


Abstract
DNA damage-induced cell cycle checkpoints serve as surveillance mechanisms to maintain genomic stability, and are regulated by ATM/ATR-mediated signaling pathways that are conserved from yeast to humans. Trypanosoma brucei, an early divergent microbial eukaryote, lacks key components of the conventional DNA damage-induced G2/M cell cycle checkpoint and the spindle assembly checkpoint, and nothing is known about how T. brucei controls its cell cycle checkpoints. Here we discover a kinetochore-based, DNA damage-induced metaphase checkpoint in T. brucei. MMS-induced DNA damage triggers a metaphase arrest by modulating the abundance of the outer kinetochore protein KKIP5 in an Aurora B kinase- and kinetochore-dependent, but ATM/ATR-independent manner. Overexpression of KKIP5 arrests cells at metaphase through stabilizing the mitotic cyclin CYC6 and the cohesin subunit SCC1, mimicking DNA damage-induced metaphase arrest, whereas depletion of KKIP5 alleviates the DNA damage-induced metaphase arrest and causes chromosome mis-segregation and aneuploidy. These findings suggest that trypanosomes employ a novel DNA damage-induced metaphase checkpoint to maintain genomic integrity.
INTRODUCTION
To maintain genomic integrity in eukaryotes, two surveillance mechanisms, the DNA damage response (DDR) and the spindle assembly checkpoint (SAC), function to ensure the fidelity of genome replication and segregation. The DDR recognizes DNA damage and arrests the cell cycle, allowing cells to repair DNA damage before they enter the next cell cycle stage. The signaling pathway for DDR has been well characterized in yeast and animals (1,2). DNA damage induces DNA replication folk stalling, thereby activating the PI3K-like kinases, ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and Rad3-related (ATR). ATM phosphorylates the checkpoint kinase Chk2, which activates p53 and p21 for the two proteins to inhibit the Cyclin E/Cdk2 complex, thus leading to G1/S cell cycle arrest. ATR phosphorylates another checkpoint kinase named Chk1, which, together with Chk2, arrests the cell cycle at the intra-S phase or the G2/M boundary by inhibiting the Cdc25 phosphatase, which activates the G1 cyclin-dependent kinase (CDK) Cdk2 and the mitotic CDK Cdk1 by removing the inhibitory phosphorylation in the activation loop of these CDKs (3). Additionally, Chk1 can activate the Wee1 kinase, which phosphorylates Cdk1 in the activation loop, thereby inhibiting Cdk1 and preventing the G2/M cell cycle transition (4).
While the DDR activates G1/S, intra-S phase and G2/M checkpoints (3), the SAC delays anaphase onset in response to kinetochore-microtubule attachment errors through inhibiting Cdc20, a co-activator of the anaphase-promoting complex/cyclosome (APC/C), an E3 ubiquitin ligase that targets crucial cell cycle regulators for degradation during the metaphase-anaphase transition (5). When the SAC is activated, the mitotic checkpoint complex (MCC), composed of Mad2, BubR1 and Bub3, binds to Cdc20 and prevents Cdc20 from activating APC/C. Once kinetochore-microtubule attachment errors are corrected, the SAC is inactivated and Cdc20 is released from the MCC, thereby activating the APC/C to promote anaphase onset (6,7). APC/C promotes the degradation of securin (Pds1 in yeast), an inhibitor of the separase protease, thus activating separase, and the activated separase then cleaves the cohesin subunit SCC1, leading to sister chromatid separation (8). APC/C also targets the mitotic B-type cyclin for degradation to promote anaphase onset (9,10) and mitotic exit (11). The signal for SAC activation is generated from unattached kinetochores, to which the SAC components are recruited through binding to the outer kinetochore component KNL1 (12–14), an essential subunit of the KMN complex that also includes the Mis12 sub-complex and the Ndc80 sub-complex (15). Targeting of SAC components to kinetochores requires the Aurora B kinase, which phosphorylates Ndc80 for recruitment of the SAC component Mps1 initially and other SAC components subsequently (16).
Although the DDR and the SAC sense different perturbations, extensive studies have demonstrated synergistic interactions and cross-talk between these two checkpoints in yeast and animals (for a review, see (17)). In addition to responding to kinetochore-microtubule attachment errors, the SAC can also be triggered by DNA damage, and the DNA damage-induced metaphase arrest depends on the SAC and both the ATM and ATR kinases, but is independent of kinetochores (18–20). DNA damage can activate the SAC in a kinetochore-dependent, but ATM/ATR-independent manner to arrest meiosis metaphase I in mouse oocytes (21). The DDR and the SAC regulate a common target, the anaphase inhibitor securin/Pds1, to control anaphase onset. Pds1 is phosphorylated and stabilized by DNA damage checkpoint in a SAC-independent manner (22,23), whereas the SAC stabilizes Pds1 through inhibition of APC/C (24,25).
The molecular components and the signaling pathways of the DDR and the SAC are conserved across evolution from yeast to humans (26,27). However, distinctions in the DDR are observed in plants (28), and certain components of the DDR and the SAC are not identifiable in the lineage of the flagellated protozoa kinetoplastids, which include Trypanosoma brucei, Trypanosoma cruzi and Leishmania spp. and belong to the Excavata supergroup of eukaryotes (29). Trypanosoma brucei appears to lack the homologs of all but Mad2 of the SAC machinery, and the Mad2 homolog (TbMad2) localizes to the flagellar basal body region instead of kinetochores (30). T. brucei also lacks the homologs of the Cdc25-family protein phosphatase (31) and the checkpoint kinases Chk1 and Chk2 (32), key components of the ATM/ATR-mediated DNA damage checkpoint (3). Moreover, T. brucei appears to lack the mitosis-cytokinesis checkpoint to monitor the faithful transition from mitosis to cytokinesis in the procyclic (insect) life cycle stage (33–35). Trypanosoma brucei may employ distinct cell cycle checkpoint pathways during its cell cycle.
Here, we report that DNA damage induces a metaphase checkpoint in the procyclic form of T. brucei through modulating the abundance of the outer kinetochore protein KKIP5. This KKIP5-mediated checkpoint operates in an ATM/ATR-independent manner, but requires functional kinetochores and the Aurora B kinase. This finding suggests that trypanosomes utilize an unusual DNA damage-induced metaphase checkpoint to maintain genomic integrity.
MATERIALS AND METHODS
Trypanosome cell culture and RNA interference
The procyclic trypanosome Lister 427 strain and the 29-13 cell line (36), which expresses the T7 RNA polymerase and the tetracycline repressor, were used in this work. The Lister 427 strain was maintained at 27°C in SDM-79 medium supplemented with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals, Inc.). The 29-13 cell line was cultured at 27°C in SDM-79 medium containing 10% heat-inactivated fetal bovine serum, 15 μg/ml G418, and 50 μg/ml hygromycin B. Cell density was maintained between 106 to 107 cells/ml by routine dilutions with fresh medium.
To generate RNAi cell lines, a 479-bp DNA fragment (nucleotides 104–582) of the KKIP5 gene, a 581-bp DNA fragment (nucleotides 1250–1830) of the ATM gene, a 500-bp DNA fragment of the ATR gene (nucleotides 407–906), a 560-bp DNA fragment (nucleotides 296–855) of the KKIP1 gene, and a 610-bp DNA fragment (nucleotides 1–610) of the KKT8 gene were each cloned into the pZJM vector (37). To construct the ATM-ATR double RNAi plasmid, the same DNA fragments of ATM and ATR genes used for single gene knockdown above were ligated in tandem into the pZJM vector. The resulting plasmids were linearized by restriction digestion with NotI, and then transfected into the 29-13 cell line by electroporation. Transfectants were selected with 2.5 μg/ml phleomycin, and cloned by limiting dilution in 96-well plates containing SDM-79 medium supplemented with 20% fetal bovine serum and appropriate antibiotics. The TbAUK1 RNAi cell line was generated previously (38,39).
Epitope tagging of proteins at the endogenous locus
For epitope tagging of proteins at the endogenous locus, the PCR-based epitope tagging approach (40) was used. KKIP5 was tagged with a triple HA epitope at the C-terminus, Kif13-1, KKT2, ATM, ATR, KKIP1, KKT8, TbSCC1 and TbAUK1 were each tagged with a PTP epitope at the C-terminus, and CYC6 was tagged with an N-terminal PTP epitope. PCR products were transfected into the Lister427 strain, certain RNAi (KKIP5 RNAi, ATM RNAi, ATR RNAi, ATM-ATR double RNAi, KKIP1 RNAi, KKT8 RNAi or TbAUK1 RNAi) cell lines, or the KKIP5 overexpression cell line. Transfectants were selected with 1 μg/ml puromycin or 10 μg/ml blasticidin, and were further cloned by limiting dilution as described above. To confirm that epitope tagging did not affect KKIP5 function, we knocked out the other allele of KKIP5 in the cell line expressing endogenously KKIP5-3HA, and the resulting cell line (KKIP5-3HA+/KKIP5−) grew at a similar rate as the wild-type (KKIP5+/KKIP5+) and the KKIP5-3HA cell line (KKIP5+/KKIP5-3HA+) (Supplementary Figure S1).
Yeast two-hybrid library screening and directional yeast two-hybrid assays
Yeast two-hybrid library screening using TbAUK1 as the bait was performed by Hybrigenics Services (https://www.hybrigenics-services.com). The full-length TbAUK1 coding sequence was cloned in the pGADT7 vector (38), and the T. brucei yeast two-hybrid genomic library, containing 7.5 million independent genomic DNA fragments (41), was used for screening. A total of 67.4 million interactions with TbAUK1 were tested, and positive clones were selected on medium lacking Leu, Trp and His.
Directional yeast two-hybrid assays were carried out essentially as described previously (38). KKIP5 was cloned in the pGBKT7 vector, and was expressed in yeast strain Y187 (mating type α). TbAUK1 was cloned in the pGADT7 vector, and was expressed in yeast stain AH109 (mating type a). Yeast mating was carried out by mixing the Y187 and AH109 strains in YPDA medium at 30°C for 24 h and then plating on SD medium lacking Leu and Trp for selection of clones carrying both plasmids. The diploid stain thus obtained was spotted in four 10-fold serial dilutions onto the SD medium plate lacking Leu and Trp and the SD medium plate lacking Leu, Trp and His. Yeast strains containing the empty vector were used as negative controls. The interaction between p53 and SV40 was used as the positive control.
