

Abstract
SIRT6 is critical for activating transcription of Nuclear factor (erythroid-derived 2)-like 2 (NRF2) responsive genes during oxidative stress. However, while the mechanism of SIRT6-mediated silencing is well understood, the mechanism of SIRT6-mediated transcriptional activation is unknown. Here, we employed SIRT6 separation of function mutants to reveal that SIRT6 mono-ADP-ribosylation activity is required for transcriptional activation. We demonstrate that SIRT6 mono-ADP-ribosylation of BAF170, a subunit of BAF chromatin remodeling complex, is critical for activation of a subset of NRF2 responsive genes upon oxidative stress. We show that SIRT6 recruits BAF170 to enhancer region of the Heme oxygenase-1 locus and promotes recruitment of RNA polymerase II. Furthermore, SIRT6 mediates the formation of the active chromatin 10-kb loop at the HO-1 locus, which is absent in SIRT6 deficient tissue. These results provide a novel mechanism for SIRT6-mediated transcriptional activation, where SIRT6 mono-ADP-ribosylates and recruits chromatin remodeling proteins to mediate the formation of active chromatin loop.
INTRODUCTION
Cells respond to oxidative stress, metabolic stress, or bacterial invasion by inducing a wide array of sophisticated stress-response pathways (1–3). One of such host defense mechanism, the NRF2 system, is involved in oxidative-stress response, metabolism, and innate immunity, and the deregulation this response is associated with multiple human diseases, including tumor development and aging (4). Accumulation of reactive oxygen species (5) in the cell is prevented through the actions of small antioxidant molecules such as Glutathione (GSH) and vitamins, and antioxidant enzymes such as heme oxygenase 1 (HO-1), catalase, quinone oxidoreductase 1 (NQO1), thioredoxin reductase (TXNRD1) and superoxide dismutase (SOD) (4). Such concerted gene induction of the antioxidant response occurs through common antioxidant responsive elements (ARE) that are located in the promoter regions of these genes and are bound by the nuclear factor erythroid 2–related factor 2 (NRF2) (5–7). Interestingly, the upregulation of these genes has been shown to extend lifespan in flies and worms (4,7,8).
The gene expressing HO-1 is one of the NRF2-targeted, ARE-containing genes, where HO-1 catabolizes heme into equimolar amounts of labile Fe, carbon monoxide (CO), and biliverdin (1). The cytoprotective effect of HO-1 is then mediated by down-modulation of NF-κB and tumor necrosis factor (TNF), leading to suppression of inflammation or inhibition of apoptosis (9). It is worth mentioning that the presence of two distal enhancers, E1 and E2, that are located far upstream of the transcription start site (TSS) of HO-1 gene makes it an ideal model for studying transcriptional enhancement and long-range chromatin interactions.
Sirtuins are a family of NAD+ dependent protein deacetylases involved in stress resistance and metabolic homeostasis. In mammals, there are seven members of this family (SIRT1–7) (10). SIRT6 is an NAD+ dependent histone and protein deaceylase and mono-ADP ribosyltransferase enzyme, which regulates an array of cellular processes, including DNA repair (11–13), repression of repetitive elements (14), telomere maintenance (15), inflammation (16), stem cell maintenance (17,18) and metabolism (19–21). Well characterized SIRT6 deacetylation targets on chromatin include H3K9, H3K18 and H3K56. Deacetylation of these histones leads to chromatin condensation and gene suppression. Fewer SIRT6 mono-ADP-ribosylation targets are known. These include PARP1 (13), modification of which promotes DNA repair under oxidative stress, and Kap1 which is involved in silencing of LINE1 elements (14). One major characteristic of SIRT6 knockout cells is their sensitivity to oxidative stress, a feature reminiscent of Nrf2 deficiency (4). Consistent with this, Pan et al. (17) has shown that SIRT6-mediated NRF2 activation is required for safeguarding mesenchymal stem cells from deleterious effect of ROS. The above observations prompted us to investigate the mechanism by which SIRT6 cooperates with NRF2 to activate anti-oxidant genes during stress.
Here, we define the mechanism of NRF2 target gene activation by SIRT6. We show that mono-ADP-ribosylation activity of SIRT6 is critical for activation of NRF2 target HO-1. To identify the targets of SIRT6 mono-ADP-ribosylation activity mediating HO-1 activation we employed mass spectrometry combined with phospho-peptide enrichment strategy since the phosphate groups of ribosylated peptides were shown to be enriched by TiO2 beads (22). We identified SIRT6-depenedent mono-ADP-ribosylation of BRG/BRM-associated factor (BAF) chromatin remodeler subunit BAF170. Remarkably, earlier studies have shown involvement of BAF complex in antioxidant response (23). BAF chromatin remodeling complex associated with the HO-1 promoter in a SIRT6-dependent manner. Furthermore, BAF complex enrichment was concomitant with a chromatin looping event leading to transcriptional activation.
MATERIALS AND METHODS
Cell lines and treatments
We obtained mouse embryonic fibroblasts (MEFs) from SIRT6 wild type (WT) and knockout (KO) embryos. MEFs were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with sodium pyruvate, nonessential amino acids, antibiotics (penicillin and streptomycin) and 15% fetal bovine serum. The media was replaced with minimum essential Eagle's medium (MEM) supplemented with nonessential amino acids, antibiotics (penicillin and streptomycin), and 15% fetal bovine serum before adding the hydrogen peroxide. Oxidative stress was induced by treating cells with 250 μM paraquat for 10 h. All compounds were purchased from Sigma. Paraquat was dissolved in PBS, the other in DMSO.
Generation of SIRT6 G60A knock-in mouse cells
For G60A CRISPR edit, low passage (PD = 5) mouse skin fibroblasts were transfected with the pX459 Cas9 and sgRNA vector (sgRNA Sequence: 5′-GCCGGCATCAGCACCGCCTC-3′) along with the single-stranded donor oligonucleotide (5′-GGCCCGGCTAATGTGGCAGTCCTCCAGCGTGGTTTTCCACACGGGCGCCGGCATCAGCACCGCCAGCGCTATCCCCGACTTCAGGTCGGTGAACAATGAGGGGAAACTGAGGCAGGGCTCTTGCTGATACACCTC-3′) using the Lonza 2B Nucleofector. Twenty four hours post-transfection, puromycin was added to plates to remove untransfected cells. Fresh media was added 72 h post-transfection. Cells were split on the fifth day and plated at low density to promote single colony formation. Colonies were screened for correct edit via PCR and verified with sequencing.
Western blot and Antibodies
Cells were harvested at growing stage by regular trypsinization. Cell pellets were re-suspended in 1× RIPA buffer (cell signaling technology) with the volume proportional their cell number followed by vortex, then another volume of 2× Laemmli buffer (Bio-Rad) were added to the cell lysate and boiled for 10 min. The following antibodies were used in this study: anti-RNA polymerase II CTD (ab5131), anti-Nrf2 (ab62352), anti BRG1 (ab110641), anti-H3 tri methyl (ab8580), anti-SIRT6 (ab62739 and ab191385) from Abcam. Anti SIRT6 (12486) anti BAF170 (8829, 12760) from cell signaling. Anti- H3K56Ac from active motif, 39281.
Plasmids
pCMV BAF170-Flag vector was purchased from Addgene (Cat # 19142). The mutagenesis was performed using Q5 Site Directed Mutagensis Kit (NEB Cat # E0554S) with primers designed against K312. For mammalian expression 5 μg of plasmid was incubated with Fugene 6 reagent and the mix was used for transfection into 293 cells. The cell lysate was then used for purification by Anti-flag M2 Immunoprecipitation kit (M8823) according to manufacturer's protocol.
Protein pull-down assay
Protein immunoprecipitations were performed as previously (13). Briefly, MEF cells were harvested and lysed in lysis buffer (20 mM HEPES pH 8.0, 0.2 mM EDTA, 5% glycerol, 150 mM NaCl and 1% NP40) at 4°C for 10 min and sonicated 30 s at 50% duty. The cleared lysate was incubated overnight with 5 μg of antibodies against the indicated proteins, added 25 μl of protein A agarose beads and rotated for another 1 h. After washing 5 times with the same buffer, the IP was eluted with 2× sample buffer (Laemmli buffer:beta-mercapto ethanol = 950:50) by boiling for 10 min. The eluted samples were spun down and the supernatants were collected for western blotting with the indicated antibodies.
Chromatin immunoprecipitation and qPCR Detection
ChIP assays were performed using ChIP kit (Abcam 500) according to manufacturer's protocol. Approximately 3 × 106 cell equivalents were used for each immunoprecipitation. Of the sample, 1.7% was removed for use as an input control. ChIP was performed using antibodies toward H3K4Me3 (Abcam ab8580), H3K4Me2 (Abcam ab7766), BAF170 (Cell Signaling 12760), BRG1 (Abcam 110641), Nrf2 (Cell Signaling 12721), phosphorylated RNA polymerase C-terminal domain (Abcam ab5131) or a control rabbit IgG (Abcam ab46540), H3K27Me3 (Abcam ab6002), H3K27Ac (Abcam ab4729), H3K56Ac (Active Motif, 39281). ChIP-qPCR was performed using Green Master mix (BIORAD 1725270). Primers for b-actin are listed in Supplementary Table S1. Primers that targeted the promoter, TSS and E1 and E2 regions of HO-1, were also used (see Supplementary Table S1 for primer sequences). Standard curves were generated for each amplicon using purified mouse genomic DNA. Each experiment was performed at least in triplicate.
RNA purification and RT-qPCR
Total RNA was isolated from cultured cells at 70% confluency using QIAGEN RNasey mini kit according to manufacturer's instruction. cDNA was then synthesized using iscript reverse transcriptase (BIORAD 1708840). Real-time quantitative PCR analysis was performed in 20 μl reaction using SYBR Green Master mix (BIORAD 1725270) and CFX Real Time PCR system (BIORAD). Primer efficiency was verified by linear regression to standard curve using 2, 1, 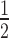 , ¼, 1/8, 1/16, 1/32, 1/64 and 1/128 dilution. Reactions were carried out in quadruplicate and values were standardized to housekeeping gene actin (Supplementary Table S1). To normalize values obtained in the samples, control actin Ct values were subtracted from the target gene Ct values for each sample (ΔCT) then, ΔCt of the unstimulated sample was subtracted from ΔCT of the stimulated sample (ΔΔCT). The relative levels of target gene were calculated as 2 –ΔΔCT.
