

ABSTRACT
Background
Early-life iron deficiency (ID) impairs hippocampal energy production. Whether there are changes in glucose transporter (GLUT) expression is not known.
Objective
The aim of this study was to investigate whether early-life ID and the treatment iron dose alter brain regional GLUT expression in adult rats and mice.
Methods
In Study 1, ID was induced in male and female Sprague Dawley rat pups by feeding dams a 3-mg/kg iron diet during gestation and the first postnatal week, followed by treatment using low-iron [3–10 mg/kg; formerly iron-deficient (FID)-10 group], standard-iron (40-mg/kg; FID-40 group), or high-iron (400-mg/kg; FID-400 group) diets until weaning. The control group received the 40 mg/kg iron diet. GLUT1, GLUT3, hypoxia-inducible factor (HIF)-1α, and prolyl-hydroxylase-2 (PHD2) mRNA and protein expression in the cerebral cortex, hippocampus, striatum, cerebellum, and hypothalamus were determined at adulthood. In Study 2, the role of hippocampal ID in GLUT expression was examined by comparing the Glut1, Glut3, Hif1α, and Phd2 mRNA expression in adult male and female wild-type (WT) and nonanemic hippocampal iron-deficient and iron-replete dominant negative transferrin receptor 1 (DNTfR1−/−) transgenic mice.
Results
In Study 1, Glut1, Glut3, and Hif1α mRNA, and GLUT1 55-kDa protein expression was upregulated 20–33% in the hippocampus of the FID-10 group but not the FID-40 group, relative to the control group. Hippocampal Glut1 mRNA (−39%) and GLUT1 protein (−30%) expression was suppressed in the FID-400 group, relative to the control group. Glut1 and Glut3 mRNA expression was not altered in the other brain regions in the 3 FID groups. In Study 2, hippocampal Glut1 (+14%) and Hif1α (+147%) expression was upregulated in the iron-deficient DNTfR1−/− mice, but not in the iron-replete DNTfR1−/− mice, relative to the WT mice (P < 0.05, all).
Conclusions
Early-life ID is associated with altered hippocampal GLUT1 expression in adult rodents. The mouse study suggests that tissue ID is potentially responsible.
Keywords: iron deficiency, hippocampus, glucose transporter-1, glucose transporter-3, hypoxia-inducible factor, prolyl-hydroxylase, iron toxicity, transferrin receptor
Introduction
Iron deficiency (ID) during the fetal and early postnatal periods (early-life ID) has a negative impact on hippocampal development and function (1, 2). The structural and functional hippocampal deficits persist at adulthood despite correction of ID and irrespective of the iron dose used for treatment (3–5). Early-life ID impairs oxidative ATP production in the hippocampus (3, 6–9), raising the possibility that persistent abnormalities in hippocampal energy metabolism could be responsible for the long-term impairments.
Decreased ATP production due to ID leads to a compensatory increase in glucose uptake and glycolysis in cell cultures and hepatic tissue (10, 11). Studies in nonhuman primate models of early-life ID anemia (IDA) suggest that a similar response potentially occurs in the brain (12, 13). The brain has minimal glucose stores and thus requires the continuous delivery of glucose from plasma. Brain glucose transport involves specific glucose transporters (GLUTs). GLUT1 is the primary GLUT at the blood–brain barrier (BBB) and glia, whereas GLUT3 is the primary neuronal GLUT (14). Two GLUT1 isoforms are expressed: a 55-kDa isoform in the endothelium of the BBB and a 45-kDa isoform in nonendothelial cells, including the astrocytes and oligodendrocytes (15, 16). The same gene encodes both GLUT1 isoforms. In rats, GLUT1 expression remains low until postnatal day (P) 14, doubles between P14 and P21, and then doubles again to reach the adult levels by P30 (17). There are no interregional variations in GLUT1 expression during normal development. GLUT3 expression is also low during the first postnatal week and then increases steadily to reach the adult levels by P21–P30 (17). Unlike GLUT1, GLUT3 expression demonstrates regional variations, paralleling the neuronal maturation and synaptogenesis in the region (17).
We have previously demonstrated that GLUT1 expression is upregulated in the cerebral cortex and hippocampus of neonatal rats during the period of early-life IDA (18, 19). The long-term changes in GLUT expression in the brain regions, especially in the context of the iron dose used for treatment, are not known. The objective of the present study was to determine the long-term effects of early-life ID and iron treatment dose on GLUT1, GLUT3, hypoxia-inducible factor (HIF)-1α, and prolyl hydroxylase-2 (PHD2) expression in the brain regions of formerly iron-deficient (FID) adult rats and mice. HIF-1α is a transcription factor involved in cellular metabolic adaptation to hypoxia (20). Under normoxia, the HIF-1α subunit is hydroxylated soon after its synthesis at the 2 proline residues by PHD2, followed by degradation (20, 21). Iron is an essential cofactor in the enzymatic action of PHD2. Under hypoxia or ID, the PHD2 action is inhibited, leading to HIF-1α stabilization and transcription of HIF target genes, including GLUT1, GLUT3, vascular endothelial growth factor (VEGF), lactate dehydrogenase (LDH), and transferrin receptor (TfR) (20, 22). Two experiments were performed: in Study 1, the effects of early-life IDA and iron treatment dose on GLUT1 and GLUT3 expression in the different brain regions were determined using a rat model of diet-induced early-life IDA treated using different doses of iron. In Study 2, the role of tissue ID in early-life ID–induced GLUT changes was examined using a mouse model of nonanemic hippocampus-specific neuronal ID.
Methods
Animal preparation
Experiments were approved by the Institutional Animal Care and Use Committees at the University of Michigan and University of Minnesota and conformed to the NIH guidelines for care and use of animals. Animals were maintained under standard laboratory conditions on a 12-h light/dark cycle and allowed to consume food and water ad libitum. Animals of both sexes were used in the experiments.
