

Abstract
CDKL5 deficiency disorder (CDD) is a neurodevelopmental disorder characterized by a severe global developmental delay and early-onset seizures. Notably, patients show distinctive visual abnormalities often clinically diagnosed as cortical visual impairment. However, the involvement of cerebral cortical dysfunctions in the origin of the symptoms is poorly understood. CDD mouse models also display visual deficits, and cortical visual responses can be used as a robust biomarker in CDKL5 mutant mice. A deeper understanding of the circuits underlying the described visual deficits is essential for directing preclinical research and translational approaches. Here, we addressed this question in two ways: first, we performed an in-depth morphological analysis of the visual pathway, from the retina to the primary visual cortex (V1), of CDKL5 null mice. We found that the lack of CDKL5 produced no alteration in the organization of retinal circuits. Conversely, CDKL5 mutants showed reduced density and altered morphology of spines and decreased excitatory synapse marker PSD95 in the dorsal lateral geniculate nucleus and in V1. An increase in the inhibitory marker VGAT was selectively present in V1. Second, using a conditional CDKL5 knockout model, we showed that selective cortical deletion of CDKL5 from excitatory cells is sufficient to produce abnormalities of visual cortical responses, demonstrating that the normal function of cortical circuits is dependent on CDKL5. Intriguingly, these deficits were associated with morphological alterations of V1 excitatory and inhibitory synaptic contacts. In summary, this work proposes cortical circuit structure and function as a critically important target for studying CDD.
Introduction
In humans, mutations in CDKL5 cause a devastating disorder of neurodevelopment-designated CDKL5 deficiency disorder (CDD). Patients are characterized by a severe global developmental delay, early-onset seizures and a distinctive visual impairment (23,35) often clinically diagnosed as cortical visual impairment (CVI). CVI, also designated as cerebral visual impairment, is generally due to alterations involving the retrochiasmal visual tracts in the brain with non-existent or minimal ocular morbidity (18). However, little is known about the neural structures involved in CVI in CDD patients. During the past years, CDD mouse models have become available (1,14,24,41), paving the way to mechanistic and preclinical investigations on possible therapies. These studies have produced a plethora of preclinical data; however, the efficacy assessment of these potential treatments in a clinical trial critically depends on the availability of non-invasive biomarkers with high sensitivity. Unfortunately, these tools are still missing. Previous studies demonstrated that visual cortical responses assessed by visual evoked potential (VEP) recordings or intrinsic optical signal (IOS) imaging (a technique to dynamically image the activity-dependent blood oxygenation-dependent signal) can be used to monitor the progression of the symptoms in a CDD mouse model (22). Sensitivity analysis revealed that visual responses were able to correctly classify mice as mutant or wild-type (WT) with >93% accuracy and 92% sensitivity proving the robustness of this technique in phenotype assessment. Defects in sensory processing have also been reported using auditory evoked potentials (41). These observations raise the possibility that visual alterations could represent a biomarker of high-translational potential for CDD. To gain a deeper understanding of the circuits underlying visual deficits induced by CDKL5 deficiency, we characterized the morphofunctional defects present at different levels along the retino-cortical projection in CDKL5 mutant mice. Our data show that, although CDKL5 is expressed in the retina, its deletion does not alter any of its major neuronal populations. By contrast, neurons in the visual thalamic relay nucleus [the dorsal lateral geniculate nucleus (dLGN)] and in the visual cortex showed reduced spine density and PSD95 synaptic staining in CDKL5 germline knockout mice. To understand whether the impairment in cortical visual responses present in CDKL5 mutants originates from alterations occurring at the thalamic level, we used IOS imaging to record the amplitude of primary visual cortical activation in response to visual stimulation in conditional CDKL5 mutant mice with preserved CDKL5 expression in subcortical nuclei. We found that the selective deletion of CDKL5 from cortical excitatory and some glial cells is sufficient to produce abnormalities of visual cortical responses, demonstrating that CDKL5 plays an essential role in establishing or maintaining a normal function of cortical circuitry.
Results
CDKL5 null mice show normal retinal morphology
CDKL5 deletion results in structural and functional abnormalities in the visual cortex (22,27). In order to assess whether CDKL5 deletion also affects upstream structures of the visual system, we first analyzed the retina of mice with CDKL5 deletion (CDKL5−/y) (1). We first performed a western blot analysis on adult male CDKL5−/y (N = 4) and WT (CDKL5+/yN = 4) retinal homogenates to demonstrate CDKL5 retinal expression. Our data (Fig. 1A) indicate that CDKL5 is expressed at the protein level in the retina, in accordance with RNA sequencing data showing CDKL5 in NRL-positive rods (16). No signal was observed at the expected molecular weight in the retina of CDKL5 null mice confirming the specificity of the CDKL5 antibody. This prompted us to assess the effect of the lack of CDKL5 on retinal organization. We stained retinal tissue of P28 male CDKL5−/y (N = 3) and CDKL5+/y (N = 3) mice with an extensive set of markers for diverse cell types and markers of connectivity. Staining for cone photoreceptors (cone arrestin positive cells) and rod outer segments (Rhodopsin positive cells) in retinal sections (Fig. 1B and C) revealed that the overall structural organization of photoreceptor cells is identical in the two experimental groups. We then proceeded to assess the total number of selected cell types (cones, cholinergic amacrine cells and ganglion cells) using whole-mount preparations. No statistical difference between CDKL5−/y and CDKL5+/y mice was present in the number of cone photoreceptors (cone arrestin positive cells; Fig. 1D–F; two-tailed Student’s t-test P = 0.435), retinal ganglion cells (RGCs; RNA-binding protein with multiple splicing; RNA-binding protein with multiple splicing (RBPMS) positive cells; Fig. 1G–I; two-tailed Student’s t-test P = 0.098) and cholinergic amacrine cells [GAD67 and choline acetyltransferase (ChAT) double positive cell] both in the inner nuclear layer (INL; Supplementary Material, Fig. S2C–F; two-tailed Student’s t-test P = 0.782) and in the ganglion cell layer (GCL; Supplementary Material, Fig. S2C–F; two-tailed Student’s t-test P = 0.932). To further characterize how the structural organization of the retina is influenced by the lack of CDKL5, we performed immunohistochemical experiments for specific markers of different retinal subpopulations on vertical sections of CDKL5−/y (N = 3) and CDKL5+/y (N = 3) retinas. This analysis reveals cellular morphology, layering and connectivity allowing an in-depth assessment of possible abnormalities. We investigated the following cell types: cholinergic amacrine cells (GAD67 and ChAT double positive cells; Supplementary Material, Fig. S2A and B), horizontal and amacrine cells (calbindin D positive cells) together with type 2 cone bipolar cells and axonal endings of type 6 cone bipolar cells (ZNP1/synaptotagmin staining; Supplementary Material, Fig. S1A and B). Rod bipolar cells [protein kinase Cα (PKCα) positive cells] were analyzed in combination with a marker of synaptic ribbons [C-terminal-binding protein 2 (CtBP2/RIBEYE) staining] in the outer and inner plexa of the retina (Supplementary Material, Fig. S1C and D). Finally, we labeled Müller glial cells (glutamine synthetase positive cells) in combination with a gap-junction subunit (Connexin36) abundant in the inner plexiform layer of the retina (Supplementary Material, Fig. S1E and F). We found no qualitative alterations in the morphology and distribution of any of these retinal markers. In summary, this series of experiments indicate that the lack of CDKL5 does not alter the morphological organization of retinal circuits.
Figure 1.
