

Abstract
Folate receptor alpha (αFR) is overexpressed in 90% of ovarian cancers, one of the most lethal gynecologic cancers. Recent studies have suggested that natural killer (NK) cells may be better chimeric antigen receptor (CAR) drivers because of their favorable innate characteristics, such as directly recognizing and killing tumor cells. However, the therapeutic effects of CAR-engineered NK cells targeting αFR in ovarian cancer have not been reported. In this research, 3 generations of anti-αFR CAR were constructed, namely αFR-ζ (first generation), αFR-28ζ (second generation), and αFR-28BBζ (third generation), and were highly expressed on the surface of NK-92 cells by lentivirus gene transfection. Three anti-αFR CAR-engineered NK-92 cells can specifically kill αFR-positive tumor cells in vitro, especially ovarian cancer cells with high αFR expression. Compared with NK-92 cells expressing αFR-ζ or αFR-28ζ, NK-92 cells expressing αFR-28BBζ showed not only higher antigen-specific cytotoxicity and proliferation but also lower antigen-induced apoptosis. Moreover, stronger degranulation and cytokine secretion were detected in NK-92 cells expressing αFR-28BBζ cocultured with αFR-positive tumor cells. Real-time cell analysis and live cell imaging recorded the process of NK-92 cells expressing αFR-28BBζ killing ovarian cancer cells in vitro. Furthermore, NK-92 cells expressing αFR-28BBζ can effectively eliminate cancer cells in a mouse xenograft model of ovarian cancer and significantly prolong the survival of tumor-bearing mice. These results demonstrate that the anti-αFR CARs redirect NK-92 cells with specific antitumor activity, and the third-generation anti-αFR CAR-engineered NK-92 cells display more potent cytotoxicity against αFR-positive ovarian cancer, laying the foundation for future clinical research.
Key Words: ovarian cancer, folate receptor alpha, immunotherapy, chimeric antigen receptor, natural killer cells
Ovarian cancer is the most common and most lethal among gynecologic tumors.1 Because of its insidious onset and lack of effective early diagnostic methods, >60% of ovarian cancer patients are diagnosed in the advanced stage. Moreover, although the treatment regimens for ovarian cancer have been improved, the 5-year survival rate of patients has not been significantly improved.2 Therefore, new methods are urgently needed in the clinical treatment for ovarian cancer.
Numerous studies have confirmed that chimeric antigen receptor (CAR)-engineered immune cells represent a powerful approach for tumor therapy.3–6 The structure of CAR is typically composed of a single-chain variable fragment (scFv) from an antibody that targets tumor antigens and different intracellular signaling domains from lymphocyte-activated receptors.5 On the basis of this novel structure, CAR-engineered immune cells can specifically recognize and kill target cells independent of MHC restriction and antigen presentation.3 Presently, several clinical trials have confirmed that treatment with CAR-engineered T cells (CAR-T cells) is effective for various tumors such as leukemia and lymphoma and, therefore, has great potential for clinical application.4,7
Before constructing a CAR, it is first necessary to find a suitable tumor antigen. Folate receptor alpha (αFR) is highly expressed in 90% of ovarian cancers but is not expressed in normal tissues or is restricted to the apical surface of polarized epithelial cells.8,9 In addition, αFR expression is not affected by previous chemotherapy.10 Thus, αFR represents an ideal tumor antigen for targeted treatment of ovarian cancer. Kershaw and colleagues first constructed CAR-T cells targeting αFR using the murine MOv18 scFv and signaling domain of the Fc receptor γ chain and used the CAR-T cells to treat 8 patients with ovarian cancer. Although the CAR-T cells did not show the desired therapeutic effects and induced human anti-mouse antibodies (HAMA) in the recipients, the results confirmed that CAR-T cells targeting αFR can be safely administered to patients, encouraging many researchers to conduct related studies.11 In the follow-up studies, the researchers replaced the murine anti-αFR scFv in the CAR structure with an anti-αFR scFv derived from the human antibody C4 and confirmed that this CAR can also functionally redirect T cells with specific antitumor activity to αFR-positive ovarian cancer cells in preclinical experiments.12
Recently, many studies have suggested that natural killer (NK) cells may be better CAR drivers.13,14 However, primary NK cells have similar defects as primary T cells. For example, the expansion capacity of NK cells from peripheral blood varies greatly among different patients, and the efficiency of gene transfection is low.15,16 Furthermore, the survival time of primary NK cells is limited.17 However, the human NK cell line NK-92 may address these limitations. NK-92 cells can be effectively expanded in a Good Manufacturing Practice (GMP)-compliant process and are also more susceptible to genetic manipulation by viral or nonviral methods.17,18 More importantly, early clinical trials have demonstrated the safety of NK-92 cells as an allogeneic cell therapeutic in patients with advanced malignancies.19,20 Currently, CAR-engineered NK-92 cells targeting CD19, CD133, human epidermal growth factor receptor 2 (Her2), epidermal growth factor receptor (EGFR), and EGFR variant III mutant (EGFRvIII) have been reported, and anti-Her2 CAR-engineered NK-92 cells are ready for phase I clinical trials.21–24 However, no report has been published on anti-αFR CAR-engineered NK-92 cells (NK-92-αFR-CAR).
In the present study, we constructed all 3 generations of fully humanized anti-αFR CAR (αFR-CAR) on the basis of the scFv fragment derived from human antibody C4, the first generation containing the CD3ζ signaling domain (αFR-ζ), the second generation containing the composite CD28-CD3ζ signaling domain (αFR-28ζ), and the third generation containing the composite CD28-CD137 (4-1BB)-CD3ζ signaling domain (αFR-28BBζ). These 3 αFR-CARs were expressed in NK-92 cells by lentiviral gene transfer. In addition, we systematically confirmed the cytotoxicity of the NK-92-αFR-CAR cells against αFR-positive ovarian cancer cells in vitro and in vivo by the LDH cytotoxicity assay, cytokine release assay, real-time cell analysis, live cell imaging, and mouse xenograft model of ovarian cancer. The antitumor activity, antigen-specific proliferation, and antigen-induced apoptosis of different generations of αFR-CAR-engineered NK-92 cells were also compared. Our results indicated that it is feasible to construct NK-92-αFR-CAR cells and that the composite CD28-CD137-CD3ζ domain can yield stronger activation signals for NK-92 cells, providing the rationale for the development of NK-92 cell-based therapy for αFR-positive ovarian cancer.
MATERIALS AND METHODS
Cell Lines and Culture Conditions
The human ovarian cancer cell line SK-OV-3 and human epidermoid carcinoma cell line A-431 were kindly provided by Stem Cell Bank, Chinese Academy of Sciences (Shanghai, China). The human ovarian cancer cell line A2780 was purchased from Sigma-Aldrich (St Louis). The human colorectal cancer cell line HCT 116 was kindly provided by Jian Yu, University of Pittsburgh. All cells were cultured in the recommended culture media supplemented with 10% fetal bovine serum (FBS). The human NK cell line NK-92 was presented by Jianhua Yu, Ohio State University. NK-92 cells were maintained in RPMI-1640 supplemented with 20% FBS and 150 IU/mL of recombinant human interleukin-2 (IL-2) (Genscript, Nanjing, China). All the above cell lines were cultured at 37°C in a 5% CO2 incubator. All stocks of the above cell culture medium and FBS were purchased from Gibco (Grand Island).