, ¼, 1/8, 1/16, 1/32, 1/64 and 1/128 dilution. Reactions were carried out in quadruplicate and values were standardized to housekeeping gene actin (Supplementary Table S1). To normalize values obtained in the samples, control actin Ct values were subtracted from the target gene Ct values for each sample (ΔCT) then, ΔCt of the unstimulated sample was subtracted from ΔCT of the stimulated sample (ΔΔCT). The relative levels of target gene were calculated as 2 –ΔΔCT.
Ribosylation assay
In vitro mono-ADP ribosylation assay was conducted as reported previously with minor modification (13). Briefly, 1 μg of bacterially purified SIRT6 was incubated with 0.5 μg of the mammalian purified target Flag-BAF170 wild type and K312A substrate for 30 minutes at 37°C.
siRNA and plasmid transfection
ON-TARGETplus SMARTpool siRNA against human Nrf2 (L-003755-00-0005) were purchased from Dharmacon. Final siRNA concentrations of 100 nM were used for 72 h for silencing of Nrf2 transcripts in human skin fibroblast cells. Mouse embryonic fibroblast (MEF) cells were plated at 500 000 cells per well in 10 cm dishes and grown for at least 48 h in Eagle's minimum essential medium (EMEM) with Earl's salts supplemented with 15% fetal calf serum, non-essential amino acids, 1 mM sodium pyruvate, and penicillin–streptomycin. Transfection was performed using Amaxa Nucleofector machine (Lonza) and the NHDF transfection solution. At 24 h post transfection, the medium was aspirated and replaced with MEM with Earl's salts supplemented with 15% fetal calf serum, non-essential amino acids, and penicillin-streptomycin. At 16 h after media replacement, cells were treated with paraquat for 8–10 h times. Experiments were performed in triplicate.
For siRNA transfection MEF cells were plated at 500 000 cells per well in 10 cm dishes and grown for at least 48 h in EMEM with Earl's salts supplemented with 15% fetal calf serum, non-essential amino acids, 1 mM sodium pyruvate, and penicillin-streptomycin. Cells at 70% confluency were then transfected with 25 nM of indicated siRNA using Amaxa Nucleofector machine (Lonza) and the NHDF transfection solution. All siRNAs were purchased from Dharmacon. Second round of transfection was performed at 48 h. After 24 h, the medium was aspirated and replaced with MEM with Earl's salts supplemented with 15% fetal calf serum, non-essential amino acids, and penicillin-streptomycin. At 16 h after media replacement, cells were treated with paraquat for 8–10 h. Experiments were performed in triplicate.
DNase hypersensitivity assay
Real-time, quantitative DNAse I sensitivity assays were performed as previously described (24). The HO-1 promoter and enhancers primers and cycling conditions were the same as for the ChIP assays (see Supplementary Table S1 for primer sequences).
Titanium oxide enrichment of ADP-ribosylated peptides
We previously found that SIRT6 mono-ADP-ribosylation activity appears to be as critical as its deacetylase activity in promoting DNA repair (13). Due to their low abundance, the Leung and Hottiger groups have employed macrodomains to initially enrich for ADP-ribosylated peptides in a ‘shotgun’-type of approach from whole cells extracts (25,26). A potential limitation of this approach is that the macrodomain may impart a bias due to its specificity for only certain residues being modified. Given that mono-ADP ribosylation may occur on several residues, and that a SIRT6 target bias has not been defined, we wanted to employ a method that would be robust in its capacity for enrichment, yet not be biased in ADP-ribosylation sites that could be identified. We chose to employ a phospho-peptide enrichment strategy since the phosphate groups of ribosylated peptides were already show to be enriched by TiO2 beads (22). We employed TiO2 beads to enrich for both phosphorylated (and mono-ADP-ribosylated) peptides as previously described in (27) with slight modification. We employed mouse embryonic fibroblast (MEF) cells derived from either wild type or SIRT6 knock out mice since any SIRT6-dependent mono-ADP-ribosylation would be absent in the latter. Four T150 flasks grown to approximately 75% confluency were rinsed twice with PBS before trypsin to be sure to avoid carry-over from growth media. Cells from 4 flasks were removed/lysed directly in a total of 3 ml of lysis buffer (75 mM NaCl, 3% SDS, 1 mM NaF, 1 mM beta-glycerophosphate, 1 mM sodium orthovanadate, 10 mM sodium pyrophosphate, 1 mM PMSF and 1× Roche Complete Mini EDTA free protease inhibitors) in 50 mM HEPES, pH 8.5, and removal from the flask was facilitated by a cell scraper. Lysis was enhanced by passing through a 21-gauge needle 20 times followed by homogenization in a Dounce homogenizer, all the while keeping the samples on ice. Next, lysates were sonicated (Branson) with 5 × 20 s pulses at 40% output keeping on ice for one min in between each pulse. Cellular debris was removed by centrifugation at 14 000 rpm for 10 min. The protein concentration of the cell lysates was determined using the BCA assay (Thermo Scientific) after the samples were diluted 10-fold to lower the SDS concentration. Next, extracted proteins were reduced by adding freshly prepared DTT to 10 mM final concentration and samples were incubated at 50°C for 1 h. Proteins were allowed to cool briefly on ice and then alkylated by adding iodoacetamide to 15 mM final concentrations and incubated in the dark (wrapped in foil and placed in drawer) for 1 h at room temperature (RT). Unreacted iodoacetamide was quenched by adding and additional 10 mM DTT. Next, proteins were precipitated (and SDS removed) with methanol–chloroform (28). Precipitated pellets to air-dry overnight in hood at RT. Proteins were resuspended in 1 ml of 50 mM ammonium bicarbonate buffer (pH 7.0) by vortexing for several minutes in 30 min incremental incubations at 55°C until the pellet appeared to be largely dissolved by the loss of visual aggregates (3 h). Samples were allowed to remain at 4°C overnight to help them dissolve further. Proteins were digested with 50 μg of MS-grade trypsin (Thermo-Pierce 100 μg dried trypsin was resuspended in 100 μl of 50 mM acetic acid and 50 μl was added) and incubated on a rotator at 37°C overnight (16–24 h). The following morning an additional 50 μg of trypsin was added and incubate continued at 37°C for an additional 8 h. Trypsin was quenched by adding 6 ul of 100% TFA (tri-fluoroacetic acid). Peptides were desalted by dividing the sample into 3 480 mg Sep-pak C-18 columns (Waters) to be sure of the capacity. After elution from Sep-pak column with 70% acetonitrile (ACN), peptides were dried in a speedvac in low-bind tubes and care was taken not to over-dry the samples to avoid loss.
Peptides were resuspended in 0.5 ml of 5% ACN made with MS-grade H2O and vortexed for 5 min at RT. The yield of peptides was determined using a peptide fluorometric quantitation assay (Thermo Pierce). Approximately 5 mg was employed for the TiO2 enrichment. Samples were adjusted to ∼1.8 ml with a final concentration of 2 M lactic acid and 50% ACN by adding concentrated stocks of each and vortexed for 5 min at RT. Samples were centrifugated at 13 000 rpm for 5 min and supernatant was taken leaving behind ∼20 μl of sample. Prior to adding to the extract, TiO2 beads were prepared by washing sequentially with 1.5 ml of binding buffer: (2 M lactic acid, 50% ACN), elution buffer (50 mM KHxPO4, pH 10) followed by a final wash with binding buffer. A 4:1 ratio of TiO2 beads:peptide was employed for each enrichment. After peptides were mixed with the TiO2 beads, the samples were vortexed for 1 h at RT. Samples were centrifuged at 3000 rpm for 2 min and the unbound fraction (supernatant) was removed and saved. Next, samples were washed three times each in 0.5 ml of binding buffer, then 50% ACN:0.1% TFA and vortexed for 5 min at RT. Each time the samples were centrifuged at 3000 rpm and the supernatant was removed. ADP-ribosylated and phosphopheptides were eluted with two consecutive 0.5 ml washes with elution buffer and they were pooled. Samples were desalted again with a single SepPak C-18 column (480 mg) and dried by speed vac as previously. Peptides were resuspended in 5% ACN, 0.1% formic acid (29) and subsequently analyzed by nano-electropsray LC/MS/MS on Q-Exactive instrument (Thermo). A 90 min ACN gradient was applied with data-dependent acquisition. Raw files were converted to MGF files and analyzed with Protein Prospector (UCSF) (30). Phospho-serine, -tyrosine, -threonine and default ADP ribosylation (of lysine, arginine, cysteine, glutamate, asparagine and serine) as well as ADP-ribose (by manually adding the formula C5H8O6P- denoted as P5-H-mADPr in Supplementary Figure S3) were included in the search criteria. Otherwise search criteria employed default parameters with 50 ppm parental and 0.2 Da MS2 fragment error cut off. For TiO2 enrichment from the spleen, two spleens removed from 129S1/SvImJ mice were rapidly frozen in liquid nitrogen and stored at –80°C until extraction. The extraction buffer and protocol was essentially identical to the one described above for cells, except that SDS was 7% during the initial extraction and the tissue was homogenized in the extraction buffer by 20 strokes with a Dounce homogenizer on ice. Next, the SDS was removed and samples digested with trypsin using an S-Trap™ Midi Spin Column (Protifi; Huntington, NY, USA) using the manufacturer's recommended protocol. Following the S-Trap column, approximately 5 mg of tryptic peptides were enriched using the TiO2 beads, and samples were analyzed by electrospray LC/MS/MS on and Orbitrap Fusion Tribrid Lumos MS instrument (ThermoFisher). Data were analyzed with Protein Prospector (UCSF) as described above.
Chromosome conformation capture and urea-ChIP-loop assay.
