


Abstract
The identification of transcription factor binding sites and cis-regulatory motifs is a frontier whereupon the rules governing protein–DNA binding are being revealed. Here, we developed a new method (DEep Sequence and Shape mOtif or DESSO) for cis-regulatory motif prediction using deep neural networks and the binomial distribution model. DESSO outperformed existing tools, including DeepBind, in predicting motifs in 690 human ENCODE ChIP-sequencing datasets. Furthermore, the deep-learning framework of DESSO expanded motif discovery beyond the state-of-the-art by allowing the identification of known and new protein–protein–DNA tethering interactions in human transcription factors (TFs). Specifically, 61 putative tethering interactions were identified among the 100 TFs expressed in the K562 cell line. In this work, the power of DESSO was further expanded by integrating the detection of DNA shape features. We found that shape information has strong predictive power for TF–DNA binding and provides new putative shape motif information for human TFs. Thus, DESSO improves in the identification and structural analysis of TF binding sites, by integrating the complexities of DNA binding into a deep-learning framework.
INTRODUCTION
Transcription factors (TFs) control gene expression by binding to specific DNA sequences, known as transcription factor binding sites (TFBSs) (1) with aligned profiles of TFBSs referred to as cis-regulatory motifs (2). The binding or release of TFs promotes or suppresses transcription to guarantee that the target genes are expressed at the proper time and in the appropriate amount according to particular cell states and circumstance (3). Substantial computational efforts have been invested in the studies of TF binding specificities and motif prediction, resulting in the development of numerous algorithms, computational tools and databases (4–6). However, the understanding of TF–DNA binding mechanisms remains fragmented. Improved computational approaches are needed to allow integration of nuanced biophysical information across large datasets and thus, remains a considerable challenge in systems biology (7–9).
Beyond the sequence level, recent studies have highlighted the essential role of DNA structure in influencing TF–DNA binding specificity both in vitro and in vivo across diverse TF families (10–14). Owing to the advances in DNA structure elucidation, four distinct DNA shape features (i.e. Minor Groove Width (MGW), Propeller Twist (ProT), Helix Twist (HelT) and Roll) can be computationally derived from DNA sequences by Monte Carlo simulation (15). These features can be considered as shape motifs and thereby an extension of traditional sequence-based cis-regulatory motifs. As a result, features in DNA sequences in combination with shapes determine the TF binding in a more sophisticated way than originally thought (16,17).
ChIP-sequencing (ChIP-seq) provides a view of genome-wide interactions between DNA and DNA-associated proteins and enables new insights into gene regulation. An extensive amount of ChIP-seq data has been generated and is available in the public domain, including ∼6000 human datasets from the ENCODE database (18). Additionally, new methods are expanding the available data on TF–TFBS interactions. Protein binding microarrays (PBMs) have enabled genome-scale characterization of TFBSs in a high-throughput manner (19). ChIP in combination with lambda exonuclease digestion before high-throughput sequencing (ChIP-exo), can identify almost single-nucleotide-resolution binding sites of TFs (20). Furthermore, selective microfluidics-based ligand enrichment followed by sequencing (SMiLE-seq) (21) is a newly-developed technology for protein–DNA interaction characterization that can efficiently characterize DNA binding specificities of TF monomers, homodimers and heterodimers. These data provide an unprecedented opportunity to predict motifs, identify TFBSs and capture more features affecting TF binding (22). Although a variety of popular methods have been developed for ChIP-seq data mining and modeling, computational challenges, including high-dimensional and heterogeneous data properties, remain for the accurate and exhaustive identification of motifs (23,24). For example, a current computational challenge is to integrate information from these diverse methods with structural and biophysical constraints on TF–TFBS interactions.
Deep learning (DL) has achieved unprecedented performance for capturing motif patterns and elucidating complex regulatory mechanisms based on large-scale ChIP-Seq datasets (25). For example, the convolutional neural network (CNN) improved the state-of-the-art performance in TF–DNA binding specificity prediction through optimizing the position-weight-matrix-like motif detectors (26); and the motif patterns predicted by DeepBind have been well mapped to documented motifs by Alipanahi et al. based on available human TFBS databases (25). However, these methods focused on predicting TF–DNA binding specificity and failed to identify motifs and TFBSs accurately.
Overall, the demand for the following data analysis and interpretation is significant: (i) how to improve the performance of capturing sequence-specific motifs; and (ii) how the non-sequence-specific TFBSs contribute to TF–DNA binding in alternative ways. In this study, we proposed a novel DL framework, named DESSO (DEep Sequence and Shape mOtif), using the CNN model to predict motifs and identify TFBSs in both sequence and regional DNA shape features. For the first time, these DNA shape features were integrated into DL models to explore how these features quantitatively contribute to TF–DNA binding, even though conserved shape patterns (or shape motifs) encoded in the human genome are still not well modeled (27). The identified motifs were evaluated using the documented motifs in JASPAR (28) and TRANSFAC (29), then compared with DeepBind (25), Basset (30), MEME-ChIP (31), KMAC (32) and gkm-SVM (33). Further analyses were conducted by integrating multiple types of biological information including TF binding domain types, chromatin accessibility, phylogenetic conservation, protein–protein interactions (PPI), etc. For the first time, rather than determine TFBSs using a subjective cutoff as previously reported (34), we integrated the binomial distribution into DESSO to optimize the TFBSs identification based on identified motif patterns (35). DESSO and the analyses it enabled will continue to improve our understanding of how gene expression is controlled by TFs and the complexities of DNA binding.
MATERIALS AND METHODS
Overview of the DESSO (DEep Sequence and Shape mOtif) pipeline
The DESSO framework is composed of (i) a CNN model for extracting motif patterns from given ChIP-seq peaks, and (ii) a statistical model based on the binomial distribution for optimizing the identification of motif instances (i.e. TFBSs). This framework can accept both DNA sequences and DNA shape features as input to identify sequence and shape motifs, respectively. DESSO enables the extraction of more complex motif patterns compared to existing motif prediction methods owing to its multi-layer network architecture. We designed a binomial-based model in DESSO to identify all the significant TFBSs under the statistical hypothesis that the number of random sequence segments that contain the motif of interest in the human genome is binomially distributed.
The first layer of the CNN model contains multiple convolutional filters (Figure 1A), which were used to identify low-level features from given ChIP-seq peaks. A subsequent max pooling layer and a fully connected layer were used to extract high-level features based on the output from the convolutional layer. Specifically, the CNN model takes DNA sequences centered on the ChIP-seq peaks as input query sequences and learns motif patterns using convolutional filters (denoted as motif detectors) (36). Then, a large set of background sequences was selected from the human genome, considering GC content, CpG frequency and promoter and repeat overlaps to eliminate biases created by these features (37). Both the query and background sequences were then aligned as sequence matrices, where each row represents a distinct sequence. For each optimized motif detector, two motif signal matrices were derived by sliding the detector along the query sequence matrix and background sequence matrix, respectively (Figure 1B). Each element of a signal matrix represents the occurrence probability of the corresponding motif detector on a sequence segment in the corresponding sequence matrix. These two motif signal matrices were then used to generate motif candidates by varying a motif instance signal cutoff in a predefined interval. For each value of the motif signal cutoff, the motif instance candidates in the query sequence matrix and background sequence matrix were obtained and then used to calculate a P-value according to the binomial distribution (Figure 1C). The optimal motif instances for a motif detector were finally determined as the motif instance candidates in the query sequence matrix that correspond to the minimum P-value. More details regarding the datasets, the CNN model, and the statistical model used in our study can be found in the following sections.
Figure 1.

Schematic overview of DESSO framework. (A) The CNN model for optimizing motif detectors. (B) Determination of optimal motif instances recognized by each motif detector. Both the query data (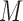 ) from the corresponding ChIP-seq dataset and the background data (
) from the corresponding ChIP-seq dataset and the background data (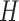 ) were fed into the convolutional layer in the trained model. For each motif detector, two motif signal matrices
) were fed into the convolutional layer in the trained model. For each motif detector, two motif signal matrices 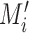 and
and 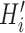 representing the probability of motif occurrence at each position in
representing the probability of motif occurrence at each position in 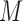 and
and 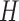 were derived, respectively. (C) Construction of the optimized motif profile.
were derived, respectively. (C) Construction of the optimized motif profile. 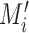 and
and 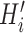 were then used to determine motif instance sets
were then used to determine motif instance sets 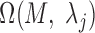 and
and 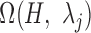 in
in 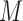 and
and 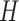 by varying the cutoff
by varying the cutoff 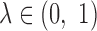 . For each
. For each 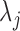 , a P-value was approximated by the binomial distribution based on the number of motif instances in
, a P-value was approximated by the binomial distribution based on the number of motif instances in 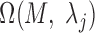 and
and 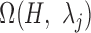 . The motif instances
. The motif instances 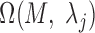 corresponding to the minimum P-value were used to generate the motif logo.
corresponding to the minimum P-value were used to generate the motif logo.
The 690 ChIP-seq datasets
The 690 ChIP-Seq datasets of uniform TFBS based on March 2012 ENCODE data freeze were downloaded from the ENCODE Analysis Database at UCSC (https://genome.ucsc.edu/ENCODE/downloads.html). These datasets contained 161 TFs and cover 91 human cell types (38). Each dataset contained a number of peaks (ranging from 101 to 92 358), ranked in the decreasing order of their signal scores. These peaks were derived from the SPP peak caller (39) and de-noised by the Irreproducible Discovery Rate (40) based on signal reproducibility among biological replicates. The average length of the de-noised peaks is 300 bps.
