

ABSTRACT
EGFR mutations display striking organ-site asymmetry and heterogeneity. We have shown that structurally diverse extracellular mutations, typical of glioblastomas, converge to a similar intermediate conformation, which can be synergistically targeted extra- and intracelullarly by antibody mAb806 and type-II kinase inhibitors. Our findings reveal convergence behind heterogeneity, paving the way for allostery-based co-targeting.
KEYWORDS: EGFR, ectomutations, convergence, mAb806, kinase inhibitors
EGFR mutational heterogeneity and organ-site specificity
EGFR is one of the main oncogenes and drug targets for cancer therapies, but the puzzling organ-site asymmetry observed for its mutations,1 has remained a mystery. While in tumors like lung cancer, mutations tend to focus on the kinase domain (KD), in brain glioblastomas (GBM), strikingly heterogeneous missense mutations and deletions concentrate at the ligand-binding ectodomain (ECD).2 This remarkable tissue-specific asymmetry is linked to different sensitivities for small tyrosine kinase inhibitors (TKIs) (Figure 1), with lung KD mutations responding better to type-I TKIs, which bind the active asymmetric KD dimer (aKD), and GBM mutations, paradoxically displaying higher sensitivity to type-II TKIs, which bind the inactive symmetric KD dimer (sKD).3 Regardless of this differential preference for inhibitors, both intra- and extracellular mutations are known to result in oncogenic ligand-independent activation.
Figure 1.
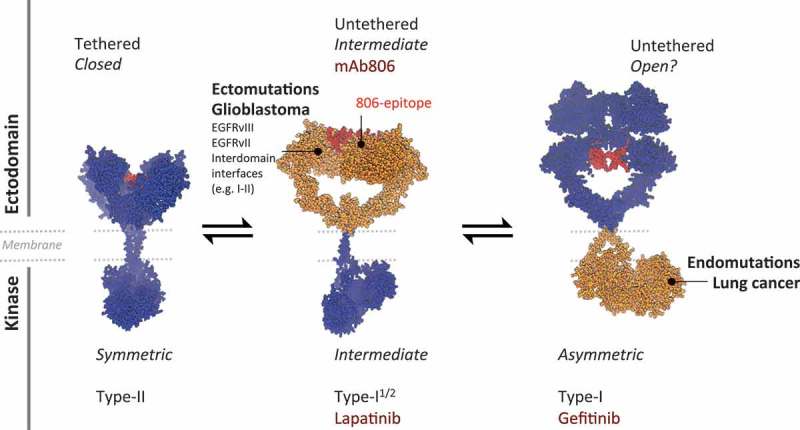
Organ asymmetry and mAb806-convergence of heterogeneous mutations affecting the EGFR ectodomain. Current evidences indicate that EGFR has two main dimeric states: fully inactive (left), with a tethered ectodomain (ECD) and a symmetric kinase domain (sKD); and fully active (e.g. upon EGF binding, right), with an untethered ECD and an asymmetric KD (aKD). The cancer-specific antibody-806 (mAb806) cannot bind any of them (note the buried cryptic 806-epitope, red), but was known to detect a transient conformer as they interconvert. Oncogenic EGFR mutations concentrate either extra- or intra-celullarly depending on the tissue and in relation with differential sensitivities for Tyrosine Kinase Inhibitors (TKIs). In lung cancer, mutations focus on the kinase (right, orange) favoring an aKD that binds type-I TKIs (e.g. gefitinib), while in glioblastomas (GBM), a plethora of activating missense mutations and deletions target the ECD, paradoxically responding better to “inactive” inhibitors like lapatinib. Our work has revealed that in spite of structural heterogeneity, GBM mutants converge to an intermediate state (center, orange), in which part of the ECD is displaced or removed to activate the kinase in an sKD-like arrangement, which is recognized extracellularly by mAb806. In contrast, the aKD is coupled to an ECD with a buried 806-epitope of unknown configuration. Given 806-convergence, tumors carrying the main ectomutations are all sensitive to mAb806; WT-EGFR, unresponsive to mAb806, can also be allosterically sensitized by lapatinib-induced conversion to the 806-intermediate. Models based on 1NQL and 3NJP simulations (see ref.5).