CAR Construction and Lentivirus Production
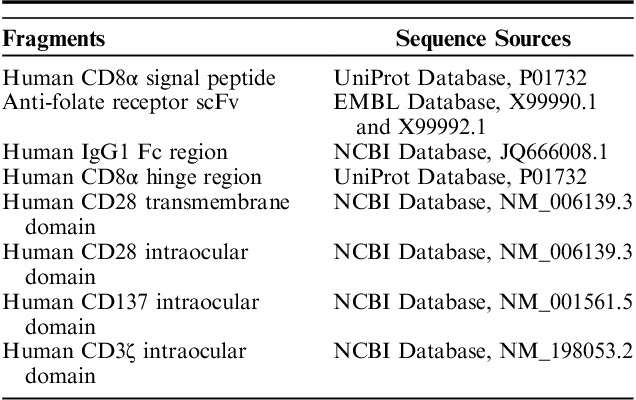
All 3 generations of αFR-CAR have a CD8α signal peptide, an anti-αFR scFv derived from human antibody C4, a human IgG1 Fc fragment, a CD8α hinge region, and a CD28 transmembrane region. In addition, αFR-ζ contains a CD3ζ intracellular signaling domain, αFR-28ζ contains a composite CD28-CD3ζ intracellular signaling domain, and αFR-28BBζ contains a composite CD28-CD137-CD3ζ intracellular signaling domain. The sequence information of these fragments is shown in Table 1. The codon-optimized CAR sequence was synthesized and ligated into the lentiviral expression plasmid pLenti (OBiO, Shanghai, China), downstream of the elongation factor 1 alpha (EF1α) promoter. High-titer mock lentiviral particles, as well as lentiviral particles expressing green fluorescent protein (GFP), red fluorescent protein (RFP), αFR-ζ, αFR-28ζ, αFR-28BBζ, αFR-28BBζ with GFP, or firefly luciferase, were produced by OBiO (Shanghai, China).
TABLE 1.
The Sequence Information of αFR-CAR
Cell Transfection and Analysis of αFR-CAR Expression
The transfection procedures were modified from a previously published protocol.25 After transfection, stable transfectants were selected for 2 weeks in the relative complete growth medium containing 5 mg/mL of puromycin dihydrochloride (Beyotime, Beijing, China). The expression of different αFR-CAR mRNAs was detected by quantitative real-time polymerase chain reaction (qPCR), which was performed as described previously.26 The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as an endogenous control. The primers for αFR-CARs were 5′-TCCCAGTTACCGCCCTTCTCCT-3′ (sense) and 5′-ATGCTCTGTCCAGGAGACCCAGAC-3′ (anti-sense). The primers for GAPDH were 5′-GCACCGTCAAGGCTGAGAAC-3′ (sense) and 5′-TGGTGAAGACGCCAGTGGA-3′ (antisense). The expression of αFR-CARs on the cell surface was analyzed by flow cytometry, which was conducted as described previously.24 FITC-conjugated goat anti-human IgG Fc antibody (Jackson ImmunoResearch, West Grove) and FITC-conjugated goat IgG isotype control (Abcam, Cambridge, UK) were used in flow cytometry, which was performed using a BD FACS Canto II flow cytometer (BD Biosciences, San Jose), and the data were analyzed using CellQuest Pro software (BD Biosciences).
αFR Expression and LDH Cytotoxicity Assay
αFR expression on the surface of cancer cells was investigated by flow cytometry using PE-conjugated mouse anti-human FOLR1 antibody (R&D SYSTEMS, Minneapolis) and PE-conjugated mouse IgG isotype control (BD Biosciences). Flow cytometry was performed using a BD FACS Canto II flow cytometer (BD Biosciences), and the data were analyzed using CellQuest Pro software (BD Biosciences). The cytotoxicity of the effector cells (NK-92 cells and transfected NK-92 cells) was measured using the LDH cytotoxicity assay kit (DOJINDO, Tokyo, Japan). After coculture of the target cells and the effector cells for 18 hours in 96-well plates at different effector-to-target (E/T) ratios, the supernatants were collected. According to the manufacturer’s instructions, the LDH content in the supernatants was detected, and the specific cell lysis of the effector cells at different E/T ratios was calculated.
Analysis of Cell Proliferation and Apoptosis
To detect the proliferation of effector cells, the wells of a 96-well plate were coated with 5 μg/mL of recombinant human αFR protein (Ybio, Shanghai, China) overnight, and then the wells were washed with phosphate-buffered saline (PBS), followed by the addition of 100 μL of medium containing 2000 effector cells without IL-2. After 48 hours of culture, Cell Counting Kit-8 (CCK-8) (Beyotime) was used to assess the viable cells in each well according to the manufacturer’s instructions. For apoptosis assays, the effector cells were cocultured with SK-OV-3 or A-431 cells at an E/T ratio of 2:1. After 48 hours, the effector cells were harvested, and apoptotic cells were stained with the Annexin V-FITC apoptosis detection kit (KeyGen Biotech, Nanjing, China) according to the manufacturer’s instructions. The stained cells were analyzed using the BD FACS Canto II flow cytometer and CellQuest Pro software (BD Biosciences).
Real-time Cell Analysis (RTCA)
The antitumor activity of effector cells was also evaluated by RTCA using the xCELLigence RTCA TP system (ACEA, Hangzhou, China). Before seeding the target cells, 50 μL of the cell culture medium was added to the wells of an E-plate 16, which was placed onto the instrument to examine the background readings of each well. The E-plate 16 was then removed, and 50 μL of medium containing the appropriate number of target cells (SK-OV-3 or A-431: 4000) was added to the wells and kept still for 30 minutes. The E-plate 16 was then returned to the instrument, and the assay program was continued. The effector cells were added to the wells at different E/T ratios 24 hours later, the total volume of each well was supplemented to 200 μL with the corresponding medium, and the assay program was continued.
Degranulation Assay and Analysis of Cytokine Secretion
Degranulation of the effector cells was analyzed by detecting the cell-surface expression of CD107a using the BD FastImmune CD107a Kit (BD Biosciences). Briefly, different effector cells were cocultured with target cells for 1 hour at an E/T ratio of 10:1 in the presence of APC-conjugated mouse anti-human CD107a antibody. Next, 1 μL of GolgiStop (BD Biosciences) was added to the medium, followed by further incubation for 4 hours. At the end of the coculture, the effector cells were collected and washed, and cell-surface expression of CD107a was analyzed using the BD FACS Canto II flow cytometer and CellQuest Pro software (BD Biosciences). Cytokine secretion of the effector cells was determined by enzyme-linked immunosorbent assay (ELISA). To detect interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α), the effector cells were cocultured with the target cells for 18 hours at an E/T ratio of 10:1, and then IFN-γ and TNF-α in the supernatant of the culture medium were determined using the corresponding ELISA kits (BOSTER, Wuhan, China) according to the manufacturer’s instructions. To detect IL-2, the effector cells were cultured in αFR-coated (5 μg/mL) or uncoated wells of 48-well plates using medium without IL-2. After 18 hours of incubation, IL-2 in the supernatant of the culture medium was determined using the IL-2 ELISA kits (BOSTER) according to the manufacturer’s instructions.
Live Cell Imaging
The process of effector cells killing target cells in vitro was observed using the IXplore live cell imaging system (Olympus, Tokyo, Japan). RFP-expressing SK-OV-3 (SK-OV-3-RFP) cells were seeded at 4000 per well in 96-well plates. After overnight culture, NK-92-GFP cells or NK-92-αFR-28BBζ-GFP cells were cocultured with the SK-OV-3 cells at an E/T ratio of 10:1. After 30 minutes of incubation, the 96-well plate was placed in the live cell imaging system incubator and analyzed, as described in the manufacturer’s instructions. The time-lapse images were recorded every 30 minutes for a duration of 4 hours. All imaging data were collected and analyzed by cellSens software (Olympus).
