

Abstract
Proteus syndrome is a mosaic, progressive overgrowth disorder caused by a somatic activating variant c.49G > A p.(E17K) in AKT1. The presentation in affected individuals is variable, with a diversity of tissues demonstrating abnormalities. Common manifestations include skin and bony overgrowth, vascular malformations (VMs), cysts and benign tumors. We used two methods to create mouse models that had endogenously-regulated mosaic expression of the Proteus syndrome variant. Variant allele fractions (VAFs) ranged from 0% to 50% across numerous tissues in 44 Proteus syndrome mice. Mice were phenotypically heterogeneous with lesions rarely observed before 12 months of age. VMs were the most frequent finding with a total of 69 found in 29 of 44 Proteus syndrome mice. Twenty-eight cysts and ectasia, frequently biliary, were seen in 22 of 44 Proteus syndrome mice. Varying levels of mammary hyperplasia were seen in 10 of 16 female Proteus syndrome mice with other localized regions of hyperplasia and stromal expansion noted in several additional animals. Interestingly, 27 of 31 Proteus syndrome animals had non-zero blood VAF that is in contrast to the human disorder where it is rarely seen in peripheral blood. Identification of variant-positive cells by green fluorescent protein (GFP) staining in chimeric Proteus syndrome mice showed that in some lesions, hyperplastic cells were predominantly GFP/Akt1E17K-positive and showed increased pAKT signal compared to GFP-negative cells. However, hyperplastic mammary epithelium was a mixture of GFP/Akt1E17K-positive and negative cells with some GFP/Akt1E17K-negative cells also having increased pAKT signal suggesting that the variant-positive cells can induce lesion formation in a non-cell autonomous manner.
Introduction
Proteus syndrome (OMIM #176920) is a rare disorder of disproportionate, asymmetric overgrowth that can affect virtually any organ or tissue. It is caused by a heterozygous, mosaic c.49G > A, p.(E17K) mutation in the serine threonine kinase AKT1 (1), a key node in the phosphoinositide 3-kinase (PI3K)/AKT signaling network. The disease has remarkable phenotypic heterogeneity and can be severe, with a near 25% mortality by 22 years of age (2). Some patients have few overgrowths while others have nearly all systems affected. The mechanism of the overgrowth is also heterogeneous and can comprise hyperplasia, expansion of the extracellular matrix (ECM), ectasia or combinations of these processes (3). Common manifestations include skeletal overgrowth, epidermal and cerebriform connective tissue nevi (CCTN), dysregulation of adipose tissue and vascular malformations (VMs) (4,5). Other findings include visceral overgrowth, cysts and benign tumors such as ovarian cystadenomas and parotid adenomas. Many other manifestations have been seen less frequently in affected individuals (6). A key aspect of the disorder is its progressive nature—which is best exemplified by the CCTN. This lesion typically appears in one or a few areas of the foot and then spreads to encompass the entire sole of the foot (and sometimes the toes) in many patients (7) over a 10–15 year period. It has never been clear as to whether this increase or spreading is due to nascent, mutation-positive cells that later manifest the trait (primarily ECM-mediated) or whether a subset of mutant cells induces a pathologic level of AKT signaling in mutation-negative cells. The rarity, severity, progressive nature and variability make the disorder challenging to study in the human.
Studies using mutagenized or myristoylated AKT have advanced our understanding of the effect of constitutive activation of AKT in defined, in vitro, systems. Activated forms of AKT have also been used in animal studies to address its role in the development of several tissues. Some examples include epidermis (8–10), cartilage (11), endothelial cells (12,13) and mammary tissue (14,15). Interpreting these studies is challenging because of the restricted lineage/tissue expression of activated AKT1 and because of their use of artificial promoters. While these studies are valuable for understanding specific aspects of AKT function, they do not address the broad question of how mosaicism throughout the animal causes the overgrowth of Proteus syndrome. Specifically, this is a question as to whether the progressive or spreading nature of this or other mosaic disorders are cell-autonomous (the pathology limited to mutation-positive cells) or non-cell-autonomous (the pathology is induced in mutation-negative cells as well) (16). Therefore, we set out to create a new mouse model that had endogenously-regulated, mosaic expression of the Akt1 c.49G > A, p.(E17K) variant that could generate randomly distributed variant-positive cells modeling the human trait.
Results
Generation of a mouse model for Proteus syndrome
To model Proteus syndrome, we engineered mice that would allow organism-wide, random, mosaic expression of the Akt1 c.49G > A p.(E17K) variant under the control of its endogenous regulatory elements. Since we did not know how many variant cells a mouse could tolerate, or would be needed to produce a variant phenotype, we pursued two strategies with differing attributes to generate Proteus syndrome mice. The first relied on a conditional Proteus syndrome allele, Akt1flx, that remains inactive due to the presence of a floxed βGeo cassette that contained a transcription termination signal and was inserted upstream of the Proteus syndrome variant in Akt1 (Fig. 1A and Supplementary Material, Fig. S1A). This allele will only be expressed when CRE is present to mediate recombination of the βGeo cassette. To test that this Akt1flx allele would be activated by our system, ES cells were transfected with Cre to remove the βGeo cassette and RNA was isolated. cDNA sequencing of the transfected Akt1Δneo3 ES cells showed expression of both Akt1WT and Akt1E17K alleles, whereas the untransfected Akt1–18 ES cells had only Akt1WT transcripts (Supplementary Material, Material, Fig. S1C).
Figure 1.
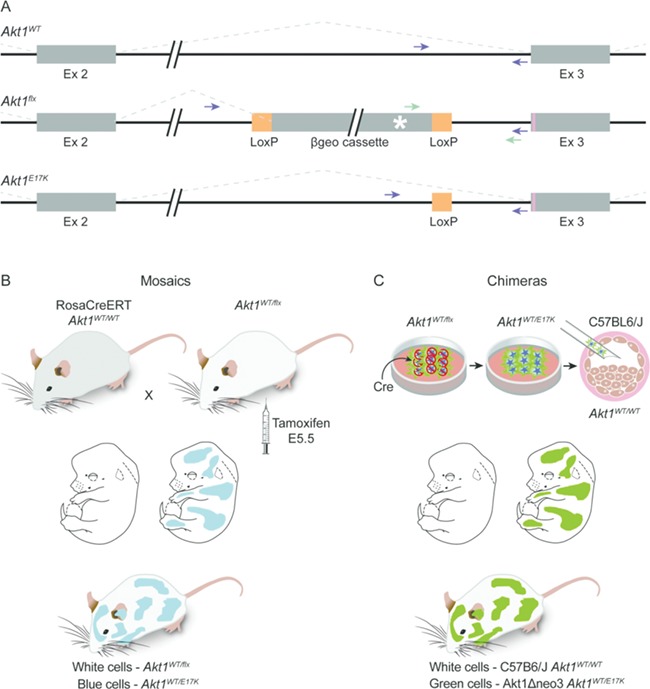
Strategy for making mosaic Proteus syndrome mice. (A) Diagrams of the wild type (top), conditional (middle) and p.(E17K)-expressing (bottom) Akt1 alleles. Exons are represented by the thick boxes; introns are shown by thin lines. The Akt1flx allele contains the 5 Kb floxed βgeo cassette inserted 124 bp upstream of the start of exon 3. Orange boxes represent LoxP sites. Splicing patterns are indicated by the dotted lines. The p.(E17K) variant is indicated by the pink line in exon 3 and the poly A signal that terminates transcription in the βgeo cassette is indicated by the white asterisk. After CRE-mediated recombination of Akt1flx, one LoxP site remains in intron 2 in the Akt1E17K allele as depicted in the bottom drawing. Green arrows mark the location of primers used for identifying Akt1flx. Purple arrows mark the location of primers used to identify Akt1WT and Akt1E17K, which can be distinguished by their size difference. Note, because of the βgeo cassette, Akt1flx does not amplify with primers denoted by the purple arrows using conditions to amplify the other alleles. (B) Diagram of the mosaic strategy. Pregnant females from timed matings between RosaCreERT-expressing and Akt1WT/flx animals were dosed with tamoxifen at E5.5 to activate CRE and the Akt1 conditional allele. Offspring are mosaic for Akt1E17K. (C) Diagram of the chimeric strategy. Akt1WT/flx embryonic stem (ES) cells were transfected with Cre to activate the conditional allele. One to three Akt1WT/E17K ES cells were injected into blastocysts, which were then transferred to pseudo-pregnant mothers for gestation, resulting in animals that were chimeric for Akt1E17K.
To make mosaic animals, Akt1WT/flx mice were crossed to 129-t (ROSA)26Sortm1(cre/ERT)Nat/J mice, which are homozygous for the ubiquitously-expressed, tamoxifen-inducible CreER fusion protein (Fig. 1B). Because some patients with Proteus syndrome have affected tissues that are derived from multiple germ layers, we hypothesized that their somatic mutation occurred before gastrulation. Therefore, we chose embryonic day 5.5 (E5.5) to deliver a single dose of tamoxifen to pregnant females. We used three concentrations of tamoxifen (1.7, 4 and 8 μg/g body weight) to activate varying numbers of Akt1E17K-positive cells in the embryos. Tail snip genotyping determined that 28 of 59 pups born from this cross carried the Akt1flx allele. Activation of the conditional allele in these animals resulted in true mosaics in that the only difference between mutant, Akt1WT/E17K, cells and wild-type, Akt1WT/flx, cells was the expression of the Akt1 c.49G > A p.(E17K) variant.
The second method used to generate mice that would model Proteus syndrome was to make chimeras. We accomplished this by injecting wild-type host C57BL/6 J blastocysts with Akt1Δneo3 ES cells in which the Akt1E17K allele had already been activated by Cre-mediated excision of the βGeo gene (Fig. 1C). These animals were chimeras and not mosaics in that the variant-positive Akt1WT/E17K, and variant-negative, Akt1WT/WT, cells came from different, albeit highly similar, strains of mice. For clarity, we refer to mice that were generated by partial Akt1E17K gene activation by tamoxifen as mosaic Proteus syndrome mice and those generated by blastocyst injection of recombined Akt1E17K ES cells as chimeric Proteus syndrome mice.
