologs are also the rhizobial NodU proteins, carbamoyltransferases involved in the chemical decoration of Nod factors. Therefore, the phylogeny of actinobacterial hydroxymethyl cephem carbamoyltransferases and alpha-proteobacterial carbamoyl transferases was examined (fig. 6).
Fig. 6.

—Carbamoyl transferase phylogeny. Rhizobial strain designations are given in red, actinobacterial strain designations in blue. The sequence from Cppng1_Ca_nod is highlighted in gray. The phylogram shows that rhizobial NodU proteins form a distinct clade, while the non-NodU carbamoyltransferases group in two clades, one of which also includes all actinobacterial sequences examined. *denotes nodes with >90 bootstrap support; **denotes nodes with 100 bootstrap support.
The phylogenetic tree (fig. 6) consisted of three major clades each with high bootstrap support. One of the three clades included all known rhizobial NodU proteins with one rhizobial protein that had not explicitly been described as NodU. All actinobacterial sequences including the Candidatus Frankia meridionalis Cppng1 carbamoyl transferase belonged to another clade, which was the only one of the three clades that included both rhizobial and actinobacterial sequences. Thus, the Cppng1 gene does not have the same origin as rhizobial nodU genes. However, based on its linkage to nodB2’C, we are calling the Cppng1 gene “nodU-like” (nltU) in this article.
Expression of Nod Genes in Symbiosis
Previous studies had shown that nodB1A and nodB2C are expressed in nodules of D. glomerata while nodH1 and nodH2 are not (Persson et al. 2015; Nguyen et al. 2016). Therefore, the expression of nod genes was examined in nodules induced on Cea. thyrsiflorus by Cv1, nodules of C. myrtifolia induced by Cj1, nodules of C. nepalensis induced by Dg1, and in nodules of C. arborea and C. terminalis induced by Cppng1, as well as in nodules of D. glomerata induced by Dg2. The results are depicted in figure 7. NodB1A and nodB2C were expressed in all host plants from the Northern Hemisphere, while nodC and nltU were expressed in nodules of C. arborea and C. terminalis. NodH was expressed in nodules of Cea. thyrsiflorus. Thus, nodH expression seemed to be host-specific in that the gene was not expressed in nodules of D. glomerata (Datiscaceae, Cucurbitales) induced by Dg2 which contains nodH, but was expressed in nodules of Cea. thyrsiflorus (Rhamnaceae, Rosales) induced by Cv1.
Fig. 7.

—Nod gene expression in nodules. Relative expression levels were determined based on IF-3 as reference gene (Alloisio et al. 2010) and presented at log10 scale. Expression levels of the nitrogenase structural gene nifH were included in the analysis as a further point of comparison. Results are listed based on initials of host plant-name of inoculum-name of gene. Host plants were Coriaria myrtifolia (Cm), C. nepalensis (Cn), C. terminalis (Cote), C. arborea (Ca), Ceanothus thyrsiflorus (Ceth), and Datisca glomerata (Dg). The figure was prepared using R (R Core Team 2019).
What Is the Function of Frankia Cluster-2 Nod Genes?
Two facts point toward a role of Frankia nod genes in determining host specificity: first, nodH shows host-specific expression, and second, all Frankia cluster-2 metagenomes from North America, that is, from the area of host plants from the Rosales order, contain nodH. With the exception of the Southern Hemisphere metagenome Cppng1_C1_nod, the nod gene regions display various signs of earlier transposition events (figs. 3 and 4). Nevertheless, the nod regions of Eurasian strains show strong sequence conservation, while the nod regions of North American strains show less conservation; the latter fact could be linked to the increased transposase abundance in North American compared with Eurasian (meta-)genomes (fig. 2). The facts that 1) the two nod operons nodA’B1A and nodB2CnltIJ are part of transposable units and that 2) both operons are linked in two of the North American metagenomes (Dg2_Dg_vc and Dd1_Dg_vc) and in one of the metagenomes based on inoculum Dg1 from Pakistan (Dg1_Dg_nod1) suggest that in the common progenitor of Frankia cluster-2 all nod genes were linked.