We performed the chromatin conformation capture (3C) assay essentially as described in (31,32). We combined ChIP and the 3C assay to perform the ChIP-loop assay with few modifications. For in vivo crosslinking we incubated 0.05 g of minced spleen tissue obtained from 20 days old SIRT6 wild type and knock out mice in culture medium with 1% buffered formaldehyde at 4°C for 2 h. After crosslinking, we washed the cells twice with ice-cold PBS and then lysed them overnight in a solution of 4% SDS in 10 mM Tris, pH 8.0 and 1 mM EDTA. For urea gradient ultracentrifugation, crosslinked DNA and proteins were purified from uncrosslinked free proteins by centrifugation at 30 000 r.p.m. for 16 h, 4°C in a Beckman SW41 rotor, through a gradient of 5–8 M urea prepared in 10 mM Tris (pH 8.0) and 1 mM EDTA. We collected crosslinked chromatin at the bottom of the urea gradient and dialyzed it overnight against 10 mM Tris–HCl (pH 8.0), 1 mM EDTA, 0.5 mM EGTA and 5% glycerol. To perform immunoprecipitation 60 μg DNA of crosslinked chromatin was digested with HindIII in 500 μl of reaction buffer at 37°C overnight. We used 20 μl of the sample to purify DNA and confirmed by agarose gel electrophoresis that undigested high-molecular weight chromatin was absent. We added 1% Nonidet P-40 (NP-40) to the remaining sample of digested chromatin and precleared the sample by incubation (1 h at 4°C with rotation) with 50 μl of a 50% suspension of protein A Sepharose 4B beads (Sigma). We removed beads by brief centrifugation and repeated preclearing of crosslinked chromatin by adding 10 μl of preimmune serum and incubating at 4°C for 1 h, followed by incubation with 50 μl of protein A-Sepharose 4B beads at 4°C for 1 h with rotation. After centrifugation, we incubated precleared chromatin with preimmune rabbit serum with anti-Brg1 or anti-CTCF at 4°C for 4 h and then incubated the mixture with protein A Sepharose 4B beads at 4°C overnight with rotation. We washed beads four times with 1.0% NP-40 in PBS and two times with washing buffer (10 mM Tris–HCl (pH 8.0), 0.25 M LiCl, 0.5% NP-40, 0.5% DOC and 1 mM EDTA). DNA was ligated by resuspending the beads with ChIP DNA in 50 μl ligation buffer and incubated the mixture overnight at 16°C with T4 DNA ligase. To reverse crosslinking the samples were digested sequentially with 100 μg/ml RNase A and 250 μg/ml proteinase K, treated them at 68°C for 6 h to reverse crosslinking and subjected them to phenol/chloroform extraction followed by ethanol precipitation.
PCR amplification was performed as follows. We designed forward primers in each of HindIII DNA fragments 1, 5–10, as well as a reverse primer in DNA fragment 1–5. Primers were designed less than 150 bp from a HindIII restriction site. Sequences for these primers are shown in Supplementary Table S1. Using a combination of forward and backward primers from different DNA fragments, we amplified ligated DNA with SYBR Green Master mix (BIORAD 1725270) and CFX Real Time PCR system (BIORAD) as follows: one cycle at 95°C for 3 min; 35–40 cycles at 95°C for 30 s, 55°C for 40 s and 72°C for 30 s; followed by one cycle at 72°C for 5 min. The purification of crosslinked chromatin through urea gradient centrifugation allows more quantitative digestion with HindIII. Therefore, the pattern of ligation products remains unchanged even after an additional 10–20 cycles of PCR amplification.
To correct for differences in crosslinking, ligation, PCR amplification efficiencies or sample loading, we prepared a control template set with DNA fragments from the HO-1 locus. For internal ChIP-loop control we designed primers for the Ints4 locus which does not show transcriptional enhancement during our experimental setting. For preparing the HO-1 locus control template, we digested the mouse BAC clone (290L7, Invitrogen) containing DNA derived from the HO-1 locus with HindIII and religated them with T4 ligase at a DNA concentration of 100 ng/μl. All possible ligation products were present in the sample (data not shown). To prepare the Ints4 locus control template, we cloned a DNA fragment from the mouse Ints4 locus using one and four primers with a TA cloning kit (Invitrogen). We used additional primers, 2 and 3, from two closely located neighboring fragments in the Ints4 locus so that the PCR amplification occurs reproducibly (Supplementary Figure S9). The HindIII fragments between primers 2 and 3 are separated by 686 bp. As a control, we mixed equimolar amounts of INTS4 with HO-1 BAC control DNA templates, followed by digestion and re-ligation. To correct for the amount of genomic DNA used for the 3C assay, we added INTS4 primers S2 and S3 in re-ligated DNA templates derived from spleen tissue crosslinked chromatin. For the ChIP-loop assay, we added the plasmid DNA containing the Ints4 locus in the original IP DNA to correct for the amount of the starting DNA template used. For the 3C assay, we measured the signal of each reaction from MEF-derived and control re-ligated DNA templates (described above), and calculated the relative crosslinking frequency using the formula illustrated in Supplementary Figure S10 which corrects for any differences in PCR amplification efficiencies, crosslinking and ligation efficiencies and the amounts of the templates. We used 20 ng of DNA for the 3C assay and ∼1 ng of IP DNA for the ChIP-loop assay. For the ChIP-loop assay, relative crosslinking frequency was calculated in a similar manner as for the 3C assay. All experiments performed for the 3C assay and ChIP-loop assay were repeated a minimum of three times, and the data shown are the mean of multiple experiments. Standard errors were within 10% of the values shown in the figures.
RESULTS
Activation of NRF2 target genes is dependent on SIRT6 mono-ADP ribosylase activity
It was recently reported that SIRT6 is required for activation of NRF2 target genes (17). Here we set out to understand the contribution of the SIRT6 enzymatic activities in this function. First, we tested the levels mRNA transcripts of ARE-containing promoters in SIRT6 WT and SIRT6 knockout (SIRT6 KO) cells. SIRT6 KO cells showed inefficient induction of NRF2 transcriptional targets, upon oxidative stress induced by paraquat, compared to the WT cells (Figure 1A). As all the tested NRF2 target genes showed similar dependence on SIRT6, for the subsequent experiments we focused on HO-1. Within 8 hours post paraquat treatment, HO-1 gene expression was enhanced in the WT but not in the SIRT6 KO cells (Figure 1B), indicating that SIRT6 deficiency confers lack of responsiveness to oxidative stress, which is consistent with the previous report (17). Re-expression of the WT SIRT6, but not catalytically dead allele SIRT6 H133Y, rescued the phenotype of SIRT6 KO cells (Figure 1C).
Figure 1.
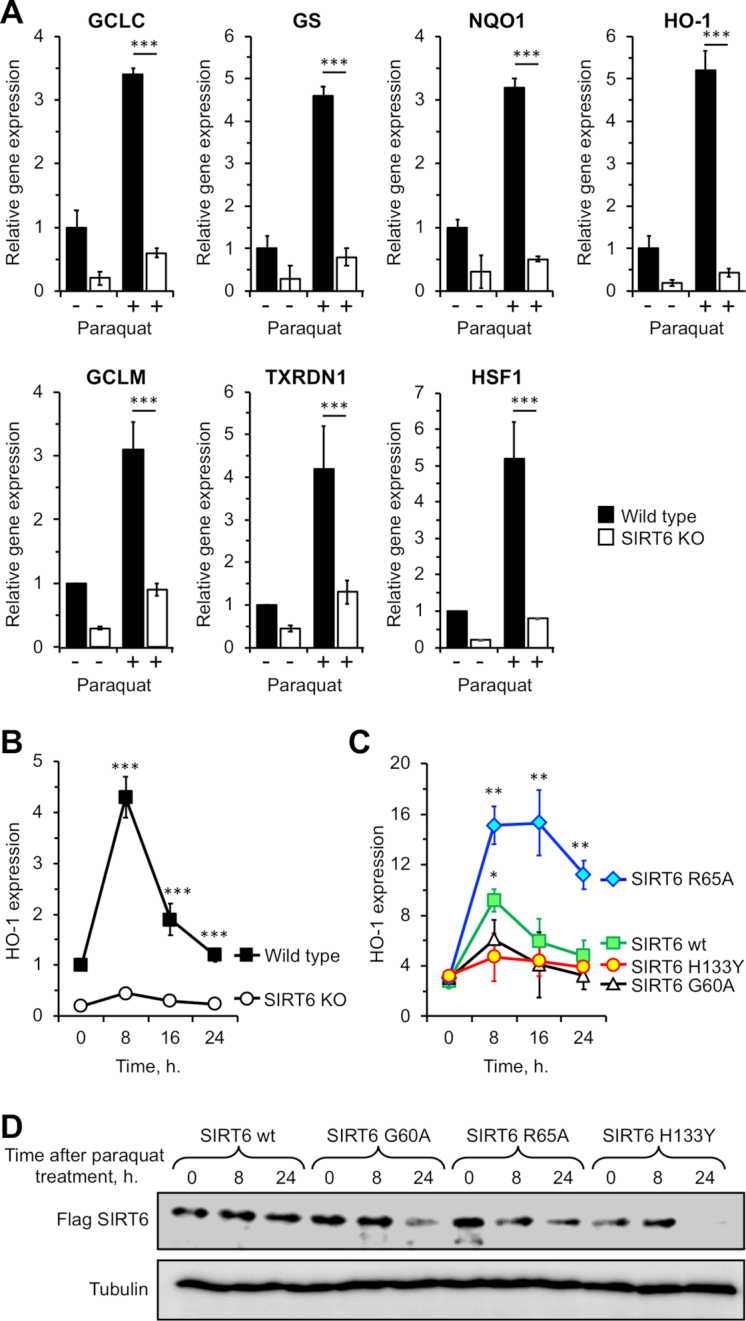
SIRT6 mono-ADP ribosylation activity is involved in transcriptional activation of Nrf-2 target genes. (A) Nrf2 target genes transcriptional enhancement depends on SIRT6. RT-qPCR analysis of GSH synthetase (GS), glutamate-cysteine ligase catalytic subunit (GCLC), NAD(P)H quinone dehydrogenase 1 (NQO1), heme oxygenase (HO-1), glutamate-cysteine ligase modifying subunit (GCLM), thioredoxin reductase 1 (TXNRD1), heat shock factor (HSF1) under basal conditions and 8 h after treatment with paraquat. Data were obtained from three independent experiments. Error bars represent s.d. Statistical analyses were made using Student's t-test. Paraquat treated and untreated samples were compared. ***P < 0.001. **P < 0.01. (B) HO-1 transcription peaks within 8 h after addition of paraquat and diminishes by 24 h. RT-qPCR analysis of heme oxygenase (HO-1) at different time points upon addition of paraquat. mRNA values are normalized to beta-actin. Error bars represent s.d., n = 3. Statistical analyses were made using Student's t-test. WT cells were compared to SIRT6 KO cells. ***P < 0.001. (C) in the presence of ribosyl transferase active SIRT6 allele (R65A), HO-1 transcription shows an enhanced and prolonged expression pattern. RT-qPCR analysis of heme oxygenase (HO-1) at different time points upon addition of paraquat in SIRT6 KO cells rescued with different alleles of SIRT6. Error bars represent s. d., n = 3. Statistical analyses were made using Student's t-test. Cells transfected with SIRT6 R65A allele or SIRT6 WT are compared to the cells transfected with enzymatically dead SIRT6 H133Y. **P < 0.001. *P < 0.01. (D) Immunoblot analysis of cells transfected with different flag-SIRT6 alleles used in (C). SIRT6 KO cells were transfected with different SIRT6 alleles (WT, G60A, R65A, H133Y) and were harvested for immunoblot analysis at indicated timepoints.
