

Abstract
Although the ramifications associated with polybrominated diphenyl ethers (PBDEs) exposures during human pregnancy have yet to be determined, increasing evidence in humans and animal models suggests that these compounds cause neurodevelopmental toxicity. Human embryonic stem cells (hESCs) models can be used to study the effects of environmental chemicals throughout the successive stages of neuronal development. Here, using a hESC differentiation model, we investigated the effects of common PBDE congeners (BDE-47 or -99) on the successive stages of early neuronal development. First, we determined the points of vulnerability to PBDEs across 4 stages of in vitro neural development by using assays to assess for cytotoxicity. Differentiated neural progenitors were identified to be more sensitive to PBDEs than their less differentiated counterparts. In follow-up investigations, we observed BDE-47 to inhibit functional processes critical for neurogenesis (eg, proliferation, expansion) in hESC-derived neural precursor cells (NPCs) at sub-lethal concentrations. Finally, to determine the mechanism(s) underlying PBDE-toxicity, we conducted global transcriptomic and methylomic analyses of BDE-47. We identified 589 genes to be differentially expressed due to BDE-47 exposure, including molecules involved in oxidative stress mediation, cell cycle, hormone signaling, steroid metabolism, and neurodevelopmental pathways. In parallel analyses, we identified a broad significant increase in CpG methylation. In summary our results suggest, on a cellular level, PBDEs induce human neurodevelopmental toxicity in a concentration-dependent manner and sensitivity to these compounds is dependent on the developmental stage of exposure. Proposed mRNA and methylomic perturbations may underlie toxicity in early embryonic neuronal populations.
Keywords: polybrominated diphenyl ethers, human, embryonic stem cells, neurogenesis, neurotoxicity, transcriptome, methylome, alternative model, in vitro, endocrine disruptors
Polybrominated diphenyl ethers (PBDEs) are used globally as flame retardants in a variety of consumer products (McDonald, 2002) and are of major concern for wildlife and human populations due to significant body burdens (Hites, 2004; Mazdai et al., 2003). PBDE exposures begin early on in embryonic development, with fetal exposures exceeding maternal levels after crossing the placenta barrier (Schecter et al., 2007; Zota et al., 2018). In total 209 PBDE congeners exist in the environment and are detected globally in biological matrices (Frederiksen et al., 2009). Multiple congeners are detected in maternal and fetal sera (Mazdai et al., 2003), with BDE-47 documented to be at the high levels in the majority of U.S. mothers and/or their offspring (Frederiksen et al., 2009; Main et al., 2007; Woodruff et al., 2011). Though PBDEs are being phased out from use human exposure is persistent, with recent studies showing levels of BDE-47 at 0.17 and 0.22 ng/g in maternal and cord serum, respectively (Zota et al., 2018).
Epidemiological and experimental evidence (Costa and Giordano, 2007; Dingemans et al., 2011) strongly suggest that early life exposures to PBDEs alters neurodevelopmental outcomes, including measures of attention (Cowell et al., 2015; Eskenazi et al., 2013) and cognition (Chen et al., 2014; Lam et al., 2017). Although unresolved, a number of studies have focused on the link between PBDE-induced neurotoxicity and alterations in thyroid hormone (TH) homeostasis (eg, as reviewed in Costa et al., 2014; Dingemans et al., 2011; Herbstman and Mall, 2014). TH signaling is a critical factor in neurological development, including neuron and oligodendrocyte proliferation, differentiation, and maturation (Pascual and Aranda, 2012; Patel et al., 2011; Stenzel and Huttner, 2013). PBDEs accumulate within neuronal cell populations (Mundy et al., 2004; Viberg et al., 2003) and potentially mimic THs, thyroxine, and triodothyrionine, to interfere with TH production/regulation, receptor binding, or transport (Herbstman et al., 2008; Kojima et al., 2009; Li et al., 2010). PBDEs may also alter neuronal development (Dingemans et al., 2007; Schreiber et al., 2010) through other mechanisms (Costa and Giordano, 2007) including oxidative stress, disruption of cell signaling, inflammation, and epigenetic modifications—any of which may contribute to neurodevelopmental alterations.
Human embryonic stem cells (hESCs) derived from the inner cell mass of the human blastocyst possess an unlimited potential for self-renewal and the capacity to differentiate into multiple somatic cell types, including neurons, glial cells, and astrocytes (Evans and Kaufman, 1981). Thus, hESC models are applied in research to study early in vivo human neurodevelopmental events in vitro, including basic cellular functions (eg, self-renewal, proliferation, differentiation) and specific features of neurogenesis and neurulation. With these inherent characteristics, hESCs serve as a valuable model to evaluate environmental effects on aspects of early human neurodevelopment that are otherwise difficult to study in vivo. Protocols for murine (Theunissen et al., 2013) and human (Colleoni et al., 2011; Kleinstreuer et al., 2011; Krtolica et al., 2009; Stummann et al., 2009) ESC model systems have been established for developmental toxicological investigations, which employ a combination of morphological, functional, and molecular parameters as endpoints (Pal et al., 2011).
Although toxicological evidence suggests a significant relationship between early life PBDE exposures and alterations in neurodevelopment, studies utilizing human-derived model systems are lacking (Costa and Giordano, 2007) which can complicate species comparisons (Clancy et al., 2001; Watson et al., 2006). Furthermore, sensitivity to chemical exposures vary between species and during development (Bal-Price et al., 2012) and limited information exists regarding the windows of vulnerability to PBDE exposures during early life. Toxicological comparisons spanning the differentiation of ESCs to neural derivatives have been proposed to account for chemical sensitivity during early development on a cell- and/or timing basis (Fritsche et al., 2018; Lim et al., 2009). Therefore in this study, using a multi-stage hESC neural differentiation model, we characterized the concentration-dependent effects of 2 commonly used PBDE congeners, BDE-47 and -99, across stages of neuronal development using functional and transcriptomic approaches. In this context, we propose functional and molecular targets which may underlie PBDE’s developmental neurotoxicity.