DNA shape feature generation
DNA shape features (i.e. HelT, MGW, ProT and Roll) provide three-dimensional structure information of the corresponding DNA sequences and play an essential role in TF–DNA recognition (17). Such features were obtained by the Monte Carlo simulation and can be applied to any given nucleotide sequences by a sliding-window method (15). A recent method designed for DNA shape analysis and its R implementation, DNAshapeR, was used to generate DNA shape features of each of the query/positive and background/negative sequences (10,41). All the resulting feature vectors were normalized to [0, 1].
CNN model construction
Our CNN model requires binary vectors as input, in which each input DNA sequence was first converted to an 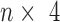 matrix
matrix 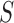 in one-hot format with A = [1, 0, 0, 0], C = [0, 1, 0, 0], G = [0, 0, 1, 0] and T = [0, 0, 0, 1] (Supplementary Figure S1A) and where
in one-hot format with A = [1, 0, 0, 0], C = [0, 1, 0, 0], G = [0, 0, 1, 0] and T = [0, 0, 0, 1] (Supplementary Figure S1A) and where 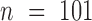 (25). This was sufficient for the convolutional filters to operate on sequence (S) alone. To incorporate DNA shape into DESSO, shape vectors of each DNA sequence were generated and represented by
(25). This was sufficient for the convolutional filters to operate on sequence (S) alone. To incorporate DNA shape into DESSO, shape vectors of each DNA sequence were generated and represented by 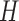 (HelT),
(HelT), 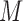 (MGW),
(MGW), 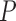 (ProT) and
(ProT) and 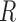 (Roll) (Supplementary Figure S1A). The input of this CNN model could be (i)
(Roll) (Supplementary Figure S1A). The input of this CNN model could be (i) 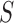 , (ii)
, (ii) 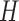 , (iii)
, (iii) 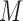 , (iv)
, (iv) 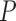 , (v)
, (v)  and (vi)
and (vi)  , where (vi) represents the combination of four DNA shape features. Each input was first fed into the convolutional layer to get the activation score of each convolutional filter as shown in formula 1 (Supplementary Figure S1B):
, where (vi) represents the combination of four DNA shape features. Each input was first fed into the convolutional layer to get the activation score of each convolutional filter as shown in formula 1 (Supplementary Figure S1B):
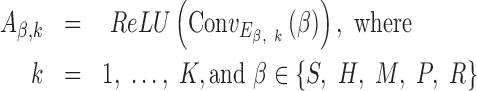 |
(1) |
Here, 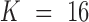 indicates the total number of convolutional filters, and
indicates the total number of convolutional filters, and 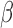 indicates a different input format. Concretely,
indicates a different input format. Concretely, 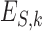 represents convolutional filters corresponding to
represents convolutional filters corresponding to 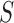 , each of which is an
, each of which is an 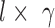 weight matrix with
weight matrix with 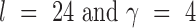 and can be interpreted as a sequence motif detector.
and can be interpreted as a sequence motif detector. 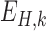 ,
, 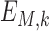 ,
, 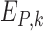 and
and 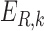 indicate convolutional filters corresponding to
indicate convolutional filters corresponding to  ,
,  ,
,  and
and 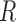 , respectively, each of which is an
, respectively, each of which is an 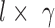 weight matrix with
weight matrix with 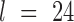 and
and 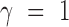 and can be interpreted as a shape motif detector. The
and can be interpreted as a shape motif detector. The 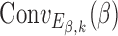 represents the convolution between
represents the convolution between 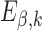 and
and 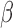 at each position
at each position  , which can be calculated using formula 2:
, which can be calculated using formula 2:
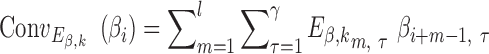 |
(2) |
The  indicates rectified linear unit, which is a widely used activation function in DL. Specifically,
indicates rectified linear unit, which is a widely used activation function in DL. Specifically, 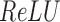 can avoid gradient vanishing problem and has better convergence performance. The max pooling layer enforced downsampling to the activation score vectors by selecting the maximum value in each
can avoid gradient vanishing problem and has better convergence performance. The max pooling layer enforced downsampling to the activation score vectors by selecting the maximum value in each  for
for  and
and 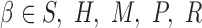 . This max pooling layer has two benefits: (i) it can reduce the dimension of the input data to make the CNN model more computationally efficient; and (ii) it enables motif translation invariance, which allows the motif of interest to always be captured regardless of its location.
. This max pooling layer has two benefits: (i) it can reduce the dimension of the input data to make the CNN model more computationally efficient; and (ii) it enables motif translation invariance, which allows the motif of interest to always be captured regardless of its location.
The concatenation of the output from the max pooling layer was represented by 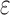 and finally fed into a fully connected layer. This layer has 32 hidden neurons and used the
and finally fed into a fully connected layer. This layer has 32 hidden neurons and used the 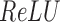 activation function as above. The output layer containing only one neuron was used to predict the TF–DNA binding specificity, which ranges from 0 to 1 (formula 3):
activation function as above. The output layer containing only one neuron was used to predict the TF–DNA binding specificity, which ranges from 0 to 1 (formula 3):
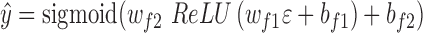 |
(3) |
where 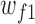 and
and 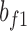 represent the weights and bias units in the fully connected layer, while
represent the weights and bias units in the fully connected layer, while 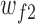 and
and 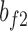 are the weights and bias units in the output layer. The
are the weights and bias units in the output layer. The 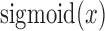 is a sigmoid function, where
is a sigmoid function, where 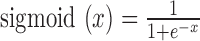 .
.
To further investigate the impact of model complexity on shape features, we also extended this CNN model with two convolutional layers. The network architecture can be found in Supplementary Table S1, where the local max pooling layer aims to select the maximum value at each of the three adjacent positions with a step size of three in  (42).
(42).
CNN model training
The same strategy in DeepBind (25) was used to split the peaks in each ChIP-seq dataset into training data and test data in this study. Specifically, for a ChIP-Seq dataset, the 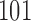 -bp-long sequences centered on each peak summit was defined as positive sequences, each of which has a label of ‘1’. To overcome overfitting problems in the model training, for those datasets with fewer than 10 000 peaks, we generated complementary random peaks until we had 10 000 sequences. Unlike previous studies that used dinucleotide-preserving shuffled sequences (25), regions near the transcription start sites (35), or flanking regions of ChIP-seq peaks (43,44), we picked the same number of 101-bp-long genomic sequences with the same GC content from the GENCODE to generate negative sequences (45). These negative sequences have matched GC-content to positive sequences and did not have overlap with any peaks in the positive dataset. Each negative sequence was labeled as ‘0’, which has less chance to be bound by the target TF.
-bp-long sequences centered on each peak summit was defined as positive sequences, each of which has a label of ‘1’. To overcome overfitting problems in the model training, for those datasets with fewer than 10 000 peaks, we generated complementary random peaks until we had 10 000 sequences. Unlike previous studies that used dinucleotide-preserving shuffled sequences (25), regions near the transcription start sites (35), or flanking regions of ChIP-seq peaks (43,44), we picked the same number of 101-bp-long genomic sequences with the same GC content from the GENCODE to generate negative sequences (45). These negative sequences have matched GC-content to positive sequences and did not have overlap with any peaks in the positive dataset. Each negative sequence was labeled as ‘0’, which has less chance to be bound by the target TF.
For each dataset, its training data was then used to train the CNN model by minimizing the following loss function (formula 4):
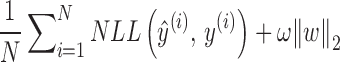 |
(4) |
where 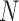 is the size of the sample set from training data (i.e. the number of sequences in the training set),
is the size of the sample set from training data (i.e. the number of sequences in the training set), 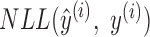 is the negative log-likelihood between prediction
is the negative log-likelihood between prediction 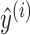 and target
and target 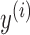 ,
, 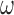 is a regularization parameter to leverage the trade-off between the goal of fitting and the goal of the generalizability of the trained model, and
is a regularization parameter to leverage the trade-off between the goal of fitting and the goal of the generalizability of the trained model, and 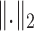 indicates the
indicates the 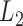 norm.
norm.
The loss function was optimized by mini-batch gradient descent with momentum, using a comparatively small batch size of 64 to avoid the generalization drop of the trained models (46). The backpropagation algorithm was used for gradient calculating (47), and exponential decay was applied to the learning rate with a decay rate equal to 0.95. The learning rate, momentum, regularization parameter 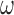 and the standard deviation of the initial weights in the neural network were randomly selected from the pre-prepared intervals
and the standard deviation of the initial weights in the neural network were randomly selected from the pre-prepared intervals 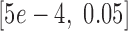 ,
, 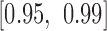 ,
, 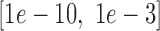 and
and 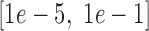 , respectively, and the dropout rate was taken from
, respectively, and the dropout rate was taken from 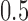 ,
, 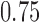 or
or 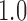 (48). These hyper-parameters were sampled ten times, and three-fold cross-validation was performed on the training data to select the hyper-parameter set which corresponded to the highest average AUC (i.e. the area under the receiver operating characteristic curve). The optimal hyper-parameter set was then applied to the whole training data for the final model training. Each model mentioned above was trained for 30 epochs maximally, and an early stopping strategy was applied to prevent overfitting. The average running time for the training of each model was ∼10 min. Our model was implemented on a single Tesla K80 GPU with 11GB memory using TensorFlow, which is the most widely used DL framework in the public domain (49).