Animal models
Study 1. Dietary IDA rat model
Sprague Dawley rats were used to create the early-life IDA model used in our previous studies (4, 5). The overall study design is shown in Figure 1. We have previously reported the dietary composition and the characteristics of the model (4). Briefly, 8-wk-old female rats were purchased from Harlon Laboratories, started being fed a 40 mg/kg iron diet (TD89300; Harlan Teklad) upon arrival, and were mated after 7–10 d of acclimatization. After confirmation of pregnancy, dams assigned to the ID group started being fed a low-iron diet (TD80396; Harlan Teklad; iron concentration, 3 mg/kg diet) from gestational day 5 to P7 to induce early-life IDA in the pups. Dams assigned to the control group were maintained on the 40 mg/kg iron diet. The litter size was culled to 10 pups within 24 h of delivery. All the pups were cross-fostered on P8 to other lactating dams that had been fed a specific iron diet since gestational day 5. Pups in the control group were cross-fostered to other dams fed the 40 mg/kg iron diet. Pups in the ID group were cross-fostered to dams fed either 1) the 40 mg/kg iron diet to generate the FID standard-dose iron treatment (FID-40) group; 2) a 400 mg/kg iron diet (TD02545; Harlan Teklad) to generate the FID high-dose iron treatment (FID-400) group; or 3) the 3 mg/kg iron diet until P15, then a 10 mg/kg iron diet (TD01094; Harlan Teklad) to maintain litter viability, to generate the FID low-dose iron treatment (FID-10) group. All the pups were weaned on P21 to the 40 mg/kg iron diet, except the FID-10 group, which was weaned to the 10 mg/kg iron diet. The groups remained on their respective postweaning diets until adulthood.
FIGURE 1.
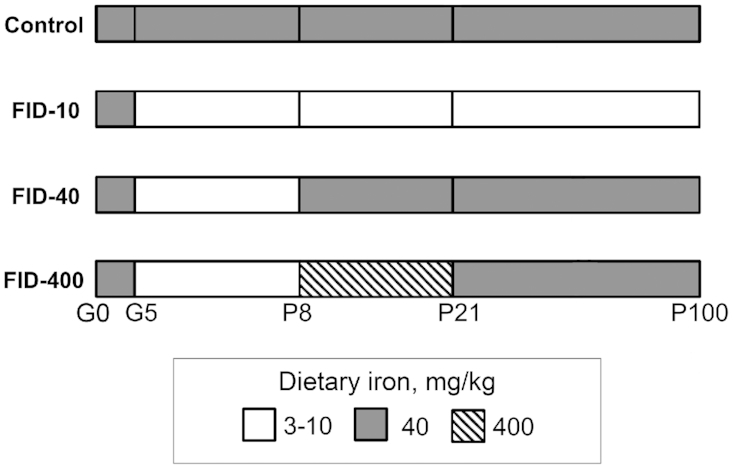
Design of Study 1. FID, formerly iron deficient; G, gestational day; P, postnatal day.
We have previously reported the hematocrit and tissue iron concentration in the brain regions on P21 and P100 in this model (4) (Supplemental Table 1). Briefly, on P21, the hematocrit was 50% lower and tissue iron concentration in the cerebral cortex, hippocampus, striatum, and cerebellum was 40–50% lower in the FID-10 group than in the control group (4). On P100, the FID-10 group continued to have 10% lower hematocrit and 20% lower iron concentration in the hippocampus and cerebellum than the control group. Both the 40 mg/kg iron and the 400 mg/kg iron diets corrected anemia and normalized iron concentration in the brain regions on P21 and P100, except for a 14% lower hippocampal iron concentration in the FID-400 group on P100 (4). Our prior studies demonstrated that, as compared with the control group, all 3 FID groups had abnormal neurochemistry in the brain regions on P100 with the FID-10 and FID-400 groups also demonstrating behavioral deficits at this age (4, 5).
Study 2. Nonanemic hippocampus-specific neuronal ID mouse model
The transgenic mouse model has been described previously (23, 24). Briefly, these mice express a tetracycline transactivator–regulated, dominant negative TfR (DNTfR1−/−) localized to hippocampal pyramidal neurons using a calcium calmodulin kinase-II Cre driver that activates at embryonic day 18.5. The mutant TfR disrupts normal TfR-mediated neuronal iron uptake beginning at that time. Normal iron status can be rapidly restored by administering doxycycline through diet (23). Mutants with uncorrected ID have impaired hippocampal structure and function at adulthood (23, 24). Restoration of tissue iron status via dietary doxycycline treatment at P21 leads to structural and functional hippocampal recovery at adulthood (23). Adult male and female wild-type (WT) and hippocampal iron-deficient DNTfR1−/− and iron-replete DNTfR1−/− mice were used in the experiments.
Tissue preparation
Animals were killed using an overdose of pentobarbital (100 mg/kg, intraperitoneally). The brain was removed and the cerebral cortex, hippocampus, striatum, cerebellum, and hypothalamus were dissected (25, 26) from the rats in Study 1 (n = 8 per group). Only the hippocampus was collected from the mice in Study 2 (n = 7 per group). Tissue samples were flash-frozen using liquid nitrogen and stored at −80°C until analysis.
Transcript expression
Transcript expression was determined using the real-time qRT-PCR method according to the Minimum Information for Publication of Quantitative Real-Time PCR Experiments guidelines (27) and using the 2−ΔΔCT method as in our previous studies (25, 26). Briefly, the RNA was extracted and cDNA was generated using commercial kits (RNAqueous Total RNA Isolation Kit and High-Capacity RNA-to-cDNA kit; Thermo Fisher Scientific). The expression of Glut1, Glut3, Hif1α, Vegfa, Ldhb, and Tfrc mRNA in rats, and Glut1, Glut3, Hif1α, and Phd2 mRNA in mice was determined using FastStart Universal Probe Master (Sigma Aldrich) and TaqMan gene expression probes (Life Technologies; Supplemental Table 2). Samples were assayed in duplicate using ribosomal protein s18 as control.
