

Chronic active and slowly expanding/evolving lesions with smouldering inflammation are neuropathological correlates of progressive multiple sclerosis. Elliott et al. report that T1-weighted measures of chronic lesion activity predict clinically progressive multiple sclerosis, may represent a longitudinal neuroimaging correlate of smouldering demyelination and axonal loss, and are reduced by ocrelizumab.
Keywords: MS: imaging, MS: biomarkers, MS: clinical trials, neuroinflammation, white matter lesion
Abstract
Chronic active and slowly expanding lesions with smouldering inflammation are neuropathological correlates of progressive multiple sclerosis pathology. T1 hypointense volume and signal intensity on T1-weighted MRI reflect brain tissue damage that may develop within newly formed acute focal inflammatory lesions or in chronic pre-existing lesions without signs of acute inflammation. Using a recently developed method to identify slowly expanding/evolving lesions in vivo from longitudinal conventional T2- and T1-weighted brain MRI scans, we measured the relative amount of chronic lesion activity as measured by change in T1 volume and intensity within slowly expanding/evolving lesions and non-slowly expanding/evolving lesion areas of baseline pre-existing T2 lesions, and assessed the effect of ocrelizumab on this outcome in patients with primary progressive multiple sclerosis participating in the phase III, randomized, placebo-controlled, double-blind ORATORIO study (n = 732, NCT01194570). We also assessed the predictive value of T1-weighted measures of chronic lesion activity for clinical multiple sclerosis progression as reflected by a composite disability measure including the Expanded Disability Status Scale, Timed 25-Foot Walk and 9-Hole Peg Test. We observed in this clinical trial population that most of total brain non-enhancing T1 hypointense lesion volume accumulation was derived from chronic lesion activity within pre-existing T2 lesions rather than new T2 lesion formation. There was a larger decrease in mean normalized T1 signal intensity and greater relative accumulation of T1 hypointense volume in slowly expanding/evolving lesions compared with non-slowly expanding/evolving lesions. Chronic white matter lesion activity measured by longitudinal T1 hypointense lesion volume accumulation in slowly expanding/evolving lesions and in non-slowly expanding/evolving lesion areas of pre-existing lesions predicted subsequent composite disability progression with consistent trends on all components of the composite. In contrast, whole brain volume loss and acute lesion activity measured by longitudinal T1 hypointense lesion volume accumulation in new focal T2 lesions did not predict subsequent composite disability progression in this trial at the population level. Ocrelizumab reduced longitudinal measures of chronic lesion activity such as T1 hypointense lesion volume accumulation and mean normalized T1 signal intensity decrease both within regions of pre-existing T2 lesions identified as slowly expanding/evolving and in non-slowly expanding/evolving lesions. Using conventional brain MRI, T1-weighted intensity-based measures of chronic white matter lesion activity predict clinical progression in primary progressive multiple sclerosis and may qualify as a longitudinal in vivo neuroimaging correlate of smouldering demyelination and axonal loss in chronic active lesions due to CNS-resident inflammation and/or secondary neurodegeneration across the multiple sclerosis disease continuum.
Introduction
While acute multiple sclerosis plaques predominate in patients with early relapsing multiple sclerosis and are the likely substrate of clinical attacks, chronic active or smouldering plaques are more prominent in patients with progressive multiple sclerosis (Kutzelnigg et al., 2005; Frischer et al., 2015) and may expand as a result of sustained inflammatory processes driven by a rim of iron-laden microglia/macrophages (Kutzelnigg et al., 2005; Frischer et al., 2009, 2015; Bramow et al., 2010; Correale et al., 2017).
Pathological studies have shown that smouldering demyelination occurs in a similar extent in primary progressive multiple sclerosis (PPMS) and secondary progressive multiple sclerosis (SPMS), and is associated with incomplete remyelination resulting in irreparable myelin loss (Bramow et al., 2010). In vivo, analysis of the natural history of new multiple sclerosis lesions using 7-T MRI has shown that a persistent phase rim (which may reflect both smouldering inflammation and the presence of iron-laden microglia/macrophages) predicts poor tissue outcome with lower quantitative T1 signal intensity over time (Absinta et al., 2016a, b, 2018). These observations are consistent with the concept of slowly expanding demyelination as a pathological correlate of clinically progressive multiple sclerosis (Prineas et al., 2001).
Recently, we developed a method for automatic detection of chronic active or slowly expanding/evolving lesions (SELs) on conventional brain MRI as a potential read-out of ‘smouldering’ or chronic active plaques (Elliott et al., 2018a, b). We defined SELs as contiguous regions of pre-existing T2 lesions that show local constant and concentric expansion as assessed by the Jacobian determinant of the non-linear deformation between reference and follow-up scans. SELs were shown to be devoid of T1 gadolinium (Gd)-enhancement, had a lower mean T1 signal intensity at baseline and showed a more severe pattern of further progressive decrease in T1 intensity over time, as compared with non-SEL areas of pre-existing lesions (Elliott et al., 2018a, b). The constant decrease in T1 signal intensity of SELs was consistent with the expected T1-weighted MRI behaviour of smouldering plaques, as the core of such lesions is typically characterized by severe accumulation of axonal damage (Kornek et al., 2000; Frischer et al., 2009). Thus, SELs may qualify as a potential read-out for progressive accumulation of irreversible neural tissue damage and especially axonal loss (van Walderveen et al., 1998; Filippi et al., 2012). These observations are also consistent with pathological and proton magnetic resonance spectroscopy studies showing that the magnitude of tissue destruction and, in particular, axonal loss, was reflected by the decrease in T1-weighted signal intensity within multiple sclerosis lesions (van Waesberghe et al., 1999; van Walderveen et al., 1999; Filippi et al., 2012). Several observational cohort studies suggested that cross-sectional multiple sclerosis lesion burden and longitudinal change in overall lesion counts and volume may predict long-term disability accumulation in patients with PPMS (Sastre-Garriga et al., 2005; Khaleeli et al., 2008; Rocca et al., 2017). However, those studies relied on relatively limited sample sizes and did not address the underlying role of chronic multiple sclerosis lesion activity.
Here we examined the respective contributions of new acute lesion formation versus chronic lesion activity to the accrual of T1 hypointense lesion volume, in the PPMS study population of the ORATORIO trial. We assessed the effect of ocrelizumab versus placebo on the accrual of T1 hypointense lesion volume related to chronic lesion activity. Finally, we evaluated whether T1-weighted conventional brain MRI measures of chronic lesion activity predicted disability outcomes along the natural history of PPMS disease course in the placebo arm from ORATORIO.
Materials and methods
Trial design, clinical endpoints and MRI acquisition specifications
All analyses were performed in the phase III, randomized, placebo-controlled, double-blind, multicentre ORATORIO trial (NCT01194570). ORATORIO study details have been reported previously (Montalban et al., 2017). Key eligibility criteria included an age of 18 to 55 years, diagnosis of PPMS (2005 revised McDonald criteria) (Polman et al., 2005) and an Expanded Disability Status Scale (EDSS) score of 3.0 to 6.5 at screening. Patients (n = 732) were randomized (2:1) to receive either ocrelizumab 600 mg by intravenous infusion every 24 weeks or placebo every 24 weeks for ≥120 weeks until a prespecified number of 12-week confirmed disability progression events occurred. After the primary data-cut, patients remained on randomized treatment (controlled treatment phase) until the outcome of the trial was evaluated and patients were eligible for open-label ocrelizumab treatment (open-label extension phase). Patients who completed the controlled treatment phase received randomized treatment for at least 144 weeks and up to 240 weeks. All clinical assessments in the double-blind controlled period of ORATORIO trial were performed 12-weekly. As described in the ORATORIO primary outcome results (Montalban et al., 2017), the assessments of 12-week confirmed disability progression endpoints were performed in a time-to-first event analysis, in which disability increase had to be sustained on subsequent visits for at least 12 weeks. A composite measure of disability progression was used in primary intent for the analysis of the clinical predictive value of chronic versus acute white matter lesion activity and brain atrophy. Composite disability progression outcome includes measures of hand/arm function and ambulation speed and provides a more comprehensive capture of change in the totality of disability features (Bosma et al., 2009; Cadavid et al., 2017), especially in progressive forms of multiple sclerosis. Furthermore, composite disability progression has recently been used as a primary endpoint in randomized controlled trials in patients with PPMS (Lublin et al., 2016) and SPMS (Kapoor et al., 2018).