MATERIALS AND METHODS
Chemicals
PBDE congeners BDE-47 (2, 2'4, 4'-tetrabromodiphenyl ether, >99%, No. 5436-43-1, AccuStandard) and BDE-99 (2, 2'4, 4', 5 pentabromodiphenyl ether, No. 60348-60-9, AccuStandard), bisphenol A (BPA; >99%, No. 80-05-7, Sigma), perfluorooctanoic acid (PFOA, >96%; No. 335-67-1, Sigma), all-trans-retinoic acid (RA; 97%, No. 302-79-4, Acros Organics), and valproic acid (VPA) sodium salt (No. 1069-66-5, ≥98%, Santa Cruz Biotechnology) were dissolved in dimethyl sulfoxide (DMSO; Sigma) or deionized water (VPA only) to make stock solutions and serial dilutions for all experimental studies. Compounds were added at a 1:1000 (vol/vol media) dilution. Exposed cultures were evaluated in parallel with vehicle (0.1% DMSO) and media-only controls.
hESC model of neural differentiation
We employed a multi-stage hESC differentiation model to evaluate PBDE-induced neurodevelopmental toxicity, which included 5 predefined testing windows in vitro (Stages I–V) representative of human neurogenesis (Figure 1, Supplementary Figure 1). hESCs (UCSF4, NIH registry code 0044) were maintained in mTESR-defined medium (Stemcell Technologies) on Matrigel-coated plates (BD Biosciences). For Stage I experiments, hESCs were passaged every 3–5 days (passage number <60) via manual dissection and Dispase (Stemcell Technologies). PBDE exposures were introduced 2 days after plating approximately 32 895 cells/cm2. For Stage II experiments, embryoid body (EB) formation was conducted by disassociating hESCs (Accutase, Life Technologies) and placing them in an Aggrewell 400um well plates (Stemcell Technologies) containing KSR medium (Life Technologies) supplemented with 10% fetal bovine serum (FBS) and ROCK inhibitor (1 µl/ml) at a density of 500 cells/microwell. After 24 h, EBs were gently displaced into KSR medium with 10% FBS and cultured in suspension. PBDE exposures were initiated after transfer to low-adhesion plates (Costar) for 24 h. For Stage III experiments, suspended EBs were grown in KSR medium for 6 days (exchanged after 3 days), then placed in neural induction media containing DMEM-F12 with Glutamax, penicillin/streptomycin, N2 supplement (Stemcell Technologies), and FGF2 (Invitrogen, 20 ng/ml) to promote ectoderm specification for an additional 6 days (exchanged after 3 days). PBDE exposures were initiated at this step. After a total of 12 days in suspension, EBs were cultured on Cellstart (Gibco) precoated wells in neural enhancement media (NEM) consisting of Neurobasal Medium (Gibco), NEAA (2 mM), L-Glutamine (2 mM), penicillin/streptomycin, B27, FGF2 (20 ng/ml), and LIF (10 ng/ml; Millipore). Neural rosette patterning was apparent after 2–3 days in culture. For Stage IV experiments, neural precursor cells were isolated and expanded from extracted neural rosettes. NPCs were cultured on Cellstart substrate and maintained in NEM. NPCs were expanded every 3–5 days using Accutase dissociation. For PBDE exposures, NPCs were seeded at approximately 32 895 cells/cm2 for 2 days (approximately 60%–70% confluency, passages <15) prior to treatment. For Stage V experiments, neurospheres were generated by disassociating NPCs (>70% confluent) via Accutase and placing single cells (35 000 cells/ml) in low attachment plates (CoStar) suspended in complete NEM with ROCK inhibitor (1 µl/ml) for 3 days. Resulting neurospheres were collected and centrifuged at 200 rpm for 5 min, then resuspended in NEM media (without ROCK inhibitor) and for an additional 3 days. Chemical exposures were initiated during this step. After 6 days of culture, neurospheres were gently pelleted via centrifugation (200 rpm; 5 min), placed in fresh NEM with chemical and plated on Cellstart precoated wells to evaluate neural expansion. At all stages, cells were grown at 37°C in 5% CO2 and 8% O2. Stemness of hESCs and NPCs were monitored via morphology and expression of pluripotency markers (eg, NANOG and POU5F1) or early neural differentiation markers (eg, PAX6 and NES) via immunocytochemistry and/or qRT-PCR. In a previous transcriptomic study (GSE74378; Robinson et al., 2016), we demonstrated that multiple genes involved in neural differentiation are significantly induced in NPCs as compared with hESCs, supporting our ability to generate neural precursors using the described protocol.
Figure 1.

Implementing a hESC neural differentiation model to evaluate chemical toxicity sensitivity during neurogenesis. We developed a hESC model of neurogenesis representing transitions occurring during human neurogenesis to evaluate BDE-47 toxicity in vitro. The 5 stages include: (I) hESC self-renewal; (II) EB formation and initiation of the 3 germ layers; (III) EB ectoderm specification and neural rosette patterning; (IV) NPC proliferation; and (V) neurosphere expansion.