Protein expression
To determine whether altered mRNA expression in a brain region has functional significance, protein expression in the brain region was determined using commercially available primary antibodies against GLUT1 (1:100; Santa Cruz Biotechnology), GLUT3 (1:400; Abcam), PHD2 (1:500; Novus Biologicals), and β-actin (1:5000; Abcam) (Supplemental Table 2) and using published methods (28, 29). The specificity of the PHD2 primary antibody has been shown previously (30). Although we expected the GLUT1 and GLUT3 primary antibodies to be specific based on the size of the protein band observed on western blots, we were not able to find any data to indicate that the specificity of the antibodies had been rigorously evaluated. After incubation with an appropriate fluorescent secondary antibody, the membranes were imaged (Odyssey Infrared Imaging System; LI-COR Biosciences) and the intensity of the target protein relative to β-actin was determined. All bands in the 45-kDa and 55-kDa regions were included in the densitometry analysis of GLUT1.
Statistical analysis
The effect of group on transcript and protein expression in a brain region was determined using 1-factor ANOVA. Intergroup differences were determined whenever indicated by a significant F ratio using Bonferroni-adjusted t tests. A statistical computer program (GraphPad Prism 7; https://www.graphpad.com/scientific-software/prism/) was used. Data are presented as mean ± SEM. Significance was set at P < 0.05.
Results
Study 1
Effect of early-life ID and iron treatment dose on brain regional GLUT1 expression in the FID adult rats
There was an effect of group on Glut1 mRNA expression in the hippocampus (P < 0.001; Figure 2A). Compared with the control group, expression was 33% higher in the FID-10 group (P < 0.02; Figure 2A) and was accompanied by 20% higher GLUT1 55-kDa protein expression (P < 0.05; Figure 2C). GLUT1 45-kDa protein expression did not differ between the control and FID-10 groups (P = 0.6; Figure 2D). Glut1 mRNA (P = 0.5) and GLUT1 55-kDa protein (P = 0.9) expression in the hippocampus of the FID-40 group did not differ from the control group (Figure 2A, C). Compared with the control and FID-10 groups, GLUT1 45-kDa protein expression was 41% lower in the FID-40 group (P < 0.05; Figure 2D). Compared with the control and FID-10 groups, Glut1 mRNA (Figure 2A) and GLUT1 protein (Figure 2C, D) expression was lower in the FID-400 group (P < 0.05). Both the 55-kDa and 45-kDa GLUT1 isoforms were suppressed. Glut1 mRNA expression in the other brain regions was not altered in any of the FID groups (Table 1).
FIGURE 2.

Glut1 mRNA and GLUT1 protein expression in the hippocampus of control and FID adult rats in Study 1. (A) Quantification of Glut1 mRNA expression. (B) Representative Western blots of GLUT1 protein. (C) Quantification of the GLUT1 55-kDa protein isoform relative to β-actin. (D) Quantification of the GLUT1 45-kDa protein isoform relative to β-actin. Values are mean ± SEM percentage. Changes in mRNA expression (A) or protein expression (C, D) relative to the control group, n = 8 for transcript and n = 3–4 for protein expression. Dietary groups without a common letter differ, P < 0.05. FID, formerly iron deficient; GLUT1, glucose transporter-1; M, reference protein marker.
TABLE 1.
Glut1 mRNA expression in the brain regions of adult rats in the control and FID groups treated using low-iron, standard-iron, and high-iron diets in Study 11
| Brain region | Control group | FID-10 group | FID-40 group | FID-400 group |
|---|---|---|---|---|
| Hippocampus | 100 ± 11b | 133 ± 9a | 80 ± 5b | 61 ± 8b |
| Cerebral cortex | 100 ± 10 | 109 ± 8 | 92 ± 9 | 93 ± 16 |
| Striatum | 100 ± 7 | 108 ± 9 | 114 ± 8 | 78 ± 12 |
| Cerebellum | 100 ± 18 | 122 ± 10 | 107 ± 10 | 133 ± 9 |
| Hypothalamus | 100 ± 17 | 82 ± 23 | 85 ± 15 | 84 ± 6 |
Values are mean ± SEM percentage changes relative to control, n = 8 per group. Labeled means in a row without a common letter differ, P < 0.05. FID, formerly iron-deficient; FID-10, FID group treated using low-iron (10 mg/kg) diet; FID-40, FID group treated using standard-iron (40 mg/kg) diet; FID-400, FID group treated using high-iron (400 mg/kg) diet; Glut1, glucose transporter-1.
Effect of early-life ID and iron treatment dose on brain regional GLUT3 expression in the FID adult rats
There was an effect of group on Glut3 mRNA expression in the hippocampus (P < 0.001). Compared with the control group, expression was 29% higher in the FID-10 group [control group, 1.00 ± 0.04 arbitrary units (au); FID-10 group, 1.29 ± 0.09 au, P < 0.05]. However, GLUT3 protein expression was not altered (Supplemental Figure 1). Glut3 mRNA expression in the hippocampus of the FID-40 group did not differ from the control group (control group, 1.00 ± 0.04; FID-40 group, 0.97 ± 0.06, P = 0.7). Neither low-dose iron nor standard-dose iron treatment altered Glut3 mRNA expression in the cerebral cortex, cerebellum, striatum, or hypothalamus (Supplemental Table 3). Similarly, compared with the control, FID-10, and FID-40 groups, Glut3 mRNA and GLUT3 protein expression was not altered in the hippocampus of the FID-400 group (Supplemental Figure 1).
Effect of low-dose iron and standard-dose iron treatment on Hif1α mRNA, PHD2 protein, and their target gene mRNA expression in the hippocampus of FID adult rats
There was an effect of group on Hif1α mRNA expression in the hippocampus (P < 0.05). Relative to the control group, expression was 23% higher in the FID-10 group (1.00 ± 0.07 au compared with 1.23 ± 0.07 au; P < 0.05). PHD2 protein expression in the FID-10 group did not differ from the control group (Supplemental Figure 2). Vegfa and Tfrc expression was upregulated in the FID-10 group, whereas there was no effect on Ldhb expression (Table 2). Hif1α mRNA expression in the hippocampus of the FID-40 group was comparable with the control group (1.00 ± 0.07 au compared with 1.03 ± 0.06 au; P = 0.7). Based on the lack of changes in Glut1 and Hif1α mRNA expression, PHD2 protein expression in the hippocampus of the FID-40 group was not determined.