Standardized conventional brain MRI was performed at baseline and Weeks 24, 48 and 120 in the ORATORIO trial. Axial 3 mm T1-weighted slices (3D spoiled gradient-echo, repetition time = 28–30 ms, echo time = 5–11 ms, flip angle = 27–30) were acquired pre- and post-Gd injection (0.1 mmol/kg, 10-min post-injection delay). Axial 3 mm T2-weighted slices were acquired with 2D fast spin-echo, repetition time = 4000–6190 ms, echo time = 74–91 ms and echo train length = 7–11. The original 1 mm × 1 mm × 3 mm image resolution was resampled into a 1-mm isotropic space as a first step of the SEL Jacobian analysis detection pipeline (Nakamura et al., 2013), which was described previously (Elliott et al., 2018a, b). Both T1-weighted and T2-weighted images were used for detection of SELs. The T1-weighted signal was normalized using least trimmed squares (LTS) over time for a given patient, followed by tissue-based normalization, where 0 and 1 values represent median T1 signal intensities of normal-appearing grey and white matter, respectively.
Identification of overall T2 hyperintense lesions, T1 hypointense lesions and new focal T2 lesions
Overall, T2 lesions were identified on baseline scans using a semi-manual approach, where an initial automatic segmentation of T2 hyperintense lesions (Francis, 2004) was manually corrected by trained readers. The automatic detection of T2-hyperintense lesions was performed using a Bayesian classifier that provides probabilistic tissue classification at each voxel based on multi-sequence MRI intensities and on atlas-derived prior probabilities of healthy tissue classes (white matter, grey matter and CSF) and T2-hyperintense lesion, based on spatial location in a standard template [International Consortium for Brain Mapping (ICBM)] space (Francis, 2004). T1 hypointense lesions were identified as the subset of T2 hyperintense lesions that (i) were more T1 hypointense than median grey matter based on voxel-level analysis of absolute T1 signal intensity; and (ii) did not show Gd-enhancement on a post-contrast T1-weighted scan. New or enlarging T2 lesions were identified based on differences in T2 lesion segmentations at reference and follow-up time points, with post-processing to remove spurious difference due to segmentation variability and misregistration, and on subsequent manual review by trained readers. New or enlarging T2 lesion detection provides a marker for acute focal inflammation in multiple sclerosis. The algorithm used to detect ‘enlargement’ in ‘new or enlarging’ T2 lesion segmentation was designed to detect new foci of activity confluent with pre-existing T2-lesion, as well as obvious concentric lesion growth during the acute stage of new lesion formation. More subtle and gradual chronic enlargement/evolution within pre-existing T2 lesions was intentionally not captured.
Identification of slowly expanding/evolving lesions
The method for the identification of SELs using the T1-weighted and T2-weighted images simultaneously has been described previously (Elliott et al., 2018a, b). Briefly, SELs are identified (based on MRI scans obtained at baseline and at post-baseline Weeks 24, 48 and 120) as areas of pre-existing T2 lesions of at least 10 contiguous voxels (voxel size is ∼3 mm3) showing gradual and constant concentric expansion. Identification of SELs was carried out using a two-stage process. First, the Jacobian determinant of the non-linear deformation field is computed between the reference and follow-up scans, to quantify subtle and gradual change in pre-existing T2 lesions (Nakamura et al., 2013). Contiguous regions of the baseline pre-existing T2 lesions undergoing local expansion are identified as SEL candidates, which are subsequently heuristically scored, to favour those undergoing concentric and constant change, consistent with gradual inside-out radial expansion. Results pertaining to the analyses of SELs will be presented for all SEL candidates (regions of pre-existing T2 lesion undergoing local expansion, regardless of heuristic score; ‘SELs’), and high-confidence SELs (with a heuristic score ≥0; ‘high-confidence SELs’) (Elliott et al., 2018a, b). SEL identification and all T1-weighted measures related to SELs and non-SELs were performed by NeuroRx Research who remained blinded to all study patient-level and treatment assignment information.
Normalization of T1 signal intensity
Prior to measuring T1 intensity change over time, T1-weighted images were normalized in a two-stage process: (i) LTS was used to normalize all subsequent (in time) T1-weighted images of a given subject to the baseline T1-weighted scan; and (ii) T1-weighted images for a given subject were linearly normalized by mapping the median grey matter T1 intensity at baseline to a value of zero, and mapping the median normal-appearing white matter intensity at baseline to 1. LTS performs linear regression between co-registered sequential scans using the 50% of voxels whose least squares fit possesses the smallest sum of squared residuals (Rousseeuw et al., 2006). This has the effect of normalizing intensities within a given subject based only on the subset of voxels that remain relatively unchanged over time.
The first stage of normalization ensures a common intensity space across time for a given subject, while the second stage provides comparable measures of T1 intensity change across different subjects.
Statistical analysis
The statistical analysis of SEL data was exploratory and included all patients from ORATORIO with no missing or non-evaluable MRI scans (SEL analysis population). Mean changes in T1 and T2 lesion volume were calculated as power means of cube root transformed data. No imputation of missing data was performed, except for the analysis of the change of T1 hypointense volume, where for patients from the SEL analysis population who did not have any SELs, the SEL-associated T1 hypointense volume and its change from baseline were imputed as zero.
A Wilcoxon rank-sum test for continuous variables and Fisher’s exact test for categorical variables were applied to compare baseline characteristics between two treatment groups (ocrelizumab and placebo) for both the intention-to-treat (ITT) and SEL analytical populations.
Identification of SELs and the change of longitudinal predictive factors were calculated based on data from the first 120 weeks after randomization. To avoid misinterpretation in inferring the direction of causal dependence, clinical outcomes were re-baselined at Week 120 and only data after Week 120 until the end of the controlled period was used to evaluate the effect of chronic lesion activity, acute lesion activity and whole brain atrophy on disability progression outcomes. The survival analysis of predictive factors for clinical endpoints employed Cox regression models, including the predictive factors of interest, and stratified for region (rest of world, US) and age (<45 years, ≥45 years). The comparison of continuous variables by lesion sector (SELs, non-SELs, total T2 burden and new T2 lesions) was performed using the Van Elteren test, stratified for treatment group (ocrelizumab, placebo) and baseline T2 lesion volume category based on tertiles (≤3.013 cm3, <3.013 cm3 to ≤11.122 cm3, and >11.122 cm3). Longitudinal analyses of lesion counts and volumes used negative binomial regression and linear mixed models for repeated measures, respectively. Statistical tests were two-sided and conducted at the 5% significance level without adjustment for multiplicity.