To reduce the chance that the entire chimeric embryo would develop from the activated ES cells that was predicted to be lethal (17), only one to three cells were used for blastocyst injection. Because the HG3 129x B6 hybrid ES cell line expresses ubiquitin C-driven green fluorescent protein (GFP) and produces the agouti coat color, chimeras were identified by expression of GFP in their skin and patches of agouti fur. Both of these markers differentiate ES-derived cells from those of the host C57Bl6/J blastocyst, which has no GFP expression and produces black coats. Of the 319 pups born using this method, 45 had GFP expression; however, 15 of them died within 2 days of birth and were not studied further.
We next set out to evaluate the phenotype and variant distribution of Proteus syndrome mice by performing a natural history study, hypothesizing that the phenotype would develop primarily postnatally, as is the case in humans. Tissues were collected at necropsy from 24 mosaic and 20 chimeric animals when any observed growths approached 2 cm, pain or distress could not be medically managed, they reached at least 20 months of age, or to address specific questions that arose during the study (Table 1). The remaining Proteus syndrome animals were not included in the analyses because they did not meet euthanasia criteria and were still alive (7), they died unexpectedly and the tissue was too degraded for full analyses (5), or they were sacrificed to examine their skeletons with only limited soft tissue collected (2). The variant allele fraction (VAF) in each tissue was measured (Fig. 2 and Supplementary Material, Tables S1 and S2). Since the mutation occurs in the heterozygous state and variant-positive cells also contain a wild-type allele, if all cells were heterozygous (i.e. the animal was not mosaic and had a constitutional or germline variant) the VAF level would be 50%. The VAFs ranged from 0% to 50% across all tissues assayed from Proteus syndrome animals.
Table 1.
Phenotype of Proteus syndrome mice
| Mice | Method (tamoxifen dose)* | Average VAF (%) | Age at necropsy (months) | Reason for sacrifice | Explanation of reason for sacrifice | VM gross/microscopic** | Cysts/Ectasia | Mammary overgrowth*** | Hyperplasia/stromal expansion | Total number of lesions |
|---|---|---|---|---|---|---|---|---|---|---|
| B-12 | Chimera | 18 | 4 | Died after anesthesia | 0/0 | NA | 0 | |||
| CF2–5 | Mosaic (8) | 28 | 4 | Died after anesthesia | 0/0 | No | 0 | |||
| GF2–6 | Mosaic (8) | 34 | 4 | Died after anesthesia | 0/0 | NA | 0 | |||
| E-2 | Chimera | 40 | 8 | Mass on flank | VM connective tissue | 3/2 | NA | Stromal expansion—peritoneal wall; epidermal hyperplasia—ear; interstitial cell hyperplasia—testis | 8 | |
| M-11 | Chimera | 39 | 11 | Distended abdomen | VM lymph node | 1/1 | Biliary ductule ectasia | NA | 3 | |
| A-17 | Chimera | 24 | 12 | Swelling on head | Pituitary adenoma | 0/1 | NA | Lymphoid hyperplasia—spleen; epithelial cell hyperplasia—testis | 3 | |
| E-4 | Chimera | 34 | 13 | Mass on neck | VM lymph node | 1/0 | NA | Dermal expansion—ear | 2 | |
| RSC-11 | Mosaic (4) | 13 | 13 | Look for lesions | 0/0 | No | 0 | |||
| RSC-17 | Mosaic (8) | 23 | 13 | Look for lesions | 0/0 | No | 0 | |||
| CF2–11 | Mosaic (4) | 27 | 14 | Mass on neck | VM lymph node | 2/0 | Yes | 3 | ||
| RSC-6 | Mosaic (4) | 4 | 14 | Look for lesions | 0/0 | NA | 0 | |||
| RSC-15 | Mosaic (8) | 18 | 14 | Look for lesions | 0/0 | Ectopic nasal epithelial cyst | NA | 1 | ||
| C-1 | Chimera | 12 | 15 | Anal prolapse | 0/0 | Yes | 1 | |||
| D-1 | Chimera | 25 | 15 | Distended abdomen | Enlarged seminal vesicle | 0/2 | Pancreatic ductal ectasia | NA | 3 | |
| E-1 | Chimera | 21 | 15 | Abdominal mass | VM lymph node | 1/1 | Biliary ductule ectasia | Yes | 4 | |
| E-3 | Chimera | 21 | 15 | Abdominal mass | Biliary cysts | 0/1 | Spinal cord cyst biliary cyst† | NA | 3 | |
| A-9 | Chimera | 22 | 17 | Anal prolapse | 5/5 | Pancreatic ductal ectasia | Yes | Stromal expansion—middle ear; endothelial cell hyperplasia—kidney | 14 | |
| B-13 | Chimera | 27 | 17 | Enlarged foot | Not found | 0/1 | Meibomian gland cyst; biliary cyst† | NA | 3 | |
| GF2–3 | Mosaic (8) | 22 | 17 | Abdominal mass | VM lymph node | 1/0 | Biliary ductule ectasia | NA | Epithelial cell hyperplasia—pancreas | 3 |
| DF1–18 | Mosaic (4) | 10 | 18 | Possible abdominal mass | VM spleen | 1/1 | NA | 2 | ||
| DF1–21 | Mosaic (4) | 1 | 18 | Possible abdominal mass | Not present | 0/0 | NA | 0 | ||
| F-5 | Chimera | 30 | 19 | Abdominal mass | Not found; small VM and anasarca were present | 4/3 | Yes | Islet cell hyperplasia—pancreas | 9 | |
| H-1 | Chimera | 27 | 19 | Found dead | Compression of brain and hemorrhage from pituitary adenoma | 5/1 | Biliary cyst | Yes | 8 | |
| A-10 | Chimera | 0.55 | 20 | Head tilt | 0/0 | NA | 0 | |||
| A-11 | Chimera | 34 | 20 | Age | 1/0 | Biliary cyst† | NA | 2 | ||
| A-12 | Chimera | 26 | 20 | Age | 0/2 | Biliary cyst† | NA | 3 | ||
| A-5 | Chimera | 32 | 20 | Age | 0/4 | Parathyroid cyst; biliary cyst† | Yes | 7 | ||
| CF1–28 | Mosaic (1.7) | 8 | 20 | Breathing difficulty | Not found | 1/0 | Spinal cord cyst | Yes | 3 | |
| CF1–29 | Mosaic (1.7) | 5 | 20 | Age | 0/1 | Clitoral gland ectasia | Yes | 3 | ||
| CF2–15 | Mosaic (4) | 30 | 20 | Enlarged organs | Sarcoma | 1/3 | Ankle joint synovial cyst | NA | 5 | |
| CF2–4 | Mosaic (8) | 12 | 20 | Age | 0/0 | No | 0 | |||
| GF2–1 | Mosaic (8) | 19 | 20 | Age | 0/1 | Yes | 2 | |||
| GF2–5 | Mosaic (8) | 17 | 20 | Age | 0/1 | NA | 1 | |||
| J-5 | Chimera | 38 | 21 | Hunched with tremors | CNS hemorrhages; no cause seen | 0/1 | Biliary cyst†; stomach glandular cysts | NA | 3 | |
| DF1–13 | Mosaic (1.7) | 13 | 21 | Distended abdomen | Biliary cysts | 0/0 | Pancreatic cyst biliary cysts† | NA | 2 | |
| BF1–18 | Mosaic (1.7) | 12 | 22 | Age | 0/0 | NA | 0 | |||
| DF1–14 | Mosaic (1.7) | 4 | 23 | Age | 0/1 | Biliary cysts | NA | 2 | ||
| DF1–7 | Mosaic (1.7) | 13 | 23 | Age | 0/1 | No | 1 | |||
| DF1–8 | Mosaic (1.7) | 5 | 23 | Age | 0/1 | Biliary ductule ectasia | No | 2 | ||
| I-5 | Chimera | 35 | 24 | Swelling in abdomen | Enlarged spleen | 0/4 | Biliary ductule ectasia | NA | 5 | |
| J-6 | Chimera | 39 | 24 | Age | 0/2 | Biliary cysts†; spinal cord cyst | NA | 4 | ||
| G-2 | Chimera | 6 | 25 | Age | 0/0 | Biliary cyst† | NA | 1 | ||
| RSC-3 | Mosaic (4) | 2 | 27 | Age | 0/1 | NA | 1 | |||
| RSC-5 | Mosaic (4) | 4 | 27 | Age | 0/0 | NA | 0 |
*For mosaic animals, the dose of tamoxifen (μg/g body weight) used to induce CRE activation is indicated in parentheses. **Number of VM seen by gross or microscopic examination. Details of VM are in Table S3. ***Presence or absence of mammary changes. Details of mammary changes are in Table S4. †Seen by gross examination. VM, vascular malformation. NA, not applicable.
Figure 2.