(48). These hyper-parameters were sampled ten times, and three-fold cross-validation was performed on the training data to select the hyper-parameter set which corresponded to the highest average AUC (i.e. the area under the receiver operating characteristic curve). The optimal hyper-parameter set was then applied to the whole training data for the final model training. Each model mentioned above was trained for 30 epochs maximally, and an early stopping strategy was applied to prevent overfitting. The average running time for the training of each model was ∼10 min. Our model was implemented on a single Tesla K80 GPU with 11GB memory using TensorFlow, which is the most widely used DL framework in the public domain (49).
Sequence motif prediction
Without loss of generality, the above CNN model, trained using DNA sequences, was used as an example to illustrate how to predict motifs based on our statistical model. Let 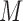 represent the 101-bp-long sequences from the top-
represent the 101-bp-long sequences from the top-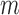 peaks in each dataset, where each sequence is centered at its corresponding peak summit and
peaks in each dataset, where each sequence is centered at its corresponding peak summit and 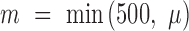 , where
, where 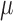 is the total number of peaks in that dataset. Define the motif signal matrix
is the total number of peaks in that dataset. Define the motif signal matrix 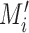 as the activation values between a motif detector
as the activation values between a motif detector 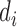 (each has length
(each has length 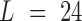 ) and
) and 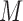 by feeding
by feeding 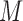 into the convolutional layer in its corresponding trained model, and define
into the convolutional layer in its corresponding trained model, and define 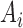 to be the maximum value in
to be the maximum value in 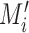 . A sequence segment (
. A sequence segment (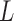 bps) in each sequence is defined as an activation segment if its activation score is larger than an activation cutoff
bps) in each sequence is defined as an activation segment if its activation score is larger than an activation cutoff 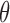 . A motif instance set, denoted as
. A motif instance set, denoted as 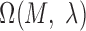 , contains all activation segments with
, contains all activation segments with 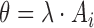 in
in 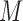 . The value of
. The value of 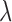 ranges from 0 to 1, and it can be optimized through exhaustively identifying the minimal P-value of
ranges from 0 to 1, and it can be optimized through exhaustively identifying the minimal P-value of 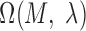 . It is noteworthy that the P-value of a motif instance set can be derived based on the assumption that the number of activation segment containing sequences follows a binomial distribution when using random selection with replacement in the human genome. To estimate the ‘success’ probability
. It is noteworthy that the P-value of a motif instance set can be derived based on the assumption that the number of activation segment containing sequences follows a binomial distribution when using random selection with replacement in the human genome. To estimate the ‘success’ probability 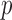 of each random selection, the human genome was divided into non-overlapping bins with length 101 bp, and
of each random selection, the human genome was divided into non-overlapping bins with length 101 bp, and 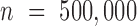 bins were selected as a background sequence set
bins were selected as a background sequence set 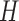 (37).
(37).
Let 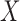 be a random variable representing the number of activation segment containing bins with
be a random variable representing the number of activation segment containing bins with 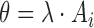 in
in 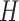 ,
, 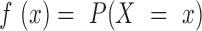 be the probability function, and
be the probability function, and 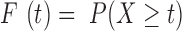 be the cumulative distribution function. It was assumed that
be the cumulative distribution function. It was assumed that 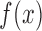 can be approximated by a binomial distribution
can be approximated by a binomial distribution 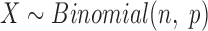 , where
, where 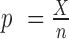 is a maximum likelihood estimate. Therefore, the P-value of
is a maximum likelihood estimate. Therefore, the P-value of 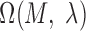 is given by formula 5:
is given by formula 5:
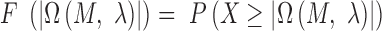 |
(5) |
For each motif detector 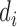 , the optimal motif instance
, the optimal motif instance 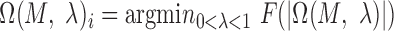 and the corresponding P-value can be obtained. Only
and the corresponding P-value can be obtained. Only 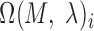 with the P-value <
with the P-value < 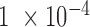 ,
, 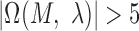 and with at least three positions having information content (IC) larger than 1 were retained in our study, which assumes that motif should be conserved and observed more frequently in
and with at least three positions having information content (IC) larger than 1 were retained in our study, which assumes that motif should be conserved and observed more frequently in 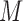 . The derived motif instances were aligned as motif profiles and visualized using WebLogo 2.8.2 (50).
. The derived motif instances were aligned as motif profiles and visualized using WebLogo 2.8.2 (50).
Comparison with five existing sequence motif finding tools
DESSO is closely related to DeepBind (25) and Basset (30), which are two well-known DL-based methods for motif identification. However, rather than a subjective motif signal cutoff for motif instance identification, DESSO used a binomial distribution model to optimize the motif prediction (see details above on motif prediction). Specifically, for each motif detector, sequence fragments with the maximum activation score in each query sequence were aligned to obtain motifs in DeepBind, while Basset aligned sequence fragments with activation scores larger than half of each motif detector's maximum value. For a fair comparison in this study, DeepBind and Basset used the same motif detectors, query sequences and corresponding motif signal matrices as DESSO to identify DNA motifs. As a highly cited web server in this field, MEME-ChIP identifies motifs from ChIP-seq peaks by integrating two complementary motif discovery algorithms, i.e. MEME (51) and DREME (52). KMAC innovatively used k-mer set memory for motif representation in order to capture the contribution of nucleotides dependency and flanking k-mers in TF–DNA binding (32). Compared with other state-of-the-art motif finding methods (e.g. HOMER (35) and ChIPMunk (53)), KMAC achieved the best performance in discovering known motifs from ChIP-seq datasets (32). Gkm-SVM was selected in the comparison as it significantly outperforms traditional kmer-SVM methods by using gapped k-mers for accurately and efficiently identifying longer motifs, which are hard to model as k-mers. Specifically, the C++ implementation, gkm-SVM-2.0, was used in this study (33). DESSO, DeepBind and Basset were evaluated on all peaks in each ChIP-seq dataset. Following the same settings in DeepBind, the top 500 peaks were used for the evaluation of MEME-ChIP limited by its computational complexity. Additionally, the top 5000 and top 10 000 peaks were used in KMAC and gkm-SVM as suggested by the corresponding papers, respectively. Besides the maximum motif length and the maximum number of output motifs being set to 24 and 16, respectively, default parameters in MEME-ChIP and gkm-SVM were used. KMAC was run with options ‘–k_win 101 –k_min 5 –k_max 13’, where ‘–k_min 5 –k_max 13’ was suggested by the authors.
RESULTS
DESSO accurately predicts motifs from ChIP-seq data
We began by making similarity comparisons between motifs predicted by DESSO from 690 ENCODE TF ChIP-seq datasets and experimentally validated motifs in the human JASPAR and TRANSFAC databases using TOMTOM (54). These comparisons were extended to other five existing methods in this field, i.e. DeepBind, Basset, MEME-ChIP, KMAC and gkm-SVM. The results showed that DESSO significantly improved the motif prediction performance on 161 TFs in 91 cell lines, covered by the above ChIP-seq datasets.
First, to assess the similarity of query motifs against validated motifs, TOMTOM was used to compare the statistical significance (i.e. E-value, P-value and q-value) across JASPAR and TRANSFAC for motifs that were predicted by all the six methods in comparison. The 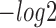 (E-value),
(E-value), 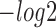 (P-value) and
(P-value) and 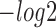 (q-value) of DESSO were significantly larger than the values for the other methods (Wilcoxon test P-values < 5 × 10−4, Figure 2A). DeepBind, Basset and MEME-ChIP achieved comparable performance across the three measurements and significant performance over KMAC and gkm-SVM. Hence, the DL frameworks were able to learn motif patterns from DNA sequences more accurately than k-mer-based strategies and strategies combining expectation maximization algorithm with regular expression. Most importantly, these results highlight the advantage of DESSO’s binomial model over strategies used by DeepBind and Basset and demonstrate that DESSO reduces both false positive and false negative rates.
(q-value) of DESSO were significantly larger than the values for the other methods (Wilcoxon test P-values < 5 × 10−4, Figure 2A). DeepBind, Basset and MEME-ChIP achieved comparable performance across the three measurements and significant performance over KMAC and gkm-SVM. Hence, the DL frameworks were able to learn motif patterns from DNA sequences more accurately than k-mer-based strategies and strategies combining expectation maximization algorithm with regular expression. Most importantly, these results highlight the advantage of DESSO’s binomial model over strategies used by DeepBind and Basset and demonstrate that DESSO reduces both false positive and false negative rates.
Figure 2.

Performance comparison of sequence motif identification accuracy. (A) The 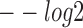 (E-value),
(E-value), 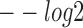 (P-value) and
(P-value) and 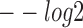 (q-value) derived from TOMTOM for all methods. The Wilcoxon test P-values of the above three scores between DESSO and other five methods. (B) A total of 435 motifs (known motifs) from DESSO can be matched to the JASPAR or TRANSFAC, and 248 motifs (undocumented motifs) do not have any matches (pie chart). For these known motifs, 388 of them are in the JASPAR and 283 are in the TRANSFAC database (bar plot).
(q-value) derived from TOMTOM for all methods. The Wilcoxon test P-values of the above three scores between DESSO and other five methods. (B) A total of 435 motifs (known motifs) from DESSO can be matched to the JASPAR or TRANSFAC, and 248 motifs (undocumented motifs) do not have any matches (pie chart). For these known motifs, 388 of them are in the JASPAR and 283 are in the TRANSFAC database (bar plot).