Data availability
Qualified researchers may request access to individual patient level data through the clinical study data request platform (www.clinicalstudydatarequest.com). Further details on Roche’s criteria for eligible studies are available at https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Roc he.aspx. Further details on Roche’s Global Policy on the sharing of clinical information and how to request access to related clinical study documents, are available at https://www.roche.com/research_and_development/who_we_are_how_we_work/cl inical_trials/our_commitment_to_data_sharing.htm.
Results
Baseline demographics and characteristics of the ORATORIO PPMS study population
Baseline demographics and MRI characteristics were well balanced between the ITT and SEL analytical populations (Table 1). There was a minor imbalance in the mean number of T1 Gd-enhancing lesions in the ITT population between the ocrelizumab and placebo treatment groups, which is slightly larger in the SEL analytical population. Overall, T2 and T1 lesion volume was higher in the ocrelizumab-treated patients in both the ITT and SEL analytical populations, with the difference being more pronounced in the SEL analytical population. Importantly, there was no numerical imbalance between the T1/T2 lesion volume ratio between the two treatment arms. All between-group differences were non-significant in the ITT and SEL analytical populations (all P-values >0.05; Table 1).
Table 1.
Baseline demographics and disease characteristics for the ITT and SEL analytical populations of the ORATORIO trial
| Baseline demographics and disease characteristics | ITT population (n = 732) | SEL analytical population (n = 555) | ||
|---|---|---|---|---|
| Placebo (n = 244) | Ocrelizumab (n = 488) | Placebo (n = 171) | Ocrelizumab (n = 384) | |
| Age, mean (SD), years | 44.4 (8.3) | 44.7 (7.9) | 45.0 (8.1) | 44.9 (7.8) |
| Female, n (%) | 124 (50.8) | 237 (48.6) | 88 (51.5) | 188 (49.0) |
| Time since multiple sclerosis symptom onset, mean (SD), years | 6.1 (3.6)a | 6.7 (4.0)b | 5.9 (3.3)c | 6.6 (3.9)d |
| EDSS, mean (SD) | 4.7 (1.2) | 4.7 (1.2)e | 4.6 (1.2) | 4.6 (1.2)f |
| MRI | ||||
| Number of T1 Gd-enhancing lesions, mean (SD) | 0.6 (1.5)g | 1.2 (5.1)h | 0.4 (1.2) | 1.2 (5.5)f |
| Proportion of patients with ≥1 T1 Gd-enhancing lesion (%) | 24.7g | 27.5h | 21.1 | 27.2f |
| Brain T2 hyperintense lesion volume, median (range), cm3 | 6.2 (0–81.1)g | 7.3 (0–90.3)i | 5.5 (0–59.2) | 7.4 (0–82.4)f |
| Brain non-enhancing T1 hypointense lesion volume, median (range), cm3 | 1.7 (0–35.4)g | 2.0 (0–64.4)h | 1.5 (0–35.4) | 2.1 (0–53.5)f |
| T1/T2 lesion volume ratio, median (range) | 0.32g (0; 0.69) | 0.32h (0; 0.77) | 0.31 (0; 0.69) | 0.32 (0; 0.77) |
| Normalized brain volume, mean (SD), cm3 | 1469.9 (88.7)g | 1462.9 (84.0)j | 1469.2 (86.3) | 1459.3 (84.5)k |
| Cortical grey matter volume, mean (SD), cm3 | 542.1 (48.9)g | 534.9 (55.1)j | 543.7 (47.7) | 534.6 (55.7)k |
| White matter volume, mean (SD), cm3 | 786.7 (54.8)g | 787.9 (52.1)j | 784.1 (51.8) | 784.9 (50.0)k |
All between-group differences were tested non-significant (all P-values > 0.05, Wilcoxon rank-sum test for continuous data and Fisher’s exact test for categorical data). SD = standard deviation.
an = 237. bn = 474. cn = 165. dn = 373. en = 487. fn = 383. gn = 243. hn = 484. in = 486. jn = 482. kn = 379.
Increase in non-enhancing T1 hypointense lesion volume occurs mostly within pre-existing T2 lesions
An increase in non-enhancing T1 hypointense lesion volume was seen in both the ocrelizumab arm and the placebo arm, with a reduced T1 lesion volume increase from baseline to Week 120 with ocrelizumab as compared with placebo (P < 0.001) (Fig. 1A). In the ocrelizumab group, we observed a continued increase in non-enhancing T1 hypointense lesion volume from baseline to Week 120 (Fig. 1A), despite a decrease in T2 lesion volume (Fig. 1B) and near absence of new focal T2 lesion formation (Fig. 1C). In the placebo group, T2 lesion volume increased from baseline to Week 120 (Fig. 1B), and although there was continued formation of new T2 lesions, the overall absolute increase in non-enhancing T1 hypointense lesion volume was larger by more than 2-fold that of T2 lesion volume from baseline to Week 120 (cf.Fig. 1A and B). These findings show that T1 hypointense lesion volume increase in the ORATORIO PPMS population was mostly driven by chronic lesion activity in T2 lesions pre-existing at baseline, with a minimal impact of new or enlarging T2 lesions in both treatment groups (Fig. 3). Chronic brain tissue damage as measured by increasing non-enhancing T1 hypointense lesion volume may largely occur independently from new T2 lesion formation, thereby reflecting chronic T1 signal intensity decrease within pre-existing baseline T2 lesions, as illustrated by the visual example shown in Fig. 1D.
Figure 1.

Chronic brain tissue damage as measured by increasing non-enhancing T1 hypointense lesion volume is reduced by ocrelizumab and may occur independently from new T2 lesion formation. (A) Change in total non-enhancing T1 lesion volumea. (B) Change in total T2 lesion volume. (C) New focal T2 lesion formation. (D) Chronic brain tissue damage mostly occurs within pre-existing T2 lesions. An animated version of D is more appropriate for data visualization and is available in the Supplementary material. OCR = ocrelizumab; T1w = T1-weighted. aThe analysis of total non-enhancing T1 lesion volume was based on a T1 hypointense lesion threshold definition (as per pre-specified ORATORIO study protocol) that was subsequently optimized for all T1 lesion volume analyses performed. Estimates and P-values are from a mixed-effect model of repeated measures using an unstructured covariance matrix. bRates and P-values are from a negative binomial model adjusted for baseline T2 lesion count, geographic region (rest of world, US) and age (≤45 years, >45 years).
Figure 3.
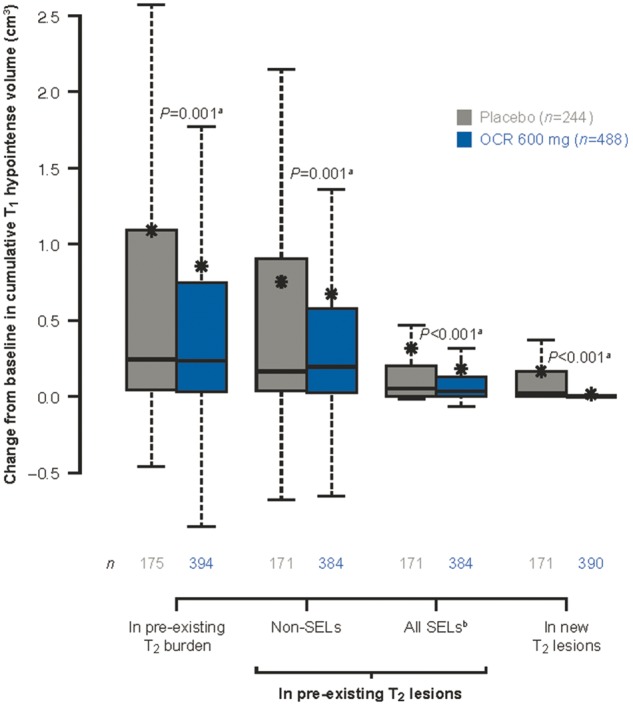
T1 hypointense lesion volume change from baseline to Week 120 by lesion type and treatment group. Box-and-whisker plots: box displays the quartiles; asterisks represent the mean values; bars represent the median values; whiskers extend from the lowest datum ≥ 1.5 × lower quartile − IQR to the highest datum ≤ 1.5 IQR upper quartile + IQR; range of values in placebo/ocrelizumab, respectively: pre-existing T2 burden: −0.46–12.90/−6.23–24.54; non-SELs: −0.68–8.71/−6.61–17.65; all SELs: −0.02–5.29/−0.07–6.89; new T2 lesions: 0–3.36/0–1.18. IQR = interquartile range; OCR = ocrelizumab. aVan Elteren test stratified by treatment group (ocrelizumab, control) and baseline T2 lesion volume category based on tertiles. n = the number of patients evaluable at Week 120. bFor each patient the sum change from baseline in T1 volume of evaluable lesions was calculated. Missing changes from baseline in volume are imputed to zero.
