


Abstract
Cas9-assisted targeting of DNA fragments in complex genomes is viewed as an essential strategy to obtain high-quality and continuous sequence data. However, the purity of target loci selected by pulsed-field gel electrophoresis (PFGE) has so far been insufficient to assemble the sequence in one contig. Here, we describe the μLAS technology to capture and purify high molecular weight DNA. First, the technology is optimized to perform high sensitivity DNA profiling with a limit of detection of 20 fg/μl for 50 kb fragments and an analytical time of 50 min. Then, μLAS is operated to isolate a 31.5 kb locus cleaved by Cas9 in the genome of the plant Medicago truncatula. Target purification is validated on a Bacterial Artificial Chromosome plasmid, and subsequently carried out in whole genome with μLAS, PFGE or by combining these techniques. PacBio sequencing shows an enrichment factor of the target sequence of 84 with PFGE alone versus 892 by association of PFGE with μLAS. These performances allow us to sequence and assemble one contig of 29 441 bp with 99% sequence identity to the reference sequence.
INTRODUCTION
The emergence of third generation sequencing technologies with long-read capabilities has raised great interest to speed up the production of quality sequencing data (1,2). However, de novo assembly of plant genomes remains a feat due to their complexity in terms of size, polyploidy or high percentage of repetitive elements (3). In situations where whole genome sequencing is not necessarily required, sequence capture methods appear to offer a relevant solution to obtain reliable and very high quality sequence assembly in large genotype panels with advantages with respect to time to results and computational cost. This statement is particularly true for crop improvement, in which the key challenge consists in linking a phenotype to a genomic region mapped with great precision, i.e. with no gaps or misassemblies (4).
For sequences of less than 10 kb, specific enrichment is mostly achieved using polymerase chain reaction amplification or by array capture (5). Yet, the typical size of e.g. a human gene is 10–15 kb (6), requiring the use of multiple amplification reactions that can only be performed in documented genomic regions. For longer tracks with a high degree of polymorphism (7), the multifunction CRISPR/Cas9 technology (8–10) has been viewed as a unique opportunity to overcome some limitations of current methods, which require whole-genome clone libraries and haplotype-specific iterative sequencing (11,12). This approach, called CATCH (Cas9-Assisted Targeting of CHromosome segments) or CISMR (CRISPR-mediated Isolation of Specific Megabase-size Regions of the genome), has been implemented to isolate a gene target of 100 kb in bacteria (13,14), as well as a 200 kb target containing BRCA1 gene in the human genome (15) or a 2.3 Mb region in mouse (16). After Cas9-directed restriction, the target region is isolated according to its size using pulsed field gel electrophoresis (PFGE). Direct sequencing of the purified DNA fragment, which was characterized by a maximal enrichment of ∼200-fold, enabled mapping and exploration of structural diversity but the assembly of the sequence in one contig remains to be reported.
We recently introduced the DNA processing technology μLAS (17), which allows us to perform the operations of separation and concentration by tuning electro-hydrodynamic actuation in viscoelastic (VE) liquids (18). In the size range 100–1500 bp, this technology reached good detection performances down to 10 fg/μl (19) and proved to be efficient to detect the expansion of genes implicated in neurodegenerative diseases (20). Briefly, when DNA molecules conveyed in a Poiseuille flow are subjected to counter electrophoresis, a transverse force oriented toward the capillary walls appears (Figure 1A). Because this transverse force builds up with DNA molecular weight, long molecules are dragged closer to the wall than smaller ones, which are transported at a faster hydrodynamic velocity (Figure 1B). Hence μLAS separation is operated in continuous flow. We thus reasoned that it was well-suited for DNA purification and collection of target fragments defined by CRISPR/Cas9 restriction. To prove this assertion, we first established the settings to separate, concentrate, and detect DNA molecules of high molecular weight (HMW) spanning 3–200 kb. We report a limit of detection (LOD) of 20 fg/μl for fragments of 50 kb, equivalently 360 molecules/μl, for an analytical time of 50 min. We also demonstrate the isolation, sequencing and assembly of a locus of 31.5 kb fragment extracted from a BAC clone and from genomic DNA (gDNA) of the plant Medicago truncatula (2,21). In comparison to PFGE alone, μLAS selection enables us to increase the enrichment factor of the target fragment by a factor of 4- to 10-fold. We finally discuss the relevance of our technological development for HMW DNA isolation and its usefulness for targeted sequencing in complex genomes.
Figure 1.

Mechanism of DNA separation by μLAS. (A) The panel shows the parabolic profile of the Poiseuille flow in blue and the counter electrophoretic force in red, which are responsible for DNA transport. In a viscoelastic fluid, a transverse force oriented toward the capillary walls occurs. (B) The amplitude of transverse viscoelastic forces is size-dependent and DNA molecules are transported along different streamlines according to their molecular weight.
MATERIALS AND METHODS
Chemical and instrumentation
Molecular biology grades chemicals were purchased from Sigma-Aldrich (France). The buffer of the experiments was TBE 1× (Tris–HCl 89 mM, boric acid 89 mM, ethylenediaminetetraacetic acid (EDTA) 1 mM, pH = 8.3) supplemented with different PVP polymers purchased from Sigma-Aldrich, BASF or custom-synthesized by the IMRCP laboratory. Quick-Load® 1 kb Extend DNA Ladder (# N3239S) was purchased from New England Biolabs and used at 1 ng/μl in separation experiments by μLAS. The Lambda Ladder PFG Marker (New England Biolabs) was used for PFGE experiments. The intercalating dye (Picometrics Technologies, France, # 16-BB-DNA1K/01) was used at a 2× final concentration in the buffer solution. All the solutions were filtered at 0.22 μm.
Measurements and sample purification were performed with an Agilent 1600CE system (Germany) equipped with a Zetalif LED 480 nm detector (Picometrics Technologies, France). Fused silica capillaries were purchased from Polymicro Technologies (Phoenix, USA). Two different capillary systems have been used in this study. Capillary system 1 consists of a single capillary of 50 μm in diameter and 1 m in length. Capillary system 2 is an assembly of a large capillary of 320 μm in diameter with two smaller capillaries of 50 μm, as described in (19). Electrokinetic injection was used at 100 V/cm for 10 s in separation experiments. Hydrodynamic injection at 50 mBar during 180 s was used for concentration and collection experiments.