Ectopic overexpression of KKIP5
The full-length coding sequence of KKIP5 was cloned into the pLew100-3HA vector (42), which was modified from the pLew100 vector (36). The resulting plasmid pLew100-KKIP5-3HA was linearized with NotI and transfected into the 29-13 cell line by electroporation. Transfectants were selected under 2.5 μg/ml phleomycin, and then cloned by limiting dilution in a 96-well plate as described above. To induce KKIP5-3HA overexpression, cells were incubated with 1.0 μg/ml tetracycline.
Treatment of cells with the proteasome inhibitor MG-132 and DNA damage-inducing agents
Cells expressing endogenously triple HA-tagged KKIP5 were incubated with 50 μM MG-132 for up to 6 h, and time-course samples were collected every 2 h for western blotting and immunofluorescence microscopy with anti-HA monoclonal antibody. For treatment with DNA damage-inducing agents, the following cell lines were used: the Lister 427 cell line expressing KKIP5-3HA; the ATM RNAi cell line expressing KKIP5-3HA; the ATR RNAi cell line expressing KKIP5-3HA; the ATM-ATR double RNAi cell line expressing KKIP5-3HA; the KKIP1 RNAi cell line expressing KKIP5-3HA; the KKT8 RNAi cell line expressing KKIP5-3HA; and the TbAUK1 RNAi cell line expressing KKIP5-3HA. Cells were incubated with 0.3 μM hydroxyurea (HU), 0.0003% (v/v) methyl methanesulfonate (MMS), 0.1% (v/v) ethyl methanesulfonate (EMS), 28 μM Camptothecin (CPT), 5 μM 4-nitroquinoline-1-oxide (4NQO), 100 μM 5-fluorouracil (5-FU), or 1 μg/ml Zeocin for 16 h, and then collected for western blotting and immunofluorescence microscopy with anti-HA monoclonal antibody. The KKIP5 RNAi cells were incubated with 0.0003% (v/v) MMS for a total of 120 h for analysis of growth defects or for 16 h for immunofluorescence microscopic analysis with the anti-Rad51 polyclonal antibody and for cell counting to assess cell cycle defects.
Quantitative RT-PCR
Trypanosome Lister 427 cells were treated with MMS for 16 h, and cells were collected and wash once with PBS for RNA extraction. Total RNA was isolated using TRIzol Reagent (Ambion–Life technology) and chloroform. RNA was then treated with DNAse I to remove all DNA contamination. RNA was washed with 100 μl phenol/chloroform, precipitated with isopropanol, and then washed twice with 70% ethanol. RNA was dried and re-suspended in DEPC-treated water. RNA concentration was determined using a NanoDrop spectrometer (Thermo Scientific). One microgram of total RNA was then subjected to cDNA synthesis by using ProtoScript II Reverse Transcriptase (BioLabs, New England). qRT-PCR experiment was carried out using FastStart SYBR Green master mix (Bio-Rad) according to manufacturer's instruction with primers 5′- ACCAGAAGAGCCAGTCTCCA-3′ and 5′-CCCACAGGCTCCTATGAGTT-3′ for KKIP5 and primers 5′-ATGGCAGTAGTGGAGATTG-3′ and 5′-CAGGCGGTTCCAACAAGTG-3′ for actin gene as an internal control. The fluorescence from DNA-SYBR green complex was monitored with CFX-Connect Real-time System (Bio-Rad). The level of KKIP5 mRNA, relative to the mean of the control gene actin was calculated by comparative Ct method.
Measurement of protein degradation kinetics by cycloheximide chase analysis
To monitor the degradation kinetics of CYC6, KKIP5 and TbSCC1, cycloheximide chase analysis was carried out. The following cell lines were used: the Lister 427 cell line expressing PTP-CYC6; the Lister427 cell line expressing TbSCC1-PTP; the Lister 427 cell line expressing KKIP5-3HA; the TbAUK1 RNAi cell line expressing KKIP5-3HA; the KKIP5 overexpression (OE) cell line expressing PTP-CYC6 or TbSCC1-PTP. Cells were treated without or with 0.0003% (v/v) MMS for 16 h, and then treated with 100 μg/ml cycloheximide for up to 8 h. Time-course samples were collected every 2 h for western blotting. TbAUK1 RNAi cells were induced for RNAi with 1.0 μg/ml tetracycline for 8 h before treated with MMS, whereas KKIP5 OE cells were induced for KKIP5 overexpression with 1.0 μg/ml tetracycline for 8 h before they were treated with MMS.
For quantitative western blotting, an equal number (5 × 105) of cells were lysed, and cell lysate was fractionated on SDS-PAGE, transferred onto a PVDF membrane, and immunoblotted with the primary antibodies for 1 h at room temperature. The following primary antibodies were used: anti-HA monoclonal antibody (mAb) (1:1000 dilution, Sigma-Aldrich, Clone HA-7), anti-Protein A polyclonal antibody (pAb) (1:1000 dilution, Sigma-Aldrich), and anti-TbPSA6 pAb (1:1000 dilution) (43). After washing three times with TBST, the membrane was incubated with FITC-conjugated anti-mouse IgG (1:1000 dilution, Sigma-Aldrich), FITC-conjugated anti-rabbit IgG (1:1000 dilution, Sigma-Aldrich), or IRDye® 680LT anti-rabbit IgG (1:2500 dilution, Li-Cor Cooperate), and western blot signals were captured using the Bio-Rad ChemiDoc™ MP imaging system, which allows fluorescent western blot imaging and quantitative analysis of protein bands. Protein band intensity was normalized with the loading control, and plotted against time of cycloheximide treatment.
Analysis of chromosome segregation defects
Two molecular markers were used to analyze the defects in chromosome segregation in KKIP5 RNAi and OE cells. The mitotic MCAK-type kinesin, Kif13-1, which localizes to nucleus during interphase and to the spindle during mitosis (44,45), was used as the spindle marker. Kif13-1 was endogenously tagged with a C-terminal PTP epitope in KKIP5 RNAi cell line and KKIP5 OE cell line. The inner kinetochore protein KKT2, which localizes to the kinetochores throughout the cell cycle (46), was used to monitor kinetochore segregation. KKT2 was endogenously tagged with a C-terminal PTP epitope in KKIP5 RNAi cells. Tagging of Kif13-1 and KKT2 was carried out by the PCR-based tagging approach (40).
Co-immunoprecipitation and western blotting
For co-immunoprecipitation of TbAUK1 and KKIP5, a T. brucei cell line expressing PTP-tagged TbAUK1 and triple HA-tagged KKIP5 from their respective endogenous locus was generated by PCR-based epitope tagging method (40). As negative controls, T. brucei cell lines expressing TbAUK1-PTP and KKIP5-3HA alone were also generated using the PCR-based epitope tagging method. Cells were lysed by incubating in 1 ml immunoprecipitaiton buffer (25 mM Tris–Cl, pH 7.6, 100 mM NaCl, 1 mM DTT, 1% NP-40 and protease inhibitor cocktail) for 30 min on ice, and lysed cells were cleared by centrifugation at 18407 × g for 10 min at 4°C. Cleared cell lysate was incubated with 20 μl settled anti-HA agarose beads (Sigma-Aldrich) at 4°C for 1 h, and the anti-HA beads were washed six times with the immunoprecipitation buffer. Proteins bound to the beads were eluted with 10% SDS, separated by SDS-PAGE, transferred onto a PVDF membrane, and immunoblotted with anti-Protein A pAB (Sigma-Aldrich) and anti-HA mAb to detect TbAUK1-PTP and KKIP5-3HA, respectively.
Immunofluorescence microscopy
Cells were collected by centrifugation at 5000 × g for 5 min and washed once in PBS. Cells were then attached onto glass coverslips for 30 min at room temperature and fixed in cold (–20°C) methanol for 30 min. Coverslips were rehydrated in PBS for 10 min and blocked with 3% BSA in PBS for 30 min. Fixed cells were incubated with primary antibodies diluted in blocking buffer for 1 h. The following primary antibodies were used: FITC-conjugated anti-HA mAb (1:400 dilution, Sigma Aldrich, Clone HA-7), anti-Protein A pAb (1:400 dilution, Sigma Aldrich), anti-γH2A pAb (1:2000 dilution) (47) and anti-Rad51 pAb (1:2000 dilution) (48). After washing with PBS three times, cells were incubated with the Cy3-conjugated anti-rabbit IgG (1:400 dilution, Sigma Aldrich) for one hour at room temperature. After washing three times in PBS, coverslips were mounted with DAPI-containing VectaShield mounting medium (Vector Labs) and imaged using an inverted fluorescence microscope (Olympus IX71) equipped with a cooled CCD camera (Hamamatsu, Japan) and a PlanApo N 60× 1.42 NA oil lens. Images were acquired with the Slidebook5 software.
RESULTS
MMS-induced DNA damage inhibits metaphase-to-anaphase transition in T. brucei
We investigated whether DNA damage exerts any effect on cell cycle progression in T. brucei. Treatment of the procyclic form of T. brucei cells with various concentrations of DNA damage-inducing agent, Methyl methanesulfonate (MMS), resulted in moderate to severe growth defects (Figure 1A). Given the toxicity of higher concentrations of MMS to T. brucei cells, we used 0.0003% (v/v) of MMS for cell cycle analysis. At this concentration, MMS treatment caused a significant increase of cells containing one nucleus and two kinetoplasts (1N2K), followed by a significant decrease of cells containing two nuclei and two kinetoplasts (2N2K) (Figure 1B). T. brucei cells at different cell cycle stages can be distinguished by the numbers of nucleus and kinetoplast, the cell's unique mitochondrial DNA complex. G1 and S-phase cells contain one nucleus and one kinetoplast (1N1K), G2 and metaphase cells contain one nucleus and two kinetoplasts (1N2K), and anaphase and telophase cells contain two nuclei and two kinetoplasts (2N2K). Given the increase of 1N2K cells and a corresponding decrease of 2N2K cells, MMS-induced DNA damage appears to inhibit cell cycle transition from metaphase to anaphase in the procyclic form of T. brucei.
Figure 1.