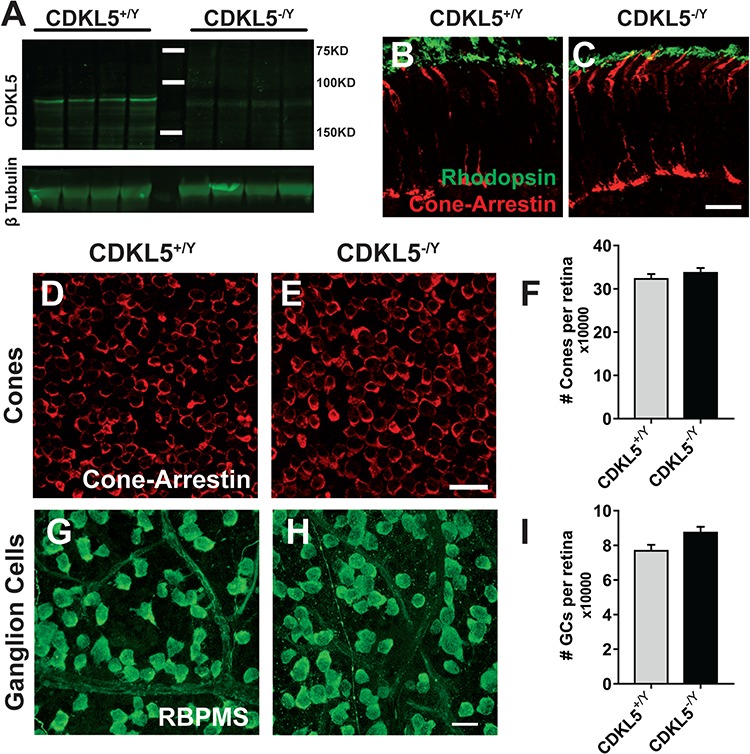
CDKL5 null mice show normal retinal morphology. (A) Western blot analysis on adult male CDKL5−/y and WT (CDKL5+/y) retinal homogenates showing CDKL5 expression. (B and C) Retinal sections stained for cone photoreceptors (cone arrestin, red) and rod outer segments (rhodopsin, green). The overall structural organization of photoreceptor cells is identical in the two experimental groups. (D and E) Representative images from retinal whole-mount preparations of CDKL5+/y (D) and CDKL5−/y (E) stained for cone arrestin as a cone marker. (F) Quantitation of the total number of cones showed no statistical significance between the two groups (two-tailed Student’s t-test P = 0.435). (G and H) Representative images from retinal whole-mount preparations of CDKL5+/y (G) and CDKL5−/y (H) stained for RBPMS as a RGCs marker. (I) Quantitation of the total number of cones (I) showed no statistical significance between the two groups (two-tailed Student’s t-test P = 0.098). Scale bars are equal to 20 μm. Error bars represent standard error of mean (SEM).
CDKL5−/y mice show synaptic alterations in the lateral geniculate nucleus and in the primary visual cortex
CDKL5 is also expressed in the mouse thalamus (30). For this reason, we next asked whether the lack of CDKL5 could affect the circuits of the dLGN, the next station of the visual information pathway from the retina to the primary visual cortex (V1). Since CDKL5 is known to regulate spine formation and stability in the cortex and hippocampus (8,38), we assessed density and morphology of dendritic spines in Golgi-stained slices (Fig. 2A) of the dLGN of CDKL5−/y (N = 4) and CDKL5+/y (N = 5) control littermates. CDKL5−/y mice showed a reduction in spine density (Fig. 2B; two-tailed Student’s t-test P = 0.026). To evaluate spine morphology, we classified spines as immature (filopodia, thin-shaped and stubby-shaped) or mature (mushroom and cup-shaped) (20,39). CDKL5 null mice had a greater proportion of immature spines (Fig. 2C; χ2 test P = 0.0027) compared to the controls. Furthermore, the distribution of spines in classes was significantly skewed toward a more immature state in mice lacking CDKL5 (Fig. 2D; χ2 test P = 0.0009). To corroborate this result we performed immunohistochemistry (IHC), staining for PSD95, a postsynaptic marker of excitatory synapse maturation (32). Consistently with Golgi-staining data, CDKL5−/y mice (N = 6) showed a strong decrease in the density of PSD95 positive puncta with respect to CDKL5+/y (N = 6; two-tailed Student’s t-test P < 0.0001; Fig. 2E and F). Moreover, to assess the impact of the lack of CDKL5 onto inhibitory circuitry, we stained for VGAT, a presynaptic marker of inhibitory synapses (5). The results showed no significant alterations in VGAT positive puncta suggesting that the lack of CDKL5 produces synaptic alterations in the dLGN occurring at the level of excitatory connections.
Figure 2.
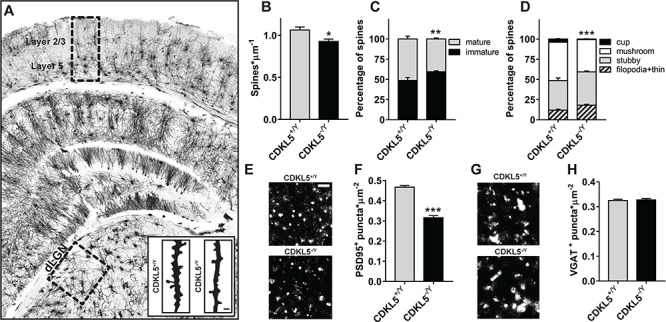
Morphological synaptic abnormalities in CDKL5 KO dLGN. (A) Representative image of a Golgi-stained coronal section containing the dLGN and showing the cortical layering that has been used in sections containing V1. (B) Quantitation of spine density in Golgi-stained dLGN slices showing a reduction in CDKL5−/y mice (two-tailed Student’s t-test P = 0.0259). (C) Quantitation of the relative proportion of mature and immature spines in Golgi-stained dLGN slices showing an increased fraction of immature spines in CDKL5−/y mice (χ2 test P = 0.0027). (D) Quantitation of spine morphology in Golgi-stained dLGN slices showing a pattern of decreased morphological maturity in CDKL5−/y mice (χ2 test P = 0.0009). (E) Representative images of PSD95 punctate staining from CDKL5−/y and CDKL5+/y dLGN. (F) Quantitation of PSD95+ puncta density showing a reduction in CDKL5−/y mice (two-tailed Student’s t-test P < 0.0001). (G) Representative images of VGAT punctate staining from CDKL5−/y and CDKL5+/y dLGN. (H) Quantitation of VGAT+ puncta density showing no difference in CDKL5−/y mice (two-tailed Student’s t-test P = 0.706). *P < 0.05; **P < 0.01; ***P < 0.001. Scale bars (A, insets) are equal to 1 μm; (E and G), 2 μm. Error bars represent SEM.
Previous in vivo studies (8) showed that CDKL5 knockout mice demonstrated decreased dendritic spine stability in cortical neurons in the somatosensory cortex. We, therefore, asked whether a spine density deficit was present in V1. This possibility was suggested by findings showing downregulation of the density of PSD95 positive puncta in this area (27). Golgi-staining in V1 coronal slices of CDKL5 KO mice unveiled a decreased spine density in layer 2/3 (Fig. 3) and layer 5 (Supplementary Material, Fig. S3) apical dendrites of pyramidal neurons in CDKL5−/y animals (N = 4) with respect to CDKL5+/y controls (Fig. 3A; N = 5; two-tailed Student’s t-test P < 0.003; Supplementary Material, Fig. S3A). Furthermore, analyzing the distribution of spines in morphological classes, we found a lower proportion of mature spines (Fig. 3B; Fisher’s exact test P < 0.001; Supplementary Material, Fig. S3B) and an increased fraction of filopodia-like, thin and stubby spines (Fig. 3C; χ2 test P < 0.001; Supplementary Material, Fig. S3C). These morphological abnormalities were coupled to a reduction in the PSD95+ puncta in CDKL5−/y (N = 6) versus CDKL5+/y (Fig. 3D and E; N = 6; two-tailed Student’s t-test P < 0.001) animals, confirming previous results (27). Interestingly, we also found an increase in the density of VGAT+ puncta (Fig. 3F and G; two-tailed Student’s t-test P = 0.0135). Taken together, these results confirm that the lack of CDKL5 leads to a complex dysregulation of synaptic morphological markers. Excitatory spines are consistently reduced in both the dLGN and in the visual cortex, while V1 shows an additional specific increase in inhibitory connections.