Quantification of resolution, DNA size, concentration and limit of detection
Resolution between two peaks is defined by 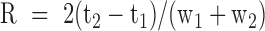 with
with 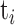 and
and 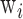 the time of passage and width of band i. R50 corresponds to the resolution between the 20 and 50 kb bands in the ladder. Quantification and sizing precision were estimated using the kb extend DNA ladder as reference with the methods described in (19). The LOD was calculated by linear extrapolation of the DNA concentration corresponding to a signal to noise ratio of three from the four dilutions. Noise was estimated with Open LAB CDS (Agilent Technologies) software using a lag of 1 min at the beginning and the end of the experiment (Supplementary Figure S1).
the time of passage and width of band i. R50 corresponds to the resolution between the 20 and 50 kb bands in the ladder. Quantification and sizing precision were estimated using the kb extend DNA ladder as reference with the methods described in (19). The LOD was calculated by linear extrapolation of the DNA concentration corresponding to a signal to noise ratio of three from the four dilutions. Noise was estimated with Open LAB CDS (Agilent Technologies) software using a lag of 1 min at the beginning and the end of the experiment (Supplementary Figure S1).
BAC and genomic DNA agarose plug production
The Mth2–17015 (CR931730) M. truncatula BAC clone containing the 31.5 kb region was isolated from the Mtr-B-mth2 M. truncatula BAC library available at the CNRGV (https://cnrgv.toulouse.inra.fr/library/genomic_resource/Mtr-B-%20mth2). The BAC clone was extracted with the NucleoBond® Xtrac Midi Plus kit (Macherey-Nagel) following the manufacturer's instructions using 100 ml of LB media with a chloramphenicol selection at 12.5 μg/ml. Agarose plugs with BAC DNA were produced by mixing 40 μl of an agarose solution InCert® (Lonza) at 1.5% with 40 μl of BAC DNA at 50 ng/μl during 10 min at 45°C. The plugs were then casted in CHEF Mapper® XA System 50-Well Plug Mold (BioRad) placed on ice and incubated at 4°C for 30 min. The plugs were stored in a buffer containing 10 mM Tris–HCl (pH = 7.4) and EDTA 1 mM (TE) at 4°C.
Agarose plugs with genomic DNA were produced from 40 g of fresh A17 M. truncatula leaves at the three-leaf seedling stage pretreated in the dark for 2 days, following an adjusted version of the protocol published by Zhang et al. (22). The main modification is the step of nuclei centrifugation and washing performed at 2400 g in order to take into account nuclei and genome sizes. The resulting material was dissolved in 40 agarose plugs of 80 μl at an agarose concentration of 0.75%. gDNA was extracted in the plugs with the lysis buffer of (22), washed and treated with 0.1 mM Phenylmethylsulfonyl fluoride. The plugs were finally stored in TE at 4°C.
Cas9 digestion
The sgRNA were designed using the Crispor V4.3. software (23; http://crispor.tefor.net/), their target sequence being GTTTGTTAATTCTCGACCGG(AGG) and CTGCTCGACAATTTCACCAG(AGG) in Medtr5g005120.1 and Medtr5g005050.2 (the 3 nucleotides in parenthesis correspond to the PAM sequence). The RNA guides provided by Integrated DNA Technologies were rehydrated and complexed using IDT Nuclease-Free Duplex Buffer following the supplier recommendations at 300 ng/μl. Before starting the Cas9 digestion reaction, the plugs were washed in fresh TE during 1 h at room temperature with agitation at 450 rpm, as recommended in (14). Each plug was then cut into 32 pieces and pre-incubated in 2 ml of 1× Cas9 nuclease reaction buffer (New England Biolabs) at room temperature with agitation at 450 rpm during 30 min. In parallel, 12 μg of each sgRNA was pre-incubated with 40 pmol of EnGen Cas9 NLS (20 μM, New England Biolabs) in 1× Cas9 reaction buffer at 25°C during 30 min. The two sgRNA complexes were finally incubated with gDNA in the plugs during 2 h at 37°C. To stop Cas9 digestion, 240 μg of Proteinase K (20 mg/ml) was added and incubated during 3 h at 43°C. The plugs were eventually rinsed in 1× TE by overnight incubation at 4°C.
PFGE, electroelution and FEMTO Pulse® analysis
PFGE was used for (i) removing small DNA from the gDNA plugs, (ii) quality control or (iii) separation and isolation of the fragment of interest. (i) Small and medium MW DNA were removed from the plugs by running a short PFGE cycle of 1 h in a 1% agarose gel in 0.25× TBE at 12°C with an electric field of 9 V/cm, 120° tilt angle, 0.5 s initial switch time, 1.5 s final switch time, linear ramping (CHEF Mapper® XA Pulsed Field Electrophoresis System (BioRad). The plugs were eventually removed from the PFGE stage and stored in TE at 4°C. (ii) Quality control of HMW DNA plugs was carried out by running a long PFGE cycle of 24 h. One half of a plug was placed on a 1% agarose gel in 0.25× TBE and the electric field was set to 6 V/cm, 120° tilt angle, pulse time 60–120 s, linear rampling at 12°C (Supplementary Figure S2). In order to evaluate DNA concentration in the plugs, one half of a plug was melted at 70°C for 2 min and digested by 1 unit of Agarase (Thermo Fisher Scientific) during 45 min at 43°C. DNA concentration was determined with the Qubit™ dsDNA BR Assay Kit and the associated Qubit 3.0 Fluorometer (Invitrogen) after 10 min of sonication. (iii) The separation of the fragment of interest from the bulk of gDNA was performed in a 1% agarose 0.25× TBE PFGE gel was casted with a low melting window following Peterson and collaborators protocol (24). The NEB Lambda Ladder PFG Marker and the BAC DNA digested by Cas9 were used as size makers. The Cas9 digested plug pieces were run for 16 h with the following conditions: 6 V/cm, 120° tilt angle, pulse time 5–15 s, linear ramping at 12°C. A band corresponding at the 30 kb fragment size was cut from the gel. The DNA was eluted in about 100 μl of TBE 1× from the plug pieces using the BioRad 422 Electro-Elutor for 2 h following the manufacturer's recommendations. The DNA fragments were analyzed on the FEMTO Pulse® Automated Pulsed-Field CE Instrument (Advanced Analytical Technologies) with the gDNA 165 kb methods.