Cytotoxicity assays
We evaluated the ability of chemicals to induce cytotoxicity within 4 stages of hESC neural development. In adherent cultures of hESCs (Stage I) or NPCs (Stage IV), we exposed cells to BDE-47 (0.1–25 µM), BDE-99 (0.1–25 µM), BPA (0.01–100 µM), or PFOA (0.1–1000 µM) for 24 or 48 h durations, and employed the neutral red (NR) uptake assay (Borenfreund and Puerner, 1985; Repetto et al., 2008) to determine relative cell viability based on lysosomal accumulation of NR dye (40 ng/ml, VWR). Briefly, PBDE-containing media was removed and wells were gently washed with PBS to eliminate residual chemical compound. Fresh media containing NR was added to each plate and incubated for 2 h at 37°C. Wells were washed with PBS and a 50% ethanol/1% acetic acid solution was added to release cellular NR. In adherent cultures of hESCs (Stage I), neural rosettes (Stage III), or NPCs (Stage IV) and suspended EB cultures (Stages II), we evaluated cell death after 24 or 48 h exposures to BDE-47 or -99 (0.1–25 µM) by measuring lactate dehydrogenase (LDH) activity using the Cytotoxicity Detection Kit (Roche) following the manufacturer’s protocol. Absorbance readings were obtained via a spectrophotometer (Biotek Epoch) for both assays (590 nm, LDH; 540 nM, NR). We determined relative viability/death by comparing absorbance ratios between exposure groups and the media-only control (=100%), which were computed for each experiment, and then averaged across experiments. ANOVA and Dunnett’s t tests were applied to determine significant effects due to PBDE exposure (p < .05).
Cell proliferation assay
The effects of PBDEs on cell proliferation were evaluated using the Cell Proliferation Elisa, BrdU (chemiluminescent) kit (Roche) following the manufacturer’s protocol. In brief, plates were precoated with Cellstart and NPCs were cultured at a density of approximately 32 895 cells/cm2. After 1 day, cells were exposed to BDE-47 or -99 (0.1–25 µM) in parallel with vehicle and media-only controls as well as blank wells (no cells). Eight hours into the exposure protocol (approximately 36 h total), BrdU (10 µM) labeling solution was added to each well. After a 24 h exposure duration, cells were fixed and incubated with antiBrdU-peroxidase solution for 90 min, washed 3×, and substrate solution containing luminol and 4-iodophenol was applied for 10 min at 20°C. Chemiluminescence readings were obtained using a luminometer (Biotek Epoch 2) and average background readings (blank wells) were subtracted. Ratios between control (media-only) and exposure groups, including the vehicle control, were calculated for each experiment and averaged across experiments (n = 6). ANOVA and student t tests were applied to determine significant effects due to PBDE exposure (p < .05).
Neurosphere expansion assay
Suspended neurospheres were exposed for 3 days to BDE-47 (1 or 5 µM) or control conditions (0.1% DMSO or media only), plated on CellStart, and then, reexposed to the same experimental conditions, evaluated for neurosphere expansion at 1, 24 h, and/or 72 h. Experiments were conducted in parallel with subcytotoxic concentrations of neurodevelopmental toxicants, RA (20 µM) and VPA (250 µM), as experimental controls. We quantified the total cell expansion area in exposed and control neurosphere cultures after 1, 24, and 72 h postplating. At 1 h, phase images of neurospheres were acquired using an inverted microscope (10×; Leica). Coordinates (x, y) were stored and successive images were acquired at 24 and 72 h at the same location for each respective neurosphere. Phase images were transformed into binary pictures and total neurosphere expansion area was quantified (Image J). Ratios were determined between 24 or 72 h in comparison with 1 h measurements. Within each experimental group (x—= 12.1 neurospheres per group; minimum > 5 neurospheres) the average percent change in expansion area was determined between the 2 time points, then averaged across experiments (n = 4; 1 and 24 h; n = 3; 72 h). ANOVA and student t tests were applied to determine significant effects due to PBDE exposure (p < .05) at each timepoint.
RNA isolation for gene expression analyses
Total RNA was isolated from cells (NPCs or neurosphere cultures) using the RNAeasy Micro Plus RNA Isolation Kit (Qiagen) following the manufacturer’s protocol for adherent cells. Extracts were collected in RTL Cell Lysis Buffer and stored at −80°C until further use. Genomic DNA was removed from cell lysates and following column purification, RNA was eluted with 20 µl of nuclease-free water and stored at −80°C. RNA concentration and quality was estimated (260/280 nm = 1.9–2.1) by using a Nanodrop spectrometer (Nanodrop Technologies Inc., Wilmington, Delaware). Samples destined for microarray analyses were assessed for quality (RIN > 9) using the Agilent RNA 6000 Nano LabChip Kit and Bioanalyzer 2100 system (Agilent Technologies, Palo Alto, California). Nine samples were used for microarray analyses (Supplementary Table 1).