Xenograft Model of Ovarian Cancer and Bioluminescence Imaging
The xenograft ovarian cancer model was established using 6-week-old to 8-week-old female NOD-Prkdcscid IL2rgtm1/Bcgen mice (B-NDG mice) (BIOCYTOGEN, Beijing, China). The mice were inoculated intraperitoneally (IP) with 1×106 SK-OV-3 cells expressing firefly luciferase (fLuc+ SK-OV-3 cells). Two weeks after peritoneal inoculation, the mice bearing well-established SK-OV-3 tumors were randomized into 4 groups to receive treatments with different effector cells or PBS at the indicated doses and time intervals. The mice were euthanized upon signs of distress such as ruffled fur, increased abdominal girth, difficulty breathing, impaired ambulation, inability to remain upright, decreased response to stimuli, or evidence of being moribund. All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee of Daping Hospital and Research Institute of Surgery. Bioluminescence imaging of tumor-bearing mice was performed using the FUSION FX imaging system (Vilber Lourmat, Paris, France). After anesthetizing the mice with 10% chloral hydrate, D-luciferin potassium salt (150 mg/kg) (Beyotime) dissolved in PBS was injected IP, and bioluminescence imaging was performed 5 to 10 minutes later. All images were collected and analyzed by Fusion software (Vilber Lourmat).
Statistical Analysis
The data are presented as means±SEM. SPSS software (version 20.0, SPSS Inc., Chicago) was utilized for statistical analysis. For normally distributed endpoints, 1-way analysis of variance was used to compare among 3 or more independent groups. In addition, for in vivo bioluminescence signal intensity, which is not normally distributed, the Kruskal-Wallis test was used to compare the median of the NK-92-αFR-28BBζ group to that of the NK-92, NK-92-EV, and PBS-treated groups. Survival curves were plotted according to the Kaplan-Meier method and were compared using the log-rank test. The statistical significance level was set at P<0.05.
RESULTS
Construction of CAR and Lentiviral Transfection of NK-92 Cells
The structures of all 3 generations of αFR-CAR are shown in Figure 1A. These αFR-CARs all have a CD8α signal peptide, an anti-αFR scFv derived from human antibody C4, a human IgG1 Fc fragment, a CD8α hinge region, and a CD28 transmembraneTM region. In addition, αFR-ζ contains a CD3ζ intracellular signaling domain, αFR-28ζ contains a composite CD28-CD3ζ intracellular signaling domain, and αFR-28BBζ contains a composite CD28-CD137-CD3ζ intracellular signaling domain. Different αFR-CAR sequences were respectively inserted into the pLenti lentiviral vector backbone, which was subsequently packaged into the corresponding lentiviral particles. NK-92 cells were infected with the lentiviral particles to obtain NK-92-αFR-ζ cells, NK-92-αFR-28ζ cells, and NK-92-αFR-28BBζ cells. The transfected NK-92 cells were selected by 2 weeks of puromycin treatment to obtain the above 3 different homogenous αFR-CAR-expressing NK-92 cell populations. The expression of different αFR-CAR in the transfected NK-92 cells was detected by qPCR and flow cytometry. As shown in Figure 1B, αFR-CAR mRNA was only highly expressed in NK-92-αFR-ζ cells, NK-92-αFR-28ζ cells, and NK-92-αFR-28BBζ cells but not in NK-92 cells transfected with empty vector (NK-92-EV cells) and uninfected NK-92 cells. Similarly, high-level surface expression of CAR was observed on NK-92-αFR-ζ cells, NK-92-αFR-28ζ cells, and NK-92-αFR-28BBζ cells by flow cytometry using the antibody against the IgG1 Fc fragment of different αFR-CARs but was not detected on NK-92-EV cells or NK-92 cells (Fig. 1C). These results indicated that all of the above 3 αFR-CARs can be highly expressed in NK-92 cells by lentiviral transfection.
FIGURE 1.
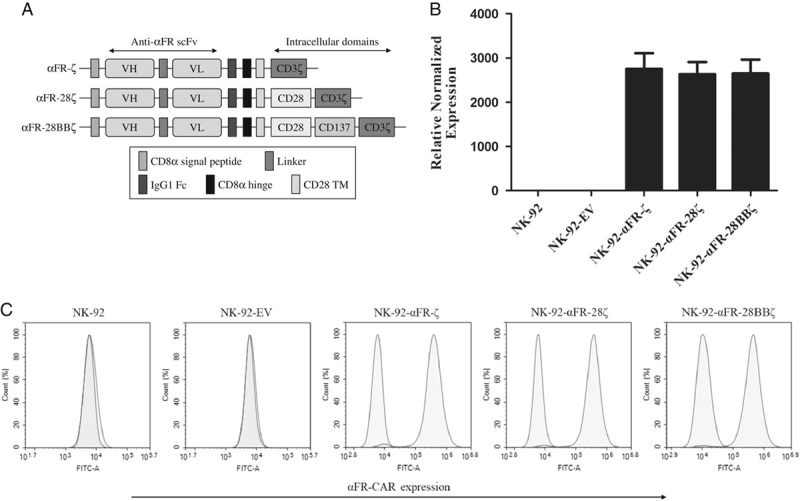
Construction and expression of αFR-CARs in NK-92 cells. A, Schematic representation of αFR-CARs. All 3 generations of αFR-CAR contained a CD8α signal peptide, an anti-αFR scFv, an IgG1 Fc fragment, a CD8α hinge region, and a CD28 transmembrane (TM) region. Moreover, αFR-ζ contained a CD3ζ intracellular signaling domain, αFR-28ζ contained a composite CD28-CD3ζ intracellular signaling domain, and αFR-28BBζ contained a composite CD28-CD137-CD3ζ intracellular signaling domain. B, The relative expression levels of different αFR-CAR mRNAs normalized to GAPDH mRNA expression in NK-92 cells were detected by qPCR. The data are expressed as the means±SEM of triplicate samples. C, Surface expression of αFR-CARs was analyzed by flow cytometry. The filled green histograms indicate isotype control, whereas the filled red histograms indicate the αFR-CARs expression.
Specific Cytotoxicity of NK-92-αFR-CAR Cells Against αFR-positive Cancer Cells In Vitro
αFR was highly expressed in most ovarian cancer cell lines, making them suitable to evaluate the function of the effector cells. In addition, it was necessary to select tumor cell lines that expressed αFR at a low or undetectable level. Therefore, a panel of established human cancer cell lines was analyzed for αFR expression by flow cytometry. As shown in Figures 2A and B, αFR was highly expressed on the surface of human ovarian cancer cell lines SK-OV-3 and A2780, was low expressed on the human colorectal cancer cell line HCT 116, and was not expressed on the human epidermoid carcinoma cell line A-431, all of which are suited for our functional assays.
FIGURE 2.
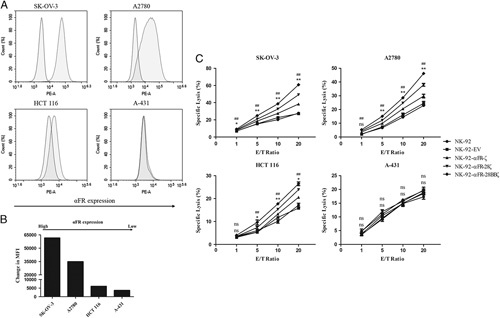
Specific cytotoxicity of NK-92-αFR-CAR cells against αFR-positive cancer cells. A, Surface expression of αFR on the established human cancer cell lines was analyzed by flow cytometry. The filled green histograms indicate isotype control, whereas the filled red histograms indicate αFR expression. B, Surface expression of αFR, as measured by the change in the mean fluorescence intensity (MFI). C, Cell killing by NK-92 cells (filled circles), NK-92-EV cells (filled squares), NK-92-αFR-ζ cells (filled triangles), NK-92-αFR-28ζ cells (filled inverted triangles), and NK-92-αFR-28BBζ cells (filled rhombus) was investigated in the LDH cytotoxicity assay after coculture with target cells at the indicated E/T ratios. All data are expressed as the means±SEM of triplicate samples. Statistical analysis was shown for NK-92-αFR-28BBζ cells versus NK-92-αFR-ζ cells (the upper labels, represents significant difference) or NK-92-αFR-28ζ cells (the nether labels, *represents significant difference). ##P<0.01; **P<0.01; *P<0.05; ns, P≥0.05.