Figure 3.
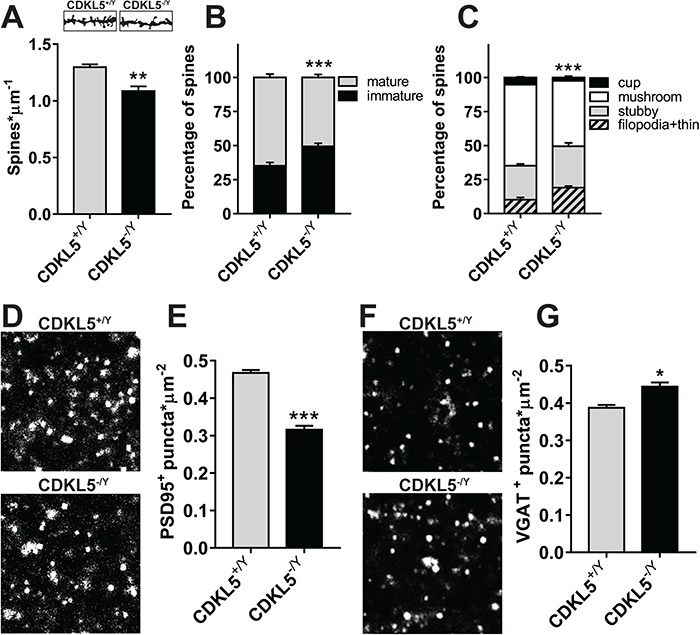
Morphological synaptic abnormalities in CDKL5 KO V1. (A) Quantitation of spine density in Golgi-stained V1 slices showing a reduction in CDKL5−/y mice (two-tailed Student’s t-test P = 0.026). In the insets are shown representative images of dendritic spines after Golgi staining from CDKL5−/y and CDKL5+/y V1 neurons. (B) Quantitation of the relative proportion of mature and immature spines in Golgi-stained V1 slices showing an increased fraction of immature spines in CDKL5−/y mice (Fisher’s exact test P < 0.001). (C) Quantitation of spine morphology in Golgi-stained V1 slices showing a pattern of decreased morphological maturity in CDKL5−/y mice (χ2 test P < 0.001). (D) Representative images of PSD95 punctate staining from CDKL5−/y and CDKL5+/y V1. (E) Quantitation of PSD95+ puncta density showing a reduction in CDKL5−/y mice (two-tailed Student’s t-test P < 0.001). (F) Representative images of VGAT punctate staining from CDKL5−/y and CDKL5+/y dLGN. (G) Quantitation of VGAT+ puncta density showing an increase in CDKL5−/y mice (two-tailed Student’s t-test P = 0.0135). *P < 0.05; **P < 0.01; ***P < 0.001. Scale bars (D and F) are equal to 2 μm. Error bars represent SEM.
Impaired visual responses in mice with deletion of CDKL5 in cortical excitatory neurons
CDKL5 mutant mice present a deficit in the processing of visual stimuli that can be assessed with a remarkably high discrimination power by IOS imaging of visually evoked responses (22). These abnormal visual responses, however, could arise from defects in upstream visual areas with no intrinsic dysfunction present in visual cortical circuits. In order to assess the specific contribution of the visual cortex, we used a conditional KO mouse model in which the removal of the floxed CDKL5 allele in mutant mice (CDKL5flox/y) was achieved by crossing these mice with mice expressing CRE-recombinase driven by the emx1 promoter (emx1-CRE+/−). As a result, CDKL5flox/y;emx1-CRE+/− animals (cKO) lack CDKL5 only in cortical excitatory neurons and in some glial cells (11), thus sparing all the subcortical visual areas (Supplementary Material, Fig. S4). The expression of CRE-recombinase driven with the emx1 promoter is known to induce recombination as early as at embryonic day (E) 10.5 (11), a time point in which the expression of all the most abundant isoforms of CDKL5 in the mouse brain is still very low (12). This ensures that the phenotypic effects observable in adult mice are the result of an altered development or maturation, closely resembling CDKL5 null mice and patients.
To explore the role of cortical CDKL5 in the pathogenesis of visual deficits in CDKL5 mutants, we first asked whether the morphological alterations previously identified in germline CDKL5 null mice were present in cKO mice in which CDKL5 is normally present in subcortical visual nuclei. Since the retina is unaffected in CDKL5 germline mutants, we began evaluating the presence of such alterations in the visual pathway of cKO mice from the dLGN. Spine density and morphology (N = 4 per group), PSD95+ puncta (N = 3 per group) and VGAT+ puncta (N = 4 per group) were analyzed in cKO and WT littermates. The results showed that spine density in the dLGN of cKO mice was not different from WT control mice (Fig. 4A; two-tailed Student’s t-test P = 0.528). Moreover, both the proportion of mature and immature spines (Fig. 4B; χ2 test P = 0.061) and their morphology (Fig. 4C; χ2 test P = 0.163) were unaltered. Finally, the density of PSD95+ and VGAT+ puncta did not differ between WT and cKO mice (Fig. 4D–G; two-tailed Student’s t-test P = 0.533 for PSD95 and P = 0.812 for VGAT). Thus, as expected from the pattern of expression of Cre recombinase, the cKO model preserves the integrity of the subcortical retino-cortical pathway, allowing to study the role of cortical CDKL5 in the genesis of visual alterations.
Figure 4.
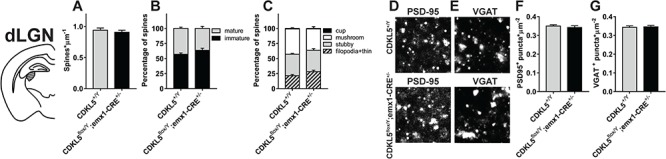
Synaptic morphology in the dLGN and V1 of cKO mice with deletion of CDKL5 in cortical excitatory neurons. (A) Quantitation of spine density in Golgi-stained dLGN slices from cKO (CDKL5flox/y;emx1-CRE+/−) and CDKL5+/y mice showing no significant difference between the two groups (two-tailed Student’s t-test P = 0.528). (B) Quantitation of the relative proportion of mature and immature spines in Golgi-stained dLGN slices showing no difference in the fraction of mature and immature spines in cKO mice compared to WT controls (χ2 test P = 0.0612). (C) Quantitation of spine morphology in Golgi-stained dLGN slices showing no difference in morphological maturity in cKO mice (χ2 test P = 0.163). (D) Representative images of PSD95 punctate staining from cKO and CDKL5+/y dLGN. (E) Representative images of VGAT punctate staining from cKO and CDKL5+/y dLGN. (F) Quantitation of PSD95+ puncta density showing no difference (two-tailed Student’s t-test P = 0.533). (G) Quantitation of VGAT+ puncta density showing no difference (two-tailed Student’s t-test P = 0.812). *P < 0.05; **P < 0.01; ***P < 0.001. Calibration bar is equal to 2 μm. Error bars represent SEM.