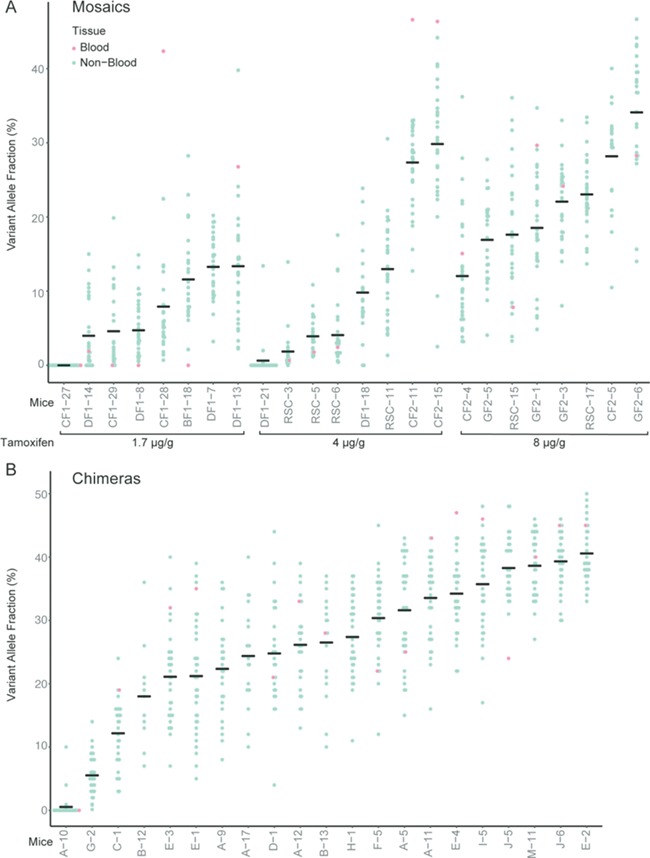
VAF of tissues collected from Proteus syndrome animals. The VAFs of all tissues except blood from the tamoxifen mosaics (A) and chimeras (B) are indicated by the blue dots. Red dots represent the VAF in blood DNA. The horizontal line in each column marks the average VAF across all tissues tested for each mouse and represents the overall average VAF for that animal. The tamoxifen dose per gram of body weight used to induce CRE activation is shown below the graph in (A).
Tissues from 23 littermates of the chimeras that were sacrificed as potential control animals were also assayed for Akt1E17K to ensure that they did not harbor low levels of the variant in any tissue. One animal, A-10, did have four tissues, testis, heart, pancreas and spleen with detectable Akt1E17K levels of 10%, 4%, 0.9% and 0.2%, respectively and was therefore evaluated as a chimeric Proteus syndrome mouse. Variant-positive tissues from A-10 immunostained for GFP showed that developing sperm, endothelial and hematopoietic cells were responsible for the positive VAF (Supplementary Material, Fig. S2). As these cell types are known to arise in the yolk sac and then migrate into the embryo during development (18–20), we postulate that the ES cell from which they were derived contributed primarily to extraembryonic tissue rather than the embryo proper. With the exception of mouse A-10, all tissues tested in the chimeric animals had measurable levels of Akt1E17K.
In the mosaic mice, 16 of 24 animals had non-zero VAF in all tissues tested. One mouse from a tamoxifen-injected dam, CF1–27, had no detectable Akt1E17K in any tissue assayed. To determine whether there was an error in the original genotyping of the tail DNA from CF1–27, two additional tissues were tested for the presence of the conditional Akt1 allele. All three DNAs were positive for Akt1flx, indicating that the failure to detect Akt1E17K was likely due to a lack of tamoxifen activation of CRE in the embryo. Since mosaicism could not be confirmed in this mouse, it was not included in the phenotype analysis.
To assess the effectiveness of each method to model Proteus syndrome and to estimate the overall variant level in each animal, the average (mean) VAF was calculated across all tissues assayed for each mouse (Fig. 2 and Supplementary Material, Tables S1 and S2). In mosaic animals, the average VAF in offspring of mice given 1.7 μg/g of tamoxifen ranged from 0% to 13%, with 0 of 8 mosaics having an average VAF > 20%. In offspring of mice given 4 μg/g tamoxifen, the average VAF ranged from 0.63% to 30% with two of eight having an average VAF > 20%, and in offspring of mice given 8 μg/g tamoxifen, the average VAF was 12–34% with four of eight having an average VAF > 20%. In the chimeric mice, the average VAF was 0.55–40% with 17 of the 21 chimeras having an average VAF > 20%. The increased number of mice with average VAF > 20% in the chimeras (P = 0.0004) suggested that with the parameters used in this study, the chimeric method was more likely to generate animals with higher overall variant levels.
In contrast to patients with Proteus syndrome where the Akt1 p.(E17K) mutation is rarely present in peripheral blood, 12 of 15 mosaics and 15 of 16 chimeras, had non-zero VAFs in blood DNA of 0.67–47% (Fig. 2 and Supplementary Material, Tables S1 and S2). The VAFs in blood showed a moderate correlation (R2 = 0.6) to the overall (non-blood) VAF burden of each animal (Supplementary Material, Fig. S3).
Proteus syndrome mice model overgrowth seen in patients with Proteus syndrome
The phenotypes in the Proteus syndrome mice were heterogeneous. Of the 44 Proteus syndrome mice (28 male; 16 female) that were phenotyped, 21 developed lesions or distress that required them to be euthanized (Table 1). Of these 21, only 2, E-2 and M-11, required sacrifice before 1 year of age. Four animals died unexpectedly and were necropsied before their tissues were autolyzed. Fifteen mice were sacrificed because they reached at least 20 months of age. Finally, four were sacrificed at 13–14 months to determine if lesions could be identified before having an observable overgrowth. Thirty-two control animals (19 male; 13 female) were also phenotyped (Table 2). Eleven were littermates of chimeras that had no detectable Akt1E17K in any tissue assayed, 12 were littermates of mosaic animals that were positive for Cre and Akt1WT/WT, 7 were Cre-negative and Akt1WT/flx and 2 were Cre-negative and Akt1WT/WT. Ten of the 32 control animals were sacrificed because of distress or the development of lesions, with the remaining animals euthanized because of their advanced age or they were cage mates of Proteus syndrome animals that required sacrifice.
Table 2.
Phenotype of control animals
| Mice | Type | Age at necropsy (months) | Reason for sacrifice | Explanation of reason for sacrifice | VM gross/microscopic* | Cysts/Ectasia | Mammary overgrowth** | Hyperplasia/stromal expansion | Total number of lesions |
|---|---|---|---|---|---|---|---|---|---|
| A-14 | Chimera littermate | 12 | Cage mate*** | 0/0 | NA | 0 | |||
| A-16 | Chimera littermate | 12 | Cage mate | 0/0 | NA | 0 | |||
| T2C-57 | Cre− Akt1WT/WT | 12 | Cage mate | 0/0 | No | 0 | |||
| RSC-12 | Cre+ Akt1WT/WT | 13 | Cage mate | 0/0 | No | 0 | |||
| CF2–10 | Cre+ Akt1WT/WT | 14 | Cage mate | 0/0 | No | 0 | |||
| RSC-16 | Cre+ Akt1WT/WT | 14 | Cage mate | 0/0 | NA | 0 | |||
| T2C-29 | Cre− Akt1WT/flx | 14 | Cage mate | 0/0 | NA | 0 | |||
| T2C-55 | Cre− Akt1WT/WT | 16 | Cage mate | 0/0 | NA | 0 | |||
| A-13 | Chimera littermate | 17 | Pelvic mass | Enlarged bladder | 0/0 | NA | Stromal expansion—seminal vesicle (mild) | 1 | |
| DF1–20 | Cre+ Akt1WT/WT | 18 | Enlarged organs | Lymphoma | 0/0 | NA | 0 | ||
| F-4 | Chimera littermate | 19 | Abdominal mass | Sarcoma | 0/0 | No | 0 | ||
| CF2–8 | Cre+ Akt1WT/WT | 19 | Distended abdomen | Sarcoma | 0/0 | NA | 0 | ||
| CF2–9 | Cre+ Akt1WT/WT | 19 | Cage mate | 0/0 | NA | 0 | |||
| EF4–2 | Cre− Akt1WT/flx | 19 | Distended abdomen | Pancreatic cyst | 0/0 | Pancreatic cyst† | NA | 1 | |
| T2C-39 | Cre− Akt1WT/flx | 19 | Cage mate | 0/0 | NA | 0 | |||
| CF2–12 | Cre+ Akt1WT/WT | 20 | Possible enlarged organs | Not enlarged | 0/0 | NA | 0 | ||
| PXCRT-40 | Cre− Akt1WT/flx | 20 | Age | 0/0 | NA | 0 | |||
| PXCRT-42 | Cre− Akt1WT/flx | 20 | Age | 0/0 | NA | 0 | |||
| B-14 | Chimera littermate | 21 | Distended abdomen | Sarcoma | 0/0 | NA | 0 | ||
| B-5 | Chimera littermate | 21 | Labored breathing | Severe renal disease | 0/0 | Yes | 1 | ||
| T2C-81 | Cre− Akt1WT/flx | 21 | Age | 0/0 | No | 0 | |||
| BF1–19 | Cre+ Akt1WT/WT | 22 | Age | 0/0 | NA | 0 | |||
| DF1–6 | Cre+ Akt1WT/WT | 23 | Age | 0/0 | Biliary ductule ectasia | Yes | 2 | ||
| DF1–9 | Cre+ Akt1WT/WT | 23 | Neck mass; distended abdomen | Neoplasm | 0/1 | Biliary ductule ectasia | Yes | 3 | |
| C-3 | Chimera littermate | 24 | Rigid abdomen | Not found | 0/1 | No | 1 | ||
| C-4 | Chimera littermate | 24 | Age | 0/0 | No | 0 | |||
| C-5 | Chimera littermate | 24 | Age | 0/0 | No | 0 | |||
| I-2 | Chimera littermate | 24 | Age | 0/1 | NA | 1 | |||
| G-1 | Chimera littermate | 25 | Age | 0/0 | Epithelial cyst—ear; epididymal cyst; ectasia of rete testis | NA | Chondroid metaplasia and hyperplasia—femur periosteum | 4 | |
| DF1–16 | Cre+ Akt1WT/WT | 25 | Age | 1/1 | Biliary cysts† | Yes | 4 | ||
| PXC-4 | Cre− Akt1WT/flx | 25 | Age | 0/0 | NC | 0 | |||
| DF1–12 | Cre+ Akt1WT/WT | 26 | Age | 0/0 | Biliary cysts† | NA | 1 |
*Number of VM seen by gross or microscopic examination. Details of VM are in Table S3. **Presence or absence of mammary changes. Details of mammary changes are in Table S4. ***cage mate—animals that were sacrificed because a Proteus syndrome cage mate required euthanasia. †Seen by gross examination. VM, vascular malformation. NC, not collected. NA, not applicable.