TABLE 2.
Expression of HIF-1α target genes in the hippocampus of adult rats in the control and FID groups treated using low-iron diet in Study 11
| Gene | Control group | FID-10 group |
|---|---|---|
| Vegf | 100 ± 12b | 133 ± 8a |
| Tfrc | 100 ± 6b | 170 ± 14a |
| Ldh | 100 ± 5 | 96 ± 3 |
Values are mean ± SEM percentage changes relative to control, n = 8 per group. Given the lack of Hif1α mRNA changes, HIF-1α target gene expression was not determined in the hippocampus of the FID-40 group. Labeled means in a row without a common letter differ, P < 0.05. FID, formerly iron-deficient; FID-10, FID group treated using low-iron (10 mg/kg) diet; HIF-1α, hypoxia inducible factor-1α; Ldh, lactate dehydrogenase mRNA; Tfrc, transferrin receptor 1 mRNA; Vegf, vascular endothelial growth factor mRNA.
Study 2: effect of nonanemic hippocampal neuronal ID and iron restoration on Glut1, Glut3, Hif1α, and Phd2 mRNA expression in the adult mouse hippocampus
There was a main effect of group on Glut1 and Hif1α mRNA expression in the adult mouse hippocampus (P < 0.05). Compared with the WT mice, Glut1 (+14%) and Hif1α (+147%) expression was higher in the iron-deficient DNTfR1−/− mice (P < 0.05; Table 3). Glut3 and Phd2 mRNA expression was not altered (Table 3). Glut1 and Hif1α mRNA expression in the iron-replete DNTfR1−/− mice was comparable with the WT group (Table 3).
TABLE 3.
Glucose transporter, HIF-1α, and PDH2 mRNA expression in the hippocampus of WT and hippocampal iron-deficient and iron-replete DNTfR1−/− adult mice in Study 21
| Gene | WT mice | Iron-deficient DNTfR1−/− mice | Iron-replete DNTfR1−/− mice |
|---|---|---|---|
| Glut1 | 100 ± 4b | 114 ± 3a | 94 ± 4b |
| Glut3 | 100 ± 6 | 104 ± 4 | 100 ± 3 |
| Hif1α | 100 ± 18b | 247 ± 25a | 168 ± 28b |
| Pdh2 | 100 ± 5 | 104 ± 2 | 94 ± 1 |
Values are mean ± SEM percentage changes relative to WT, n = 7 per group. Labeled means in a row without a common letter differ, P < 0.05. DNTfR1−/−, dominant negative transferrin receptor; Glut1, glucose transporter-1 mRNA; Glut3, glucose transporter-3 mRNA; Hif1α, hypoxia inducible factor-1α mRNA; Pdh2, prolyl-hydroxylase-2 mRNA; WT, wild-type.
Discussion
An increased reliance on glucose as a metabolic substrate is a well-known phenomenon in ID (31, 32). A prior study demonstrated that IDA is associated with a selective increase in glucose transport across the BBB in adult rats (33). The present study demonstrates that upregulation of GLUT1 55 kDa, which is primarily expressed in the microvasculature and responsible for glucose transport across the BBB, may be the mechanism through which increased glucose transport is achieved. The results also demonstrate that long-term alteration in GLUT1 expression associated with early-life ID is mainly confined to the hippocampus and influenced by the iron dose used for treatment.
We have previously demonstrated that the IDA rat model used in the present study leads to mild anemia (10% lower hematocrit) and 20% lower iron concentration in the hippocampus of the FID-10 group at adulthood (Supplemental Table 1) (4). The upregulation of Tfrc mRNA in the hippocampus of the FID-10 group supports the presence of hippocampal tissue ID in the present study. Thus, systemic hypoxia due to anemia, as well as hippocampal tissue ID could have led to the GLUT1 upregulation in the hippocampus of the FID-10 group. Tissue ID likely had a greater role, because systemic hypoxia due to anemia should have also led to GLUT1 upregulation in other brain regions (34), which was not the case. Glut1 mRNA upregulation in the nonanemic hippocampal iron-deficient DNTfR1−/− mice in Study 2 supports this contention. Iron chelation studies also support that intracellular ID is the primary driver of GLUT1 upregulation (35). Nevertheless, because we did not determine tissue iron concentrations in the present study, this possibility remains conjectural.
The GLUT1 55-kDa protein isoform was upregulated, whereas the GLUT1 45-kDa protein isoform remained unaltered. This pattern is similar to the one seen with chronic hypoglycemia (36, 37), a condition that is also associated with impaired ATP production in the hippocampus (38, 39). Collectively, the 2 studies suggest that regulation of hippocampal glucose uptake in response to suboptimal oxidative energy production is primarily at the level of the BBB. In chronic hypoglycemia, GLUT1 upregulation is accompanied by enhanced regional glucose uptake (36, 37). A similar response may be present in the FID-10 hippocampus, but requires confirmatory studies. Surprisingly, GLUT1 expression was not altered in the cerebellum of the FID-10 group, despite the presence of a degree of tissue ID comparable with that in the hippocampus we had previously demonstrated in this animal model (4) (Supplemental Table 1). Glucose utilization rate influences GLUT1 expression in a brain region (40). Thus, lack of GLUT1 changes may reflect unchanged glucose utilization in the cerebellum despite the presence of ID. It is noteworthy that relative to the hippocampus, baseline glucose utilization and GLUT1 expression are 30–60% lower in the cerebellum of adult humans and rats (40, 41).