We then asked whether cortical CDKL5 plays a necessary role in the normal physiology of cortical visual processing. To this purpose, we performed IOS imaging, recording visually evoked responses in male cKO animals (N = 8) and, as a control, in parental mouse lines expressing CRE (N = 5; emx1-CRE+/−) or carrying a CDKL5 floxed allele (N = 9; CDKL5flox/y; Fig. 5C and D). IOS response amplitude to visual stimulation was significantly different between groups [Fig. 5A; one-way analysis of variance (ANOVA) p = 0.004]. cKO animals showed a decreased response amplitude with respect to both CRE expressing mice (post hoc Holm–Sidak’s multiple comparisons test; emx1-CRE+/− versus cKO; P = 0.0342) and CDKL5 floxed mice (post hoc Holm–Sidak’s multiple comparisons test; CDKL5flox/y versus cKO; P = 0.004). Such a reduced response is also evident in the average time course of the cortical reflectance for each group (Fig. 4C). In order to further characterize the precise impact of cortical CDKL5 onto the visual deficit, we also recorded IOS responses from germline mutant mice. As expected from previously published data (22), this cohort recapitulated the impairment in the amplitude of visual responses (Fig 5B; two-tailed Student’s t-test P < 0.001). Interestingly, by quantitatively measuring the magnitude of the impairment using Cohen’s d as an effect size estimator (6), we noticed that the effect produced by the germline deletion of CDKL5 was bigger (Cohen’s d = 2.78) than the one produced by conditional deletion from excitatory cortical cells (Cohen’s d = 1.73), although the effect size can be regarded as very large for both CDKL5 null mice and cKO (31). Thus, the effect size analysis suggests a major role for the cortex in generating the visual phenotype although it cannot be excluded an enhancement of the deficit by defects in other brain areas or cell types.
Figure 5.
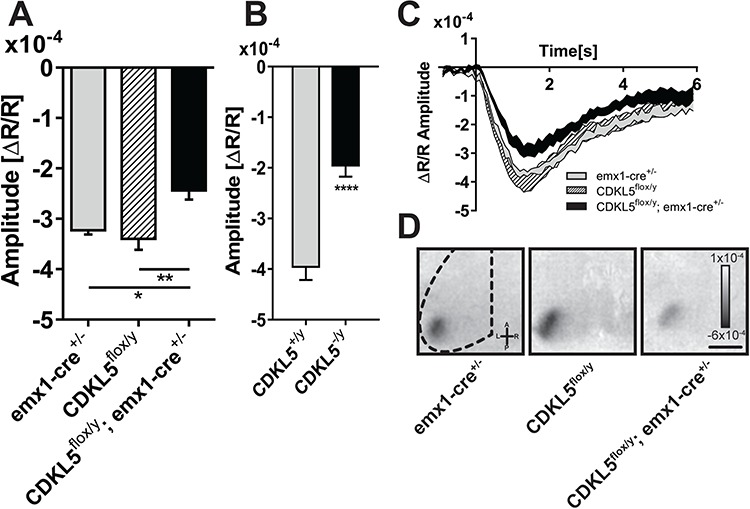
Cortical visual responses are impaired in cKO mice with deletion of CDKL5 in cortical excitatory neurons. (A) Quantitation of the average amplitude of the intrinsic signal between the experimental groups, cKO, floxed controls (CDKL5flox/y) and emx1-CRE driver controls (emx1-CRE+/−), showing a significant reduction of the evoked visual responses in cKO mice with respect to both control groups (one-way ANOVA P = 0.004; post hoc Holm–Sidak’s multiple comparisons test, emx1-CRE+/− versus cKO, P = 0.0342; CDKL5flox/y versus cKO, P = 0.0043). (B) Quantitation of the average amplitude of the intrinsic signal in CDKL5 null animals showing a significant reduction with respect to controls (two-tailed Student’s t-test P < 0.001). (C) Average time course of the intrinsic signal in the V1 after visual stimulation for the experimental groups in (A). Each area represents mean ± SEM for all the mice in the group. (D) Representative images of a typical intrinsic signal response to visual stimulation localized in the V1. Dark areas represent active portions of brain tissue. *P < 0.05; **P < 0.01; ***P < 0.001. Scale bar (C) is equal to 1.8 mm. Error bars represent SEM.
cKO animals lack CDKL5 from only excitatory cortical neurons and some glial cells. To characterize the structural abnormalities underlying such functional deficit, we performed a series of anatomical analysis in the V1 of cKO animals. Surprisingly, dendritic protrusion density in Golgi-stained dendrites of layer 2/3 (Fig. 6) and layer 5 (Supplementary Material, Fig. S3) cortical cells was increased in cKO animals compared to controls (Fig. 6A; two-tailed Student’s t-test P = 0.0014; Supplementary Material, Fig. S3D). This increase, however, was accompanied by a marked shift in dendritic spine morphology toward a more immature state (Fig 6B, χ2 test P < 0.001; Fig 6C, χ2 test P < 0.001; Supplementary Material, Fig. S3E and F). In accordance with the increase in spine density, also the density of PDS95+ puncta, assessed in layer 2/3, was increased (Fig. 6D and F; two-tailed Student’s t-test P = 0.0085) in the cKO animals. Intriguingly, despite the fact that CDKL5 deletion happened only in excitatory cells, we also found an increase in the density of layer 2/3 VGAT+ puncta (Fig. 6E and G; two-tailed Student’s t-test P < 0.001) suggesting that a complex interaction between excitatory and inhibitory cells, possibly involving non-cell-autonomous effects, might be induced by CDKL5 mutations.
Figure 6.
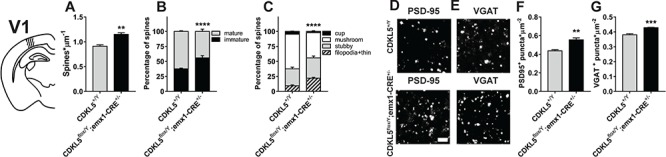
(A) Quantitation of spine density in Golgi-stained V1 slices from cKO (CDKL5flox/y;emx1-CRE+/−) and CDKL5+/y mice showing an increase in cKO mice (two-tailed Student’s t-test P = 0.0014). (B) Quantitation of the relative proportion of mature and immature spines in Golgi-stained V1 slices showing an increased fraction of immature spines in cKO mice compared to WT controls (χ2 test P < 0.001). (C) Quantitation of spine morphology in Golgi-stained V1 slices showing a pattern of decreased morphological maturity in cKO mice (χ2 test P < 0.001). (D) Representative images of PSD95 punctate staining from cKO and CDKL5+/y V1. (E) Representative images of VGAT punctate staining from cKO and CDKL5+/y V1. (F) Quantitation of PSD95+ puncta density showing an increase in cKO mice (two-tailed Student’s t-test P = 0.0085). (G) Quantitation of VGAT+ puncta density showing an increase in cKO mice (two-tailed Student’s t-test P < 0.001). *P < 0.05; **P < 0.01; ***P < 0.001. Calibration bar is equal to 2 μm. Error bars represent SEM.