The most frequent abnormality found in mosaic or chimeric Proteus syndrome mice were VMs. Twenty-nine of 44 Proteus syndrome mice had at least one VM per animal compared with only 4 of 32 controls (P < 0.0001) (Tables 1 and 2). VMs are also a frequent finding in humans with Proteus syndrome (5,21). In a recent study, superficial VMs were found in 68% of the cohort with 26% of them also having a visceral VM (22). The number of VMs per Proteus syndrome mouse ranged from 1 to 10 with 26 visible grossly and 43 additional VMs found by microscopic examination (Supplementary Material, Table S3). Thirty-four VMs were in lymph nodes, 13 in adipose tissue, 11 in spleen and 11 distributed among other locations. The four control mice with VMs had a total of five VMs with only one visible on gross examination (Supplementary Material, Table S3). Grossly visible VMs at necropsy appeared as dark, blood-filled, often multi-lobulated cystic masses, a few of which reached 1–3 cm in size (Supplementary Materials, Fig. S4 and Table S3). These VMs were characterized as venous-capillary mixed-type malformations, as noted by the absence of a muscularis, with ectatic channels and often containing a lymphatic component and no atypia. Numerous, variably-sized, thin-walled ectatic vessels, often separated by increased stroma were seen in these lesions. Lymph, blood and thrombin were commonly identified. VMs in adipose tissue were comprised of multiple thin-walled vessels, few of which were visible by gross examination. These VMs were remarkably similar to those seen in patients with Proteus syndrome as shown in Figure 3. In many cases, the cells lining these vascular channels stained with antibodies to CD31 and LYVE-1 suggesting endothelial lined spaces that demonstrate a mixed phenotypic expression of both vascular and lymphatic cell-surface markers (Supplementary Material, Figure S5).
Figure 3.
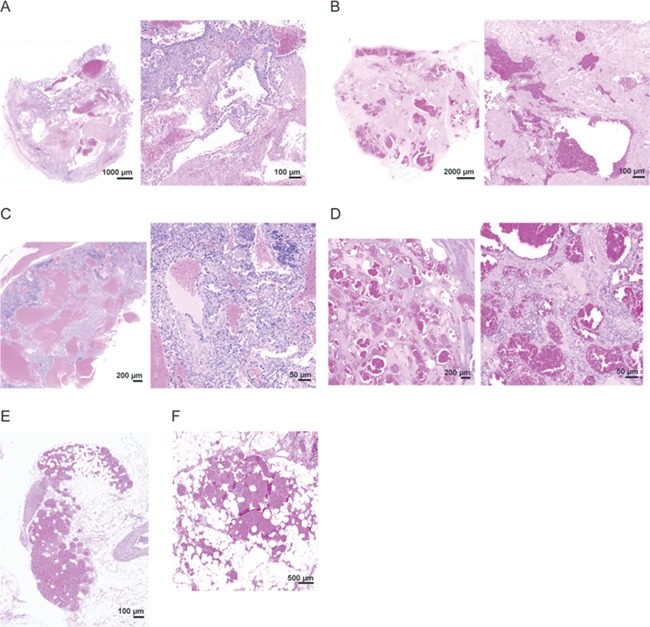
Examples of VMs in mice and patients with Proteus syndrome. (A and B) VMs found in the abdominal connective tissue of chimera E-2 (A) and a patient with Proteus syndrome (B). (C and D) VMs found in the spleen of mosaic DF1–18 (C) and a patient with Proteus syndrome (D). All showed variably-sized thin-walled ectatic vessels separated by increased stroma. (E and F) VMs found in adipose tissue of chimera A-9 (E) and a patient with Proteus syndrome (F) that were comprised of numerous, small, thin-walled, ectatic channels.
As in the human disorder where a wide variety of cysts have been identified (6) both mosaic and chimeric Proteus syndrome mice also developed cysts or ductule ectasia. Several studies in our patient cohort have identified ovarian cysts (44%), ovarian and paraovarian cystadenomas (38%), testicular enlargement with cysts (22%), epididymal cysts (35%) and testicular or paratesticular cystadenomas (32%) [K.M. Keppler-Noreuil, personal communication]. Cysts, also described as bullae in the lung, are common in humans with Proteus syndrome, with 17 of 51 (33%) patients in our cohort having lung involvement [K.M. Keppler-Noreuil, personal communication]. Hepatic cysts occurred in 4 of 34 (12%) individuals with Proteus syndrome, compared with 1% in adults in the general population. Cerebral and white matter cysts were described in 4 of 20 individuals in our cohort with brain imaging (23). Porencehalic, subarachnoid and periventricular cysts have also been reported in individuals with Proteus syndrome (6). In our mouse cohort, 22 of 44 Proteus syndrome mice developed a total of 28 cystic lesions or dilations whereas only 6 of 32 control animals developed a total of 8 cysts or ectatic ducts (P = 0.0077) (Tables 1 and 2). These cysts and ectasia were exclusive of three additional types of cysts, ovarian, uterine and kidney, which were found to occur at similar rates in the Proteus syndrome and control groups (ovarian—7/16 in Proteus syndrome versus 6/13 in control; uterine—13/16 in Proteus syndrome versus 12/13 in control; kidney—6/44 in Proteus syndrome versus 6/32 in control). Therefore, the impact of the Proteus syndrome variant on the development of these three types of cysts could not be assessed, and they were not further evaluated. Nearly all of the remaining cysts or ectatic ducts were simple, fluid-filled dilations with an epithelial lining. One exception to this was a cyst that formed in the ankle of mosaic mouse CF2–15 that was lined with synovial tissue. Biliary cysts were the most frequent with nine Proteus syndrome mice having grossly visible biliary cysts (Fig. 4). Microscopic examination showed smaller biliary cysts in 2 other Proteus syndrome mice, and 5 instances of biliary hyperplasia with ductule ectasia bringing the total of biliary findings to 16 in the Proteus syndrome animals. This is in contrast to the control animals where only two were found to have grossly visible cysts; two others had microscopically-detected biliary hyperplasia with ductule ectasia (P = 0.0333). Other, less common cysts or ductal ectasia are listed in Tables 1 and 2 and examples are shown in Figure 4.
Figure 4.
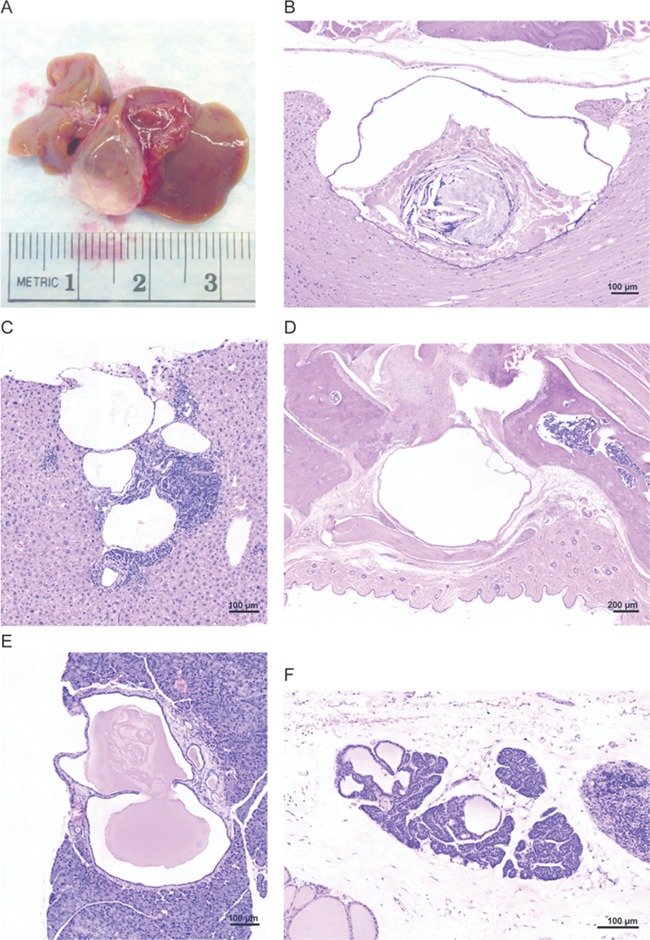
Examples of cysts and ductal ectasia seen in Proteus syndrome mice. (A) A large biliary cyst seen in chimera E-3. (B) A cyst found in the spinal cord of mosaic animal CF1–28. (C) Biliary ductule ectasia seen in mosaic animal DF1–8. (D) A synovial cyst found in the ankle of mosaic animal CF2–15. (E) A pancreatic cyst seen in mosaic animal DF1–13. (F) A parathyroid cyst seen in chimera A-5.
In addition to cyst formation, other types of proliferative changes were observed. Mammary changes were noted in 10 of 16 female Proteus syndrome mice (Table 1 and Supplementary Material, Table S4). Findings included alveolar hyperplasia, ductal ectasia and expansion of the supporting stroma with tissue appearing similar to what would be expected if these animals were retired breeders, instead of being aged nulliparous animals (Fig. 5A–C and E). The secretions in many of the mammary ducts appeared more fatty and proteinaceous than the homogenous eosinophilic fluid that was present in the ducts of females without mammary changes, which is also what would be predicted if these animals were nursing mothers. In addition, four chimeras had unusual polypoid projections into mammary ducts that appeared to be inversions of disorganized fatty and stromal tissue (Fig. 5F and G). Four of 12 female nulliparous control animals had some mammary changes as compared to other aged wild-type females that had never been bred (Fig. 5D, Table 2 and Supplementary Material, Table S4). While the difference between the number of mice with mammary changes in each group was not statistically significant, the extent of abnormalities in the Proteus syndrome mice was greater than in the control group, suggesting that overgrowth of breast tissue is a consequence of AKT1 activation. This is consistent with numerous examples of benign breast overgrowth seen in humans with Proteus syndrome (3,24,25).