GLUT1 upregulation in the FID-10 hippocampus was likely mediated by the HIF-1α/PHD2 mechanism. Upregulation of other HIF-1 target genes, Vegfa and Tfrc, in the FID-10 group supports this possibility. Lack of Ldhb upregulation is not surprising given that LDH upregulation occurs only with severe IDA (42). It is noteworthy that a prior study found only a trend towards LDH upregulation in the brain of adult rats, despite a more severe IDA (65% lower hematocrit) than the present study (10% lower hematocrit) (43). The magnitude of Hif1α mRNA upregulation in the hippocampus was greater in the iron-deficient DNTfR1−/− mice than in the FID-10 rats (147% compared with 23%), likely because of a more severe hippocampal tissue ID (85% compared with 20% in the FID-10 group) (4, 22). The higher Hif1α mRNA upregulation did not lead to a comparable magnitude of Glut1 mRNA upregulation in the hippocampus of iron-deficient DNTfR1−/− mice. A similar discordance between HIF-1α and GLUT1 expression has been described in cell cultures and whole brain preparations after exposure to hypoxia (44–46), although the reasons for the discordant expression remain to be determined.
Whereas Glut3 mRNA expression was upregulated, GLUT3 protein expression was not altered in the hippocampus of the FID-10 group. Although protein amounts are largely determined by transcript concentrations under steady-state conditions, during dynamic conditions, such as during cellular differentiation or stress response, posttranscriptional processes (delayed synthesis and/or degradation of the protein) may lead to discordance between mRNA and protein expression (47). This may explain the discordance between Glut3 mRNA and GLUT3 protein expression in the present study. Discordant Glut3 mRNA and GLUT-3 protein expression in the context of hypoxia indicates an insignificant role for GLUT-3 in hypoxia-induced adaptations in glucose transport (48). A similar effect may be present in early-life ID. Some known effects of early-life ID may explain the lack of GLUT3 protein upregulation in the hippocampus of the FID-10 group. 1) Neuronal glucose oxidation is coupled to glutamatergic neurotransmission (49). Both ID and chronic hypoxia suppress glutamatergic neurotransmission in the hippocampus (7, 50). Lack of GLUT3 protein changes may be reflective of the lower neuronal glucose oxidation and thus a lower glucose requirement in the FID-10 hippocampus. 2) ID reduces glycolytic capacity in hippocampal neurons (8). Excess neuronal glucose delivery under such conditions can generate reactive oxygen species by diverting glucose into the pentose phosphate pathway (51, 52). Lack of GLUT3 upregulation may be an adaptive response to prevent glucose-induced oxidative stress. Without confirmatory studies, all these possibilities remain conjectures. It is noteworthy that chronic hypoglycemia also does not alter regional GLUT3 expression in adult rats (37).
The iron dose used for treatment had a differential effect on hippocampal GLUT1 expression. We have previously demonstrated that treatment using the 40 mg/kg iron diet corrects anemia and normalizes tissue iron concentration by P21 in this model (4) (Supplemental Table 1). Paralleling this effect, hippocampal Glut1 mRNA and GLUT1 55-kDa protein expression in the FID-40 group was found to be comparable with the control group (Figure 2). Normalization of Glut1 mRNA expression in the iron-replete DNTfR1−/− mice suggests that correction of tissue ID was likely responsible for normalization of GLUT1 expression in the FID-40 group. However, correction of hypoxia, secondary to the resolution of anemia, also may have played a role (34). The expression of the GLUT1 45-kDa protein isoform was lower in the FID-40 group. Because the GLUT1 45-kDa isoform is primarily expressed in astrocytes and oligodendrocytes (15), this may suggest lower glial number and/or function. Consistent with this possibility, our previous study demonstrated evidence of abnormal myelin synthesis, an oligodendrocyte-mediated process, in the hippocampus of the FID-40 group (5).
Unlike the standard iron dose, treatment using high-iron diet from P8 to P21 led to hippocampal GLUT1 suppression at adulthood. We have previously demonstrated that both the standard and the high iron doses have equivalent efficacy in correcting anemia and achieving tissue iron sufficiency in this model, and that the high-dose iron does not lead to increased tissue iron concentration in the brain regions (4) (Supplemental Table 1). However, unlike the control, FID-10, and FID-40 groups, hippocampal tissue iron concentration does not increase beyond P21 in the FID-400 group, leading to 14% lower iron concentration at P100 (4). The mechanism of this effect has yet to be determined. Nevertheless, the effect appears specific to the hippocampus because tissue iron concentration is not affected in the other brain regions. In this respect, the FID-400 group is comparable with the FID-10 group, with the hippocampus demonstrating tissue ID in both conditions, albeit not to the same degree (14% lower in the FID-400 group and 21% lower in the FID-10 group) (4). Hippocampal neurochemistry and performance in hippocampus-mediated behaviors also are impaired in the 2 groups (4, 5). The changes in hippocampal GLUT1 expression were not identical in the 2 groups, however. Whereas GLUT1 expression was upregulated in the FID-10 group, it was suppressed in the FID-400 group. A loss of hippocampal tissue is unlikely to explain the lower GLUT1 expression. A prior study did not find neuronal or glial loss in the hippocampus of the FID-400 group (5). We posit that GLUT1 suppression in the FID-400 group represents a programming effect of excess iron delivery on the ontogeny of hippocampal GLUT1 expression during development. Such an effect has been described in other adverse perinatal conditions (53, 54). Early-life ID upregulates iron transporter expression in the developing hippocampus (55), thus increasing the potential for excess iron delivery to this brain region. As mentioned, hippocampal GLUT1 expression doubles between P14 and P21 (17). Exposure to high iron during this period may arrest this process, leading to lower GLUT1 expression at adulthood. Additional studies are necessary to confirm this possibility. Unlike the situation with FID-10, both 55-kDa and 45-kDa GLUT1 isoforms were suppressed in the FID-400 group, suggesting that glucose uptake is impaired at both the BBB and glia. Decreased GLUT1 expression in a brain region is associated with lower glucose uptake, lipid synthesis, and behavioral deficits (56, 57). Consistent with this possibility, our prior studies have demonstrated abnormal phospholipid synthesis in the hippocampus, as well as hippocampus-mediated behavioral deficits in the FID-400 group (4, 5).