The fact that BMG5.1 and BMG5.30 do not contain the canonical nod genes (Gtari et al. 2015; Gueddou et al. 2019) shows that nod gene-free strains can be part of cluster-2 assemblages. This is not surprising given the fact that the nod genes are present on transposable units in all Northern Hemisphere (meta-)genomes sequenced so far; indeed, it seems likely that all cluster-2 strain assemblages would contain nod gene-free member(s). BMG5.1 and BMG5.30 can nodulate C.myrtifolia (Gtari et al. 2015; Gueddou et al. 2019). Thus, the lack of nod genes is no obstacle to nodulation of Coriaria sp.
However, the fact that all Northern hemisphere (meta-) genomes described in this study, nine of which were propagated in D. glomerata nodules, contain the canonical nod genes seems to imply that the nod gene-free strains do not compete very well with nod gene-containing strains when it comes to infection of D. glomerata. In our hands, BMG5.1 could not nodulate D. glomerata while it could nodulate C. myrtifolia; this contradicts the results of Gtari et al. (2015), who reported nodulation of both species by BMG5.1. The discrepancy might be explained by the fact that Gtari et al. (2015) studies were performed in a Mediterranean climate which D. glomerata is adapted to.
A role of cluster-2 Frankia nod genes in the nodulation of Datisca sp. and of the North American host plants, but not of Coriaria sp., would also be consistent with the fact that the Southern hemisphere metagenome Cppng1_Ca_nod, which does not contain functional copies of nodA and nodB, could nodulate C. arborea and C. terminalis but was unable to nodulate Datisca sp. However, the fact that Cppng1_Ca_nod nodC and nltU were expressed in nodules of C. arborea could be interpreted to mean that the function of Frankia cluster-2 nod genes is not, or not only, the synthesis of LCOs, but, for example, the modification of N-acetylglucosamine-containing phospholipids that occur in actinobacteria (Sun et al. 2015). In any case, the fact that also in all nod gene containing cluster-2 strains including Cppng1_Ca_nod, the nod2 region includes nltIJ, the operon encoding part of an ABC transport system, suggests that the enzymes encoded by the nod genes are involved in producing molecules for export. Altogether, further research is needed to clarify the function of Frankia cluster-2 nod genes, but the data available thus far support a function in the nodulation of all cluster-2 host plants except for Coriariaceae.
Evolution of Frankia Cluster-2 Symbioses
All plants able to enter a root nodule symbiosis with nitrogen-fixing soil bacteria go back to a common ancestor (Soltis et al. 1995), and there are two hypotheses on the evolution of root nodule symbioses. In the first one, ∼100 Ma the common ancestor of the Rosales, Cucurbitales, Fagales, and Fabales acquired a predisposition based on which a root nodule symbiosis could evolve, and the evolution of such symbioses then took place in several lineages for Frankia, and in several lineages for rhizobia as microsymbionts (Soltis et al. 1995; Doyle 2016). The second hypothesis assumes that the common ancestor of the four orders evolved a symbiosis with nitrogen-fixing soil bacteria, which in turn was lost in the majority of lineages; this hypothesis is supported by phylogenomic analyses (Griesmann et al. 2018; van Velzen et al. 2018) and therefore will be the basis of this discussion. Since the distribution of the Fagales has been studied in detail, leading to the conclusion that the order evolved in Gondwana (Cook and Crisp 2005), it can be concluded that the common ancestor of plants forming a nitrogen-fixing root nodule symbiosis (Fagales, Fabales, Rosales, and Cucurbitales) evolved in the Gondwana supercontinent.