Chronic lesion activity measured by T1 lesion volume change in SELs and non-SEL areas
We have shown previously (Elliott et al., 2018a, b) that a proportion of total pre-existing T2 lesion volume in the ORATORIO study population could be identified as SEL candidates and high-confidence SELs, while the rest of pre-existing T2 lesions were defined as non-SEL areas. Hence, we introduced a three-compartment model to further advance the understanding of the origin of overall T1 hypointense lesion volume accumulation, which may stem either from ‘chronic’ lesion activity in (i) SELs; (ii) non-SEL areas within boundaries of pre-existing T2 lesions (Fig. 2); or (iii) from ‘acute’ lesion activity by new/enlarging T2 lesion formation outside of the baseline T2 lesion boundaries.
Figure 2.

T1 hypointense volume accumulation may occur in SELs showing longitudinal expansion and in non-SEL areas of pre-existing baseline T2 lesions. In this example, effectively all T1 hypointense lesion volume accumulation in this slice occurs within baseline T2 lesions. An animated version of this figure is more appropriate for data visualization and is available in the Supplementary material. T1w = T1-weighted; T2w = T2-weighted.
SEL candidates were identified as 5.1% and 7.1% of pre-existing T2 lesion volume in ocrelizumab- and placebo-treated patients, respectively, while high-confidence SELs (with a score ≥0) represented 2.5% and 3.4% of pre-existing T2 lesion volume for ocrelizumab and placebo patients, respectively. While accounting for a relatively small fraction of pre-existing T2 lesions, proportionally, SELs accounted for a much higher amount of T1 hypointense lesion volume accumulation from baseline to Week 120 in both the ocrelizumab and placebo populations (Fig. 3).
Longitudinal profile of T1-weighted measures of chronic lesion activity
The longitudinal by-patient (volume-normalized) analysis of change from baseline to Week 120 in normalized T1 signal intensity showed a lower T1 intensity at baseline (Supplementary Fig. 5) and a greater decrease in T1 intensity in SEL regions as compared with non-SEL regions (Fig. 4) both in ocrelizumab and placebo-treated PPMS patients. High-confidence SELs (with a score ≥0) also showed a slightly greater T1 intensity decrease than all SEL candidates (Fig. 4) in both treatment arms.
Figure 4.

T1-weighted intensity measures of chronic white matter lesion activity from baseline to Week 120. (A) Total baseline T2 lesion burden. (B) All SEL candidates. (C) SELs (with a score ≥0). (D) Non-SELs. Asterisks represent the median values. OCR = ocrelizumab. aVan Elteren test stratified by treatment group (ocrelizumab, control) and baseline T2 lesion volume category based on tertiles. n = the number of patients with at least one SEL at each visit. Volume normalization is calculated as: sum(SEL_volume × SEL_intensity) / sum(SEL_volume): SEL volume is based on baseline T2 volume of SELs. For each patient, the sum relative change from baseline in T1 lesion volume of SELs was calculated and this value was then summarized for the ITT population. bn-values at Week 120 for all panels. Reproduced from Elliott et al. (2018a).
Chronic lesion activity in pre-existing T2 lesions predicts clinical PPMS progression
Table 2 shows a comparison of the predictive value of baseline T1 lesion volume burden versus chronic lesion activity measured by longitudinal change in T1-weighted lesion volume in pre-existing lesions (SELs and non-SELs), respectively, from study baseline and from Week 120 to the end of the controlled treatment period in the placebo group of the ORATORIO trial. Baseline T1 lesion volume only predicted clinical progression at the level of upper extremity function defined by a 12-week confirmed ≥20% increase in 9-Hole Peg Test (9HPT) time [hazard ratio (95% confidence interval, CI): 1.75 (1.08, 2.84); P = 0.024] (Table 2, Supplementary Fig. 1A, G, M and S). T1 lesion volume re-baselined at Week 120 had no significant effect for any of the disability endpoints but showed a similar pattern of higher hazard ratio for 9HPT progression (data not shown). By contrast, longitudinal chronic lesion activity from baseline to Week 120 as measured by T1 lesion volume accumulation in (i) total pre-existing T2 lesions (SELs and non-SELs); (ii) all SEL candidates; (iii) SELs (with a score ≥0); and (iv) non-SELs, significantly predicted clinical progression (from Week 120 to end of study) on 12-week confirmed composite disability progression [12-week confirmed disability progression as measured by EDSS, or 12-week confirmed ≥20% increase in Timed 25-Foot Walk (T25FW) time, or 12-week confirmed ≥20% increase in 9HPT time] [hazard ratio (95% CI): (i) 2.32 (1.38,3.90), P = 0.001; (ii) 1.98 (1.20,3.28), P = 0.008; (iii) 1.87 (1.13,3.08), P = 0.015; (iv) 2.20 (1.32,3.67), P = 0.003] with consistent numerical trends on all components of the disability burden (EDSS, T25FW and 9HPT) (Table 2 and Supplementary Fig. 1). In comparison, longitudinal whole brain volume loss from baseline to Week 120 did not predict clinical progression from Week 120 to end of study on either component of the disability burden nor on composite confirmed disability progression, with only a numerical trend observed for progression of T25FW (Supplementary Fig. 2). Similarly, acute white matter lesion activity from baseline to Week 120 as measured by T1 lesion volume accumulation in new focal T2 lesions did not predict clinical progression from Week 120 to end of study on either component of the disability burden nor on composite confirmed disability progression, with only a numerical trend observed for progression of 9HPT (Table 2 and Supplementary Fig. 1).
Table 2.