Next, we evaluated the antitumor activity of NK-92-αFR-ζ cells, NK-92-αFR-28ζ cells, and NK-92-αFR-28BBζ cells in vitro using the LDH cytotoxicity assay. When cocultured with SK-OV-3 cells or A2780 cells, NK-92-αFR-ζ cells, NK-92-αFR-28ζ cells, and NK-92-αFR-28BBζ cells showed significantly higher cytotoxicity than NK-92 or NK-92-EV cells at all tested E/T ratios, with higher cytotoxicity at higher E/T ratios. Among them, the killing activity of NK-92-αFR-28BBζ cells against SK-OV-3 cells and A2780 cells was notably stronger than that of NK-92-αFR-ζ cells and NK-92-αFR-28ζ cells, especially at high E/T ratios. However, the effector cells cocultured with HCT 116 cells, NK-92-αFR-ζ cells, and NK-92-αFR-28ζ cells showed significantly higher cytotoxicity only at E/T ratios of 10:1 and 20:1. Moreover, NK-92-αFR-28BBζ cells can markedly kill HCT 116 cells at E/T ratios of 5:1, 10:1, and 20:1, and the killing efficacy was better than that of NK-92-αFR-ζ cells and NK-92-αFR-28ζ cells. The above NK-92-αFR-CAR cells could not kill A-431 more effectively than NK-92 cells or NK-92-EV cells. Notably, all cancer cell lines were intrinsically sensitive to NK-92 cells and NK-92-EV cells (Fig. 2C). These results demonstrated that 3 different NK-92-αFR-CAR cells can specifically kill αFR-positive cancer cells, and with a stronger killing effect for higher αFR expression. Particularly, NK-92-αFR-28BBζ cells containing the CD28, CD137, and CD3ζ intracellular signaling domains showed the strongest antitumor activity.
Enhanced Antigen-specific Proliferation and Attenuated Antigen-induced Apoptosis of NK-92-αFR-28BBζ Cells
To assess antigen-specific proliferation, the above effector cells were cultured in recombinant human αFR protein-coated or uncoated culture plates using a medium without IL-2. Cell proliferation was detected by CCK-8. As shown in Figure 3A, no significant difference was found in the absorbance values of different effector cells when they were cultured in αFR uncoated culture plates. When the effector cells were cultured in αFR-coated culture plates, the absorbance values of the 4 NK-92-αFR-CAR cells were significantly higher than those of NK-92 cells and NK-92-EV cells. In addition, the absorbance values of NK-92-αFR-28BBζ cells were markedly higher than those of NK-92-αFR-ζ cells and NK-92-αFR-28ζ cells. These results suggested that there is no distinct difference in the proliferation of effector cells without antigen stimulation. However, when stimulated with the antigen, the 3 NK-92-αFR-CAR cells showed significantly stronger proliferative activity, especially NK-92-αFR-28BBζ cells.
FIGURE 3.
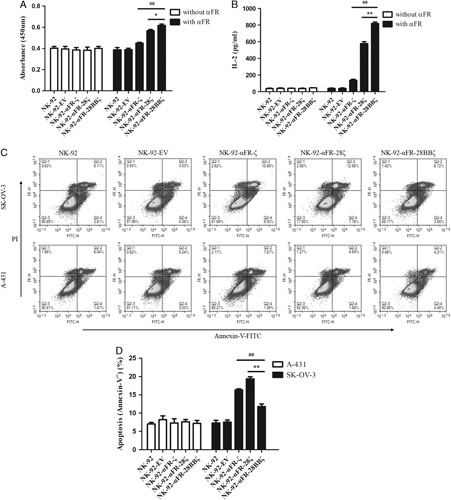
Antigen-specific expansion and antigen-induced apoptosis of NK-92-αFR-CAR cells. A, Expansion of the effector cells in the presence or absence of αFR protein using the CCK-8 assay, as described in the Materials and methods section. B, IL-2 release of the effector cells in the presence or absence of αFR protein using the ELISA assay, as described in the Materials and Methods section. C, Apoptosis of the effector cells induced by SK-OV-3 cells or A-431 cells was analyzed by flow cytometry. D, Apoptosis ratio of the effector cells induced by SK-OV-3 cells or A-431 cells. The E/T ratios are indicated in the Materials and Methods section. All the data are means±SEM of triplicate samples. Statistical analysis is shown for NK-92-αFR-28BBζ cells versus NK-92-αFR-ζ cells (represents significant difference) or NK-92-αFR-28ζ cells (*represents significant difference). ##P<0.01; **P<0.01; *P<0.05.
Because the proliferation of NK-92 cells depends on the stimulation of IL-2, it was determined by the above culture medium and ELISA whether the effector cells are capable of antigen-specific secretion of IL-2. When the effector cells were cultured in αFR-coated plates, significantly higher levels of IL-2 were detected in the supernatants of the 3 NK-92-αFR-CAR cells, and the level of IL-2 in the supernatants of NK-92-αFR-28BBζ cells was higher than that in the supernatants of NK-92-αFR-ζ cells and NK-92-αFR-28ζ cells. However, high levels of IL-2 were not detected in the supernatants when the effector cells were cultured in αFR uncoated culture plates (Fig. 3B). These results demonstrated that αFR protein can specifically induce the IL-2 secretion of the 3 NK-92-αFR-CAR cells, and the IL-2 secretion level of NK-92-αFR-28BBζ cells was the highest, possibly promoting the proliferation of corresponding effector cells.
Furthermore, antigen-induced apoptosis of the effector cells was also detected. After coculture of the effector cells with SK-OV-3 cells or A-431 cells for 48 hours, apoptosis of the effector cells was analyzed by the Annexin V-FITC apoptosis detection kit and flow cytometry. As shown in Figures 3C and D, the percentages of apoptotic cells (Annexin-V+) in the 3 NK-92-αFR-CAR cells were significantly higher than those in NK-92 cells and NK-92-EV cells after coculture with SK-OV-3 cells. However, in the 3 NK-92-αFR-CAR cells, the percentage of apoptotic cells in NK-92-αFR-28BBζ cells was lower than that in NK-92-αFR-ζ cells and NK-92-αFR-28ζ cells. Moreover, when the effector cells were cocultured with A-431 cells, there was no marked difference in the percentage of apoptotic cells among the different effector cells. These results indicated that the apoptosis of the 3 NK-92-αFR-CAR cells was more obvious when cocultured with αFR-positive tumor cells, but the apoptosis level of NK-92-αFR-28BBζ cells was relatively low, possibly related to the CD137 molecule in the CAR structure.