Global RNA expression profiling of BDE-47-exposed NPCs
We applied the Affymetrix Human Gene 2.0 ST platform to conduct microarray analysis of BDE-47 exposed (1 or 10 µM) or vehicle control (0.1% DMSO) NPCs following a 24 h exposure duration. Although 10 µM BDE-47 did not produce significant effects in cytotoxicity at 24 h in our assessments, a modest decline in cell viability (approximately 18%) and increase in LDH activity were noted. Sample processing and hybridization was performed by the UCSF Gladstone Institute as previously described (Winn et al. 2007). Affymetrix CEL files were processed using the Affymetrix Expression Console and Transcriptome Analysis Console (TAC) software packages. Raw values were normalized via the Robust Multi-array Average (RMA) algorithm. Datasets were deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus GSE123458. We removed probes corresponding with log 2 probe intensity values < 3.0 across all arrays or without a corresponding Official Gene Symbol (OGS). Data were imported into BRBarray tools (NCI) and a fixed-effects linear model (ANOVA) was applied to identify differentially expressed (DE) genes due to BDE-47 exposure (+ controlling for batch effects). Average fold change (FC) values were determined by calculating the ratio of average log 2 intensities between each of the BDE-47 exposure groups and the average vehicle control. We defined DE genes due to BDE-47 exposure by applying a cutoff of unadjusted p ≤ .01 (ANOVA) and an absolute FC ≥ 1.5 between BDE-47 and vehicle control in at least 1 of the 2 concentration groups. In cases of multiple probes per gene, the one with the lowest p-value, ie, the most significantly altered due to BDE-47, was used for downstream analyses. Using this approach, we examined 22, 568 unique genes. Within DE BDE-47 genes, we conducted a post hoc paired t test of DE BDE-47 genes to determine significant changes with 1 or 10 µM BDE-47 versus vehicle control (p < .05). Hierarchical clustering of FC values was completed by using average linkage and Euclidean distance (TIGR MEV; Saeed et al., 2006). Functional enrichment analysis of Gene Ontology (GO) terms (Biological Processes, Level 4) was evaluated via DAVID (Huang et al., 2007) and significant criteria of a Bonferroni corrected p < .05, and a minimum/maximum of 12 or 200 targets per GO term (Supplementary Table 2). Terms were grouped based on GO classification (Gene Ontology Consortium, 2015).
Targeted validation of PBDE DE genes
Using independent NPC samples, we converted purified RNA to cDNA using ISCRIPT Universal TaqMan (Bio-Rad), and performed qRT-PCR via TaqMan primers (Supplementary Table 4) mixed with TaqMan Universal Master Mix II, no UNG (Life Technologies). We conducted qRT-PCR to investigate the concentration-dependent effects of BDE-47 or -99 in NPCs (24 h) on RNA expression of specific targets: CDK1, GRIA1, HMOX1, DHCR7, NFE2L2, NRCAM, PAX6, THRB, and THRA. Differential expression between PBDE-exposed and controls was calculated via the ΔΔCT method, which entails: (1) normalization to geometric mean of housekeeping genes (GAPDH and ACTB); and (2) adjustment to the vehicle control (0.1% DMSO) for each experiment. Reactions were carried out for 40 cycles. A minimum of 3 technical replicates were analyzed for all comparisons. To determine significant changes across concentrations or between specific concentrations and control, we conducted ANOVA or student t tests as appropriate. FC values were expressed as average log 2 ratios between each exposure group and the vehicle control.
DNA isolation of BDE-47 exposed NPCs
Following exposure to BDE-47 (1, 10 µM) or vehicle for 24 h, NPCs were washed with PBS and collected using a cell scraper. Cells were suspended in 10 ml of PBS, pelleted (800 rpm, 5 min), washed 2X with PBS and stored at −80°C. To extract genomic DNA, NPCs were digested with 1 mg/ml proteinase K in lysis buffer (50 mM Tris, pH 8.0, 1 mM EDTA pH 8.0, 0.5% SDS) overnight at 55°C. After RNase treatment, DNA was isolated using the Phenol-Chloroform Isoamyl Alcohol method, followed by precipitation with ethanol, and resuspended in TE. DNA quality was evaluated via Nanodrop and the Agilent RNA 6000 Nano LabChip Kit and Bioanalyzer 2100 system. DNA was extracted from exposed and control cultures from 3 independent experiments (approximately 1 million cells/sample).
Methylation profiling of BDE-47 exposed NPCs
Downstream processing of genomic DNA bisulfite conversion was performed using the EZ DNA Methylation Kit (ZymoResearch) and Infinium HumanMethylation450 bead arrays (Illumina) following the manufacturer’s protocols. Methylation data were processed via Illumina standard background subtraction and control probe normalization, and converted to M values, using the minfi package and BRB Array Tools (Simon et al., 2007). Probes that mapped to the Y chromosome were eliminated from the analysis (UCSF4 line is XX). We utilized a fixed effect multivariate model (using M-value = ratio of methylated probe vs. unmethylated probe intensities) to identify differentially methylated (DM) CpGs due to exposure, while controlling for experimental differences (batch effects). Beta values, which reflect the proportion of methlyated probes (ranging from 0 to 1) were determined via logit transformation [log2(M/(M + 1)] of M-values (Du et al., 2010). The change in methylation was determined by subtracting β values between BDE-47 exposure groups and the respective vehicle control within each experiment. Significant DM CpGs were identified using a p ≤ .005 (unadjusted) and average absolute β ≥ 0.05 between BDE-47 and DMSO for either of the 2 concentrations of BDE-47 tested. We evaluated enrichment of DM CpGs by genomic region or chromosome location (Fisher’s exact test). We interrogated genes in proximity to DM CpGs for functional relevance using DAVID (Biological Level 4). These analyses were also conducted for the subset of DM CpGs located within promoter regions of genes, defined to be located < 1500 b upstream of the TSS, 5’ untranslated region (UTR), or first Exon. Raw and normalized data were deposited in the NCBI GEO repository (GSE125465). We examined correlations between mRNA expression and DM CpGs by aligning the datasets based on associated OGS annotation. For DM CpGs in proximity of promoter gene regions, we analyzed the effect of BDE-47 exposures on expression in comparison with the change in methylation at each independent CpG island.
RESULTS
A Multi-Stage hESC Model to Evaluate Toxicity During Neurogenesis
Using a hESC neural differentiation model, we evaluated the effects of BDE-47 within 5 independent stages of neuronal development in vitro (Figure 1; Supplementary Figure 1). The predefined stages encompass: (I) hESC self-renewal/proliferation; (II) EB formation and initiation of the 3 germ layers; (III) EB ectoderm specification and neural rosette patterning; (IV) NPC expansion; and (V) neurosphere expansion—representative stages of cellular transitions that occur during early embryonic neurodevelopment in vivo.