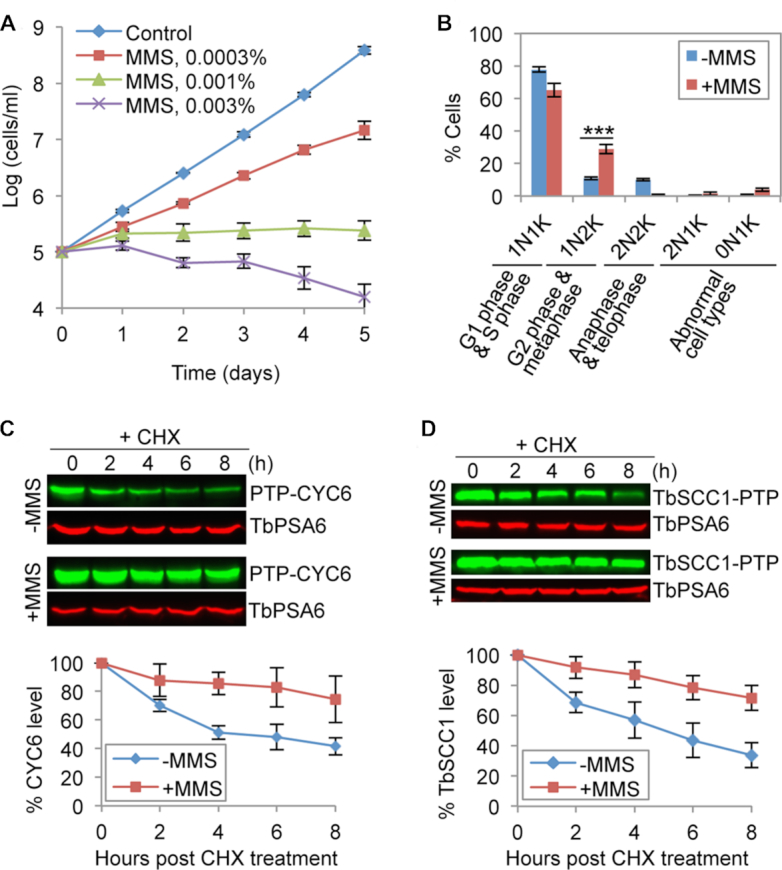
MMS-induced DNA damage inhibits anaphase onset in T. brucei. (A) MMS treatment caused growth defects in the procyclic form of T. brucei. Error bars indicate S.D. calculated from three independent experiments. (B) Analysis of cell cycle progression in control and MMS-treated cells. Cells were treated with 0.0003% (v/v) MMS, and cells were stained with DAPI for counting of cells with different numbers of nucleus (N) and kinetoplast (K). Error bars indicate S.D. from three independent experiments. ***P < 0.001. (C) Effect of MMS treatment on CYC6 stability. Shown is the cycloheximide (CHX) chase analysis. CYC6 was endogenously tagged with a PTP epitope and detected by anti-Protein A pAb. TbPSA6, the 26S proteasome α-6 subunit, was detected by anti-TbPSA6 pAb and served as a loading control. The histogram below the western blots is the quantitation of CYC6 protein band intensity, which was normalized with that of TbPSA6. Error bars represent S.D. from three independent experiments. (D) Effect of MMS treatment on TbSCC1 stability. Shown is the cycloheximide (CHX) chase analysis. TbSCC1 was endogenously tagged with a PTP epitope and detected by anti-Protein A pAb. TbPSA6 served as a loading control. The histogram shown below the western blots is the quantitation of TbSCC1 protein band intensity, which was normalized with that of TbPSA6. Error bars indicate S.D. from three independent experiments.
To understand the molecular basis of MMS-induced metaphase defect, we monitored the degradation kinetics of the mitotic B-type cyclin CYC6, since its degradation is required for metaphase-to-anaphase transition (10). Cycloheximide chase analysis showed that MMS treatment significantly slowed down CYC6 degradation, increasing its half-life from ∼4 to >8 h (Figure 1C). In response to DNA damage, yeast cells stabilize securin/Pds1, an inhibitor of separase, to delay anaphase onset (22). Since the securin/Pds1 homolog in T. brucei has not been identified (49), we examined the degradation kinetics of the cohesin subunit SCC1, which is a separase substrate and whose homolog, named TbSCC1, has been identified in T. brucei (50). Cleavage of SCC1 by separase and subsequent degradation of SCC1 by the proteasome are necessary for anaphase onset (50,51). Treatment with MMS significantly slowed down TbSCC1 degradation, causing an increase of its half-life from ∼5 to >8 h (Figure 1D). Together, these results provided the molecular evidence for the MMS-induced inhibition of anaphase onset in the procyclic form of T. brucei.
MMS-induced DNA damage modulates the abundance of the outer kinetochore protein KKIP5
The finding that MMS treatment inhibited anaphase onset in T. brucei (Figure 1) suggests a potentially novel DNA damage-induced metaphase checkpoint. In animals, DNA damage is known to trigger the SAC concurrently (18–20), thereby inhibiting the metaphase-to-anaphase transition. It is thus tempting to wonder whether MMS-induced anaphase delay is attributed to the concurrently activated SAC in T. brucei. A bona fide SAC has not been identified in T. brucei, and this parasite expresses a close homolog of Mad2 (TbMad2), but not other SAC components (30,49). In wild-type cells, TbMad2 localizes to the flagellar basal body region, but not kinetochores (30). In MMS-treated cells, TbMad2 remained at the basal body region (Supplementary Figure S2A). Remarkably, TbMad2 was not responsive to the perturbation of spindle microtubules (Supplementary Figure S2B), which is known to trigger the SAC by targeting Mad2 to kinetochores in other organisms (7), ruling out the possibility that TbMad2 plays a role in the SAC in T. brucei. Given that MCC components transiently associate with kinetochores when SAC is activated (12–14), we reasoned that certain outer kinetochore protein(s), in particular those proteins that associate with the outer kinetochore transiently, might participate in the DNA damage-induced metaphase checkpoint. Out of the seven outer kinetochore proteins identified in T. brucei, KKIP5 and KKIP7 associate with kinetochores transiently during the cell cycle (52), suggesting a potential regulatory role for KKIP5 and KKIP7.
KKIP5 was of particular interest, because it was only detectable in ∼12% of the total cell population and was expressed only in cells from S phase to metaphase (Figure 2A, B), suggesting that KKIP5 likely is not a constitutive component of the outer kinetochore and that KKIP5 protein level is under stringent control. Treatment of cells with the proteasome inhibitor MG-132 significantly increased the population of KKIP5-positive cells (Figure 2A, B). In MG-132-treated cells, KKIP5 was detectable in cells of all cell cycle stages except post-metaphase cells, because MG-132 treatment arrested the cells at metaphase and thus no anaphase and telophase (2N2K) cells were available for examination of KKIP5 localization (Figure 2C). Further, MG-132 treatment also significantly increased the level of KKIP5 protein (Figure 2D). These results demonstrated that KKIP5 expression is tightly controlled at the post-translational level.
Figure 2.

Inhibition of proteasome activity by MG-132 stabilizes KKIP5. (A) Immunofluorescence microscopy of KKIP5 localization in control and MG-132-treated cells. KKIP5 was endogenously tagged with a triple HA epitope and immunostained with FITC-conjugated anti-HA mAb. DNA (blue) was counterstained with DAPI. 1N1eK, one nucleus and one elongating kinetoplast. Scale bar: 5 μm. (B) Effect of MG-132 treatment on KKIP5 localization to kinetochores. Two hundred cells were counted for each time point. Error bars indicate S.D. from three independent experiments. ***P < 0.001. (C) Effect of MG-132 treatment on cell cycle progression. Two hundred cells were counted for each time point. Error bars indicate S.D. from three independent experiments. (D) KKIP5 protein level increased by MG-132 treatment. Endogenously 3HA-tagged KKIP5 was detected by anti-HA mAb. TbPSA6 was detected by anti-TbPSA6 pAb and served as a loading control.
We tested whether KKIP5 may respond to DNA damage. Treatment of cells with MMS significantly increased the population of KKIP5-positive cells (Figure 3A and Supplementary Figure S3, A, B) and the protein level of KKIP5 (Figure 3B), but not KKIP7 and other outer kinetochore proteins (Supplementary Figure S4). Treatment with other DNA damage agents, Ethyl methanesulfonate (EMS) and hydroxurea (HU), caused similar effects on KKIP5 (Figure 3A, B and Supplementary Figure S3B). To explore the potential response of KKIP5 to various types of DNA damages, we treated the cells with a variety of DNA damage-inducing agents, Camptothecin (CPT), 4-nitroquinoline-1-oxide (4NQO), 5-fluorouracil (5-FU), and Zeocin, which induced distinct DNA damages in eukaryotes, and these DNA damage agents also caused the increase of KKIP5-positive cells and KKIP5 protein level (Figure 3C, D). However, treatment with a variety of microtubule inhibitors did not affect KKIP5 protein level (Supplementary Figure S3C). These results suggest that KKIP5 responds to DNA damage, but not perturbations of spindle microtubules, and that KKIP5 is not a component of the SAC in T. brucei.
Figure 3.

DNA damage increases KKIP5 protein level through inhibiting KKIP5 degradation. (A) Treatment with DNA damage-inducing agents MMS, EMS and HU increased the population of KKIP5-positive cells. Two hundred cells were counted for each cell sample. Error bars represent S.D. calculated from three independent experiments. ***P < 0.001. (B) KKIP5 protein level increased in response to DNA damage agents. Endogenous KKIP5-3HA was detected by anti-HA mAb, and TbPSA6 was detected by anti-TbPSA6 pAb to serve as a loading control. (C) Treatment with a variety of DNA damage-inducing agents that cause distinct DNA damages increased the population of KKIP5-positive cells. Two hundred cells were counted for each cell sample. Error bars represent S.D. calculated from three independent experiments. **P < 0.01; ***P < 0.001. (D) KKIP5 protein level increased in response to DNA damage agents. Endogenous KKIP5-3HA was detected by anti-HA mAb, and TbPSA6 was detected by anti-TbPSA6 pAb to serve as a loading control. (E) MMS treatment stabilized KKIP5 protein. Shown is the cycloheximide (CHX) chase analysis. KKIP5 was tagged with a triple HA epitope, and detected by anti-HA antibody. TbPSA6 served as a loading control. (F) Quantitation of the KKIP5 protein level in panel E. KKIP5 protein band intensity was normalized with that of TbPSA6 and plotted against time of CHX treatment. Error bars represent S.D. from three independent experiments. (G) Co-immunofluorescence of KKIP5 with phospho-histone H2A (γH2A) and KKT2. KKIP5-3HA was detected by anti-HA mAb, and histone γH2A was detected by anti-γH2A pAb. KKIP5-PTP was detected by anti-Protein A pAb, and KKT2-3HA was detected by anti-HA mAb. Scale bar: 5 μm. (H) Quantitation of γH2A/KKIP5-positive and -negative cells in control and MMS-treated cells. γH2A+, γH2A-positive cells; γH2A-, γH2A-negative cells; KKIP5+, KKIP5-positive cells; KKIP-, KKIP5-negative cells. Two hundred cells were counted for each cell sample. Error bars represent S.D. calculated from three independent experiments. ***P < 0.001.