Figure 5.
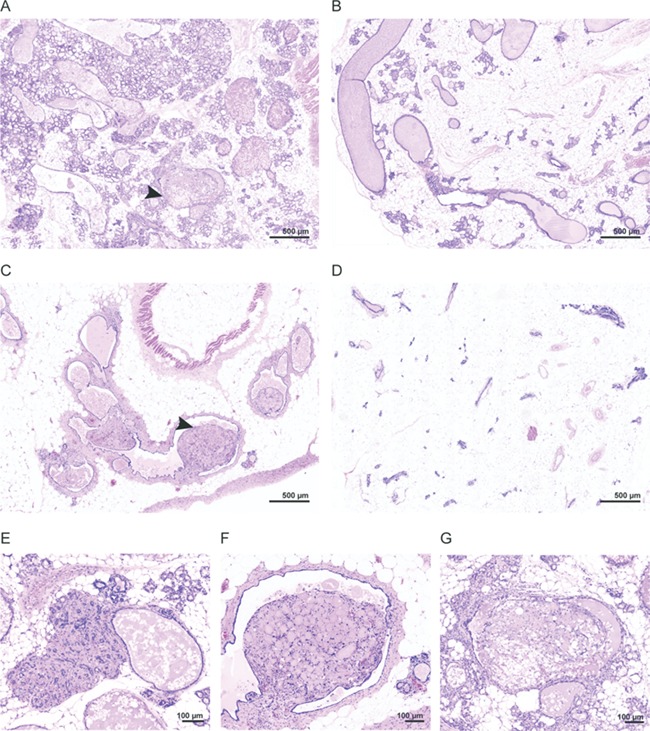
Range of mammary overgrowth seen in Proteus syndrome mice. (A) An area in chimera A-9 with multiple areas of alveolar hyperplasia and moderate ductal ectasia. (B) An area of milder alveolar hyperplasia and ductal ectasia in mosaic animal CF1–28. (C) An area of moderate ductal ectasia and stromal expansion in chimeric animal H-1. (D) An area in control animal DF1–6 with mild focal hyperplasia and ectasia shown for comparison. (E) An area of stromal expansion in chimera A-9. (F–G) Higher magnification of polypoid projections seen in chimeric animals H-1 and A-9 marked by arrows in (A) and (C).
Seven additional instances of hyperplasia were noted in 5 of 44 Proteus syndrome animals whereas only 1 was noted in the control group of 32 animals (Tables 1 and 2). Chimera E-2 had an area of epidermal hyperplasia similar to what is seen in epidermal nevi that are often found in humans with Proteus syndrome (Fig. 6A and B). Other areas of hyperplasia included pancreatic epithelial and islet cells, splenic lymphoid cells, kidney endothelial cells surrounding a VM and testicular epithelial and interstitial cell hyperplasia. Finally, in addition to the increased stroma seen in some VMs and mammary lesions, an expansion of stromal tissue was seen in three isolated instances in the Proteus syndrome mice and one in a control animal. In the peritoneal wall of chimera E-2, and in the middle ear of chimera A-9, which in addition to stromal expansion also had cholesterol clefts, osseous metaplasia, connective tissue mineralization and cysts, the increased stroma appeared to be a combination of increased number of fibroblasts and increase in the amount of ECM present (Fig. 6C–F). However, the stromal expansion in the dermis of the ear in chimera E-4 was predominantly due to an accumulation of ECM (Fig. 6G and H) that is characteristic of the CCTN seen in patients with Proteus syndrome. The number of mice with isolated instances of hyperplasia or stromal expansion was not statistically different between the Proteus syndrome and control animals, however, given the extensive list of less frequently associated manifestations in the human disorder, we speculate that these uncommon proliferative changes in the Proteus syndrome mice are due to the presence of the Akt1E17K-positive cells.
Figure 6.
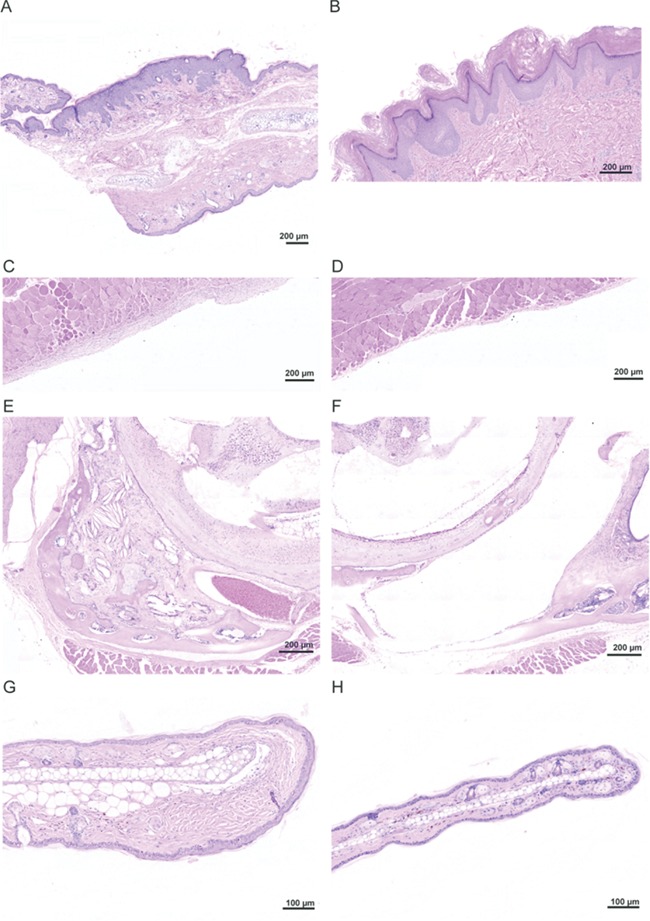
Hyperplasia and stromal expansion in Proteus syndrome mice. (A) A region of epidermal hyperplasia on the ear of chimera E-2. The upper edge showed expanded epidermis whereas the lower edge epidermis was normal thickness. (B) A linear epidermal nevus from a patient with Proteus syndrome. Note the similarity to the epidermal hyperplasia shown in chimera E-2 shown in panel A. (C) Expansion of the stroma lining the peritoneal wall in chimera E-2. (D) Normal thickness of stroma in an adjacent region of the peritoneal wall of this animal. (E) Overgrowth of multiple tissues in the left middle ear of chimera A-9. Expanded stroma, osseous metaplasia, cholesterol clefts, and several small cysts and areas of mineralization were seen. (F) Normal right middle ear of that same animal for comparison. (G) Increased collagen in the dermis at the tip of the ear in chimera E-4. (H) A normal ear from control T2C-81 for comparison.
Malignancies are an infrequent manifestation of human Proteus syndrome (benign tumors such as cystadenomas and meningiomas are common). To date, only three patients have been reported with a malignant phenotype. One patient had a mesothelioma of the tunic vaginalis (26), another had a ductal carcinoma in situ of the breast (25) and a third had a papillary thyroid carcinoma (3). In Proteus syndrome mice, lymphoma was present in 3, while histiocytic sarcoma was found in 2 additional animals, out of 44 examined. These neoplasms are common to aged C57BL/6 and 129S4/Sv animals and were also found in 7 of the 32 control animals in this study (27–29). Therefore, it is unlikely that expression of Akt1E17K was the driver of these malignancies in the mosaic animals. Pituitary tumors were also seen in the cohort. Adenomas of the pars distalis consisted of a monomorphic population of cells that compressed adjacent pituitary structures and often contained angiectasia and hemorrhages. These were distinct from pituitary adenocarcinomas, which share features with adenomas, but are characterized by cellular pleomorphism and atypia, and have definitive infiltration outside of the pituitary (30). Pituitary adenomas are relatively common in mice while adenocarcinomas are more rare (28). Three study animals (2 of 44 Proteus syndrome and 1 of 32 controls) had tumors consistent with a diagnosis of adenoma, while two additional Proteus syndrome animals, A-5 and F-5, had tumors that had begun to invade the surrounding tissue and were deemed to be adenocarcinomas (Supplementary Material, Fig. S6). We hypothesize that Akt1E17K contributed to the formation of pituitary adenocarcinomas, which deserves future study to confirm.
Because of the heterogenous nature of a mosaic disease, the overall severity in an individual is challenging to quantify. To develop a simplified approach, we summed the number of findings per mouse and used that number as a proxy for phenotypic impact of the Akt1E17K variant. Thirty-three of 44 Proteus syndrome mice had a total of 117 lesions with a range of 1–14 per animal. For the controls, 10 of 32 mice had a total of 19 lesions with a range of 1–4 per animal. The difference in number of animals with lesions (regardless of how many lesions they had) was statistically significant (P = 0.0002). Within the Proteus syndrome animals, 19 of 21 chimeras had a total of 86 lesions, whereas 14 of 23 mosaics had a total of 31 lesions (P = 0.0365) suggesting that the chimeric method generated a more severe phenotype. A comparison of the number of lesions versus the average VAF in each animal showed a weak, but positive correlation (R2 = 0.2) (Supplementary Material, Fig. S7). As noted above, the overall level of severity was also correlated with the peripheral blood VAF. For the 11 Proteus syndrome animals with no detectable lesions, there was a moderate inverse correlation of the age at necropsy to the average VAF for each animal (R2 = 0.6) (Supplementary Material, Fig. S8). This inverse correlation suggests that the Proteus syndrome mouse is a valid model for the progressive nature of the human disorder. Mice with lower overall VAF would be expected to have fewer phenotypic consequences and therefore more likely necropsied because they reached the end of the study rather than morbidity, whereas those animals with higher VAF but necropsied at a younger age for reasons other than the presence of lesions, may not have had sufficient time to develop any manifestations of Proteus syndrome.