The SIRT6 protein has deacetylase, deacylase and mono-ADP-ribosyl transferase enzymatic activities (13). To assess which activity of SIRT6 is required for transcriptional enhancement, we expressed SIRT6 separation of function mutants in SIRT6 KO fibroblasts: G60A (deacetylase and deacylase active but mono-ADP-ribosylase defective) and R65A (mono-ADP-ribosyl transferase active but deacetylase/deacylase defective) SIRT6 alleles (13). The catalytically dead (H133Y) and G60A alleles of SIRT6 did not significantly enhance HO-1 expression upon stress, suggesting that deacetylase and deacylase activities of SIRT6 are not involved in transcriptional activation. In contrast, WT and ribosylation active (R65A) alleles enhanced HO-1 expression by 10- and 30-fold, respectively (Figure 1C). Remarkably, the activation of transcription conferred by R65A allele was persistent over time. While upon expression of WT SIRT6 HO-1 transcription declined rapidly, expression of SIRT6 R56A allele conferred expression that lasted longer than 24 h (Figure 1C). These results suggest that SIRT6 ribosylation activity is involved in transcriptional activation of HO-1, while the deacetylation activity may be involved in tuning-off the stress response. In our earlier in vitro studies involving separation of function SIRT6 mutants (13) we did not observe antagonistic effects between deacetylation and mono-ADP ribosylation activities of SIRT6. Therefore, we hypothesize that the two enzymatic activities act on different substrates. Mono-ADP ribosylation activity may be responsible for activating transcription by recruiting Pol II (17) and chromatin remodelers, while deacetylation activity may shut down NRF2 response by deacetylating SIRT6 histone targets.
To further examine the contribution of mono-ADP ribosylation activity into oxidative stress response we generated knock-in SIRT6 G60A mouse cells using CRSPR/CAS9 technology. Under basal conditions, HO-1 expression was similar between the WT and SIRT6 G60A cells, while under oxidative stress, HO-1 induction was strongly attenuated (Supplementary Figure S1). The mild activation of transcription conferred by SIRT6 G60A allele may be due to the scaffolding effect of SIRT6 protein on chromatin independent of the enzymatic activity, since similar activation was conferred by the catalytically dead H133Y allele (Figure 1C). This result indicates that SIRT6 mono-ADP ribosylation activity plays important role in the transcriptional activation of HO-1.
Besides the different enzymatic activities of SIRT6 alleles, we also observed a difference in protein stability. Specifically, SIRT6 WT had the highest stability as evident at 24 h-time point, whereas H133Y alleles showed the lowest (Figure 1D). We also noticed a difference in the stability of G60A and R65A alleles at 8 h-time point (Figure 1D). Importantly, although R65A had lower stability, it still enhanced HO-1 expression 3–4-fold higher than the SIRT6 G60A allele.
SIRT6 maintains the HO-1 locus in transcriptionally permissive mode
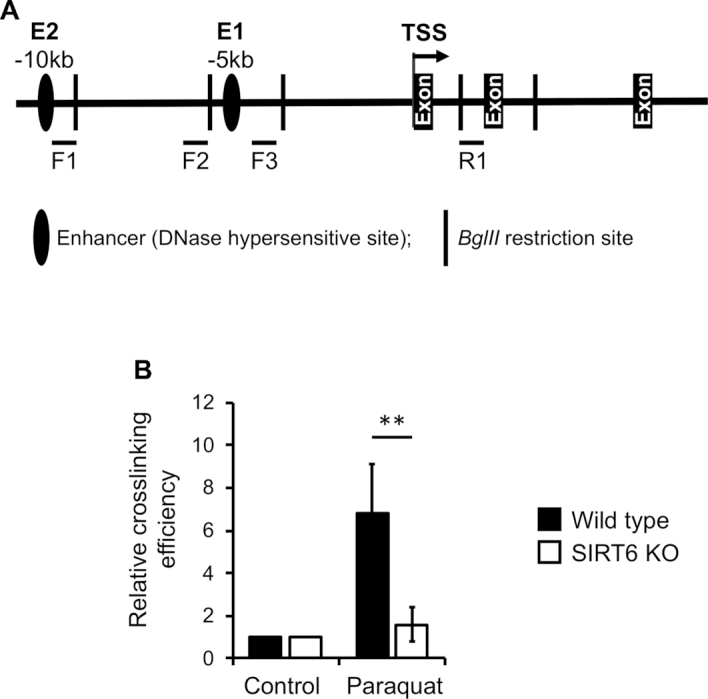
Given the failure of SIRT6KO cells to upregulate NRF2 target genes upon oxidative stress, we hypothesized that inefficient histone dynamics might prevent transcriptional enhancement. To test this, we set out to study chromatin status in both WT and SIRT6 KO cells. The HO-1 locus has been characterized in considerable detail and serves as a model for studying transcriptional regulatory mechanisms and the relationship between NRF2 and chromatin modifying factors (4). Previous studies have reported the existence of two DNase I hypersensitive sites upstream of the HO-1 promoter region. The enhancer regions contain multiple antioxidant responsive elements (ARE), which are common in the promoter regions of anti-oxidant genes and act as a recognition sites for NRF2 transcription factor (4). The basal level of expression of HO-1 gene in the cells is dependent on the enhancer regions, which consist of two sites spread over 3–4 kb of DNA and located 4–15 kb upstream of the HO-1 transcription start site (TSS) (Figure 2A). Consistent with this, DNase sensitivity assay showed that in the WT cells, the HO-1 enhancers are DNase-I hypersensitive under basal conditions but become DNase-I resistant in SIRT6KO cells (Figure 2B). This suggests the lack of nucleosome hypermobility sites in the absence of SIRT6.
Figure 2.

SIRT6 is required for chromatin conditioning on the HO-1 locus. (A) Schematic of the HO-1 locus containing transcription start site (TSS) and upstream enhancers E1 and E2 (black ovals). (B) HO-1 locus is compact in SIRT6 KO cells. DNase Hyper sensitivity (DHS) assay was performed on WT and KO cells and undigested DNA templates were quantified using qPCR. Experiments were repeated three times and error bars show s.d., *P < 0.05. (C) Lack of nucleosome dynamics in SIRT6 KO cells. WT and KO cells were left untreated or treated with paraquat and locus occupancy by H3 was analyzed by ChIP using H3 antibody. (D) E1 and E2 enhancers are pre-marked independent of SIRT6. WT and KO cells were harvested and locus occupancy by H3K4me2 were analyzed by ChIP using H3K4me2 antibody. Control refers to the region one kb upstream of TSS. (E) Higher repressive chromatin mark deposition in the absence of SIRT6. WT and KO cells were harvested and locus occupancy by H3K27me3 was analyzed by ChIP using H3K27me3 antibody. (F) SIRT6 is required for the maintenance and enhancement of H3K27 acetylation after stress. WT and KO cells were treated with paraquat or left untreated and harvested and locus occupancy by H3K27me3 was analyzed by ChIP using H3K27Ac antibody. (G) SIRT6 is recruited to the HO-1 locus post oxidative stress. WT cells were treated with paraquat, harvested and locus occupancy by SIRT6 was analyzed by ChIP using SIRT6 antibody. Control refers to the region one kb upstream of TSS. (H) Hyper-acetylation of H3K56 in the absence of SIRT6. WT and KO cells were treated with paraquat or left untreated and harvested and locus occupancy by H3K56Ac was analyzed by ChIP using H3K56Ac antibody. (C–H) Data were from three independent ChIP experiments, normalized to control IgG; error bars show s.d. Significance was determined using Student's t-test. *P < 0.05, **P < 0.01.
To determine whether the reduced DNase-I sensitivity of mutant cells is correlated with higher nucleosome density, we performed ChIP assays for total histone H3 before and after oxidative stress. Three different regions of the HO-1 locus were analyzed: 5′ enhancer 1 (E1), 5′ enhancer 2 (E2), and transcription start site (TSS). A ChIP quantitative PCR (qPCR) assay showed significantly higher nucleosome occupancy on the E2 in the SIRT6 KO cells compared to the WT (Figure 2C), indicating that the DNase-I insensitivity is correlated with increased histone density in the SIRT6 KO cells. To investigate the kinetics of nucleosome dynamics, we then studied nucleosome occupancy upon oxidative stress. Nucleosome density was reduced on E1 region upon oxidative stress, but it did not change in SIRT6 KO cells (Figure 2C). Together these results suggest that SIRT6 is required to maintain HO-1 locus in transcriptionally permissive state.
A bivalent-like state of the HO-1 locus in the absence of SIRT6
Having observed lack of chromatin accessibility and enhanced nucleosome density in the absence of SIRT6 we set out to study the HO-1 locus anatomy in detail. Towards this aim, we characterized repressive as well as permissive histone marks on the enhancer regions as well as on the promoter. Enhancers display enrichment of histone H3 lysine 4 di-methylation H3K4me2 (33). Therefore, we studied H3K4me2 on the enhancer E1 and E2 regions under basal conditions. Interestingly, in both WT and SIRT6 KO cells, the enhancer regions were pre-marked with H3K4me2 (Figure 2D).