Enhanced Antigen-specific Degranulation and Cytokine Secretion of NK-92-αFR-28BBζ Cells
In the previous experiments, compared with NK-92-αFR-ζ cells and NK-92-αFR-28ζ cells, NK-92-αFR-28BBζ cells showed not only higher antigen-specific cytotoxicity and proliferation but also lower antigen-induced apoptosis. Therefore, NK-92-αFR-28BBζ cells were selected for follow-up studies. To further analyze the activation status of NK-92 cells, NK-92-EV cells and NK-92-αFR-28BBζ cells were cocultured with the tumor cells; the degranulation level and cytokine secretion level of these effector cells were examined. When these effector cells were cocultured with SK-OV-3 cells or A-431 cells for 5 hours, only the NK-92-αFR-28BBζ cells cocultured with SK-OV-3 cells showed a significant increase in CD107a expression. αFR-negative A-431 cells did not induce CD107a expression in NK-92-αFR-28BBζ cells that was higher than that in NK-92 cells and NK-92-EV cells (Figs. 4A, B). Moreover, the secretion levels of IFN-γ and TNF-α in these effector cells were detected by ELISA. As shown in Figures 4C and D, significantly higher levels of IFN-γ and TNF-α were detected in the supernatant of NK-92-αFR-28BBζ cells cocultured with αFR-positive tumor cells, and the levels in the supernatant of the NK-92-αFR-28BBζ cells cocultured with SK-OV-3 cells or A2780 cells were higher than those in the supernatant of cocultured HCT 116 cells. In addition, the levels of IFN-γ or TNF-α in the supernatant of different effector cells cocultured with A-431 cells were roughly equal. These results demonstrated that αFR-positive tumor cells can cause stronger degranulation and cytokine secretion of NK-92-αFR-28BBζ cells, depending on the αFR level in tumor cells.
FIGURE 4.
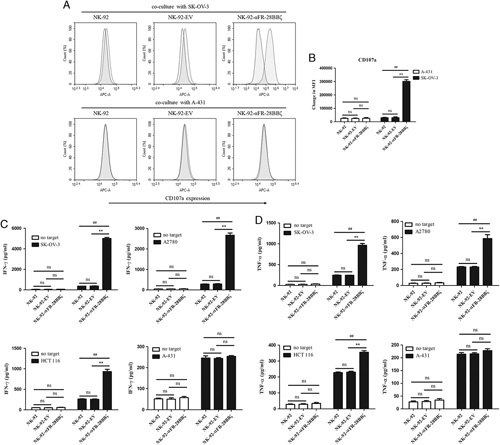
Antigen-specific degranulation and cytokine secretion of NK-92-αFR-28BBζ cells. A, CD107a expression represents the degranulation of NK cells that were analyzed by flow cytometry. The filled green histograms indicate the isotype control, whereas the filled red histograms indicate CD107a expression. B, Surface expression of CD107a, as measured by MFI. C and D, IFN-γ (C) and TNF-α (D) release of NK-92 cells, NK-92-EV cells, and NK-92-αFR-28BBζ cells in the presence or absence of SK-OV-3 cells, A2780 cells, HCT 116 cells, or A-431 cells using the ELISA assay. The E/T ratios are indicated in the Materials and Methods section. All the data are expressed as the means±SEM of triplicate samples. Statistical analysis is shown for NK-92-αFR-28BBζ cells versus NK-92 cells or NK-92-EV cells (*represents significant difference), and NK-92-EV cells versus NK-92 cells. ##P<0.01; **P<0.01; ns, P≥0.05.
Real-time Analysis and Live Cell Imaging of NK-92-αFR-28BBζ Cells Killing αFR-positive Cancer Cells In Vitro
Most other studies have used endpoint detection to assess the killing effects of CAR-engineered immune cells on tumor cells. In this research, the specific cytotoxicity of NK-92-αFR-28BBζ cells was also analyzed by RTCA. SK-OV-3 and A-431 cells were seeded in the assay plates. One day later, NK-92 cells, NK-92-EV cells, and NK-92-αFR-28BBζ cells were added to the coculture with SK-OV-3 cells or A-431 cells at different E/T ratios. As shown in Figure 5A, the cell index curve of SK-OV-3 cells cocultured with the effector cells at the E/T ratios of 5:1, 10:1, and 20:1 showed a decrease during the 100-hour test period. Among them, the cell index curve of SK-OV-3 cells cocultured with NK-92-αFR-28BBζ cells decreased first, indicating a stronger inhibition effect of the NK-92-αFR-28BBζ cells on SK-OV-3 cells. Although the cell index curve of SK-OV-3 cells cocultured with NK-92 cells and NK-92-EV cells also decreased, it was significantly delayed compared with that of the NK-92-αFR-28BBζ cell treatment group, indicating weaker effects of the NK-92 cells and NK-92-EV cells on SK-OV-3 cells. In addition, no decrease in the cell index curve of SK-OV-3 cells was observed at an E/T ratio of 1:1, suggesting that the growth inhibition effects of the effector cells was not significant at this dose. As regards A-431 cells cocultured with the effector cells, a decrease in the cell index was observed at the E/T ratios of 5:1, 10:1, and 20:1 for all 3 effector cells and at a similar time, which was significantly delayed compared with SK-OV-3 cells cocultured with NK-92-αFR-28BBζ cells. In addition, no decrease in the cell index of A-431 cells was observed at the E/T ratio of 1:1 (Fig. 5B). These results confirmed once again that the antitumor activity of NK-92-αFR-28BBζ cells depends on the surface expression of αFR on tumor cells.
FIGURE 5.
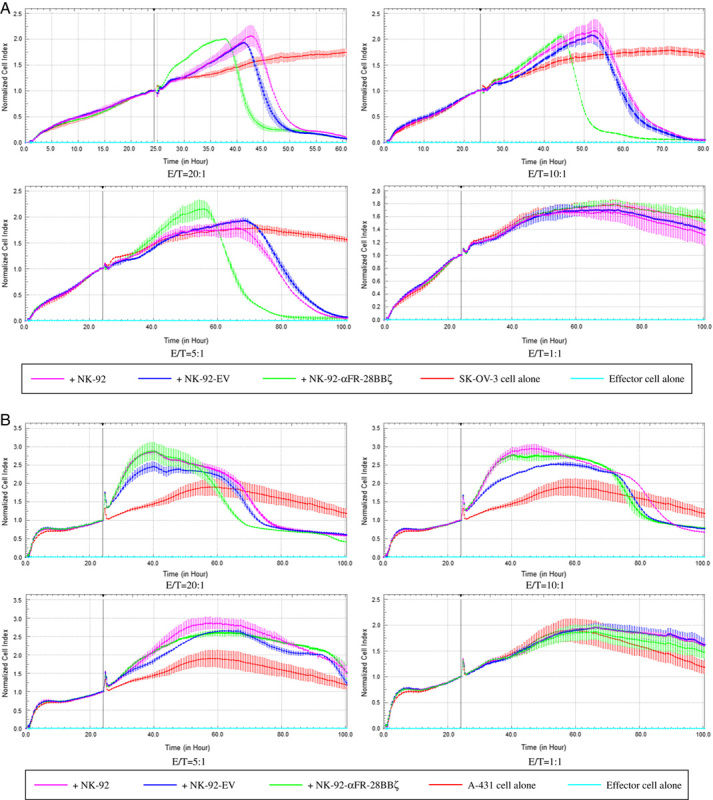
Analysis of NK-92-αFR-28BBζ cell antitumor activity by RTCA. A, Cytotoxic effect of NK-92 cells, NK-92-EV cells, and NK-92-αFR-28BBζ cells on SK-OV-3 cells. The E/T ratios are indicated in the figure. B, Cytotoxic effects of NK-92 cells, NK-92-EV cells, and NK-92-αFR-28BBζ cells on A-431 cells. The E/T ratios are indicated in the figure. All the data are expressed as the means±SEM of triplicate samples.