PBDEs Induce Cytotoxicity in a Developmental Stage-Dependent Manner
We examined the ability of PBDE exposures to induce cytotoxicity during 4 proposed stages (I–IV) of hESC-neural development in vitro. We conducted concentration-response assessments on endpoints corresponding with cell viability (neutral red uptake) and death (LDH activity in the media). In this context, we compared toxicological potencies between BDE-47 and another common PBDE congener, BDE-99. In undifferentiated hESCs (Stage I), BDE-47 (0.1–25 µM) did not cause significant changes in cell viability at 24 or 48 h (ANOVA, p > .05; Figure 2A). BDE-99 induced a significant decrease in viability at 24 h (p = .05); however, changes were generally modest in magnitude (maximum ↓13% with 25 µM). Significant effects with BDE-99 were not observed at 48 h. In general, these results mirrored LDH activity. BDE-47 or -99 did not induce significant changes in LDH activity as compared with the vehicle control (p > .05; Figure 2B). In contrast, in NPCs (Stage IV) significant effects on cell viability or death were observed with BDE-47 or -99 exposures in a concentration-dependent manner (ANOVA, p < .05; Figs. 2C and 2D). Maximal alterations were observed with 25 µM BDE-47 or -99, which reduced viability by 52% or 61% or induced LDH activity by 384% or 352%, respectively at 24 h, as compared with the vehicle control.
Figure 2.

PBDEs exposures and cytotoxicity in a hESC model of neurogenesis. Concentration-dependent effects of BDE-47 or -99 on cell viability or LDH in hESCs (Stage I; A, B) or NPCs (Stage IV; C, D) at 24 or 48 h. Relative percentages were calculated as ratios between each exposure group and the media-only control. Significant effects due to PBDE exposures (p < .05) are indicated by double-cross (‡, ANOVA) or asterisk (t test). BMC-response modeling of compound-induced cytotoxicity across stages of hESC-neural differentiation. Estimated BMC50 (cell viability, E) or BMC200 (LDH activity, F) for each testing compound and exposure duration. Additional endocrine disruptors, BPA and PFOA, were included as part of these assessments. All experiments were conducted in 3 or more independent experiments unless noted ( ^; n = 2). Due to a lack of response, ie, cytotoxicity, to the tested dose range, in specific cases, negative (neg) BMCs were identified.
To determine relative potencies of BDE-47 and -99, we performed logistic regression of concentration-response data and calculated benchmark concentrations (BMCs) equivalent to a 50% (viability; Figure 2E) or 200% (LDH activity; Figure 2F) change. BPA and PFOA, widespread endocrine-disrupting compounds commonly detected in biological matrices, were also evaluated (Meeker, 2012). These analyses confirmed BDE-47 and/or -99 to be more potent in neural derivatives (Stages III and IV) as compared with undifferentiated (Stage I) or early differentiating hESCs (Stage II). For example, BMC200 corresponding with BDE-47-induced LDH activity during Stages III or IV ranged from 14.5 to 21.1 µM, while Stages I or II had BMC200 >232.1 µM (or could not be modeled due to a lack of response, “neg”). Based on BMC values, BDE-99 tended to be slightly more potent than BDE-47. In NPCs (Stage IV) PBDE congeners were identified to be more potent than BPA or PFOA by approximately an order of magnitude. These results suggest that PBDEs are potent toxicants in embryonic cells transitioning to neurons; however, sensitivity is dependent on concentration and developmental stage.
BDE-47 Impairs Neural Progenitor Cell and Neurosphere Expansion
In follow-up experiments, we focused our analyses of BDE-47 exposures during proposed developmental stages of high sensitivity by evaluating processes critical for neurogenesis in vitro: NPC proliferation (Stage IV) and neurosphere expansion (Stage V). In a concentration-dependent manner, BDE-47 significantly inhibited NPC proliferation after a 24 h exposure duration (ANOVA, p < .05; Figure 3). Pairwise comparisons revealed significant impairment with 10 and 25 µM of BDE-47 (p < .05). When compared with the vehicle control, 10 or 25 µM BDE-47 reduced NPC proliferation by 43% or 68%, respectively. Similar to BDE-47, BDE-99 also significantly reduced cell proliferation (ANOVA, p < .05). However, pairwise analyses suggested only significance with the highest concentration (25 µM). These data suggest BDE-47 and -99 can inhibit NPC proliferation.
Figure 3.
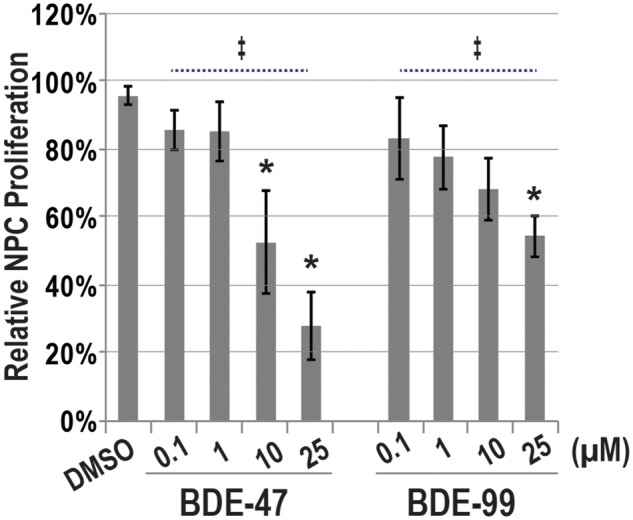
PBDE exposures and NPC proliferation. Relative BrDU uptake, a measure of cell proliferation, in NPCs exposed to BDE-47, BDE-99, or 0.1% DMSO (Veh) for 24 h. Percentages are expressed as average ratios in fluorescence intensity between each exposure group and the media-only control (=100%; n = 6 independent experiments). Significant differences (p < .05) due to PBDE exposure are indicated by double-cross (‡, ANOVA) or asterisk (t test).
