

Nowadays, p53 is like a Dreamliner airplane, riding the high skies of cancer research with dignity and pride. You do not have to be a card-bearing p53 researcher to know about p53, care about it, and respect it. Yet, this was not always so. As a matter of fact, in the first years of p53 research, from its discovery in the late 1970’s to about the end of the 1980’s, p53 was more like a kite: one moment high up in the air, the next moment being dragged on the ground.
I joined Arnie’s lab in the summer of 1978. These were times of great excitement, right after the discovery of p53 (then called 54K by the Levine lab) and before the iconic paper of Linzer and Levine (1979) was published in Cell. The year 1979 was a memorable year for p53: it was reported independently by several groups, most prominently in the Nature paper of David Lane and Lionel Crawford (Lane and Crawford, 1979) and the Linzer–Levine Cell paper, but also in a number of publications of lower visibility (DeLeo et al., 1979; Kress et al., 1979; Melero et al., 1979; Smith et al., 1979; Rotter et al., 1980), which have since been somewhat pushed into the backstage.
In 1979, p53 was only one of many topics explored in Arnie’s lab. Notably, Arnie was most excited at that time by the differentiation of embryonal carcinoma cells, which he was hoping to employ toward gaining new insights into fundamental biological processes. In addition, there were also SV40, adenoviruses, herpesvirus, and more, all of whom strongly attracted Arnie’s curiosity. In fact, Arnie was then known primarily as a molecular virologist. This was also what attracted me originally to Arnie’s lab, being a direct continuation of my previous interest in SV40. For more than a year, I tried in vain to establish a cell-free system for SV40 DNA replication. Arnie was always supportive and encouraging, despite the fact that nothing really seemed to work. It was only a year later that he finally advised me to give up and instead join the p53 team, which had already grown to three members: Daniel Linzer (graduate student), Warren Maltzman (post-doc), and Archie—a joyful Puerto Rican student whom Arnie brought to Princeton after having met him in a course that he taught in Puerto Rico earlier on. For me, this turned out to be a blessed advice, which has charted the entire course of my subsequent scientific career (Figure 1).
Figure 1.

A picture of Levine lab in 1979 (Arnie is not in the picture). The bearded guy in the center is Daniel Linzer.
However, in 1980, the bright future of p53 was still far from being within sight. The subsequent several years were rather frustrating for the Levine p53 team. As a matter of fact, they were rough years for the entire (very small!) international p53 community. For quite a while, no significant progress seemed to be made in understanding what p53 was good for and why it was accumulated in many cancer cell types. In particular, our efforts to clone p53, eventually expanded to a team of three post-docs (Kaoru Segawa, myself, and my spouse Rachel), repeatedly met with failure. Likewise, none of the other p53 projects in the lab managed to really take off and fly high. I recall vividly a day in 1981 (at that time already in StonyBrook, to where Arnie moved from Princeton to become Chair of the Department of Microbiology) when Arnie assembled all of us in his office. With an unusually sad face, he brought up for discussion the question whether we should abandon p53 research altogether, because it seemed to be going nowhere. Luckily, not only for us in Arnie’s lab but also for the entire p53 field, Arnie’s conclusion was that we should not give up. And indeed, the rest is history.
Eventually, we succeeded to clone p53 (Oren and Levine, 1983) and so did several other labs. Now the road was open to study the functions of p53 and understand what it was doing in cancer. Very logically, the expectation was that it would turn out to be an oncogene. After all, why else would cancer cells want to overexpress this protein? And, reassuringly enough, several groups were indeed able to show that overexpressed p53 could drive cell transformation and even promote tumor growth in vivo, fulfilling the basic criteria for being an oncogene (Eliyahu et al., 1984, 1985; Jenkins et al., 1984a, b; Parada et al., 1984). So all seemed good, and p53 was soon accepted as a legitimate member of the growing family of cellular oncogenes. Mind you, not a senior member of the family. I recall those times in the Oncogene Meeting in Frederick, Maryland, when a couple of short p53 presentations would characteristically be squeezed into the last part of a big session dedicated to Myc; this was prompted by the common understanding that p53 was some sort of a Myc-like oncogene, but far less important and less interesting than the great famous Myc itself.