Discussion
Visually evoked responses are widely used for assessment of visual function in humans. They can be measured rapidly and non-invasively from the scalp by extracting and averaging evoked signals from electroencephalographic activity recorded during a passive fixation task. In particular, the VEP waveform is fully mature by the end of the first year of life, allowing for longitudinal studies throughout development in children (19). These features make the VEP, or functional imaging, an ideal translational tool for functional assessment in children with neurodevelopmental disorders. Indeed, previous studies showed abnormal visual responses in animal models of CDD (22) and other closely related disorders (3,9). Importantly, a standardized analysis of visual responses was found to be useful to reveal stage-specific alterations in Rett syndrome patients (17). However, a dissection of the abnormalities in the visual circuitry responsible for the origin of VEP alterations remains elusive. Indeed, damage anywhere along the visual path may affect the signal in the cortex. Our study provides evidence supporting a cortical origin of the visual response deficit occurring in a mouse model of CDD. Indeed, visual cortical neuronal morphology in the visual cortex was strongly affected by CDKL5 mutation, and deletion of CDKL5 from excitatory cells of the visual cortex was sufficient to elicit a significant impairment of visual responses. Moreover, although CDKL5 was found to be present in the retina, no differences in retinal laminar organization, morphology and fine structural features were observed. This analysis was performed using a battery of specific antibodies labeling cellular components of either the retinal vertical pathway of information flow (specifically, photoreceptors, rod and cone bipolar cells and ganglion cells) or horizontal modulatory elements (horizontal and amacrine cells). In particular, no abnormalities were evident in the photoreceptors or GCLs, which undergo major degeneration in a number of diseases, such as, respectively, retinitis pigmentosa and glaucoma (7,34). Furthermore, no signs of glial hyperreactivity (a generalized indicator of tissue damage) were found, and inner retinal layers, whose structure and distribution of synaptic proteins remodel in inherited photoreceptor degeneration (36,40) and diabetic retinopathy (2), were also intact. Cholinergic amacrines, a sensitive marker of the retinal patterning and laminar positioning were also normal (28). Hence, although subtle molecular differences cannot be totally ruled out, we can conclude that retinal morphology in CDKL5 null mice is indistinguishable from that of age-matched, WT animals.
Our analysis of visual pathway integrity in CDKL5 null mice continued with the study of the LGN, a thalamic nucleus relaying visual information from the retina to the visual cortex. CDKL5 is known to be expressed in the thalamus and we found that LGN neurons from CDKL5 null mice displayed reduced spine density and PSD95 synaptic staining. CDKL5 is highly concentrated in dendritic spines (29,42) where it interacts with essential protein components of mature synapses like PSD95 and NGL-1, respectively, a major scaffold protein in the postsynaptic density and a cell adhesion molecule crucial in synapse formation and maturation (15,29,42). Its absence determines a reduction in dendritic complexity, spine density and stability both in vitro and in vivo (8,10,25,38). These mechanisms have not been addressed specifically in thalamic cells; however, they could be involved in the deficits observed in LGN neurons.
At the functional level, thalamic deficits induced by CDKL5 absence seems to be dispensable for visual response deficits; indeed, conditional deletion of CDKL5 from cortical excitatory cells is sufficient to reduce visual responses even if LGN dendritic spines and synaptic PSD95 staining are normal.
The use of the emx1 promoter to drive Cre recombinase expression and CDKL5 deletion also preserves CDKL5 expression in inhibitory neurons. Global CDKL5 KO cortex has altered inhibitory synaptic labeling and increase in parvalbumin positive cells (27). CDKL5 deletion from inhibitory cells has been previously shown to result in a partial phenotype (1,33); however, visual function was not tested in these models. Our data suggest that CDKL5 deficiency in inhibitory cells is not strictly necessary for the reduction of visual responses, although we cannot exclude that visual deficits could be induced by CKDL5 deletion from inhibitory cells. An indirect involvement of inhibitory cells in the visual impairment of CDKL5 cKO could be hypothesized considering the results of our morphological analysis showing dramatic rearrangements occurring both in excitatory and inhibitory synaptic contacts in the cKO mouse. This non-cell-autonomous phenotype is reminiscent of the one observed in the hippocampus of CDKL5 mutants with deletion restricted to excitatory neurons (37) where an increase in the frequency of miniature inhibitory synaptic currents was observed. These indirect effects of CDKL5 mutation could be relevant for the understanding of the circuit deficits present in CDD patients that are often a mosaic of normal and mutant cells. In summary, our study demonstrates a key role for CDKL5 in the physiology of postretinal visual pathways. Previous work showed that visual impairment is present in CDD patients and represents a biomarker of high accuracy and sensitivity to classify CDD mice (22). Our data provide a neurobiological background for the use of visual responses for assessment in CDD.
Materials and Methods
Mice housing and handling
Animals were maintained in rooms at 22°C with a standard 12 h light–dark cycle. During the light phase, a constant illumination <40 lux (measured at the cage) from fluorescent lamps was maintained. Food (standard diet, 4RF25 GLP Certificate, Mucedola SRL) and water were available ad libitum and changed weekly. Open-top cages (36.5 × 20.7 × 14 cm; 26.7 × 20.7 × 14 cm for up to five adult mice or 42.5 × 26.6 × 15.5 cm for up to eight adult mice) with wooden dust-free bedding were used. All the experiments were carried out in accordance with the directives the European Community Council (2011/63/EU) and approved by the Italian Ministry of Health. All necessary efforts were made to minimize both stress and the number of animals used.
The CDKL5 mutant mouse strain used in this work derives from the C57BL/6N background, has been developed in (1) and has been backcrossed in C57BL/6J for five generations. Colony founders were selected for the absence of the rd8 retinal degeneration allele spontaneously present in C57BL/6N mice (21). Male WT mice were bred with female heterozygous to obtain mutant and WT littermates. To generate conditional knock-out mice lacking CDKL5 only from pallial structures, sparing most of the subcortical regions, we crossed homozygous mice bearing a floxed version of CDKL5, described in (1), with mice expressing CRE recombinase under the control of emx1 promoter (The Jackson Laboratory; B6.129S2-Emx1tm1(cre)Krj/J, stock no. 005628) (11). Genotyping (P10–12) was performed on ear tissue. Weaning was performed at P21–23. Data analysis was performed by experimenters blind to the genotype.
Western blot
Adult (>P120) CDKL5+/y (N = 4) and CDKL5−/y (N = 4) male mice were sacrificed by decapitation, and retinas were rapidly removed from eyes. The retinal extracts were homogenized in a radioimmunoprecipitation assay (RIPA) buffer and protein concentration was determined with the Bio-Rad protein assay kit. Total lysates were boiled in SDS sample buffer, separated by Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) and blotted to nitrocellulose membrane (Bio-Rad, Milano Italy). Filters were blocked in blocking Odyssey buffer (Licor) and incubated with primary antibody for 16 h at 4°C following the instruction of Odyssey System (Licor, Lincoln, NE, USA). The following primary antibodies were used: anti-CDKL5 (S957D) 1:750 (Division of Signal Transduction Therapy at the University of Dundee) and beta-tubulin (anti-mouse 1:3000; Sigma, Milano, Italy). After washing three times with phosphate buffered saline (PBS) and blocking Odyssey buffer (Licor) 1:1, filters were incubated with secondary antibodies IRDye® 680LT/800CW Goat (polyclonal) anti-sheep or anti-mouse IgG, diluted 1:20000 (Licor) for 2 h at room temperature. Detection was performed by Odyssey System CLX.