With regard to host specificity it is interesting that the inoculum from the Southern hemisphere, Cppng1 could not only nodulate C. arborea but also the Northern Hemisphere species C. terminalis, while it failed to nodulate D. glomerata. This is to our knowledge the first report on the nodulation of a Northern Hemisphere Frankia cluster-2 host plant by an inoculum from the Southern Hemisphere. Previous attempts with Purshia sp. and Ceanothus sp. as host plants were unsuccessful (Silvester 1977; Benson and Silvester 1993), and nodulation of C. arborea by the Northern Hemisphere inoculum Dg1 failed as well (data not shown). So while the fact that Frankia cluster-2 inocula represent strain assemblages is complicating the interpretation of cross-infection studies, it is worth noting that Coriaria is the only host plant genus distributed in both hemispheres, and so far, the only successful cross-hemisphere inoculation involved a Coriaria species.
The fact that most inocula used in this study were propagated in nodules of D. glomerata might have biased the outcome, both with regard to the members of the strain assemblages that were sequenced, and with regard to the results of cross-inoculation studies. The distribution of Coriaria sp. and D.cannabina overlaps in northern India/Pakistan/Nepal and it temporarily overlapped in France since D. cannabina used to be cultivated there (Stenhouse 1856). That is, in these areas, selection could have favored cluster-2 inocula able to nodulate D. cannabina and Coriaria spp. Their distribution does/did not, however, currently or in the recorded past, overlap in Japan; yet, the Cj1 inoculum could nodulate D. glomerata.
As outlined by Nguyen et al. (2016), cluster-2 Frankia strains probably reached North America from Asia during the Eocene/Oligocene (55 − 25 Ma) with Datisca sp. over the Beringian land bridge. In the Southern hemisphere, currently cluster-2 Frankia strains can only nodulate Coriaria species. In Eurasia, currently they can be hosted by Coriaria species and also by D.cannabina. When Datisca sp. arrived in North America—allopatric speciation between D. cannabina and D. glomerata has been dated to 25 Ma (Zhang et al. 2006)—Frankia cluster-2 strains from Eurasia encountered the microsymbionts of the North American host plants, Dryadoideae and Ceanothus sp., all from the Rosales order. The phylogenetic position of the metagenomes from the North American inocula (fig. 1), one of which was isolated from Cea. thyrsiflorus, suggests that the Eurasian lineage of Frankia cluster-2 outcompeted the endogenous North American cluster-2 strains. Again, the results presented here may be biased based on the use of D. glomerata to propagate inocula. Nevertheless, the fact that the Cv1 metagenome isolated from nodules of Cea. thyrsiflorus maps in the same lineage as the three North American metagenomes that were propagated in D. glomerata, supports the hypothesis that the original Rosales lineage(s) of cluster-2 Frankia strains was/were outcompeted. Data on transposase abundance indicate that this process was associated with increased transposition rates, that is, increased genomic instability in the Datisca lineage of cluster-2 Frankia strains.
Conclusions
The analysis of 12 new (meta-)genomes of Frankia cluster-2 strains based on two previously used and five novel inocula showed that cluster-2 inocula represent groups of strains. This might explain their wide host range. A comparison of (meta-)genomes based on DNA isolated from whole nodules versus DNA isolated from symbiotic bacterial structures (vesicle clusters) showed that strains that are strongly underrepresented in vesicle clusters may still be able to colonize nodules.
The analysis of transposases in the available (meta-)genomes showed that North American strains contain more transposases than Eurasian strains. An analysis of the evolution and distribution of host plants indicates that bursts of transposition may have coincided with the expansion of the host range while outcompeting the endogenous cluster-2 strains.
All novel (meta-)genomes contained the canonical nod genes nodABC; the North American ones also contained the sulfotransferase gene nodH. An analysis of the synteny of the nod regions indicated that they were located on mobile genetic elements. This would lead to the expectation that groups of cluster-2 strains would also contain members that had lost the canonical nod genes, like the only cultured cluster-2 strain F. coriariae BMG5.1 (Gtari et al. 2015).