Additionally, of the six methods, DESSO achieved the best performance for identifying validated motifs in the JASPAR and TRANSFAC datasets (Supplementary Figure S2A). Five validated motifs were exclusively covered by DESSO, which are recognized by NR3C2, FOS::JUN (var. 2), RFX3, HIC2 and DUXA, respectively (Supplementary Figure S2B). Specifically, NR3C2 plays an essential role in mediating ion and waster transport (55), and DUXA is closely associated with Facioscapulohumeral Muscular Dystrophy 1. FOS::JUN (var. 2) is one of the JUN-FOS heterodimers (56), which was first discovered by SMiLE-seq (21).
To explore the sequence motifs identified by DESSO we performed clustering using similarity scores from TOMTOM (Figure 2B and Supplementary Method S1). The most significant motif in terms of the binomial P-value in each cluster was defined as the representative sequence motif. Of the 683 representative sequence motifs 435 were known motifs, supported by the JASPAR or TRANSFAC databases. The other 248 were undocumented motifs and retained for additional analysis owing to their statistical significance (Figure 2B). The motif patterns, corresponding genomic features and ChIP-seq peak enrichment analyses of the 435 and 248 motifs can be found in Supplementary KnownMotif file and UndocumentedMotif file, respectively.
Analysis of 683 representative sequence motifs identified by DESSO
All the human TFs or TF complexes that recognize the 435 known motifs, identified by DESSO, were grouped into 24 classes based on their structural information (Supplementary Table S2) (57). All structural classes were showcased in terms of their AUC performance on the corresponding ChIP-seq datasets and the IC of the identified motifs (Figure 3A). Notably, the majority of structural classes, which tend to be sequence-specific in TF–DNA binding, demonstrated competitive AUC performance (more details can be found in Supplementary Figure S3A). This can be further demonstrated based on the significant positive AUC-IC correlation among some structural classes, e.g. TATA, Fork head and C2H2 ZF (58) (Supplementary Figure S3B). It is relatively intuitive that some TF classes with highly conserved motifs produced high prediction AUC performance. But more interestingly, for 5 of the 24 structural classes, DESSO’s AUCs were not predictable by IC, suggesting more complicated binding mechanisms extending beyond linear sequence recognition. Specifically, Leucine-rich repeat, CXXC ZF and TEA, had AUCs that were significantly lower than others structural classes in of similar IC ∼0.50. Whereas while both TATA and C2CH THAP-type ZF achieved relatively high AUCs, despite their mean ICs were only 0.30 and 0.34. Thus, our results revealed that the TFs belonging to these five structural classes may bind to DNA using more complicated mechanisms which goes beyond sequence-level recognition. In addition, for more than half of the 24 structural classes, the number of distinct known motifs are significantly larger than the number of TFs (Supplementary Table S3), indicating additional motifs of the TFs, other than the ChIP-ed TF, can be identified in a ChIP-Seq dataset. This phenomenon suggested that co-factor and tethering binding activities are prevalent in the human TF–DNA regulations.
Figure 3.

Detailed view of identified sequence motifs. (A) The ICs of 435 know sequence motifs identified by DESSO and the AUC performance of TFs in the 690 ChIP-seq datasets, showcased in terms of the 24 structural classes in the TFClass system. (B) The phylogram tree of 88 enriched sequence motifs according to their similarity derived from TOMTOM. The inner circle indicates the TFs and their structural classes (background color is the same as (A), while the TFs that are not covered by any of these 24 structural classes were indicated by the white background color) of the corresponding validated motifs either in the JASPAR or TRANSFAC. The outer circle represents the motif logo of each enriched sequence motif, including CTCF (red star) and MYCN (green star). (C) The heat map of per-nucleotide DNase I cleavage and the corresponding mean value; (D) The heat map of per-nucleotide vertebrate conservation and the corresponding mean value of CTCF’s TFBSs as well as 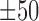 bps flanking regions within the A549 cell line. Each row in the heat map represents a motif instance. (E) The red curve indicates the enrichment score of CTCF on its corresponding ChIP-seq peaks. Vertical black lines indicate the presence of ChIP-seq peaks that contain at least one TFBS of CTCF. The motif logo of CTCF identified by DESSO is shown below.
bps flanking regions within the A549 cell line. Each row in the heat map represents a motif instance. (E) The red curve indicates the enrichment score of CTCF on its corresponding ChIP-seq peaks. Vertical black lines indicate the presence of ChIP-seq peaks that contain at least one TFBS of CTCF. The motif logo of CTCF identified by DESSO is shown below.
To extend DESSO’s predictive power, a total of 88 representative motifs, frequently observed in the 690 ChIP-seq ENCODE datasets, were derived from these 435 known motifs using the motif comparison tool TOMTOM and hierarchical clustering (Figure 3B). Various computational validations based on additional biochemical data have been carried out for the 88 motifs. Here we analyze those data to interrogate the overall quality of the motifs in the ENCODE ChIP-seq data identified by DESSO (see details in DESSO website). Among the 88 motifs, the CTCF motif was selected for further interrogation as the example for the following functional analyses, as it plays an important role in modulating chromatin structure (59) and was the most enriched ChIP-ed TF in our study (in ∼15% ChIP-seq datasets). The DNase I Digital Genomic Footprinting (60) and evolutionary conservation (phastCons scores (61)) of CTCF’s TFBSs within the A549 cell line were collected (Supplementary Method S2 and 3). CTCF’s TFBSs were more susceptible to DNase I enzyme (Figure 3C), revealing the binding preference of CTCF to accessible chromatin, which showed significant evolutionary conservation compared to the flanking regions (Figure 3D), illustrating strong phylogenetic conservation of the identified CTCF motif. To investigate the occurrence of CTCF motif in the corresponding ChIP-seq peaks, which are ranked by peak signal, an enrichment score was calculated using GSEA (Gene Set Enrichment Analysis software) (62) (Figure 3E and Supplementary Method S4). The enrichment score curve clearly showed the dramatic left-skewed trend, indicating that the DESSO-identified CTCF motif was more enriched in top-ranked peaks. This is consistent with the fact that peaks with higher peak signal also have a higher probability to be bound by the ChIP-ed TF. The strong functionality conservation of another TF, MAX, and its left-skewed enrichment in the K562 cell line can be found in Supplementary Figure S4.
An extended investigation of the 248 undocumented motifs showed that they are likely to be cis-regulatory elements having similar functionalities as known motifs, but have not been experimentally validated. Seventy-eight distinctly enriched motifs were collected from the 248 motifs based on similar clustering analyses as above (Supplementary Figure S5A) (63). The functionality and enrichment analysis of these motifs also demonstrated strong DNase footprint patterns and evolutionary conservation, revealing the potential role of this motif in transcriptional regulation (Supplementary Figure S5B–D). Using TOMTOM, we further compared the 248 undocumented motifs with the computationally predicted and inferred human motifs in the Cis-BP database (64). We found that 86 motifs have statistically significant matches in the database (Supplementary Table S4). These results support the functional conservation of both known motifs and undocumented motifs identified by DESSO and indicated the distinguished ability and potential of DESSO in identifying regulatory code in the human genome.
DESSO infers indirect bindings from the 100 TFs expressed in the K562 cell line
Sequence motifs identified by DESSO in each of the 690 ENCODE datasets may also contain motifs that were bound by TFs associated with a partner, rather than a direct interaction and picked up in the ChIP-ed TFs. To investigate this phenomenon for human TFs, the 100 TFs in the K562 cell line were examined by analyzing their DESSO-identified sequence motifs (Supplementary Method S5 and Table S5). Among the 100 TFs, 75 of them are DNA-binding proteins involved in RNA polymerase II transcription (65), which were referred to as sequence-specific TFs. The remaining 25 TFs are non-sequence-specific since no significant motifs identified and are thought to interact with DNA by PPI with other DNA-binding proteins. Among the 75 sequence-specific TFs, 67 had known canonical motifs specifically recognized by their DNA-binding domains (66). A likely reason for this is that rather than bind to DNA sequences directly, some sequence-specific TFs can also tether to DNA by interacting with other DNA-binding proteins (67). Such indirect binding is abundant in human TFs, e.g. the estrogen receptor 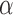 is enabled to regulate gene expression by interacting with Runx1 in breast cancer cells (68) and interact with c-Fos/c-Jun heterodimers at TFBSs of AP-1 in ER/AP-1-dependent transcription (69). To interrogate this possibility, each ChIP-seq dataset containing the canonical motif of the ChIP-ed TF were defined as direct-binding peaks (
is enabled to regulate gene expression by interacting with Runx1 in breast cancer cells (68) and interact with c-Fos/c-Jun heterodimers at TFBSs of AP-1 in ER/AP-1-dependent transcription (69). To interrogate this possibility, each ChIP-seq dataset containing the canonical motif of the ChIP-ed TF were defined as direct-binding peaks (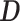 ) and the others were defined as indirect-binding peaks (
) and the others were defined as indirect-binding peaks (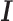 ). Fifty-three of the 75 TFs have known canonical motifs, indicating DESSO was able to identify 80% of the canonical motifs. About 48% of the ChIP-seq peaks for these 53 TFs belonged to
). Fifty-three of the 75 TFs have known canonical motifs, indicating DESSO was able to identify 80% of the canonical motifs. About 48% of the ChIP-seq peaks for these 53 TFs belonged to 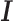 on average, and this proportion (72% average) was observed across all the 100 TFs (blue bars in Figure 4).
on average, and this proportion (72% average) was observed across all the 100 TFs (blue bars in Figure 4).