IHC on retinal tissue
Tissue preparation and histology
Adult (>p120) male mice were anesthetized with intraperitoneal injections of Avertin (3-bromo-ethanol in 1% tert-amyl alcohol; 0.1 ml/5 g body weight). Eyes, which were previously labeled on the dorsal pole with a surgical skin marker (Secureline Surgical Skin Marker, Aspen Surgical, USA) were quickly enucleated and immersed in 0.1 M PB, followed by removal of the anterior segments (cornea, iris and lens). Mice were sacrificed by quick cervical dislocation. Eyecups were immediately fixed in 4% paraformaldehyde (PFA) in 0.1 M PB, pH 7.4, for 30/60 min at room temperature. Repeated (5 × 5 min) washes were performed in 0.1 M PB, followed by incubation in 30% w/w sucrose dissolved in 0.1 M PB overnight at 4°C and infiltration in Tissue-Tek O.C.T. compound (4583; Sakura Olympus, Italy). Sample freezing in plastic cryo-molds was achieved using isopentane cooled down with dry ice; samples were stored at −20°C until further use. Frozen, vertical sections were cut at a cryostat at 12 μm thickness and collected on Superfrost slides for IHC. For whole-mount preparations, defrosted eyecups were extensively washed in PBS; the retina was gently detached from the sclera and flattened by making four radial cuts toward the optic nerve head maintaining a reference on the dorsal pole. For IHC, both retinal whole mounts and sections were incubated in blocking solution (0.03% Triton X-100 + 5% serum from the species in which the secondary antibody was generated in 0.01 M PBS), for 2 h at room temperature if sections and overnight at 4°C if whole mounts; primary antibody (Ab) solution (diluted in PBS + 0.01% Triton X-100 + 1% serum), overnight at 4°C for sections and 3 days at 4°C for whole mounts. The antigen-antibody complex was revealed by incubation in fluorescent secondary Ab solution—diluted as the primary Ab—for 2 h at room temperature for sections and 2 days at 4°C for retinal whole mounts.
Antibodies
Mouse monoclonal primary antibodies used on retinal specimens were C-terminal binding protein-2 (CtBP2/RIBEYE; 612044; BD Biosciences; diluted 1:800), postsynaptic density-95 (PSD95; ab13552; Abcam Ltd, Cambridge, UK; diluted 1:500), ZNP-1/synaptotagmin-II (ZNP-1; kind gift from Professor Erica Fletcher, developed at the Zebrafish International Research Centre, University of Oregon, US; diluted 1:1000) and glutamic acid decarboxylase-67 (GAD67; MAB5406; Merck-Millipore, Italy; diluted 1:500).
Rabbit polyclonal primary antibodies were cone arrestin (AB15282; Merck-Millipore; diluted 1:5000), calbindin D (CB38a; Swant Ltd, Switzerland; diluted 1:500), PK Cα (PKCα; P4334, Sigma-Aldrich, Italy; diluted 1:800) and RBPMS (1830-RBPMS; PhosphoSolutions, Colorado; diluted 1:500).
A goat polyclonal ChAT (AB144P; Merck-Millipore, Italy; 1:500) was also used.
Antibodies against cone arrestin and RBPMS were also used in whole-mount preparations, at 10× the concentrations used for sections.
Secondary antibodies were donkey anti-mouse Alexa Fluor 488 (715-547-003) and donkey anti-rabbit Rhodamine Red X (711-295-152) from Jackson ImmunoResearch Laboratories, Inc., USA; these were diluted 1:800 both for retinal sections and whole-mount preparations.
After multiple washing steps in PBS, specimens were mounted in Vectashield Antifade Mounting Medium (H-1000; Vector Laboratories, Burlingame, CA), coverslipped and sealed with nail polish.
Imaging and data analysis
Retinal whole mounts were imaged with a Zeiss Axioplan light microscope equipped with a Zeiss Axiocam color camera using a Plan Neofluar 1.25×/0.035 objective to measure retinal surface prior and postconfocal microscopy. Image acquisition for morphometric observations in whole mounts and vertical sections was performed with a Leica TCS-SL confocal microscope equipped with 488 and 453 lasers, using a Plan Apochromat 40×/1.40 oil objective, employing identical laser and acquisition settings for all samples. For cell counts in retinal whole mounts, four fields (250 × 250 μm) were acquired at each eccentricity of the retina, altogether representing 1/15 of the total area of the tissue. Images were saved as TIFF files and transferred to a MetaMorph image analyzer, where semi-automatic cell counts were performed. The total number of cells/retina was calculated.
dLGN and V1 morphological analysis
Tissue preparation and histology
Male mice (2–3 months old) were euthanized with isoflurane (2% in pure oxygen) and sacrificed by cervical dislocation. Brains were quickly removed and cut along the midline. Right hemispheres were processed for IHC, while left hemispheres were Golgi stained as described below. All steps of sectioning, imaging and data analysis were conducted blindly to genotype.
For PSD95 and VGAT IHC, right hemispheres were fixed by immersion in 4% PFA in 0.1 M PB pH 7.4, stored in fixative for 48 h, kept in 20% sucrose w/w dissolved in 0.1 PB for an additional 24 h and then frozen with cold ice. Right hemispheres were cut with a freezing microtome into 30 μm thick coronal sections that were serially collected. One out of four free-floating sections from the LGN and from the V1 were incubated for 24 h at 4°C with the following primary antibodies: a rabbit polyclonal anti-PSD95 antibody (1:1000; Abcam Ltd Cambridge UK; ab 18258) or rabbit polyclonal anti-VGAT antibody (1:600; Synaptic System, Göttingen, Germany). Sections were then incubated at room temperature for 2 h with a CY3-conjugated goat anti-rabbit IgG (1:200; Jackson Immuno Research Laboratories, Inc. PA, USA).
For Golgi staining, left hemispheres were Golgi-stained using the FD Rapid Golgi Stain TM Kit (FD NeuroTechnologies, MD, USA). Briefly, hemispheres were immersed in the impregnation solution containing mercuric chloride, potassium dichromate and potassium chromate and stored at room temperature in darkness for 2–3 weeks. Hemispheres were cut with a microtome in 100 μm thick coronal sections that were directly mounted on gelatin-coated slides and were air dried at room temperature in the dark for an additional 2–3 days. After drying, sections were rinsed with distilled water and subsequently stained in the developing solution of the kit.
Imaging and data analysis
All analyses were carried out in cortical layers 2/3 and 5 from the V1 and in the LGN identified according to the Paxinos mouse brain atlas (26). For quantification of PSD95 and VGAT immunoreactive puncta, images were acquired using a LEICA TCS SL confocal microscope (Leica Microsystems; 63× oil immersion objective, NA 1.32; zoom factor, 8). Three to four sections per animal were analyzed and puncta counts were performed on a single plan, 1024 × 1024 pixel images. Counting was carried out using Image Pro Plus software (Media Cybernetics, Silver Spring, MD, USA) and the number of PSD95 and VGAT immunoreactive puncta was expressed per μm2. For the quantification of dendritic spines, images were acquired using a Leica light microscope (Leica Microsystems; 100× oil immersion objective, NA 1.4). Dendritic spine density was measured by manually counting the number of dendritic spines on dendritic segments in the LGN and on secondary dendrites of pyramidal neurons of layer 2/3 and layer 5 of the V1. In each mice, 15 dendritic segments (segment length, 10–30 μm) from each zone were analyzed and the linear spine density was calculated by dividing the total number of counted spines by the length of the sampled dendritic segment. Based on their morphology, dendritic spines can be divided into five different classes (immature spines: filopodium-like, thin- and stubby-shaped; mature spines: mushroom- and cup-shaped), which also reflect their state of maturation. The total number of spines was expressed per μm and the number of spines belonging to each class was counted and expressed as a percentage.