To test whether MMS induced KKIP5 expression at the transcriptional level, quantitative RT-PCR was performed, and we found no significant difference in KKIP5 mRNA level between control and MMS-treated cells (Supplementary Figure S3D). Since KKIP5 is regulated at the post-translational level (Figure 2), we examined whether the increase in KKIP5 protein level was due to its defective degradation by using cycloheximide chase analysis to monitor the degradation kinetics of KKIP5. In MMS-untreated control cells, KKIP5 was gradually degraded and its half-life was estimated to be ∼3 h (Figure 3E, F). In MMS-treated cells, however, degradation of KKIP5 was significantly slowed down and its half-life was increased to >8 h (Figure 3E, F). This result suggests that MMS-induced DNA damage stabilizes KKIP5 protein. To further corroborate the correlation of DNA damage with KKIP5 expression, we immunostained the cells for the co-existence of KKIP5 and γH2A, a phosphorylated histone H2A and one of the earliest markers of DNA damage in eukaryotes including T. brucei (47). Without MMS treatment, the majority (∼89%) of cells were both γH2A-negative and KKIP5-negative (Figure 3G, H). However, when treated with MMS for 16 h, the majority (∼74%) of cells were both γH2A-positive and KKIP5-positive (Figure 3G, H), confirming that expression of KKIP5 correlates with DNA damage. It is noteworthy that the KKIP5 foci did not overlap with the γH2A foci, but co-localized with the kinetochore protein KKT2 (46) (Figure 3G), indicating that KKIP5 remained on the kinetochores and did not accumulate at the sites of DNA damage.
MMS-induced stabilization of KKIP5 is independent of ATM and ATR, but requires kinetochore function
We investigated whether the response of KKIP5 to DNA damage is mediated by the ATM and ATR kinases, both of which have close homologs in T. brucei (53). To this end, we generated an ATM RNAi cell line, an ATR RNAi cell line, and an ATM-ATR double RNAi cell line, and then tagged KKIP5 with a C-terminal triple HA epitope at its endogenous locus (Figure 4A). These cell lines were induced for RNAi for 56 h and then treated with or without MMS for an additional 16 h for a total time of 72 h. In either MMS-untreated cells or MMS-treated cells, depletion of ATM, ATR, or both ATM and ATR by RNAi affected neither the protein level of KKIP5 nor the number of KKIP5-positive cells (Figure 4A, B). Taken together, these results demonstrated that the response of KKIP5 to MMS-induced DNA damage is independent of the ATM and ATR kinases.
Figure 4.

MMS-induced stabilization of KKIP5 is independent of ATM and ATR kinases. (A) MMS-induced increase in KKIP5 protein level was not affected by depletion of ATM, ATR or both ATM and ATR. KKIP5 was endogenously tagged with a triple HA epitope, whereas ATM and ATR were each tagged with a PTP epitope. TbPSA6 served as a loading control. (B) MMS-induced increase in KKIP5-positive cells was not affected by depletion of ATM, ATR, or ATM and ATR. Two hundred cells were counted for each cell sample. Error bars indicate S.D. from three independent experiments. ns, no statistical significance.
Since KKIP5 was identified as a component of the outer kinetochore (52), we asked whether its response to MMS-induced DNA damage depends on the integrity of the kinetochore structure. We knocked down the outer kinetochore protein KKIP1 (52) and the inner kinetochore protein KKT8 (46) by RNAi, and tested the effect on the response of KKIP5 to MMS. Cells were induced for RNAi for 8 or 32 h and then treated with or without MMS for an additional 16 h for a total time of 24 and 48 h, respectively. Knockdown of KKIP1 or KKT8 reduced the protein level of KKIP5 in MMS-treated cells after RNAi induction for 48 h (Figure 5A). Consistent with the reduction on KKIP5 protein level, knockdown of KKIP1 or KKT8 significantly decreased the number of KKIP5-positive cells among the MMS-treated cell population (Figure 5B). These results suggest that knockdown of KKIP1 or KKT8 may prevent KKIP5 from assembling onto the kinetochore and subsequently cause KKIP5 degradation. Indeed, a previous study showed that KKIP1 knockdown impaired KKIP5 localization to kinetochores (52), and we showed that knockdown of KKT8 also disrupted KKIP5 localization to kinetochores (Figure 5C, D). Together, these results suggest that MMS-induced stabilization of KKIP5 protein requires functional kinetochores.
Figure 5.

DNA damage-induced stabilization of KKIP5 requires functional kinetochores. (A) MMS-induced increase in KKIP5 protein level depends on KKIP1 and KKT8. KKIP5 was endogenously tagged with a triple HA epitope, whereas KKIP1 and KKT8 were each tagged with a PTP epitope. TbPSA6 served as a loading control. (B) MMS-induced increase of KKIP5-positive cells requires KKIP1 and KKT8. Two hundred cells were counted for each cell sample. –Tet, RNAi non-induction; +Tet, RNAi induction. Error bars represent S.D. from three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001. (C, D) KKT8 RNAi impaired KKIP5 localization. Shown are the immunofluorescence microscopy of KKIP5-3HA (C) and quantitation of KKIP5-3HA-positive cells in control and KKT8 RNAi cells (D). KKIP5-3HA was detected by anti-HA mAb. 200 cells were counted for each time point. Error bars represent S.D. from three independent experiments. *P < 0.05; ***P < 0.001. Scale bar: 5 μm.
MMS-induced stabilization of KKIP5 requires Aurora B kinase activity
KKIP5 was recently identified as an interacting protein of TbAUK1, the Aurora B kinase homolog in T. brucei (42), in a yeast two-hybrid screen. Directional yeast two-hybrid assay confirmed the interaction between KKIP5 and TbAUK1 (Figure 6A). Co-immunoprecipitation showed that KKIP5 and TbAUK1 interact in vivo in trypanosomes (Figure 6B), and co-immunofluorescence microscopy showed that they co-localize at kinetochores from S phase to metaphase in the ∼12% cells that express KKIP5 under normal growth conditions (Figure 6C). These results suggest that KKIP5 may be regulated by TbAUK1.
Figure 6.

DNA damage-induced stabilization of KKIP5 depends on Aurora B kinase. (A) KKIP5 interacts with TbAUK1 in yeast cells. Shown is the yeast two-hybrid experiment. TbAUK1 was expressed as a GAL4 activation domain (AD)-fusion protein, and KKIP5 was expressed as a GAL4 DNA binding domain (BD)-fusion protein in yeast cells. Yeast cells expressing TbAUK1-AD alone or KKIP5-BD alone served as controls. Yeast cells containing the empty vectors or expressing p53-AD/SV40-BD served as a negative control and a positive control, respectively, for protein-protein interactions. (B) KKIP5 interacts with TbAUK1 in trypanosome cells. Shown is the co-immunoprecipitation (co-IP) of PTP-tagged TbAUK1 and 3HA-tagged KKIP5. Cells were treated with MMS to enrich KKIP5 before co-IP. IB: immunoblotting. (C) KKIP5 co-localizes with TbAUK1 at kinetochores during S phase to metaphase. KKIP5 was tagged with a triple HA epitope, and TbAUK1 was tagged with a PTP epitope. 1N1eK, one nucleus and one elongating kinetoplast. Scale bar: 5 μm. (D) MMS-induced increase in KKIP5 protein level requires TbAUK1. KKIP5 was endogenously tagged with a triple HA epitope, and TbAUK1 was tagged with a PTP epitope. TbSA6 served as a loading control. (E) MMS-induced increase in KKIP5-positive cells depends on TbAUK1. Two hundred cells were counted for each cell sample. –Tet, TbAUK1 RNAi non-induction; +Tet, TbAUK1 RNAi induction. Error bars represent S.D. calculated from three independent experiments. ***P < 0.001. (F) KKIP5 protein stability in TbAUK1 RNAi cells treated with MMS. Shown is the cycloheximide (CHX) chase analysis. TbAUK1 RNAi was induced for 48 h. MMS was incubated for 16 h. KKIP5-3HA was detected by anti-HA mAb, and TbPSA6 was detected by anti-TbPSA6 pAb to serve as a loading control. (G) Quantitation of KKIP5 protein level in panel F. KKIP5 protein band intensity was normalized with that of TbPSA6 and plotted against time of CHX treatment. Error bars indicate S.D. from three independent experiments. (H) Effect of TbAUK1 RNAi on KKIP5 protein level. KKIP5 was endogenously tagged with a triple HA epitope, and detected by anti-HA mAb. Quantitation of KKIP5 protein band intensity was shown under the western blots. (I, J) Depletion of TbAUK1 impaired KKIP5 localization. Shown are the immunofluorescence microscopy of KKIP5-3HA (I) and quantitation of KKIP5-3HA-positive cells in control and TbAUK1 RNAi cells (J). Three hundred cells were counted for each time point. Error bars represent S.D. from three independent experiments. ***P < 0.001. Scale bar: 5 μm.
We asked whether the response of KKIP5 to MMS-induced DNA damage requires TbAUK1 function. We knocked down TbAUK1 by RNAi, and tested the effect on KKIP5 protein level in MMS-treated cells. Cells were induced for TbAUK1 RNAi for 8 or 32 h and then treated with MMS for an additional 16 h for a total time of 24 and 48 h, respectively. Depletion of TbAUK1 did not cause detectable change in KKIP5 protein level at 24 h but significantly reduced the level of KKIP5 protein at 48 h in MMS-treated cells (Figure 6D). However, immunofluorescence microscopy showed that in MMS-treated cells after TbAUK1 RNAi for 24 and 48 h, the number of KKIP5-positive cells was significantly decreased (Figure 6E). These results suggest that TbAUK1 depletion may either prevent KKIP5 from localizing to the kinetochores or remove KKIP5 from the kinetochores to the cytosol, leading to KKIP5 degradation after prolonged RNAi induction. To corroborate this finding, we tested the effect of inhibiting TbAUK1 activity on the response of KKIP5 to MMS-induced DNA damage. Cells were treated with Hesperadin, a potent inhibitor of the Aurora B kinase in a vast variety of organisms including T. brucei (54,55), for 8 h and then incubated with MMS for an additional 16 h. Treatment with Hesperadin significantly reduced the level of KKIP5 protein and the number of KKIP5-positive cells (Supplementary Figure S5). These results suggest that DNA damage-induced stabilization of KKIP5 requires Aurora B kinase activity.