The live cell imaging system is a powerful tool that is widely used to observe many physiological or pathologic processes. To more clearly and intuitively investigate the process of NK-92-αFR-28BBζ cell killing of ovarian cancer cells, SK-OV-3-RFP cells, NK-92-GFP cells, and NK-92-αFR-28BBζ-GFP cells were constructed. The effector cells were cocultured with the SK-OV-3-RFP cells and were continuously observed using the live cell imaging system for 4 hours. SK-OV-3-RFP cells cultured alone survived normally without abnormal changes during the observation period (Fig. 6A). In the coculture of SK-OV-3-RFP cells with NK-92-GFP cells, although some NK-92-GFP cells were observed in the vicinity of SK-OV-3-RFP cells, the survival of SK-OV-3-RFP cells seemed to be not affected because the morphology and number of SK-OV-3-RFP cells showed no significant change (Fig. 6B). It is interesting to note that, in the coculture of SK-OV-3-RFP cells with NK-92-αFR-28BBζ-GFP cells, the live cell imaging system clearly recorded the process of SK-OV-3 cells killed by NK-92-αFR-28BBζ-GFP cells (as indicated by the arrow in Fig. 6C). It can be seen that several NK-92-αFR-28BBζ-GFP cells surrounded this SK-OV-3-RFP cell. Over time, this SK-OV-3-RFP cell showed a decrease in the cell volume (1 h 0 min) and increase in cytoplasmic density (1 h 30 min), followed by vesicle formation in the cell membrane (3 h 0 min), and finally cell rupture (4 h 0 min). Another SK-OV-3-RFP cell above appears to just begin undergoing similar changes. These results provided image evidence for the specific cytotoxicity of NK-92-αFR-28BBζ cells against the ovarian cancer cell line SK-OV-3.
FIGURE 6.
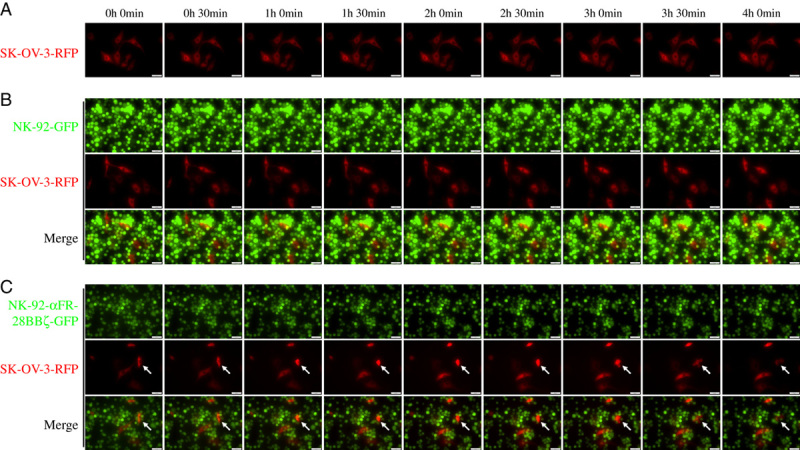
Live cell imaging of NK-92-αFR-28BBζ-GFP cell killing of SK-OV-3-RFP cells. A, SK-OV-3-RFP cells were cultured alone. B, SK-OV-3-RFP cells cocultured with NK-92-GFP cells. C, SK-OV-3-RFP cells cocultured with NK-92-αFR-28BBζ-GFP cells. The E/T ratios are indicated in the Materials and methods section. Scale bar: 50 μm. The arrows indicate SK-OV-3-RFP cells killed by NK-92-αFR-28BBζ-GFP cells.
Potent Antitumor Activity of NK-92-αFR-28BBζ Cells In Vivo
Because NK-92-αFR-28BBζ cells showed significant antitumor activity in vitro, its cytotoxicity was evaluated in a mouse xenograft model of ovarian cancer. The fLuc+ SK-OV-3 cells were constructed to allow in vivo monitoring of tumor growth by bioluminescence imaging. As shown in Figure 7A, B-NDG mice were inoculated IP with 1×106 fLuc+ SK-OV-3 cells. Two weeks after the implantation, these mice began receiving effector cell infusion (defined as day 0). On day 0, day 4, and day 8, these mice were injected IP with 1×106 NK-92 cells, NK-92-EV cells, or NK-92-αFR-28BBζ cells. Mice in the control group were injected IP with PBS. Ovarian cancer xenografts in these mice were observed by bioluminescence imaging on day 0 and day 10, as shown in Figure 7B. Mice in the control group, injected with NK-92 cells or NK-92-EV cells, showed significant enhancement in bioluminescence signals in the abdominal cavity on day 10 compared with those on day 0, with the most significant increase observed in the control group, while the intraperitoneal bioluminescence signals of mice receiving NK-92-αFR-28BBζ cells injection significantly decreased on day 10 (Fig. 7C). In addition, the survival time was significantly longer than that of the control group, NK-92 group, and NK-92-EV group (Fig. 7D). These results suggested that NK-92-αFR-28BBζ cells can effectively eradicate tumor cells, inhibit the development of ovarian cancer in vivo, and prolong the survival of the tumor-bearing mice.
FIGURE 7.
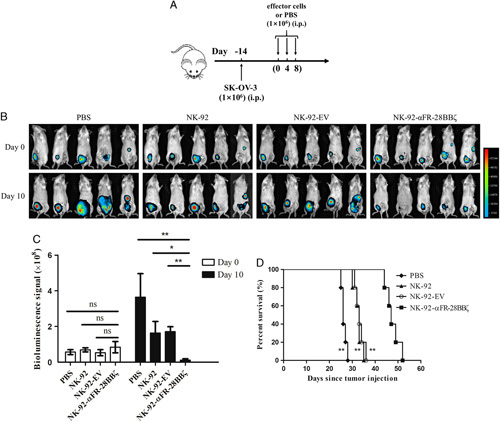
In vivo antitumor activity of NK-92-αFR-28BBζ cells. A, Schematic representation of the construction of mouse xenograft model of ovarian cancer and treatment procedures. B, Ovarian cancer development was monitored by in vivo bioluminescence imaging. Images taken at day 0 and day 10 of the experiment are shown. C, Quantification summary of bioluminescence signal intensity in each group shown in (B). The data are expressed as the means±SEM of 5 mice for all groups. *P<0.05; **P<0.01; ns, P≥0.05. D, Kaplan-Meier survival curves of tumor-bearing mice treated with PBS, NK-92 cells, NK-92-EV cells, or NK-92-αFR-28BBζ cells. **P<0.01, compared with the NK-92-αFR-28BBζ group.
DISCUSSION
Ovarian cancer is not only one of the most common gynecologic tumors but also one of the most common causes of cancer-related death in women.1 Although surgical treatment, chemotherapy and radiotherapy for ovarian cancer are constantly improving, the statistical data of the past 30 years show that the 5-year survival rate of patients with ovarian cancer has not significantly improved. By contrast, chemotherapy resistance, a high recurrence rate, and other problems have emerged.2 Thus, it is urgent to find more effective treatment methods. αFR is a glycophosphatidylinositol-anchored membrane protein that is highly expressed in many types of human tumors, such as ovarian cancer, breast cancer, lung cancer, and colorectal cancer.27 In addition, αFR is not expressed in normal tissues or restrictedly expressed in the apical surface of polarized epithelial cells wherein the circulating drugs can hardly reach.9,12 These characteristics make αFR an excellent target for cancer therapy. Previous studies have reported the use of several anti-αFR monoclonal antibodies to treat ovarian cancer, but no significant effect was observed.28 Recently, researchers have constructed different CAR T cells, which showed promising efficacy and specificity to kill αFR-positive ovarian cancer cells in vitro and in vivo.11,12,27 Moreover, several phase Ι clinical trials have been conducted to assess the safety of CAR-T cells targeting αFR.11 Therefore, we chose αFR as a target to construct CAR-engineered NK-92 cells.