So far, the presence of nodABC in a cluster-2 Frankia (meta-)genome is correlated with the ability to nodulate D. glomerata, and nodH was found only in genomes originating in North America. NodABC expression was found in all nodules examined, while nodH was expressed only in Cea. thyrsiflorus (Rhamnaceae, Rosales), not in D. glomerata (Datiscaceae, Cucurbitales). However, more data are needed to confirm the hypothesis that cluster-2 Frankia nod genes are involved in host specificity.
The first metagenome of an inoculum from the Southern hemisphere was obtained, Cppng1_Ca_nod. The strain represents a novel cluster-2 Frankia species and does not contain the canonical nod genes nodAB, but still contains nodC. It could nodulate a northern hemisphere Coriaria species, C. terminalis.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We are very grateful to Susanne Renner (LMU Munich, Germany) for helpful discussions about the evolution and systematics of Cucurbitales, to Michael Lovave and Tiberius Jimbo (Papua New Guinea Forest Research Institute, PNG) and Axel Dalberg Poulsen (Royal Botanic Garden Edinburgh, Scotland) for providing the Cppng1 inoculum and to Matthew L. Carlson (University of Alaska at Anchorage, AK) for providing the Dd1 inoculum. We thank Warwick Silvester (Waikato, New Zealand) for kindly providing C.arborea seeds, Amir Ktari (University of Tunis, Tunisia) for the gift of C.myrtifolia seeds and Alexander Nieto (LMU Munich, Germany) for expert technical support. Last not least, we thank Martin Parniske (LMU Munich, Germany) for the idea of sequencing total nodule DNA. This project was supported by a grant from the Swedish Research Council Vetenskapsrådet (VR 2012-03061) to KP. The bioinformatics support of the BMBFfunded project “Bielefeld-Gießen Center for MicrobialBioinformatics”-BiGi and the BMBF grant FKZ 031A533 within the German Network for Bioinformatics Infrastructure (de.NBI) are gratefully acknowledged. Microscopy and nodule structure researchwas financially supported by the Russian Science Foundation (grant no. 16-16-00089) and performed using equipment of the Core Facility of Cell and Molecular Technologies in Plant Science at the Komarov Botanical Institute (Saint-Petersburg, Russia).
Data deposition: This project has been deposited at EMBL/GenBank/DDBJ under the accession PRJEB19438 - PRJEB19449.
Literature Cited
- Aharonowitz Y, Cohen G, Martin JF.. 1992. Penicillin and cephalosporin biosynthetic genes: structure, organization, regulation, and evolution. Annu Rev Microbiol. 46:461–495. [DOI] [PubMed] [Google Scholar]
- Alloisio N, et al. 2010. The Frankia alni symbiotic transcriptome. Mol Plant Microbe Interact. 23(5):593–607. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ.. 1990. Basic local alignment search tool. J Mol Biol. 215(3):403–410. [DOI] [PubMed] [Google Scholar]
- Altschul SF, et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25(17):3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ara I, Bakir MA, Kudo T.. 2008. Transfer of Catellatospora koreensis Lee et al 2000 as Catelliglobosispora koreensis gen. nov., comb. nov. and Catellatospora tsunoense Asano et al. 1989 as Hamadaea tsunoensis gen. nov., comb. nov., and emended description of the genus Catellatospora Asano and Kawamoto 1986 emend. Lee and Hah 2002. Int J Syst Evol Microbiol. 58:1950–1960. [DOI] [PubMed]
- Barabote RD, et al. 2009. Complete genome of the cellulolytic thermophile Acidothermus cellulolyticus 11B provides insights into its ecophysiological and evolutionary adaptations. Genome Res. 19(6):1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battenberg K, et al. 2017. The influence of the host plant is the major ecological determinant of the presence of nitrogen-fixing root nodule symbiont cluster II Frankia species in soil. Appl Environ Microbiol. 