Figure 4.

Indirect binding of the 100 TFs analyzed in the K562 cell line. The names of the 100 TFs are indicated around the inner circle, and a ribbon connects two TFs which have predicted tethering binding association. The thickness of the ribbon is proportional to the ratio of peaks, linking wide-sided 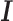 -TF’s (indirect) with the narrow-sided
-TF’s (indirect) with the narrow-sided 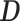 -TF’s (direct). The blue bar and red bar in the outer circle indicate the ratio of
-TF’s (direct). The blue bar and red bar in the outer circle indicate the ratio of 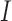 and the ratio of
and the ratio of 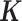 (indecipherable—cannot be resolved as tethering/binding) in each TF’s ChIP-Seq dataset, respectively. Four examples of tethering binding association are showcased around the outer circle, each of which indicates that one TF (lavender ball) interacts with DNA by binding to another DNA-binding TF (orange ball).
(indecipherable—cannot be resolved as tethering/binding) in each TF’s ChIP-Seq dataset, respectively. Four examples of tethering binding association are showcased around the outer circle, each of which indicates that one TF (lavender ball) interacts with DNA by binding to another DNA-binding TF (orange ball).
To further investigate DESSO’s ability to predict tethering and pairwise binding among these 100 TFs, we calculate the proportion of 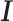 peaks of one TF that are consistent with the
peaks of one TF that are consistent with the 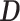 peaks of another TF (see Supplementary Table S6). A total of 61 predicted tethering associations were discovered (the links in the inner ring of Figure 4), including two known tethering binding mechanisms (i.e. ATF3-USF1 (66) and NFE2-MAX (60)) and some potential interactions which have been observed in recent studies, such as USF2-MAX and SP2-NFYB (60) (in the four corners of Figure 4).
peaks of another TF (see Supplementary Table S6). A total of 61 predicted tethering associations were discovered (the links in the inner ring of Figure 4), including two known tethering binding mechanisms (i.e. ATF3-USF1 (66) and NFE2-MAX (60)) and some potential interactions which have been observed in recent studies, such as USF2-MAX and SP2-NFYB (60) (in the four corners of Figure 4).
Notably, our results reported that 45 TFs have tethering interactions with MAX, of which 30 were TFs that had sequence-specific motifs and the remaining 15 were non-sequence-specific TFs (Supplementary Table S5). Out of these 45 tethering interactions, 7 (15.6%) of them were documented interactors with MAX, as supported by the PPIs in the BIOGRID database and were documented MAX-associated binders (70). As a basic helix-loop-helix zipper (bHLHZ) TF, the biological function of MAX (71) can only be activated by forming dimers/complexes with other proteins. Importantly, MAX was always the DNA binder, reinforcing the idea that it serves as the tethering sites for many other TFs. The most well-known MAX-associated complex is the MYC/MAX/MAD network, including MYC-MAX and MAD-MAX heterodimers, which are widely recognized to play an important role in cell proliferation, differentiation and neoplastic disease (72). Our observation revealed that not only does MAX specifically dimerize with proteins in the MYC family (71), MAX also extensively interact with other sequence-specific and non-sequence-specific TFs from diverse protein families.
For each ChIP-seq dataset of the 100 TFs, the peaks in 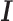 that are not involved in any tethering binding interactions were classified as indecipherable peaks (
that are not involved in any tethering binding interactions were classified as indecipherable peaks (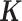 ), indicating the peaks that cannot be deciphered based on direct DNA binding and tethering binding mechanisms. These peaks composed about 49% of all ChIP-seq peaks in these 100 datasets (red bars in the outer ring of Figure 4). Furthermore, no statistically significant sequence motifs were identified by DESSO in a total of 51 of the 690 ChIP-seq datasets. Taken together, these analyses implied that sequence motifs still have considerable limitations in elucidating TF–DNA recognition in human, and that more sophisticated mechanisms may occur beyond sequence-level TF–DNA interactions should be considered. In addition to indirect binding, DNA shape is emerging as another factor influencing TF–DNA interactions.
), indicating the peaks that cannot be deciphered based on direct DNA binding and tethering binding mechanisms. These peaks composed about 49% of all ChIP-seq peaks in these 100 datasets (red bars in the outer ring of Figure 4). Furthermore, no statistically significant sequence motifs were identified by DESSO in a total of 51 of the 690 ChIP-seq datasets. Taken together, these analyses implied that sequence motifs still have considerable limitations in elucidating TF–DNA recognition in human, and that more sophisticated mechanisms may occur beyond sequence-level TF–DNA interactions should be considered. In addition to indirect binding, DNA shape is emerging as another factor influencing TF–DNA interactions.
DESSO recognized DNA shape features as contributors to TF–DNA binding specificity
To investigate the importance of DNA structure in human TF–DNA recognition, DESSO was used to infer the power of DNA shape in elucidating TF–DNA binding specificity across the 690 ChIP-seq datasets. For each dataset, the 101-bp sequences centered at their peak summits were defined as positive sequences. Additionally, the corresponding negative sequences were selected from the human genome, provided that they do not have any overlaps with peaks in the positive dataset and they have the same GC-content as the positive sequences. The HelT, MGW, ProT and Roll of each positive and negative sequence were then generated by DNAshapeR and used to train DESSO.
DESSO was then applied to HelT, MGW, ProT, Roll and the combination of these four shape features (referred to as DNA shape combination) was used to classify the positive and negative sequences in each dataset (Figure 5A and Supplementary Figure S6). For the five kinds of inputs, their performance was evaluated using AUC. DESSO predicts that DNA shape factors HelT and Roll have more significant predictive power than the other two shape factors in identifying TFBS. Specifically, HelT, MGW, ProT and Roll achieved an average AUC of 0.72, 0.66, 0.69 and 0.75, respectively (Figure 5B). It is clear that HelT and Roll surpassed the classification performance of MGW and ProT, which may stem from the fact that HelT and Roll were calculated by the two central base pair steps within a sliding-pentamer window (secondary structure information), while MGW and ProT were calculated by only the central base pair. We also observed a weak positive correlation between the AUC performance of sequence and shape features (Supplementary Figure S7). This observation indicated that these two kinds of features are not strongly correlated in predicting TF–DNA binding and are not one-to-one matched to each other, which means that different DNA sequences can encode the same shape feature (15). More specifically, some conserved shape motifs could disperse into completely un-conserved patterns when the DL model takes sequence motifs as features, thus these shape motifs cannot be detected and contribute to the prediction model.
Figure 5.

The performance of DNA shape in predicting TF–DNA binding specificity. (A) DESSO was applied to five different inputs, i.e. HelT, MGW, ProT, Roll and DNA shape combination. (B) The AUC of the five inputs above using single and two convolutional layers based on the 690 ChIP-seq datasets. The Wilcoxon test P-values between one-layer and two-layer model. (C) The contribution of HelT (32%), MGW (9%), ProT (22%) and Roll (37%) in DNA shape combination in predicting TF–DNA binding specificity. (D) The heat map is a more detailed analysis of diagram (C), indicating the contribution of each DNA shape feature on the 690 datasets, where each column represents a dataset. Those columns were organized by hierarchical clustering based on Pearson correlation and complete linkage. The structural class of ChIP-ed TF in each dataset was showcased at the bottom. (E) A performance comparison between sequence and the combination of sequence and shape (Sequence + DNA Shape) against 24 structural classes in terms of AUC. The two red boxes indicate the classes with the most significant AUC improvement when combining Sequence and Shape compared to Sequence only.
Compared to individual DNA shape features, the performance of the combined DNA shape features had significantly improved predictive power (AUC of 0.81 average) (Figure 5B). However, the DNA shape combination was inferior to the DNA sequence for predictive performance (Supplementary Figure S8A). To investigate the influence of the model complexity on the predictive power of DNA shape features, we further extended DESSO with two convolutional layers. Our results revealed that the performance of four individual DNA shape features can significantly benefit from two-layer models (all achieving Wilcoxon test P-values < 5 × 10−6, Figure 5B). Similar improvement was also observed on the combination of DNA shape features (Figure 5B). This is different from DNA sequence which has little or negative, effects on the predictive performance by using multi-convolutional layers (42). Clearly, our finding suggested that DNA shape and sequence may use different mechanisms in TF–DNA binding. Notably, performing motif identification based on multi-layer models can dramatically degrade the interpretability of the learned features, which is limited by their complex network architectures (73). Hence, all the following studies were carried out with a one-layer model unless otherwise specified.
To evaluate the extent to which each DNA shape feature contributed to the remarkable detection of TF–DNA binding, we analyzed the fraction of the average motif signal from the max pooling layer for each shape feature across the 690 ENCODE datasets. Here, HelT, MGW, ProT and Roll contributed 32, 9, 22 and 37%, respectively (Figure 5C). To assess the common occurrences of DNA shape factors across individuals from the 690 datasets, we clustered the DESSO prediction results (Figure 5D). HelT and Roll were the most important contributors, often with both present, but one predominating. ProT appeared predicative within only for a small clave of samples. Thus, DNA shape factors tended to frequently contribute simultaneously to TF binding and have dominant roles in different datasets (Figure 5D). Surprisingly, the ChIP-ed TFs had no apparent predictive power, as seen by a lack of clustering (Figure 5D), on the dominant shape factor. This suggests that there are more rules or additional shape factor and structural information to be uncovered across TFs.
