

ABSTRACT
Telomeres and sirtuins are independently implicated in causing disease and aging, but how they cooperate is not well understood. A recent study demonstrates that telomere shortening represses sirtuins and increasing sirtuin activity stabilizes telomeres and improves telomere-dependent disease, suggesting that these two pathways are tightly intertwined.
Keywords: Telomeres, p53, sirtuins, NAD(+), aging, disease, damage
Telomeres are repetitive nucleoprotein sequences at the ends of chromosomes that are synthesized by the specialized RNA-dependent DNA polymerase, telomerase. The majority of human cells lack or have insufficient telomerase activity and undergo progressive telomere shortening that is strongly implicated in driving pathologies and aging. Although our understanding of the pathogenic mechanisms underlying telomere-dependent disease and aging is still incomplete, studies in various model organisms have established an important role for the DNA damage response and its main transducer, tumor protein p53 (TP53, known as p53), in these processes through the induction of cellular senescence, growth arrest and apoptosis. Recent studies have also suggested that telomere shortening can cause cellular dysfunction by compromising metabolism via a p53-dependent repression of mitochondrial biogenesis and function that results in fewer and dysfunctional mitochondria.1
In a recent study, we have further characterized how telomere dysfunction and p53 impact metabolism through the regulation of sirtuins. The sirtuin family comprises a class of Nicotinamide adenine dinucleotide ((NAD(+))-dependent enzymes that have known deacetylase activity. They regulate many metabolic processes and are heavily implicated in multiple diseases and in aging.2 Previous studies have shown that sirtuins localize to telomeres and regulate telomere length. In yeast, telomeres serve as a reservoir for Sir2p, the homolog of mammalian Sirt1, where it is involved in the telomere silencing effect on neighboring genes, but does not regulate telomere length.3,4 Sirt1 is also initially dispensable for telomere length maintenance in young mice, although it is required for maintaining telomere length during aging. Furthermore, Sirt1 overexpression increases telomere length in mice, suggesting that telomere length is modulated by Sirt1 in a time- and dosage-dependent manner.5 The moderate Sirt1 effect on telomeres contrasts with the severe telomere length defect in mice deficient for Sirt6 that develop pronounced telomere dysfunction, marked genomic instability, and premature aging syndrome.6 While these studies indicate that Sirt1 and Sirt6 bind and regulate telomere length, it remains unclear how telomere shortening in turn impacts sirtuin expression, and what the functional relevance of sirtuins as downstream mediators of telomere-dependent dysfunction is.
In this respect, our recent study in telomerase knockout mice has shed new light on how telomere shortening impacts sirtuins and demonstrated that telomere shortening and p53 are upstream regulators of sirtuins (Figure 1). This is based on the finding that short telomeres induce the repression of all seven sirtuins in liver tissue, a highly metabolic organ where telomeres and sirtuins play an important role in normal physiology and disease. The global sirtuin repression is accompanied by hyper-acetylation of their targets including Forkhead box protein O1 (FoxO1), Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Pgc-1α), p53 and histones. The observed hyperacetylation indicates that decreased expression of sirtuins is paralleled by diminished sirtuin deacetylase activity that affects their multiple targets. This telomere shortening-induced suppression of sirtuins is cell-autonomous and reversible based on the observation that sirtuins are also downregulated in mouse embryonic fibroblasts and reintroduction of telomerase into livers of mice with short telomeres normalizes their expression levels. Mechanistically, the repression of sirtuins is dependent on p53 as liver specific deletion of p53 in telomerase knockout mice restores sirtuin expression and activity.2 While previous studies have shown that p53 represses Sirt1 post-transcriptionally through miRNA-34a, the p53-mediated repression of all seven sirtuins in the setting of short telomeres is remarkable and points to a wider role of p53 in sirtuin regulation. The repression of several sirtuins has also been reported in cells lacking the DNA repair Cockayne syndrome group A/B proteins (CSA/CSB) indicating that other DNA–damage associated processes can impact several sirtuins.7,8 The identification of telomeres and p53 as upstream regulators of sirtuins raises the possibility that both might be involved in the downregulation of sirtuins in different contexts, including diseases and aging. The telomere and p53-mediated repression of sirtuins also suggests that the simultaneous repression of multiple sirtuins could drive pathologies and aging and it remains to be established whether, in addition to the well-recognized decrease of Sirt1 expression during aging and many pathological conditions, other sirtuins are simultaneously repressed. In the context of dysfunctional telomeres, the importance of other sirtuins is supported by the finding that the beneficial effect of NAD(+) supplementation on liver fibrosis is only partially Sirt1-dependent, indicating that other sirtuins or, alternatively, other NAD(+)-dependent enzymes such as Poly (ADP-ribose) polymerases (PARPs) are functionally relevant (Figure 1).2
Figure 1.