Structural convergence of GBM mutations: missense mutations and deletions reduced to one large class recognized by mAb806
Our work aimed to understand how the most frequent GBM missense mutations (I-II, see below) activate EGFR, leading to important mechanistic and therapeutically relevant insights.4,5 The ECD consists of four subdomains (I-IV), which are held in a compact and inactive conformation by an inter-domain tether (II-IV). Upon ligand binding, the tether breaks and the ECD opens, releasing a dimerization arm that forms inter-receptor interactions in the active dimer. Most GBM mutations cluster at interdomain interfaces (I-II, II-IV and II-III). While II-IV tether mutations clearly favour untethering, the mechanism of I-II mutations, located at an interface away from both the tether and the ligand-binding site, was unclear.6 Our I-II mutant simulations revealed that these mutations also promote untethering towards a not fully open but intermediate state,4 which unexpectedly, exposes a cryptic epitope recognized by the cancer-specific antibody mAb806, raised against the main GBM variant, the large deletion EGFRvIII. Although it was known that this peculiar antibody recognized a transitional conformer as EGFR activates,7 different from both the closed and open crystallographic structures, it had eluded structural determination. Remarkably, we observed that the region displaced in our I-II mutant simulations is the same deleted in EGFRvIII. This suggested a surprising “structural equivalence” of two extremely different variants (point changes versus a large deletion), and hence, potential convergence to activate EGFR in a similar way, which could explain how such heterogeneous variations share the same TKI sensitivity. Using small angle X-ray scattering (SAXS), along with Fluorescence Activated Cell Sorting (FACS), cell and mouse GBM models, we have validated this hypothesis,5 demonstrating that GBM mutations, representative of the main structural classes in patients, all converge to a similar intermediate state, detected by mAb806. Exposure of the 806-epitope would be then the hallmark of an “unrestrained” ECD, where an inhibitory region has been either displaced or deleted to switch on tissue-preferred signalling pathways. These findings also provide key evidence suggesting that EGFR activates in pre-formed dimers by removing a steric block.8 On a side note, our SAXS data also revealed for the first time that the deglycosylated ECD untethers spontaneously, providing a clear example of how altered glycans patterns (e.g. upon overexpression) can disrupt flexibility and function.
Overall, our results expand the therapeutical utility of mAb806 far beyond EGFRvIII and EGFR amplification, the two primary biomarkers for clinical trials. Recently, we presented the first application of mAb806 for the most aggressive GBM mutation, A289V.9 Now we dramatically extend mAb806 spectrum based on the convergence of ECD mutations, by demonstrating in mice models that low-dose mAb806 treatment triggers tumour regression of all the main GBM mutation classes, including less frequent ones like EGFRvII. These findings indicate that, as happens for EGFR-KD mutations, the sole presence of ECD mutations could predict positive responses to anti-EGFR therapy targeting the main GBM conformation; on a wider perspective, they also rationalize mutational heterogeneity in evolutionary-biochemical terms, suggesting that tissue-specificity can be a useful hallmark of convergence in drug responses.
Allosteric coupling as basis for synergistic EGFR targeting: synching the ectodomain and the kinase
The second important finding from our study is the tight allosteric coupling between the ECD and the KD, and how it can be exploited for rational co-targeting. The mAb806-ECD mutant convergence naturally raised the question whether such mutations also would share the same KD conformation as previously suggested,3 that is, whether both mAb806 and type-II TKIs target the same EGFR conformational state in GBMs. Using FACS, we showed that the accessibility of the 806-epitope is tightly coupled to the KD conformation stabilized by TKIs: type-I TKIs decrease binding to mAb806 while type-II and particularly lapatinib (type-I1/2, with intermediate active-inactive features) increase it (Figure1) i.e. TKI stabilization of an intermediate sKD conformation allosterically changes the ECD configuration and its epitope accessibility. Significantly, KD mutations are known to favour the aKD dimer,3,10 which we show is coupled to an ECD with a buried 806-epitope, possibly explaining their poorer responses to mAb806. Based on this coupling, we proved that lapatinib co-treatment sensitizes unresponsive WT-EGFR to mAb806, resulting in synergistic tumour inhibition by simultaneous extra-intracelullar blockade of the same conformer. The demonstration of how allosteric coupling can convert the WT-ECD to the state bound by mAb806 with conformation-specific and synergic TKIs expands the applicability of this antibody to EGFR-altered tumors independent of their mutational status.