We further investigated the complementary role of sequence and shape information in predicting TF–DNA binding specificity by combining them together and found that the average AUC performance was not improved significantly by incorporating DNA shape features (Supplementary Figure S8A). This observation is in contrast to previous studies claiming that the combination of sequence and shape information outperforms sequence alone by using traditional classification models (e.g. SVM and GradientBoostingClassifier) (10,11,14,74). To explore whether shape information can contribute to TFs belonging to specific structural classes, we then compared the AUC performance of two different inputs (i.e. sequence and the combination of sequence and shape) across TFClass (57) (Figure 5E). Among the 24 structural classes, 10 (i.e. Runt, Other alpha, TEA, RHR, Fork head, Other C4 ZF, Nuclear receptor, C2CH THAP-type ZF, TATA and CXXC ZF) demonstrated improved motif detection by incorporating shape information. It is noteworthy that TEA achieved a significant performance boost with 0.96 AUC by considering both of sequence and shape, while the average AUCs were only 0.85 and 0.83 by considering sequence and shape alone, respectively (Supplementary Figure S8B). This phenomenon of TEA has not been previously reported and suggests that TFs with different structures exhibit different shape recognition preference when they bind to DNA.
Overall, these results demonstrated the remarkable predictive performance of DNA shape features alone in TF–DNA binding specificity prediction. This finding implies that the underlying conserved DNA shape patterns (or shape motif) are also encoded in the human genome and may involve in TF-shape readouts recognition.
DESSO predicts novel DNA shape motifs
DESSO was used to determine if the human genome contained regions of evolutionarily conserved shape motifs (Supplementary Method S6). We sought to have DESSO discover shape motifs based on the same strategy that was used to discover sequence motifs (Figure 1). This approach added HelT, MGW, ProT, Roll or their combinations to discover four kinds of shape motifs within the 690 datasets from ENCODE (named HelT motif, MGW motif, ProT motif and Roll motif). DESSO identified 1257 HelT motifs, 84 MGW motifs, 885 ProT motifs and 478 Roll motifs, with 598 out of the 690 ENCODE datasets having at least one shape motif. A shape motif can be represented by a vector of shape features describing the mean of the corresponding motif instances. Using the same strategy as in Figure 5D, and counting the shape motifs belonging to each dataset, we found that the distribution of shape motifs across the datasets were enriched for HelT motifs and ProT motifs (Figure 6A). It was surprising that given the large fraction of Roll shape factors and low abundance of ProT shape factors (Figure 5C and D), so many ProT motifs were identified (Figure 6A). Overall, these results demonstrate that DESSO was able to identify shape motifs across a large range of datasets, indicating that shape motifs are abundant in the human genome.
Figure 6.

A comprehensive analysis of the identified shape motifs. (A) The number of datasets in the 690 ENCODE ChIP-seq datasets that were covered by shape motifs of HelT (446), MGW (43), ProT (392) and Roll (141). (B) IC of underlying sequences of the identified sequence motifs and shape motifs. (C) Each entry in the heatmap indicates the ratio of peaks covered by the four kinds of shape motifs which were identified in the 51 TFs within GM12878, K562 and HepG2 cell lines, while the black entries represent missing values. Four representative shape motif logos were listed at the left side, where each of them represents the shape motif profile and 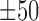 bps flanking regions using a bold orange curve. The two boundary curves of the blue region represent upper and lower bounds of shape features in the corresponding motif instances. (D) The enrichment score of MAX’s Roll motif in its corresponding ChIP-seq peaks in the K562 cell line. Black ticks indicate the occurrence of ChIP-seq peaks that contain at least one instances of MAX’s Roll motif. (E) The heat map of per-nucleotide DNase I cleavage of TFBSs of MAX’s Roll as well as
bps flanking regions using a bold orange curve. The two boundary curves of the blue region represent upper and lower bounds of shape features in the corresponding motif instances. (D) The enrichment score of MAX’s Roll motif in its corresponding ChIP-seq peaks in the K562 cell line. Black ticks indicate the occurrence of ChIP-seq peaks that contain at least one instances of MAX’s Roll motif. (E) The heat map of per-nucleotide DNase I cleavage of TFBSs of MAX’s Roll as well as 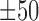 bps flanking regions, where each row represents a motif instance. The orange curve represents mean DNase I cleavage. (F) Twelve histone marks of
bps flanking regions, where each row represents a motif instance. The orange curve represents mean DNase I cleavage. (F) Twelve histone marks of 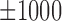 bps around the summits of Max's Roll motif. (G) The average ration (31%) of ChIP-seq peaks that cannot be explained by the 100 TFs within the K562 cell line.
bps around the summits of Max's Roll motif. (G) The average ration (31%) of ChIP-seq peaks that cannot be explained by the 100 TFs within the K562 cell line.
Given that the shape features were derived from conserved DNA sequences, we predicted that the newly identified shape motifs should have a high probability of coinciding with shape features within the sequence motifs in their respective datasets. To examine this hypothesis, the underlying DNA sequences of each shape motif were aligned as a sequence motif profile, which we defined as a shape-sequence-motif. The IC of each shape motif class was then computed across the shape-sequence-motifs (Figure 6B). Compared with the sequence motifs identified by DESSO (Figure 1), shape-sequence-motifs have significantly lower IC (Figure 6B). We also measured the similarity between each shape-sequence-motif and validated motifs in JASPAR and TRANSFAC using TOMTOM. Only 66% of shape-sequence-motifs could be matched to JASPAR or TRANSFAC. These two results indicate that shape motifs are less conserved at the sequence level and their conservedness is largely independent from sequence motifs. Indeed, even the TFs in the same structural class demonstrated different preferences for the sequence and shape information (Supplementary Figure S9).
To investigate the enrichment of shape motifs and whether they are cell-line-specific or DNA-binding-domain-specific, the peaks covered by each kind of shape motifs among the 51 TFs within GM12878, K562 and HepG2 cell lines were analyzed. The majority of peaks in ZNF274’s datasets can be accounted for by its shape motifs (Figure 6C), even though no peaks were explained by direct TF–DNA binding and tethering (Figure 4). CTCF coherently recognizes HelT and ProT motifs among the three aforementioned cell lines, while SP1 is dominated explicitly by ProT motifs within the HepG2 cell line. Also, the Roll motif is prevalent in TFs which have basic helix-loop-helix (bHLH) structure, implying that Roll is recognized by structurally conserved DNA binding domains (Figure 6C). To explore the occurrence of shape motifs in their corresponding ChIP-seq peaks, enrichment analysis of Max's Roll motif in the K562 cell line was performed. As opposed to its sequence motif, Max's Roll motif is more enriched in the low-ranked peaks, which is consistent with the recent observation that TFs bind to peaks with low peak signal by recognizing their preferred shape profiles (13) (Figure 6D). We also found that Max's Roll motif also prefers histone-depleted regions similar to its sequence motifs by analyzing DNase I Digital Genomic Footprinting and 12 histone marks surrounding its TFBSs (75), indicating that Max's Roll motif is functionally conserved (Figure 6E and F).
Considering ChIP-seq peaks covered by shape motifs, the average ratio, 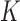 of the 100 TFs within the K562 cell line (Figure 4) was decreased to 31% (Figure 6G). This suggested that human TFs are capable of recognizing shape motifs in the genome, which contributes to explaining the ChIP-Seq peaks that cannot be interpreted by direct TF–DNA interaction and tethering binding.
of the 100 TFs within the K562 cell line (Figure 4) was decreased to 31% (Figure 6G). This suggested that human TFs are capable of recognizing shape motifs in the genome, which contributes to explaining the ChIP-Seq peaks that cannot be interpreted by direct TF–DNA interaction and tethering binding.
DISCUSSION
We developed a DL-based motif finding framework, named DESSO, combined with a new statistical method for motif profile construction, followed by its application on the 690 human ChIP-seq datasets within ENCODE. This work lead to the first comprehensive analyses of newly identified sequence and shape motifs. The identified motifs provide a set of novel human regulatory lexicons and can be used to construct a gene regulatory atlas for the human genome. Specifically, shape motifs can potentially contribute to the interpretation of indiscriminate binding behavior of human TFs (76). Co-regulated gene groups, revealed by identification of motifs may define cell-type specific regulons and thus, provide critical biological insights to the cell heterogeneity mechanisms (77). In addition, motifs can be linked to single-cell gene expression data to overcome dropout issues by removing genes lacking motif support and optimize the characterization of cell state. Another scenario is to combine motif identification with single nucleotide polymorphisms to identify disease-associated genetic variants. For example, an A/G mutation in the globin cluster (positioning at 209 709 in human chromosome 16) may create a motif for GATA1 binding that disrupts the original globin cluster promoters. Meanwhile, a T/A mutation in the BCL-2 promoter (position 60 988 353 on human chromosome 18) can cause the loss of GATA4 binding function and potentially disrupt the formation of ovarian granulosa tumors. Such variants provide clues for diagnosing diseases, developing therapeutic targets and elucidating disease etiology (25).