In summary, early-life ID that is inadequately treated, or treated using higher than the standard iron dose, alters GLUT1 expression in the hippocampus of adult rats and mice. That GLUT changes are limited to the hippocampus highlights the vulnerability of this region during early-life ID. Because well-regulated glucose delivery is essential for optimal hippocampal function (58), altered hippocampal GLUT1 expression may partially explain the previously reported hippocampal functional deficits in this model (4, 5). From a translational standpoint, early-life ID predisposes to ID in childhood (59, 60), a situation modeled by the FID-10 group in the present study. Similarly, daily iron delivery to the pups in the FID-400 group (6 mg · kg−1 · d−1) (4, 5) is the same as the iron dose recommended for treating IDA in infants and children (61). The potential for long-term changes in GLUT expression suggests the need for rethinking treatment strategies for early-life ID.
Supplementary Material
Acknowledgments
We acknowledge the technical assistance of Amy Hurst, Roshny Vijayakar, and Laura Dahl in the experiments. The authors’ responsibilities were as follows—RR: designed the research and had primary responsibility for final content; KE, BF, and RR: conducted the research; KE and RR: analyzed the data; KE, MKG, and RR: wrote the paper; and all authors: read and approved the final manuscript.
Notes
Supported by Eunice Kennedy Shriver National Institute of Child Health and Human Development of the NIH award numbers P01HD39386, R01HD29241 (to MKG), and R21HD54490 (to MKG).
Author disclosures: KE, BF, MKG, and RR, no conflicts of interest.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Supplemental Tables 1–3 and Supplemental Figures 1 and 2 are available from the “Supplementary data” link in the online posting of the article and from the same link in the online table of contents at https://academic.oup.com/jn/.
Abbreviations used: au, arbitrary units; BBB, blood–brain barrier; DNTfR1, dominant negative transferrin receptor; FID, formerly iron-deficient; GLUT, glucose transporter; HIF, hypoxia inducible factor; ID, iron deficiency; IDA, iron deficiency anemia; LDH, lactate dehydrogenase; P, postnatal day; PHD2, prolyl hydroxylase-2; TfR, transferrin receptor; VEGF, vascular endothelial growth factor; WT, wild-type.
References
- 1. Lozoff B, Beard J, Connor J, Barbara F, Georgieff M, Schallert T. Long-lasting neural and behavioral effects of iron deficiency in infancy. Nutr Rev. 2006;64:S34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Siddappa AM, Georgieff MK, Wewerka S, Worwa C, Nelson CA, Deregnier RA. Iron deficiency alters auditory recognition memory in newborn infants of diabetic mothers. Pediatr Res. 2004;55:1034–41. [DOI] [PubMed] [Google Scholar]
- 3. Rao R, Tkac I, Schmidt AT, Georgieff MK. Fetal and neonatal iron deficiency causes volume loss and alters the neurochemical profile of the adult rat hippocampus. Nutr Neurosci. 2011;14:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Unger EL, Hurst AR, Georgieff MK, Schallert T, Rao R, Connor JR, Kaciroti N, Lozoff B, Felt B. Behavior and monoamine deficits in prenatal and perinatal iron deficiency are not corrected by early postnatal moderate-iron or high-iron diets in rats. J Nutr. 2012;142:2040–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rao R, Tkac I, Unger EL, Ennis K, Hurst A, Schallert T, Connor J, Felt B, Georgieff MK. Iron supplementation dose for perinatal iron deficiency differentially alters the neurochemistry of the frontal cortex and hippocampus in adult rats. Pediatr Res. 2013;73:31–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. deUngria M, Rao R, Wobken JD, Luciana M, Nelson CA, Georgieff MK. Perinatal iron deficiency decreases cytochrome c oxidase (CytOx) activity in selected regions of neonatal rat brain. Pediatr Res. 2000;48:169–76. [DOI] [PubMed] [Google Scholar]
- 7. Rao R, Tkac I, Townsend EL, Gruetter R, Georgieff MK. Perinatal iron deficiency alters the neurochemical profile of the developing rat hippocampus. J Nutr. 2003;133:3215–21. [DOI] [PubMed] [Google Scholar]
- 8. Bastian TW, von Hohenberg WC, Mickelson DJ, Lanier LM, Georgieff MK. Iron deficiency impairs developing hippocampal neuron gene expression, energy metabolism, and dendrite complexity. Dev Neurosci. 2016;38:264–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tran PV, Dakoji S, Reise KH, Storey KK, Georgieff MK. Fetal iron deficiency alters the proteome of adult rat hippocampal synaptosomes. Am J Physiol Regul Integr Comp Physiol. 2013;305:R1297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oexle H, Gnaiger E, Weiss G. Iron-dependent changes in cellular energy metabolism: influence on citric acid cycle and oxidative phosphorylation. Biochim Biophys Acta. 1999;1413:99–107. [DOI] [PubMed] [Google Scholar]
- 11. Dongiovanni P, Valenti L, Ludovica Fracanzani A, Gatti S, Cairo G, Fargion S. Iron depletion by deferoxamine up-regulates glucose uptake and insulin signaling in hepatoma cells and in rat liver. Am J Pathol. 2008;172:738–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rao R, Ennis K, Lubach GR, Lock EF, Georgieff MK, Coe CL. Metabolomic analysis of CSF indicates brain metabolic impairment precedes hematological indices of anemia in the iron-deficient infant monkey. Nutr Neurosci. 2018;21:40–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rao R, Ennis K, Oz G, Lubach GR, Georgieff MK, Coe CL. Metabolomic analysis of cerebrospinal fluid indicates iron deficiency compromises cerebral energy metabolism in the infant monkey. Neurochem Res. 2013;38:573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vannucci SJ, Maher F, Simpson IA. Glucose transporter proteins in brain: delivery of glucose to neurons and glia. Glia. 1997;21:2–21. [DOI] [PubMed] [Google Scholar]