Another histone mark catalyzed by trithorax-group and associated with active genes is H3K4me3 (33). Given the association of this mark with transcription, we next examined whether the patterns of H3K4me3 are different in the presence or absence of SIRT6 (Supplementary Figure S2A). To our surprise, there was no difference in the basal level of H3K4me3 deposition on the promoter of HO-1 between the WT and SIRT6 KO cells, however, H3K4me3 was strongly enriched in the WT cells upon oxidative stress. To further confirm that equal level of H3K4me3 corresponds to the equal initiating RNA polymerase II association on the TSS, we performed ChIP using an antibody which targets RNA Polymerase II phospho Ser5. Consistent with H3K4 methylation, there was no difference in initiating RNA polymerase II levels between WT HO-1 TSS and SIRT6 KO under basal conditions (Supplementary Figure S2B). This data suggests that SIRT6 is dispensable for establishment of active chromatin marks on the HO-1 locus.
To investigate the mechanism of chromatin condensation in the absence of SIRT6, we studied enrichment of repressive mark H3K27me3. H3K27me3 is catalyzed by Polycomb-group proteins and is associated with gene silencing and formation of facultative heterochromatin (34). Strikingly, we observed significantly higher levels of H3K27me3 on the proximal enhancer E1 of the HO-1 gene region in the absence of SIRT6 (Figure 2E). This region also showed a higher histone density compared to the WT (Figure 2C). A higher density of H3K27me3 could also be observed on the distal enhancer E2 as well as on the TSS in the SIRT6 KO cells (Figure 2E). Our results show that HO-1 promoter in the absence of SIRT6 acquired both repressive and activating histone modifications. Under such circumstances, the H3K27me3 is generally dominant over the ubiquitous H3K4me2/3 activity resulting in lower gene expression compared to the locus which only carries H3K4me2/3 (34).
Thus, in the SIRT6 KO cells, the HO-1 locus has characteristics of a bivalent (poised or paused) gene which carries both activating and repressive marks. Normally, bivalent genes are is expressed at low levels but are poised for rapid activation. In the absence of SIRT6, however, HO-1 gene could not be activated even in the presence of oxidative stress (Figure 1). To be activated, bivalent genes depend on removal of H3K27me3 and an ATP-dependent process which requires recruitment of BAF remodeling complex (35,36) to evict PRC2 from the locus and to provide chromatin accessibility (37).
SIRT6 is required for global histone H3 acetylation on the HO-1 locus
A hallmark feature associated with active enhancers is H3K27 acetylation (H3K27ac) (38). Therefore, we examined this H3K27Ac marks on the HO-1 enhancer region. The levels of H3K27Ac were different under basal conditions between the SIRT6 WT and KO cells (Figure 2F). After treatment with paraquat in the WT cells E1 and E2 regions experienced 6- and 3-fold increase in H3K27Ac marks, respectively. Strikingly, there was no such increase in the H3K27 acetylation on the enhancer regions in SIRT6 KO cells (Figure 2F).
The total level of histone H3 acetylation on a given gene was shown to correlate directly with the transcription status, where transcriptionally active regions are associated with hyperacetylated histones, whereas transcriptionally silent regions are associated with hypoacetylated histones (39). Thus, for better understanding of the role of acetylation in HO-1 regulation, we investigated total histone acetylation at the HO-1 locus by using ChIP assays with anti-acetylated histone H3 antibody which can recognize acetyl K9, K14, K18, K23, K27. The results of this study showed that under basal conditions, the HO-1 locus is less acetylated in SIRT6 KO cells. Furthermore, while in the WT cells, H3 acetylation increased following oxidative stress, there was no increase in H3 acetylation levels after paraquat treatment in SIRT6 KO cells (Supplementary Figure S2C). This result indicates that SIRT6 is required for enhancement of chromatin acetylation on HO-1 gene following oxidative stress.
SIRT6 is a histone deacetylase enzyme best known for its activity on H3K9Ac, H3K56Ac, and as recently reported, H3K18Ac (40,41). To address a direct link between SIRT6 and HO-1 locus, we performed ChIP against SIRT6 and observed that SIRT6 is recruited to HO-1 locus only after oxidative stress (Figure 2G). Interestingly, a higher density of SIRT6 was detected on E1 and E2 regions compared to the TSS (Figure 2G). This prompted us to examine if there is a correlation between acetylation of the SIRT6 substrates and HO-1 activation via a ChIP against H3K56Ac. Consistent with SIRT6 role as a deacetylase, we found that H3K56Ac at the HO-1 enhancers and promoter are hyperacetylated in SIRT6 KO cells under basal conditions (Figure 2H). Besides, in the WT cells H3K56Ac levels on the enhancer regions were lower than those in the KO cells and these levels further decreased post-oxidative stress (Figure 2H). This result is somewhat paradoxical as H3K56 acetylation is associated with active transcription. We can speculate that, while SIRT6 is recruited to HO-1 locus and deacetylates H3K56, this does not impact activation of HO-1 transcription, indicating that another SIRT6 activity is responsible for transcriptional activation.
SIRT6 is not required for NRF2 recruitment
As we found that SIRT6 is recruited to HO-1 locus upon oxidative stress, we set out to test if SIRT6 is required for the recruitment of NRF2. We observed no difference in NRF2 binding to the ARE elements of the E1 and E2 enhancer sites between SIRT6 WT and KO cells under basal conditions (Supplementary Figure S2D). Interestingly, we found more NRF2 bound to E1 region in SIRT6 KO cells after oxidative stress (Supplementary Figure S2D). This data shows that the lack of HO-1 transcriptional enhancement in SIRT6 KO cells is not due to inefficient recruitment of NRF2.
We next tested if NRF2 recruitment and histone acetylation in the WT cells is concomitant with polymerase II assembly. ChIP-qPCR analysis showed that compared to SIRT6 KO cells, in the WT cells there was a drastic increase in polymerase II initiation complex on the TSS post oxidative stress (Supplementary Figure S2B) which is in agreement with the transcriptional activation of HO-1 gene and hyper acetylation of enhancers that we observed post oxidative stress only in the WT cells.
Chromatin remodelers function to alter the conformation and position of nucleosomes and promote enhanceosome-like-complex assembly to initiate and enhance transcription (42,43). Since SIRT6 deficient cells failed to alter HO-1 chromatin structure and to enhance transcription after stress, despite efficient NRF2 recruitment, we hypothesized that SIRT6-mediated transcriptional enhancement is mediated by chromatin remodeling enzymes.
SIRT6 mono-ADP ribosylation promotes recruitment of BRG/BRM-associated factor (BAF) chromatin remodeling complex to the HO-1 enhancer upon oxidative stress
As chromatin dynamics is impaired in the absence of SIRT6, we set out to identify a chromatin remodeler involved in HO-1 activation after oxidative stress. Since our data suggest that SIRT6 mono-ADP-ribosylation is important for HO-1 activation, we sought to identify the substrate for SIRT6 mono-ADP-ribosylation. Towards this aim, we treated the WT and SIRT6 KO mouse embryonic fibroblasts with paraquat, total proteins were extracted and then enriched for mono-ADP-ribosylated peptides along with phosphopeptides using titanium oxide (TiO2). The eluted samples were subjected to high-resolution bottom-up mass spectroscopy. We found that K312 (residues numbered according to the human protein product; Uniprot, Q8TAQ2) of BAF170 was modified by a ribose-phosphate group only in the WT but not in SIRT6 KO cells (Supplementary Figure S3A) which is an evidence for mono-ADP-ribosylation. Similarly, we observed ribosylation of the same residue of BAF170 from samples obtained from wild type spleen tissue (Supplementary Figure S3B). Remarkably, BAF complex is the major chromatin remodeling complex which responds to oxidative stress and activates NRF2 targets (23).
To investigate whether BAF complex is associated with HO-1 promoter, we performed ChIP using antibody against ATPase subunit of BAF complex, BRG1. BAF ATPase subunit BRG1 could be detected on the distal enhancer (E2) of the HO-1 promoter in a SIRT6-independnet manner (Figure 3A). Proximal enhancer E1, however, showed a different pattern. BRG1 was not associated with the promoter under basal conditions in the absence of SIRT6. Under oxidative stress, however, there was a very strong increase in BRG1 binding to this region in the WT, but only a slight increase in the KO cells (Figure 3A). To further confirm whether BAF complex is involved in HO-1 transcriptional enhancement, we knocked down its core subunit BAF170, and observed that it completely abolished HO-1 transcription (Figure 3B). These data suggest that BAF complex is involved in transcriptional enhancement of NRF2 target gene HO-1 and is consistent with the previous report (23).
Figure 3.

SIRT6 mediates HO-1 transcriptional enhancement through mono-ADP ribosylation of BAF170. (A) SIRT6 is required for recruitment of BAF complex to E1 region. ChIP analysis of the HO-1 locus in WT and SIRT6 KO cells untreated and post paraquat treatment using antibody against BAF complex ATPase subunit BRG1. Error bars denote s.d., n = 3; *P < 0.05. (B) BAF170 is critical for HO-1 transcriptional enhancement. Total RNA was isolated from WT MEF cells transfected with siBAF170, and treated with paraquat. BAF170 mRNA level in control cells and those treated with paraquat was analyzed using qPCR. (C) SIRT6 is a component of BAF complex and interacts with BAF170. BAF170 was immunoprecipitated from nuclear extracts of MEFs using SIRT6 antibody, and then immunoblotted for BAF170. (D) SIRT6 mono-ADP ribosylates BAF170. Purified flag-BAF170 was incubated with SIRT6 and 32P-NAD+ for 30 minutes. To decrease random interactions between SIRT6 and BAF170 and to enhance the specificity of modification, antibodies against BAF170 and flag were added to specified reactions. Ribosylated proteins were detected by autoradiography. SIRT6 and BAF170 were detected using antibodies against SIRT6 and Flag, respectively. (E) BAF170 overexpression enhances HO-1 transcription in WT MEFs. MEF WT and SIRT6 KO cells were transfected with a plasmid encoding flag-BAF170. Forty eight hours post transfection cells were treated with paraquat, and the HO-1 transcripts were analyzed using RT-qPCR and normalized to beta actin. Error bars show s.d., n = 3, **P < 0.01. (F) BAF170 mutation abrogates HO-1 transcriptional enhancement. WT MEFs were transfected a plasmid expressing flag-BAF170-K312A. 48 hours post transfection cells were treated with and the HO-1 transcripts and analyzed by RT-qPCR. Error bars show s.d., n = 3, **P < 0.01. (G) SIRT6 is required for BAF170 recruitment to E1 region after stress. WT MEFs were treated with paraquat for 8 h and ChIP assay was run using antibody against BAF170. Error bars show s.d., n = 3, **P < 0.01; ***P < 0.001. (H) SIRT6-dependent ribosylation of BAF170 enhances BAF170 binding to chromatin. WT MEFs were transfected with flag-BAF170 or flag-BAF170-K312A, and treated with paraquat 48 h post transfection. Eight hours post treatment BAF170 recruitment was analysed by ChIP assay using anti-Flag antibody. *P < 0.05.
