


Abstract
Recently, a few Na+-specific RNA-cleaving DNAzymes were reported, where nucleobases are likely to play critical roles in catalysis. The NaA43 and NaH1 DNAzymes share the same 16-nt Na+-binding motif, but differ in one or two nucleotides in a small catalytic loop. Nevertheless, they display an opposite pH-dependency, implicating distinct catalytic mechanisms. In this work, rational mutation studies locate a catalytic adenine residue, A22, in NaH1, while previous studies found a guanine (G23) to be important for the catalysis of NaA43. Mutation with pKa-perturbed analogs, such as 2-aminopurine (∼3.8), 2,6-diaminopurine (∼5.6) and hypoxanthine (∼8.7) affected the overall reaction rate. Therefore, we propose that the N1 position of G23 (pKa ∼6.6) in NaA43 functions as a general base, while that of A22 (pKa ∼6.3) in NaH1 as a general acid. Further experiments with base analogs and a phosphorothioate-modified substrate suggest that the exocyclic amine in A22 and both of the non-bridging oxygens at the scissile phosphate are important for catalysis for NaH1. This is an interesting example where single point mutations can change the mechanism of cleavage from general base to general acid, and it can also explain this Na+-dependent DNAzyme scaffold being sensitive to a broad range of metal ions and molecules.
INTRODUCTION
DNAzymes are DNA sequences with catalytic activities (1–3). A variety of RNA-cleaving DNAzymes require various monovalent (e.g. Na+ (4,5), Ag+ (6)), divalent (e.g., Pb2+ (1), Zn2+ (7), Cu2+ (8), UO22+ (9), Hg2+ (10), Cd2+ (11)) and trivalent (e.g. lanthanides (12)) metal ions for catalysis. DNAzymes are highly attractive for their excellent stability, programmability and cost-effectiveness, leading to interesting applications in biosensing and therapeutics (13–16). However, our understanding of DNAzyme catalysis is still limited compared to the substantial knowledge in ribozyme catalysis due to their rich structural biology data. Most self-cleaving ribozymes (e.g. the Rzb hammerhead (17,18), env25 pistol (19,20) and twister ribozymes (21)) use a general base mechanism, where a guanine helps activate the 2′-OH nucleophile. Adenines or cytosines can play the general acid role in the pistol (19), twister (21) and hepatitis delta virus (HDV) ribozymes (22).
The involvement of polyvalent metal ions may complicate data analysis. For mechanistic studies, monovalent-metal-dependent enzymes are more attractive. The first Na+-dependent DNAzyme, NaA43 was reported in 2015 by Lu et al. (4). Interestingly, its conserved sequence is quite similar to a lanthanide-dependent DNAzyme named Ce13d (23–25). Their overlapping sequence was revealed to be part of a Na+-binding aptamer, which is responsible for their Na+ specificity (25,26). Extensive studies have been performed on the aptamer part to understand specific Na+ binding (27,28).
Recently, we reported the NaH1 DNAzyme, which differs from the NaA43T DNAzyme (a mutated shorter version of NaA43) by only two nucleotides in the enzyme loop (29). An important guanine in NaA43 was replaced by an adenine in NaH1, and NaH1 also requires Na+ for activity. An interesting difference between them is their pH-dependency: the optimal pH being 7 for NaA43 and 6 for NaH1. Such DNAzymes are ideal for mechanistic studies since no polyvalent metals are involved, and catalysis is likely achieved via nucleobases. In this work, we performed careful biochemical and spectroscopic experiments, providing compelling evidence that on the same DNAzyme scaffold, a simple point mutation can switch the mechanism from being general base (for NaA43) to general acid (for NaH1) catalysis. The tolerance of different mechanisms may also explain the activity of this scaffold (e.g. Ce13d) with a diverse range of metal ions and even non-metals.
MATERIALS AND METHODS
Chemicals
All the DNA samples used in this work were purchased from Integrated DNA Technologies (Coralville, IA). Sodium chloride, lithium chloride, potassium chloride, acetate acid, 2-(N-morpholino)ethanesulfonic acid (MES), 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES), and 3-(N-morpholino)propanesulfonic acid (MOPES) were from Mandel Scientific (Guelph, ON, Canada). Lithium hydroxide was purchased from Alfa Aesar. All of the solutions and buffers used in this work were prepared with Milli-Q water. All the DNA sequences used in this work are listed in Supplementary Table S1.
Activity assays
The FAM-labeled substrate (2 μM) was respectively hybridized with each enzyme or their mutants (3 μM) in the reaction buffer to form the DNAzyme complexes. The mixtures were annealed by heating at 93°C for 1 min and then gradually cooling down to 4°C for over 30 min. The DNAzyme complexes were then diluted in reaction buffer (6 μl) to 0.3 μM. A small volume (1 μl) of NaCl was then added to initiate the cleavage reaction with a final concentration of 10 mM Na+. At each time point, a 7 μl reaction solution was quenched with 8 μl of 8 M urea. The cleavage products were quantified by 15% denaturing polyacrylamine gel electrophoresis (dPAGE) and analyzed by a Bio-Rad Chemi-Doc MP imaging system. Reaction buffer A (50 mM MES, pH 6.0, 25 mM LiCl) and buffer B (50 mM MOPES, pH 7.0, 25 mM LiCl) were used.
