

Abstract
DEPDC5 is now recognized as one of the genes most often implicated in familial/inherited focal epilepsy and brain malformations. Individuals with pathogenic variants in DEPDC5 are at risk for epilepsy, associated neuropsychiatric comorbidities and sudden unexplained death in epilepsy. Depdc5flox/flox-Syn1Cre (Depdc5cc+) neuronal-specific Depdc5 knockout mice exhibit seizures and neuronal mTORC1 hyperactivation. It is not known if Depdc5cc+ mice have a hyperactivity/anxiety phenotype, die early from terminal seizures or whether mTOR inhibitors rescue DEPDC5-related seizures and associated comorbidities. Herein, we report that Depdc5cc+ mice were hyperactive in open-field testing but did not display anxiety-like behaviors on the elevated-plus maze. Unlike many other mTOR-related models, Depdc5cc+ mice had minimal epileptiform activity and rare seizures prior to seizure-induced death, as confirmed by video-EEG monitoring. Treatment with the mTORC1 inhibitor rapamycin starting after 3 weeks of age significantly prolonged the survival of Depdc5cc+ mice and partially rescued the behavioral hyperactivity. Rapamycin decreased the enlarged brain size of Depdc5cc+ mice with corresponding decrease in neuronal soma size. Loss of Depdc5 led to a decrease in the other GATOR1 protein levels (NPRL2 and NPRL3). Rapamycin failed to rescue GATOR1 protein levels but rather rescued downstream mTORC1 hyperactivity as measured by phosphorylation of S6. Collectively, our data provide the first evidence of behavioral alterations in mice with Depdc5 loss and support mTOR inhibition as a rational therapeutic strategy for DEPDC5-related epilepsy in humans.
Introduction
DEPDC5 is among the most commonly identified genes associated with epilepsy (1). Pathogenic loss-of-function variants in DEPDC5 have been identified in autosomal dominant familial focal epilepsy (OMIM# 604364) (2–4), sporadic focal epilepsy (5), infantile spasms (6) and focal epileptic brain malformations (7–9). Indeed, the phenotypic range now associated with pathogenic variants in DEPDC5 is broad and continues to expand. In a recent series of 63 patients with DEPDC5 variants, drug-resistant seizures occurred in over 50% of patients, sudden unexplained death in epilepsy (SUDEP) occurred in 13% of families, and psychiatric comorbidities such as attention deficit hyperactivity disorder were also common (10). Current treatments are not targeted and often lack efficacy; thus, it is critical to identify targeted treatments for DEPDC5-related conditions.
Only recently has the role of DEPDC5 been studied in the brain. The protein product of DEPDC5, DEP domain-containing protein 5 (DEPDC5), is a crucial regulator of mTOR complex 1 (mTORC1) (11). DEPDC5 together with NPRL2 and NPRL3 form the GATOR1 complex, which is crucial for mTORC1-mediated nutrient sensing (12,13). In vitro cell lines missing a single GATOR1 component have reduced expression of remaining GATOR1 proteins and hyperactive mTORC1 (11). DEPDC5 is ubiquitously and constitutively expressed in the developing and adult brain, and its expression is largely restricted to neurons (2). Knockdown of Depdc5 or Nprl3 in mouse neural precursor cells in vitro increases neuronal soma size and increases mTORC1 activation as measured by the downstream phosphorylation of the ribosomal protein S6 (p-S6 Ser240/244) (14). mTORC1 activity is the final common pathway for upstream signals regulated by either the tuberous sclerosis complex (TSC) or the GATOR1 complex (15). mTOR dysregulation is associated with many neurological and psychiatric disorder and represents an exciting new target to address in the development of rational treatment (16). The mTORC1 inhibitors rapamycin and an analogue, everolimus, have successfully treated seizures in children with TSC (17,18). We recently demonstrated rapamycin treatment prior to 6 weeks of age rescued cellular and behavioral phenotypes in a Tsc1 mouse model (19). mTORC1 inhibitors may be potential treatments for DEPDC5-related conditions.
Depdc5 knockout models have only recently been generated. Germline homozygous knockout Depdc5 rat and mouse models are embryonic lethal, while the germline heterozygous Depdc5 rodents have minimal pathology and no seizures (20,21). Zebrafish Depdc5 knockdown models have demonstrated seizure-like activity, increased mTOR activity (22) and mTOR-dependent hyperactive motor behavior (23). We and two other groups independently developed brain-specific Depdc5 knockout rodent models (24–26). Depdc5 brain-specific knockout models demonstrate increased mTOR activity in dysplastic and enlarged cortical neurons, similar to resected brain tissue from a patient with DEPDC5-related epilepsy and brain malformation (7). Behavioral measures including hyperactivity have yet to be done in these models. In addition, the hypothesized therapeutic potential of mTORC1 inhibitors to reduce the increased mTOR signaling and other phenotypes associated with DEPDC5 loss has yet to be tested.
This study aims to determine if mTORC1 is a rational therapeutic target for DEPDC5-related conditions. We found that Depdc5cc+ mice exhibit hyperactive behaviors and die from terminal seizures. Long-term treatment with rapamycin rescued the increase in brain size, cortical thickness and neuronal soma size seen after Depdc5 loss. Rapamycin treatment reduced downstream mTORC1 hyperactivity but did not rescue the GATOR1 complex protein levels after Depdc5 loss. Finally, rapamycin prolonged the survival and partially rescued the behavioral hyperactivity of Depdc5cc+ mice. Taken together, we demonstrated that chronic rapamycin treatment rescued the behavioral deficits from neuronal Depdc5 loss and normalized neuronal mTORC1 hyperactivation.
Results
Neuronal Depdc5 loss results in early mortality from seizure-induced death
Our neuronal-specific Depdc5 knockout mouse model has a conditional allele and expresses Cre under neuron-specific synapsin-1 promoter, which is expressed in most differentiated neuronal populations (24). We previously demonstrated that loss of DEPDC5 results in early mortality and mTORC1 hyperactivation. To clarify the reduced survival, we performed video-electroencephalography (EEG) recordings to determine whether seizures led to early mortality. Depdc5cc+ mice with selective neuronal loss of DEPDC5 exhibit early mortality, similar to our prior results (Fig. 1A). The median age of spontaneous death of Depdc5cc+ mice (n = 45) was 123 days old (interquartile range 105–139) and was not different between male and female mice. No unexpected deaths occurred in littermate control mice (n = 43). We suspected the early mortality of Depdc5cc+ mice may be a result of spontaneous seizures.
Figure 1.
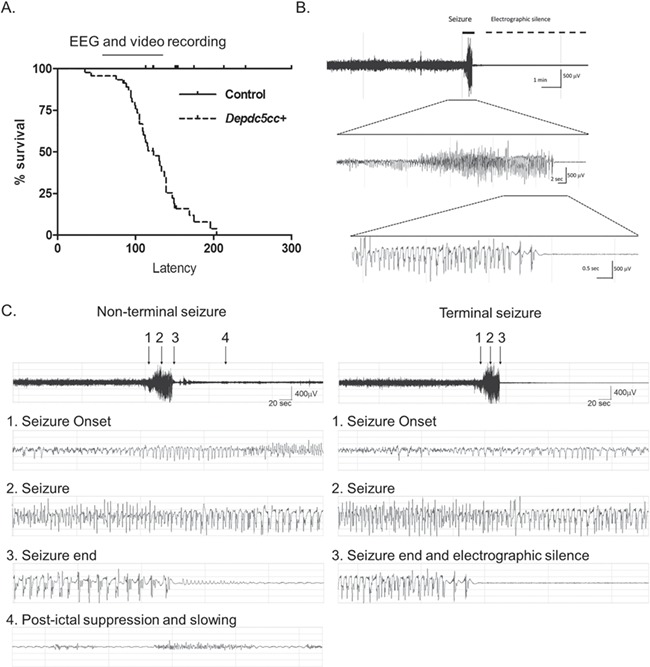
Neuronal Depdc5 loss in mice results in seizure-induced death. (A) Neuronal-specific Depdc5 knockout mice (Depdc5cc+) have a shortened lifespan compared to littermate controls (Depdc5cc+ n = 45, median survival = 123 days; littermate controls n = 43). The bar over the survival curve represents the range of ages of which video and/or EEG telemetry was recorded. (B) EEG tracing of a terminal seizure in a Depdc5cc+ mouse demonstrating a short 35 s seizure followed by death and electrographic silence, as seen in Video 1. (C) EEG tracing of a non-terminal and terminal seizure in a single animal. The initial seizure occurred 24 h prior to the terminal event. Note the electrographic similarities between the two events at (1) seizure onset, (2) middle of the seizure and (3) seizure termination. Only the survival event has persistent electrographic activity as manifested by (4) postictal slowing and suppression.