PacBio sequencing
The 31.5 kb fragments isolated from BAC or gDNA were amplified using the whole genome amplification (WGA) kit Illustra GenomiPhi V2 DNA (GE Healthcare) starting from 2 μl of material. The amplified fragments were pooled for the construction of a SMRT® library using the standard Pacific Biosciences preparation protocol for 10 kb library with Barcoded Adapters. It was sequenced in one SMRT cell using the P6 polymerase with C4 chemistry. Sequencing was performed on a PacBio RSII sequencer at the INRA sequencing platform GeT-PlaGe in Toulouse, France (http://get.genotoul.fr/la-plateforme/get-plage/) or the NGI Platform in Uppsala, Sweden (http://snpseq.medsci.uu.se/about-us/ngi/).
Bioinformatics analysis
The data were demultiplexed and filtered, low quality reads (read quality <0.75 and read length <500 bp) were removed. As WGA generates chimeric reads, filtered reads were then mapped against their corresponding reference -BAC clone (GenBank CR931730.1) for the BAC sample, and the sequence of the v4.0 M. truncatula genome for gDNA samples- with minimap2 once with option -cx map-pb –secondary = n to generate PAF format output and once with the option -ax map-pb –secondary = n to generate SAM format output (25). This step allows the splitting of chimeric reads, and non-chimeric corresponding fastq sequences were generated using homemade python scripts. Those sequences were then assembled and corrected with the pipeline of the CANU assembler (26). Using samtools-1.8 (27), the SAM files were sorted and indexed with function sorted and index, then depth coverage of the target region and the rest of the matrix were estimated with the function bedcov. Enrichment was calculated as the ratio of the mean depth of the targeted region over the mean depth of the other part of the genome:
 |
(1) |
Coverage along the genome was calculated using the function depth and plots coverage were generated using R-3.4.3 (R Core Team 2017, https://www.r-project.org/).
RESULTS
Viscoelastic fluid formulation for high molecular weight DNA separation by μLAS
DNA separation and concentration by μLAS is dictated by transverse migration in VE liquids (18). Viscoelastic properties are conferred to the separation buffer by addition of the neutral polymer Polyvinylpyrrolidone (PVP). For DNA molecules of less than 1 kb (19), we used PVP chains of HMW and a volume concentration of 5%, which was associated to a viscosity of 30 mPa.s and an elasticity of 0.7 Pa (18). The isolation of longer fragments of ∼30 kb required the definition of an appropriate VE fluid formulation and the associated flow actuation conditions. Eight different PVP polymers with average molar mass (Mn) spanning 10 to 360 kDa have been purchased or custom-synthesized ((28), Table 1). For each PVP, we carried out series of separation experiments on the capillary system 1 (see ‘Materials and Methods’ section) with a ladder containing 13 bands in the range 0.5–50 kb using the resolution R50 between the bands of 20 and 50 kb as readout (see the definition in ‘Materials and Methods’ section).
Table 1.
Characteristics of PVP polymers
| PVP | Sigma 10 kDa | Custom 12.5 kDa | BASF k30 (†) | Custom30 kDa | Sigma 40 kDa (**) | Custom 43 kDa | BASF k90 (†) | Sigma 360 kDa |
|---|---|---|---|---|---|---|---|---|
| Mn (kDa) | 9.3 | 12.5 | 14 | 30.3 | 26.5 | 43.6 | 325 | 427 |
| Mw (kDa) | 16.4 | 13.5 | 50 | 41.1 | 59.3 | 66.2 | 1400 | 740 |
| Concentration for a viscosity of 2.6 mPa.s | 10% | 8% | 5% | 4% | 5% | 3% | 2.5% | 0.7% |
| Resolution R50 | 1.5 | 2.8 | 1.5 | 2.1 | 2.3 | 3.0 | 1.3 | 1.0 |
Eight PVP formulations are compared according to their number average molecular weight Mn, average molecular weight Mw, mass:volume concentration corresponding to a viscosity of 2.6 mPa.s and separation performance based on resolution between the 20 and 50 kb fragments. Structural properties are inferred from supplier information (marked with †) or chromatography, as described in (28). The Sigma 40 kDa marked with two asterisks corresponds to the reference formulation in our study.
We first selected PVP of 40 kDa as reference for benchmarking the polymer library (Table 1), and optimized HMW DNA separation by tuning its concentration, the flow velocity and the electric field. PVP 40 kDa was thus dissolved in a buffer for electrophoresis at four different concentrations spanning 2–20%, which corresponded to viscosities of 1.4–18.6 mPa.s, respectively. The chromatograms shown in Figure 2A were obtained for a mean flow velocity vm set to 1.5 mm/s, and electric fields adjusted to optimize R50 as reported in Supplementary Figure S3. We noticed that R50 increased from 0.7 to 2.3 as the concentration of PVP decreased from 20 to 5%, and then decreased to 1.7 for a concentration of 2%. Notably, vm was set to 1.5 mm/s because we obtained an optimal R50 of 2.3 with a minimal separation time of 15 min in a parameter space defined by a flow velocity in the range 0.6 to 20 mm/s and an electric field spanning 10 to 50 V/cm (Figure 2B and C). This first set of experiments therefore showed that HMW DNA separation required a formulation of PVP characterized by a viscosity 15-fold lower, as well as settings for the flow velocity and electric field 10-fold lower than the conditions to separate molecules of 0.1–1 kb (19).
Figure 2.

HMW DNA separation by μLAS. (A) The four chromatograms correspond to the kb extend DNA ladder separated by μLAS using four different concentrations of PVP 40 kDa, as indicated in inset. The flow velocity vm is set to 1.5 mm/s. (B) Using 5% PVP 40 kDa, the graph presents the separation of the same sample for four different flow velocities vm ranging from 0.6 to 10 mm/s as indicated in inset. (C) The contour plot shows the resolution R50 as a function of the electric field and flow velocity with a buffer containing 5% PVP 40 kDa. (D) Chromatogram of the kb extend DNA ladder with the custom-synthetized PVP 43 kDa dissolved at a volumic concentration of 3%. (E) Separation of a custom DNA ladder containing four bands of 50, 100, 150 and 210 kb at 0.5 mm/s and 9 V/cm with the custom-synthetized PVP 43 kDa dissolved at a volumic concentration of 3%. (F) Resolution of the separation in the size range 3–210 kb.