We also assessed the potential of BDE-47 to disrupt NPC expansion (Stage V). At 24 h, we identified no differences in neurosphere size between BDE-47 exposed and vehicle control (Figure 4A). At 72 h a significant approximately 26% reduction in neurosphere area was observed with BDE-47 5 µM (t test; p < .05). Similarly, VPA impaired neurosphere projections at 72 h (22% decline) while RA did not. We also examined projection rates as the average ratio between each neurosphere at 24 or 72 h as compared with the initial plating size (1 h) (Figure 4B). In pairwise analyses significant changes in rates were not observed at 24 or 72 h with BDE-47; however, a modest correlation at 72 h (p = .08) was observed at 5 µM as compared with vehicle (↓249%). Our analyses suggest BDE-47 can inhibit neurosphere expansion in a concentration-dependent manner.
Figure 4.

BDE-47 exposures and neurosphere expansion. (A) Average neurosphere size (µm2) in cultures exposed to BDE-47, 0.1% DMSO (VEH) or media only (CON). Suspended neurospheres (preexposed for 3 days) were plated on Cellstart. At 1, 24, and 72 h, successive phase images of single neurospheres (x—= 12.1 neurospheres per group; minimum ≥ 5) were acquired. Total neurosphere area was quantified (ImageJ) and averaged across experiments (n = 4; 1 and 24 h; n = 3; 72 h). B, Average neurosphere expansion rates at 24 or 72 h in comparison with 1 h measurements. In each experiment, expansion rate was determined, and then, averaged across experiments. Significant differences (p < .05) due to exposure as compared with the vehicle control are indicated by double-crosses (‡, ANOVA) or asterisks (t test).
Global Gene Expression Alterations by BDE-47
In NPCs, we investigated the effects of BDE-47 on global gene expression at 24 h using Affymetrix microarrays. In total, we observed 589 genes to be DE due to BDE-47 exposure (“DE BDE-47 genes”; ANOVA, p < .01, absolute FC > 1.5; Figure 5A). In general, genes within this subset were up (356 total) or down-regulated (233 total) with BDE-47. For the majority of transcripts, the greatest effects were observed with 10 versus 1 µM. We further examined the concentration-dependent relationship on gene expression by conducting a post hoc analysis (t test) between BDE-47 (10 or 1 µM) as compared with the vehicle control within genes of this subset. Approximately 95.4% (562/589) of genes were altered with 10 µM, whereas approximately 4.2% (25/589) were altered with 1 µM (p < .05; Figure 5B), suggesting 10 µM to be approximately 22× more effective in disrupting mRNA expression. We identified the 50 most differentially regulated genes due to BDE-47 exposure (Figure 5C). AXNA1, ANXA3, DDIT3, EGR1, LAMP3, MXD1, SERPINE1, SGCG, SLFN5, SPP1, TAC1, UNC5B, UPP1, and WIPI1, were identified as some of the most upregulated targets; and CDCA3, CDCA8, SPAG5, HIST2H3D, CYP26A1, HIST1H2BL, KIF20A, PVALB. MKI67, TXNIP, and HIST1H1T, were identified as some of the most downregulated targets with BDE-47. Our results suggest that BDE-47 perturbs expression of the transcriptome in NPCs across a diversity of targets.
Figure 5.

Concentration-dependent effects of BDE-47 on global gene expression in NPCs. A, Genes identified to be DE due to BDE-47 exposure (589 total; ANOVA; p < .01, abs FC > 1.5). Expression profiles display patterns in expression with 1 or 10 µM (as compared with the respective vehicle control) across the 3 independent experiments. Clusters I and II indicated up and downregulated clusters of DE genes. Within this DE subset, we performed post hoc analyses for significance (t test) between vehicle and 1 or 10 µM BDE-47. B, Distribution of genes DE by each concentration. C, The top 25 most up (left) and down (right) regulated genes due to BDE-47 exposure.
Functional Enrichment of Genes Perturbed by BDE-47
We conducted functional enrichment analysis of genes identified to be significantly DE by BDE-47 via DAVID. We identified 153 GO Biological Processes to be overrepresented (p < .05). GO terms were grouped into 16 functional categories and plotted with their respective: (1) enrichment scores [−log(p)]; (2) number of DE genes per GO term (size); and 3) tendency of patterning within gene subset (color scale) (Figure 6A). Within each grouping, we calculated the geometric mean of enrichment (black bars). Specific enriched terms related to cell cycle (ie, cell cycle process, mitotic cell cycle, mitotic nuclear division) and cellular organization (ie, organelle fission, sister chromatid fission, mitotic sister chromatid segregation), were the most significantly enriched categories (p < 5E-13); predominately consisting of downregulated targets (77%–97%). In ascending order of mean enrichment, overrepresented categories included: apoptosis, cytoskeleton, response to stimuli, transferase activity, signal transduction, differentiation, general metabolism, lipid metabolism, development, biosynthesis, immune, migration, communication, and neurodevelopment. With the exception of cell cycle or organizational related processes, categorical gene subsets tended to include more upregulated versus downregulated targets.
Figure 6.