IOS imaging
Surgery
Surgery for IOS imaging was performed as described in (22). Mice were anesthetized isoflurane (3% induction; 1% maintenance) and head fixed on a stereotaxic frame using ear bars. Body temperature was controlled using a heating pad and a rectal probe to maintain the animals’ body at 37°C. Local anesthesia was provided using subcutaneous lidocaine (2%) injection and the eyes were protected with a dexamethasone-based ointment (Tobradex, Alcon Novartis). The scalp was thoroughly cleaned with 70% ethanol, then it was removed and the skull was carefully cleaned with saline. The skin was secured to the skull using cyanoacrylate. A thin layer of cyanoacrylate was poured over the exposed skull to attach a custom-made metal ring (9 mm internal diameter) centered over the binocular visual cortex. When the glue dried off, a drop of transparent nail polish was spread over the area to ameliorate optical access. After surgery, the animals were allowed to recover fully in a heated box and monitored to ensure the absence of any sign of discomfort. Before any other experimental procedure, mice were left to recover for 24/48 h. During this period, paracetamol (5 mg/ml) was administered in the water as antalgic therapy.
Imaging and data analysis
IOS recordings were performed under isoflurane anesthesia (3%). Images were visualized using a custom Leica microscope (Leica Microsystems). Red light illumination was provided by eight individually addressable LEDs (WS2812) attached to the objective (Leica Z6 APO coupled with a Leica PlanApo 2.0× 10447178) by a custom 3D printed conical holder. Visual stimuli were generated using Matlab Psychtoolbox and presented on a gamma corrected 24 in monitor (C24F390FHU).
Horizontal sine wave gratings were presented in the binocular portion of the visual field enclosed in a Gaussian envelope spanning −10 to +10 degrees of azimuth and −5 to +60 (full monitor height) degrees of altitude, with a spatial frequency of 0.03 cycles per degree, mean luminance 20 cd/m2 and a contrast of 90%. The stimulus consisted in the abrupt contrast reversal of a grating with a temporal frequency of 4 Hz for 1 s, time locked with a 12-bit depth acquisition camera (PCO edge 5.5) using a parallel port trigger. Interstimulus time was 13 s. Frames were acquired at 30 fps with a resolution of 540 × 640 pixels. The signal was averaged for at least 4 groups of 20 trials and downsampled in time to 10 fps and in space to 270 × 320 pixels. Fluctuations of reflectance (R) for each pixel were computed as the normalized difference from the average baseline (ΔR/R). For each recording, an image representing the mean evoked response was computed by averaging frames between 0.5 and 2.5 s after stimulation. The mean image was then low-pass filtered with a 2D average square spatial filter (7 pixels). To select the binocular portion of the V1 for further analysis, a region of interest (ROI) was automatically calculated on the mean image of the response by selecting the pixels in the lowest 20% ΔR/R of the range between the maximal and minimal intensity pixel (4). To weaken background fluctuations a manually selected polygonal region of reference (ROR) was subtracted. The ROR was placed where no clear response, blood vessel artifact or irregularities of the skull were observed (13) Mean evoked responses were quantitatively estimated as the average intensity inside the ROI.
Statistical analysis
Statistical analysis was carried out in Graphpad Prism 7. For each result, the statistical tests used are specified in the figure legends and in the respective result section. Briefly, when two different normally distributed experimental groups were compared, we used a two-tail Student’s t-test. When the proportion of observations in mutually exclusive classes was compared we used chi-square test. When more than two normally distributed groups were compared we used a one-way ANOVA test.
Conflict of Interest statement. None declared.
Funding
International Foundation for CDKL5 Research; Fondazione Telethon (GGP15098); University of Pennsylvania Orphan Disease Center on behalf of LouLou Foundation; European Union Research and Innovation Programme, Marie Sklodowska-Curie (674901, ‘switchboard’ to A.S. and E.S.).
Supplementary Material
References
- 1. Amendola E., Zhan Y., Mattucci C., Castroflorio E., Calcagno E., Fuchs C., Lonetti G., Silingardi D., Vyssotski A.L., Farley D. et al. (2014) Mapping pathological phenotypes in a mouse model of CDKL5 disorder. PLoS One, 9(5), e91613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barber A.J. and Baccouche B. (2017) Neurodegeneration in diabetic retinopathy: potential for novel therapies. Vision Res., 139(October), 82–92. [DOI] [PubMed] [Google Scholar]
- 3. Boggio E.M., Pancrazi L., Gennaro M., Lo Rizzo C., Mari F., Meloni I., Ariani F., Panighini A., Novelli E., Biagioni M. et al. (2016) Visual impairment in FOXG1-mutated individuals and mice. Neuroscience, 324(June), 496–508. [DOI] [PubMed] [Google Scholar]
- 4. Cang J., Kalatsky V.A., Löwel S. and Stryker M.P. (2005) Optical imaging of the intrinsic signal as a measure of cortical plasticity in the mouse. Vis. Neurosci., 22(5), 685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chaudhry F.A., Reimer R.J., Bellocchio E.E., Danbolt N.C., Osen K.K., Edwards R.H. and Storm-Mathisen J. (1998) The vesicular GABA transporter, VGAT, localizes to synaptic vesicles in sets of glycinergic as well as GABAergic neurons. J. Neurosci., 18(23), 9733–9750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cohen J. (2013) Statistical Power Analysis for the Behavioral Sciences. Routledge, London UK. [Google Scholar]
- 7. Cuenca N., Fernández-Sánchez L., Campello L., Maneu V., De la Villa P., Lax P. and Pinilla I. (2014) Cellular responses following retinal injuries and therapeutic approaches for neurodegenerative diseases. Prog. Retin. Eye Res., 43(November), 17–75. [DOI] [PubMed] [Google Scholar]
- 8. Della Sala G., Putignano E., Chelini G., Melani R., Calcagno E., Michele Ratto G., Amendola E., Gross C.T., Giustetto M. and Pizzorusso T. (2016) Dendritic spine instability in a mouse model of CDKL5 disorder is rescued by insulin-like growth factor 1. Biol. Psychiatry, 80(4), 302–311. [DOI] [PubMed] [Google Scholar]
- 9. Durand S., Patrizi A., Quast K.B., Hachigian L., Pavlyuk R., Saxena A., Carninci P., Hensch T.K. and Fagiolini M. (2012) NMDA receptor regulation prevents regression of visual cortical function in the absence of Mecp2. Neuron, 76(6), 1078–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fuchs C., Gennaccaro L., Trazzi S., Bastianini S., Bettini S., Martire V.L., Ren E., Medici G., Zoccoli G., Rimondini R. and Ciani E. (2018) Heterozygous CDKL5 knockout female mice are a valuable animal model for CDKL5 disorder. Neural Plast., 2018(May), 9726950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gorski J.A., Talley T., Qiu M., Puelles L., Rubenstein J.L.R. and Jones K.R. (2002) Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci., 22(15), 6309–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hector R.D., Dando O., Landsberger N., Kilstrup-Nielsen C., Kind P.C., Bailey M.E.S. and Cobb S.R. (2016) Characterisation of CDKL5 transcript isoforms in human and mouse. PLoS One, 11(6), e0157758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heimel J.A., Hartman R.J., Hermans J.M. and Levelt C.N. (2007) Screening mouse vision with intrinsic signal optical imaging. Eur. J. Neurosci., 25(3), 795–804. [DOI] [PubMed] [Google Scholar]