To confirm seizure-induced death, we implanted seven Depdc5cc+ and four littermate control mice with wireless EEG telemetry prior to the median age Depdc5cc+ mice died (Table 1). No littermate control animals died from surgical complications or exhibit seizures during EEG recording. Two of the seven (29%) Depdc5cc+ mice died due to surgical complications with EEG transmitter implantation. The remaining five Depdc5cc+ mice all died due to witnessed spontaneous seizures with a median age of death of 133 days (range 105–144). Unfortunately, only video was recorded at the time of death in two animals because the wireless EEG telemetry battery died during the monitoring period. One animal died shortly after a cage change before the monitor was turned back on, but the witnessed terminal seizure was less than 30 s in duration and similar to those captured on video. The terminal seizure duration and clinical presentation of the seizures were similar across all five animals (median seizure duration 22 s, range 19–36 s). A representative EEG from the animals with EEG and video at time of death demonstrated high-amplitude rhythmic discharges during a generalized tonic seizure with hind limb extension followed by electrographic silence and death (Fig. 1B, Video 1). The entire video-EEG recording prior to terminal seizures was reviewed across all animals (median duration 6 days, range 1–36 days). Non-terminal seizures were rare with a single seizure event detected in three animals the day prior to the terminal seizure. One of these non-terminal seizure events occurred in a cluster of two short tonic seizures and intermittent freezing behavior lasted a total of 210 s before returning to baseline. The other two events were single tonic seizures. The median duration was equivalent between non-terminal and terminal seizures (25 versus 22 s, respectively) (Fig. 1C, Video 1). Taken together, Depdc5cc+ mice experience early mortality from a terminal seizure that is similar in duration to rare non-terminal seizures.
Table 1.
Depdc5cc+ and control mice monitored for seizures with video and/or implanted wireless EEG telemetry
| Genotype | Number (male/female) | Died in surgery or euthanized N (median age; range) | Age recording started (median; range) | Monitoring duration (median days; range) | Age of spontaneous death (median; range) | Seizure-induced death witnessed or video N (% of recorded mice) | Seizure-induced death on EEG and video N (% of recorded mice) | Seizure-induced death total N (% of recorded mice) |
|---|---|---|---|---|---|---|---|---|
| Depdc5cc+ | 7 (4/3) | 2 (98; 91–105)a | 104 (91–138) | 6 (1–36) | 133 (105–144) | 3 (60%) | 2 (40%) | 5 (100%) |
| Control | 4 (2/2) | 4 (113; 97–200)b | 105 (94–135) | 9 (3–65) | n/a | 0 (0%) | 0 (0%) | 0 (0%) |
aOne died during surgery and one euthanized a day after surgery due to illness.
bNo surgical complications and mice were euthanized after several days of EEG monitoring without clinical seizures or signs of illness.
Neuronal Depdc5 loss results in hyperactivity but not anxiety in mice
In addition to terminal seizures, it is not known whether Depdc5cc+ mice exhibit behavioral deficits, such as hyperactivity, which was identified in a zebrafish Depdc5 knockdown model (23). We quantified locomotor activity using the open-field assay. Adult Depdc5cc+ mice (n = 8) exhibited increased locomotor activity throughout the testing period compared to littermate controls (n = 13) and heterozygous Depdc5cw+ mice (n = 6) (Fig. 2A, P < 0.0001, one-way analysis of variance (ANOVA) with Tukey’s post hoc test). Heterozygous Depdc5cw+ mice performed similarly when compared to littermate controls. We utilized the elevated plus-maze test as a measure of anxiety-related behaviors. We noted a trend towards a difference in both closed and open arm entries, but no measure rose to the level of significance across genotypes (Fig. 2B). Taken together, these data indicate that neuronal Depdc5 loss in mice results in behavioral hyperactivity but not anxiety.
Figure 2.
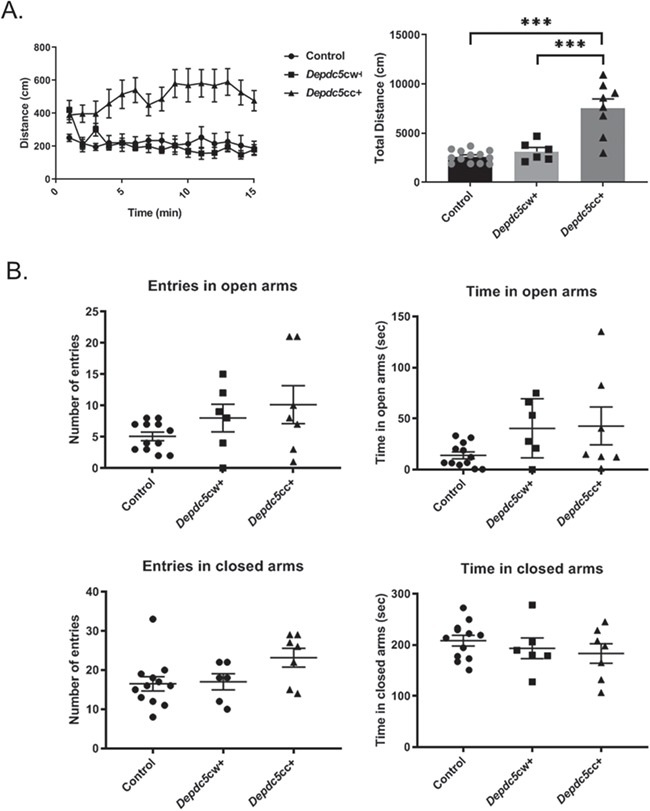
Neuronal Depdc5 loss results in hyperactivity but not anxiety in mice. (A) Left: Depdc5cc+ (n = 8) have increased distance traveled throughout the 15-min open-field paradigm compared to littermate heterozygous Depdc5cw+ (n = 6) or control (n = 13) mice. Right: Depdc5cc+ cumulative distance traveled is increased [F (2,24) = 25.57, P < 0.0001, one-way ANOVA; Tukey’s post hoc test ***P < 0.001]. (B) Elevated plus-maze entries and duration into the open arms closed and open arms were measured in littermate Depdc5cc+ (n = 7), Depdc5cw+ (n = 6), control (n = 12) mice. All mice were male and female littermates >100 days old. Error bars represent SEM.
Chronic rapamycin decreases brain and neuronal size of Depdc5cc+ mice without changes in body weight
Loss of Depdc5 increases mTORC1 activity in mouse cortical neurons (24). We tested if chronic inhibition of mTORC1 by rapamycin treatment rescues the behavioral and biochemical alterations from neuronal Depdc5 loss. Littermates from five consecutive litters in each genotype were randomly assigned treatment with 6 mg/kg rapamycin or vehicle control 3 days a week for up to 7 months (Table 2). We began treatment with rapamycin in early adulthood at 1 month of age (3–5 weeks). Rapamycin treatment dosage was based upon our prior pharmacokinetic study of rapamycin (27). The rapamycin treatment timing was similar to our recent study in Tsc1 conditional knockout mice (19). Vehicle-treated Depdc5cc+ mice weighed less than vehicle-treated littermate controls (Fig. 3A, two-way ANOVA with Tukey’s post hoc test P = 0.0123). In contrast, vehicle- and rapamycin-treated Depdc5cc+ mice weights were not different. No mice in any treatment group exhibited overt signs of illness.
Table 2.
Animals treated with 6 mg/kg of rapamycin every other day starting 1 month of age
| Vehicle | Rapamycin (6 mg/kg) every other day | |||||
|---|---|---|---|---|---|---|
| Control | Depdc5cw+ | Depdc5cc+ | Control | Depdc5cw+ | Depdc5cc+ | |
| N per group | 10 | 4 | 6 | 9 | 7 | 9 |
| Male/Female | 6/4 | 2/2 | 3/3 | 5/4 | 4/3 | 7/2 |
| Sacrificed prior to 200 days | 3 | 2 | 3a | 4b | 3 | 3 |
| Survival to 200 days N (M/F) | 7 (4/3) | 2 (1/1) | 0 | 5 (4/1) | 4 (2/2) | 5 (3/2) |
| Spontaneous death prior to 200 days N (% total) | 0 | 0 | 3 (50%) | 0 | 0 | 1 (11.1%) |
| Median age of spontaneous death (range) | − | − | 126 (93–128) | − | − | 163 (n/a) |
aOne vehicle-treated Depdc5cc+ and bone rapamycin-treated littermate control was sacrificed due to fighting wounds at 58 and 71 days, respectively. After 50% of the vehicle-treated Depdc5cc+ died, the litters with remaining vehicle-treated Depdc5cc+ mice were sacrificed for pathology and biochemical experiments.