Kinetic data were fit with the first-order equation, 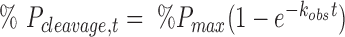 , where %Pmax is the maximum cleavage yield at the end of the reaction and
, where %Pmax is the maximum cleavage yield at the end of the reaction and 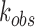 is the cleavage rate constant. For pH-rate profiles, acetate buffer (50 mM) was used for pH 4.0 and 4.5; MES buffer (50 mM) for pH 5.5–6.5; and MOPES buffer (50 mM) for pH 7.0–8.0. All the buffer solutions were adjusted by 1 M LiOH to eliminate background Na+. The pH-rate profiles of NaH1 and NaA43 were fit with equations
is the cleavage rate constant. For pH-rate profiles, acetate buffer (50 mM) was used for pH 4.0 and 4.5; MES buffer (50 mM) for pH 5.5–6.5; and MOPES buffer (50 mM) for pH 7.0–8.0. All the buffer solutions were adjusted by 1 M LiOH to eliminate background Na+. The pH-rate profiles of NaH1 and NaA43 were fit with equations  and
and 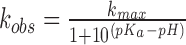 respectively, where
respectively, where 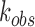 is the observed rate constant, and
is the observed rate constant, and  is the maximal observed rate constant.
is the maximal observed rate constant.
Phosphorothioate substitution
The FAM-labeled PS-modified substrates, Rp and Sp, were separated using HPLC as described previously (11). The substrates were annealed with the DNAzymes with a ratio of 1:1.5. Cleavage kinetics was measured in the presence of 10 mM NaCl for 1 h in reaction buffer A for NaH1 or buffer B for NaA43T. In the metal rescue assays, CdCl2 was introduced in the reaction with a final concentration of 2 mM.
Fluorescence spectroscopy
The DNAzyme complex was prepared by annealing the 2-aminopurine (2AP)-modified enzyme and non-modified substrate with a ratio of 1:2 in buffer A (50 mM MES, 25 mM LiCl, pH 6.0). The mixture was heated to 93°C for 1 min followed by gradual cooling down to 4°C. With excitation at 310 nm, the emission was measured from 360 to 450 nm. Small volumes of chloride salts were gradually titrated into the sample to achieve a final concentration up to 45 mM. After a quick mix, the fluorescence signal was immediately measured in a 1 × 1 cm quartz fluorescence cuvette using a Cary Eclipse fluorometer. The emission intensity enhancement at 370 nm was normalized by F/F0, where F0 and F represent the fluorescence signal before and after the addition of Na+, respectively. The Kd value was obtained by fitting into the one-site binding equation: . [M+] is the metal concentration, and
. [M+] is the metal concentration, and  is the fluorescence change when [M+] = ∞.
is the fluorescence change when [M+] = ∞.
RESULTS AND DISCUSSION
The NaA43 and NaH1 DNAzymes
The secondary structures of the NaA43T and NaH1 DNAzymes are shown in Figure 1A and B, respectively. NaA43T is a truncated version of NaA43 with retained specificity for Na+ (25). These DNAzymes have the same substrate that contained a single RNA linkage (rA for ribo-adenine) serving as the cleavage site. NaA43T and NaH1 have the same 16-nt Na+-binding motif. The catalytically important regions are drawn in red. Our previous study indicated that the G23 residue in NaA43 is highly conserved (25), but no guanine is present in the corresponding region of NaH1. Therefore, these two DNAzymes might have distinct catalytic mechanisms.
Figure 1.

The secondary structures of two Na+-specific RNA-cleaving DNAzymes named (A) NaA43T and (B) NaH1. The key catalytic motifs are the small loops in red. (C) A general RNA cleavage reaction requires the nucleophilic attack from 2′-OH resulting in the leaving of the 5′-OH group, which can be accelerated by the general acid/base catalysis.
The catalysis of many small self-cleaving ribozymes has been thoroughly studied (30–32). In general, the RNA cleavage reaction is initiated by a nucleophilic attack from the 2′-OH of the ribose to the scissile phosphate, resulting in the leaving of the 5′-oxygen of the next nucleotide (Figure 1C). In self-cleaving ribozymes, the cleavage reaction is often accelerated by a general base (e.g., a guanine or a metal bound water after deprotonation) (19,33) to help deprotonate the 2′-OH, or by a general acid (e.g. adenine or cytosine) (22,34) to neutralize the developing negative charge on the 5′-oxygen (32). This study intends to compare these two Na+-specific DNAzymes for mechanistic insights taking advantage that no polyvalent metal ions are involved.