Evidence for cell-intrinsic and -extrinsic effects of Akt1E17K
In humans with Proteus syndrome, little is known about which cells within an affected tissue drive the phenotype as there is only a weak correlation of bulk tissue VAF and tissue affection status (1,3). Expression of GFP in cells derived from the Akt1Δneo3 ES cells in the chimeras can be used as a proxy to identify the Akt1E17K cells and begin to investigate this question. Staining of multiple tissues from several chimeras showed varying patterns and numbers of GFP-positive cells across tissues indicating that most cell types are capable of expressing Akt1E17K (data not shown). We assumed that lesional cells would be Akt1E17K-positive and indeed, the epithelial cells lining several biliary cysts were predominantly GFP-positive with elevated pAKT staining compared with adjacent GFP-negative cells, suggesting a correlation of epithelial VAF, AKT signaling and subsequent cyst development (Fig. 7A). However, in mammary tissue, hyperplastic epithelia and stromal tissue were comprised of both GFP-positive and negative cells (Fig. 7B). In some areas, nearly all of the hyperplastic epithelia were negative for GFP (Supplementary Material, Fig. S9A and B). These data suggest that Akt1E17K-positive cells may be signaling neighboring variant-negative cells to proliferate. Elevated pAKT staining in hyperplastic GFP-negative alveolar epithelia in these areas supports this hypothesis. Even in VMs where the endothelial lining of ectatic vessels was largely GFP-positive, areas of GFP-negative endothelia were positive for pAKT (Fig. 7C and Supplementary Material, Fig. S9C and D). Increased pAKT signal in Akt1E17K-negative lesional cells suggests that the Proteus syndrome variant can drive overgrowth in a non-cell autonomous manner.
Figure 7.
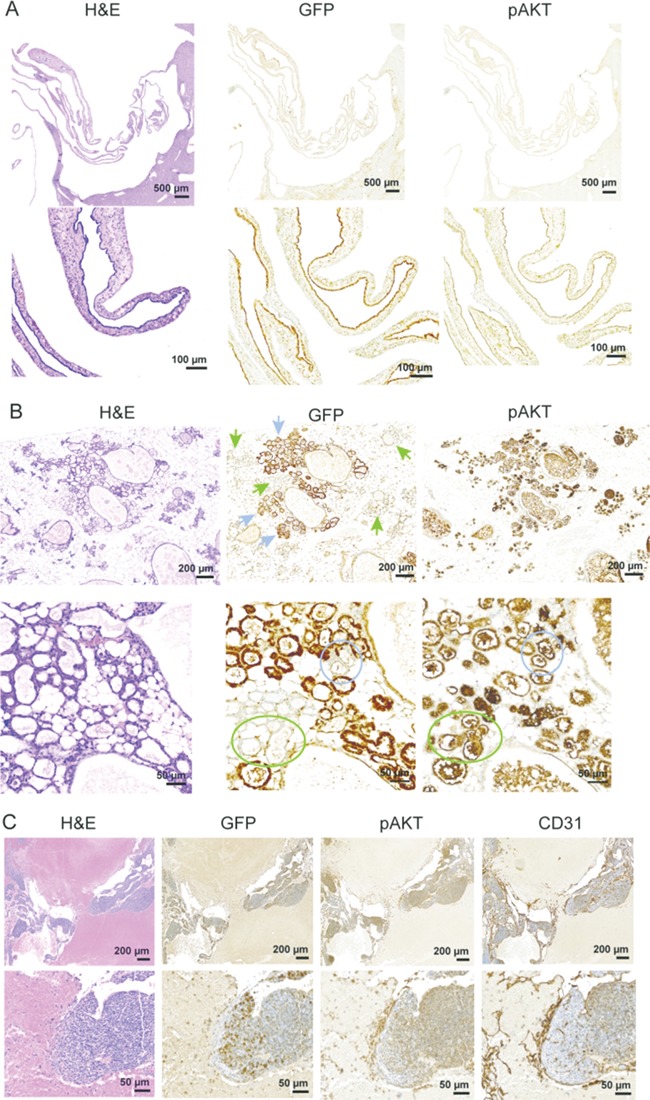
Identification of Akt1E17K-positive cells and increased AKT signaling in lesional tissue of chimeric Proteus syndrome mice. Hematoxylin and eosin staining and immunostaining with antibodies to green fluorescent protein (GFP), pAKT, and CD31 are shown. (A) A collapsed biliary cyst from chimera E-3 showed that the epithelial cells lining the cysts were predominantly GFP (Akt1E17K)-positive. Phospho-AKT staining correlated with the GFP staining in the cyst. (B) Immunostaining showed hyperplastic mammary alveolar epithelia were comprised of both GFP-positive (blue arrows) and GFP-negative (green arrows) cells. In the higher magnification images, hyperplastic GFP-negative alveolar epithelia that stained positive with the pAKT antibody can be seen (compare color-matched circles). (C) CD31-positive endothelial cells lining the ectatic channels of a VM found in a peri-ovarian lymph node in A-9 were largely GFP-positive. Importantly, substantial numbers of GFP-negative endothelial cells were found that stained positive for pAKT.
Discussion
While no animal model can replicate all aspects of a given disease, creating a system that models the pathophysiology of the disease under the control of the animal’s endogenous regulatory and developmental program is important for making physiologically-relevant discoveries. That Proteus syndrome is both remarkably heterogeneous and extremely rare is a further impetus to develop a faithful animal model for this severe and often fatal disease. To that end, we used two strategies for the development of a mouse model for Proteus syndrome. The chimera method used blastocyst injection of Akt1E17K-positive ES cells. Mosaic mice were created by crossing Akt1WT/flx conditional mice with mice ubiquitously expressing an inducible Cre allele and activating CRE by a single dose of tamoxifen at E5.5. While both methods generated Proteus syndrome animals that manifested several of the characteristic types of overgrowth seen in the human disease including VMs, cysts, hyperplasia and stromal expansion, the chimeric method appeared more successful as measured by the number of animals with VAF > 20% and the number of animals with lesions. Higher doses and earlier injection of tamoxifen would likely increase the VAF in mosaic animals and potentially yield more severely affected mice, although this might also increase their prenatal lethality.
AKT1 is considered oncogenic and has shown transforming ability in vitro (31), however, AKT1 variants in tumors are relatively infrequent. It is most frequently associated with breast cancer where it is expressed in ~ 3% of tumors. Mosaic mice did not develop mammary cancer, but they did exhibit hyperplasia and ductal ectasia that was quite extensive in some. These changes were similar to those seen in a study using MMTV-driven expression of Akt1E17K (15). However, the extent of the hyperplasia appeared greater in several of our Proteus syndrome mice compared with those with MMTV-driven expression of Akt1E17K and was more similar to what was seen when HER2 was also overexpressed in the mammary tissue (15). In addition, some Proteus syndrome animals developed polypoid projections of disorganized mixed adipose and stromal tissue that invaginated into the mammary ducts, which was not reported in the MMTV-Akt1E17K study. It is possible that if mice with MMTV-driven Akt1E17K were allowed to age longer than 1 year, the hyperplasia would reach levels seen in some of the Proteus syndrome animals. Alternatively, in the MMTV transgenic model, Akt1E17K expression was restricted to cells where the MMTV promoter was expressed, whereas any cell type in the Proteus syndrome mice could harbor Akt1E17K and therefore non-MMTV expressing cells could contribute to the increased hyperplasia seen in the Proteus syndrome animals.
Previous studies have shown that the VAF in tissues from patients with Proteus syndrome did not correlate well with the histopathology of that tissue (1,3). Due to clinical considerations, human samples used for VAF determination are usually a small percentage of the organ or tissue being evaluated and therefore may not accurately represent the overall mutation burden of that tissue. However, in the single example of an autopsy of an individual with Proteus syndrome, more tissue could be collected and assayed and there was still poor correlation of VAF and histopathological effect (3). We hypothesize that disease manifestations in Proteus syndrome are due to a constitutive AKT signaling in a small subset of key cells within a tissue and conversely, that unless there are enough of these critical Akt1E17K-positive driver cells, a lesion will not form. This would explain how an apparently unaffected tissue could have a high VAF, and a highly affected tissue could have a low VAF. Identification of Akt1E17K-positive cells tissues from the chimeras was an important first step in addressing this issue.
The chimera method of modeling Proteus syndrome provided a window into this relationship as the Akt1 mutational status of the cells could be distinguished, using GFP expression. Multiple tissues from several chimeras showed apparently random patterns of GFP expression that encompassed many cell types in unaffected tissue. This suggests that the simple presence or absence of Akt1E17K-positive cells in a particular cell type is not sufficient for overgrowth to occur, though it does not rule out the idea that a certain threshold of local concentration of mutant cells is necessary to begin or maintain a lesion. Whether a single cell type is sufficient for lesion formation could be tested using tissue specific Cre alleles that can force Akt1E17K expression in all cells of a given type. However, this simplistic view is not likely to be the case in all types of Proteus syndrome overgrowth. The evidence of paracrine signaling in the mouse mammary and vascular tissue suggests that intercellular signals may also play a role in lesion formation.