Figure 3.
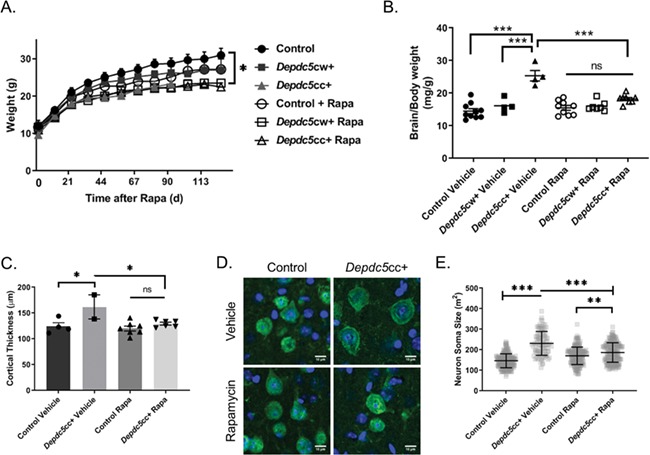
Chronic rapamycin treatment normalizes the cortical thickness and neuron size in Depdc5cc+ mice brains. (A) Animal weights after initiation of rapamycin or vehicle treatment demonstrate a difference across genotypes and treatments [Treatment: F (1,36) = 7.258, P = 0.0107; Genotype: F (2,36) = 7.612, P = 0.0017, two-way ANOVA]. *Tukey’s post hoc analysis: vehicle-control and vehicle-treated Depdc5cc+, P = 0.012; vehicle-control and rapamycin-treated Depdc5cc+ (P = 0.0013). (B) Brain to body weight measurements were compared between Depdc5cc+ and control littermates treated with vehicle or rapamycin [Genotype: F (2,36) = 36.42, P < 0.0001; Treatment: F (1,36) = 9.007, P = 0.0049; Interaction: F (2,36) = 13.34, P < 0.0001, two-way ANOVA; Tukey’s post hoc test ***P < 0.0001 vehicle Depdc5cc+ compared to all other groups, **P = 0.0094 vehicle control versus rapamycin-treated Depdc5cc+]. Symbols represent each animal in a group. Error bars are +/− SEM. (C) Cortical thickness was averaged per animal by measuring at least six independent locations per section of matched frontal cortex from each group [two-way ANOVA, Genotype: F (1,15) = 11.16, P = 0.0045; Treatment: F (1,15) = 6.723, P = 0.0204; Interaction: F (1,15) = 3.948, P = 0.0655; Tukey’s post hoc test *P = 0.0291 vehicle control versus Depdc5cc+, *P = 0.0496 Depdc5cc+ vehicle versus rapamycin-treated]. Symbols represent the average of each animal in a group. Error bars are +/− SEM. (D) NeuN-stained pyramidal cortical neurons from matched locations of M1 motor cortex obtained by confocal microscopy. Scale = 10 μm. (E) Quantified neuronal soma area by ImageJ from 100 layer V NeuN+ cortical neurons from matched locations of M1 motor cortex per genotype (n = 4 per genotype, except n = 2 for vehicle-treated Depdc5cc+). Two-way ANOVA Interaction: F (1,696) = 95.27, P < 0.0001, Genotype: F (1,696) = 8.155, P = 0.0044, Treatment: F (1,696) = 204.7, P < 0.0001; Tukey’s post hoc test ***P < 0.0001, **P = 0.0020. Error bars are +/− SD.
Depdc5cc+ mice have increased brain size compared to littermate controls (24). We measured if chronic rapamycin treatment started after 3 weeks of age could normalize increase in the brain size of Depdc5cc+ mice. All rapamycin-treated animals were treated for a minimum of 8 weeks before brain weight measurements. To avoid bias of the body weight effects of rapamycin, we normalized brain weight to body weight. Both treatment and genotype influenced brain size [Genotype: F (2,36) = 36.42, P < 0.0001; Treatment: F (1,36) = 9.007, P = 0.0049; Interaction: F (2,36) = 13.34, P < 0.0001, two-way ANOVA]. Brains of vehicle-treated Depdc5cc+ mice were larger than vehicle-treated littermate controls (Fig. 3B, P < 0.0001, two-way ANOVA with Tukey’s post hoc test). Rapamycin treatment of Depdc5cc+ mice significantly reduced the brain weight compared to vehicle-treated Depdc5cc+ mice (P < 0.0001, two-way ANOVA with Tukey’s post hoc test) and was not significantly different from rapamycin-treated control mice.
We evaluated if changes in cortical thickness corresponded to the brain size after rapamycin treatment. Measurements of cortical thickness were taken from sections of motor cortex using matched sections across all animals. As expected, vehicle-treated Depdc5cc+ mice cortical thickness was increased compared to vehicle-treated littermate controls (Fig. 3C, P = 0.0291, two-way ANOVA with Tukey’s post hoc test). Rapamycin-treatment of Depdc5cc+ mice reduced the cortical thickness back to vehicle-treated control size. There was no difference between vehicle and rapamycin-treated littermate control mice.
We previously reported that the increase in cortical thickness of Depdc5cc+ mice was due to increased neuronal cell size but not increased cell density (24). Thus, it is possible that rapamycin reduction in brain size and cortical thickness of Depdc5cc+ mice may be due to decreasing neuronal cell size. We measured the cell soma from layer V pyramidal neurons from the motor cortex using matched sections in each treatment group. Neurons from the vehicle-treated Depdc5cc+ cortex were significantly larger than vehicle-treated controls (Fig. 3D and E, P < 0.0001, two-way ANOVA with Tukey’s post hoc test). Rapamycin treatment of Depdc5cc+ mice reduced the neuronal soma size compared to vehicle-treated Depdc5cc+ mice both qualitatively (Fig. 3D) and quantitatively (Fig. 3E, P < 0.0001, two-way ANOVA with Tukey’s post hoc test). However, rapamycin-treated Depdc5cc+ neurons were not completely normalized in cell size compared to control neurons (P < 0.0001 compared to vehicle-treated controls, P = 0.002 compared to rapamycin-treated controls, two-way ANOVA with Tukey’s post hoc test).
Chronic rapamycin rescues hyperactive mTORC1 signaling in the brain but does not alter GATOR1 complex protein levels after DEPDC5 loss
Neuronal DEPDC5 loss results in hyperactive mTORC1 signaling, as measured by the increased phosphorylation of downstream ribosomal S6 (p-S6 on Ser240/244) (14,24). Therefore, we tested if rapamycin also rescued the increased p-S6 in Depdc5cc+ cortical neurons. p-S6 levels of vehicle-treated Depdc5cc+ cortical lysates were increased [Fig. 4A; F (3,14) = 178.6, P < 0.0001, one-way ANOVA with Tukey’s post hoc test], similar to our prior results (24). Importantly, rapamycin treatment of Depdc5cc+ mice reduced p-S6 levels in cortical lysates back to vehicle-treated control levels (Fig. 4A).
Figure 4.