We also tested whether BAF complex is involved in activation of other NRF2 target genes. BAF170 depletion abolished transcriptional activation of HO-1 (Figure 3B) and HSF1 during oxidative stress, but not GCLC, GS, GCLM and TXRDN1 (Supplementary Figure S4). Intriguingly, NQO-1 had a significantly reduced level of transcripts compared to scrambled control samples under basal conditions. This result suggests that BAF170 is required for enhanced transcription of a subset of NRF2 targets including HO-1 and HSF1.
Co-immunoprecipitation assays with SIRT6 revealed that BAF170 interacts with SIRT6 both under basal conditions and upon oxidative stress. Interestingly, this interaction was DNA independent (Figure 3C). We next examined if SIRT6 ribosylates BAF170 by performing in vitro ribosylation assay. Based on mass spectroscopy data (Supplementary Figure S3), SIRT6 mono-ADP-ribosylates BAF170 on K312. SIRT6 ribosylated BAF170, but ribosylation of BAF170 K312A mutant was attenuated (Figure 3D). Overexpression of BAF170 did not rescue the induction of HO-1 expression in SIRT6 KO cells under oxidative stress (Figure 3E). WT mouse cells overexpressing BAF170 K312A mutant failed to induce HO-1 expression upon treatment with paraquat, in contrast to the cells overexpressing BAF170 WT (Figure 3F). This result suggests that modification of K312 is required for the enhancement of HO-1 transcription during stress.
We next tested whether the difference in BAF complex enrichment accounts for higher enhancement of HO-1 transcription upon stress. We first examined endogenous BAF170 enrichment on HO-1 locus under basal conditions and post oxidative stress. BAF170 enrichment on both E1 and E2 regions required SIRT6 (Figure 3G).
Next, we examined the recruitment of flag-tagged BAF170 WT and BAF170 K312A to the HO-1 locus (Figure 3H). BAF170 WT showed significantly higher recruitment to the E1 region compared to BAF170 K312A mutant. There was also a trend towards higher recruitment of the BAF170 WT to the E2 region but it did not reach statistical significance, possibly, due to the attenuated function of the flag-tagged proteins.
Next, we performed a ChIP experiment to examine BRG1 association with chromatin under basal conditions and after oxidative stress in the presence of different SIRT6 alleles. Interestingly, we found higher association of BRG1 with HO-1 enhancer E1 region in KO cells rescued with the WT and R65A alleles (Supplementary Figure S5). These results suggest that mono-ADP-ribosylation of BAF170 by SIRT6 promotes recruitment of BAF complex to the HO-1 promoter region, and that SIRT6-mediated mono-ADP-ribosylation of BAF170 is a prerequisite for HO-1 transcriptional enhancement.
NRF2 is required for SIRT6 recruitment to the enhancer region of the HO-1 locus
NRF2 binding to chromatin ensures maintenance of the ARE element in a transcriptionally permissive mode. To test whether depletion of NRF2 also impacts SIRT6 association with the HO-1 locus, we knocked down Nrf2 gene in human fibroblasts (Supplementary Figure S6A) and examined the enrichment of SIRT6, BAF170 and H3 before and after oxidative stress. Depletion of NRF2 significantly reduced SIRT6 enrichment on the enhancer regions E1 and E2 (Supplementary Figure S6B). However, the level of SIRT6 bound to TSS was not affected by NRF2 depletion. The level of BAF170 on enhancer regions E1 and E2 was also significantly reduced after NRF2 depletion (Supplementary Figure S6C), which was consistent with the lack of H3 dynamics on the E2 region in Nrf2 knock down cells (Supplementary Figure S6D).
These data further confirm the role of NRF2 as a pioneer transcription factor which is critical for establishment of a transcriptionally permissive and marked chromatin. The observation that SIRT6 enrichment on E1 and E2 depends on presence of NRF2 but the recruitment of SIRT6 to TSS is independent of NRF2 suggests that SIRT6 might be a component of two independent complexes (namely BAF complex and RNA pol lI complex) which act independently of each other at the recruitment step but converge on the transcription initiation step.
SIRT6 promotes loop formation on HO-1 locus during oxidative stress
To confirm the potential interaction between the –5.0 kb HO-1 proximal enhancer region and the transcription start site, chromosome conformation capture (3C) assay was performed using the relevant restriction enzyme BglII as described previously (44) (Figure 4A). A specific qPCR product from primer pairs Forward primer 3 (F3) and Reverse primer 1 (R1) was detected in samples in which HO-1 expression was induced with paraquat (Figure 4B). Real-time PCR demonstrated that paraquat-treated SIRT6 WT samples had crosslinking efficiency that was significantly higher than that of vehicle-treated samples (Figure 4B) with the F3/R1 primer pairs. These results suggest that the two DNA fragments, one containing the regulatory promoter sequences at –5.0 kb region and the other locating next to the TSS, are potentially interacting in a SIRT6-dependent manner. The other primer pairs specific for the distal enhancer (located at –9.2 kb) (F1/R1) did not show any difference between the WT and SIRT6 KO cells before and after paraquat treatment, indicating that only proximal enhancer looping with TSS during oxidative stress is SIRT6-dependent. HO-1-containing BAC DNA was used as a positive control and genomic DNA obtained from human skin fibroblast cells without crosslinking was used as a negative control in these experiments.
Figure 4.
SIRT6-dependent long-range chromatin interaction on the proximal enhancer in HO-1 locus. Chromosome conformation capture (3C) assay was performed as described in the Experimental Procedures. 100 ng of the sample was amplified by qPCR. A bacterial artificial chromosome containing human HO-1 locus (HO-1 BAC, Genbank accession number Z82244) clone was used as a positive control and non-crosslinked genomic DNA from primary skin fibroblast cells was used as a negative control. Reverse-crosslinked DNA was then amplified with primers (bars indicated by F1–3, R1). (A) Schematic representation of the HO-1 promoter and gene with the positions of BglII restriction sites (vertical bars), DNase I–hypersensitive sites (ovals). (B) Relative crosslinking frequencies between fragment F3 and fragment R1 of the locus. The graph shows the ligation products by qPCR produced by the 3C assay with untreated and stressed WT and SIRT6 cells 8 h after treatment with 0.5 M paraquat. The 3C assay was performed to determine the interaction between enhancer and promoter using BglII restriction enzyme. Interaction between the fragment containing –4.0 kb proximal enhancer and that next to the TSS was identified from the PCR product of F3 and R1 primer pairs. DNA from the PCR product of paraquat-treated sample with the F3/R1 primer pairs was extracted, TOPO TA-cloned, and then sequenced. Error bars show s.d., n = 3, **P < 0.01.
SIRT6 promotes BRG1-mediated chromatin loop formation on HO-1 locus in mouse spleen
To further characterize the SIRT6-associated loop structure in vivo in the WT and SIRT6 KO mice, we explored the binding status of the two major players in the chromatin organization, BRG1 and CTCF, at the specific positions in the 50-kb locus indicated in Supplementary Figure S7A–C. towards this aim we isolated the spleen from WT and SIRT6 KO mice. Based on the available RNAseq datasets, spleen has the highest HO-1 expression compared to other organs. This is consistent with the fact that spleen is the major organ involved in Heme recycling, a HO-1 dependent pathway performed mainly in spleen-resident macrophages. The level of HO-1 expression was first quantified using RT-qPCR. After confirming that HO-1 is significantly lower in the SIRT6 KO spleen tissue (Supplementary Figure S8), we performed real-time PCR and urea-ChIP-loop assay, using the same primer sets shown in Supplementary Table S2, on urea gradient-purified chromatin.
HO-1 is located downstream of a very low transcribing gene TOM1 and upstream of MCM5 which has a similar transcription profile (45). The region is also marked by two major CTCF binding motifs, at the promoter of TOM1 gene (17) and one upstream of MCM5 promoter which had a similar expression than HO-1. Consistent with low expression, TOM1 gene is devoid of any H3K4me1 and H3K4me3 on the promoter region, further making it an ideal reference point which has a differential expression pattern of HO-1 and MCM5 (Supplementary Figure S9). Therefore, we used the HindIII restriction site downstream of TOM1 gene which includes a CTCF binding site. We used a battery of reverse primers which covered the first 100 bp downstream of each HindIII restriction site and analyzed up to 10 fragments scattered across the HO-1 locus. Interestingly, we found that BRG1 was bound to multiple sites within the 50-kb region (TOM1 and MCM5), including all enhancers, promoter and upstream (fragment 6) and downstream (fragment 7, 8 and 10) sites and its binding pattern was not significantly different from the observed pattern for activated paraquat-treated WT skin cells, as well as MEF cells (Figure 4B, and Supplementary Figure S7B). Such a pattern, however, was absent in the spleen tissue obtained from SIRT6 KO mouse (Supplementary Figure S7C). Similar to BRG1, CTCF bound at multiple positions (fragment 5 and 7) throughout the 50-kb region in the WT spleen tissue. However, the interaction that marks the end of HO-1 locus (fragment 7) was missing in the SIRT6 KO spleen tissue. The most enhanced binding to fragment 1 and 10 in the WT cells was reduced in the spleen tissue obtained from SIRT6 KO mice (Supplementary Figure S7C). These results show that SIRT6 is required to fold chromatin into a three-dimensional, active chromatin configuration required for transcriptional enhancement of the target gene HO-1.