The finding of the Akt1E17K variant in the peripheral blood of Proteus syndrome mice was an unexpected result. In humans with Proteus syndrome, detectable levels of the AKT1 c.49G > A variant in peripheral blood have been reported in only 2 individuals out of more than 40 evaluated ((1) and Lindhurst et al., unpublished observations). The two patients that did show evidence of peripheral blood mosaicism for this variant were deceased at the time of the analysis (the experiment was performed on their banked blood sample) and thus the result could not be confirmed by collecting a second sample. We conclude either that (1) the observed mosaicism was due to sample contamination and that detectable levels of AKT1 c.49G > A in peripheral blood is not observed in humans or (2) that detectable levels of AKT1 c.49G > A in peripheral blood is an uncommon occurrence. We hypothesize that the human AKT1 c.49G > A variant is lethal to hematopoietic cells, or drives hematopoietic stem cell progenitors to lineage commitment, which would exhaust the pool of mutant progenitor cells. That a detectable VAF was seen in 27 of 31 Proteus syndrome mice is remarkably different than the human observation. We propose two alternative models to explain this observation. The first is that the role of the AKT-PI3K pathway in murine hematopoiesis is distinct from that in humans and that constitutive activation of this pathway in the mouse does not exhaust the mutant progenitor pool. The second explanation is that the lineage exhaustion process occurs over time and the mice in our experiments were not old enough to demonstrate this. Further work is warranted to address these hypotheses.
Understanding mosaic disease is challenging because not only does one have to understand the regulation and mutation mechanism of the causative gene, but also one has to account for the heterogenous nature of the presentation in each individual and the variation in the spatial and temporal expression of the mutation among patients with Proteus syndrome. The mouse model manifests several key aspects of the disease and has given us new insights into how Proteus syndrome overgrowth develops and will be a valuable tool for future studies and therapeutic treatments designed to leverage a system that relies on both the endogenous regulation of Akt1 and precise mutation mechanism found in the disease.
Materials and Methods
Targeting construct
The targeting vector pBLAkt1MutFL was constructed using standard molecular biology techniques in a multistep process (Supplementary Material, Fig. S1A). First, a 1.5 Kb fragment containing Akt1 exon 3 was amplified from the 129Sv bMQ BAC library clone 278p07 using a primer that introduced a LoxP site at the 5′ end of the fragment, ligated into a pBluescript vector, and mutagenized to create the c.49G > A p.(E17K) variant. The insert was then excised from pBluescript and ligated into the plasmid pSP72-T3-IRES-βGeo downstream of the poly A signal sequence adding a loxP site and the Akt1 mutagenized sequence 3′ to the βGeo gene (fusion of the LacZ and neomycin resistance genes). Prior to ligation of the Akt1 sequence, pSP72-T3-IRES-βGeo was modified by introducing a splice acceptor site upstream of the IRES. Next, primers were used to amplify a 1.6 Kb fragment from 278p07 immediately adjacent to the amplified exon 3-containing fragment used for mutagenesis. For this amplicon, a LoxP site was introduced at the 3′ end so that ligation of this fragment 5′ to the splice acceptor site in the pSP72 plasmid would result in a LoxP flanked βGeo cassette. To extend the region of homology for targeting, 3.3 Kb and 3.2 Kb fragments were amplified and ligated to the 5′ and 3′ ends of the pSP72 plasmid insert, respectively. The entire insert was excised from pSP72 and ligated into pBL253 to create the targeting construct, pBLMutAkt1FL. All coding and junctional regions were sequenced verified in the final construct. Primers used for amplifying Akt1 are listed in Supplementary Material, Table S5.
pBLMutAkt1FL was linearized with PmeI and transfected into HG3 ES cells (passage 7 C57BL/6 J-Tg (UBC-GFP)30Scha/J X 129SvEvTac) B6/129 hybrid ES cells that expresses GFP from the ubiquitin C promoter. Electroporation of HG3 cells were performed by standard methods (32). Long PCR (AccuPrime™ Taq DNA Polymerase kit, Thermo Fisher Scientific Inc., Waltham, MA) was used to screen ES clones for homologous recombination using primers listed in Supplementary Material, Table S5. One primer of each pair bound to sequence outside of the targeted region and the other to sequence in the βGeo cassette. Southern blot analyses confirmed homologous recombination in seven ES clones (Supplementary Figure, S1B) and one, Akt1–18, was chosen for microinjection into C57BL/6 J 8-cell embryos. Five chimeras had germline transmission of the conditional Akt1 allele (Akt1flx) and were used for generation of mosaic animals or to establish the B6.129Akt1tmfl/Lind conditional line. A second ES cell line was created by transfection of a plasmid containing Cre into Akt1–18 ES cells. Individual colonies were isolated, grown in the presence of G418 and tested for recombination at the loxP sites. One colony, Akt1Δneo3, was correct and was expanded for further studies.
Mouse procedures
All procedures were approved by the NHGRI Animal Care and Use Committee under the protocol G-14-1. Mosaic animals were created by timed matings of chimeras carrying the Akt1flx allele or F1 offspring from the B6.129Akt1tmfl/Lind conditional line to 129-Gt (ROSA)26Sortm1(cre/ERT)Nat/J mice (Jackson Laboratory stock #004847). A stock solution of 4-hydroxytamoxifen (product #H7904, Sigma-Aldrich Inc., St. Louis, MO) dissolved in ethanol at 20–40 mg/ml and then diluted with an equal volume of Kolliphor® EL (product #C5135, Sigma-Aldrich Inc.) was made and then further diluted with saline to make a final concentration of 1.7, 4 or 8 μg tamoxifen/g body weight in 100–300 μL. Pregnant females were dosed at E5.5 by intraperitoneal injection. Chimeric animals were created by injection of Akt1Δneo3 ES cells into C57BL/6 J blastocysts using 1–3 cells for injection.
Mosaic animals and control littermates were allowed to age and monitored for signs of overgrowth or abnormalities as well as discomfort or distress. Animals were euthanized by CO2 inhalation when distress could not be medically managed, growths approached 2 cm, or they reached 20–27 months of age, whichever occurred first. Necropsies were performed to identify gross changes and collect tissues for microscopic evaluation and mutation detection. For most organs, one-fifth to one-half of the organ was frozen for mutation detection with the rest fixed for histology. For smaller paired organs, e.g. lymph nodes, ears, ovaries or testes, one was frozen and the other fixed. A small piece of dorsal and ventral skin, back muscle, peritoneal wall and omental fat was collected for mutation detection with adjacent pieces fixed for histology. Muscle was evaluated in the limbs that were fixed intact. Formalin-fixed tissues were embedded, sectioned and stained with hematoxylin and eosin (Histoserv Inc. Germantown, MD). Immunohistochemistry was performed using standard protocols. Briefly, after rehydration steps, slides were steamed for 20 min in 1X Hier solution and then washed with Tris-buffered saline plus 0.05% Tween 20 (TBST). Endogenous enzymes and proteins were blocked for 12 min using Bloxall (Vector Laboratories, Inc., Burlingame, CA) followed by washing in TBST and incubating in protein block (product #X0909, Agilent Technologies, Inc., Santa Clara, CA or product #15019, Cell Signaling Technology, Inc., Danvers, MA). Primary antibodies were diluted as follows: GFP 1:1000 (product #2555), pAKT(S473) 1:25 (product #3787), CD31 1:200 (product #77699) from Cell Signaling Technology, Inc. and LYVE-1 1:1000 (product #ab14917, Abcam plc., Cambridge, MA) and hybridized overnight at 4°C. After washing, slides were incubated for 30 min with SignalStain® Boost (product #8114) washed and developed using the SignalStain® DAB Substrate kit (product #8509) from Cell Signaling Technology. Slides were counterstained with hematoxylin, dehydrated and mounted with CytoSeal 60 mounting medium (Thermo Fisher Scientific Inc., Waltham, MA).
DNA and RNA isolation, genotyping and sequencing
DNA was isolated from frozen tissue by phenol/chloroform/isoamyl alcohol extraction followed by ethanol precipitation. RNA and DNA were isolated from ES cells using the AllPrep DNA/RNA/miRNA Universal kit (Qiagen Sciences, Inc., Germantown, MD). Genotyping was performed using two separate PCR reactions. Primers mAkt1N2-MF1 and mAkt1N2-MR16 (Supplementary Material, Table S5) amplify a 193 bp fragment from the wild-type allele and a 225 bp fragment from the mutant allele because of the 32 bp LoxP site that remains after Cre recombination. The Akt1flx allele was identified using primers SV40-MF1 and mAkt1N2-MR3 and Cre was identified using primers Cre-MF1 and Cre-MR1. At least two tissues from control animals that carried a Cre gene were tested to ensure the absence of the Akt1flx allele. Conversely, for control animals that carried the Akt1flx allele, at least two tissues were tested to ensure the absence of Cre. For cDNA, amplification and sequencing was performed using the OneStep PCR kit (Qiagen Science Inc.) and BigDyeTerminator cycle sequencing kit (Thermo Fisher Scientific Inc., Waltham, MA) and separated using an ABI 3130 Genetic Analyzer (Thermo Fisher Scientific Inc.).
VAF detection
The VAF levels for the tamoxifen mosaic animal tissue DNAs were determined using the genotyping assay for the mutant and wild-type alleles described above with the following modifications. A FAM-labeled mAKT1N2-MR16 primer was used for amplification and the resulting fragments were separated on the ABI 3130 Genetic Analyzer and processed using GeneMapper software (Thermo Fisher Scientific, Waltham, MA). The VAF levels were calculated by dividing the area under the mutant peak by the combined areas of the mutant and wild-type peaks. The VAF levels in for the chimera tissue DNAs were determined by droplet digital PCR (ddPCR) using a custom assay for the Akt1 c.49G > A p.(E17K) variant and the QX200 ddPCR system (Bio-Rad Laboratories, Inc., Hercules, CA). Littermates of chimeric animals were uses as controls if the VAF determined by the ddPCR custom assay was zero in all amplifiable tissue. The number of tissues tested per control animal ranged from 22 to 33. VAF data was visualized in R (33) using ggplot2 (34) and ggbeeswarm packages https://CRAN.R-project.org/package=ggbeeswarm.