However, cracks soon started to appear in this seemingly clear picture. Notably, in parallel with the cloning effort that was successfully completed at the Weizmann Institute (Oren and Levine, 1983), Arnie engaged in a collaboration with Genentech, where Diane Pennica cloned mouse p53 cDNA from the F9 mouse embryonal carcinoma cell line (Pennica et al., 1984). Frustratingly, despite repeated attempts, the Levine lab could not detect any transforming activity when they overexpressed their cloned p53 in rodent cells. In an attempt to figure out what was wrong and why his clone was not performing as nicely as those of the other labs, Arnie eventually ended up sending one of his graduate students, Ida Deichaite, to my laborarory at the Weizmann Institute. Sure enough, Ida was able to reproduce our results with the expression plasmids that we generated, but again the F9-derived plasmid stubbornly refused to elicit transformation.
The puzzle was resolved only a few months later. At that time, I was visiting Arnie’s lab to further discuss the differences between our results and theirs. In a memorable moment, Phil Hinds—then a senior graduate student of Arnie’s—came into Arnie’s office with a pile of computer printouts and comparisons. He had carefully aligned all the available mouse and human p53 cDNA sequences and the corresponding predicted amino acid sequences. The conclusions from Phil’s analysis were unequivocal: none of the clones were identical in their amino acid sequences! As a matter of fact, they differed from each other by single nucleotides, within a region that is highly conserved between mouse and human p53 (at that time, we had no clue that this is the DNA-binding domain: the biochemical functions of p53 were still a total mystery). Together, this suggested that at least the majority of the clones obtained by different labs and reported in the literature might carry one or another mutation in the p53 coding region. But what then is the correct sequence of the non-mutated, wild-type p53? Fresh analysis of mouse genomic DNA, performed in parallel in Arnie’s and our labs (Eliyahu et al., 1988; Finlay et al., 1988), yielded an unequivocal answer: of all the clones that had been subjected to functional assays, the Pennica clone was the only one that had faithfully retained the wild-type p53 sequence! In retrospect, we appreciate that this was predictable: the first step in all p53 cDNA cloning efforts was to identify a cell line that makes copious amounts of the p53 protein (determined by the very good antibodies that we already had at that time), on the logical assumption that these cells would also have abundant p53 messenger RNA (mRNA); we realized only later that the copious amounts of p53 were due to protein stabilization, rather than to mRNA overexpression. Thus, it eventually became clear that, unlike its cancer cell-derived mutated versions, wild-type p53 does not harbor transforming activity! It did not take long to realize that not only did wild-type p53 fail to transform, but also it actually was capable of robustly suppressing the transforming activity of otherwise very potent oncogenes (Eliyahu et al., 1989; Finlay et al., 1989). Hence, at least in cell culture, wild-type p53 behaved like a tumor suppressor.
In perfect timing, independent work carried out by Bert Vogelstein and coworkers, primarily by his graduate students Susan Baker and Janice Nigro, revealed that the p53 gene was frequently mutated in human colorectal tumors; often, the remaining wild-type allele was lost, resulting in total absence of wild-type p53 protein (Baker et al., 1989; Nigro et al., 1989). This was precisely the type of behavior expected of a well-behaved tumor suppressor gene. Thus, the combination of Vogelstein’s human cancer genetic analysis and the rodent p53 expression experiments by Arnie’s lab and ours firmly established p53 as a tumor suppressor gene. As a matter of fact, the clues had already been there several years earlier: previous work from both Varda Rotter at the Weizmann Institute and the Toronto team led by Sam Benchimol and Alan Bernstein had identified a complete loss of p53 expression in a human cancer cell line and in mouse leukemias (Mowat et al., 1985; Wolf and Rotter, 1985; Rovinski et al., 1987), but these had been dismissed by most of the p53 community members as rare exceptions to the general oncogenic function of p53.
Remarkably, within a short time, it was realized that many types of human cancer carry p53 mutations, often at a remarkably high frequency. Accordingly, by the early 1990’s, p53 had already rapidly risen to prominence, drawing broad interest and excitement all throughout the cancer research community and beyond. In fact, in 1993, p53 was already crowned as Molecule of the Year by Science magazine, and since then, p53 has become the most extensively studied of all human genes.
Thus, after being considered an oncogene for several years, p53 finally was canonized as a star tumor suppressor.
But what about the old experiments, where a variety of p53 mutants displayed transforming activity? If such mutations merely abolish wild-type p53’s tumor suppressor function, they would be expected to have no effect in transformation assays, neither positive nor negative. This clearly was not the case. Yet, overtaken by the importance of p53 as a tumor suppressor, the field was not eager to give much attention to those early experiments. But bit by bit, evidence started to accumulate that cancer-derived p53 mutants could exert a variety of oncogenic activities, extending beyond what was initially observed in the early rodent cell transformation experiments. Finally, in 1993, Arnie came up with the outright statement that such mutants harbor oncogenic gain-of-function (GOF) (Dittmer et al., 1993). The concept of mutant p53 GOF gradually increased its grip, finally becoming widely embraced when Gigi Lozano and Tyler Jacks reported compelling in vivo experiments showing that knock-in of a mutant p53 allele can cause mice to develop more aggressive and more metastatic tumors than p53-null mice (Lang et al., 2004; Olive et al., 2004). Since then, much more was learned about the various molecular mechanisms and biological processes that underpin mutant p53 GOF, practically constituting a whole new field of p53-related research.