Prediction of clinical progression in PPMS
| Total n | <Median | ≥Median | HRa | 95% CI | P-value | KM curve layout | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | Events | n | Events | |||||||
| Clinical progression outcomes by baseline total T1 lesion volume | ||||||||||
| 12-w composite CDP | Placebo (median = 1.734) | 243 | 121 | 90 | 122 | 90 | 0.93 | (0.69, 1.25) | 0.61 | Supplementary Fig. 1A |
| 12-w CDP | Placebo (median = 1.734) | 243 | 121 | 51 | 122 | 55 | 1.01 | (0.69, 1.49) | 0.95 | Supplementary Fig. 1G |
| 12-w 9HPT progression | Placebo (median = 1.734) | 243 | 121 | 27 | 122 | 46 | 1.75 | (1.08, 2.84) | 0.024 | Supplementary Fig. 1M |
| 12-w T25FW progression | Placebo (median = 1.734) | 243 | 121 | 76 | 122 | 73 | 0.89 | (0.64, 1.23) | 0.49 | Supplementary Fig. 1S |
| Clinical progression outcomes from study Week 120 to end of controlled period by change in T1 lesion volume from baseline to Week 120 within pre-existing T2 lesions (SELs and non-SELs) | ||||||||||
| 12-w composite CDP | In total pre-existing T2 lesions (SELs + non-SELs), cm3; placebo (median = 0.243) | 167 | 82 | 24 | 85 | 45 | 2.32 | (1.38, 3.90) | 0.001 | Supplementary Fig. 1C |
| In all SEL candidates, cm3; placebo (median = 0.052) | 167 | 82 | 26 | 85 | 43 | 1.98 | (1.20, 3.28) | 0.008 | Supplementary Fig. 1D | |
| In SELs (with a score ≥0), cm3; placebo (median = 0.017) | 167 | 82 | 26 | 85 | 43 | 1.87 | (1.13, 3.08) | 0.015 | Supplementary Fig. 1E | |
| In non-SELs, cm3; placebo (median = 0.166) | 167 | 82 | 25 | 85 | 44 | 2.20 | (1.32, 3.67) | 0.003 | Supplementary Fig. 1F | |
| 12-w CDP | In total pre-existing T2 lesions (SELs + non-SELs), cm3; placebo (median = 0.243) | 167 | 82 | 15 | 85 | 26 | 1.87 | (0.98, 3.57) | 0.058 | Supplementary Fig. 1I |
| In all SEL candidates, cm3; placebo (median = 0.052) | 167 | 82 | 15 | 85 | 26 | 2.01 | (1.06, 3.83) | 0.033 | Supplementary Fig. 1J | |
| In SELs (with a score ≥0), cm3; placebo (median = 0.017) | 167 | 82 | 14 | 85 | 27 | 2.07 | (1.07, 3.98) | 0.030 | Supplementary Fig. 1K | |
| In non-SELs, cm3; placebo (median = 0.166) | 167 | 82 | 15 | 85 | 26 | 1.87 | (0.98, 3.57) | 0.058 | Supplementary Fig. 1L | |
| 12-w 9HPT progression | In total pre-existing T2 lesions (SELs + non-SELs), cm3; placebo (median = 0.243) | 162 | 80 | 4 | 82 | 13 | 3.39 | (1.10, 10.47) | 0.034 | Supplementary Fig. 1O |
| In all SEL candidates, cm3; placebo (median = 0.052) | 162 | 80 | 5 | 82 | 12 | 2.53 | (0.88, 7.25) | 0.083 | Supplementary Fig. 1P | |
| In SELs (with a score ≥0), cm3; placebo (median = 0.017) | 162 | 80 | 7 | 82 | 10 | 1.46 | (0.55, 3.85) | 0.45 | Supplementary Fig. 1Q | |
| In non-SELs, cm3; placebo (median = 0.166) | 162 | 80 | 6 | 82 | 11 | 1.93 | (0.71, 5.24) | 0.20 | Supplementary Fig. 1R | |
| 12-w T25FW progression | In total pre-existing T2 lesions (SELs + non-SELs), cm3; placebo (median = 0.243) | 164 | 80 | 14 | 84 | 27 | 1.97 | (1.02, 3.81) | 0.044 | Supplementary Fig. 1U |
| In all SEL candidates, cm3; placebo (median = 0.052) | 164 | 80 | 17 | 84 | 24 | 1.42 | (0.75, 2.69) | 0.28 | Supplementary Fig. 1V | |
| In SELs (with a score ≥0), cm3; placebo (median = 0.017) | 164 | 80 | 16 | 84 | 25 | 1.66 | (0.87, 3.18) | 0.13 | Supplementary Fig. 1W | |
| In non-SELs, cm3; placebo (median = 0.166) | 164 | 80 | 13 | 84 | 28 | 2.26 | (1.16, 4.43) | 0.017 | Supplementary Fig. 1X | |
| Clinical progression outcomes from study Week 120 to end of controlled period by change in T1 lesion volume from baseline to Week 120 in new focal T2 lesions | ||||||||||
| 12-w composite CDP | Placebo (median = 22.89) | 167 | 82 | 30 | 85 | 39 | 1.47 | (0.89, 2.42) | 0.13 | Supplementary Fig. 1B |
| 12-w CDP | Placebo (median = 22.89) | 167 | 82 | 16 | 85 | 25 | 1.43 | (0.75, 2.71) | 0.28 | Supplementary Fig. 1H |
| 12-w 9HPT progression | Placebo (median = 22.89) | 162 | 79 | 4 | 83 | 13 | 2.68 | (0.84, 8.56) | 0.095 | Supplementary Fig. 1N |
| 12-w T25FW progression | Placebo (median = 22.89) | 164 | 80 | 20 | 84 | 21 | 0.99 | (0.52, 1.89) | 0.97 | Supplementary Fig. 1T |
Baseline T1 hypointense lesion volume burden versus longitudinal T1-weighted measures of chronic lesion activity and acute lesion activity as measured by the change in T1 hypointense lesion volume in pre-existing lesions (SELs and non-SELs) and new focal T2 lesions, respectively; analysis of the ORATORIO placebo group up until the end of the controlled period. Results are obtained from a Cox model including region (rest of world, US) and age (<45 years, ≥45 years) as stratification factors. 12-w = 12-week; CDP = confirmed disability progression; KM = Kaplan–Meier.
aHazard ratio (≥median versus <median).
It is important to emphasize that all placebo group analyses performed to evaluate the clinical predictive value of imaging phenotypes were strictly devoid of ocrelizumab influence since the clinical outcome analysis was performed from Week 120 to the end of the controlled period. A switch of all placebo-treated patients to ocrelizumab treatment only happened beyond the end of the controlled period at the beginning of the open-label extension phase, which was not integrated to the current analysis. Hence, all the findings presented are not confounded by unblinding or ocrelizumab treatment-related changes.
Accumulation of T1 hypointense lesion volume in pre-existing T2 lesions is reduced by ocrelizumab
While an increase in T1 hypointense lesion volume was seen in both the ocrelizumab and the placebo arm, T1 lesion volume increase from baseline to Week 120 was reduced with ocrelizumab compared with placebo (P < 0.001) (Fig. 1A). Accumulation of T1 hypointense volume was reduced with ocrelizumab as compared with placebo in both pre-existing T2 lesions (P = 0.001) (Fig. 3) and in acute new T2 lesions (P < 0.001) (Fig. 3). While the greatest relative effect of ocrelizumab compared with placebo was seen on T1 hypointense lesion volume increase associated with acute new T2 lesion formation (P < 0.001), proportionally, this was a minor contributor to overall T1 hypointense lesion volume accumulation (Fig. 3). If we consider SEL and non-SEL regions of pre-existing T2 lesion separately, ocrelizumab showed a reduction in T1 volume increase for both compartments of chronic lesion activity (non-SEL: P = 0.001, SEL: P < 0.001) (Fig. 3). The analysis of the treatment effect over time showed a significant separation favouring ocrelizumab versus placebo in terms of accumulation of T1 hypointense volume as of Week 48 within all SEL candidates, high-confidence SELs (with a score ≥0), and non-SELs (Supplementary Fig. 3). To confirm that the effect of ocrelizumab on SELs was irrespective of the heuristic threshold for high-confidence SELs, we performed a lesion-level analysis that showed consistent benefits of ocrelizumab with respect to T1 hypointense volume accumulation in SEL candidates above all tested thresholds of heuristic SEL score distribution (Supplementary Fig. 4).