83:e02661–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DR, Silvester WB.. 1993. Biology of Frankia strains, actinomycete symbionts of actinorhizal plants. Microbiol Rev. 57(2):293–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DR, Stephens DW, Clawson ML, Silvester WB.. 1996. Amplification of 16S rRNA genes from Frankia strains in root nodules of Ceanothus griseus, Coriaria arborea, Coriaria plumosa, Discaria toumatou, and Purshia tridentata. Appl Environ Microbiol. 62(8):2904–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom J, et al. 2009. EDGAR: a software framework for the comparative analysis of prokaryotic genomes. BMC Bioinformatics 10(1):154.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom J, et al. 2016. EDGAR 2.0: an enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 44(W1):W22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cérémonie H, Debelle F, Fernandez MP.. 1999. Structural and functional comparison of Frankia root hair deforming factor and rhizobia Nod factor. Can J Bot. 77:1293–1301. [Google Scholar]
- Chabaud M, et al. 2016. Chitinase-resistant hydrophilic symbiotic factors secreted by Frankia activate both Ca2+ spiking and NIN gene expression in the actinorhizal plant Casuarina glauca. New Phytol. 209(1):86–93. [DOI] [PubMed] [Google Scholar]
- Clawson ML, Bourret A, Benson DR.. 2004. Assessing the phylogeny of Frankia-actinorhizal plant nitrogen-fixing root nodule symbioses with Frankia 16S rRNA and glutamine synthetase gene sequences. Mol Phylogenet Evol. 31(1):131–138. [DOI] [PubMed] [Google Scholar]
- Cook LG, Crisp MD.. 2005. Not so ancient: the extant crown group of Nothofagus represents a post-Gondwanan radiation. Proc Biol Sci. 272(1580):2535–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D.. 2011. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27(8):1164–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JJ. 2016. Chasing unicorns: nodulation origins and the paradox of novelty. Am J Bot. 103(11):1865–1868. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. 1989. PHYLIP - Phylogeny Inference Package (Version 3.2). Cladistics 5: 164–166.
- Gherbi H, et al. 2008. SymRK defines a common genetic basis for plant root endosymbioses with arbuscular mycorrhiza fungi, rhizobia, and Frankia bacteria. Proc Natl Acad Sci U S A. 105(12):4928–4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghodhbane-Gtari F, et al. 2013. Draft genome sequence of Frankia sp. strain CN3, an atypical, noninfective (Nod–) ineffective (Fix–) isolate from Coriaria nepalensis. Genome Announc. 1:e0008513.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghodhbane-Gtari F, et al. 2014. Draft genome sequence of Frankia sp. strain BMG5.23, a salt-tolerant nitrogen-fixing actinobacterium isolated from the root nodules of Casuarina glauca grown in Tunisia. Genome Announc. 2:e00520–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goris J, et al. 2007. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 57(Pt 1):81–91. [DOI] [PubMed] [Google Scholar]
- Griesmann M, et al. Forthcoming 2018. Phylogenomics reveals multiple independent losses of the nitrogen-fixing root nodule symbiosis. Science. 361(6398):eaat1743. [DOI] [PubMed] [Google Scholar]
- Gtari M, et al. 2015. Cultivating the uncultured: growing the recalcitrant cluster-2 Frankia strains. Sci Rep. 5(1):13112.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueddou A, et al. 2019. Draft genome sequence of the symbiotic Frankia sp. strain BMG5.30 isolated from root nodules of Coriaria myrtifolia in Tunisia. Antonie Van Leeuwenhoek. 112(1):67–74. [DOI] [PubMed] [Google Scholar]
- Hilker R, et al. 2014. ReadXplorer – visualization and analysis of mapped sequences. Bioinformatics 30(16):2247–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilker R, et al. 2016. ReadXplorer 2 – detailed read mapping analysis and visualization from one single source. Bioinformatics 32(24):3702–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoagland DR, Arnon DT.. 1938. The water-culture method for growing plants without soil. California Agriculture Experiment Station Circular 347. Berkeley (CA: ): University of California. [Google Scholar]