So, the early conclusions were not entirely wrong: p53 can indeed be oncogenic, but only as a consequence of mutations within its coding region. And as a matter of fact, such mutations are very abundant and are not merely a lab artifact.
So now we had a clear distinction: wild-type p53 is always a tumor suppressor, while cancer-associated p53 mutants may be oncogenic. Indeed so? Not so fast, not so simple, certainly not when one deals with p53. Nowadays, we realize that this black-and-white picture has many shades of gray. A number of studies have revealed that perfectly wild-type p53 can acquire biochemical and biological features that are usually ascribed to cancer-associated GOF mutants. While such cancer-supportive features are not revealed under commonly studied lab conditions, particularly when cells are subjected to severe genotoxic stress that elicits a robust activation of canonical p53, they can be observed under quite a number of other conditions. In fact, robust clues in this direction were already provided by Jo Milner in the early days of p53 research. Essentially, Milner reported that p53 can be driven to acquire a mutant-like conformation in non-transformed cells, when these are exposed to conditions that favor their proliferation (Milner and Watson, 1990). Likewise, Al Deisseroth and coworkers showed that growth factors can enforce a mutant-like conformational switch of wild-type p53 in both normal and transformed hematopoietic cells (Zhang et al., 1992; Zhang and Deisseroth, 1994), demonstrating the existence of a ‘pseudomutant’ state of wild-type p53. More recent work by Karen Vousden’s group provided insights into a possible molecular mechanism, demonstrating that a set of molecular chaperone proteins are required in order to maintain p53 in its canonical wild-type conformation and prevent it from misfolding into a mutant-like state (Trinidad et al., 2013). Curiously, dynamic switching of p53 states is not limited only to wild-type p53: in a reciprocal manner, genetically mutant p53 can be lured into adopting a wild-type-like conformation, and even wild-type-like functionality, as shown by Varda Rotter and coworkers for embryonic stem cells (Rivlin et al., 2014). Furthermore, such reverse state switch also seems to be dependent on the action of molecular chaperones (Rivlin et al., 2014). The dynamic, signal, and context-dependent switching of p53 between alternative states is schematically illustrated in Figure 2.
Figure 2.
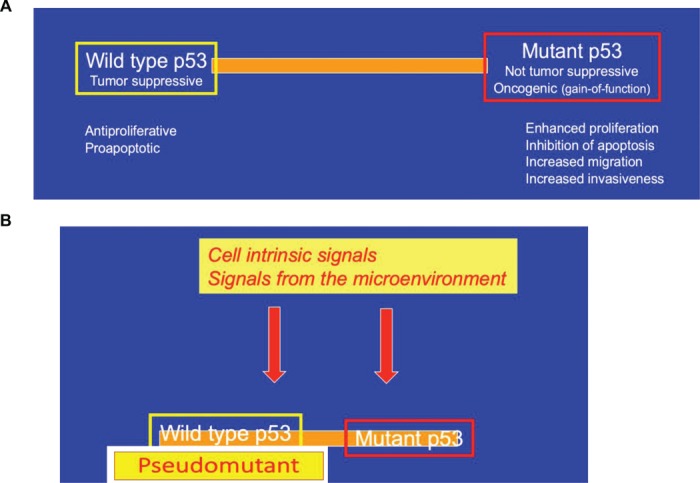
Dynamics of p53 states. (A) Under most conditions, wild-type p53 and cancer-associated mutants such as p53R175H are in very distinct functional states. (B) However, in response to cell-intrinsic signals and conditions (e.g. chaperone dysfunction) or signals from the microenvironment (e.g. growth factors, tissue damage), wild-type p53 may be toggled toward a ‘pseudomutant’ state. Likewise, genetically mutant p53 can be toggled toward a wild-type-like state by excessive chaperone activity and presumably also by signals from the microenvironment.