Rational evolution from NaA43T to NaH1
Since these two DNAzymes are highly similar, we gradually mutated one DNAzyme into the other to identify important residues. As shown in Figure 2A, the cleavage rate of NaA43T measured at pH 7 was ∼3-fold faster than that at pH 6. Before modifying the catalytic motif, we first deleted the C1 and C2, and then A19 in NaA43T (Mut1 and Mut2 in Figure 2B). Mut1 and Mut2 showed gradually decreasing activity, but their activity became similar at both pH’s. Based on Mut2, we further deleted G22 (or G23) in the small loop resulting in Mut3, which had almost no activity detected at either pH values. Based on our previous study, NaA43 was still active when G22 was mutated to A, T or C (25). However, deleting G22 inhibited the cleavage activity in this study. Therefore, G22 is likely to play a structural role (e.g. a spacer) to position the catalytically important G23 for catalysis.
Figure 2.

(A) The cleavage rate of each mutant in the presence of 10 mM NaCl at pH 6 (gray bars) and pH 7 (black bars). (B) List of the DNAzyme sequences used in this study. The A22 residue is highlighted with blue squares.
Based on Mut3, we mutated G23 into an adenine and NaH1 was obtained (this adenine is A22 in NaH1). NaH1 exhibited a higher cleavage rate under pH 6, while its activity at pH 7 remained low. We also added a guanine back to position 22 (Mut4), whose activity decreased at both pH’s, but the pH 6 activity was still higher than that at pH 7. Therefore, the activity of Mut4 is more similar to NaH1 than to NaA43T. Overall, the highest activity was at pH 7 when G23 was present, and was at pH 6 when A22 was present. Based on this experiment, we can define an enzyme to be from NaA43T if it has G23 and the activity at pH 7 is higher or comparable to that at pH 6. On the other hand, if the enzyme has A22 and its activity is higher at pH 6, it is NaH1. If these nucleotides are directly involved in catalysis, they likely have very different reaction mechanisms, since G and A can play different acid/base catalysis roles (31,32).
The A22 in NaH1 is highly conserved
To further locate important nucleotides in the catalytic loop of NaH1, point mutaions were performed (Figure 3A). The cleavage rate of each mutant was measured at pH 6 (Figure 3B). Mutating T23 to A resulted in a ∼1.6-fold reduction in rate, while mutating C21 to T or G decreased the rate by 2 to 3-fold. Besides, purine-to-purine replacement (G20A) was tolerated with ∼50% rate remaining, which was also observed for the G20 site in NaA43T (25). On the contrary, mutation of A22 to T, C or G led to a ∼100–1000-fold decrease in rate. Therefore, A22 plays a crucial role in the cleavage reaction, which is in agreement with the above rational evolution experiments.
Figure 3.

(A) The sequences used in this point mutaion study based on NaH1 from G20 to T23. (B) The cleavage rate of each mutant measured with 10 mM NaCl in pH 6 buffer.
pH-rate profiles
Catalytically important guanines often play a general base role in ribozymes such as the Rzb hammerhead (17,18), env25 pistol (19,20), and twister ribozymes (21). The pKa of the N1 position of guanine is ∼9.4, which can be lowered toward neutrality by coordinating with nearby nucleobases or metal ions, allowing its general base role at neutral pH (35,36). Adenine, on the other hand, is often known for its general acid role in stabilizing the leaving group (i.e. 5′-O) (19,21,34). To better understand the reaction mechanism, we carefully examined the pH-rate profiles. For NaA43T, we observed a gradual increase in cleavage rate with increasing pH, plateauing at pH 7.5 (Figure 4A, circles). Such a pH-rate profile implies that a single general base functions during the cleavage reaction until it is fully deprotonated. In contrast, a declining trend was observed for NaH1 between pH 5.0 to 8.0 (Figure 4A, diamonds), indicating a general acid catalysis and its protonation was inhibited by raising pH.
Figure 4.

(A) pH-rate profiles of NaH1 and NaA43T in the presence of 10 mM Na+. The pKa of the general acid catalyst in NaH1 was 6.3 ± 0.4 (R2 = 0.92), while the pKa of the general base in NaA43T was 6.6 ± 0.1 (R2 = 0.97). (B) Chemical structures and pKa values of adenine, 2-aminopurine (2AP), 2,6-diaminopurine (2,6-diAP), guanine, and hypoxanthine (I). (C) pH-rate profiles of the wild-type NaH1 and its 2,6-diAP-, 2AP- and G-substitutions in the presence of 10 mM NaCl. (D) pH-rate profiles of the wild-type NaA43T and its I- and A-substitutions in the presence of 10 mM NaCl.