Statistics
Statistical significance of lesion frequency was calculated by the Fisher’s exact test using Prism 8 software (GraphPad Software, Inc., San Diego, CA).
Supplementary Material
Acknowledgements
We thank Douglas Strathdee for the pSP72-T3-IRES- βGeo plasmid, Jorge J. Chavez and Rachel M. Fleischmann for technical assistance, Stephen Wincovitch for microscopy support and Julia Fekecs for graphics support. The authors thank Dr David Bodine for his insights into the hematopoietic findings in these animals.
Conflict of Interest statement. LGB is a member of the Illumina Ethics Advisory Board, receives royalties from Genentech, receives honoraria from Cold Spring Harbor Press, and in-kind research support from ArQule, Inc.
Funding
Intramural Research Program of the National Human Genome Research Institute (grants HG200328 13 and HG200388 05).
References
- 1. Lindhurst M.J., Sapp J.C., Teer J.K., Johnston J.J., Finn E.M., Peters K., Turner J., Cannons J.L., Bick D., Blakemore L. et al. (2011) A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N. Engl. J. Med., 365, 611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sapp J.C., Hu L., Zhao J., Gruber A., Schwartz B., Ferrari D. and Biesecker L.G. (2017) Quantifying survival in patients with Proteus syndrome. Genet. Med., 19, 1376–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Doucet M.E., Bloomhardt H.M., Moroz K., Lindhurst M.J. and Biesecker L.G. (2016) Lack of mutation-histopathology correlation in a patient with Proteus syndrome. Am. J. Med. Genet. A, 170, 1422–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Biesecker L. (2006) The challenges of Proteus syndrome: diagnosis and management. Eur. J. Hum. Genet., 14, 1151–1157. [DOI] [PubMed] [Google Scholar]
- 5. Biesecker L.G., Happle R., Mulliken J.B., Weksberg R., Graham J.M. Jr., Viljoen D.L. and Cohen M.M. Jr. (1999) Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am. J. Med. Genet., 84, 389–395. [DOI] [PubMed] [Google Scholar]
- 6. Turner J.T., Cohen M.M. Jr. and Biesecker L.G. (2004) Reassessment of the Proteus syndrome literature: application of diagnostic criteria to published cases. Am. J. Med. Genet. A, 130A, 111–122. [DOI] [PubMed] [Google Scholar]
- 7. Nathan N.R., Patel R., Crenshaw M.M., Lindhurst M.J., Olsen C., Biesecker L.G., Keppler-Noreuil K.M. and Darling T.N. (2018) Pathogenetic insights from quantification of the cerebriform connective tissue nevus in Proteus syndrome. J. Am. Acad. Dermatol., 78, 725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Segrelles C., Garcia-Escudero R., Garin M.I., Aranda J.F., Hernandez P., Ariza J.M., Santos M., Paramio J.M. and Lorz C. (2014) Akt signaling leads to stem cell activation and promotes tumor development in epidermis. Stem Cells, 32, 1917–1928. [DOI] [PubMed] [Google Scholar]
- 9. Segrelles C., Lu J., Hammann B., Santos M., Moral M., Cascallana J.L., Lara M.F., Rho O., Carbajal S., Traag J. et al. (2007) Deregulated activity of Akt in epithelial basal cells induces spontaneous tumors and heightened sensitivity to skin carcinogenesis. Cancer Res., 67, 10879–10888. [DOI] [PubMed] [Google Scholar]
- 10. Segrelles C., Moral M., Lorz C., Santos M., Lu J., Cascallana J.L., Lara M.F., Carbajal S., Martinez-Cruz A.B., Garcia-Escudero R. et al. (2008) Constitutively active Akt induces ectodermal defects and impaired bone morphogenetic protein signaling. Mol. Biol. Cell, 19, 137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rokutanda S., Fujita T., Kanatani N., Yoshida C.A., Komori H., Liu W., Mizuno A. and Komori T. (2009) Akt regulates skeletal development through GSK3, mTOR, and FoxOs. Dev. Biol., 328, 78–93. [DOI] [PubMed] [Google Scholar]
- 12. Sun J.F., Phung T., Shiojima I., Felske T., Upalakalin N., Feng D., Kornaga T., Dor T., Dvorak A.M., Walsh K. and Benjamin L.E. (2005) Microvascular patterning is controlled by fine-tuning the Akt signal. Proc. Natl. Acad. Sci. U. S. A., 102, 128–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Phung T.L., Ziv K., Dabydeen D., Eyiah-Mensah G., Riveros M., Perruzzi C., Sun J., Monahan-Earley R.A., Shiojima I., Nagy J.A. et al. (2006) Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer Cell, 10, 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blanco-Aparicio C., Perez-Gallego L., Pequeno B., Leal J.F., Renner O. and Carnero A. (2007) Mice expressing myrAKT1 in the mammary gland develop carcinogen-induced ER-positive mammary tumors that mimic human breast cancer. Carcinogenesis, 28, 584–594. [DOI] [PubMed] [Google Scholar]
- 15. Mancini M.L., Lien E.C. and Toker A. (2016) Oncogenic AKT1(E17K) mutation induces mammary hyperplasia but prevents HER2-driven tumorigenesis. Oncotarget, 7, 17301–17313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Madsen R.R., Vanhaesebroeck B. and Semple R.K. (2018) Cancer-associated PIK3CA mutations in overgrowth disorders. Trends Mol. Med., 24, 856–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Happle R. (1986) Cutaneous manifestation of lethal genes. Hum. Genet., 72, 280. [DOI] [PubMed] [Google Scholar]
- 18. Nikolic A., Volarevic V., Armstrong L., Lako M. and Stojkovic M. (2016) Primordial germ cells: current knowledge and perspectives. Stem Cells Int., 2016, 1741072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McGrath K.E., Frame J.M., Fegan K.H., Bowen J.R., Conway S.J., Catherman S.C., Kingsley P.D., Koniski A.D. and Palis J. (2015) Distinct sources of hematopoietic progenitors emerge before HSCs and provide functional blood cells in the mammalian embryo. Cell Rep., 11, 1892–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li W., Johnson S.A., Shelley W.C., Ferkowicz M., Morrison P., Li Y. and Yoder M.C. (2003) Primary endothelial cells isolated from the yolk sac and para-aortic splanchnopleura support the expansion of adult marrow stem cells in vitro. Blood, 102, 4345–4353. [DOI] [PubMed] [Google Scholar]
- 21. Nguyen D., Turner J.T., Olsen C., Biesecker L.G. and Darling T.N. (2004) Cutaneous manifestations of proteus syndrome: correlations with general clinical severity. Arch. Dermatol., 140, 947–953. [DOI] [PubMed] [Google Scholar]
- 22. Takyar V., Khattar D., Ling A., Patel R., Sapp J.C., Kim S.A., Auh S., Biesecker L.G., Keppler-Noreuil K.M. and Heller T. (2018) Characterization of the hepatosplenic and portal venous findings in patients with Proteus syndrome. Am. J. Med. Genet. A, 176, 2677–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Keppler-Noreuil K.M., Baker E.H., Sapp J.C., Lindhurst M.J. and Biesecker L.G. (2016) Somatic AKT1 mutations cause meningiomas colocalizing with a characteristic pattern of cranial hyperostosis. Am. J. Med. Genet. A, 170, 2605–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gordon P.L., Wilroy R.S., Lasater O.E. and Cohen M.M. Jr. (1995) Neoplasms in Proteus syndrome. Am. J. Med. Genet., 57, 74–78. [DOI] [PubMed] [Google Scholar]
- 25. Iqbal J., He G., Biesecker L.G., Rosen P., Duray P.H., Schwartzentruber D., Beg M. and Kahn E. (2006) Morphological characterization of the breast in Proteus syndrome complicated by ductal carcinoma in situ. Ann. Clin. Lab. Sci., 36, 469–474. [PubMed] [Google Scholar]
- 26. Malamitsi-Puchner A., Dimitriadis D., Bartsocas C. and Wiedemann H.R. (1990) Proteus syndrome: course of a severe case. Am. J. Med. Genet., 35, 283–285. [DOI] [PubMed] [Google Scholar]
- 27. Brayton C.F., Treuting P.M. and Ward J.M. (2012) Pathobiology of aging mice and GEM: background strains and experimental design. Vet. Pathol., 49, 85–105. [DOI] [PubMed] [Google Scholar]
- 28. Pettan-Brewer C. and Treuting P.M. (2011) Practical pathology of aging mice. Pathobiol. Aging Age Relat. Dis., 1, 7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Szymanska H., Lechowska-Piskorowska J., Krysiak E., Strzalkowska A., Unrug-Bielawska K., Grygalewicz B., Skurzak H.M., Pienkowska-Grela B. and Gajewska M. (2014) Neoplastic and nonneoplastic lesions in aging mice of unique and common inbred strains contribution to modeling of human neoplastic diseases. Vet. Pathol., 51, 663–679. [DOI] [PubMed] [Google Scholar]
- 30. Brandli-Baiocco A., Balme E., Bruder M., Chandra S., Hellmann J., Hoenerhoff M.J., Kambara T., Landes C., Lenz B., Mense M. et al. (2018) Nonproliferative and proliferative lesions of the rat and mouse endocrine system. J. Toxicol. Pathol., 31, 1S–95S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Carpten J.D., Faber A.L., Horn C., Donoho G.P., Briggs S.L., Robbins C.M., Hostetter G., Boguslawski S., Moses T.Y., Savage S. et al. (2007) A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature, 448, 439–444. [DOI] [PubMed] [Google Scholar]
- 32. Hogan B., Beddington R., Costantini F. and Lacy E. (1994) Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 33. R Core Team (2014) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 34. Wickham H. (2016) Elegant Graphics for Data Analysis, Springer-Verlag, New York, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.