To understand the underlying mechanism for TbAUK1-dependent response of KKIP5 to MMS-induced DNA damage, we carried out cycloheximide chase analysis to monitor the degradation kinetics of KKIP5 in TbAUK1 RNAi cells treated without or with MMS. Without MMS treatment, KKIP5 in TbAUK1 RNAi cells was degraded at a rate faster than in the non-RNAi cells, with its half-life reduced from ∼3 to ∼1.5 h (Figure 6F, G). With MMS treatment, faster degradation of KKIP5 was also detected in TbAUK1 RNAi cells than in the non-RNAi cells (Figure 6F, G). These findings indicated that depletion of TbAUK1 promoted KKIP5 degradation. To further confirm this notion, we monitored the level of KKIP5 protein in TbAUK1 RNAi cells, and we found that KKIP5 level was indeed gradually decreased from 24 h and beyond (Figure 6H). Immunofluorescence microscopy showed that KKIP5-positive cells were significantly decreased upon TbAUK1 depletion (Figure 6I, J). Altogether, these results suggest that TbAUK1 functions in stabilizing KKIP5.
Overexpression of KKIP5 causes metaphase arrest and mimics MMS-induced cell cycle defects
If KKIP5 mediates the DNA damage-induced metaphase checkpoint through its elevated protein level, one would assume that overexpression of KKIP5 should also inhibit anaphase onset, mimicking the effect of MMS treatment. We tested this hypothesis by overexpressing KKIP5 from an ectopic locus (Figure 7A). Overexpressed KKIP5 localized to kinetochores, as demonstrated by its co-localization with the kinetochore protein KKT2 (Figure 7B). Intriguingly, in metaphase cells, overexpressed KKIP5 additionally localized to the spindle (Figure 7B, C), as demonstrated by the co-immunostaining with the spindle marker Kif13-1 (44,45) (Figure 7C). Overexpression of KKIP5 caused a severe growth defect (Figure 7D), and analysis of the cell cycle showed a significant increase of 1N2K cells (Figure 7E), suggesting a defective anaphase onset. After 16 h of KKIP5 overexpression, anucleate (0N1K) cells were also significantly increased (Figure 7E), indicating that aberrant cytokinesis occurred in some mitotically deficient cells, as observed in other mitosis-defective cells (33–35). Flow cytometry analysis showed that after 8 hours of KKIP5 overexpression, G1 cells decreased from ∼56% to ∼23%, whereas G2/M cells increased from ∼20% to ∼45% (Figure 7F), further demonstrating that KKIP5 overexpression caused a mitotic defect. We next examined the molecular basis of the metaphase arrest induced by KKIP5 overexpression by monitoring the degradation kinetics of CYC6 and TbSCC1. Cycloheximide chase analysis showed that both CYC6 and TbSCC1 were stabilized in KKIP5 overexpression cells (Figure 7G-J), with the half-life of CYC6 increased from ∼4 to >8 h (Figure 7G, H) and the half-life of TbSCC1 increased from ∼5 to >8 h (Figure 7I, J). These results suggest that the inhibition of anaphase onset by KKIP5 overexpression was attributed, at least in part, to the failed degradation of CYC6 and TbSCC1.
Figure 7.

Overexpression of KKIP5 inhibits anaphase onset in T. brucei. (A) Overexpression (OE) of KKIP5 in procyclic trypanosomes. KKIP5 tagged with a triple HA epitope was overexpressed in a tetracycline-dependent manner in a cell line expressing a 3HA-tagged KKIP5 at the endogenous locus and was detected by anti-HA mAb. TbPSA6 served as a loading control. (B) Subcellular localization of overexpressed KKIP5-3HA in G2 and metaphase cells. KKT2, tagged with a PTP epitope, served as the kinetochore marker. The arrows indicate localization of KKIP5-3HA on the metaphase spindle. Scale bar: 5 μm. (C) Overexpressed KKIP5-3HA additionally localized to the metaphase spindle. PTP-tagged Kif13-1 served as the spindle marker. Scale bar: 5 μm. (D) KKIP5 overexpression (OE) caused a severe growth defect. –Tet, KKIP5 OE non-induced; +Tet, KKIP5 OE induced. (E) Effect of KKIP5 overexpression on cell cycle progression. Two hundred cells at each time point were counted for the number of kinetoplas (K) and nucleus (N). Error bars represent S.D. from three independent experiments. **P < 0.01. (F) Flow cytometry analysis of control and KKIP5 overexpression cells. The insets showed the percentage of cells at different cell cycle stages. (G) Effect of KKIP5 overexpression on the stability of CYC6. Shown is the cycloheximide (CHX) chase analysis. CYC6 was tagged with a PTP epitope and detected by anti-ProtA pAb. TbPSA6 served as a loading control. (H) Quantitation of CYC6 protein level in panel G. CYC6 protein band intensity was normalized with that of TbPSA6. Error bars represent S.D. from three independent experiments. (I) Effect of KKIP5 overepxression on the stability of TbSCC1. Shown is the cycloheximide (CHX) chase analysis. TbSCC1 was tagged with a PTP epitope, and detected by anti-ProtA pAb. TbPSA6 served as a loading control. (J) Quantitation of TbSCC1 protein level in panel I. TbSCC1 protein band intensity was normalized with that of TbPSA6. Error bars represent S.D. from three independent experiments.
Depletion of KKIP5 causes chromosome mis-segregation and aneuploidy, and alleviates the MMS-induced metaphase arrest
The findings that KKIP5 was only detectable in ∼12% of the total cell population and responded to DNA damage (Figure 3) suggest that KKIP5 might function as an effector of the DDR instead of a bona fide component of the outer kinetochore. In this scenario, we postulated that depletion of KKIP5 would alleviate the DNA damage-induced inhibition of anaphase onset and cause pre-mature chromosome segregation (or chromosome mis-segregation) and aneuploidy through bypassing the metaphase checkpoint. To test this hypothesis, we generated a procyclic-form KKIP5 RNAi cell line, in which KKIP5 was efficiently depleted after 24 h of RNAi induction (Figure 8A). Unlike in the bloodstream form (52), however, knockdown of KKIP5 in the procyclic form caused a moderate growth defect (Figure 8B). Quantitation of cells at different cell cycle stages showed that the G1/S (1N1K) cells decreased by ∼8%, whereas the abnormal 0N1K cells and 2N1K cells increased to ∼12% of the cell population (Figure 8C), indicating that KKIP5 depletion interfered with cell cycle progression in a small cell population. The anaphase and telophase (2N2K) cells were slightly decreased by ∼3% (Figure 8C), but ∼13% of the 2N2K cells underwent unequal nuclear division (Figure 8D), producing cells with a big nucleus and a small nucleus (Figure 8E, F). Formation of a mitotic spindle, which was labeled with the spindle marker Kif13-1, was not affected in these 2N2K cells (Figure 8E). Moreover, the anaphase cells containing lagging kinetochores, which were marked by KKT2, were increased to ∼50% of the total anaphase cell population (Figure 8F, G). These results suggest that depletion of KKIP5 caused chromosome mis-segregation and aneuploidy. Intriguingly, knockdown of KKIP5 caused a faster rate of TbSCC1 degradation (Figure 8H), opposite to the slower rate of TbSCC1 degradation by KKIP5 overexpression (Figure 7I, J), suggesting that modulation of KKIP5 protein level impacts TbSCC1 stability and, hence, chromosome segregation.
Figure 8.

Depletion of KKIP5 causes chromosome mis-segregation and aneuploidy. (A) RNAi-mediated depletion of KKIP5 in procyclic trypanosomes. KKIP5-3HA was detected by anti-HA mAb. TbPSA6 served as a loading control. (B) KKIP5 depletion caused a growth defect.-Tet, KKIP5 RNAi non-induced; +Tet, KKIP5 RNAi induced. (C) Effect of KKIP5 depletion on cell cycle progression. Cells with different numbers of nucleus (N) and kinetoplast DNA (K) were counted (200 cells for each time point). Error bars indicate S.D. from three independent experiments. (D) KKIP5 depletion resulted in unequal nuclear division. Shown are the percentages of 2N2K cells undergoing equal and unequal nuclear division in control and KKIP5 RNAi cells, based on the results in panel E. Two hundred cells were counted for each time point. Error bars represent S.D. from three independent experiments. (E) KKIP5 depletion caused unequal nuclear division. Shown are a non-induced control cell undergoing equal nuclear division and a KKIP5 RNAi cell undergoing unequal nuclear division. PTP-tagged Kif13-1 served as the spindle marker. sN, small nucleus; bN, big nucleus. Scale bar: 5 μm. (F) KKIP5 depletion caused defective chromosome segregation. Shown are a control cell with well-segregated kinetochores and two KKIP5 RNAi cells with either unequal kinetochore segregation or lagging kinetochores. PTP-tagged KKT2 served as the kinetochore marker. The open arrowheads indicate lagging chromosomes, whereas the solid arrowheads indicate the lagging kinetochores. Lagging chromosomes/kinetochores are defined as those chromosomes/kinetochores that are distributed between the two segregating nuclei. Scale bar: 5 μm. (G) KKIP5 depletion caused defective chromosome segregation. Shown are the percentages of anaphase cells with lagging kinetochores in control and KKIP5 RNAi cells, based on the results in panel G. Two hundred cells were counted for each cell sample. Error bars represent S.D. from three independent experiments. ***P < 0.001. (H) Effect of KKIP5 RNAi on the stability of TbSCC1 in MMS-treated cells and MMS non-treated cells. Shown is the cycloheximide (CHX) chase analysis. TbSCC1 was tagged with a PTP epitope and detected by anti-ProtA pAb. TbPSA6 served as a loading control. The histogram below the western blots shows the quantitation of TbSCC1 protein level. TbSCC1 band intensity was normalized with that of TbPSA6. Error bars indicate S.D. from three independent experiments.