Altogether, our findings allow first, to group a large number of heterogeneous mutations into a single mAb806-targetable class thanks to their molecular convergence, and second, to potentially apply this therapy to any EGFR-driven tumors by allosterically “synching” the kinase with TKIs, paving the way for rational ECD-KD co-targeting.
Funding Statement
This work was supported by the O. E. och Edla Johanssons Vetenskapliga Stiftelse.
Acknowledgments
L.O. thanks the O.E. and Edla Johanssons Stiftelse and the Knut and Alice Wallenberg Foundation for their support.
Disclosure of potential conflicts of interest
The author has no conflicts of interest.
References
- 1.Huang PH, Xu AM, White FM.. Oncogenic EGFR signaling networks in Glioma. Sci Signal. 2009;2:1–13. [DOI] [PubMed] [Google Scholar]
- 2.Furnari FB, Cloughesy TF, Cavenee WK, Mischel PS.. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat Rev Cancer. 2015;15:302–310. doi: 10.1038/nrc3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vivanco I, Ian Robins H, Rohle D, Campos C, Grommes C, Nghiemphu PL, Kubek S, Oldrini B, Chheda MG, Yannuzzi N, et al. Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2012;2:458–471. doi: 10.1158/2159-8290.CD-11-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orellana L, Hospital A, Orozco M. Oncogenic mutations of the EGF-Receptor ectodomain reveal an unexpected mechanism for ligand-independent activation. bioRxiv. 2014. doi: 10.1101/009068. [DOI] [Google Scholar]
- 5.Orellana L, Thorne AH, Lema R, Gustavsson J, Parisian AD, Hospital A, Cordeiro TN, Bernadó P, Scott AM, Brun-Heath I, et al. Oncogenic mutations at the EGFR ectodomain structurally converge to remove a steric hindrance on a kinase-coupled cryptic epitope. Proc Natl Acad Sci U S A. 2019;116:10009–10018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bessman NJ, Bagchi A, Ferguson KM, Lemmon MA, Bessman NJ, Bagchi A, Ferguson KM, Lemmon MA. Complex relationship between ligand binding and dimerization in the epidermal growth factor. CellReports. 2014;9:1306–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gan HK, Burgess AW, Clayton AHA, Res C, Onlinefirst P, Scott AM. Targeting of a conformationally exposed, tumor-specific epitope of EGFR as a strategy for cancer therapy. Cancer Res. 2012;72:2924–2930. doi: 10.1158/0008-5472.CAN-11-3898. [DOI] [PubMed] [Google Scholar]
- 8.Maruyama IN. Activation of transmembrane cell-surface receptors via a common mechanism? The “rotation model”. BioEssays. 2015;37:959–967. doi: 10.1002/bies.201500041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Binder ZA, Thorne AH, Bakas S, Wileyto EP, Bilello M, Akbari H, Rathore S, Ha SM, Zhang L, Ferguson CJ, et al. Epidermal growth factor receptor extracellular domain mutations in glioblastoma present opportunities for clinical imaging and therapeutic development. Cancer Cell. 2018;34:163–177.e7. doi: 10.1016/j.ccell.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z, Longo PA, Tarrant MK, Kim K, Head S, Leahy DJ, Cole PA. Mechanistic insights into the activation of oncogenic forms of EGF receptor. Nat Struct Mol Biol. 2011;18:1388–1393. doi: 10.1038/nsmb.2168. [DOI] [PMC free article] [PubMed] [Google Scholar]