In the mutation studies, G23 and A22 were determined to be crucial for the activity of NaA43 and NaH1, respectively. Taken together, we propose that G23 functions as a general base in NaA43T, while A22 as a general acid in NaH1 (see base analog studies below for further evideince). The pKa of the general base in NaA43T shifts from ∼9.4 (N1 position) to 6.6 ± 0.1 (Figure 4A, gray curve). For NaH1, a shifted pKa of 6.3 ± 0.4 (Figure 4A, black curve) for the catalytic adenine was observed in the presence of 10 mM Na+.
It is very interesting that on the same DNAzyme scaffold, by just mutating one or two nucleotides, the catalytic mechanism is completely changed. Considering the very small size of these DNAzymes, the tolerance of different catalytic mechanisms by point mutations has not been seen previously. The closest example is a hamerhead ribozyme, where mutating an invariant guanine to adenine retained its general base role for the catalysis (instead of general acid) (17). Meanwhile, this mutant exhibited a 13 000-fold lower rate and required a high Mg2+ concentration.
This tolerating property and switchable general acid/base catalysis mechanism have reminded us of another related DNAzyme called Ce13d, which requires both Ce3+ (or another trivalent lanthanide) and Na+ for activity. Ce13d has the same Na+ binding motif, but the small catalyitc loop is almost completely eliminated (Ce3+ to carry out the catalytic role). The Ce13d DNAzyme was extremely tolerant for various metal ions, including all the trivalent lanthanides, Y3+ (23), a low activity with Cr3+ (37), and a moderate activity with Pb2+ (23). After making a single phosphorothioate modification at the cleavage site, it became active with all thiophilic metals such as Hg2+, Cd2+, Pb2+, Tl3+ and Au3+ (38). It can even accelerate the cleavage of the PS substrate by I2, a non-metal (39). The activity with various metal ions and even elimination of polyvalent metal ions indicates that the DNAzyme scaffold is highly tolerant for catalysis. Thus, the swithcing of general acid/base mechanism might also be quite reasonable.
Base analogs further probing catalytic mechanism
To confirm the general acid role of A22 in NaH1, we tested its mutants including using base analogs. A22 has three possible positions to donate protons to the 5′-O: N1, N3 and N7. In small ribozymes, the N1 and N3 positions were reported for general acid catalysis (19,21,40). With a higher pKa (41), N1 is more likely to donate protons. In this work, a series of pKa-perturbed analogs (Figure 4B), such as 2-aminopurine (∼3.8), 2,6-diaminopurine (∼5.6), and guanine (∼9.4) were tested (Figure 4C). The pH-rate profile of the 2,6-diaminopurine substitution (2,6-diAP, circles) overlapped with that of adenosine (diamonds). Mutating A to 2-aminopurine (2AP, grey triangles) decreased the activity by ∼2 orders of magnitude, but its overall profile was parallel to that of the wild type NaH1. Therefore, the 6-amino group of A22 is important for catalytic rate, likely by forming hydrogen bonding to stabilize the transition state (31). With their N1 positions still available for protonation, these analogs displayed a similar pH-dependent activity plateauing at pH <6. When substituting A with G (red triangles), a gradual rate decrease was observed below pH 7, with its maximum activity occurring at a higher pH. Therefore, the N1 position of A22 is indeed responsible for the general acid catalysis of NaH1.
In NaA43T, substitution at G23 with hypoxanthine (I, pKa ∼ 8.7) and adenine (A, pKa ∼ 3.8) was tested to determine its catalytic role (Figure 4D). The G to I mutation (gray circles) did not affect its pH-rate dependence but gave a lower rate between pH 5.0 and 7.0. The 2′-exocyclic amino group of G23 could better stabilize the transition state likely by hydrogen bonding (42). On the contrary, mutating G23 into adenine resulted in a bell-shaped pH-rate profile with the highest activity at pH ∼5.5, similar to that of NaH1 (blue diamonds). Therefore, the N1 position of G23 is likely to directly participate in the catalysis by serving as a general base.
The above mutation studies primarily focused on general acid/base catalysis. After mutation, the loss of activity could also be due to perturbed enzyme folding or Na+ binding. The fluorescence of 2AP is highly sensitive to its nearby base stacking environment (43). Thus, 2AP has been frequently used to probe local folding of DNAzymes and ribozymes (44–46). We then substituted A22 by 2AP (Figure 5A). In activity assays, the enzyme bearing the A22–2AP mutation retained its cleavage activity with a ∼100-fold lower rate compared to the wild type (Figure 4C, gray triangles). However, titrating NaCl induced a gradual increase in its fluorescence (Figure 5B). The fluorescence at 370 nm was further plotted as a function of Na+ concentration showing binding curve with a Kd of ∼40 mM Na+ (Figure 5C). To confirm the Na+ specificity, Li+ and K+ were also titrated. The intensity barely changed (<20%) with up to ∼30 mM KCl, while a slight fluorescence decrease was observed with LiCl (Figure 5D). Therefore, the 2AP-substitution does not affect the enzyme folding upon Na+ binding. Therefore, the activity drop of the A22-to-2AP mutant was likely caused by destabilizing the hydrogen bonding on the 6-amino of A22 instead of perturbed sodium binding. An enhanced fluorescence suggests a weakened base-base stacking near the catalytic residue (43), supporting the importance of A22 to the catalysis. A22 is likely to be released from stacking interactions from the neighbor bases due to Na+ binding, and exposed to the active site. As shown in Figure 4C, the pH-rate profile of NaH1 revealed a slope close to 1 in the high pH region, indicating one proton transfer is the rate-limiting step. A similar relationship was also seen in the low pH region of NaA43T. This supports the idea that chemistry is the rate-limiting step instead of the folding.