Since T1 hypointense volume accumulation analysis, by definition, may not capture potential changes pertaining to lesion voxels maintaining a T1 intensity superior to the defined T1 hypointense threshold (median of normal-appearing grey matter), or pertaining to lesion voxels with continued decrease in T1 intensity beyond this threshold, we also assessed the effect of ocrelizumab versus placebo on chronic lesion activity as measured by the volume-normalized T1 signal intensity (mean T1 intensity of the voxels constituting a lesion). The longitudinal analysis of the absolute change from baseline to Week 120 in normalized T1 signal intensity in SELs showed a reduced decrease in T1 intensity in ocrelizumab-treated patients (ocrelizumab versus placebo: –0.20 versus –0.24, P = 0.005) (Fig. 4B). The same finding was observed in high-confidence SELs (with a score ≥0) (ocrelizumab versus placebo: –0.24 versus –0.28, P = 0.013) (Fig. 4C), and in non-SEL regions (ocrelizumab versus placebo: –0.05 versus –0.09, P = 0.003) (Fig. 4D).
In comparison with these chronic lesion activity features of SELs, the overall prevalence of SELs was not impacted greatly by ocrelizumab versus placebo. The proportion of patients with ‘all SEL candidates’ detected from baseline to Week 120 was similar in the two treatment arms (ocrelizumab versus placebo: 85.2% versus 83.6%), as was the proportion of patients with high-confidence SELs [with a score ≥0 (73.2% versus 69.0%)]. However, ocrelizumab-treated patients had a lower proportion of total pre-existing T2 lesion identified as SEL candidates (median 5.1 versus 7.1%, P < 0.001) and high-confidence SELs [with a score ≥0 (median 2.5 versus 3.4%, P = 0.044)], from baseline to Week 120.
Discussion
Our results indicate that in patients with PPMS the increase in overall T1 lesion volume burden is mostly due to chronic lesion activity leading to the accumulation of tissue damage within pre-existing lesions and that T1-weighted measures of chronic lesion activity are predictive of clinical PPMS progression. We also showed that ocrelizumab, which is highly effective in silencing acute new lesion formation in early relapsing multiple sclerosis (Hauser et al., 2017) and PPMS (Montalban et al., 2017), also reduced the relative volume of SELs and T1-weighted in vivo measures of chronic lesion activity in SELs and in non-SEL areas of pre-existing lesions in patients with PPMS.
Although smouldering inflammation with sustained macrophage/microglia activation and secondary neurodegeneration may intrinsically drive ongoing myelin/axonal damage within chronic active multiple sclerosis lesions (Frischer et al., 2015; Absinta et al., 2016a,b; Dal-Bianco et al., 2017), it is also expected that acute multiple sclerosis lesion activity could influence the CNS milieu and indirectly impact the severity of chronic lesion activity features. In the specific context of the ORATORIO trial population, as much as 73.5% of the PPMS population had MRI signs of acute multiple sclerosis lesion activity (T1 Gd-enhancing lesions and/or new focal T2 lesions) based on results from the longitudinal analysis of the placebo group from baseline to Week 120 (Wolinsky et al., 2018). Nonetheless, the ocrelizumab-mediated near complete suppression of acute multiple sclerosis lesion formation, as observed especially beyond treatment Week 48, did not prevent a continued concurrent decrease in T1 signal intensity and increase in T1 hypointense lesion volume within SEL and non-SEL regions of chronic pre-existing lesions. Those data show that chronic white matter multiple sclerosis lesion activity may evolve largely independently from acute (focal) inflammatory activity. However, the separation of T1-weighted measures of chronic lesion activity between the placebo and ocrelizumab groups was also most prominent beyond treatment Week 48. This observation suggests that the mechanism of ocrelizumab treatment effect on chronic lesion activity may be partially and indirectly explained by its potency to silence acute new lesion formation and by-product release of mediators that may participate in the activation of pro-inflammatory macrophages/microglia, the core engine of chronic active lesion pathology (Frischer et al., 2015).
This study of a large-scale PPMS clinical trial placebo dataset also showed that at the population level T1-weighted measures of chronic lesion activity predict clinical PPMS progression measured by the composite of the three major physical disability outcomes (EDSS, 9HPT and T25FW) with consistent trends on all components, while the baseline chronic lesion burden measured by total T1 lesion volume only predicted 9HPT outcome. In contrast to chronic white matter lesion activity, the measures of acute lesion activity did not predict subsequent disability progression in this PPMS clinical trial population. These results are consistent with previous retrospective analyses of observational cohorts (Sastre-Garriga et al., 2005; Khaleeli et al., 2008; Rocca et al., 2017) where the effect of acute versus chronic lesion activity was not disentangled. Importantly, longitudinal whole brain volume loss in ORATORIO did not predict composite clinical disability progression or its components, with only a numerical trend with respect to ambulation progression as measured by T25FW. ORATORIO findings further suggest that chronic lesion activity is the major driver of overall accumulation of lesion-related CNS tissue loss, and that recent chronic lesion activity may carry more clinically predictive information than recent brain atrophy or the overall baseline lesion volume, which may principally reflect disease duration and history. However, the overall cumulative burden of chronic lesion activity is obviously related to total multiple sclerosis lesion volume, and it was previously shown in another major PPMS phase III trial population that the rate of whole brain volume loss was strongly predicted by the baseline lesion volume burden but not by other factors [e.g. number of Gd-enhancing lesions at baseline (Miller et al., 2018)]. It was also shown in the latter study that a higher lesion burden predicted brain volume loss in the specific subgroup of patients with no signs of acute lesion activity at baseline (Miller et al., 2018). The value of in vivo longitudinal T1-weighted measures of chronic lesion activity in SELs and non-SELs as surrogates for clinical efficacy remains to be investigated and compared with that of measures of acute lesion activity in progressive multiple sclerosis clinical trial datasets using multivariate analysis.
A limitation of this study is that we cannot claim independence of the clinical predictive value of T1-weighted in vivo longitudinal measures of chronic lesion activity, as (apart for stratification for T2 volume) our analysis models could not include other potentially relevant predictors due to the small number of clinical events post-Week 120. The confirmation of our findings in other PPMS trial datasets will be important as false positive results cannot be excluded in our exploratory analysis that did not correct for multiple testing. Another limitation is the absence of spinal cord evaluation by MRI, which was not part of the acquisition protocol in this study. The entire dynamic of multiple sclerosis lesion formation and natural history, as well as the potential involvement of smouldering or slowly expanding demyelination at the spinal cord level, remain less characterized in multiple sclerosis pathology. However, it is known that chronic lesions in the spinal cord are more likely to be inactive and less likely to be smouldering compared with brain supratentorial lesions (Frischer et al., 2015). Another limitation is the present incapacity using exclusively conventional T1- and T2-weighted imaging data to disentangle the contribution of smouldering inflammation versus Wallerian secondary neurodegeneration with respect to T1 intensity decrease in SELs and non-SELs. A recently initiated study (CONSONANCE; NCT03523858) will further address this question by assessing the association between the SEL phenotype and paramagnetic rims on susceptibility-based MRI or with metabolic changes or axonal/neuronal damage measured by spectroscopic magnetic resonance. Efforts are ongoing to characterize the potential neuro-pathological correlates of SELs in relation to that of paramagnetic rim lesions.