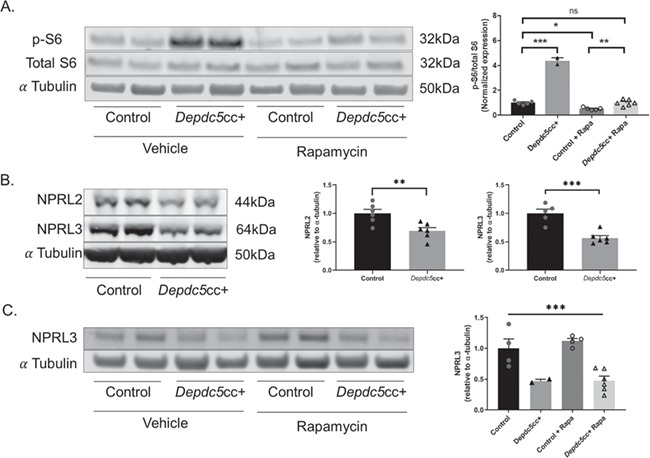
Chronic rapamycin treatment rescues hyperactive mTORC1 signaling in the brain after DEPDC5 loss but does not alter GATOR1 complex protein levels. (A) Immunoblots (left) and quantitative analysis (right) of cortical brain lysates from Depdc5cc+ and control littermates treated with vehicle or rapamycin (n ≥ 4 mice per group except n = 2 for vehicle-treated Depdc5cc+) for p-S6 (S240/244) [two-way ANOVA, Genotype effect: F (1,14) = 326.4, P < 0.0001; Treatment effect: F (1,14) = 326.2, P < 0.0001; Interaction effect: F (1, 4) = 183.7, P < 0.0001; Tukey’s post hoc analysis *P = 0.0117, **P = 0.0085, ***P < 0.0001]. (B) Immunoblots (left) and quantitative analysis (right) of cortical brain lysates from adult Depdc5cc+ and littermate control mice (n ≥ 5 per genotype) demonstrate a reduction in the other components of the GATOR1 complex, NPRL2 (unpaired t-test, t = 3.364, P = 0.0072) and NPRL3 (unpaired t-test, t = 5.255, P = 0.0005). (C) Immunoblots (left) and quantitative analysis (right) of cortical brain lysates from Depdc5cc+ and control littermates treated with vehicle or rapamycin for NPRL3 [two-way ANOVA, Genotype effect: F (1,12) = 30.08, ***P = 0.0001; Treatment effect: F (1,12) = 0.3331, P = 0.57]. Expression of levels was normalized to alpha-tubulin, and p-S6 (S240/244) was normalized to total levels of S6. All ratios for the control samples are normalized to 1. Symbols represent each brain sample. Mean ± SEM.
DEPDC5 is a critical component of the GATOR1 complex, a negative regulator of mTORC1 activity (11). In HEK cells, knockdown of DEPDC5 results in loss of the other GATOR1 complex proteins NPRL2 and NPRL3 (12). Using Depdc5cc+ mice, we asked whether neuronal DEPDC5 loss results in a reduction of the other GATOR1 complex proteins, NPRL2 and NPRL3. We found a reduction in both NPRL2 (P = 0.0072, unpaired two-tailed t-test) and NPRL3 (P = 0.0005, unpaired two-tailed t-test) levels in Depdc5cc+ cortical lysates. These data provide evidence that DEPDC5 is critical to maintain levels of the other GATOR1 complex proteins, NPRL2 and NPRL3, in the brain in vivo. Finally, we tested if rapamycin could rescue the GATOR1 complex using NPRL3 as a marker. Cortical lysates from vehicle-treated and rapamycin-treated Depdc5cc+ mice had lower NPRL3 levels compared to littermate control samples but were unchanged by rapamycin (Fig. 4C). Thus, rapamycin failed to rescue the other components of the GATOR1 complex in the absence of DEPDC5.
Chronic rapamycin rescues increased neuronal mTORC1 signaling and astrogliosis in the brains of Depdc5cc+ mice
We stained anatomically matched brain sections from each genotype and treatment to determine if the increased p-S6 due to DEPDC5 loss was isolated to neurons. In cortical sections co-stained for p-S6 and the neuronal marker, NeuN, we found intense p-S6 staining colocalized with large NeuN-positive neurons throughout the cortical layers of Depdc5cc+ mice (Fig. 5A). In comparison, the size and p-S6 intensity of rapamycin-treated Depdc5cc+ neurons were dramatically reduced and similar to littermate control neurons. GFAP+ astrocytes did not colocalize with p-S6 staining in the cortex across genotypes (Fig. 5B). GFAP+ astrocytes were sparse in the cortex of vehicle-treated control mice. Larger and more intensely GFAP-stained astrocytes were notable throughout the cortex of vehicle-treated Depdc5cc+ mice, similar to our prior results (24). After chronic rapamycin treatment, GFAP+ astrocytes were dramatically reduced in the cortex of rapamycin-treated Depdc5cc+ mice compared to vehicle-treated Depdc5cc+ mice. A decrease in GFAP+ astrocytes was also noted in rapamycin-treated control animals when compared to vehicle-treated controls.
Figure 5.
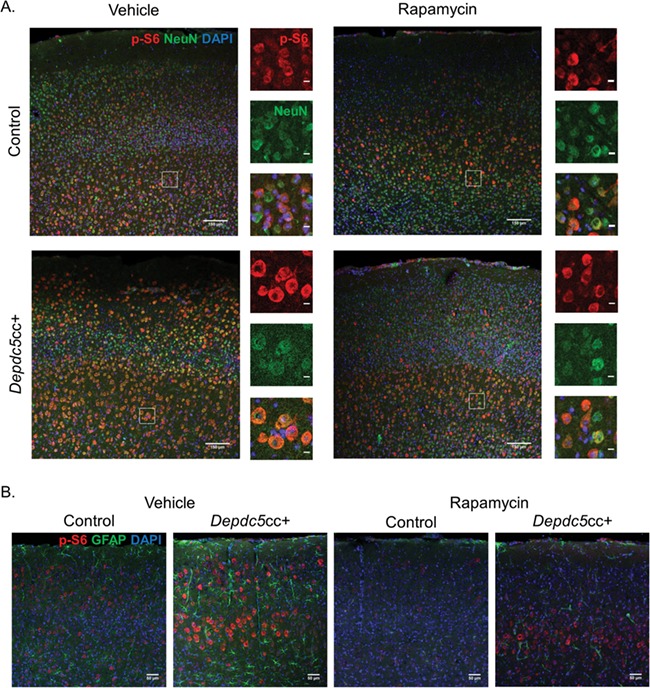
Chronic rapamycin rescues increased neuronal mTORC1 signaling and astrogliosis in the brains of Depdc5cc+ mice. (A) Anatomically matched motor cortex sections with p-S6 staining (red) in NeuN+ (green) neurons throughout the cortical layers from Depdc5cc+ and littermate control mice treated with vehicle or rapamycin for >90 days (n ≥ 4 mice per group except n = 2 for vehicle-treated Depdc5cc+, scale = 150 μm). ROI highlights the magnified area to the right of each image demonstrating the p-S6 colocalization with NeuN (scale = 10 μm). (B) GFAP+ astrocytes (green) and p-S6 (red) staining of anatomically matched cortex of Depdc5cc+ and littermate control mice treated with vehicle or rapamycin for >90 days. Note the p-S6 does not colocalize with GFAP staining. GFAP+ cells are increased in number and staining intensity in vehicle Depdc5cc+ brains and reduced in rapamycin-treated brains (n ≥ 4 mice per group except n = 2 for vehicle-treated Depdc5cc+, scale bar = 50 μm).
Chronic rapamycin treatment prolongs survival and rescues hyperactivity of Depdc5cc+ mice
We evaluated if chronic rapamycin reduces behavioral hyperactivity of Depdc5cc+ mice in the open-field assay. The open-field assay was performed between 75–90 days of age, after 5–8 weeks of rapamycin treatment. A significant difference in locomotor activity was seen across the genotypes [F (2,38) = 36.35, P < 0.0001, two-way ANOVA] and treatment groups [F (1,38) = 4.317, P = 0.0445, two-way ANOVA]. Vehicle-treated Depdc5cc+ mice had increased locomotor activity compared to all other groups (Fig. 6A, Tukey’s post hoc test P < 0.0001). Open-field activity was reduced by 40% in rapamycin-treated Depdc5cc+ mice compared to vehicle-treated Depdc5cc+ mice (P = 0.004, two-way ANOVA with Tukey’s post hoc test). Activity of rapamycin-treated Depdc5cc+ mice was reduced but not to the level of vehicle-treated controls (P = 0.023, two-way ANOVA with Tukey’s post hoc test). Rapamycin- and vehicle-treated controls were equivalent. We also measured if rapamycin altered elevated plus-maze behavior (Fig. 6B); however, no changes rose to the level of significance across genotypes or treatment groups.
Figure 6.