Figure 5.

(A) Using 2AP to probe the local environment of the catalytic adenine due to Na+ binding. (B) The fluorescence spectrum of the A22-to-2AP mutant after titrating NaCl. (C) The 2AP fluorescence at 370 nm versus the Na+ concentration. (D) Fluorescence titration with Na+, K+ or Li+.
Phosphorothioate substitution and metal rescue
In self-cleaving ribozymes, non-bridging oxygens at the scissile phosphate often play important roles in directly coordinating with catalytic metal ions or nucleobases. Phosphorothioate (PS) substitution has been widely applied in probing the importance of non-bridging oxygens (24,47). Previous biochemical studies indicate that the Na+ binding site in NaA43 is the 16-nt loop (Figure 1A), instead of the cleavage site (25). To test metal binding at the cleavage site, the activity of NaH1 with the PS-substrates was examined in the presence of 10 mM Na+. First, a racemic mixture of the PS substrate was used, which contained an equal amount of the Rp and Sp diastereomers as shown in Figure 6A.
Figure 6.

(A) The Rp and Sp phosphorothioate diastereomers at the cleavage junction. (B) The comparison between the normal PO-substrate and the PS-substrates of NaH1 in the presence of 10 mM NaCl with or without 2 mM Cd2+. (C) Cleavage activity of NaH1 using PO-, Rp- or Sp-substrates in the presence of 10 mM NaCl.
The cleavage rate with the PS-substrate was ∼68-fold slower than the PO-substrate (Figure 6B), showing a normal thio effect at 10 mM Na+. A careful examination of the kinetic data can reveal that the cleavage did not have a fast phase and the yield was very low. Normally, for metal-based interactions, if the metal binds specifically to one of the non-bridging oxygens, one of the stereoisomers should remain active (48). However, it is not the case here. In addition, adding thiophilic Cd2+ could not rescue the activity, also disapproving metal/phosphate interactions. For comparison, the lanthanide-dependent Ce13d DNAzyme has a normal thio effect which can be fully rescued by Cd2+ (24). Therefore, the role of Na+ is to bind to the aptamer loop instead of directly interacting with the scissile phosphate.
The loss of activity with PS-substrates could be due to a diminished hydrogen bonding between catalytic A22 and non-bridging oxygens caused by the sulfur substitution. The importance of the exocyclic amine of A22 has been addressed in the activity study of A22-to-2AP mutant. We further measured the activity of NaH1 with the two diastereomers Rp and Sp (Figure 6A). The cleavage rates of both Rp and Sp substrates were slower than the PO-substrate (Figure 6C). With a relatively lower activity, the pro-Rp oxygen seems to contribute more to interacting with the catalytic adenine at the cleavage site. Notably, the pro-Rp oxygen was reported to be more important in the catalysis of many RNA-cleaving ribozymes and DNAzymes including the hammerhead (49), the hepatitis delta virus (HDV) ribozymes (47,48), and the Ce13d DNAzymes (24). For example, in the HDV ribozyme, the pro-Rp oxygen directly interacts with a Mg2+ ion and is within hydrogen bonding distance of a catalytic C75 according to its crystal structure (48,50).
In addition, the activity difference between the Rp- and Sp-substitutions was only ∼3-fold, suggesting that both of the non-bridging oxygens are involved in interacting with the catalytic adenine. A similar difference was also seen in a study of the activity of Ce13d (<10-fold) with the PS-substrates (24). As a comparison, the activity of the Sp was >100-fold faster than the Rp in the hammerhead ribozyme (49,51). The importance of the 6-amino group in A22 has been addressed in the previous mutation study. Together, we propose that both of the non-bridging oxygens can form hydrogen bonding with the 6-amino of A22 in NaH1. The pro-Rp oxygen likely plays a slightly more important role than the pro-Sp oxygen. The involvement of both oxygens might also contribute to the tolerance to different catalytic mechanisms.