- Hurst SG, et al. 2014. Draft Genome sequence of Frankia sp. strain Thr, a nitrogen-fixing actinobacterium isolated from the root nodules of Casuarina cunninghamiana grown in Egypt. Genome Announc. 2:e00493–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huss-Danell K. 1997. Tansley Review No. 93. Actinorhizal symbioses and their N2 fixation. New Phytol. 136(3):375–405. [DOI] [PubMed] [Google Scholar]
- Juhas M, et al. 2009. Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol Rev. 33(2):376–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DD, Froula J, Egan R, Wang Z.. 2015. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. Peer J. 3:e1165.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28(12):1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Oh HS, Parks SC, Chun J.. 2014. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol. 64(Pt 2):346–351. [DOI] [PubMed] [Google Scholar]
- Ktari A, et al. 2017. Permanent draft genome sequence of Frankia sp. NRRL B-16219 reveals the presence of canonical nod genes, which are highly homologous to those detected in Candidatus Frankia Dg1 genome. Stand Genomic Sci. 12:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg S.. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Berry AM.. 1991. Localization and characterization of pectic polysaccharides in roots and root nodules of Ceanothus spp. during intercellular infection by Frankia. Protoplasma 163(2–3):93–101. [Google Scholar]
- Mansour SR, et al. 2014. Draft genome sequence of Frankia sp. strain CcI6, a salt-tolerant nitrogen-fixing actinobacterium isolated from the root nodule of Casuarina cunninghamiana. Genome Announc. 2:e01205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markmann K, Giczey G, Parniske M.. 2008. Functional adaptation of a plant receptor-kinase paved the way for the evolution of intracellular root symbioses with bacteria. PLoS Biol. 6(3):e68.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz VM, et al. 2014. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 42(D1):D560–D567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20(9):1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meesters TM, Van Vliet WM, Akkermans A.. 1987. Nitrogenase is restricted to the vesicles in Frankia strain EANlpec. Physiol Plant. 70(2):267–271. [Google Scholar]
- Meyer F, et al. 2003. GenDB – an open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 31(8):2187–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza MS, Hameed S, Akkermans ADL.. 1994. Genetic diversity of Datisca-compatible Frankia strains determined by sequence analysis of PCR-amplified 16S rRNA gene. Appl Environ Microbiol. 60:2371–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugnier PD, Poirel L, Nordmann P.. 2009. Functional analysis of insertion sequence ISAba1, responsible for genomic plasticity of Acinetobacter baumannii. J Bacteriol. 191(7):2414–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb WR, Wood S.. 1987. Morphogenesis and fine structure of Frankia (Actinomycetales): the microsymbiont of nitrogen-fixing actinorhizal root nodules. Int Rev Cytol. 109:1–88. [DOI] [PubMed] [Google Scholar]
- Ngom M, et al. 2016. Permanent draft genome sequence for Frankia sp. strain CeD, a nitrogen-fixing actinobacteria isolated from the root nodules of Casuarina equisetifolia grown in Senegal. Genome Announc. 4(2):e00265-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TV, et al. 2016. An assemblage of Californian cluster II Frankia strains contains the canonical nod genes and also the sulfotransferase gene nodH. BMC Genomics 17(1):796.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normand P, et al. 1996. Molecular phylogeny of the genus Frankia and related genera and emendation of the family Frankiaceae. Int J Syst Bacteriol. 46:1–9. [DOI] [PubMed] [Google Scholar]
- Normand P, et al. 2007. Genome characteristics of facultatively symbiotic Frankia sp. strains reflect host range and host plant biogeography. Genome Res. 17(1):7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normand P, et al. 2017. Proposal of Candidatus Frankia californiensis, the uncultured nitrogen-fixing symbiont associated with a phylogenetically broad group of hosts endemic to California. Int J Syst Evol Microbiol. 67(10):3706–3715. [DOI] [PubMed] [Google Scholar]