- 15. Yu S, Ding WG. The 45 kDa form of glucose transporter 1 (GLUT1) is localized in oligodendrocyte and astrocyte but not in microglia in the rat brain. Brain Res. 1998;797:65–72. [DOI] [PubMed] [Google Scholar]
- 16. Vannucci SJ, Clark RR, Koehler-Stec E, Li K, Smith CB, Davies P, Maher F, Simpson IA. Glucose transporter expression in brain: relationship to cerebral glucose utilization. Dev Neurosci. 1998;20:369–79. [DOI] [PubMed] [Google Scholar]
- 17. Vannucci SJ. Developmental expression of GLUT1 and GLUT3 glucose transporters in rat brain. J Neurochem. 1994;62:240–6. [DOI] [PubMed] [Google Scholar]
- 18. Carlson ES, Stead JD, Neal CR, Petryk A, Georgieff MK. Perinatal iron deficiency results in altered developmental expression of genes mediating energy metabolism and neuronal morphogenesis in hippocampus. Hippocampus. 2007;17:679–91. [DOI] [PubMed] [Google Scholar]
- 19. Bastian TW, Santarriaga S, Nguyen TA, Prohaska JR, Georgieff MK, Anderson GW. Fetal and neonatal iron deficiency but not copper deficiency increases vascular complexity in the developing rat brain. Nutr Neurosci. 2015;18:365–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weidemann A, Johnson RS. Biology of HIF-1α. Cell Death Differ. 2008;15:621–7. [DOI] [PubMed] [Google Scholar]
- 21. Acker T, Acker H. Cellular oxygen sensing need in CNS function: physiological and pathological implications. J Exp Biol. 2004;207:3171–88. [DOI] [PubMed] [Google Scholar]
- 22. Lok CN, Ponka P. Identification of a hypoxia response element in the transferrin receptor gene. J Biol Chem. 1999;274:24147–52. [DOI] [PubMed] [Google Scholar]
- 23. Fretham SJ, Carlson ES, Wobken J, Tran PV, Petryk A, Georgieff MK. Temporal manipulation of transferrin-receptor-1-dependent iron uptake identifies a sensitive period in mouse hippocampal neurodevelopment. Hippocampus. 2012;22:1691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barks A, Fretham SJ, Georgieff MK, Tran PV. Early-life neuronal-specific iron deficiency alters the adult mouse hippocampal transcriptome. J Nutr. 2018;148:1521–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rao R, Sperr D, Ennis K, Tran P. Postnatal age influences hypoglycemia-induced poly(ADP-ribose) polymerase-1 activation in the brain regions of rats. Pediatr Res. 2009;66:642–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rao AR, Quach H, Smith E, Vatassery GT, Rao R. Changes in ascorbate, glutathione and α-tocopherol concentrations in the brain regions during normal development and moderate hypoglycemia in rats. Neurosci Lett. 2014;568C:67–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL et al.. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–22. [DOI] [PubMed] [Google Scholar]
- 28. Rao R, Ennis K, Mitchell EP, Tran PV, Gewirtz JC. Recurrent moderate hypoglycemia suppresses brain-derived neurotrophic factor expression in the prefrontal cortex and impairs sensorimotor gating in the posthypoglycemic period in young rats. Dev Neurosci. 2016;38:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Satrom KM, Ennis K, Sweis BM, Matveeva TM, Chen J, Hanson L, Maheshwari A, Rao R. Neonatal hyperglycemia induces CXCL10/CXCR3 signaling and microglial activation and impairs long-term synaptogenesis in the hippocampus and alters behavior in rats. J Neuroinflammation. 2018;15:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zimmermann AS, Morrison SD, Hu MS, Li S, Nauta A, Sorkin M, Meyer NP, Walmsley GG, Maan ZN, Chan DA et al.. Epidermal or dermal specific knockout of PHD-2 enhances wound healing and minimizes ischemic injury. PLoS One. 2014;9:e93373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Henderson SA, Dallman PR, Brooks GA. Glucose turnover and oxidation are increased in the iron-deficient anemic rat. Am J Physiol. 1986;250:E414–21. [DOI] [PubMed] [Google Scholar]
- 32. Brooks GA, Henderson SA, Dallman PR. Increased glucose dependence in resting, iron-deficient rats. Am J Physiol. 1987;253:E461–6. [DOI] [PubMed] [Google Scholar]
- 33. Ben-Shachar D, Yehuda S, Finberg JP, Spanier I, Youdim MB. Selective alteration in blood–brain barrier and insulin transport in iron-deficient rats. J Neurochem. 1988;50:1434–7. [DOI] [PubMed] [Google Scholar]
- 34. Kanaan A, Farahani R, Douglas RM, Lamanna JC, Haddad GG. Effect of chronic continuous or intermittent hypoxia and reoxygenation on cerebral capillary density and myelination. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1105–14. [DOI] [PubMed] [Google Scholar]
- 35. Pollak Y, Mechlovich D, Amit T, Bar-Am O, Manov I, Mandel SA, Weinreb O, Meyron-Holtz EG, Iancu TC, Youdim MB. Effects of novel neuroprotective and neurorestorative multifunctional drugs on iron chelation and glucose metabolism. J Neural Transm (Vienna). 2013;120:37–48. [DOI] [PubMed] [Google Scholar]
- 36. Kumagai AK, Kang YS, Boado RJ, Pardridge WM. Upregulation of blood–brain barrier GLUT1 glucose transporter protein and mRNA in experimental chronic hypoglycemia. Diabetes. 1995;44:1399–404. [DOI] [PubMed] [Google Scholar]
- 37. Simpson IA, Appel NM, Hokari M, Oki J, Holman GD, Maher F, Koehler-Stec EM, Vannucci SJ, Smith QR. Blood–brain barrier glucose transporter: effects of hypo- and hyperglycemia revisited. J Neurochem. 1999;72:238–47. [DOI] [PubMed] [Google Scholar]