Finally, we have analyzed the cleavage product of NaH1 using mass spectrometry (Supplementary Figure S1). The 5′-fragment of the cleaved substrate terminated with a 2′,3′-cyclic phasphate. Therefore, a normal RNA cleavage mechanism was confirmed. Taken together, Figure 7 schematically illustrates the catalytic mechanism of the Na+-dependent RNA-cleaving DNAzymes from this study. In NaH1, a N1-protonated adenine (A22) with a shifted pKa of ∼6.3 serves as a general acid, which could stabilize the negative charge on the 5′-leaving oxygen. Meanwhile, 2AP- and PS-substitution assays suggested that 6′-exocyclic amine directly participates in the cleavage by forming the hydrogen bonding with both of the non-bridging oxygens. In the wild-type NaA43, the N1 position of G23 functions as a general base with its pKa shifted to ∼6.6 during the reaction. Its deprotonated form facilitates the deprotonation of the 2′-OH, which then performs the nucleophilic attack on the scissile phosphate. Due to their high sequence similarity, we propose that the Na+-dependent DNAzyme folds into a defined structure upon Na+ binding with catalytic purine bases at the active site accelerating the RNA cleavage.
Figure 7.

A model describing the general acid/base mechanism of the NaH1 and NaA43 DNAzymes in the RNA cleavage reaction.
CONCLUSIONS
The NaA43 and NaH1 DNAzymes were selected separately under different selection conditions. The pH optima of each DNAzyme matched the pH of their respective selection buffers. Despite their high sequence similarity, the minor differences near the cleavage site resulted in distinct pH preferences. In this work, rational evolution studies revealed a 4 to 5-nt catalytic motif in these DNAzymes. Furthermore, a series of mutation studies in NaH1 demonstrated that A22 is indispensable for its catalysis, and previous studies indicated that G23 plays an important role in NaA43. Combining with pH-rate profiles of their base analogs, the N1 position of A22 (pKa shifted to ∼6.3) functions as a general acid in NaH1, while N1 of G23 (pKa shifted to ∼6.6) serves as a general base in NaA43. Base analogs revealed the importance of 6-amine in A22 for the catalysis. To our knowledge, this is the first report of a shift between general acid and base catalysis mechanism by mutating only one or two nucleotides in a conserved DNAzyme scaffold. Notably, this scaffold is also active with a diverse range of polyvalent metal ions by removing the current catalytic motif. Such tolerance may explain the ability to use distinct reaction mechanisms on such a small structure. Future studies may include NMR spectroscopy to directly monitor the actual pKa values of critical residues in the DNAzymes and study kinetic isotope effects for probing proton transfer.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Natural Sciences and Engineering Research Council of Canada (NSERC). Funding for open access charge: NSERC.
Conflict of interest statement. None declared.
REFERENCES
- 1. Breaker R.R., Joyce G.F.. A DNA enzyme that cleaves RNA. Chem. Biol. 1994; 1:223–229. [DOI] [PubMed] [Google Scholar]
- 2. Ali M.M., Aguirre S.D., Lazim H., Li Y.. Fluorogenic DNAzyme probes as bacterial indicators. Angew. Chem. Int. Ed. 2011; 50:3751–3754. [DOI] [PubMed] [Google Scholar]
- 3. He S., Qu L., Shen Z., Tan Y., Zeng M., Liu F., Jiang Y., Li Y.. Highly specific recognition of breast tumors by an RNA-cleaving fluorogenic DNAzyme probe. Anal. Chem. 2015; 87:569–577. [DOI] [PubMed] [Google Scholar]
- 4. Torabi S.-F., Wu P., McGhee C.E., Chen L., Hwang K., Zheng N., Cheng J., Lu Y.. In vitro selection of a sodium-specific DNAzyme and its application in intracellular sensing. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:5903–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou W., Saran R., Chen Q., Ding J., Liu J.. A new Na+-dependent RNA-cleaving DNAzyme with over 1000-fold rate acceleration by ethanol. ChemBioChem. 2016; 17:159–163. [DOI] [PubMed] [Google Scholar]
- 6. Saran R., Liu J.. A silver DNAzyme. Anal. Chem. 2016; 88:4014–4020. [DOI] [PubMed] [Google Scholar]
- 7. Santoro S.W., Joyce G.F., Sakthivel K., Gramatikova S., Barbas C.F. III.. RNA cleavage by a DNA enzyme with extended chemical functionality. J. Am. Chem. Soc. 2000; 122:2433–2439. [DOI] [PubMed] [Google Scholar]