All the above experiments, demonstrating p53 state switching, were performed in cell culture. Conceivably, conversion of wild-type p53 into a ‘pseudomutant’ state occurs also under some physiological conditions, when accelerated cell proliferation may be advantageous and even essential. Such situation may pertain in the early phases of the response to tissue damage, when tissue integrity is disrupted and the process of wound healing is initiated. In agreement with this notion, p53 undergoes a shift toward ‘pseudomutant’ state upon intentional inactivation of core components of the Hippo signal transduction pathway (Furth et al., 2015), mimicking processes that occur when cell-cell contact is lost. While this state-transition of wild-type p53 is reversible and is presumably dismissed once tissue integrity is restored, cancer-associated p53 mutations may lock p53 chronically in its pro-proliferative state, yielding the familiar portfolio of GOF oncogenic activities. Similarly, some types of constitutive pathway deregulation, when occurring during tumor development, might trick non-mutated p53 to ‘think’ that it should switch into a ‘pseudomutant’ state. However, unlike in a normal regenerating tissue, those signals are persistent in the cancer milieu (‘a wound that does not heal’), maintaining wild-type p53 continuously in a ‘pseudomutant’ state (Figure 2). Moreover, such conversion of wild-type p53 to a ‘pseudomutant’ state during tumor progression may occur not only in the cancer cells themselves when those retain a non-mutated p53 gene, but also in their microenvironment; for example, p53 appears to undergo a non-mutational state switch in cancer-associated fibroblasts, in association with the transition of the microenvironment from tumor-suppressive to tumor-supportive (Arandkar et al., 2018).
The idea that cancer-associated pathway deregulation may tame p53 and render it cancer-supportive would seem to disagree with the prevailing dogma that excessive oncogenic signaling actually triggers activation of p53 in its canonical tumor suppressive, proapoptotic, and antiproliferative state, thereby providing a failsafe mechanism against facile oncogene-driven transformation and cancer. This seeming conundrum may be reconciled by proposing that the outcome might depend on the particular oncogenic pathway that is being deregulated, as well as on the magnitude of its deregulation. Obviously, more work is required in order to understand what determines whether p53 will switch to a pseudomutant state, or will emerge as a potent guardian of the genome and of the host’s wellbeing.
It is clear that the p53 saga is still far from complete. We keep learning as we move forward, replacing one dogma by the next one, only to be dethroned by further studies. Thank you, Arnie, for having started us on a wonderful journey that never ends!
References
- Arandkar S., Furth N., Elisha Y., et al. (2018). Altered p53 functionality in cancer-associated fibroblasts contributes to their cancer-supporting features. Proc. Natl Acad. Sci. USA 115, 6410–6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker S.J., Fearon E.R., Nigro J.M., et al. (1989). Chromosome 17 deletion and p53 gene mutations in colorectal carcinomas. Science 244, 217–221. [DOI] [PubMed] [Google Scholar]
- DeLeo A.B., Jay G., Appella E., et al. (1979). Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc. Natl Acad. Sci. USA 76, 2420–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer D., Pati S., Zambetti G., et al. (1993). Gain of function mutations in p53. Nat. Genet. 4, 42–46. [DOI] [PubMed] [Google Scholar]
- Eliyahu D., Goldfinger N., Pinhasikimhi O., et al. (1988). Meth-A fibro-sarcoma cells express 2 transforming mutant p53 species. Oncogene 3, 313–321. [PubMed] [Google Scholar]
- Eliyahu D., Michalovitz D., Eliyahu S., et al. (1989). Wild-type p53 can inhibit oncogene-mediated focus formation. Proc. Natl Acad. Sci. USA 86, 8763–8767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliyahu D., Michalovitz D., and Oren M. (1985). Overproduction of p53 antigen makes established cells highly tumorigenic. Nature 316, 158–160. [DOI] [PubMed] [Google Scholar]
- Eliyahu D., Raz A., Gruss P., et al. (1984). Participation of p53 cellular tumor antigen in transformation of normal embryonic cells. Nature 312, 646–649. [DOI] [PubMed] [Google Scholar]
- Finlay C.A., Hinds P.W., Han T.H., et al. (1988). Activating mutations for transformation by p53 produce a gene product that forms an hsc70-p53 complex with an altered half-life. Mol. Cell. Biol. 8, 531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay C.A., Hinds P.W., and Levine A.J. (1989). The p53 proto-oncogene can act as a suppressor of transformation. Cell 57, 1083–1093. [DOI] [PubMed] [Google Scholar]