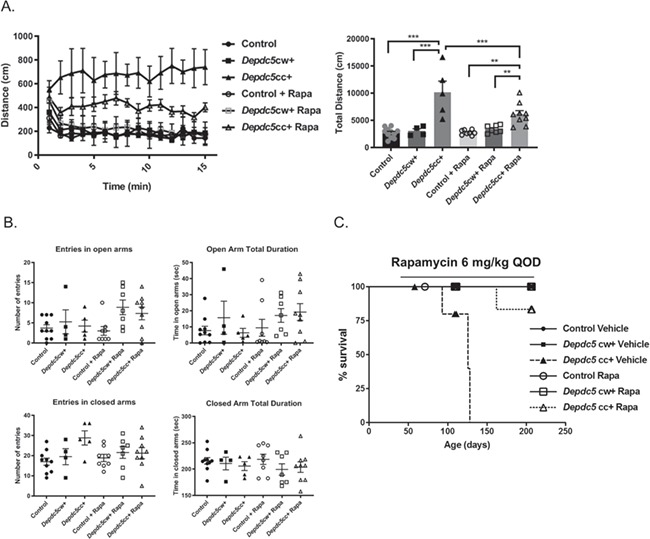
Chronic rapamycin treatment prolongs survival and decreases hyperactivity of Depdc5cc+ mice. (A) Left: distance over time was measured in open-field paradigm in mice of each group between 75–90 days of age. Right: total distance varied across the genotypes [F (2,38) = 36.35, P < 0.0001, two-way ANOVA] and treatment groups [F (1,38) = 4.317, P = 0.0445, two-way ANOVA] and the interaction between these measures [F (2,38) = 5.88, P = 0.006]. Tukey’s post hoc test **P < 0.01, ***P < 0.001. (B) Elevated plus-maze entries and duration into the open arms closed and open arms were measured across genotypes and treatment groups. All behavioral testing was performed between P75 and P90 on all animals. Each symbol represents an individual animal. Error bars are +/− SEM. (C) Survival curve of Depdc5cc+ mice and littermates treated with rapamycin of 6 mg/kg every other day or equal volume of vehicle started between 3–5 weeks of life. Note: Symbols along the lines represent censored points. Control and Depdc5cw + mice symbols overlapped at 100% survival.
Importantly, chronic rapamycin treatment started after 3 weeks of age prolonged the survival of Depdc5cc+ mice (Fig. 6C, Table 2). Half of the vehicle-treated Depdc5cc+ mice spontaneously died with a median age of death of 126 days (range 93–128). In contrast, only one (11.1%) rapamycin-treated Depdc5cc+ mouse died spontaneously at 163 days of age without obvious signs of morbidity prior to death.
Discussion
Patients with DEPDC5-related epilepsy have a high rate of refractory epilepsy and are at risk for SUDEP (10). Neuronal DEPDC5 loss results in increased mTOR activity (14,24). mTOR inhibitors have successfully reduced seizures in children with TSC (17,18). The present study demonstrated that Depdc5cc+ mice exhibit SUDEP-like terminal seizures and hyperactive behaviors as well as biochemical and behavioral rescue after chronic rapamycin treatment. Specifically, we demonstrated rapamycin treatment started after 3 weeks of age prolonged the lifespan of Depdc5cc+ mice, decreased behavioral hyperactivity, normalized neuronal size and corrected the increased mTORC1-mediated phosphorylation of S6 in Depdc5cc+ mice brains. Our preclinical data supports the potential use of mTOR inhibitors when delivered in a chronic manner to adults with DEPDC5-related epilepsy.
Although associated with autosomal dominant epilepsy, the identification of second-hit somatic DEPDC5 variants in patients suggest that a complete loss of DEPDC5 may underlie DEPDC5-related conditions (26,28). The Depdc5cc+ mouse model recapitulates many features of human DEPDC5-related conditions including large, dysplastic neurons with hyperactive mTORC1 signaling (24). Patients with DEPDC5-related conditions are at risk for SUDEP (29), thus terminal seizures in Depdc5cc+ mice provide important insight into this phenomenon. All of our animals exhibit seizure-induced death, whereas only 30% of focal Depdc5 knockout mice exhibit seizures (26). Depdc5cc+ mice animals appeared to have a heralding seizure ~24 h prior to the terminal seizure. The terminal seizures were similar in length and EEG pattern as non-terminal seizures. Overall, while Depdc5cc+ mice do not have many seizures, the infrequent seizures they do have may result in death. Together with our prior study, this suggests that the Depdc5cc+ mice may serve as a valuable model to better understand the mechanisms underlying SUDEP.
Attentional issues and hyperactivity are among the most common psychiatric comorbidities in individuals with pathogenic DEPDC5 variants (10). Here, we demonstrate Depdc5cc+ mice are hyperactive in the open-field paradigm. These results are similar to a recent zebrafish Depdc5 knockout model (23) and the hyperactivity of Tsc2-SynCre mice (30). These results suggest hyperactive behavior may have a common mechanism of action through the mTOR pathway. Collectively, we identified early mortality and behavioral hyperactivity of Depdc5cc+ mice as two important outcome measures when evaluating potential targeted therapeutic treatments.
mTOR inhibitors are potential treatments of DEPDC5-related epilepsy, as increased mTORC1 activity after DEPDC5 loss has been documented in resected brain tissue from individuals with DEPDC5-related epilepsy (7), rodent models (24–26) and in vitro (14). Therefore, we explored if the mTORC1 inhibitor rapamycin could improve survival and decrease hyperactivity of Depdc5cc+ mice. Rapamycin treatment started in early adulthood at 1 month of age improved survival and hyperactivity of Depdc5cc+ mice. Survival of Depdc5cc+ mice was significantly extended after rapamycin treatment with only one animal dying spontaneously by 200 days of age. The cause of death of the rapamycin-treated mouse was not captured, but given the lack of alternate causes identified and our direct observations in other animals, we conclude that this was most likely seizure-related death. These results are similar to Tsc1-SynCre where only one rapamycin-treated animal died (27). However, the overall impact of rapamycin treatment on seizure burden remains to be fully elucidated.
Behavioral hyperactivity of Depdc5cc+ mice was significantly reduced after rapamycin treatment. To our knowledge, this is the first study to report the response of rapamycin on locomotor activity in an mTOR-related mouse model with hyperactivity. Rapamycin only partially rescued the hyperactive phenotype of Depdc5cc+ mice, leading to the possibility of hyperactivity also being mediated by mTORC2 (31) or non-mTOR related effects (22). In other mouse models such as that of Fragile X syndrome, however, rapamycin treatment failed to improve the hyperactivity phenotype (32). Taken together, these findings suggest that mTORC1 at least in part mediates DEPDC5-related behavioral hyperactivity, which can be reduced by rapamycin treatment.
Rapamycin not only prolonged survival and hyperactivity of Depdc5cc+ mice, but also reduced brain size, cortical thickness and correspondingly neuronal soma size. These findings are similar to the response of other mTORopathies to rapamycin at both early and late ages (33). Supporting our finding, a recent study demonstrated early postnatal treatment with an alternative mTOR inhibitor everolimus decreased neuron size in a focal Depdc5 knockout rat model (25). Early developmental processes are unlikely to account for our results since the normal mouse brain is over 90% of adult size by 3 weeks of age and neuronal proliferation and migration have already ceased (34). Cell size is determined by the homeostatic balance between anabolic and catabolic processes (35). Although mTOR is a master regulator of cell growth, the precise mechanisms modulating adult neuron size are less clear (36). Rapamycin only decreased DEPDC5-deficient neuronal size and did not alter control neuron size. This suggests that the effect of rapamycin on neuronal size is specific to reducing hyperactive mTORC1 signaling in DEPDC5-deficient neurons rather than a non-specific effect.
We confirmed that rapamycin does reduce mTORC1 hyperactivity in Depdc5cc+ mice. Cortical neurons from rapamycin-treated Depdc5cc+ mice had reduced p-S6 staining in brain sections, which we also confirmed biochemically in cortical lysates. Rapamycin forms a complex with 12-kDa FK506-binding protein (FKBP12) to inhibit the phosphorylation of downstream mTORC1 substrates such as S6K1 (37). S6K1 phosphorylates S6, a reliable readout in a number of models of mTORopathies (33). However, the roles of the upstream regulators of mTORC1 in the brain are not well understood (38). As part of the GATOR1 complex, DEPDC5 regulates nutrient-sensing upstream of mTORC1 (12,13). In vitro manipulation of GATOR1, suggests that all three components, DEPDC5, NPRL3 and NPRL2, are critical for the stability of the GATOR1 complex (11). No prior studies evaluated if loss of endogenous DEPDC5 resulted in destabilization of the GATOR1 complex and loss of the other components. We provide in vivo evidence that DEPDC5 loss leads to a decrease in the other GATOR1 complex proteins, NPRL2 and NPRL3. Although basal levels of DEPDC5 are regulated by ubiquitination (39), there is no evidence that NPRL2 or NPRL3 undergo ubiquitination. Rapamycin did not rescue the loss of NPRL3. Collectively, this demonstrates that DEPDC5 is critical for GATOR1 stability independent of mTORC1 activity.