As the expression of KKIP5 responds to DNA damage, we tested whether knockdown of KKIP5 sensitizes the cells to DNA damage. Treatment of KKIP5 RNAi cells with MMS caused more severe growth defects than KKIP5 RNAi alone or MMS treatment alone (Figure 9A), demonstrating that KKIP5 is required for cells to survive in the presence of MMS. To investigate whether KKIP5 participates in DNA damage repair, we examined the expression of γH2A and Rad51, two markers of DNA damage (47,48). We found that the cells containing γH2A and Rad51 foci upon KKIP5 RNAi were not significantly different from that in the non-induced control cells, regardless of MMS treatment or non-treatment (Figure 9B). Further, we found that the KKIP5 RNAi cells had similar survival rate as the non-induced control cells (Figure 9C). These results suggest that KKIP5 likely does not participate in the repair of DNA damage. They also suggest that the sensitivity of KKIP5 RNAi cells to MMS is not attributed to the failure in DNA damage repair.
Figure 9.

Knockdown of KKIP5 sensitizes cells to DNA damage and alleviates DNA damage-induced metaphase arrest. (A) KKIP5 depletion sensitized cells to MMS-induced DNA damage. Shown are the growth curves of KKIP5 RNAi non-induced (-Tet) and KKIP5 RNAi-induced (+Tet) cells treated without MMS (–MMS) or with MMS (+MMS). Error bars indicate S.D. from three independent experiments. (B). Effect of KKIP5 knockdown on γH2A and Rad51 foci in cells treated without or with MMS. Shown is the quantitation of cells with nuclear γH2A foci or Rad51 foci. Error bars indicate S.D. from three independent experiments. -Tet, KKIP5 RNAi non-induced; +Tet, KKIP5 RNAi induced; -MMS, no MMS treatment; +MMS, MMS treatment. ns, no statistical significance. (C) Analysis of cell survival rate of non-induced control cells and KKIP5 RNAi cells (induced for 3 days) after MMS wash-off at different time (days). Error bars indicate S.D. from three independent experiments. ns, no statistical significance. (D) Effect of KKIP5 depletion on the MMS-induced metaphase arrest. Shown are the percentages of cells with different numbers of nucleus (N) and kinetoplast DNA (K). Two hundred cells were counted for each cell sample. Error bars represent S.D. from three independent experiments. –Tet, KKIP5 RNAi non-induced; +Tet, KKIP5 RNAi induced. **P < 0.01; ***P < 0.001.
Finally, we examined whether KKIP5 knockdown would relief the MMS-induced metaphase arrest. Treatment of the control cells with MMS caused a significant increase of 1N2K cells (Figure 9D), but depletion of KKIP5 in MMS-treated cells significantly reduced the number of the 1N2K cells (Figure 9D), suggesting that KKIP5 depletion alleviated the DNA damage-induced metaphase arrest. Notably, anucleate (0N1K) cells were significantly increased in MMS-treated KKIP5 RNAi cells (Figure 9D), indicating that depletion of KKIP5 in MMS-treated cells exerted greater effects on aneuploidy than the depletion of KKIP5 in non-MMS-treated cells and MMS-treated control cells.
DISCUSSION
Eukaryotic cells respond to DNA damage, which either occurs naturally or is caused by environmental agents, by arresting the cell cycle through activating the DNA damage-induced cell cycle checkpoints. These checkpoints are, mechanistically, signal transduction pathways involving the ATM and ATR kinases, and are evolutionarily conserved from yeast to humans (26). In this study, we discovered in the procyclic form of T. brucei a kinetochore-based metaphase checkpoint that responds to DNA damage by modulating the protein abundance of KKIP5, a kinetoplastid-specific outer kinetochore protein (52). KKIP5 may play dual roles in regulating chromosome segregation by serving as a component of the outer kinetochore and an effector of the DNA damage-induced metaphase checkpoint. This hypothesis was supported by the following experimental results. (i) Knockdown of KKIP5 caused chromosome mis-segregation in cells that were not exposed to DNA damage-inducing agents (Figure 8); (ii) KKIP5 expression responded to DNA damage (Figures 3–5); (iii) depletion of KKIP5 relieved DNA damage-induced metaphase arrest (Figure 9D). However, the finding that KKIP5 was only detectable in a small cell population (Figure 2) and that KKIP5 deficiency only caused a moderate defect on chromosome segregation (Figure 8C–G) suggests that its role as an outer kinetochore subunit is rather limited. As a support of this notion, knockdown of KKIP5 in the bloodstream form of T. brucei exerted little effect on cell growth (52). Further, it should be noted that expression of KKIP5 coincides with the induction of γH2A upon MMS treatment (Figure 3G, H), and that the percentage (12.2 ± 3.1%) of cells that contain γH2A foci and Rad51 foci almost equals to the percentage (11.6 ± 1.2%) of KKIP5-positive cells in the wild-type population (Figures 3A and 9B ). Thus, an alternative hypothesis could be that KKIP5 has a sole function in mediating the metaphase checkpoint in response to MMS-induced DNA damage. In this scenario, the small population of wild-type cells that express KKIP5 may contain naturally occurred DNA damage and may have activated the KKIP5-mediated metaphase checkpoint to allow these cells to repair the DNA damage. When KKIP5 was depleted, cells proceeded to anaphase and cytokinesis with their genome carrying the naturally occurred DNA damage, thus leading to aneuploidy due to chromosome mis-segregation, as observed in KKIP5 RNAi cells (Figure 8D–G). Nevertheless, no matter whether KKIP5 has dual functions or a sole function, these results demonstrated an essential involvement of KKIP5 in DNA damage-induced metaphase checkpoint.
The increase of KKIP5 protein level in response to DNA damage was attributed to its defective degradation (Figure 3E, F), indicating that DNA damage exerted an inhibitory effect on KKIP5 degradation. Although we have little information to predict the underlying mechanisms, we can conjecture about the role of DNA damage in regulating KKIP5 stability. The finding that KKIP5 was not detectable after metaphase in wild-type cells (Figure 2A, B) suggests that KKIP5 is either degraded or spread into the cytosol and thus becomes undetectable during the metaphase-anaphase transition. Given that KKIP5 is a short-lived protein (Figure 3E, F) and inhibition of proteasome activity by MG-132 stabilizes KKIP5 on kinetochores (Figure 2), we favor the hypothesis that KKIP5 is degraded after metaphase. Likewise, TbAUK1 also stabilizes KKIP5 by inhibiting its degradation (Figure 6F, G) through unknown mechanisms. Since TbAUK1 associates with and co-localizes with KKIP5 at kinetochores (Figure 6B, C), TbAUK1 may promote the interaction of KKIP5 with other outer kinetochore protein(s) or the assembly of KKIP5 onto the outer kinetochore, thereby stabilizing KKIP5. This prediction of TbAUK1 function at the kinetochore is in line with the role of the Aurora B kinase homologs in humans (56,57) and yeast (58). Another possibility could be that TbAUK1 phosphorylates KKIP5, which stabilizes KKIP5. Future work will be directed to understand the mechanism underlying DNA damage-induced KKIP5 stabilization and the mechanistic role of TbAUK1 in regulating KKIP5 stability.
The finding that MMS-induced KKIP5 expression is independent of ATM and ATR (Figure 4) is surprising, given the essential involvement of the two protein kinases in DNA-damage-induced cell cycle checkpoints in yeast and animals (3). However, this result suggests that the MMS-induced KKIP5-mediated metaphase checkpoint operates in an ATM/ATR-independent manner, and it does not rule out the possibility that ATM and ATR may still be involved in other DNA damage-induced cell cycle checkpoints in trypanosomes. It should also be noted that MMS induces heat-labile DNA damages but not double-strand DNA breaks (DSBs) in yeast, and these damages appear to be repaired by the base excision repair pathway (59). This may explain why the MMS-induced DNA damage response is not ATM/ATR dependent. In mouse oocytes, DNA damage also induces a metaphase arrest in a kinetochore-dependent, but ATM/ATR-independent manner (21). However, this DNA damage-induced metaphase checkpoint is mediated by the SAC (21), whereas KKIP5 is irresponsive to the perturbation of spindle microtubules (Supplementary Figure S3C), suggesting a distinct mechanism in trypanosomes.
The attenuation of DNA damage-induced KKIP5 response by depletion of KKIP1 and KKT8 (Figure 5A, B) demonstrated the requirement of functional kinetochores for the KKIP5-mediated DNA damage-induced metaphase checkpoint. Further, the finding that KKIP5 localization to kinetochores requires KKIP1 (52) and KKT8 (Figure 5C, D) suggests that KKIP5 may need to be assembled into kinetochores, through a process that may be regulated by TbAUK1 (see above), in order to maintain its stability. DNA damage can trigger the SAC by targeting the SAC components to kinetochores (21,60), which also requires functional kinetochores. However, KKIP5 is unlikely to function as a SAC component or a downstream factor of the SAC, due largely to its lack of response to the perturbation of spindle microtubules (Supplementary Figure S3C). Alternatively, KKIP5 may act as an upstream factor of the SAC in mediating the DDR for targeting the SAC components to kinetochores. However, whether T. brucei has a SAC is still in doubt (10,35). If there is no SAC, it is unclear how T. brucei maintains genomic stability when kinetochore-microtubule attachment errors occur. If there is a SAC in T. brucei, it is necessary to identify its components and investigate whether and how KKIP5 may regulate the SAC in response to DNA damage.
The induction of a metaphase arrest by overexpression of KKIP5 is reminiscent of the effect caused by overexpression of Mad2 in fission yeast (61). While Mad2 overexpression causes continuous inactivation of APC/C by sequestering the APC/C co-activator Cdc20 (62), the immediate target(s) of inhibition for KKIP5 overexpression remains unknown, except the inhibited degradation of CYC6 and TbSCC1 (Figure 7G–J). Since CYC6 is a substrate of APC/C (63) and APC/C is required to activate separase for the latter to cleave SCC1 (64), it is possible that the inhibited degradation of CYC6 and TbSCC1 caused by KKIP5 overexpression is attributed to APC/C inactivation. Inactivation of APC/C requires sequestration of Cdc20 through binding to Mad2 (7). However, the APC/C purified from T. brucei does not contain Cdc20 (63), and TbMad2 does not participate in the SAC (Supplementary Figure S2B), indicating a distinct mechanism for APC/C activation and inactivation in T. brucei. Overexpression of KKIP5 also mimics MMS treatment, as both block anaphase onset through inhibiting the degradation of CYC6 and TbSCC1 (compare Figures 1C, D and 7G–J). Given that MMS treatment increases KKIP5 protein level, it is tempting to speculate that MMS-induced DNA damage may inactivate the APC/C via KKIP5.