DESSO advanced the state-of-the-art in cis-regulatory motif prediction and TFBSs identification. Additionally, DESSO showcased the potential of a DL framework for identification and rationalization of results. Our results demonstrate that DESSO was able to identify a number of previously unidentified motifs and shape factors that contribute to TF–DNA binding mechanisms and infer indirect mechanisms of TF–motif interactions via tethering activities and co-factor motifs. These predictions now await experimental validation. Specifically, the role of DNA shape in explaining TF–DNA binding is still in debate (78), which mainly stems from the fact that sequence and shape features covary (15). Instead of relying on sequence motif-dependent analyses to explore shape conservation, our study investigated conserved DNA shape patterns that may be recognized by human TFs. Overall, the implementation and application of the DESSO framework provide a solid foundation for the construction of gene regulatory networks and the elucidation of TF–DNA binding mechanisms in the human genome.
Further investigations are needed to elucidate other obscure intrinsic features in gene regulation and TF binding. Specifically, in this study, 31% of the peaks in ChIP-seq data remain unexplained by sequence and shape motifs. The DL-based models provide a promising opportunity to integrate diverse data forms and quantify relationships between motifs and expression accurately. As experimental data grows and is integrated into DL-based models, the accuracy of motif prediction will increase. In-depth analysis of both sequence and shape motifs utilizing more advanced DL-techniques will facilitate inference of gene regulatory relations, and accurate modeling of the complex regulatory system in the human genome. For example, a gated CNN was proposed recently and performed competitively on benchmarks based on our preliminary analysis in Supplementary Method S7 and Figure S10.
The application of DL to motif prediction is intrinsically limited by the availability of large-scale sequencing and protein–DNA interaction data including ChIP-seq. These experiments are relative expensive and require extensive effort. This limitation can be alleviated by developing more advanced sequencing techniques or by taking advantage of other available regulatory information (e.g. chromatin accessibility and DNA methylation) to infer DNA binding motifs. For example, additional information such as epigenetic regulation measured by histone modifications, DNase-seq and ATAC-seq, can be used to predict the binding of DNA regulatory elements (79). Additionally, advanced models using matched expression data and DNA accessibility data across diverse cellular contexts can also aid in predicting the missing information, including TF subcellular localization, chromatin accessibility and gene expression (80).
Existing DL-based methods assume that each position in a motif detector contributes to TF–DNA binding affinity independently, which may underestimate the nucleotide dependencies. To further consider interdependencies between nucleotides, DNA structure information, such as DNA shape, should be also considered. For example, crystal/co-crystal structures of TF–DNA interactions (or some simplified parameters such as the geometry of binding) could be used as a layer in DESSO. Advanced mathematics and computational tools will permit the building of integrated models of gene regulatory systems and enable deliverable strategies to prevent or treat disease.
DATA AVAILABILITY
An integrated web server for DESSO is freely available at https://bmbl.bmi.osumc.edu/DESSO. The source code of DESSO and a detailed tutorial can be found at https://github.com/OSU-BMBL/DESSO.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Jennifer Xu, Shaopeng Gu, Shihan Wu and Minxuan Sun, for their assistance in language polishing, pipeline testing and web server development.
Authors’ contributions: Q.M. and B.L. conceived the basic idea and designed the overall analyses. J.Y. and Q.M. designed specific experiments and developed the DL framework. J.Y., A.M. and Y.L. carried out most of the computational analysis and data interpretation. C.W. and Y.W. designed the web server of DESSO. J.Y., A.D.H., B.L., A.M. and Q.M. wrote the manuscript.
Notes
Disclaimer: The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health and the National Science Foundation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation/EPSCoR Cooperative Agreement [#IIA-1355423]; National Institute of General Medical Sciences of the National Institutes of Health, R01 Award [GM131399–01]; National Science Foundation [ACI-1548562]; National Nature Science Foundation of China (NSFC) [61772313 to B.L. and 61572227 to Y.W.]; Young Scholars Program of Shandong University [YSPSDU, 2015WLJH19 to B.L.]; Innovation Method Fund of China [SQ2018IMC600001 to B.L.]. Shanghai Municipal Science and Technology Major Project [2018SHZDZX01 to B.L.]. Science-Technology Development Project from Jilin Province (20180414012GH to Y.W.) Funding for open access charge: National Science Foundation/EPSCoRCooperative Agreement [#IIA-1355423].
Conflict of interest statement. None declared.
REFERENCES
- 1. Mitchell P.J., Tjian R.. Transcriptional regulation in mammalian cells by sequence-specific DNA binding proteins. Science. 1989; 245:371–378. [DOI] [PubMed] [Google Scholar]
- 2. D’haeseleer P. What are DNA sequence motifs. Nat. Biotechnol. 2006; 24:423–425. [DOI] [PubMed] [Google Scholar]
- 3. Badis G., Berger M.F., Philippakis A.A., Talukder S., Gehrke A.R., Jaeger S.A., Chan E.T., Metzler G., Vedenko A., Chen X.. Diversity and complexity in DNA recognition by transcription factors. Science. 2009; 324:1720–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weirauch M.T., Cote A., Norel R., Annala M., Zhao Y., Riley T.R., Saez-Rodriguez J., Cokelaer T., Vedenko A., Talukder S.. Evaluation of methods for modeling transcription factor sequence specificity. Nat. Biotechnol. 2013; 31:126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang J., Chen X., McDermaid A., Ma Q.. DMINDA 2.0: integrated and systematic views of regulatory DNA motif identification and analyses. Bioinformatics. 2017; 33:2586–2588. [DOI] [PubMed] [Google Scholar]
- 6. Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S.. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009; 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jolma A., Yan J., Whitington T., Toivonen J., Nitta K.R., Rastas P., Morgunova E., Enge M., Taipale M., Wei G.. DNA-binding specificities of human transcription factors. Cell. 2013; 152:327–339. [DOI] [PubMed] [Google Scholar]
- 8. Barrera L.A., Vedenko A., Kurland J.V., Rogers J.M., Gisselbrecht S.S., Rossin E.J., Woodard J., Mariani L., Kock K.H., Inukai S.. Survey of variation in human transcription factors reveals prevalent DNA binding changes. Science. 2016; 351:1450–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yin Y., Morgunova E., Jolma A., Kaasinen E., Sahu B., Khund-Sayeed S., Das P.K., Kivioja T., Dave K., Zhong F.. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017; 356:eaaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou T., Shen N., Yang L., Abe N., Horton J., Mann R.S., Bussemaker H.J., Gordân R., Rohs R.. Quantitative modeling of transcription factor binding specificities using DNA shape. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:4654–4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang L., Orenstein Y., Jolma A., Yin Y., Taipale J., Shamir R., Rohs R.. Transcription factor family‐specific DNA shape readout revealed by quantitative specificity models. Mol. Syst. Biol. 2017; 13:910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mathelier A., Xin B., Chiu T.-P., Yang L., Rohs R., Wasserman W.W.. DNA shape features improve transcription factor binding site predictions in vivo. Cell Syst. 2016; 3:278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zentner G.E., Kasinathan S., Xin B., Rohs R., Henikoff S.. ChEC-seq kinetics discriminates transcription factor binding sites by DNA sequence and shape in vivo. Nat. Commun. 2015; 6:8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abe N., Dror I., Yang L., Slattery M., Zhou T., Bussemaker H.J., Rohs R., Mann R.S.. Deconvolving the recognition of DNA shape from sequence. Cell. 2015; 161:307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou T., Yang L., Lu Y., Dror I., Dantas Machado A.C., Ghane T., Di Felice R., Rohs R.. DNAshape: a method for the high-throughput prediction of DNA structural features on a genomic scale. Nucleic Acids Res. 2013; 41:W56–W62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rohs R., Jin X., West S.M., Joshi R., Honig B., Mann R.S.. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010; 79:233–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rohs R., West S.M., Sosinsky A., Liu P., Mann R.S., Honig B.. The role of DNA shape in protein–DNA recognition. Nature. 2009; 461:1248–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Consortium E.P. The ENCODE (ENCyclopedia of DNA elements) project. Science. 2004; 306:636–640. [DOI] [PubMed] [Google Scholar]
- 19. Mukherjee S., Berger M.F., Jona G., Wang X.S., Muzzey D., Snyder M., Young R.A., Bulyk M.L.J.N.G.. Rapid analysis of the DNA-binding specificities of transcription factors with DNA microarrays. Nat. Genet. 2004; 36:1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rhee H.S., Pugh B.F.. ChIP‐exo method for identifying genomic location of DNA‐binding proteins with near‐single‐nucleotide accuracy. Curr.Protoc. Mol. Biol. 2012; 100:doi:10.1002/0471142727.mb2124s100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Isakova A., Groux R., Imbeault M., Rainer P., Alpern D., Dainese R., Ambrosini G., Trono D., Bucher P., Deplancke B.. SMiLE-seq identifies binding motifs of single and dimeric transcription factors. Nat. Methods. 2017; 14:316–322. [DOI] [PubMed] [Google Scholar]
- 22. Park P.J. ChIP–seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009; 10:669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nakato R., Shirahige K.. Recent advances in ChIP-seq analysis: from quality management to whole-genome annotation. Brief. Bioinform. 2016; 18:279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu B., Yang J., Li Y., McDermaid A., Ma Q.. An algorithmic perspective of de novo cis-regulatory motif finding based on ChIP-seq data. Brief. Bioinform. 2017; 19:1069–1081. [DOI] [PubMed] [Google Scholar]
- 25. Alipanahi B., Delong A., Weirauch M.T., Frey B.J.. Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015; 33:831–838. [DOI] [PubMed] [Google Scholar]