Functional enrichment analysis of genes DE with BDE-47 in NPCs. (A). Enriched GO Biological Processes within BDE-47 DE Genes identified using DAVID (criteria: p < .01, number of DE genes associated with enriched term ≥ 7). GO enrichment scores [−log (p)] are displayed for all BDE-47 DE Genes and upregulated (Cluster I) or downregulated (Cluster II) gene subsets. The total number of DE Genes due to BDE-47 is located in parentheses (up, downregulated). Clustering of BDE-47 DE Genes associated with terms: (B) Response to Hormone, (C) Steroid Metabolism, (D) Neurogenesis, (E) Response to Oxidative Stress, and (F) Mitotic Nuclear Division. FC values represent average difference between BDE-47 and vehicle control.
We expanded our analysis to DE genes within enriched GO terms hypothesized to be linked to BDE-47 toxicity (Figs. 6B–F). Enriched biological processes and DE genes included: response to hormone (46 total, 86% ↑), eg, nuclear receptors (THRA, THRB, NRC31); steroid metabolic process (19 total, 95% ↑), eg, cholesterol biosynthesis pathway members (AKR1D1, CH25H, CYP51A1, DHCR7, HMGCR, INSIG1, LDLR, LSS, MVD, NPC1, NSDHL, and SQLE); positive regulation of neurogenesis (23 total, 74% ↑), eg, NDNF, NGF, NRCAM, and PAX6; response to oxidative stress (18 total, 78% ↑), eg, HMOX1, NFE2L2, NQO1, and CYP2B6; and mitotic nuclear division (58 total, 93% ↓), eg, major cell cycle checkpoint regulators (CCNA2, CCNB1, CCNF, CDC25C, CDCA2, CDCA3, CDCA8, and CDK1) and kinesin family members (KIF2C, KIF4A, KIF4B, KIF11, KIF15, KIF18A, KIF18B, KIF22, and KIF23). In general, with few exceptions (eg, CYP2B6), the magnitude in altered gene expression was greatest with 10 versus 1 µM BDE-47.
To validate our microarray results and extend our analysis of the concentration-response of BDE-47, and its potential similarity to BDE-99, we evaluated the effect of BDE-47 (0.01–10 µM) or -99 (10 µM) exposures on gene expression of selected DE targets in independent NPC cultures (Figure 7A). In this context, we evaluated expression of molecules with roles in oxidative stress (NFE2L2 and HMOX1), neuronal differentiation (GRIA1, NRCAM and PAX6), cell cycle regulation (CDK1), TH signaling (THRA and THRB), or cholesterol metabolism (DHCR7). We observed significant changes in all 9 transcripts with BDE-47; DHCR7, NFE2L2, NRCAM, GRIA1, HMOX1, and THRB were significantly upregulated with BDE-47; PAX6, THRA, and CDK1 were significantly downregulated with BDE-47 (p < .05). In general, responses were primarily observed with only 10 µM BDE-47; and these effects strongly correlated with 10 µM BDE-99 (R2 = 0.99, data not shown). The magnitude and directionality of expression changes between microarray and qRT-PCR were largely in agreement (R2 = 0.8) (Figure 7B).
Figure 7.

Validation studies of genes altered by BDE-47 in NPCs. A, Concentration-dependent expression alterations in proposed mRNA targets of PBDEs using qRT-PCR (n = 6 independent experiments). Expression values (−ΔΔCT) were normalized to housekeeping genes (GAPDH and ACTB) and adjusted by the respective vehicle control. Significant differences (p < .05) due to exposure as compared with the vehicle control are indicated by double-crosses (‡, ANOVA) or asterisks (t test). B, Expression changes in proposed PBDE targets between microarray and qRT-PCR experiments at 1 or 10 µM (R2 = 0.8).
Global CpG Methylation Analysis of BDE-47 Exposed NPCs
In parallel with transcriptomic analyses, we profiled global CpG methylation in BDE-47 (1, 10 µM) and vehicle exposed NPCs at 24 h. We identified 1238 CpGs to be DM with BDE-47 exposure (p ≤ .005; absolute average Δβ ≥ .05). In a concentration-dependent fashion, BDE-47 exposure significantly increased global CpG methylation as determined by Δ in median β between BDE-47 and vehicle control (Δβ = 0.4% ± 0.4%, 1 µM; 2.1% ± 0.2%, 10 µM; Figure 8A). Median changes in methylation were more pronounced when the analysis was limited to BDE-47 DM CpGs (Δβ = 1.1% ± 0.1%, 1 µM; 5.8% ± 0.1%, 10 µM). These results were consistent with observations on individual CpG levels; 99% had increased methylation (1220/1238 total) and 1% had decreased methylation (18/1238 total; Figure 8B). Post hoc analyses of BDE-47 DM CpGs revealed concentration-dependent differences between 10 and 1 µM (Figure 8C); 1, 151 of DM CpGs were altered by >5% with 10 µM; whereas, only 30 DM CpGs were altered by >5% with 1 µM. Limited single CpGs were identified with reduced methylation < −5% with either 10 µM (13 CpGs) or 1 µM (5 CpGs). Similar to mRNA profiling analyses, 10 µM was shown to be more potent in affecting CpG methylation.
Figure 8.

Global CpG methylation profiling of BDE-47-exposed NPCs. Cells were exposed to 1 or 10 µM BDE-47 or 0.1% DMSO for 24 h. We identified 1258 CpGs to be DM with BDE-47 versus 0.1% DMSO (p ≤ .005; absolute average Δβ ≥ 0.05). A, Median difference in methylation levels (Δβ) between BDE-47 exposed and vehicle control cultures in all evaluated CpGs and BDE-47 DM CpGs after 24 h. B, Hierarchical clustering of the changes in methylation in BDE-47 DM CpGs (BDE-47 vs concurrent vehicle control). C, Frequency of BDE-47 DM CpGs by changes in methylation per concentration. Enrichment of BDE-47 DM CpGs by chromosome (D) and gene regulatory region (E). Asterisks indicate over (↑) or under (↓) representation (Fisher’s test; *p ≤ .05; **p ≤ .001). F, Enrichment of GO Biological Processes within genes (# in parentheses) in proximity of BDE-47 DM CpGs.