The alleviation of the DNA damage-induced metaphase arrest by knockdown of KKIP5 (Figure 9D) is in line with the function of KKIP5 in mediating the DNA damage-induced metaphase checkpoint. In principle, depletion of KKIP5 will inactivate the metaphase checkpoint, which allows cells to proceed to anaphase without repairing damaged DNA, thus inducing chromosome mis-segregation and aneuploidy, as is evidenced by the generation of significantly more anucleate (0N1K) cells from MMS treatment and KKIP5 RNAi than from MMS treatment alone and from KKIP5 RNAi alone (Figure 9D). These anucleate cells are not able to survive. Moreover, the failure in repairing damaged DNA in the KKIP5-deficient cells likely will also cause cell death. These may explain why knockdown of KKIP5 in the MMS-treated cells caused slower cell growth than the MMS-treated cells (Figure 9A).
In summary, we have discovered in T. brucei an unusual DNA damage-induced metaphase checkpoint, which is mediated by the outer kinetochore protein KKIP5 in an Aurora B kinase-dependent, but ATM/ATR-independent manner. DNA damage induces KKIP5 stabilization by inhibiting its degradation and, consequently, leads to a significant elevation of KKIP5 protein level, which requires the activity of the Aurora B kinase homolog TbAUK1 and the function of kinetochore proteins KKT8 and KKIP1 (Figure 10). We speculate that the massive accumulation of KKIP5 protein inhibits APC/C activity through unknown mechanisms, which results in failed degradation of CYC6, thereby inhibiting the metaphase-to-anaphase transition (Figure 10). Inactivation of APC/C also leads to failed degradation of TbSCC1, likely through failed activation of separase, thereby preventing sister chromatid separation and anaphase onset (Figure 10). This KKIP5-mediated metaphase checkpoint, which requires functional kinetochores (Figure 5), but is independent of ATM and ATR (Figure 4) and is irresponsive to spindle perturbations (Supplementary Figure S3C), differs from the DNA damage-induced concurrent SAC activation, which operates in a manner that is either ATM/ATR-dependent but kinetochore-independent (18–20) or kinetochore-dependent but ATM/ATR-independent (21). Finally, the most unusual feature in this KKIP5-mediated metaphase checkpoint is the modulation of KKIP5 protein stability in response to DNA damage, as similar control mechanisms have not been discovered in any other organisms. T. brucei thus employs a novel kinetochore-based, DNA damage-induced metaphase checkpoint to maintain genomic integrity.
Figure 10.
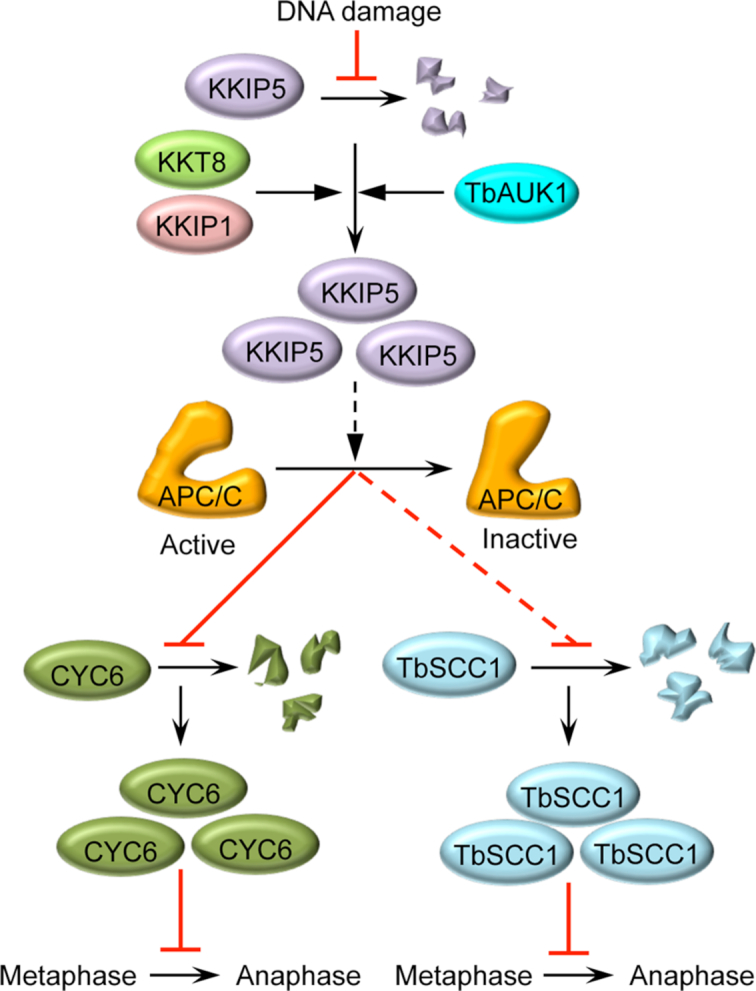
A model for the KKIP5-mediated DNA damage-induced metaphase checkpoint in the early divergent trypanosomes. DNA damage inhibits KKIP5 degradation, leading to TbAUK1- and kinetochore-dependent accumulation of KKIP5 protein at kinetochores, where KKIP5 is speculated to inhibit APC/C activity through unknown mechanisms. Inactivation of the APC/C leads to failed degradation of the mitotic cyclin CYC6, a known APC/C substrate whose degradation is required for anaphase onset, thus inhibiting the metaphase-to-anaphase transition. Inactivation of the APC/C also leads to failed degradation of the cohesin subunit TbSCC1, a substrate of the protease separase and a further downstream target of the APC/C whose degradation is required for sister chromatid separation and anaphase onset, thus inhibiting the metaphase-to-anaphase transition. Solid lines indicate the confirmed mode of actions, whereas the dashed lines indicate speculated (KKIP5 to APC/C) or indirect (APC/C to TbSCC1) mode of actions. Black lines with a solid arrow indicate positive regulation, whereas red lines indicate inhibition.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Dr David Horn of University of Dundee and Dr Richard McCulloch of University of Glasgow for providing the anti-γH2A polyclonal antibody and the anti-Rad51 polyclonal antibody, respectively. We also thank Dr Nayun Kim of the University of Texas Health Science Center at Houston for providing the DNA damage-inducing agents CPT, 4NQO, 5-FU and Zeocin.
Authors contributions: Q.Z. and Z.L. conceived and designed the project; Q.Z., K.T.M.P, H.H. and Y.K. performed experiments; Q.Z., K.T.M.P., H.H., Y.K. and Z.L. analyzed data; Z.L. wrote the original manuscript; Q.Z. and Z.L. reviewed and revised the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R01 grants AI101437 and AI118736 to Z.L.]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Ciccia A., Elledge S.J.. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010; 40:179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sirbu B.M., Cortez D.. DNA damage response: three levels of DNA repair regulation. Cold Spring Harb. Perspect. Biol. 2013; 5:a012724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shaltiel I.A., Krenning L., Bruinsma W., Medema R.H.. The same, only different - DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 2015; 128:607–620. [DOI] [PubMed] [Google Scholar]
- 4. O’Connell M.J., Walworth N.C., Carr A.M.. The G2-phase DNA-damage checkpoint. Trends Cell Biol. 2000; 10:296–303. [DOI] [PubMed] [Google Scholar]
- 5. Pines J. Cubism and the cell cycle: the many faces of the APC/C. Nat. Rev. Mol. Cell Biol. 2011; 12:427–438. [DOI] [PubMed] [Google Scholar]
- 6. Musacchio A., Salmon E.D.. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 2007; 8:379–393. [DOI] [PubMed] [Google Scholar]
- 7. Lara-Gonzalez P., Westhorpe F.G., Taylor S.S.. The spindle assembly checkpoint. Curr. Biol. 2012; 22:R966–R980. [DOI] [PubMed] [Google Scholar]
- 8. Nasmyth K., Haering C.H.. Cohesin: its roles and mechanisms. Annu. Rev. Genet. 2009; 43:525–558. [DOI] [PubMed] [Google Scholar]
- 9. Chang D.C., Xu N., Luo K.Q.. Degradation of cyclin B is required for the onset of anaphase in Mammalian cells. J. Biol. Chem. 2003; 278:37865–37873. [DOI] [PubMed] [Google Scholar]
- 10. Hayashi H., Akiyoshi B.. Degradation of cyclin B is critical for nuclear division in Trypanosoma brucei. Biol. Open. 2018; 7:bio031609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Murray A.W., Solomon M.J., Kirschner M.W.. The role of cyclin synthesis and degradation in the control of maturation promoting factor activity. Nature. 1989; 339:280–286. [DOI] [PubMed] [Google Scholar]
- 12. Yamagishi Y., Yang C.H., Tanno Y., Watanabe Y.. MPS1/Mph1 phosphorylates the kinetochore protein KNL1/Spc7 to recruit SAC components. Nat. Cell Biol. 2012; 14:746–752. [DOI] [PubMed] [Google Scholar]
- 13. Shepperd L.A., Meadows J.C., Sochaj A.M., Lancaster T.C., Zou J., Buttrick G.J., Rappsilber J., Hardwick K.G., Millar J.B.. Phosphodependent recruitment of Bub1 and Bub3 to Spc7/KNL1 by Mph1 kinase maintains the spindle checkpoint. Curr. Biol. 2012; 22:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. London N., Ceto S., Ranish J.A., Biggins S.. Phosphoregulation of Spc105 by Mps1 and PP1 regulates Bub1 localization to kinetochores. Curr. Biol. 2012; 22:900–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cheeseman I.M., Chappie J.S., Wilson-Kubalek E.M., Desai A.. The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell. 2006; 127:983–997. [DOI] [PubMed] [Google Scholar]
- 16. Krenn V., Musacchio A.. The Aurora B kinase in chromosome Bi-Orientation and spindle checkpoint signaling. Front. Oncol. 2015; 5:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lawrence K.S., Engebrecht J.. The spindle assembly checkpoint: More than just keeping track of the spindle. Trends Cell Mol. Biol. 2015; 10:141–150. [PMC free article] [PubMed] [Google Scholar]
- 18. Kim E.M., Burke D.J.. DNA damage activates the SAC in an ATM/ATR-dependent manner, independently of the kinetochore. PLos Genet. 2008; 4:e1000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dotiwala F., Harrison J.C., Jain S., Sugawara N., Haber J.E.. Mad2 prolongs DNA damage checkpoint arrest caused by a double-strand break via a centromere-dependent mechanism. Curr. Biol. 2010; 20:328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Magiera M.M., Gueydon E., Schwob E.. DNA replication and spindle checkpoints cooperate during S phase to delay mitosis and preserve genome integrity. J. Cell Biol. 2014; 204:165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lane S.I.R., Morgan S.L., Wu T., Collins J.K., Merriman J.A., ElInati E., Turner J.M., Jones K.T.. DNA damage induces a kinetochore-based ATM/ATR-independent SAC arrest unique to the first meiotic division in mouse oocytes. Development. 2017; 144:3475–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cohen-Fix O., Koshland D.. The anaphase inhibitor of Saccharomyces cerevisiae Pds1p is a target of the DNA damage checkpoint pathway. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:14361–14366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang H., Liu D., Wang Y., Qin J., Elledge S.J.. Pds1 phosphorylation in response to DNA damage is essential for its DNA damage checkpoint function. Genes Dev. 2001; 15:1361–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cohen-Fix O., Peters J.M., Kirschner M.W., Koshland D.. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev. 1996; 10:3081–3093. [DOI] [PubMed] [Google Scholar]