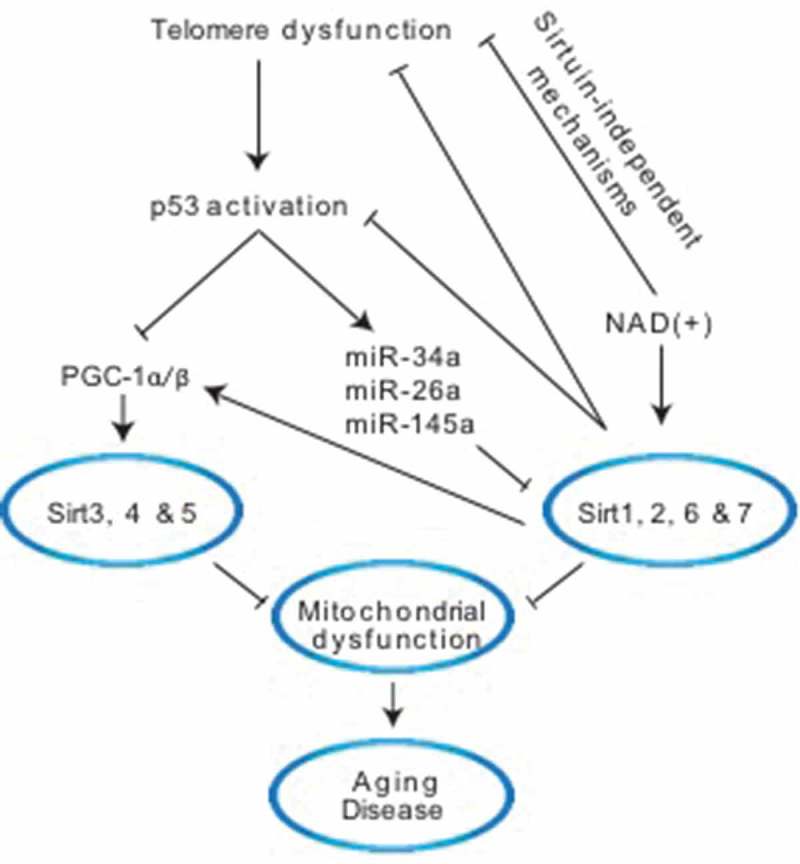
Molecular pathways linking telomeres and p53 to sirtuins. Dysfunctional telomeres activate the DNA damage response and tumor protein p53 (TP53, known as p53). P53 regulates non-mitochondrial sirtuins Sirt1, 2, 6 & 7 through miRNA-34a, 26a & 145a while it regulates the mitochondrial sirtuins Sirt3, 4 & 5 at the transcriptional level through Peroxisome proliferator-activated receptor gamma coactivator 1-alpha and beta (PGC-1α/β). Increasing Nicotinamide adenine dinucleotide ((NAD(+)) levels through NAD(+) precursors increases sirtuin activity, stabilizes telomeres and dampens p53 in a partially Sirt1-dependent manner indicating the involvement of Sirt6 or other NAD(+) – dependent enzymes. The repression of Sirt1 in the setting of short telomeres contributes to PGC-1α impairment directly as Sirt1 is known to activate PGC-1α through deacetylation. Together, these altered pathways induce mitochondrial dysfunction that is implicated in aging and disease. Other potentially important processes impacted by decreased sirtuin levels – for example, epigenetic changes through histone modifications and DNA repair pathways, among others – are likely to contribute to cellular phenotypes, and are not depicted here.