We then compared the different PVP polymers by dissolving them at a concentration that corresponded to a viscosity of 2.6 mPa.s, i.e. as for 5% PVP 40 kDa, as reported in Table 1. The flow velocity was set to 1.5 mm/s. Using HMW PVP of 360 kDa or k90 (Table 1), the resolution R50 was lower than 1.3, showing that long PVP chains were not adequate for HMW DNA separation. Optimal separation performances with an optimum R50 of 3.0 were achieved with custom-made PVP of 43 kDa (Figure 2D). The separation was performed in ∼15 min, and the theoretical number of plates per meter was ∼4.105 plates/m for the 50 kb peak. This result compared well to the best reported value of 106 plates/m obtained with nanofabricated pillar matrices (29). It was also much greater than 103 plates/m achieved with matrix-free separation based on cyclic electro-hydrodynamic actuation (30). Further, using this optimal PVP formulation, we could separate a sample containing four bands of 50, 100, 150 and 210 kb in 1 h (Figure 2E). This time to result was faster than the 4 h required by PFGE (31). Altogether, this study shows that μLAS is relevant to perform DNA separation in the size range 3–210 kb in less than an hour with a resolution in the range of 1–3 (Figure 2F).
HMW DNA concentration and determination of the limit of detection
The μLAS technology allowed us to concentrate DNA molecules of 0.1–1 kb at the junction of two capillaries of different diameters (19). For this study, we engineered the capillary system 2 composed of a large and a narrow capillary of 320 and 50 μm in inner diameter, respectively (Figure 3A). The large capillary component serves to accommodate an injected volume of ∼2 μl, i.e. 1000 times more than that of conventional CE. The long narrow capillary is dedicated to size separation. In capillary system 2, concentration operations were carried out by delivering a high electric field, which impeded the migration of DNA molecules in the separation channel and forced DNA enrichment at the constriction. The electric field was then progressively reduced in order to allow the migration of the DNA species in the separation channel. A typical concentration and separation experiment is reported in Figure 3B. The first concentration phase lasts 10 min with a flow velocity set to 1.5 mm/s and a high electric field of 165 V/cm (red curve in Figure 3B). During the next 30 min, the electric field was gradually reduced to 25 V/cm (red curve in Figure 3B), i.e. the separation conditions defined in the previous paragraph. Notably, the progressive decrease of the electric field allowed us to maintain good separation performances after the concentration phase because the resolution R50 remained equal to three with this two-step process (black curve in Figure 3B). Furthermore, we performed multiple analysis of the ladder sample and determined an accuracy in sizing of 3% and quantification of 13% for a 50 kb fragment (Supplementary Tables S1 and 2). The overall analytical process took ∼50 min, and the LOD, as measured from the analysis of serial dilutions of the kb extend DNA ladder (Supplementary Figure S1 and Table S3), was 13 and 20 fg/μl for the bands of 10 and 50 kb, respectively. For comparison, we estimated the gain in sensitivity of ∼1000-fold using the capillary system 2 versus the conventional linear capillary system 1 (not shown). Using a preconcentration strategy based on conventional electrokinetic stacking, an LOD of 2 pg/μl, i.e. two orders of magnitude higher than with μLAS, has been reported for a fragment of 50 kb (32). The LOD reported in this study is also 15-fold enhanced in comparison to state-of-the-art commercial systems, such as the FEMTO Pulse® of 300 fg/μl. The recent microfluidic technologies based on molecular rheotaxis reached better performances with an LOD of 5 fg/μl (33), yet this analytical instrument has not been reported to be adequate for DNA purification.
Figure 3.

μLAS for online concentration and separation. (A) Schematics of the capillary system 2 composed of (i) an inlet capillary of 50 μm in inner diameter and 5 cm in length, (ii) a loading chamber of 320 μm in inner diameter and 5 cm in length and (iii) an outlet capillary of 50 μm in inner diameter and 19 cm in length. The laser-induced fluorescence (LIF) detector is placed 7 cm downstream of the interconnection between the loading chamber and the outlet capillary. Collection is performed by isolating a target volume at the outlet during a calibrated time period. (B) The black curve shows the intensity versus time for the kb extend ladder at a concentration of 100 pg/μl and the red curve is the temporal evolution of the electric field. The sample is concentrated during the first 10 min of the process (dashed blue line).
Capture of a 30 kb fragment
We then decided to exploit μLAS to separate, collect and purify a fragment of ∼30 kb. Sample purification was achieved by monitoring the travel time of the sample in the separation channel and defining a temporal window for collecting the target fraction in a fresh tube placed on the carrousel of the capillary electrophoresis system, as schematized by Figure 3A. In order to calibrate the size selection window, we used a broadly distributed sample (black curve in Figure 4A) and collected fractions during 16, 8 and 2 min. Note that we performed the operation of selection by injecting ∼125 pg of material in a volume of 2 μl in the capillary system in order to avoid uncontrolled DNA leak during the concentration phase associated to the saturation of the concentrator (manuscript in preparation). Because we wished to collect a total volume of ∼25 μl to meet the requirements of downstream molecular biology operations, in particular WGA (see below), the sample collection procedure was automatically repeated ten times consecutively.
Figure 4.

Capture of a 31 kb DNA fragment from a BAC clone. (A) The chromatogram shows a broadly distributed DNA sample (black curve), and the result of its fractionation with different collection times, as indicated in the legend (red, green and blue curves). The dashed black curve corresponds to the reference DNA ladder. (B) The plot presents the DNA mass as a function of the collection time (black data points and corresponding linear fit), as inferred from the analysis of the three curves shown in panel (A). The blue line and symbol graph shows the full width at half maximum as a function of the collection time. (C) The chromatograms show the Cas9-digested BAC clone sample (black curve) with two main peaks of 31 and 65 kb, and the purified sample with one single peak of 31 kb (red curve). The dashed blue line corresponds to the end of the concentration phase. (D) The graph presents the read depth as a function of genomic position on the BAC clone. The two vertical gray lines represent the position of the sgRNA.