In the early studies involving the construction of anti-αFR CAR, an scFv derived from the murine antibody MOv18 was used. Although clinical trials have demonstrated the safety of CAR based on this scFv, CAR-T cells could not generate antitumor activity in patients due to its short duration in vivo, a finding that may be related to the induction of HAMA in the recipients.11 To solve this problem, the researchers constructed a CAR using an scFv derived from human anti-αFR antibody C4, which reduced the immunogenicity of CAR.12 Furthermore, the cell activation function of CAR depends on the appropriate intracellular signal domains. In general, the first-generation CARs contain only the CD3ζ domain, the second-generation CARs contain a costimulatory signal domain and a CD3ζ domain, and the third-generation CARs contain 2 different costimulatory signal domains and a CD3ζ domain. Common costimulatory signal domains are derived from the costimulatory molecules of immune cells, such as CD28, CD137, CD134, and CD27.3 Although the costimulatory signal provided by the CD28 domain in CAR is sufficient to activate the immune cells, the costimulatory signal from the CD137 domain is required for the immune cells to survive longer and produce a stronger antitumor response.27 In addition, signals from the CD137 domain protect CAR-engineered immune cells from CD3ζ-related activation-induced cell death.29 Zuo and colleagues constructed a CAR comprising an scFv derived from a murine anti-αFR antibody, CD28 and CD137 costimulatory signal domains, and a CD3ζ domain. The results confirmed that cytokine-induced killer cells expressing this CAR were more effective at killing αFR-positive ovarian cancer cells than the first-generation and second-generation CARs.27 In this study, we constructed all 3 generations of fully humanized αFR-CAR on the basis of scFv fragment derived from human antibody C4, with the first generation containing the CD3ζ signaling domain, the second generation containing the composite CD28-CD3ζ signaling domain, and the third generation containing the composite CD28-CD137-CD3ζ signaling domain. Next, these 3 αFR-CARs were expressed in NK-92 cells. Through the LDH cytotoxicity assay, and proliferation and apoptosis analysis, similar results as in the above studies were obtained. Compared with NK-92 cells expressing αFR-ζ or αFR-28ζ, NK-92 cells expressing αFR-28BBζ showed not only higher antigen-specific cytotoxicity and proliferation (Figs. 2C, 3A, B) but also lower antigen-induced apoptosis (Figs. 3C, D). These results demonstrate that the composite CD28-CD137-CD3ζ domain can also provide more sufficient activation signals for NK-92 cells.
CAR can be used to engineer T cells, cytokine-induced killer cells, NK cells, and macrophages, but most of the studies have concerned CAR-T cells.5,24,27,30 Recently, CAR-engineered NK cells have received increasing attention because NK cells have some favorable innate characteristics such as directly recognizing and killing tumor cells.13 However, due to individual differences in tumor patients or donors, it remains uncertain whether primary NK cells isolated and cultured in vitro can be expanded to meet clinical application criteria.15 Moreover, primary NK cells are even more difficult to genetically manipulate than primary T cells, and the survival time of primary NK cells is also short.16,17 These defects limit the further development of CAR-engineered primary NK cells. NK-92 is the most commonly used human NK cell line and can be massively amplified in a complete culture medium containing IL-2.21 While the primary NK cells can often be contaminated with other types of cells, NK-92 cells are a group of highly homogeneous cells and are thus convenient for quality control.22 In addition, NK-92 cells express many cell activation-associated receptors and do not express some inhibitory receptors that are expressed in primary NK cells.31 Furthermore, NK-92 cells are quite susceptible to transfection,18 as shown by our experimental results (Figs. 1B, C). Most importantly, NK-92 cells have been used in clinical trials, fully demonstrating their safety.19,20 Presently, an increasing number of CARs that are effective in CAR-T cells have been shown to confer NK-92 cells with highly specific cytotoxicity.21,24,32 Some CAR-engineered NK-92 cells have even completely met the clinical application requirements and are ready to enter phase I clinical trials.23 These results indicate that NK-92 cells are suitable for the construction of CAR-engineered NK cells and have good clinical application prospects. Thus, we chose to engineer NK-92 cells to express the αFR-CARs described previously.
During the application of CAR, one should always be vigilant concerning its “on-target, off-tumor” toxicity. It was previously thought that the higher the affinity of CAR for tumor antigens, the better the effect, but later observations have suggested that high affinity can also cause severe toxicity.33,34 Although the affinity of human antibody C4 to αFR was slightly lower than that of murine antibody MOv19, Song and colleagues confirmed that the antitumor activity of CAR-T cells constructed with the scFv of antibody C4 was not significantly different from that of CAR-T cells constructed with the scFv of MOv19 antibody, and T cells modified with the former secreted less cytokines when cocultured with normal cells with low expression of αFR.12 This result suggested that CAR constructed with the scFv of antibody C4 may reduce the occurrence of “on-target, off-tumor” toxicity. In addition, the cytokine secreted by activated NK-92 cells is mainly IFN-γ, which also reduces the risk of serious side effects such as cytokine release syndrome during the application of CAR-engineered NK-92 cells.35 This evidences suggests that our NK-92-αFR-CAR cells may be safer for further application in the future.
In the current experiments, qPCR and flow cytometry were used to confirm the high expression of different αFR-CARs in the transfected NK-92 cells. It was then verified that NK-92-αFR-ζ cells, NK-92-αFR-28ζ cells, and NK-92-αFR-28BBζ cells can specifically kill αFR-positive tumor cells in vitro, among which NK-92-αFR-28BBζ cells have the highest killing efficiency. Therefore, NK-92-αFR-28BBζ cells were selected for follow-up studies. Different from most other studies using endpoint detection, we observed the process of NK-92-αFR-28BBζ cells killing the target cells in real time through RTCA and the live cell imaging system. In practice, tumor cells in a patient’s body are constantly proliferating. Therefore, in our RTCA, effector cells were added when the target cells were in the logarithmic growth phase. The RTCA results from this study reflect not only the killing effect of the effector cells on the target cells (decreased cell index curve) but also the process of interaction between the effector cells and target cells. For example, the cell index curve of the coculture system increased right after the addition of the effector cells (Fig. 5), a phenomenon that may be due to the increased detection signals caused by the recognition and combination of effector cells with target cells. Of course, after the effector cells killed the target cells, the cell index curve decreased rapidly. Moreover, continuous imaging data of the effector cells killing the target cells were provided by the live cell imaging system. The results from the LDH cytotoxicity assay, RTCA, and live cell imaging fully demonstrated the specific cytotoxicity of NK-92-αFR-28BBζ cells. Another notable observation was that TNF-α was also induced in the NK-92-αFR-28BBζ cells when cocultured with αFR-positive tumor cells, although at a much lower level than IFN-γ, the main cytokine thought to be secreted by activated NK-92 cells. On the basis of potent cytotoxicity of NK-92-αFR-28BBζ cells in vitro, the xenograft model of ovarian cancer was used to evaluate the antitumor activity of NK-92-αFR-28BBζ cells in vivo, yielding exciting results. NK-92-αFR-28BBζ cells notably inhibited the growth of ovarian cancer in vivo and significantly prolonged the survival time of tumor-bearing mice.