- 25. Shirayama M., Toth A., Galova M., Nasmyth K.. APC(Cdc20) promotes exit from mitosis by destroying the anaphase inhibitor Pds1 and cyclin Clb5. Nature. 1999; 402:203–207. [DOI] [PubMed] [Google Scholar]
- 26. Melo J., Toczyski D.. A unified view of the DNA-damage checkpoint. Curr. Opin. Cell Biol. 2002; 14:237–245. [DOI] [PubMed] [Google Scholar]
- 27. Gillett E.S., Sorger P.K.. Tracing the pathway of spindle assembly checkpoint signaling. Dev. Cell. 2001; 1:162–164. [DOI] [PubMed] [Google Scholar]
- 28. Yoshiyama K.O., Sakaguchi K., Kimura S.. DNA damage response in plants: conserved and variable response compared to animals. Biology. 2013; 2:1338–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burki F. The eukaryotic tree of life from a global phylogenomic perspective. Cold Spring Harb. Perspect. Biol. 2014; 6:a016147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Akiyoshi B., Gull K.. Evolutionary cell biology of chromosome segregation: insights from trypanosomes. Open Biol. 2013; 3:130023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Szoor B. Trypanosomatid protein phosphatases. Mol. Biochem. Parasitol. 2010; 173:53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Parsons M., Worthey E.A., Ward P.N., Mottram J.C.. Comparative analysis of the kinomes of three pathogenic trypanosomatids: Leishmania major, Trypanosoma brucei and Trypanosoma cruzi. BMC Genomics. 2005; 6:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li Z., Wang C.C.. A PHO80-like cyclin and a B-type cyclin control the cell cycle of the procyclic form of Trypanosoma brucei. J. Biol. Chem. 2003; 278:20652–20658. [DOI] [PubMed] [Google Scholar]
- 34. Hammarton T.C., Clark J., Douglas F., Boshart M., Mottram J.C.. Stage-specific differences in cell cycle control in Trypanosoma brucei revealed by RNA interference of a mitotic cyclin. J. Biol. Chem. 2003; 278:22877–22886. [DOI] [PubMed] [Google Scholar]
- 35. Ploubidou A., Robinson D.R., Docherty R.C., Ogbadoyi E.O., Gull K.. Evidence for novel cell cycle checkpoints in trypanosomes: kinetoplast segregation and cytokinesis in the absence of mitosis. J. Cell Sci. 1999; 112:4641–4650. [DOI] [PubMed] [Google Scholar]
- 36. Wirtz E., Leal S., Ochatt C., Cross G.A.. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 1999; 99:89–101. [DOI] [PubMed] [Google Scholar]
- 37. Wang Z., Morris J.C., Drew M.E., Englund P.T.. Inhibition of Trypanosoma brucei gene expression by RNA interference using an integratable vector with opposing T7 promoters. J. Biol. Chem. 2000; 275:40174–40179. [DOI] [PubMed] [Google Scholar]
- 38. Li Z., Lee J.H., Chu F., Burlingame A.L., Gunzl A., Wang C.C.. Identification of a novel chromosomal passenger complex and its unique localization during cytokinesis in Trypanosoma brucei. PLoS One. 2008; 3:e2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhou Q., Gu J., Lun Z.R., Ayala F.J., Li Z.. Two distinct cytokinesis pathways drive trypanosome cell division initiation from opposite cell ends. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:3287–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shen S., Arhin G.K., Ullu E., Tschudi C.. In vivo epitope tagging of Trypanosoma brucei genes using a one step PCR-based strategy. Mol. Biochem. Parasitol. 2001; 113:171–173. [DOI] [PubMed] [Google Scholar]
- 41. Vazquez M.P., Mualem D., Bercovich N., Stern M.Z., Nyambega B., Barda O., Nasiga D., Gupta S.K., Michaeli S., Levin M.J.. Functional characterization and protein-protein interactions of trypanosome splicing factors U2AF35, U2AF65 and SF1. Mol. Biochem. Parasitol. 2009; 164:137–146. [DOI] [PubMed] [Google Scholar]
- 42. Tu X., Kumar P., Li Z., Wang C.C.. An aurora kinase homologue is involved in regulating both mitosis and cytokinesis in Trypanosoma brucei. J. Biol. Chem. 2006; 281:9677–9687. [DOI] [PubMed] [Google Scholar]
- 43. Li Z., Zou C.B., Yao Y., Hoyt M.A., McDonough S., Mackey Z.B., Coffino P., Wang C.C.. An easily dissociated 26 S proteasome catalyzes an essential ubiquitin-mediated protein degradation pathway in Trypanosoma brucei. J. Biol. Chem. 2002; 277:15486–15498. [DOI] [PubMed] [Google Scholar]
- 44. Chan K.Y., Matthews K.R., Ersfeld K.. Functional characterisation and drug target validation of a mitotic kinesin-13 in Trypanosoma brucei. PLoS Pathog. 2010; 6:e1001050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wickstead B., Carrington J.T., Gluenz E., Gull K.. The expanded Kinesin-13 repertoire of trypanosomes contains only one mitotic Kinesin indicating multiple extra-nuclear roles. PLoS One. 2010; 5:e15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Akiyoshi B., Gull K.. Discovery of unconventional kinetochores in kinetoplastids. Cell. 2014; 156:1247–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Glover L., Horn D.. Trypanosomal histone gammaH2A and the DNA damage response. Mol. Biochem. Parasitol. 2012; 183:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McCulloch R., Barry J.D.. A role for RAD51 and homologous recombination in Trypanosoma brucei antigenic variation. Genes Dev. 1999; 13:2875–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Han X., Li Z.. Comparative analysis of chromosome segregation in human, yeasts and trypanosome. Front. Biol. 2014; 9:472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gluenz E., Sharma R., Carrington M., Gull K.. Functional characterization of cohesin subunit SCC1 in Trypanosoma brucei and dissection of mutant phenotypes in two life cycle stages. Mol. Microbiol. 2008; 69:666–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Uhlmann F., Lottspeich F., Nasmyth K.. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature. 1999; 400:37–42. [DOI] [PubMed] [Google Scholar]
- 52. D’Archivio S., Wickstead B.. Trypanosome outer kinetochore proteins suggest conservation of chromosome segregation machinery across eukaryotes. J. Cell Biol. 2017; 216:379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stortz J.A., Serafim T.D., Alsford S., Wilkes J., Fernandez-Cortes F., Hamilton G., Briggs E., Lemgruber L., Horn D., Mottram J.C. et al.. Genome-wide and protein kinase-focused RNAi screens reveal conserved and novel damage response pathways in Trypanosoma brucei. PLoS Pathog. 2017; 13:e1006477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hauf S., Cole R.W., LaTerra S., Zimmer C., Schnapp G., Walter R., Heckel A., van Meel J., Rieder C.L., Peters J.M.. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol. 2003; 161:281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jetton N., Rothberg K.G., Hubbard J.G., Wise J., Li Y., Ball H.L., Ruben L.. The cell cycle as a therapeutic target against Trypanosoma brucei: Hesperadin inhibits Aurora kinase-1 and blocks mitotic progression in bloodstream forms. Mol. Microbiol. 2009; 72:442–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Emanuele M.J., Lan W., Jwa M., Miller S.A., Chan C.S., Stukenberg P.T.. Aurora B kinase and protein phosphatase 1 have opposing roles in modulating kinetochore assembly. J. Cell Biol. 2008; 181:241–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang Y., Wu F., Ward T., Yan F., Wu Q., Wang Z., McGlothen T., Peng W., You T., Sun M et al.. Phosphorylation of HsMis13 by kinase is essential for assembly of functional kinetochore. J. Biol. Chem. 2008; 283:26726–26736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Akiyoshi B., Nelson C.R., Biggins S.. The Aurora B kinase promotes inner and outer kinetochore interactions in budding yeast. Genetics. 2013; 194:785–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lundin C., North M., Erixon K., Walters K., Jenssen D., Goldman A.S., Helleday T.. Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res. 2005; 33:3799–3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Marangos P., Stevense M., Niaka K., Lagoudaki M., Nabti I., Jessberger R., Carroll J.. DNA damage-induced metaphase I arrest is mediated by the spindle assembly checkpoint and maternal age. Nat. Commun. 2015; 6:8706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. He X., Patterson T.E., Sazer S.. The Schizosaccharomyces pombe spindle checkpoint protein mad2p blocks anaphase and genetically interacts with the anaphase-promoting complex. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:7965–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schuyler S.C., Wu Y.F., Kuan V.J.. The Mad1-Mad2 balancing act–a damaged spindle checkpoint in chromosome instability and cancer. J. Cell Sci. 2012; 125:4197–4206. [DOI] [PubMed] [Google Scholar]
- 63. Bessat M., Knudsen G., Burlingame A.L., Wang C.C.. A minimal anaphase promoting complex/cyclosome (APC/C) in Trypanosoma brucei. PLoS One. 2013; 8:e59258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Peters J.M. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat. Rev. Mol. Cell Biol. 2006; 7:644–656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.