The three different fractions were subsequently analyzed by μLAS, showing the successful fractionation of the sample (compare the red and black curves in Figure 4A). As the collection decreased from 16 to 2 min, we noted that the DNA mass decreased and that the size distribution of the purified sample was narrowed down (red and blue curves in Figure 4A). Using a Gaussian fit to determine the trace amplitude and full width at half maximum (FWHM), we could show that the DNA mass increased linearly with the collection time following a slope of 11 pg/min (dashed black line in Figure 4B). Furthermore, the FWHM decreased by a factor of 3 from 15 kb to 4.4 kb for 16 and 2 min of collection. In order to reach a compromise between mass and size selection window, we chose a collection time of 6 min (red dashed vertical line in Figure 4B), which corresponded to a FWHM of 10 kb, as confirmed by the characterization shown in Supplementary Figure S4.
We then performed a model purification experiment using a 97 kb BAC clone from M. truncatula (see ‘Materials and Methods’ section), in which we identified two sgRNA for Cas9 restriction to produce two molecules of 31 512 and 65 337 bp, hereafter referred to as the 31 and 65 kb fragments. Our process of purification and cleavage was carried out by embedding 1.5 μg of BAC DNA in agarose plugs in order to emulate standard protocols for gDNA purification (see below). Quality control of the digestion reaction was performed by PFGE and μLAS, showing three bands around 100, 70 and 30 kb corresponding to the linearized BAC and the two cleaved fragments (Supplementary Figure S5). The yield of the cutting reaction was 93% according to profiling and titration with the FEMTO Pulse® system (Supplementary Figure S5C and Table S4). At last, the digested BAC DNA was electroeluted from the agarose plug, resulting in a solution of 106 μl of DNA at a concentration of 25 ng/μl, as inferred from Qubit Fluorometry. The amount of DNA after electroelution of 2.6 μg was nearly twice that of the input material, suggesting that sgRNA contributed to ∼50% of the fluorescence signal. The analysis of the electroeluted sample with μLAS confirmed this hypothesis (black curve in Figure 4C), because the detection of a strong and broad fluorescence peak during the concentration phase is very likely associated to the passage of sgRNA, which are too small to be retained at the junction during the concentration step. Furthermore, the two peaks of 31 and 65 kb were resolved during the separation, and an HMW residue that corresponded to the linearized BAC clone was detected as a shoulder after the 65 kb peak. The concentration of the 31 kb DNA fragment was 3 ± 0.4 ng/μl based on our calibration with the kb extend DNA ladder (Supplementary Figure S5), meaning that the yield of the collection process was 70 ± 9%.
Figure 4C reports the characterization of the fractionated sample, showing the discard of sgRNA and most of the 65 kb fragments. Specifically, a residual intensity signal in the 65 kb region was recorded, corresponding to ∼8 ± 1% of the total signal. We thus deduced that the purification rate was ∼92 ± 2%. The final product concentration was 4 ± 0.5 pg/μl, equivalently 100 ± 13 pg were recovered and the yield of the DNA isolation process was 67 ± 9%. The purified 31 kb fragment was then amplified using WGA in order to produce at least 8 μg of DNA for PacBio RSII long read sequencing. We obtained 6124 reads, among them 5856 mapped against the published sequence of the Mth2–17015 BAC clone (Table 2). The average read depth was 1525× for the 31 kb region of interest and of 129× for the rest of the BAC clone (Figure 4D). We calculated the enrichment factor by comparing the depth of coverage in the region of interest against the rest of the BAC clone sequence and deduced an enrichment factor of 11.9. The reproducibility of this selection strategy was confirmed by running the isolation and sequencing experiment in triplicate, showing enrichment factors of 12.7 and 10.8 for the two other repeats (Table 2). Using the CANU assembler (26), we obtained a consensus sequence with 1 contig of 31 009 bp (Supplementary Data Sequence ‘Contig_CATCH_30kb_BAC’) representing 98% of query coverage with 99% of identity and 47 gaps with the reference sequence using the Blast 2 sequences online software (34). In Figure 4D, the alignment of the sequences against the BAC reference shows a residual peak in the complementary region of the target representing ∼1/12 of the sequences, in very good agreement with our purification rate estimated from the chromatograms in Figure 4C. The average read depth of 1525× nevertheless allowed us to obtain an assembly of the sequence of interest in a unique contig.
Table 2.
The table presents sequencing data obtained with the different purification methods shown in Figure 5 and for the three repeats on BAC DNA
| Method | # bases (bp) | # reads | N50 | Mapped bases on target region | Cov target | Cov matrix | Enrichment |
|---|---|---|---|---|---|---|---|
| BAC 30 kb | 60 657 835 | 12 056 | 7401 | 74% | 1422 | 112 | 12.7 |
| 59 356 468 | 6124 | 12 665 | 80% | 1525 | 129 | 11.9 | |
| 106 274 884 | 14 161 | 8428 | 78% | 2592 | 241 | 10.8 | |
| ADNg—Method 1 | 679 211 859 | 134 435 | 5593 | 0.5% | 103 | 1.2 | 84 |
| ADNg—Method 2 | 325 120 000 | 43 863 | 8268 | 4.6% | 448 | 0.5 | 892 |
| 750 242 645 | 101 146 | 8264 | 2.2% | 498 | 1.2 | 416 | |
| ADNg—Method 3 | 43 882 868 | 9725 | 6415 | 0.3% | 4.3 | 0.08 | 56 |
Capture of a fragment of interest from plant genomic DNA
We finally focused our study on the capture of the same 31 kb fragment in the 450 Mb genome of M. truncatula (2). The fragment of interest represents a fraction of 7.10−5 of the genome. After cas9 targeting, three different capture methods have been compared (Figure 5). As a reference and for comparison to recent studies (15,16), DNA sequencing has been performed on a sample obtained by the standard method of band excision after PFGE and electro-elution (method 1, lower arrow in Figure 5). The same electro-eluted sample has been processed by μLAS to evaluate the benefit of this technology for sequencing (method 2, middle arrow in Figure 5). At last, method 3 consisted in directly performing size selection by μLAS without PFGE (upper arrow in Figure 5).