The origin and natural history of chronic multiple sclerosis lesion activity detected through the SEL phenotype based on longitudinal in vivo demonstration of chronic lesion expansion or using other definitions, such as the presence of a persistent phase rim, remains to be explored. In particular the dynamics of demyelination and potential remyelination (if at all occurring) cycles remains to be characterized in the context of chronic active white matter lesions, e.g. using magnetization transfer and diffusion-based imaging methods. It is also unknown whether newly formed multiple sclerosis lesions at some point along their lifespan may eventually and secondarily adopt the properties of chronic active lesions such as SELs. Notwithstanding that this is an assumption through generalization of observations from scattered studies, there is a consensus that all newly formed multiple sclerosis lesions are presumably having an ‘acute’ phase at inception (Absinta et al., 2016a, b; Guttmann et al., 2016). Some of those newly formed multiple sclerosis lesions are also characterized by an early phase rim, due to accumulation of paramagnetic substances possibly corresponding to myelin-breakdown compounds (Deh et al., 2018) and/or attendant infiltration by blood-derived monocytes/macrophages (Absinta et al., 2016a, b) and their related by-products. In approximately half of those new acute lesions with centripetal contrast enhancement, the early phase rim will vanish while T1 hypointensity may partially resolve, whereas in the other half, the early phase rim may persist and T1 signal intensity may continue to decrease (Absinta et al., 2016a, b). Whether some of the newly formed multiple sclerosis lesions and likely those with an early persistent phase rim may convert into chronic active lesions or be chronic active with SEL properties from their inception remains to be explored.
The observation that the depletion of CD20-expressing cells similarly reduced chronic lesion activity both in pre-existing T2 lesion areas with (SELs) or without (non-SELs) signs of slow longitudinal expansion raises questions as to the potential identity of the biological mechanisms at play in both compartments. We noted a sizeable difference in the magnitude of T1-weighted measures of chronic lesion activity in SEL and non-SEL areas of patients with PPMS, but the lower grade of chronic lesion activity in non-SELs remained significantly reduced by ocrelizumab. These data are consistent with the pathological description (Frischer et al., 2009) of the presence of inflammatory cellular infiltrates (i.e. CD20+ B cells, CD3+ T cells and HLA-D-positive macrophages and microglia cells) in chronic inactive lesions of patients with progressive multiple sclerosis, although at lower density compared with that of ‘smouldering’ or ‘slowly expanding lesions’ but at higher density than in the normal-appearing white matter (except for HLA-D-positive macrophages and microglia cells). The results of this work may indicate a potential role of CD20-expressing cells in the chronic inflammatory processes leading to ongoing tissue damage in SELs or chronic ‘active’ lesions as well as in previously termed chronic ‘inactive’ lesions or non-SELs as studied herein. Whether a continuum of chronic inflammatory pathology may also affect the normal-appearing brain tissue might be ultimately assessed using more advanced measures of tissue integrity.
Supplementary Material
Acknowledgements
We would like to thank all ORATORIO study patients, their families, and the investigators who participated in this trial. This research was funded by F. Hoffmann-La Roche Ltd, Basel, Switzerland. We are particularly grateful to Dr Alexander Kulla for his constant support at inception and throughout the lifespan of this research work. Writing and editorial assistance for this manuscript was provided by Heather Latimer, PhD of Articulate Science, UK, and funded by F. Hoffmann-La Roche.
Glossary
Abbreviations
- 9HPT
9-Hole Peg Test
- EDSS
Expanded Disability Status Scale
- ITT
intention-to-treat
- PPMS
primary progressive multiple sclerosis
- SEL
slowly expanding/evolving lesion
- T25FW
Timed 25-Foot Walk
Funding
This research was funded by F. Hoffmann-La Roche Ltd, Basel, Switzerland. F.B. is supported by the NIHR UCLH Biomedical Research Centre.
Competing interests
C.E. is an employee of NeuroRx Research and has served on an advisory board for F. Hoffmann-La Roche Ltd. S.B. was an employee of F. Hoffmann-La Roche Ltd during the completion of the work related to this manuscript. S.B. is now an employee of Biogen (Cambridge, MA, USA), which was not in any way associated with this study. J.S.W. has served on advisory boards, data monitoring or steering committees, and has consulting agreements from the following entities: AbbVie, Actelion, Alkermes, Bayer HealthCare, Biogen, Bionest, Celgene, Clene Nanomedicine, EMD Serono, Forward Pharma A/S, GeNeuro, MedDay Pharmaceuticals, Novartis Pharmaceuticals, Otsuka, PTC Therapeutics, Roche Genentech, Sanofi Genzyme, Strategic Consultants International, Takeda, and Teva Pharmaceuticals; royalties are received for out-licensed monoclonal antibodies through UTHealth from Millipore Corporation. S.L.H. serves on the board of trustees for Neurona and on scientific advisory boards for Alector, Annexon, Bionure, Molecular Stethoscope, and Symbiotix, and has received travel reimbursement and writing assistance from F. Hoffmann-La Roche Ltd for CD20-related meetings and presentations. L.K.’s institution, the University Hospital Basel, has received research support and payments that were used exclusively for research support for L.K.’s activities as principal investigator and member or chair of planning and steering committees or advisory boards for trials sponsored by Actelion, Addex, Almirall, Bayer HealthCare Pharmaceuticals, CLC Behring, F. Hoffmann-La Roche Ltd and Genentech, Inc., GeNeuro SA, Genzyme, Merck Serono, Mitsubishi Pharma, Novartis, Octapharma, Ono Pharmaceutical, Pfizer, Receptos, Sanofi, Santhera, Siemens, Teva, UCB, and XenoPort; has received licence fees for Neurostatus products; and has received research grants from the European Union, Gianni Rubatto Foundation, Novartis Research Foundation, Roche Research Foundation, Swiss Multiple Sclerosis Society, and Swiss National Research Foundation. F.B. is an editorial board member for the publications Brain, European Radiology, Multiple Sclerosis Journal, Neurology, and Radiology; has received consultancy fees from Bayer Schering, Biogen Idec, F. Hoffmann-La Roche Ltd, Genzyme, Janssen Research, Merck Serono, Novartis, Synthon, and Teva; has received grants from the Dutch MS Society (EU-FP7/Horizon 2020); and has received payments for developing educational presentations, including service on speaker bureaus, for Biogen Idec and IXICO. C.B. is a contractor for F. Hoffmann-La Roche Ltd. J.F. is an employee of F. Hoffmann-La Roche Ltd. F.M. is an employee and shareholder of F. Hoffmann-La Roche Ltd. W.W. is an employee and shareholder of F. Hoffmann-La Roche Ltd. D.L.A. has received personal fees for consulting from Acorda, Biogen, F. Hoffmann-La Roche Ltd, MedImmune, Mitsubishi, Novartis, Receptos, and Sanofi-Aventis; grants from Biogen and Novartis; and an equity interest in NeuroRx Research.