Importantly, this telomere-p53-sirtuin link might lead to a progressive deterioration of cellular function and a circulus vitiosus through feedback mechanisms. First, Sirt1 and Sirt7 have been shown to inactivate p53 and their inactivation by p53 in the setting of telomere dysfunction could further enhance p53 activity. Secondly, at the level of telomeres, the low levels of Sirt1 and Sirt6 could further destabilize telomeres and activate the DNA damage response and p53. Together, these sirtuin–telomere and sirtuin-p53 feedback mechanisms might sustain and accelerate the decline of already damaged cells, particularly in post-mitotic cells whose primary response to telomere dysfunction might not be one of the classical outcomes of apoptosis, growth arrest and senescence. This telomere-sirtuin link also provides an entry point for therapeutic modulation through the administration of the NAD(+) precursor nicotinamide mononucleotide. Our studies show that increasing NAD(+) levels stabilizes telomeres and dampens the DNA damage response and p53 in a partially Sirt1-dependent manner.2 The molecular mechanisms that lead to telomere stabilization following NAD(+) supplementation are yet to be defined, however, the stabilization of telomeres in telomerase knockout mice indicates that telomerase-independent mechanisms are involved (Figure 1). Increasing NAD(+) levels through deletion of the NAD(+)-consuming enzyme Cluster of differentiation 38 (CD38) has been reported to stabilize telomeres in aged wild type mice.9 Increasing NAD(+) might be particularly beneficial under conditions of additional exogenous damage as the decrease of NAD(+), which is mainly driven by NAD(+)-consuming DNA repair processes, is much more significant in cells with short telomeres, indicating a particular susceptibility of short-telomere bearing cells to NAD(+) decline. The molecular underpinnings of this pronounced NAD(+) decrease in cells with short telomeres in response to exogenous damage is unclear but tracks well with their marked susceptibility to additional damage such as ionizing irradiation or genotoxic drugs.
The NAD(+) supplementation associated increase in sirtuin activity in telomerase knockout mice improves mitochondrial biogenesis and function as reflected by increased expression of mitochondrial biogenesis factors, increased electron transport chain activity of complex I and IV and elevated mtDNA copy number.2 The improvement of mitochondrial function most likely results from the inactivation of p53 and its repressive effect on PGC-1α expression as well as direct activation of PGC-1α by Sirt1. Besides the improvement of mitochondrial function, Sirt1 is expected to affect several cellular processes including modulation of histones and the epigenome, generalized DNA repair pathways, all of which could act cooperatively to prevent disease and aging.10
Funding Statement
This work was supported by the Ted Nash Long Life Foundation, Edward Mallinckrodt Jr Foundation and National Institute of Aging (NIA) grant R01AG047924.
References
- 1.Sahin E, DePinho RA.. Axis of ageing: telomeres, p53 and mitochondria. Nat Rev Mol Cell Biol. 2012;13(397–404). doi: 10.1038/nrm3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amano H, Chaudhury A, Rodriguez-Aguayo C, Lu L, Akhanov V, Catic A, Popov YV, Verdin E, Johnson H, Stossi F, et al. Telomere dysfunction induces sirtuin repression that drives telomere-dependent disease. Cell Metab. 2019. doi: 10.1016/j.cmet.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guarente L. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 2000;14:1021–1026. [PubMed] [Google Scholar]
- 4.Perrod S, Gasser SM.. Long-range silencing and position effects at telomeres and centromeres: parallels and differences. Cell Mol Life Sci. 2003;60(2303–2318). doi: 10.1007/s00018-003-3246-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palacios JA, Herranz D, De Bonis ML, Velasco S, Serrano M, Blasco MA. SIRT1 contributes to telomere maintenance and augments global homologous recombination. J Cell Biol. 2010;191(1299–1313). doi: 10.1083/jcb.201005160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tennen RI, Chua KF. Chromatin regulation and genome maintenance by mammalian SIRT6. Trends Biochem Sci. 2011;36(39–46). doi: 10.1016/j.tibs.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A. 2008;105:13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scheibye-Knudsen M, Mitchell S, Fang E, Iyama T, Ward T, Wang J, Dunn C, Singh N, Veith S, Hasan-Olive M, et al. A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014;20(840–855). doi: 10.1016/j.cmet.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tarrago MG, Chini CCS, Kanamori KS, Warner GM, Caride A, de Oliveira GC, Rud M, Samani A, Hein KZ, Huang R, et al. A potent and specific CD38 inhibitor ameliorates age-related metabolic dysfunction by reversing tissue NAD(+) Decline. Cell Metab. 2018;27:1081–1095 e1010. doi: 10.1016/j.cmet.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mostoslavsky R. DNA repair, insulin signaling and sirtuins: at the crossroads between cancer and aging. Front Biosci. 2008;13:6966–6990. [DOI] [PubMed] [Google Scholar]