Figure 5.

Workflow of the CATCH method for plant genomic DNA purification and sequencing: the cartoons represent the consecutive steps for the isolation of a target region from plant genomes. We used three different strategies: (1) PFGE alone; (2) PFGE and μLAS isolation; (3) μLAS isolation only.
For this study, twenty agarose plugs were prepared, each containing ∼10 μg of gDNA. Assuming the Cas9 digestion reaction to be complete, we expect the maximal mass of 31 kb fragment to be 0.7 ng per plug. For method 3, we first analyzed the size distribution of the Cas9-digested gDNA electroeluted from the plug, hoping that the agarose meshwork would trap chromosomes and allow the migration of the 31 kb fragment. The resulting material was characterized by a broad size pattern in the range 20 to more than 100 kb without any specific signature at 30 kb (blue curve in Figure 6A). In addition to the fact that this profile was observed with or without Cas9 restriction (not shown), the total mass of Cas9-digested gDNA recovered after electroelution was 300 ng, i.e. 400 times more than the expected mass of the 31 kb fragment. This result indicated that despite careful nuclei isolation and elimination of small residues by a pre-conditioning step of PFGE (see ‘Materials and Methods’ section), gDNA degradation remained detectable in the form of an heterogeneous pool of molecules escaping from the meshwork of the gel. It was in fact consistent with the documented challenge of preparing quality gDNA from plants in particular due to the damage during the leave grinding step (22,35). The resulting material was nevertheless purified by μLAS using the selection window of 31 ± 5 kb (Supplementary Figure S5). We collected 20 μl at 4 pg/μl, as plotted by the red curve in Figure 6C. This purified sample was then amplified, sequenced, and we obtained an enrichment of 56 associated to a low coverage for the target sequence of 4.3 that was insufficient for sequence assembly (not shown).
Figure 6.

Capture of a 30 kb DNA fragment from genomic DNA. (A) The blue chromatogram corresponds to the Cas9-digested gDNA obtained according to method 3 without purification by PFGE. The black curve is the Cas9-digested BAC reaction product shown as reference. The dashed vertical line corresponds to the end of the concentration phase. (B) The red chromatogram presents the Cas9-digested gDNA sample obtained by PFGE, band excision and electro-elution (following the process flow of methods 1 and 2). The green curve is the control without Cas9 digestion, and the black one corresponds to the kb extend ladder. The dashed vertical line indicates the migration of the 31 kb fragment. (C) The blue chromatogram corresponds to the Cas9-digested sample as shown in panel (A), and the red one to the final product purified with μLAS following method 3. (D) The red chromatogram corresponds to the Cas9-digested sample as shown in panel (B), and the black one to the final product purified with μLAS (method 2).
We then moved to methods 1 and 2, which both involve the reference technology of band excision after PFGE as first step of the protocol. Band excision and electro-elusion at ∼30 kb allowed us to collect 10 ng of gDNA per plug (Supplementary Figure S6). Although this quantity still represents 15 times more than the 0.7 ng expected for the 31 kb locus, we could detect the presence of the 31 kb fragment as a spike in a broad smear pattern in the range 10–60 kb (red curve in Figure 6B). We further confirmed the presence of this 31 kb peak performing FEMTO Pulse® analysis (Supplementary Figure S6B). Notably, we detected the same smear pattern, yet without any spike, for the negative undigested control (green curve in Figure 6B). This control suggested that gDNA degradation occurred during extraction, but marginally during the cleavage reaction through Cas9 non-specific activity. We estimated that the 31 kb fragment represented ∼2.5% of the total DNA mass in the smear pattern, meaning that we collected 0.26 ± 0.03 ng of 31 kb fragment. The overall recovery yield was therefore 37 ± 5%.
We then purified the 31 kb fragment using μLAS with a size selection window centered at 31 kb with a breadth of ± 5 kb. We injected ∼0.2 ng of DNA in the capillary device and fractionated the sample 10 times consecutively to obtain 25 μl at 3 pg/μl (black curve in Figure 6D). We finally performed WGA and sequenced the product with the PacBio RSII. The control solely based on PFGE purification (method 1) allowed us to collect 139 450 reads among which 93% mapped against the published genome of M. truncatula (Table 2). The coverage was 103× for the target fragment and 1.2× for the rest of the genome, meaning that the enrichment factor was 84 (Figure 7A). This result was consistent with recent reports, in which enrichment factors of 237, 75, 174 and 39 for captured sequences of 200, 263, 610 and 2300 kb, respectively, have been obtained (15,16). A fastq file of sequences that mapped in the target region was generated and assembled with CANU into two non-overlapping contigs of 18 733 bp (Supplementary Data Sequence ‘Contig1_CATCH_30kb_ADNg_Method1’) and 7760 bp (Supplementary Data Sequence ‘Contig2_CATCH_30kb_ADNg_Method1’). Further selection of the target genomic locus with μLAS after PFGE (method 2) led to 43 863 reads of which 94% mapped against the published sequence of M. truncatula (Figure 7B). We obtained a 448× coverage of the region of interest and a final enrichment factor of 892 that was 4- to 10-fold improved in comparison to PFGE alone. A fastq file of sequences mapping in the target region was generated and assembled with CANU in one contig of 29 441 bp (Supplementary Data Sequence ‘Contig_CATCH_30kb_ADNg_Method2’) representing 93% of query coverage with 99% of identity and 11 gaps with the reference sequence according to Blast 2 software (34).
Figure 7.

Sequencing of the 31 kb DNA fragment purified by methods 1 and 2. (A) The graph presents the read depth as a function of genomic position on the first 500 kb of the Medicago truncatula chromosome V, as purified with method 1. (B) Same plot as in A based on the purified material obtained by method 2. The vertical dashed grey lines represent the position of the sgRNA.