Our study demonstrates behavioral rescue of a mouse model of DEPDC5-related epilepsy with a targeted therapeutic. We chose the current rapamycin treatment paradigm for several reasons. First, we wanted to initiate dosing at a therapeutically relevant age at initiation. Prenatal rapamycin prevented neuronal migration defects caused by focal embryonic Depdc5 loss (26); however, prenatal rapamycin treatment is clinically problematic (40–42). Second, rapamycin treatment prior to 6 weeks of age rescued cellular and behavioral phenotypes of a Tsc1 mouse model (19). The prolonged treatment course over almost 7 months provided the opportunity to evaluate the survival benefit of rapamycin treatment in Depdc5cc+ mice. However, this prolonged treatment limited the number of age-matched vehicle-treated Depdc5cc+ mice to compare against. The fact that the data from our vehicle-treated Depdc5cc+ mice was similar to our previously published results gave us confidence in the reliability and reproducibility of the current results (24).
Intriguingly, we found a dramatic reduction in astrogliosis in the cortex of rapamycin-treated Depdc5cc+ mice. Several models of mTORopathies including our prior study of Depdc5cc+ mice demonstrate reactive astrocytes (24,33,43). Astrogliosis is a known complicating factor in epilepsy (44). Reactive astrogliosis can cause the development of spontaneous seizures (45). Conversely, seizures can lead to direct injury of astrocytes causing acute vacuolization followed by astrogliosis (46). Spontaneous seizures were rare in Depdc5cc+ mice, and the increased p-S6 did not colocalize with astrocytes, suggesting alternative cell non-autonomous effects may lead to the astrogliosis. Rapamycin prevents seizure-induced astrogliosis in a kainate-induced seizure model (46). In addition to rapamycin directly reducing neuronal mTOR signaling, it is possible that prolonged survival of rapamycin-treated Depdc5cc+ mice may in part be through a reduction in epileptogenic astrogliosis. Our data provide evidence that effects of DEPDC5 loss on astrocytes may be through cell non-autonomous effects, but further studies are needed.
In summary, we provide the first preclinical evidence that rapamycin is a viable treatment option for patients with DEPDC5-related conditions. Rapamycin treatment of Depdc5cc+ mice decreased hyperactivity, normalized neuronal size, reduced epileptogenic astrogliosis and importantly, prolonged survival. We provide in vivo evidence that Depdc5 knockout leads to a decrease in the other GATOR1 complex proteins, NPRL2 and NPRL3, which is not rescued by rapamycin treatment. Rapamycin corrects the increased mTORC1-mediated phosphorylation of S6 in Depdc5cc+ mice brains, which supports the role of mTOR in the pathogenesis of DEPDC5-related epilepsy. Our study demonstrates the value of Depdc5cc+ mice as a model to test future targeted therapeutics for human DEPDC5-related epilepsy.
Materials and Methods
Mouse breeding and genotyping
All mouse procedures were performed in accordance with the Guide for the Humane Use and Care of Laboratory Animals, and the study was approved by the Animal Care and Use Committee of Boston Children’s Hospital. All mice were housed in a 12-h light-dark cycle, climate-controlled room, with access to food and water ad lib. All surgeries were performed under isoflurane anesthesia, and all efforts were made to minimize pain. Neuron-specific Depdc5 conditional knockout mice were generated as previously described (24). Briefly, Depdc5tm1c(EUCOMM) Hmgu conditional mice (referred to as Depdc5cc-) contain loxP sites flanking exon 5 of the Depdc5 gene. Depdc5cc- males were bred with female neuron-specific synapsin I cre (SynCre) allele mice from our existing colony (30). The resulting Depdc5 tm1c-SynnCre mix-strain background (129S4/SvJae, C57BL/6 and CBA) has been maintained as a distinct in-crossed line for several generations prior to experimental testing. Heterozygous females Depdc5cw+ bred with male Depdc5cc- mice generated litters of knockout mice (Depdc5cc+), heterozygous mice (Depdc5cc+) and littermate control mice (Depdc5cw- and Depdc5cc-). All experiments have been performed using littermate controls for subsequent experiments.
DNA analysis
DNA was prepared from mouse toes/tails by standard procedures for genotyping. Genotyping around exon 5 of Depdc5 allowed simultaneous analysis of both conditional and wild-type alleles (forward: 5′-CATAGACATCTTGATAAGGTCTTAGCC-3′ and reverse: 5′-TCAAGTGCAAGATCTTAAGTGATTGGC-3′). An 852 base pair (bp) band was detected for the wild-type (w), and a 1069 bp band was detected for the conditional allele (c) with the flanking loxP sites. Primers that amplify a 300 bp portion of the Cre recombinase were used to assess the presence of the SynI-Cre allele (24).
Video-EEG recording
EEG recording was performed as previously described with implanted wireless telemetry transmitters (PhysioTel ETA-F10; DSI, Data Sciences International) (47). Animals were implanted between 83 and 135 days of age and given at least 3 days to acclimate after implantation prior to recording. One-channel video-EEG was recorded differentially between the reference (right olfactory bulb) and active (left occipital lobe) electrodes, which included day and night cycles. The EEG was sampled at 1000 Hz. Video and EEGs were recorded until the primary endpoint where either a mouse died spontaneously or the mouse reached 200 days of age. In some cases, the EEG transmitter battery died prior to the primary endpoint. Video recording was continued in those cases to detect clinical seizures.
Behavioral testing
All behavioral experiments were conducted in the Neurodevelopmental Behavioral Core at Boston Children’s Hospital. Behavioral testing was initiated at P75 and completed by P90. Tests were performed blinded to genotypes in the same order and at the same time point in the circadian cycle for all mice.
To check spontaneous locomotor activity, we use the open-field paradigm. Open-field experiments were conducted with mice recorded for 15 min as they freely moved in the circular open-field arena (30). The open-field (42 × 42 cm) is enclosed in a soundproof chamber, in a room illuminated at ~30 lux. Trials were video recorded and total distance was calculated with Noldus EthoVision XT software.
To analyze mouse anxiety-related behaviors, we use the elevated plus-maze paradigm. Mice were placed in the center of the elevated plus-maze apparatus for a 5-min trial. Mouse movements were recorded by camera and analyzed with Noldus EthoVision XT software. The time spent in the open and closed arms and the number of entries into each were recorded.
Drug treatment and sample preparation
Rapamycin was obtained from LC Laboratories, dissolved at 20 mg/ml in ethanol and stored at −20°C for up to 1 month. Before each administration, rapamycin was diluted in 5% Tween 80, 5% polyethylene glycol 400 (0.5–1.5 mg/ml). Rapamycin was given at 6 mg/kg intraperitoneally. Control solution was given using the dilution vehicle at the same volume as the rapamycin. Five litters of mice were randomized to receive vehicle or rapamycin treatments every Monday, Wednesday and Friday starting at 1 month of age for a maximum duration of 7 months. Mice were weighed every other day. Mice were sacrificed early if weight loss of 20%, greatly reduced mobility or other signs of morbidity occurred.
Mice brain weights were recorded at the time of euthanasia or at the time of spontaneous death. Mice that died spontaneously in the cages were transferred immediately to 4°C at the time of discovery. Brain and body weight were measured less than 12 h after the time of death. We excluded brain weights from animals that were cannibalized or if the brain was no longer intact.
After 50% of the vehicle-treated Depdc5cc+ died, the litters with remaining vehicle-treated Depdc5cc+ mice were sacrificed for pathology and biochemical experiments. One vehicle-treated Depdc5cc+ animal and one rapamycin-treated littermate control were sacrificed early due to fighting wounds and excluded from further analysis. After anesthesia, total body weight was determined, followed by rapid removal and weighing of the brain. Half the brain was snap frozen in liquid nitrogen and stored at −80°C until use for immunoblotting. In order to collect half the brain for immunoblotting, we could not perfuse the animal for immunostaining, instead the other half was drop fixed in 4% paraformaldehyde overnight at 4°C for immunostaining.
Immunoblotting and immunostaining
To evaluate protein levels, protein extracts were prepared from dissected cortical tissue of age-matched littermate mice. Western blotting was performed using equal amounts of protein extract per lane (20–40 μg of protein) under reducing conditions in Bolt Bis-Tris 8–12% SDS polyacrylamide precast gels (ThermoFisher). Gels were transferred onto Immobilon-FL PVDF membranes (Millipore), blocked with Odyssey TBS blocking buffer (LI-COR) for 1 h at room temperature, followed by primary antibody in Odyssey blocking buffer +0.2% Tween 20 (Sigma) overnight at 4°C. Antibodies used for western blotting were as follows: NPRL2 (Santa Cruz, cat.no. sc-376 986, 1:250), NPRL3 (Abcam, cat.no. ab121346, 1:1000), phospho-S6 Ribosomal Protein (Ser 240/244) (Cell Signaling, cat.no. 5364, 1:1000), total S6 (Santa Cruz, cat.no. sc-74 459, 1:1000) and alpha-tubulin (Abcam, cat.no. 15246, 1:10 000). After washing blots three times for 10 min each in TBST, IRDye 800CW- or 680LT-conjugated secondary antibodies (1:10 000; LI-COR) in Odyssey blocking buffer, 0.2% Tween 20 and 0.01% SDS was added for 1 h at room temperature. To visualize bands, Odyssey Fc Imager was used, and densitometry analysis was performed using Image Studio Software (LI-COR). Each band was normalized to the alpha-tubulin signal per sample.