- 26. Zhou J., Troyanskaya O.G.. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods. 2015; 12:931–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stormo G.D., Roy B.. DNA structure helps predict protein binding. Cell Syst. 2016; 3:216–218. [DOI] [PubMed] [Google Scholar]
- 28. Khan A., Fornes O., Stigliani A., Gheorghe M., Castro-Mondragon J.A., van der Lee R., Bessy A., Chèneby J., Kulkarni S.R., Tan G.. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2017; 46:D260–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matys V., Fricke E., Geffers R., Gößling E., Haubrock M., Hehl R., Hornischer K., Karas D., Kel A.E., Kel-Margoulis O.V.. TRANSFAC®: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003; 31:374–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kelley D.R., Snoek J., Rinn J.L.. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 2016; 26:990–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Machanick P., Bailey T.L.. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011; 27:1696–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guo Y., Tian K., Zeng H., Guo X., Gifford D.K.J.G.R.. A novel k-mer set memory (KSM) motif representation improves regulatory variant prediction. Genome Res. 2018; 28:891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ghandi M., Mohammad-Noori M., Ghareghani N., Lee D., Garraway L., Beer M.A.. gkmSVM: an R package for gapped-kmer SVM. Bioinformatics. 2016; 32:2205–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ching T., Himmelstein D.S., Beaulieu-Jones B.K., Kalinin A.A., Do B.T., Way G.P., Ferrero E., Agapow P.-M., Xie W., Rosen G.L.. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface. 2017; 15:20170387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Park Y., Kellis M.. Deep learning for regulatory genomics. Nat. Biotechnol. 2015; 33:825–826. [DOI] [PubMed] [Google Scholar]
- 37. Mariani L., Weinand K., Vedenko A., Barrera L.A., Bulyk M.L.. Identification of human lineage-specific transcriptional coregulators enabled by a glossary of binding modules and tunable genomic backgrounds. Cell Syst. 2017; 5:187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Consortium E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kharchenko P.V., Tolstorukov M.Y., Park P.J.. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat. Biotechnol. 2008; 26:1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li Q., Brown J.B., Huang H., Bickel P.J.. Measuring reproducibility of high-throughput experiments. The annals of applied statistics. 2011; 5:1752–1779. [Google Scholar]
- 41. Chiu T.-P., Comoglio F., Zhou T., Yang L., Paro R., Rohs R.. DNAshapeR: an R/Bioconductor package for DNA shape prediction and feature encoding. Bioinformatics. 2015; 32:1211–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zeng H., Edwards M.D., Liu G., Gifford D.K.. Convolutional neural network architectures for predicting DNA–protein binding. Bioinformatics. 2016; 32:i121–i127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Arvey A., Agius P., Noble W.S., Leslie C.. Sequence and chromatin determinants of cell-type–specific transcription factor binding. Genome Res. 2012; 22:1723–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Setty M., Leslie C.S.. SeqGL identifies context-dependent binding signals in genome-wide regulatory element maps. PLoS Comput. Biol. 2015; 11:e1004271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S.. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012; 22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Keskar N.S., Mudigere D., Nocedal J., Smelyanskiy M., Tang P.T.P.. On large-batch training for deep learning: Generalization gap and sharp minima. 2016; arXiv doi: https://arxiv.org/abs/1609.04836, 15 September 2016, preprint: not peer reviewed.
- 47. LeCun Y., Boser B., Denker J.S., Henderson D., Howard R.E., Hubbard W., Jackel L.D.. Backpropagation applied to handwritten zip code recognition. Neural. Comput. 1989; 1:541–551. [Google Scholar]
- 48. Bergstra J., Bengio Y.. Random search for hyper-parameter optimization. J. Mach. Learn. Res. 2012; 13:281–305. [Google Scholar]
- 49. Abadi M., Barham P., Chen J., Chen Z., Davis A., Dean J., Devin M., Ghemawat S., Irving G., Isard M.. TensorFlow: A System for Large-Scale Machine Learning. OSDI. 2016; 16:265–283. [Google Scholar]
- 50. Crooks G.E., Hon G., Chandonia J.-M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bailey T.L., Williams N., Misleh C., Li W.W.. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006; 34:W369–W373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bailey T.L. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011; 27:1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kulakovskiy I.V., Boeva V., Favorov A.V., Makeev V.J.J.B.. Deep and wide digging for binding motifs in ChIP-Seq data. Bioinformatics. 2010; 26:2622–2623. [DOI] [PubMed] [Google Scholar]
- 54. Gupta S., Stamatoyannopoulos J.A., Bailey T.L., Noble W.S.. Quantifying similarity between motifs. Genome Biol. 2007; 8:R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Arriza J.L., Weinberger C., Cerelli G., Glaser T.M., Handelin B.L., Housman D.E., Evans R.M.. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science. 1987; 237:268–275. [DOI] [PubMed] [Google Scholar]
- 56. Shaulian E., Karin M.. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002; 4:E131–E136. [DOI] [PubMed] [Google Scholar]
- 57. Wingender E., Schoeps T., Haubrock M., Krull M., Dönitz J.. TFClass: expanding the classification of human transcription factors to their mammalian orthologs. Nucleic Acids Res. 2017; 46:D343–D347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Najafabadi H.S., Mnaimneh S., Schmitges F.W., Garton M., Lam K.N., Yang A., Albu M., Weirauch M.T., Radovani E., Kim P.M.. C2H2 zinc finger proteins greatly expand the human regulatory lexicon. Nat. Biotechnol. 2015; 33:555–562. [DOI] [PubMed] [Google Scholar]
- 59. Phillips J.E., Corces V.G.. CTCF: master weaver of the genome. Cell. 2009; 137:1194–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Neph S., Vierstra J., Stergachis A.B., Reynolds A.P., Haugen E., Vernot B., Thurman R.E., Sandstrom R., Johnson A.K., Maurano M.T.. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012; 489:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pollard K.S., Hubisz M.J., Rosenbloom K.R., Siepel A.. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010; 20:110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gu Z., Gu L., Eils R., Schlesner M., Brors B.. circlize implements and enhances circular visualization in R. Bioinformatics. 2014; 30:2811–2812. [DOI] [PubMed] [Google Scholar]
- 64. Weirauch M.T., Yang A., Albu M., Cote A.G., Montenegro-Montero A., Drewe P., Najafabadi H.S., Lambert S.A., Mann I., Cook K.J.C.. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014; 158:1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Consortium U. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017; 45:D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang J., Zhuang J., Iyer S., Lin X., Whitfield T.W., Greven M.C., Pierce B.G., Dong X., Kundaje A., Cheng Y.. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012; 22:1798–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lonard D.M., O’Malley B.W.. The expanding cosmos of nuclear receptor coactivators. Cell. 2006; 125:411–414. [DOI] [PubMed] [Google Scholar]
- 68. Stender J.D., Kim K., Charn T.H., Komm B., Chang K.C., Kraus W.L., Benner C., Glass C.K., Katzenellenbogen B.S.. Genome-wide analysis of estrogen receptor α DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol. Cell Biol. 2010; 30:3943–3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cheung E., Acevedo M.L., Cole P.A., Kraus W.L.. Altered pharmacology and distinct coactivator usage for estrogen receptor-dependent transcription through activating protein-1. PNAS. 2005; 102:559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chatr-Aryamontri A., Oughtred R., Boucher L., Rust J., Chang C., Kolas N.K., O’Donnell L., Oster S., Theesfeld C., Sellam A. et al.. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017; 45:D369–D379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Blackwood E.M., Eisenman R.N.. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science. 1991; 251:1211–1217. [DOI] [PubMed] [Google Scholar]
- 72. Nair S.K., Burley S.K.. X-ray structures of Myc-Max and Mad-Max recognizing DNA: molecular bases of regulation by proto-oncogenic transcription factors. Cell. 2003; 112:193–205. [DOI] [PubMed] [Google Scholar]
- 73. Ching T., Himmelstein D.S., Beaulieu-Jones B.K., Kalinin A.A., Do B.T., Way G.P., Ferrero E., Agapow P.M., Zietz M., Hoffman M.M. et al.. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface. 2018; 15:20170387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mathelier A., Xin B., Chiu T.P., Yang L., Rohs R., Wasserman W.W.. DNA shape features improve transcription factor binding site predictions in vivo. Cell Syst. 2016; 3:278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kundaje A., Kyriazopoulou-Panagiotopoulou S., Libbrecht M., Smith C.L., Raha D., Winters E.E., Johnson S.M., Snyder M., Batzoglou S., Sidow A.. Ubiquitous heterogeneity and asymmetry of the chromatin environment at regulatory elements. Genome Res. 2012; 22:1735–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pal S., Hoinka J., Przytycka T.M.. Co-SELECT reveals sequence non-specific contribution of DNA shape to transcription factor binding in vitro. Nucleic Acids Res. 2019; 47:6632–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Aibar S., Gonzalez-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J. et al.. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017; 14:1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Rossi M.J., Lai W.K.M., Pugh B.F.. Correspondence: DNA shape is insufficient to explain binding. Nat. Commun. 2017; 8:15643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cuellar-Partida G., Buske F.A., McLeay R.C., Whitington T., Noble W.S., Bailey T.L.. Epigenetic priors for identifying active transcription factor binding sites. Bioinformatics. 2012; 28:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Duren Z., Chen X., Jiang R., Wang Y., Wong W.H.. Modeling gene regulation from paired expression and chromatin accessibility data. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E4914–E4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
An integrated web server for DESSO is freely available at https://bmbl.bmi.osumc.edu/DESSO. The source code of DESSO and a detailed tutorial can be found at https://github.com/OSU-BMBL/DESSO.