DISCUSSION
This study identifies a novel function for the SIRT6 mono-ADP-ribosylation activity in facilitating long-range chromatin interactions and activation of oxidative stress response gene HO-1 transcription. Our data suggest a role for the SIRT6 in regulating local topology of the HO-1 locus during oxidative damage. We show that in the absence of SIRT6 HO-1 transcription cannot be activated and the locus is refractory to chromatin changes. Particularly, in SIRT6 knockout cells, the HO-1 promoter region is not accessible to DNase I and SWI/SNF chromatin remodeling complex is not enriched on the proximal enhancer during oxidative stress. Most importantly, we found that BAF170 is a new SIRT6 mono-ADP-ribosylation substrate and a critical effector for SIRT6-mediated transcriptional activation of a subset of Nrf2 targets during oxidative stress. Genes whose transcription was found to be dependent on BAF170 (HO-1, NQO-1 and HSF1) shared one common characteristic that there ARE arrays are located at a distance from the TSS. HO-1 enhancers are located far upstream of the TSS, while NQO-1 and HSF1 enhancer (s) are located downstream of the TSS in the gene body. We therefore, hypothesize that BAF complex is required for mediating long-range interactions between enhancers and TSS. Other SIRT6 effectors may be involved in the activation of Nrf2 target genes which do not possess distal enhancers.
Based on these results, we propose a model (Figure 5) in which SIRT6 nucleates the formation of a co-activator complex on the HO-1 locus and further chromatin looping. The early events in HO-1 transcriptional enhancement, including NRF2 association, happen independently of SIRT6. Enrichment of the BAF chromatin-remodeling complex, hyper-acetylation of H3K27, enhanced RNA polymerase II recruitment, and increased H3K4 tri-methylation, however, depend on the SIRT6. Notably, we showed that BAF170 ribosylation is a key event in HO-1 transcriptional enhancement. It is possible that SIRT6 mono-ADP-ribosylation on the core subunit BAF170 provides a better docking site for the rest of SWI/SNF complex to assemble on BAF170 and hence the chromatin. In eukaryotic cells, the effects of enhancers appear to be mediated, at least in part, by loop formation, and DNA looping by BAF remodeling complex proteins has been shown to regulate α-globin locus (46).
Figure 5.
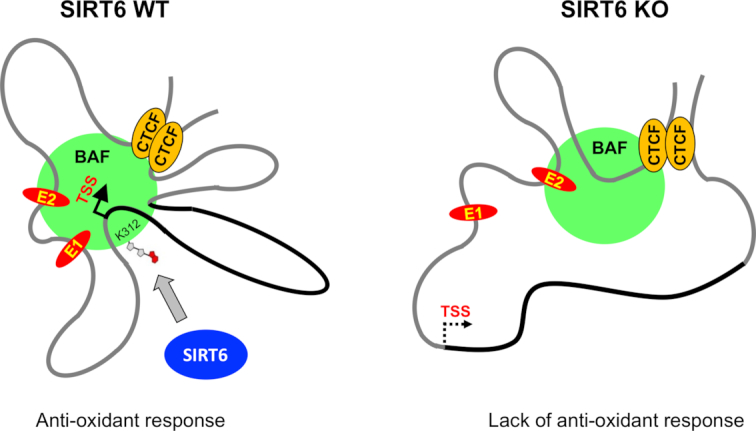
Schematic of activation-dependent looping events in HO-1 locus in SIRT6 wild type and knock out tissue. A schematic diagram based on the looping events shown in Figure 4, assuming that all looping events can occur in a single cell. In this model, all small loops converge on a common core base bound to BAF complex (green spheres) in a SIRT6- and mono-ADP ribosylation-dependent manner. As a consequence, the total physical volume of the active transcriptional complex is reduced, enhancing the accessibility of factors to genomic sites in a single transcriptionally active domain (60). HO-1 gene is highlighted in black.
This, and other studies suggest that nuclear architecture is correlated with gene expression, though the phenotypical consequences of altering 3D genome organization are not well understood. The β-globin locus control region (LCR) has been one of the most examined genes (46–49). In a landmark study, Deng et al., forced a loop between the β -globin promoter and LCR in the absence of GATA1, which is required for β-globin expression, and found this forced looping was sufficient to recruit RNA polymerase II (31). Enhancer-promoter chromatin loops are also responsible for removing repressive chromatin marks for transcriptional activation. Strikingly, Vernimmen et al. reported that in a ‘humanized’ mouse model where the human α-globin locus was inserted either in its wild type form or containing a deletion in an enhancer located 60 kb away from the promoter, the enhancer can clear polycomb proteins from the CpG island located in the α-globin promoter region and block spreading of the H3K27me3 (50). Similarly, we observed that in the absence of SIRT6, the HO-1 locus was enriched for suppressive histone mark H3K27me3. In particular, the proximal enhancer E1 region was the most affected as deposition of H3K27me3 increased up to 7–8-fold in SIRT6 KO cells as compared to the wild type controls.
There are multiple indications for a role of chromatin maintenance and nuclear structure in aging. Recent analyses of chromatin defects in the premature aging disease Hutchison-Gilford Progeria Syndrome (HGPS) have given some of the first insights into how chromatin ages (51). HGPS is an extremely rare genetic disease caused by a de novo point mutation in the Lamin A (LMNA) gene, a major structural component of the nuclear envelope (52). Analysis of the molecular mechanisms involved in bringing about chromatin defects in HGPS and old cells uncovered NRF2 as the main downstream effector of LMNA mutation. At the cellular level, HGPS cells display aberrant nuclear architecture, genome instability, and elevated ROS level. Consistent with high ROS level Kubben et al. found that NRF2 mis-localizes to the nuclear periphery due to higher affinity to the mutated form of Lamin A, called progerin (53). This suggests that progerin impairs transcriptional activation of Nrf2-target genes by sequestering of NRF2 away from its transcriptional target (53,54). It was proposed that Lamin A binds NRF2 and SIRT6, with these three proteins shaping the 3D chromatin architecture and directing transcription of antioxidant genes (54). Here we identified BAF complex as the specific chromatin remodeler that mediates the effect of SIRT6 on NRF2 target loci.
Earlier studies by Lin et al., have found the chromatin topology could be refractory to changes (55). Similarly, in the absence of SIRT6, the chromatin of HO-1 locus failed to form a far-cis interaction. The fundamental difference, however, regarding the HO-1 locus is that in the absence of SIRT6 the E1 and E2 regions are not permissive to transcription. We found this striking, because establishment of accessible chromatin is Brg1 dependent. Thus, it seems that SIRT6 is required for preparing the HO-1 locus through Brg1 to respond to Nrf2 signaling. Besides, failing to regulate the recruitment of BAF complex on chromatin could lead to polycomb-mediated gene silencing which is critical to development, and even promotion of malignancy in several cancers (35,37,43,56–58). Consistent with this we found that E1 region on the HO-1 locus is devoid of Brg1. Concomitantly, the same region was affected by H3K27me3-mediated silencing. Similar paradigm was observed in pluripotent embryonic stem cells (ESCs) in which deletion of Brg1 resulted in PRC2-mediated gene silencing of STAT3 target genes, abolishment of LIF signaling and ultimately exit from pluripotency state (2,59). Our finding that SIRT6 mediates BAF complex recruitment suggest a new mechanism for SIRT6 function in development, tumor suppression and aging.
The best understood function of SIRT6 in gene transcription is histone deacetylation and gene silencing. However, our study and the earlier study by Pan et al. (17) showed that SIRT6 serves as an activator of Nrf2-dependent gene transcription. It is interesting to test whether the activator function of SIRT6 is limited to stress-response genes and whether SIRT6 may act globally as transcriptional activator or repressor depending on the gene function and environmental conditions.