- 8. Carmi N., Balkhi H.R., Breaker R.R.. Cleaving DNA with DNA. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:2233–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee J.H., Wang Z., Liu J., Lu Y.. Highly sensitive and selective colorimetric sensors for uranyl (UO22+): development and comparison of labeled and label-free DNAzyme-gold nanoparticle systems. J. Am. Chem. Soc. 2008; 130:14217–14226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hollenstein M., Hipolito C., Lam C., Dietrich D., Perrin D.M.. A highly selective DNAzyme sensor for mercuric ions. Angew. Chem. Int. Ed. 2008; 47:4346–4350. [DOI] [PubMed] [Google Scholar]
- 11. Huang P.-J.J., Liu J.. Rational evolution of Cd2+-specific DNAzymes with phosphorothioate modified cleavage junction and Cd2+ sensing. Nucleic Acids Res. 2015; 43:6125–6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang P.-J.J., Vazin M., Matuszek Ż., Liu J.. A new heavy lanthanide-dependent DNAzyme displaying strong metal cooperativity and unrescuable phosphorothioate effect. Nucleic Acids Res. 2015; 43:461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu Y., Liu J.. Functional DNA nanotechnology: Emerging applications of DNAzymes and aptamers. Curr. Opin. Biotechnol. 2006; 17:580–588. [DOI] [PubMed] [Google Scholar]
- 14. Fichou Y., Férec C.. The potential of oligonucleotides for therapeutic applications. Trends Biotechnol. 2006; 24:563–570. [DOI] [PubMed] [Google Scholar]
- 15. Liu J., Cao Z., Lu Y.. Functional nucleic acid sensors. Chem. Rev. 2009; 109:1948–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou W., Saran R., Liu J.. Metal sensing by DNA. Chem. Rev. 2017; 117:8272–8325. [DOI] [PubMed] [Google Scholar]
- 17. Mir A., Chen J., Robinson K., Lendy E., Goodman J., Neau D., Golden B.L.. Two divalent metal ions and conformational changes play roles in the hammerhead ribozyme cleavage reaction. Biochemistry. 2015; 54:6369–6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mir A., Golden B.L.. Two active site divalent ions in the crystal structure of the hammerhead ribozyme bound to a transition state analogue. Biochemistry. 2016; 55:633–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ren A., Vušurović N., Gebetsberger J., Gao P., Juen M., Kreutz C., Micura R., Patel D.J.. Pistol ribozyme adopts a pseudoknot fold facilitating site-specific in-line cleavage. Nat. Chem. Biol. 2016; 12:702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nguyen L.A., Wang J., Steitz T.A.. Crystal structure of Pistol, a class of self-cleaving ribozyme. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:1021–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gaines C.S., York D.M.. Ribozyme catalysis with a twist: active state of the twister ribozyme in solution predicted from molecular simulation. J. Am. Chem. Soc. 2016; 138:3058–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Golden B.L. Two distinct catalytic strategies in the hepatitis delta virus ribozyme cleavage reaction. Biochemistry. 2011; 50:9424–9433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang P.-J.J., Lin J., Cao J., Vazin M., Liu J.. Ultrasensitive DNAzyme beacon for lanthanides and metal speciation. Anal. Chem. 2014; 86:1816–1821. [DOI] [PubMed] [Google Scholar]
- 24. Vazin M., Huang P.-J.J., Matuszek Z.A., Liu J. Biochemical characterization of a lanthanide-dependent DNAzyme with normal and phosphorothioate-modified substrates. Biochemistry. 2015; 54:6132–6138. [DOI] [PubMed] [Google Scholar]
- 25. Zhou W., Zhang Y., Huang P.-J.J., Ding J., Liu J.. A DNAzyme requiring two different metal ions at two distinct sites. Nucleic Acids Res. 2016; 44:354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Torabi S.-F., Lu Y.. Identification of the same Na+-specific DNAzyme motif from two in vitro selections under different conditions. J. Mol. Evol. 2015; 81:225–234. [DOI] [PubMed] [Google Scholar]
- 27. Zhou W., Ding J., Liu J.. Splitting a DNAzyme enables a Na+-dependent FRET signal from the embedded aptamer. Org. Biomol. Chem. 2017; 15:6959–6966. [DOI] [PubMed] [Google Scholar]
- 28. Zhou W., Saran R., Ding J., Liu J.. Two completely different mechanisms for highly specific Na+ recognition by DNAzymes. ChemBioChem. 2017; 18:1828–1835. [DOI] [PubMed] [Google Scholar]
- 29. Ma L., Liu J.. An in vitro-selected DNAzyme mutant highly specific for Na+ under slightly acidic conditions. ChemBioChem. 2019; 20:537–542. [DOI] [PubMed] [Google Scholar]
- 30. Cochrane J.C., Strobel S.A.. Catalytic strategies of self-cleaving ribozymes. Acc. Chem. Res. 2008; 41:1027–1035. [DOI] [PubMed] [Google Scholar]
- 31. Bevilacqua P.C., Yajima R.. Nucleobase catalysis in ribozyme mechanism. Curr. Opin. Chem. Biol. 2006; 10:455–464. [DOI] [PubMed] [Google Scholar]