For immunohistochemistry, half-brains were drop fixed in 4% paraformaldehyde overnight at 4°C followed by cryoprotection in 30% sucrose in Phosphate Buffered Saline (PBS). Coronal sections were cut at 25 μm, mounted on charged slides and stored at −80°C until staining. Anatomically matched sections from each treatment group and genotype were stained simultaneously. Sections were rehydrated in PBS for 5 min followed by permeabilization and blocking (5% goat serum +0.3% triton X-100 in PBS) for 1 h at room temperature. Overnight at 4°C, sections were incubated with primary antibody in blocking buffer: NeuN (Millipore, cat.no. MAB377, 1:500), phospho-S6 Ribosomal Protein (Ser 240/244) (Cell Signaling, cat.no. 5364, 1:1000) and GFAP (Cell Signaling, cat.no. 3670, 1:500). The following day, sections were washed 3 × 5 min in PBS then incubated in secondary antibodies (goat anti-rabbit IgG-555, Invitrogen, cat. no. #A21429 and goat anti-mouse IgG-488, Invitrogen, cat. no. #A11001, 1:1000, diluted in blocking buffer) for 1 h at room temperature. Sections were then washed in PBS for 3 × 10 min, incubated in DAPI (4',6-diamidino-2-phenylindole) for 10 min, then mounted with Fluoroumount-G (SouthernBiotech).
Images were acquired using a Zeiss LSM700 Laser Scanning Confocal Microscope equipped with Plan Apochromat ×10/0.3 and ×25/0.8 objectives. Blinded cortical thickness measurements were taken in at least six paired sites in frontal cortex per brain. The average cortical thickness per animal was reported. Cortical layers were delineated by DAPI counterstain. Area measurements of the soma layer V NeuN-positive neurons from at least 100 neurons in M1 motor cortex per animal were recorded by a blinded observer using ImageJ software.
Statistical analysis
Statistical analysis was performed by GraphPad Prism 8 software. The results are presented as mean ± standard error of mean (SEM) or standard deviation (SD). Comparisons between two groups were performed by unpaired two-tailed Student’s t-test. Comparisons between genotypes were performed by one-way ANOVA with Tukey’s multiple comparison test as post hoc analysis for parametric data and by a Kruskal–Wallis test for non-parametric data. Two-way ANOVA with Tukey’s multiple comparisons post hoc analysis was used for comparison between genotypes and treatment groups. Alpha was set at 0.05 for significance and exact P-values are reported for significant results.
Supplementary Material
Acknowledgements
We would like to thank the Boston Children’s Hospital IDDRC Cellular Imaging and Neurodevelopmental Behavioral Cores and the Experimental Neurophysiology Core at Boston Children’s Hospital for technical assistance.
Conflict of Interest statement
M.S. reports grant support from Novartis, Roche, Pfizer, Ipsen, LAM Therapeutics and Quadrant Biosciences. He has served on Scientific Advisory Boards for Sage, Roche, Celgene and Takeda. A.P. served on the Scientific Advisory Board for the Dravet Syndrome Foundation and currently serves on the Scientific Advisory Board for TevardBio. The other authors declare that they have no conflict of interest.
Funding
National Institutes of Health (2R25NS070682-07 for C.J.Y.); National Institute of Neurological Disorders and Stroke (K12NS098482 for C.J.Y.); and Boston Children’s Hospital Basic/Translational Research Executive Committee Career Development Fellowship (for C.J.Y.). U54HD090255 (for M.S.). Translational Research Program at Boston Children’s Hospital (for A.P.).
References
- 1. Epi4K Consortium and Epilepsy Phenome/Genome Project (2017) Ultra-rare genetic variation in common epilepsies: a case-control sequencing study. Lancet Neurol., 16, 135–143. [DOI] [PubMed] [Google Scholar]
- 2. Dibbens L.M., de Vries B., Donatello S., Heron S.E., Hodgson B.L., Chintawar S., Crompton D.E., Hughes J.N., Bellows S.T., Klein K.M. et al. (2013) Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat. Genet., 45, 546–551. [DOI] [PubMed] [Google Scholar]
- 3. Ishida S., Picard F., Rudolf G., Noe E., Achaz G., Thomas P., Genton P., Mundwiller E., Wolff M., Marescaux C. et al. (2013) Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat. Genet., 45, 552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lal D., Reinthaler E.M., Schubert J., Muhle H., Riesch E., Kluger G., Jabbari K., Kawalia A., Baumel C., Holthausen H. et al. (2014) DEPDC5 mutations in genetic focal epilepsies of childhood. Ann. Neurol., 75, 788–792. [DOI] [PubMed] [Google Scholar]
- 5. Perucca P., Scheffer I.E., Harvey A.S., James P.A., Lunke S., Thorne N., Gaff C., Regan B.M., Damiano J.A., Hildebrand M.S. et al. (2017) Real-world utility of whole exome sequencing with targeted gene analysis for focal epilepsy. Epilepsy Res., 131,–8. [DOI] [PubMed] [Google Scholar]
- 6. Carvill G.L., Crompton D.E., Regan B.M., McMahon J.M., Saykally J., Zemel M., Schneider A.L., Dibbens L., Howell K.B., Mandelstam S. et al. (2015) Epileptic spasms are a feature of DEPDC5 mTORopathy. Neurol Genet., 1, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scerri T., Riseley J.R., Gillies G., Pope K., Burgess R., Mandelstam S.A., Dibbens L., Chow C.W., Maixner W., Harvey A.S. et al. (2015) Familial cortical dysplasia type IIA caused by a germline mutation in DEPDC5. Ann. Clin. Transl. Neurol., 2, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. D’Gama A.M., Geng Y., Couto J.A., Martin B., Boyle E.A., LaCoursiere C.M., Hossain A., Hatem N.E., Barry B.J., Kwiatkowski D.J. et al. (2015) Mammalian target of rapamycin pathway mutations cause hemimegalencephaly and focal cortical dysplasia. Ann. Neurol., 77, 720–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Scheffer I.E., Heron S.E., Regan B.M., Mandelstam S., Crompton D.E., Hodgson B.L., Licchetta L., Provini F., Bisulli F., Vadlamudi L. et al. (2014) Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann. Neurol., 75, 782–787. [DOI] [PubMed] [Google Scholar]
- 10. Baldassari S., Picard F., Verbeek N.E., van Kempen M., Brilstra E.H., Lesca G., Conti V., Guerrini R., Bisulli F., Licchetta L. et al. (2019) The landscape of epilepsy-related GATOR1 variants. Genet. Med., 21, 398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bar-Peled L., Chantranupong L., Cherniack A.D., Chen W.W., Ottina K.A., Grabiner B.C., Spear E.D., Carter S.L., Meyerson M. and Sabatini D.M. (2013) A tumor suppressor complex with GAP activity for the rag GTPases that signal amino acid sufficiency to mTORC1. Science, 340, 1100–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wolfson R.L., Chantranupong L., Wyant G.A., Gu X., Orozco J.M., Shen K., Condon K.J., Petri S., Kedir J., Scaria S.M. et al. (2017) KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature, 543, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shen K., Huang R.K., Brignole E.J., Condon K.J., Valenstein M.L., Chantranupong L., Bomaliyamu A., Choe A., Hong C., Yu Z. et al. (2018) Architecture of the human GATOR1 and GATOR1-rag GTPases complexes. Nature, 556, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Iffland P.H. 2nd, Baybis M., Barnes A.E., Leventer R.J., Lockhart P.J. and Crino P.B. (2018) DEPDC5 and NPRL3 modulate cell size, filopodial outgrowth, and localization of mTOR in neural progenitor cells and neurons. Neurobiol. Dis., 114, 184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Saxton R.A. and Sabatini D.M. (2017) mTOR Signaling in growth, metabolism, and disease. Cell, 168, 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Crino P.B. (2016) The mTOR signalling cascade: paving new roads to cure neurological disease. Nat. Rev. Neurol., 12, 379–392. [DOI] [PubMed] [Google Scholar]
- 17. Canpolat M., Per H., Gumus H., Yikilmaz A., Unal E., Patiroglu T., Cinar L., Kurtsoy A. and Kumandas S. (2014) Rapamycin has a beneficial effect on controlling epilepsy in children with tuberous sclerosis complex: results of 7 children from a cohort of 86. Childs Nerv. Syst., 30, 227–240. [DOI] [PubMed] [Google Scholar]