- 14. Jhang C.-L., Huang T.-N., Hsueh Y.-P. and Liao W. (2017) Mice lacking cyclin-dependent kinase-like 5 manifest autistic and ADHD-like behaviors. Hum. Mol. Genet., 26(20), 3922–3934. [DOI] [PubMed] [Google Scholar]
- 15. Kim E. and Sheng M. (2009) The postsynaptic density. Curr. Biol., 19(17), R723–R724. [DOI] [PubMed] [Google Scholar]
- 16. Kim J.-W., Yang H.-J., Brooks M.J., Zelinger L., Karakülah G., Gotoh N., Boleda A. et al. (2016) NRL-regulated transcriptome dynamics of developing rod photoreceptors. Cell Rep, 17(9), 2460–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. LeBlanc J.J., DeGregorio G., Centofante E., Vogel-Farley V.K., Barnes K., Kaufmann W.E., Fagiolini M. and Nelson C.A. (2015) Visual evoked potentials detect cortical processing deficits in Rett syndrome. Ann. Neurol., 78(5), 775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lehman S.S. (2012) Cortical visual impairment in children. Curr. Opin. Ophthalmol., 23(5), 384–387. [DOI] [PubMed] [Google Scholar]
- 19. Lenassi E., Likar K., Stirn-Kranjc B. and Brecelj J. (2008) VEP maturation and visual acuity in infants and preschool children. Doc. Ophthalmol., 117(2), 111–120. [DOI] [PubMed] [Google Scholar]
- 20. Matsuzaki M., Ellis-Davies G.C., Nemoto T., Miyashita Y., Iino M. and Kasai H. (2001) Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci., 4(11), 1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mattapallil M.J., Wawrousek E.F., Chan C.-C., Zhao H., Roychoudhury J., Ferguson T.A. and Caspi R.R. (2012) The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest. Ophthalmol. Vis. Sci., 53(6), 2921–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mazziotti R., Lupori L., Sagona G., Gennaro M., Della Sala G., Putignano E. and Pizzorusso T. (2017) Searching for biomarkers of CDKL5 disorder: early-onset visual impairment in CDKL5 mutant mice. Hum. Mol. Genet., 26(12), 2290–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moseley B.D., Dhamija R., Wirrell E.C. and Nickels K.C. (2012) Historic, clinical, and prognostic features of epileptic encephalopathies caused by CDKL5 mutations. Pediatr. Neurol., 46(2), 101–105. [DOI] [PubMed] [Google Scholar]
- 24. Okuda K., Kobayashi S., Fukaya M., Watanabe A., Murakami T., Hagiwara M., Sato T., Ueno H., Ogonuki N., Komano-Inoue S. et al. (2017) CDKL5 controls postsynaptic localization of GluN2B-containing NMDA receptors in the hippocampus and regulates seizure susceptibility. Neurobiol. Dis., 106(October), 158–170. [DOI] [PubMed] [Google Scholar]
- 25. Okuda K., Takao K., Watanabe A., Miyakawa T., Mizuguchi M. and Tanaka T. (2018) Comprehensive behavioral analysis of the Cdkl5 knockout mice revealed significant enhancement in anxiety- and fear-related behaviors and impairment in both acquisition and long-term retention of spatial reference memory. PLoS One, 13(4), e0196587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paxinos G. (2007) Atlas of the Developing Mouse Brain: E17.5, PO, and P6. Academic Press, Amsterdam NL. [Google Scholar]
- 27. Pizzo R., Gurgone A., Castroflorio E., Amendola E., Gross C., Sassoè-Pognetto M. and Giustetto M. (2016) Lack of Cdkl5 disrupts the organization of excitatory and inhibitory synapses and parvalbumin interneurons in the primary visual cortex. Front. Cell. Neurosci., 10, 261. 10.3389/fncel.2016.00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reese B.E. and Keeley P.W. (2016) Genomic control of neuronal demographics in the retina. Prog. Retin. Eye Res., 55(November), 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ricciardi S., Ungaro F., Hambrock M., Rademacher N., Stefanelli G., Brambilla D., Sessa A., Magagnotti C., Bachi A., Giarda E. et al. (2012) CDKL5 ensures excitatory synapse stability by reinforcing NGL-1–PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat. Cell Biol., 14(9), 911–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rusconi L., Salvatoni L., Giudici L., Bertani I., Kilstrup-Nielsen C., Broccoli V. and Landsberger N. (2008) CDKL5 expression is modulated during neuronal development and its subcellular distribution is tightly regulated by the C-terminal tail. J. Biol. Chem., 283(44), 30101–30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sawilowsky S.S. (2009) New effect size rules of thumb. J. Mod. Appl. Stat. Methods, 8(2), Article 26, 10.22237/jmasm/1257035100. [DOI] [Google Scholar]
- 32. Scannevin R.H. and Huganir R.L. (2000) Postsynaptic organisation and regulation of excitatory synapses. Nat. Rev. Neurosci., 1(2), 133–141. [DOI] [PubMed] [Google Scholar]
- 33. Schroeder E., Yuan L., Seong E., Ligon C., Nicholas D.K., Gurumurthy C.B. and Arikkath J. (2018) Neuron-type specific loss of CDKL5 leads to alterations in mTOR signaling and synaptic markers. Mol. Neurobiol., 56(6), 4151–4162, 10.1007/s12035-018-1346-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Soto F. and Kerschensteiner D. (2015) Synaptic remodeling of neuronal circuits in early retinal degeneration. Front. Cell. Neurosci., 9(October), 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stalpers X.L., Spruijt L., Yntema H.G. and Verrips A. (2012) Clinical phenotype of 5 females with a CDKL5 mutation. J. Child Neurol., 27(1), 90–93. [DOI] [PubMed] [Google Scholar]
- 36. Strettoi E. (2015) A survey of retinal remodeling. Front. Cell. Neurosci., 9(December), 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tang S., Wang I.-T.J., Yue C., Takano H., Terzic B., Pance K., Lee J.Y., Cui Y., Coulter D.A. and Zhou Z. (2017) Loss of CDKL5 in glutamatergic neurons disrupts hippocampal microcircuitry and leads to memory impairment in mice. J. Neurosci., 37(31), 7420–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Trazzi S., De Franceschi M., Fuchs C., Bastianini S., Viggiano R., Lupori L., Mazziotti R., Medici G., Lo Martire V., Ren E. et al. (2018) CDKL5 protein substitution therapy rescues neurological phenotypes of a mouse model of CDKL5 disorder. Hum. Mol. Genet., 27(9), 1572–1592. [DOI] [PubMed] [Google Scholar]
- 39. Trazzi S., Fuchs C., Viggiano R., De Franceschi M., Valli E., Jedynak P., Hansen F.K., Perini G., Rimondini R., Kurz T., Bartesaghi R. and Ciani E. (2016) HDAC4: a key factor underlying brain developmental alterations in CDKL5 disorder. Hum. Mol. Genet., 25(18), 3887–3907. [DOI] [PubMed] [Google Scholar]
- 40. Vidal-Sanz M., Salinas-Navarro M., Nadal-Nicolás F.M., Alarcón-Martínez L., Valiente-Soriano F.J., Miralles de Imperial J., Avilés-Trigueros M., Agudo-Barriuso M. and Villegas-Pérez M.P. (2012) Understanding glaucomatous damage: anatomical and functional data from ocular hypertensive rodent retinas. Prog. Retin. Eye Res., 31(1), 1–27. [DOI] [PubMed] [Google Scholar]
- 41. Wang I.-T.J., Allen M., Goffin D., Zhu X., Fairless A.H., Brodkin E.S., Siegel S.J., Marsh E.D., Blendy J.A. and Zhou Z. (2012) Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice. Proc. Natl. Acad. Sci. U. S. A., 109(52), 21516–21521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhu Y.-C., Li D., Wang L., Bin L., Zheng J., Zhao S.-L., Zeng R. and Xiong Z.-Q. (2013) Palmitoylation-dependent CDKL5–PSD-95 interaction regulates synaptic targeting of CDKL5 and dendritic spine development. Proc. Natl. Acad. Sci. U. S. A., 110(22), 9118–9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.