At last, we noted that the enrichment of the target sequence was critically dependent on the quality of gDNA prior to size selection operations. We indeed performed another independent experiment for which the 31 kb fragment did not step out from the smear pattern of the gDNA signal after purification by PFGE (Supplementary Figure S7). Because gDNA concentration was 1.1 ng/μl, i.e. nearly five times more than the previous sample characterized in Figure 6B, and since we used the same input for both experiments, we concluded that the absence of the 31 kb peak stemmed from an excess of degradation prior to its processing. Using μLAS to isolate the 31 kb fragment from the smear, we obtained an enrichment factor of 416, i.e. 2.1 times lower than with the other sample (second line of Method 2 in Table 2), yet still higher than in every study based on PFGE alone (15,16). Keeping the same bioinformatics settings to process the resulting sequence data, we obtained two contigs instead of one. Hence, quality sequence data based on size selection procedures (PFGE or μLAS) rely on minimal damage during the purification of genomic DNA. Although our two experiments cannot be considered as duplicates per se, the 2.1-fold difference in enrichment rate that we report appears to be comparable to that obtained from a triplicate with the CISMR technology, which showed 2.2-fold difference in enrichment (16).
CONCLUSION
We presented a simple and versatile technology to perform the operations of DNA concentration, separation and purification. We first optimized the formulation of the viscoelastic solution for HMW DNA separation for fragments of up to 50 kb. We then engineered a capillary system to concentrate DNA samples and reach a LOD of ∼10 fg/μl for the 50 kb fragment. At last, taking advantage of the fact that μLAS technology is operated without separation matrix, we developed an assay to capture a fragment of interest of 30 ± 5 kb. The technology was subsequently applied to Cas9 directed targeted sequencing of a 31.5 kb fragment from the plant M. truncatula. Compared to PFGE alone, our method increases the target sequence enrichment by a factor of ∼10 and enables the assembly of a unique contig of the sequence of interest with 99% of identity to the reference sequence.
Due to the presence of residual plant gDNA degradation products, we also report that μLAS has to be performed in conjunction with PFGE in order to obtain optimal results. Because the residues can hardly be eliminated by size selection technologies alone, we suggest that the sensitivity of μLAS constitutes an additional advantage for targeted sequencing. Indeed, high sensitivity sizing is suited to guarantee an optimal purity based on rigorous quality control of virtually every step of the purification, digestion and capture process. In some sense, μLAS complements the efforts in enzyme engineering to improve Cas9 digestion efficiency and reduce off-target restriction (36). Given that human genomic DNA is generally seen as simpler to purify than that of plants, it would be interesting to test whether μLAS improves targeted sequencing of target human genes. We could start with high quality references, such as NA12878, CHM1, CHM13, HG002. Depending on these results, target regions of extremely low heterozygosity or a segmental duplication, which are particularly difficult to solve by conventional sequencing methods, should be targeted due to their impact in molecular medicine.
Future technological developments consist in defining the conditions to manipulate molecules of 100 kb or more and in processing 1 μg of target DNA in order to avoid artifacts associated to WGA and to feature performances more competitive with PFGE. The first objective appears manageable, because we showed that molecules of 100, 150 and 210 kb could be sorted in a monocapillary at the expense of longer separation times of 1.5 h instead of 15 min. The second one is more challenging because capillary systems are notoriously known to be undersized to process volumes larger than 2 μl (37). It may be overcome with arrays of capillaries, which have been fabricated to increase the throughput of analytical systems.
Supplementary Material
ACKNOWLEDGEMENTS
We are thankful to Mathias Destarac (Paul Sabatier University, Toulouse) for the synthesis of custom PVP. We thank Claudette Icher, Fabrice Devoilles and the TPMP platform for the growing of the plants. We are thankful to Kit-Sum Wong and William Amoyal from Advanced and Analytical who performed the FEMTO Pulse® analysis. We thank the platforms GeT-PlaGe and NGI for the Pacbio sequencing. We are grateful to the genotoul bioinformatics platform Toulouse Midi-Pyrenees (Bioinfo Genotoul) for access to computing resources. This project is funded by four grants, one from European Union, two from Occitanie Pyrénées-Méditerranée region and one from the Plant2Pro Carnot Institute.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Commission; Institut Carnot; Région Midi-Pyrénées–Occitanie; ANR μLAS (ANR-16-CE18-0028-01). Funding for open access charge: 2 Midi Pyrénées–Occitanie Regional Grants; European Grant; French National Carnot Grant.
Conflict of interest statement. Nicolas Milon, Laure Saïas, Audrey Boutonnet and Frédéric Ginot belong to Picometrics Technologies, a French SME which has developped the automated platform to perform DNA analysis.