- Normand P, et al. 2018. Frankia canadensis sp. nov., isolated from root nodules of Alnus incana subspecies rugosa. Int J Syst Evol Microbiol. 68(9):3001–3011. [DOI] [PubMed] [Google Scholar]
- Nouioui I, et al. 2013. Draft genome sequence of Frankia sp. strain BMG5.12, a nitrogen-fixing actinobacterium isolated from Tunisian soils. Genome Announc. 1:e00468–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouioui I, et al. 2014. Absence of cospeciation between the uncultured Frankia microsymbionts and the disjunct actinorhizal Coriaria species. BioMed Res Int. 2014:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouioui I, et al. 2017. Frankia inefficax sp. nov., an actinobacterial endophyte inducing ineffective, non nitrogen-fixing, root nodules on its actinorhizal host plants. Antonie Van Leeuwenhoek. 110(3):313–320. [DOI] [PubMed] [Google Scholar]
- Nouioui I, et al. 2017. Frankia asymbiotica sp. nov., a non-infective actinobacterium isolated from Morella californica root nodule. Int J Syst Evol Microbiol. 67(12):4897–4901. [DOI] [PubMed] [Google Scholar]
- Nouioui I, et al. 2019. Frankia torreyi sp. nov., the first actinobacterium of the genus Frankia Brunchorst 1886, 174AL isolated in axenic culture. Antonie Van Leeuwenhoek. 112(1):57–65. [DOI] [PubMed] [Google Scholar]
- Nouioui I, Ghodhbane-Gtari F, Rohde M, Klenk HP, Gtari M.. 2017. Frankia coriariae sp. nov., an infective and effective microsymbiont isolated from Coriaria japonica. Int J Syst Evol Microbiol. 67(5):1266–1270. [DOI] [PubMed] [Google Scholar]
- Okonechnikov K, Golosova O, Fursov M.. 2012. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28(8):1166–1167. [DOI] [PubMed] [Google Scholar]
- Oshone R, et al. 2016. Permanent draft genome sequence of Frankia sp. strain Allo2, a salt-tolerant nitrogen-fixing actinobacterium isolated from the root nodules of Allocasuarina. Genome Announc. 4:e00388-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons R, Silvester WB, Harris S, Gruijters WTM, Bullivant S.. 1987. Frankia vesicles provide inducible and absolute oxygen protection for nitrogenase. Plant Physiol. 83(4):728–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlowski K, Demchenko KN.. 2012. The diversity of actinorhizal symbiosis. Protoplasma 249(4):967–979. [DOI] [PubMed] [Google Scholar]
- Persson T, et al. 2011. Genome sequence of “Candidatus Frankia datiscae” Dg1, the uncultured microsymbiont from nitrogen-fixing root nodules of the dicot Datisca glomerata. J Bacteriol. 193(24):7017–7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson T, et al. 2015. Candidatus Frankia datiscae Dg1, the actinobacterial microsymbiont of Datisca glomerata, expresses the canonical nod genes nodABC in symbiosis with its host plant. PLoS One 10(5):e0127630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi AC, et al. 2018. Robust Frankia phylogeny, species delineation and intraspecies diversity based on Multi-Locus Sequence Analysis (MLSA) and Single-Locus Strain Typing (SLST) adapted to a large sample size. Syst Appl Microbiol. 41(4):311–323. [DOI] [PubMed] [Google Scholar]
- Pujic P, et al. 2015. Genome sequence of the atypical symbiotic Frankia R43 strain, a nitrogen-fixing and hydrogen-producing actinobacterium. Genome Announc. 3(6):e01387-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2019). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing, Vienna, Austria: https://www.R-project.org/, last accessed December 12, 2018. [Google Scholar]
- Raeside C, et al. 2014. Large chromosomal rearrangements during a long-term evolution experiment with Escherichia coli. MBio 5(5):e01377-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M, Rosselló-Móra R.. 2009. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 106(45):19126–19131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ried MK, Antolín-Llovera M, Parniske M.. 2014. Spontaneous symbiotic reprogramming of plant roots triggered by receptor-like kinases. eLife 3:e03891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp O, et al. 2015. The structure of the Cyberlindnera jadinii genome and its relation to Candida utilis analyzed by the occurrence of single nucleotide polymorphisms. J Biotechnol. 211:20–30. [DOI] [PubMed] [Google Scholar]
- Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30(14):2068–2069.24642063 [Google Scholar]
- Sen A, et al. 2013. Draft genome sequence of Frankia sp. strain QA3, a nitrogen-fixing actinobacterium isolated from the root nodule of Alnus nitida. Genome Announc. 1:e0010313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen A, et al. 2014. Phylogeny of the class Actinobacteria revisited in the light of complete genomes. The orders ‘Frankiales’ and Micrococcales should be split into coherent entities: proposal of Frankiales ord. nov., Geodermatophilales ord. nov., Acidothermales ord. nov. and Nakamurellales ord. nov. Int J Syst Evol Microbiol. 64:3821–3832. [DOI] [PubMed] [Google Scholar]
- Siguier P, Pérochon J, Lestrade L, Mahillon J, Chandler M.. 2006. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 35:D32–D36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvester WB. 1977. Dinitrogen fixation by plant associations excluding legumes In: Hardy RWF, Gibson AH, editors. A treatise on dinitrogen fixation. New York: John Wiley and Sons; p. 141–190. [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM.. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31(19):3210–3212. [DOI] [PubMed] [Google Scholar]
- Sobhy H, Colson P.. 2012. Gemi: pCR primers prediction from multiple alignments. Comp Funct Genomics. 2012:783138.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis DE, et al. 1995. Chloroplast gene sequence data suggest a single origin of the predisposition for symbiotic nitrogen fixation in angiosperms. Proc Natl Acad Sci U S A. 92:2647–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenhouse J. 1856. Examination of select vegetable products from India. Philos Trans R Soc Lond. 146:141–157. [Google Scholar]
- Sun HM, et al. 2015. Tenggerimyces mesophilus gen. nov., sp. nov., a member of the family Nocardioidaceae. Int J Syst Evol Microbiol. 65(10):3359–3364. [DOI] [PubMed] [Google Scholar]
- Swanson E, et al. 2015. Permanent draft genome sequence of Frankia sp. strain ACN1ag, a nitrogen-fixing actinobacterium isolated from the root nodules of Alnus glutinosa. Genome Announc. 3:e01483-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson E, et al. 2017. Permanent draft genome sequence for Frankia sp. strain Cc1.17, a nitrogen-fixing actinobacterium isolated from root nodules of Colletia cruciata. Genome Announc. 5(24):e00530-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisa LS, et al. 2015. Draft genome sequence of Frankia sp. strain DC12, an atypical, noninfective, ineffective isolate from Datisca cannabina. Genome Announc. 3(4):e00889-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Velzen R, et al. 2018. Comparative genomics of the nonlegume Parasponia reveals insights into evolution of nitrogen-fixing rhizobium symbioses. Proc Natl Acad Sci U S A. 115(20):E4700–E4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall LG, et al. 2013. Draft genome sequence of Frankia sp. strain BCU110501, a nitrogen-fixing actinobacterium isolated from nodules of Discaria trinervis. Genome Announc. 1:e00503-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse RM, et al. 2018. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol. 35(3):543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, McGinnis S, Madden TL.. 2006. BLAST: improvements for better sequence analysis. Nucleic Acids Res. 34(Web Server issue):W6–W9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LB, Simmons MP, Kocyan A, Renner SS.. 2006. Phylogeny of the Cucurbitales based on DNA sequences of nine loci from three genomes: implications for morphological and sexual system evolution. Mol Phylogenet Evol. 39(2):305–322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.