- 38. Rao R, Ennis K, Long JD, Ugurbil K, Gruetter R, Tkac I. Neurochemical changes in the developing rat hippocampus during prolonged hypoglycemia. J Neurochem. 2010;114:728–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cardoso S, Santos RX, Correia SC, Carvalho C, Santos MS, Baldeiras I, Oliveira CR, Moreira PI. Insulin-induced recurrent hypoglycemia exacerbates diabetic brain mitochondrial dysfunction and oxidative imbalance. Neurobiol Dis. 2013;49:1–12. [DOI] [PubMed] [Google Scholar]
- 40. Zeller K, Rahner-Welsch S, Kuschinsky W. Distribution of Glut1 glucose transporters in different brain structures compared to glucose utilization and capillary density of adult rat brains. J Cereb Blood Flow Metab. 1997;17:204–9. [DOI] [PubMed] [Google Scholar]
- 41. Blin J, Ray CA, Chase TN, Piercey MF. Regional cerebral glucose metabolism compared in rodents and humans. Brain Res. 1991;568:215–22. [DOI] [PubMed] [Google Scholar]
- 42. Stangl GI, Kirchgessner M. Effect of different degrees of moderate iron deficiency on the activities of tricarboxylic acid cycle enzymes, and the cytochrome oxidase, and the iron, copper, and zinc concentration in rat tissues. Zeitschrift fur Ernahrungswissenschaft. 1998;37:260–8. [DOI] [PubMed] [Google Scholar]
- 43. Tanaka M, Kariya F, Kaihatsu K, Nakamura K, Asakura T, Kuroda Y, Ohira Y. Effects of chronic iron deficiency anemia on brain metabolism. Jpn J Physiol. 1995;45:257–63. [DOI] [PubMed] [Google Scholar]
- 44. Zapata-Morales JR, Galicia-Cruz OG, Franco M, Martinez y Morales F. Hypoxia-inducible factor-1α (HIF-1α) protein diminishes sodium glucose transport 1 (SGLT1) and SGLT2 protein expression in renal epithelial tubular cells (LLC-PK1) under hypoxia. J Biol Chem. 2014;289:346–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jones NM, Bergeron M. Hypoxic preconditioning induces changes in HIF-1 target genes in neonatal rat brain. J Cereb Blood Flow Metab. 2001;21:1105–14. [DOI] [PubMed] [Google Scholar]
- 46. Bergeron M, Gidday JM, Yu AY, Semenza GL, Ferriero DM, Sharp FR. Role of hypoxia-inducible factor-1 in hypoxia-induced ischemic tolerance in neonatal rat brain. Ann Neurol. 2000;48:285–96. [PubMed] [Google Scholar]
- 47. Liu Y, Beyer A, Aebersold R. On the dependency of cellular protein levels on mRNA abundance. Cell. 2016;165:535–50. [DOI] [PubMed] [Google Scholar]
- 48. Wood IS, Wang B, Lorente-Cebrian S, Trayhurn P. Hypoxia increases expression of selective facilitative glucose transporters (GLUT) and 2-deoxy-D-glucose uptake in human adipocytes. Biochem Biophys Res Commun. 2007;361:468–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Patel AB, de Graaf RA, Mason GF, Kanamatsu T, Rothman DL, Shulman RG, Behar KL. Glutamatergic neurotransmission and neuronal glucose oxidation are coupled during intense neuronal activation. J Cereb Blood Flow Metab. 2004;24:972–85. [DOI] [PubMed] [Google Scholar]
- 50. Raman L, Tkac I, Ennis K, Georgieff MK, Gruetter R, Rao R. In vivo effect of chronic hypoxia on the neurochemical profile of the developing rat hippocampus. Brain Res Dev Brain Res. 2005;156:202–9. [DOI] [PubMed] [Google Scholar]
- 51. Suh SW, Gum ET, Hamby AM, Chan PH, Swanson RA. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J Clin Invest. 2007;117:910–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Suh SW, Aoyama K, Matsumori Y, Liu J, Swanson RA. Pyruvate administered after severe hypoglycemia reduces neuronal death and cognitive impairment. Diabetes. 2005;54:1452–8. [DOI] [PubMed] [Google Scholar]
- 53. Vingan RD, Dow-Edwards DL, Riley EP. Cerebral metabolic alterations in rats following prenatal alcohol exposure: a deoxyglucose study. Alcohol Clin Exp Res. 1986;10:22–6. [DOI] [PubMed] [Google Scholar]
- 54. Eckstein LW, Shibley IA Jr, Pennington JS, Carver FM, Pennington SN. Changes in brain glucose levels and glucose transporter protein isoforms in alcohol- or nicotine-treated chick embryos. Brain Res Dev Brain Res. 1997;103:59–65. [DOI] [PubMed] [Google Scholar]
- 55. Siddappa AJ, Rao RB, Wobken JD, Casperson K, Leibold EA, Connor JR, Georgieff MK. Iron deficiency alters iron regulatory protein and iron transport protein expression in the perinatal rat brain. Pediatr Res. 2003;53:800–7. [DOI] [PubMed] [Google Scholar]
- 56. Marin-Valencia I, Good LB, Ma Q, Duarte J, Bottiglieri T, Sinton CM, Heilig CW, Pascual JM. Glut1 deficiency (G1D): epilepsy and metabolic dysfunction in a mouse model of the most common human phenotype. Neurobiol Dis. 2012;48:92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hargrave SL, Davidson TL, Lee TJ, Kinzig KP. Brain and behavioral perturbations in rats following Western diet access. Appetite. 2015;93:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McNay EC, Sherwin RS. Effect of recurrent hypoglycemia on spatial cognition and cognitive metabolism in normal and diabetic rats. Diabetes. 2004;53:418–25. [DOI] [PubMed] [Google Scholar]
- 59. Demirchyan A, Petrosyan V, Sargsyan V, Hekimian K. Prevalence and determinants of anaemia among children aged 0–59 months in a rural region of Armenia: a case-control study. Public Health Nutr. 2016;19:1260–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gessner BD. Early childhood hemoglobin level is a strong predictor of hemoglobin levels during later childhood among low-income Alaska children. Int J Circumpolar Health. 2009;68:459–70. [DOI] [PubMed] [Google Scholar]
- 61. Baker RD, Greer FR; The Committee on Nutrition. Diagnosis and prevention of iron deficiency and iron-deficiency anemia in infants and young children (0–3 years of age). Pediatrics. 2010;126:1040–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.