- Furth N., Bossel Ben-Moshe N., Pozniak Y., et al. (2015). Down-regulation of LATS kinases alters p53 to promote cell migration. Genes Dev. 29, 2325–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins J.R., Rudge K., and Currie G.A. (1984a). Cellular immortalization by a cDNA clone encoding the transformation-associated phosphoprotein p53. Nature 312, 651–654. [DOI] [PubMed] [Google Scholar]
- Jenkins J.R., Rudge K., Redmond S., et al. (1984b). Cloning and expression analysis of full length mouse cDNA sequences encoding the transformation associated protein p53. Nucleic Acids Res. 12, 5609–5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress M., May E., Cassingena R., et al. (1979). Simian virus-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J. Virol. 31, 472–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane D.P., and Crawford L.V. (1979). T-antigen is bound to host protein in SV40-transformed cells. Nature 278, 261–263. [DOI] [PubMed] [Google Scholar]
- Lang G.A., Iwakuma T., Suh Y.A., et al. (2004). Gain of function of a p53 hot spot mutation in a mouse model of Li–Fraumeni syndrome. Cell 119, 861–872. [DOI] [PubMed] [Google Scholar]
- Linzer D.I.H., and Levine A.J. (1979). Characterization of a 54K Dalton cellular SV40 tumor antigen present in SV40 transformed cells and uninfected embryonal carcinoma cells. Cell 17, 43–52. [DOI] [PubMed] [Google Scholar]
- Melero J.A., Stitt D.T., Mangel W.F., et al. (1979). Identification of new polypeptide species (48-55K) immunoprecipitable by antiserum to purified large T antigen and present in SV40-infected and transformed cells. Virology 93, 466–480. [DOI] [PubMed] [Google Scholar]
- Milner J., and Watson J.V. (1990). Addition of fresh medium induces cell cycle and conformation changes in p53, a tumour suppressor protein. Oncogene 5, 1683–1690. [PubMed] [Google Scholar]
- Mowat M., Cheng A., Kimura N., et al. (1985). Rearrangements of the cellular p53 gene in erythroleukaemic cells transformed by Friend virus. Nature 314, 633–636. [DOI] [PubMed] [Google Scholar]
- Nigro J.M., Baker S.J., Preisinger A.C., et al. (1989). Mutations in the p53 gene occur in diverse human tumour types. Nature 342, 705–708. [DOI] [PubMed] [Google Scholar]
- Olive K.P., Tuveson D.A., Ruhe Z.C., et al. (2004). Mutant p53 gain of function in two mouse models of Li–Fraumeni syndrome. Cell 119, 847–860. [DOI] [PubMed] [Google Scholar]
- Oren M., and Levine A.J. (1983). Molecular cloning of a cDNA specific for the murine p53 cellular tumor antigen. Proc. Natl Acad. Sci. USA 80, 56–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parada L.F., Land H., Weinberg R.A., et al. (1984). Cooperation between the gene encoding p53 tumor antigen and ras in cellular transformation. Nature 312, 649–651. [DOI] [PubMed] [Google Scholar]
- Pennica D., Goeddel D.V., Hayflick J.S., et al. (1984). The amino acid sequence of murine p53 determined from a c-DNA clone. Virology 134, 477–482. [DOI] [PubMed] [Google Scholar]
- Rivlin N., Katz S., Doody M., et al. (2014). Rescue of embryonic stem cells from cellular transformation by proteomic stabilization of mutant p53 and conversion into WT conformation. Proc. Natl Acad. Sci. USA 111, 7006–7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotter V., Witte O.N., Coffman R., et al. (1980). Abelson-murine leukemia virus induced tumors elicit antibodies against a host cell protein, p50. J. Virol. 36, 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovinski B., Munroe D., Peacock J., et al. (1987). Deletion of 5′-coding sequences of the cellular p53 gene in mouse erythroleukemia: a novel mechanism of oncogene regulation. Mol. Cell. Biol. 7, 847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A.E., Smith R., and Paucha E. (1979). Characterization of different tumor antigens present in cells transformed by simian virus 40. Cell 18, 335–346. [DOI] [PubMed] [Google Scholar]
- Trinidad A.G., Muller P.A., Cuellar J., et al. (2013). Interaction of p53 with the CCT complex promotes protein folding and wild-type p53 activity. Mol. Cell 50, 805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D., and Rotter V. (1985). Major deletions in the gene encoding the p53 tumor antigen cause lack of p53 expression in HL-60 cells. Proc. Natl Acad. Sci. USA 82, 790–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., and Deisseroth A.B. (1994). Conformational change of p53 protein in growth factor-stimulated human myelogenous leukemia cells. Leuk. Lymphoma 14, 251–255. [DOI] [PubMed] [Google Scholar]
- Zhang W., Hu G.Y., Estey E., et al. (1992). Altered conformation of the p53 protein in myeloid leukemia cells and mitogen-stimulated normal blood cells. Oncogene 7, 1645–1647. [PubMed] [Google Scholar]