- 32. Ren A., Micura R., Patel D.J.. Structure-based mechanistic insights into catalysis by small self-cleaving ribozymes. Curr. Opin. Chem. Biol. 2017; 41:71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen J., Ganguly A., Miswan Z., Hammes-Schiffer S., Bevilacqua P.C., Golden B.L.. Identification of the catalytic Mg2+ ion in the hepatitis delta virus ribozyme. Biochemistry. 2013; 52:557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Košutić M., Neuner S., Ren A., Flür S., Wunderlich C., Mairhofer E., Vušurović N., Seikowski J., Breuker K., Höbartner C.. A mini-twister variant and impact of residues/cations on the phosphodiester cleavage of this ribozyme class. Angew. Chem. Int. Ed. 2015; 54:15128–15133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bevilacqua P.C. Mechanistic considerations for general acid-base catalysis by RNA: revisiting the mechanism of the hairpin ribozyme. Biochemistry. 2003; 42:2259–2265. [DOI] [PubMed] [Google Scholar]
- 36. Frankel E.A., Bevilacqua P.C.. Complexity in pH-dependent ribozyme kinetics: dark pKa shifts and wavy rate-pH profiles. Biochemistry. 2017; 57:483–488. [DOI] [PubMed] [Google Scholar]
- 37. Zhou W., Vazin M., Yu T., Ding J., Liu J.. In vitro selection of chromium-dependent DNAzymes for sensing chromium(III) and chromium(VI). Chem. Eur. J. 2016; 22:9835–9840. [DOI] [PubMed] [Google Scholar]
- 38. Huang P.-J.J., Liu J.. Sensing parts-per-trillion Cd2+, Hg2+, and Pb2+ collectively and individually using phosphorothioate DNAzymes. Anal. Chem. 2014; 86:5999–6005. [DOI] [PubMed] [Google Scholar]
- 39. Jimmy Huang P.-J., Moon W.J., Liu J.. Instantaneous iodine-assisted DNAzyme cleavage of phosphorothioate RNA. Biochemistry. 2019; 58:422–429. [DOI] [PubMed] [Google Scholar]
- 40. Kuzmin Y.I., Da Costa C.P., Cottrell J.W., Fedor M.J.. Role of an active site adenine in hairpin ribozyme catalysis. J. Mol. Biol. 2005; 349:989–1010. [DOI] [PubMed] [Google Scholar]
- 41. Kapinos L.E., Operschall B.P., Larsen E., Sigel H.. Understanding the acid-base properties of adenosine: the intrinsic basicities of N1, N3 and N7. Chem. Eur. J. 2011; 17:8156–8164. [DOI] [PubMed] [Google Scholar]
- 42. Salter J., Krucinska J., Alam S., Grum-Tokars V., Wedekind J.E.. Water in the active site of an all-RNA hairpin ribozyme and effects of Gua8 base variants on the geometry of phosphoryl transfer. Biochemistry. 2006; 45:686–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jones A.C., Neely R.K.. 2-aminopurine as a fluorescent probe of DNA conformation and the DNA-enzyme interface. Q. Rev. Biophys. 2015; 48:244–279. [DOI] [PubMed] [Google Scholar]
- 44. Zhou W., Ding J., Liu J.. A highly specific sodium aptamer probed by 2-aminopurine for robust Na+ sensing. Nucleic Acids Res. 2016; 44:10377–10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. He Y., Chen D., Huang P.-J.J., Zhou Y., Ma L., Xu K., Yang R., Liu J.. Misfolding of a DNAzyme for ultrahigh sodium selectivity over potassium. Nucleic Acids Res. 2018; 46:10262–10271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ballin J.D., Bharill S., Fialcowitz-White E.J., Gryczynski I., Gryczynski Z., Wilson G.M.. Site-specific variations in RNA folding thermodynamics visualized by 2-aminopurine fluorescence. Biochemistry. 2007; 46:13948–13960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thaplyal P., Ganguly A., Hammes-Schiffer S., Bevilacqua P.C.. Inverse thio effects in the hepatitis delta virus ribozyme reveal that the reaction pathway is controlled by metal ion charge density. Biochemistry. 2015; 54:2160–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Thaplyal P., Ganguly A., Golden B.L., Hammes-Schiffer S., Bevilacqua P.C.. Thio effects and an unconventional metal ion rescue in the genomic hepatitis delta virus ribozyme. Biochemistry. 2013; 52:6499–6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang S., Karbstein K., Peracchi A., Beigelman L., Herschlag D.. Identification of the hammerhead ribozyme metal ion binding site responsible for rescue of the deleterious effect of a cleavage site phosphorothioate. Biochemistry. 1999; 38:14363–14378. [DOI] [PubMed] [Google Scholar]
- 50. Chen J.-H., Yajima R., Chadalavada D.M., Chase E., Bevilacqua P.C., Golden B.L.. A 1.9 Å crystal structure of the HDV ribozyme precleavage suggests both Lewis acid and general acid mechanisms contribute to phosphodiester cleavage. Biochemistry. 2010; 49:6508–6518. [DOI] [PubMed] [Google Scholar]
- 51. Koizumi M., Ohtsuka E.. Effects of phosphorothioate and 2-amino groups in hammerhead ribozymes on cleavage rates and magnesium binding. Biochemistry. 1991; 30:5145–5150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.