REFERENCES
- 1. Huddleston J., Ranade S., Malig M., Antonacci F., Chaisson M., Hon L., Sudmant P. H., Graves T.A., Alkan C., Dennis M. Y. et al.. Reconstructing complex regions of genomes using long-read sequencing technology. Genome Res. 2014; 24:688–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pecrix Y., Staton S.E., Sallet E., Lelandais-Brière C., Moreau S., Carrère S., Blein T., Jardinaud M.-F., Latrasse D., Zouine M.. Whole-genome landscape of Medicago truncatula symbiotic genes. Nat. Plants. 2018; 4:1017–1025. [DOI] [PubMed] [Google Scholar]
- 3. Schatz M.C., Witkowski J., McCombie W.R.. Current challenges in de novo plant genome sequencing and assembly. Genome Biol. 2012; 13:243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bevan M.W., Uauy C., Wulff B.B., Zhou J., Krasileva K., Clark M.D.. Genomic innovation for crop improvement. Nature. 2017; 543:346–354. [DOI] [PubMed] [Google Scholar]
- 5. Mamanova L., Coffey A.J., Scott C.E., Kozarewa I., Turner E.H., Kumar A., Howard E., Shendure J., Turner D.J.. Target-enrichment strategies for next-generation sequencing. Nat. Methods. 2010; 7:111–118. [DOI] [PubMed] [Google Scholar]
- 6. Strachan T., Read A.P.. Human Molecular Genetics. 1996; BIOS Scientific Publishers; 596. [Google Scholar]
- 7. Kidd J.M., Cooper G.M., Donahue W.F., Hayden H.S., Sampas N., Graves T., Hansen N., Teague B., Alkan C., Antonacci F.. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008; 453:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E.. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lander E.S. The heroes of CRISPR. Cell. 2016; 164:18–28. [DOI] [PubMed] [Google Scholar]
- 10. Tian P., Wang J., Shen X., Rey J.F., Yuan Q., Yan Y.. Fundamental CRISPR-Cas9 tools and current applications in microbial systems. Synth. Syst. Biotechnol. 2017; 2:219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bellec A., Courtial A., Cauet S., Rodde N., Vautrin S., Beydon G., Arnal N., Gautier N., Fourment J., Prat E. et al.. Long read sequencing technology to solve complex genomic regions assembly in plants. Next Generat. Seq. & Applic. 2016; 3:1000128–1000141. [Google Scholar]
- 12. Jain M., Olsen H.E., Turner D.J., Stoddart D., Bulazel K.V., Paten B., Haussler D., Willard H.F., Akeson M., Miga K.H.. Linear assembly of a human centromere on the Y chromosome. Nat. Biotechnol. 2018; 36:321–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiang W., Zhao X., Gabrieli T., Lou C., Ebenstein Y., Zhu T.F.. Cas9-assisted targeting of CHromosome segments CATCH enables one-step targeted cloning of large gene clusters. Nat. Commun. 2015; 6:8101–8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jiang W., Zhu T.F.. Targeted isolation and cloning of 100-kb microbial genomic sequences by Cas9-assisted targeting of chromosome segments. Nat. Protoc. 2016; 11:960–975. [DOI] [PubMed] [Google Scholar]
- 15. Gabrieli T., Sharim H., Fridman D., Arbib N., Michaeli Y., Ebenstein Y.. Selective nanopore sequencing of human BRCA1 by Cas9-assisted targeting of chromosome segments (CATCH). Nucleic Acids Res. 2018; 46:e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bennett-Baker P.E., Mueller J.L.. CRISPR-mediated isolation of specific megabase segments of genomic DNA. Nucleic Acids Res. 2017; 45:e165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ranchon H., Malbec R., Picot V., Boutonnet A., Terrapanich P., Joseph P., Leïchlé T., Bancaud A.. DNA separation and enrichment using electro-hydrodynamic bidirectional flows in viscoelastic liquids. Lab. Chip. 2016; 16:1243–1253. [DOI] [PubMed] [Google Scholar]
- 18. Chami B., Socol M., Manghi M., Bancaud A.. Modeling of DNA transport in viscoelastic electro-hydrodynamic flows for enhanced size separation. Soft Matter. 2018; 14:5069–5079. [DOI] [PubMed] [Google Scholar]
- 19. Andriamanampisoa C.-L., Bancaud A., Boutonnet-Rodat A., Didelot A., Fabre J., Fina F., Garlan F., Garrigou S., Gaudy C., Ginot F.. BIABooster: online DNA concentration and size profiling with a limit of detection of 10 fg/μL and application to high-sensitivity characterization of circulating cell-free DNA. Anal. Chem. 2018; 90:3766–3774. [DOI] [PubMed] [Google Scholar]
- 20. Malbec R., Chami B., Aeschbach L., Buendía G.A.R., Socol M., Joseph P., Leïchlé T., Trofimenko E., Bancaud A., Dion V.. μLAS: Sizing of expanded trinucleotide repeats with femtomolar sensitivity in less than 5 minutes. Sci. Rep. 2019; 9:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Young N.D., Debellé F., Oldroyd G.E., Geurts R., Cannon S.B., Udvardi M.K., Benedito V.A., Mayer K.F., Gouzy J., Schoof H.. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature. 2011; 480:520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang M., Zhang Y., Scheuring C.F., Wu C.-C., Dong J.J., Zhang H.-B.. Preparation of megabase-sized DNA from a variety of organisms using the nuclei method for advanced genomics research. Nat. Protoc. 2012; 7:467–478. [DOI] [PubMed] [Google Scholar]
- 23. Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J. B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J.. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016; 17:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Peterson D.G., Tomkins J.P., Frisch D.A., Wing R.A., Paterson A.H.. Construction of plant bacterial artificial chromosome (BAC) libraries: an illustrated guide. J. Agricultural Genomics. 2000; 5:1–100. [Google Scholar]
- 25. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018; 34:3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Koren S., Walenz B.P., Berlin K., Miller J.R., Bergman N.H., Phillippy A.M.. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017; 27:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.. The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guinaudeau A., Mazières S., Wilson D.J., Destarac M.. Aqueous RAFT/MADIX polymerisation of N-vinyl pyrrolidone at ambient temperature. Polym. Chem. 2012; 3:81–84. [Google Scholar]
- 29. Kaji N., Tezuka Y., Takamura Y., Ueda M., Nishimoto T., Nakanishi H., Horiike Y., Baba Y.. Separation of long DNA molecules by quartz nanopillar chips under a direct current electric field. Anal. Chem. 2004; 76:15–22. [DOI] [PubMed] [Google Scholar]
- 30. Zheng J., Yeung E.S.. Mechanism for the separation of large molecules based on radial migration in capillary electrophoresis. Anal. Chem. 2003; 75:3675–3680. [DOI] [PubMed] [Google Scholar]
- 31. Birren B., Lai E.. Rapid pulsed field separation of DNA molecules up to 250 kb. Nucleic Acids Res. 1994; 22:5366–5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Osbourn D.M., Weiss D.J., Lunte C.E.. On-line preconcentration methods for capillary electrophoresis. ELECTROPHORESIS. 2000; 21:2768–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Friedrich S.M., Burke J.M., Liu K.J., Ivory C.F., Wang T.-H.. Molecular rheotaxis directs DNA migration and concentration against a pressure-driven flow. Nat. Commun. 2017; 8:1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 35. Xin Z., Chen J.. A high throughput DNA extraction method with high yield and quality. Plant Methods. 2012; 8:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zischewski J., Fischer R., Bortesi L.. Detection of on-target and off-target mutations generated by CRISPR/Cas9 and other sequence-specific nucleases. Biotechnol. Adv. 2017; 35:95–104. [DOI] [PubMed] [Google Scholar]
- 37. Paegel B.M., Blazej R.G., Mathies R.A.. Microfluidic devices for DNA sequencing: sample preparation and electrophoretic analysis. Curr. Opin. Biotechnol. 2003; 14:42–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.