In the previous in vivo experiments, as in other studies, we used nonirradiated CAR-engineered NK-92 cells to treat xenograft ovarian cancer model mice and observed satisfactory antitumor effects.21,24 In the previous clinical trials, the application of NK-92 cells required irradiation to limit the potential leukemic effects.19,20 However, this safety improvement is based on the irradiation significantly attenuating the proliferation activity of NK-92 cells. Thus, compared with CAR-T cells with antigen-specific proliferative capacity, the irradiated CAR-engineered NK-92 cells will not be efficiently and specifically amplified in vivo, weakening their efficacy. Recent studies have shown that excessive proliferation and activation of CAR-T cells can be well controlled by the suicide gene system, suggesting that we can construct controllable CAR-engineered NK-92 cells on the basis of the suicide gene system.36,37 When sufficient therapeutic effects are obtained or side effects occur, the suicide gene system can be used to clear the controllable CAR-engineered NK-92 cells in patients. In this way, CAR-engineered NK-92 cells can exert complete antitumor activity, and their safety in clinical applications is also improved. We have conducted some related research, and the results showed that the survival of CAR-engineered NK-92 cells can be controlled with the suicide gene system (data not shown), suggesting that it is feasible to construct the controllable CAR-engineered NK-92 cells on the basis of the suicide gene system.
In summary, both in vitro and in vivo experiments confirmed that the anti-αFR CARs redirect NK-92 cells with specific antitumor activity, and the third-generation anti-αFR CAR-engineered NK-92 cells display more potent cytotoxicity against αFR-positive ovarian cancer. Our results provide the rationale for NK-92-αFR-CAR cells to enter next-stage research and application.
ACKNOWLEDGMENTS
The authors acknowledge Jianhua Yu (Ohio State University, USA) and Jian Yu (University of Pittsburgh, USA) for their generous help with the present study.
Conflicts of Interest/Financial Disclosures
Supported by the National Key R&D Program (2018YFC1313400), the National Natural Science Foundation of China (NSFC, NO. 81502434), and the Precision Medicine Foundation of Chongqing (No. cstc2016shms-ztzx10006-7).
All authors have declared that there are no financial conflicts of interest with regard to this work.
Footnotes
X.A. and Y.Y. contributed equally to this work.
X.X. and J.G. are co-corresponding authors.
REFERENCES
- 1.Henderson JT, Webber EM, Sawaya GF. Screening for ovarian cancer: updated evidence report and systematic review for the US preventive services task force. JAMA. 2018;319:595–606. [DOI] [PubMed] [Google Scholar]
- 2.Vaughan S, Coward JI, Bast RJ, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011;11:719–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379:64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.June CH, O’Connor RS, Kawalekar OU, et al. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–1365. [DOI] [PubMed] [Google Scholar]
- 5.Chang ZL, Chen YY. CARs: synthetic immunoreceptors for cancer therapy and beyond. Trends Mol Med. 2017;23:430–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Owens GL, Sheard VE, Kalaitsidou M, et al. Preclinical assessment of CAR T-cell therapy targeting the tumor antigen 5T4 in ovarian cancer. J Immunother. 2018;41:130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kochenderfer JN, Somerville R, Lu T, et al. Long-duration complete remissions of diffuse large B cell lymphoma after anti-CD19 chimeric antigen receptor T cell therapy. Mol Ther. 2017;25:2245–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalli KR, Oberg AL, Keeney GL, et al. Folate receptor alpha as a tumor target in epithelial ovarian cancer. Gynecol Oncol. 2008;108:619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelemen LE. The role of folate receptor alpha in cancer development, progression and treatment: cause, consequence or innocent bystander. Int J Cancer. 2006;119:243–250. [DOI] [PubMed] [Google Scholar]
- 10.Despierre E, Lambrechts S, Leunen K, et al. Folate receptor alpha (FRA) expression remains unchanged in epithelial ovarian and endometrial cancer after chemotherapy. Gynecol Oncol. 2013;130:192–199. [DOI] [PubMed] [Google Scholar]
- 11.Kershaw MH, Westwood JA, Parker LL, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12:6106–6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song DG, Ye Q, Poussin M, et al. A fully human chimeric antigen receptor with potent activity against cancer cells but reduced risk for off-tumor toxicity. Oncotarget. 2015;6:21533–21546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leslie M. New cancer-fighting cells enter trials. Science. 2018;361:1056–1057. [DOI] [PubMed] [Google Scholar]
- 14.Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer. 2016;16:7–19. [DOI] [PubMed] [Google Scholar]
- 15.Boissel L, Betancur M, Wels WS, et al. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk Res. 2009;33:1255–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sutlu T, Nystrom S, Gilljam M, et al. Inhibition of intracellular antiviral defense mechanisms augments lentiviral transduction of human natural killer cells: implications for gene therapy. Hum Gene Ther. 2012;23:1090–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suck G, Odendahl M, Nowakowska P, et al. NK-92: an “off-the-shelf therapeutic” for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol Immunother. 2016;65:485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boissel L, Betancur M, Lu W, et al. Comparison of mRNA and lentiviral based transfection of natural killer cells with chimeric antigen receptors recognizing lymphoid antigens. Leuk Lymphoma. 2012;53:958–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arai S, Meagher R, Swearingen M, et al. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy. 2008;10:625–632. [DOI] [PubMed] [Google Scholar]
- 20.Tonn T, Schwabe D, Klingemann HG, et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy. 2013;15:1563–1570. [DOI] [PubMed] [Google Scholar]
- 21.Oelsner S, Friede ME, Zhang C, et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy. 2017;19:235–249. [DOI] [PubMed] [Google Scholar]
- 22.Klapdor R, Wang S, Hacker U, et al. Improved killing of ovarian cancer stem cells by combining a novel chimeric antigen receptor-based immunotherapy and chemotherapy. Hum Gene Ther. 2017;28:886–896. [DOI] [PubMed] [Google Scholar]
- 23.Schonfeld K, Sahm C, Zhang C, et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol Ther. 2015;23:330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han J, Chu J, Keung CW, et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep. 2015;5:11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chu J, Deng Y, Benson DM, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28:917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hao XY, Cai JP, Liu X, et al. EYA4 gene functions as a prognostic marker and inhibits the growth of intrahepatic cholangiocarcinoma. Chin J Cancer. 2016;35:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zuo S, Wen Y, Panha H, et al. Modification of cytokine-induced killer cells with folate receptor alpha (FRalpha)-specific chimeric antigen receptors enhances their antitumor immunity toward FRalpha-positive ovarian cancers. Mol Immunol. 2017;85:293–304. [DOI] [PubMed] [Google Scholar]
- 28.Vergote I, Armstrong D, Scambia G, et al. A randomized, double-blind, placebo-controlled, phase III study to assess efficacy and safety of weekly farletuzumab in combination with carboplatin and taxane in patients with ovarian cancer in first platinum-sensitive relapse. J Clin Oncol. 2016;34:2271–2278. [DOI] [PubMed] [Google Scholar]
- 29.Zhao Y, Wang QJ, Yang S, et al. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol. 2009;183:5563–5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alvey CM, Spinler KR, Irianto J, et al. SIRPA-inhibited, marrow-derived macrophages engorge, accumulate, and differentiate in antibody-targeted regression of solid tumors. Curr Biol. 2017;27:2065–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maki G, Klingemann HG, Martinson JA, et al. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J Hematother Stem Cell Res. 2001;10:369–383. [DOI] [PubMed] [Google Scholar]
- 32.Zhang C, Burger MC, Jennewein L, et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J Natl Cancer Inst. 2016;108:djv375. [DOI] [PubMed] [Google Scholar]
- 33.Morgan RA, Yang JC, Kitano M, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klingemann H. Are natural killer cells superior CAR drivers. Oncoimmunology. 2014;3:e28147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gargett T, Brown MP. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol. 2014;5:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minagawa K, Al-Obaidi M, Di Stasi A. Generation of suicide gene-modified chimeric antigen receptor-redirected T-cells for cancer immunotherapy. Methods Mol Biol. 2019;1895:57–73. [DOI] [PubMed] [Google Scholar]