References
- Absinta M, Sati P, Fechner A, Schindler MK, Nair G, Reich DS. Identification of chronic active multiple sclerosis lesions on 3T MRI. AJNR Am J Neuroradiol 2018; 39: 1233–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Absinta M, Sati P, Reich DS. Advanced MRI and staging of multiple sclerosis lesions. Nat Rev Neurol 2016a; 12: 358–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Absinta M, Sati P, Schindler M, Leibovitch EC, Ohayon J, Wu T et al. Persistent 7-tesla phase rim predicts poor outcome in new multiple sclerosis patient lesions. J Clin Invest 2016b; 126: 2597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosma LV, Kragt JJ, Brieva L, Khaleeli Z, Montalban X, Polman CH et al. The search for responsive clinical endpoints in primary progressive multiple sclerosis. Mult Scler J 2009; 15: 715–20. [DOI] [PubMed] [Google Scholar]
- Bramow S, Frischer JM, Lassmann H, Koch-Henriksen N, Lucchinetti CF, Sørensen PS. Demyelination versus remyelination in progressive multiple sclerosis. Brain 2010; 133: 2983–98. [DOI] [PubMed] [Google Scholar]
- Cadavid D, Cohen JA, Freedman MS, Goldman MD, Hartung HP, Havrdova E et al. The EDSS-Plus, an improved endpoint for disability progression in secondary progressive multiple sclerosis. Mult Scler 2017; 23: 94–105. [DOI] [PubMed] [Google Scholar]
- Correale J, Gaitán MI, Ysrraelit MC, Fiol MP. Progressive multiple sclerosis: from pathogenic mechanisms to treatment. Brain 2017; 140: 527–46. [DOI] [PubMed] [Google Scholar]
- Dal-Bianco A, Grabner G, Kronnerwetter C, Weber M, Höftberger R, Berger T et al. Slow expansion of multiple sclerosis iron rim lesions: pathology and 7T magnetic resonance imaging. Acta Neuropathol 2017; 133: 25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deh K, Ponath GD, Molvi Z, Parel GT, Gillen KM, Zhang S et al. Magnetic susceptibility increases as diamagnetic molecules breakdown: myelin digestion during multiple sclerosis lesion formation contributes to increase on QSM. J Magn Reson Imaging 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott C, Wolinsky JS, Hauser SL, Kappos L, Barkhof F, Model F et al. Ocrelizumab may reduce tissue damage in chronic active lesions as measured by change in T1 hypointensity of slowly evolving lesions in patients with primary progressive multiple sclerosis. Poster presented at AAN; Poster 376, April 24, 2018; Los Angeles, CA, USA; 2018a. [Google Scholar]
- Elliott C, Wolinsky JS, Hauser SL, Kappos L, Barkhof F, Bernasconi C et al. Slowly expanding/evolving lesions as a magnetic resonance imaging marker of chronic active multiple sclerosis lesions. Mult Scler J 2018b, doi: 10.1177/1352458518814117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippi M, Rocca MA, Barkhof F, Brück W, Chen JT, Comi G et al. Association between pathological and MRI findings in multiple sclerosis. Lancet Neurol 2012; 11: 349–60. [DOI] [PubMed] [Google Scholar]
- Francis SJ. Automatic lesion identification in MRI of multiple sclerosis patients. Master of Science thesis. Division of Neuroscience, McGill University, 2004. [Google Scholar]
- Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009; 132: 1175–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischer JM, Weigand SD, Guo Y, Kale N, Parisi JE, Pirko I et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol 2015; 78: 710–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttmann CR, Rousset M, Roch JA, Hannoun S, Durand-Dubief F, Belaroussi B et al. Multiple sclerosis lesion formation and early evolution revisited: a weekly high-resolution magnetic resonance imaging study. Mult Scler 2016; 22: 761–9. [DOI] [PubMed] [Google Scholar]
- Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung HP, Hemmer B et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med 2017; 376: 221–34. [DOI] [PubMed] [Google Scholar]
- Kapoor R, Ho PR, Campbell N, Chang I, Deykin A, Forrestal F et al. Effect of natalizumab on disease progression in secondary progressive multiple sclerosis (ASCEND): a phase 3, randomised, double-blind, placebo-controlled trial with an open-label extension. Lancet Neurol 2018; 17: 405–15. [DOI] [PubMed] [Google Scholar]
- Khaleeli Z, Ciccarelli O, Manfredonia F, Barkhof F, Brochet B, Cercignani M et al. Predicting progression in primary progressive multiple sclerosis: a 10-year multicenter study. Ann Neurol 2008; 63: 790–3. [DOI] [PubMed] [Google Scholar]
- Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T et al. Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol 2000; 157: 267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, Bergmann M et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005; 128: 2705–12. [DOI] [PubMed] [Google Scholar]
- Lublin F, Miller DH, Freedman MS, Cree BAC, Wolinsky JS, Weiner H et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet 2016; 387: 1075–84. [DOI] [PubMed] [Google Scholar]
- Miller DH, Lublin FD, Sormani MP, Kappos L, Yaldizli Ö, Freedman MS et al. Brain atrophy and disability worsening in primary progressive multiple sclerosis: insights from the INFORMS study. Ann Clin Transl Neurol 2018; 5: 346–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med 2017; 376: 209–20. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Guizard N, Fonov VS, Narayanan S, Collins DL, Arnold DL. Jacobian integration method increases the statistical power to measure gray matter atrophy in multiple sclerosis. Neuroimage Clin 2013; 4: 10–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polman CH, Reingold SC, Edan G, Filippi M, Hartung HP, Kappos L et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald criteria”. Ann Neurol 2005; 58: 840–6. [DOI] [PubMed] [Google Scholar]
- Popescu BF, Frischer JM, Webb SM, Tham M, Adiele RC, Robinson CA et al. Pathogenic implications of distinct patterns of iron and zinc in chronic MS lesions. Acta Neuropathol 2017; 134: 45–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prineas JW, Kwon EE, Cho ES, Sharer LR, Barnett MH, Oleszak EL et al. Immunopathology of secondary-progressive multiple sclerosis. Ann Neurol 2001; 50: 646–57. [DOI] [PubMed] [Google Scholar]
- Rocca MA, Sormani MP, Rovaris M, Caputo D, Ghezzi A, Montanari E et al. Long-term disability progression in primary progressive multiple sclerosis: a 15-year study. Brain 2017; 140: 2814–9. [DOI] [PubMed] [Google Scholar]
- Rousseeuw P, Driessen K. Computing LTS regression for large data sets. Data Min Knowl Discov 2006; 12: 29–45. [Google Scholar]
- Sastre-Garriga J, Ingle GT, Rovaris M, Téllez N, Jasperse B, Altmann DR et al. Long-term clinical outcome of primary progressive MS: predictive value of clinical and MRI data. Neurology 2005; 65: 633–5. [DOI] [PubMed] [Google Scholar]
- van Waesberghe JH, Kamphorst W, De Groot CJ, van Walderveen MA, Castelijns JA, Ravid R et al. Axonal loss in multiple sclerosis lesions: magnetic resonance imaging insights into substrates of disability. Ann Neurol 1999; 46: 747–54. [DOI] [PubMed] [Google Scholar]
- van Walderveen MA, Barkhof F, Pouwels PJ, van Schijndel RA, Polman CH, Castelijns JA. Neuronal damage in T1-hypointense multiple sclerosis lesions demonstrated in vivo using proton magnetic resonance spectroscopy. Ann Neurol 1999; 46: 79–87. [DOI] [PubMed] [Google Scholar]
- van Walderveen MA, Kamphorst W, Scheltens P, van Waesberghe JH, Ravid R, Valk J et al. Histopathologic correlate of hypointense lesions on T1-weighted spin-echo MRI in multiple sclerosis. Neurology 1998; 50: 1282–8. [DOI] [PubMed] [Google Scholar]
- Wolinsky JS, Montalban X, Hauser SL, Giovannoni G, Vermersch P, Bernasconi C et al. Evaluation of no evidence of progression or active disease (NEPAD) in patients with primary progressive multiple sclerosis in the ORATORIO trial. Ann Neurol 2018; 84: 527–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Qualified researchers may request access to individual patient level data through the clinical study data request platform (www.clinicalstudydatarequest.com). Further details on Roche’s criteria for eligible studies are available at https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Roc he.aspx. Further details on Roche’s Global Policy on the sharing of clinical information and how to request access to related clinical study documents, are available at https://www.roche.com/research_and_development/who_we_are_how_we_work/cl inical_trials/our_commitment_to_data_sharing.htm.