- 18. French J.A., Lawson J.A., Yapici Z., Ikeda H., Polster T., Nabbout R., Curatolo P., de Vries P.J., Dlugos D.J., Berkowitz N. et al. (2016) Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet, 388, 2153–2163. [DOI] [PubMed] [Google Scholar]
- 19. Tsai P.T., Rudolph S., Guo C., Ellegood J., Gibson J.M., Schaeffer S.M., Mogavero J., Lerch J.P., Regehr W. and Sahin M. (2018) Sensitive periods for cerebellar-mediated autistic-like behaviors. Cell Rep., 25, 357–367 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hughes J., Dawson R., Tea M., McAninch D., Piltz S., Jackson D., Stewart L., Ricos M.G., Dibbens L.M., Harvey N.L. et al. (2017) Knockout of the epilepsy gene Depdc5 in mice causes severe embryonic dysmorphology with hyperactivity of mTORC1 signalling. Sci. Rep., 7, 12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marsan E., Ishida S., Schramm A., Weckhuysen S., Muraca G., Lecas S., Liang N., Treins C., Pende M., Roussel D. et al. (2016) Depdc5 knockout rat: a novel model of mTORopathy. Neurobiol. Dis., 89, 180–189. [DOI] [PubMed] [Google Scholar]
- 22. Swaminathan A., Hassan-Abdi R., Renault S., Siekierska A., Riche R., Liao M., de Witte P.A.M., Yanicostas C., Soussi-Yanicostas N., Drapeau P. et al. (2018) Non-canonical mTOR-independent role of DEPDC5 in regulating GABAergic network development. Curr. Biol., 28, 1924–1937. [DOI] [PubMed] [Google Scholar]
- 23. de Calbiac H., Dabacan A., Marsan E., Tostivint H., Devienne G., Ishida S., Leguern E., Baulac S., Muresan R.C., Kabashi E. et al. (2018) Depdc5 knockdown causes mTOR-dependent motor hyperactivity in zebrafish. Ann. Clin. Transl. Neurol., 5, 510–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yuskaitis C.J., Jones B.M., Wolfson R.L., Super C.E., Dhamne S.C., Rotenberg A., Sabatini D.M., Sahin M. and Poduri A. (2018) A mouse model of DEPDC5-related epilepsy: neuronal loss of Depdc5 causes dysplastic and ectopic neurons, increased mTOR signaling, and seizure susceptibility. Neurobiol. Dis., 111, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu S., Knowlton R.C., Watson B.O., Glanowska K.M., Murphy G.G., Parent J.M. and Wang Y. (2018) Somatic Depdc5 deletion recapitulates electroclinical features of human focal cortical dysplasia type IIA. Ann. Neurol., 84, 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ribierre T., Deleuze C., Bacq A., Baldassari S., Marsan E., Chipaux M., Muraca G., Roussel D., Navarro V., Leguern E. et al. (2018) Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J. Clin. Invest, 128, 2452–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Meikle L., Pollizzi K., Egnor A., Kramvis I., Lane H., Sahin M. and Kwiatkowski D.J. (2008) Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J. Neurosci., 28, 5422–5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baulac S., Ishida S., Marsan E., Miquel C., Biraben A., Nguyen D.K., Nordli D., Cossette P., Nguyen S., Lambrecq V. et al. (2015) Familial focal epilepsy with focal cortical dysplasia due to DEPDC5 mutations. Ann. Neurol., 77, 675–683. [DOI] [PubMed] [Google Scholar]
- 29. Bagnall R.D., Crompton D.E., Petrovski S., Lam L., Cutmore C., Garry S.I., Sadleir L.G., Dibbens L.M., Cairns A., Kivity S. et al. (2016) Exome-based analysis of cardiac arrhythmia, respiratory control, and epilepsy genes in sudden unexpected death in epilepsy. Ann. Neurol., 79, 522–534. [DOI] [PubMed] [Google Scholar]
- 30. Yuan E., Tsai P.T., Greene-Colozzi E., Sahin M., Kwiatkowski D.J. and Malinowska I.A. (2012) Graded loss of tuberin in an allelic series of brain models of TSC correlates with survival, and biochemical, histological and behavioral features. Hum. Mol. Genet., 21, 4286–4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Carson R.P., Fu C., Winzenburger P. and Ess K.C. (2013) Deletion of Rictor in neural progenitor cells reveals contributions of mTORC2 signaling to tuberous sclerosis complex. Hum. Mol. Genet., 22, 140–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sare R.M., Song A., Loutaev I., Cook A., Maita I., Lemons A., Sheeler C. and Smith C.B. (2017) Negative effects of chronic rapamycin treatment on behavior in a mouse model of fragile X syndrome. Front. Mol. Neurosci., 10, 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Switon K., Kotulska K., Janusz-Kaminska A., Zmorzynska J. and Jaworski J. (2017) Molecular neurobiology of mTOR. Neuroscience, 341, 112–153. [DOI] [PubMed] [Google Scholar]
- 34. Chen C.Y., Noble-Haeusslein L.J., Ferriero D. and Semple B.D. (2013) Traumatic injury to the immature frontal lobe: a new murine model of long-term motor impairment in the absence of psychosocial or cognitive deficits. Dev. Neurosci., 35, 474–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lloyd A.C. (2013) The regulation of cell size. Cell, 154, 1194–1205. [DOI] [PubMed] [Google Scholar]
- 36. Takei N. and Nawa H. (2014) mTOR signaling and its roles in normal and abnormal brain development. Front. Mol. Neurosci., 7, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kang S.A., Pacold M.E., Cervantes C.L., Lim D., Lou H.J., Ottina K., Gray N.S., Turk B.E., Yaffe M.B. and Sabatini D.M. (2013) mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science, 341, 1236566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lipton J.O. and Sahin M. (2014) The neurology of mTOR. Neuron, 84, 275–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen J., Ou Y., Yang Y., Li W., Xu Y., Xie Y. and Liu Y. (2018) KLHL22 activates amino-acid-dependent mTORC1 signalling to promote tumorigenesis and ageing. Nature, 557, 585–589. [DOI] [PubMed] [Google Scholar]
- 40. Tsai P.T., Greene-Colozzi E., Goto J., Anderl S., Kwiatkowski D.J. and Sahin M. (2013) Prenatal rapamycin results in early and late behavioral abnormalities in wildtype C57BL/6 mice. Behav. Genet., 43, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hennig M., Fiedler S., Jux C., Thierfelder L. and Drenckhahn J.D. (2017) Prenatal mechanistic target of rapamycin complex 1 (m TORC1) inhibition by rapamycin treatment of pregnant mice causes intrauterine growth restriction and alters postnatal cardiac growth, morphology, and function. J. Am. Heart Assoc., 6, e005506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Way S.W., Rozas N.S., Wu H.C., McKenna J. 3rd, Reith R.M., Hashmi S.S., Dash P.K. and Gambello M.J. (2012) The differential effects of prenatal and/or postnatal rapamycin on neurodevelopmental defects and cognition in a neuroglial mouse model of tuberous sclerosis complex. Hum. Mol. Genet., 21, 3226–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wong M. and Crino P.B. (2012) Tuberous sclerosis and epilepsy: role of astrocytes. Glia, 60, 1244–1250. [DOI] [PubMed] [Google Scholar]
- 44. Henneberger C. (2017) Does rapid and physiological astrocyte-neuron signalling amplify epileptic activity? J. Physiol., 595, 1917–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kimbrough I.F., Robel S., Roberson E.D. and Sontheimer H. (2015) Vascular amyloidosis impairs the gliovascular unit in a mouse model of Alzheimer’s disease. Brain, 138, 3716–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guo D., Zou J. and Wong M. (2017) Rapamycin attenuates acute seizure-induced astrocyte injury in mice in vivo. Sci. Rep., 7, 2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dhamne S.C., Silverman J.L., Super C.E., Lammers S.H.T., Hameed M.Q., Modi M.E., Copping N.A., Pride M.C., Smith D.G., Rotenberg A. et al. (2017) Replicable in vivo physiological and behavioral phenotypes of the Shank3B null mutant mouse model of autism. Mol. Autism, 8, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.