In summary, our findings suggest that SIRT6 protects cells from oxidative stress through regulation of BAF complex recruitment to the NRF2 regulated HO-1 gene promoter. This provides a new mechanism for SIRT6 involvement in oxidative stress resistance and longevity.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
US National Institutes of Health (to A.S. and V.G.). Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Dhakshinamoorthy S., Jain A.K., Bloom D.A., Jaiswal A.K.. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J. Biol. Chem. 2005; 280:16891–16900. [DOI] [PubMed] [Google Scholar]
- 2. Wang H., Rojo de la Vega M., Zhang D.D., Yu S., Zheng H.. Response to comment on “NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis”. Sci. Transl. Med. 2016; 8:349lr341. [DOI] [PubMed] [Google Scholar]
- 3. Kroemer G., Marino G., Levine B.. Autophagy and the integrated stress response. Mol. Cell. 2010; 40:280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kensler T.W., Wakabayashi N., Biswal S.. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007; 47:89–116. [DOI] [PubMed] [Google Scholar]
- 5. Orr W.C., Radyuk S.N., Prabhudesai L., Toroser D., Benes J.J., Luchak J.M., Mockett R.J., Rebrin I., Hubbard J.G., Sohal R.S.. Overexpression of glutamate-cysteine ligase extends life span in Drosophila melanogaster. J. Biol. Chem. 2005; 280:37331–37338. [DOI] [PubMed] [Google Scholar]
- 6. Bishop N.A., Guarente L.. Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature. 2007; 447:545–549. [DOI] [PubMed] [Google Scholar]
- 7. Tullet J.M., Hertweck M., An J.H., Baker J., Hwang J.Y., Liu S., Oliveira R.P., Baumeister R., Blackwell T.K.. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell. 2008; 132:1025–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sykiotis G.P., Bohmann D.. Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila. Dev. Cell. 2008; 14:76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gozzelino R., Sole C., Llecha N., Segura M.F., Moubarak R.S., Iglesias-Guimarais V., Perez-Garcia M.J., Reix S., Zhang J., Badiola N. et al.. BCL-XL regulates TNF-alpha-mediated cell death independently of NF-kappaB, FLIP and IAPs. Cell Res. 2008; 18:1020–1036. [DOI] [PubMed] [Google Scholar]
- 10. Kugel S., Mostoslavsky R.. Chromatin and beyond: the multitasking roles for SIRT6. Trends Biochem. Sci. 2014; 39:72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tasselli L., Zheng W., Chua K.F.. SIRT6: novel mechanisms and links to aging and disease. Trends Endocrinol. Metab. 2017; 28:168–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kaidi A., Weinert B.T., Choudhary C., Jackson S.P.. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science. 2010; 329:1348–1353. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13. Mao Z., Hine C., Tian X., Van Meter M., Au M., Vaidya A., Seluanov A., Gorbunova V.. SIRT6 promotes DNA repair under stress by activating PARP1. Science. 2011; 332:1443–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Van Meter M., Kashyap M., Rezazadeh S., Geneva A.J., Morello T.D., Seluanov A., Gorbunova V.. SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat. Commun. 2014; 5:5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Michishita E., McCord R.A., Berber E., Kioi M., Padilla-Nash H., Damian M., Cheung P., Kusumoto R., Kawahara T.L., Barrett J.C. et al.. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008; 452:492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jiang H., Khan S., Wang Y., Charron G., He B., Sebastian C., Du J., Kim R., Ge E., Mostoslavsky R. et al.. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature. 2013; 496:110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pan H., Guan D., Liu X., Li J., Wang L., Wu J., Zhou J., Zhang W., Ren R., Zhang W. et al.. SIRT6 safeguards human mesenchymal stem cells from oxidative stress by coactivating NRF2. Cell Res. 2016; 26:190–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang H., Diao D., Shi Z., Zhu X., Gao Y., Gao S., Liu X., Wu Y., Rudolph K.L., Liu G. et al.. SIRT6 controls hematopoietic stem cell homeostasis through epigenetic regulation of wnt signaling. Cell Stem Cell. 2016; 18:495–507. [DOI] [PubMed] [Google Scholar]
- 19. Kawahara T.L., Michishita E., Adler A.S., Damian M., Berber E., Lin M., McCord R.A., Ongaigui K.C., Boxer L.D., Chang H.Y. et al.. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. 2009; 136:62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sebastian C., Zwaans B.M., Silberman D.M., Gymrek M., Goren A., Zhong L., Ram O., Truelove J., Guimaraes A.R., Toiber D. et al.. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 2012; 151:1185–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhong L., D’Urso A., Toiber D., Sebastian C., Henry R.E., Vadysirisack D.D., Guimaraes A., Marinelli B., Wikstrom J.D., Nir T. et al.. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010; 140:280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Matic I., Ahel I., Hay R.T.. Reanalysis of phosphoproteomics data uncovers ADP-ribosylation sites. Nat. Methods. 2012; 9:771–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang J., Ohta T., Maruyama A., Hosoya T., Nishikawa K., Maher J.M., Shibahara S., Itoh K., Yamamoto M.. BRG1 interacts with Nrf2 to selectively mediate HO-1 induction in response to oxidative stress. Mol. Cell Biol. 2006; 26:7942–7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McArthur M., Gerum S., Stamatoyannopoulos G.. Quantification of DNaseI-sensitivity by real-time PCR: quantitative analysis of DNaseI-hypersensitivity of the mouse beta-globin LCR. J. Mol. Biol. 2001; 313:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martello R., Leutert M., Jungmichel S., Bilan V., Larsen S.C., Young C., Hottiger M.O., Nielsen M.L.. Proteome-wide identification of the endogenous ADP-ribosylome of mammalian cells and tissue. Nat. Commun. 2016; 7:12917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vivelo C.A., Leung A.K.. Proteomics approaches to identify mono-(ADP-ribosyl)ated and poly(ADP-ribosyl)ated proteins. Proteomics. 2015; 15:203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Villen J., Gygi S.P.. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat. Protoc. 2008; 3:1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wessel D., Flugge U.I.. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984; 138:141–143. [DOI] [PubMed] [Google Scholar]
- 29. Haigis M.C., Mostoslavsky R., Haigis K.M., Fahie K., Christodoulou D.C., Murphy A.J., Valenzuela D.M., Yancopoulos G.D., Karow M., Blander G. et al.. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006; 126:941–954. [DOI] [PubMed] [Google Scholar]
- 30. Chalkley R.J., Baker P.R., Medzihradszky K.F., Lynn A.J., Burlingame A.L.. In-depth analysis of tandem mass spectrometry data from disparate instrument types. Mol. Cell. Proteomics: MCP. 2008; 7:2386–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deng W., Lee J., Wang H., Miller J., Reik A., Gregory P.D., Dean A., Blobel G.A.. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 2012; 149:1233–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Horike S., Cai S., Miyano M., Cheng J.F., Kohwi-Shigematsu T.. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 2005; 37:31–40. [DOI] [PubMed] [Google Scholar]
- 33. Ruthenburg A.J., Allis C.D., Wysocka J.. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol. Cell. 2007; 25:15–30. [DOI] [PubMed] [Google Scholar]
- 34. Margueron R., Reinberg D.. The Polycomb complex PRC2 and its mark in life. Nature. 2011; 469:343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stanton B.Z., Hodges C., Calarco J.P., Braun S.M., Ku W.L., Kadoch C., Zhao K., Crabtree G.R.. Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin. Nat. Genet. 2017; 49:282–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K. et al.. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006; 125:315–326. [DOI] [PubMed] [Google Scholar]
- 37. Ho L., Miller E.L., Ronan J.L., Ho W.Q., Jothi R., Crabtree G.R.. esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating polycomb function. Nat. Cell Biol. 2011; 13:903–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maston G.A., Landt S.G., Snyder M., Green M.R.. Characterization of enhancer function from genome-wide analyses. Annu. Rev. Genomics Hum. Genet. 2012; 13:29–57. [DOI] [PubMed] [Google Scholar]
- 39. Plank J.L., Dean A.. Enhancer function: mechanistic and genome-wide insights come together. Mol. Cell. 2014; 55:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Finkel T., Deng C.X., Mostoslavsky R.. Recent progress in the biology and physiology of sirtuins. Nature. 2009; 460:587–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tasselli L., Xi Y., Zheng W., Tennen R.I., Odrowaz Z., Simeoni F., Li W., Chua K.F.. SIRT6 deacetylates H3K18ac at pericentric chromatin to prevent mitotic errors and cellular senescence. Nat. Struct. Mol. Biol. 2016; 23:434–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee J.H., Skalnik D.G.. Wdr82 is a C-terminal domain-binding protein that recruits the Setd1A Histone H3-Lys4 methyltransferase complex to transcription start sites of transcribed human genes. Mol. Cell Biol. 2008; 28:609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ghisletti S., Barozzi I., Mietton F., Polletti S., De Santa F., Venturini E., Gregory L., Lonie L., Chew A., Wei C.L. et al.. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010; 32:317–328. [DOI] [PubMed] [Google Scholar]
- 44. Deshane J., Kim J., Bolisetty S., Hock T.D., Hill-Kapturczak N., Agarwal A.. Sp1 regulates chromatin looping between an intronic enhancer and distal promoter of the human heme oxygenase-1 gene in renal cells. J. Biol. Chem. 2010; 285:16476–16486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T., Sandstrom R., Ma Z., Davis C., Pope B.D. et al.. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014; 515:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Engel J.D., Tanimoto K.. Looping, linking, and chromatin activity: new insights into beta-globin locus regulation. Cell. 2000; 100:499–502. [DOI] [PubMed] [Google Scholar]
- 47. Wang W., Cote J., Xue Y., Zhou S., Khavari P.A., Biggar S.R., Muchardt C., Kalpana G.V., Goff S.P., Yaniv M. et al.. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 1996; 15:5370–5382. [PMC free article] [PubMed] [Google Scholar]
- 48. Kim S.I., Bresnick E.H., Bultman S.J.. BRG1 directly regulates nucleosome structure and chromatin looping of the alpha globin locus to activate transcription. Nucleic Acids Res. 2009; 37:6019–6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim S.I., Bultman S.J., Kiefer C.M., Dean A., Bresnick E.H.. BRG1 requirement for long-range interaction of a locus control region with a downstream promoter. PNAS. 2009; 106:2259–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vernimmen D., Lynch M.D., De Gobbi M., Garrick D., Sharpe J.A., Sloane-Stanley J.A., Smith A.J., Higgs D.R.. Polycomb eviction as a new distant enhancer function. Genes Dev. 2011; 25:1583–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pegoraro G., Misteli T.. The central role of chromatin maintenance in aging. Aging. 2009; 1:1017–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shumaker D.K., Dechat T., Kohlmaier A., Adam S.A., Bozovsky M.R., Erdos M.R., Eriksson M., Goldman A.E., Khuon S., Collins F.S. et al.. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. PNAS. 2006; 103:8703–8708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kubben N., Zhang W., Wang L., Voss T.C., Yang J., Qu J., Liu G.H., Misteli T.. Repression of the antioxidant NRF2 pathway in premature aging. Cell. 2016; 165:1361–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gorbunova V., Rezazadeh S., Seluanov A.. Dangerous entrapment for NRF2. Cell. 2016; 165:1312–1313. [DOI] [PubMed] [Google Scholar]
- 55. Shachar S., Pegoraro G., Misteli T.. HIPMap: a high-throughput imaging method for mapping spatial gene positions. Cold Spring Harb. Symp. Quant. Biol. 2015; 80:73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schuettengruber B., Chourrout D., Vervoort M., Leblanc B., Cavalli G.. Genome regulation by polycomb and trithorax proteins. Cell. 2007; 128:735–745. [DOI] [PubMed] [Google Scholar]
- 57. Jones P.A., Baylin S.B.. The epigenomics of cancer. Cell. 2007; 128:683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Min J., Zaslavsky A., Fedele G., McLaughlin S.K., Reczek E.E., De Raedt T., Guney I., Strochlic D.E., Macconaill L.E., Beroukhim R. et al.. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat. Med. 2010; 16:286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Toiber D., Erdel F., Bouazoune K., Silberman D.M., Zhong L., Mulligan P., Sebastian C., Cosentino C., Martinez-Pastor B., Giacosa S. et al.. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol. Cell. 2013; 51:454–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bernhart S.H., Kretzmer H., Holdt L.M., Juhling F., Ammerpohl O., Bergmann A.K., Northoff B.H., Doose G., Siebert R., Stadler P.F. et al.. Changes of bivalent chromatin coincide with increased expression of developmental genes in cancer. Sci. Rep. 2016; 6:37393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.