Within the BDE-47 DM CpG subset, we evaluated enrichment based on chromosome (Figure 8D) and gene location (Figure 8E). Across each of the chromosomes, a larger proportion of increased versus decreased methylated CpGs was observed. We identified significant enrichment of DM CpGs on chromosomes 1 and 6 and an underrepresentation of DM CpGs on chromosome 7, 18, 19, and X. Concentration-dependent changes in methylation of CpGs (10 > 1 µM) were observed at the chromosome level (see example of Chromosome 1; Supplementary Figure 2). Nonpromoter regions (intergenic sequences, 3’UTR regions, and gene bodies) were overrepresented, while those CpGs more proximal to the promoter (TSS1500, TS200, 5’UTR, and first Exon) were underrepresented.
GO analyses of genes in proximity of BDE-47 DM CpGs (809 total) suggested enrichment of processes involved in inflammation, cell migration, cell signaling, cytoskeleton organization, nervous system development, and cell differentiation (Figure 8F). Genes associated with DM CpGs proximal to promoter regions (287 total) tended to be more associated with processes related to inflammation and cell migration. Although our analyses suggested overlap between mRNA and CpG methylation on a functional level for specific associations (eg, nervous system development), poor correlation was observed between changes in mRNA expression and CpG methylation in promoter regions (Pearson coefficient = −0.05, not shown). Four genes found to be DE with BDE-47 were identified to contain DM CpGs in the promoter site; however, none of these targets followed an inverse trend between expression and methylation (Supplementary Figure 3). Our results suggest BDE-47 generally increases CpG methylation in specific sites linked to DNA regions which control pathways related to neural cell function and stress (eg, inflammation), but these changes are not coordinated with mRNA levels at 24 h.
DISCUSSION
Although legislative efforts are resulting in the gradual phasing out of PBDEs in consumer products, their continued presence in the environment and bio-persistence presents a contemporary threat to wildlife and humans. PBDEs are identified during human pregnancy (Herbstman et al., 2010) with recent studies serum show levels of BDE-47 at 0.17 and 0.22 ng/g in maternal and cord serum, respectively (Zota et al., 2018). Multiple studies in rodent and humans (as reviewed Costa and Giordano, 2007; Dingemans et al., 2011; Herbstman and Mall, 2014) suggest that these compounds negatively affect neurodevelopment and other developmental/reproductive endpoints. Questions remain regarding the underlying mechanisms in which PBDE cause neurotoxicity and the vulnerable periods in development to PBDE exposures. Using a hESC model representative of cellular transitions occurring during neurogenesis, we evaluated a concentration range (0.01–25 µM) of BDE-47 and -99, levels that are physiologically relevant for human exposures during pregnancy (Park et al., 2014) and observed as toxic in human embryonic/fetal cells (Robinson et al., 2018; Schreiber et al., 2010). Our results demonstrate that PBDEs induce neurodevelopmental toxicity in a concentration- and neurodevelopmental stage-dependent manner. For example, the BMCs of BDE-47 or -99 (100% increase in LDH activity) were >10× higher in NPCs (Stage IV) versus hESCs (Stage I), suggesting that PBDEs are cytotoxic to early embryonic neural cell populations and sensitivity may depend on developmental timing or differentiation (Figure 2; Li et al., 2013). Our findings of higher sensitivity to PBDE exposures in human neural progenitors compared with pluripotent hESCs agrees with previous studies investigating stage-dependent toxicity, such as in lead (Senut et al., 2014). Although the reasons for this differential vulnerability were not tested, potential factors which could heighten hESC sensitivity to PBDEs after differentiation may include the presence or absence of: (1) vesicular transporters (Bradner et al., 2013); (2) nuclear receptors relevant to the toxic mechanism of action (eg, TH) (Kojima et al., 2009); (3) metabolizing enzymes (Fathi et al., 2011); or (4) physical cellular characteristics (eg, tight junctions in hESCs; Pieters and van Roy, 2014) known to differ between NPCs and hESCs.
Environmental compounds can alter fundamental processes underlying neurodevelopmental deficits, such as neuronal migration, proliferation, and differentiation (Courchesne and Pierce, 2005; Rice and Barone, 2000; Wegiel et al., 2010). In our analyses, BDE-47 significantly inhibited both NPC proliferation and expansion (Figs. 3 and 4). Our data corroborate previous in vitro studies in human fetal neurons, where PBDEs inhibited both migration and proliferation via disruption of TH-dependent signaling (Schreiber et al., 2010). However PBDEs can cause neurotoxicity through diverse mechanisms (Costa et al., 2014; Costa and Giordano, 2007; Dingemans et al., 2011). To determine potential targets of BDE-47 toxicity in NPCs, we profiled the transcriptome in cells exposed to 1 or 10 µM BDE-47 and determined DE genes as compared with vehicle controls. We observed dramatic, concentration-dependent changes on the transcriptome due to BDE-47 (Figure 5). Our transcriptomic results suggest that PBDEs disrupt multiple pathways to induce toxicological and functional changes in human neural progenitors.
PBDEs are known disruptors of the TH system (Meerts et al., 2000; Richardson et al., 2008) and our data provide further evidence of dysregulated expression with genes related to hormone response and steroid metabolism (Figs. 6B and 6C). Of note, we observed significant alterations in the 2 primary human TH receptors responsible for mediating TH-signaling, THRA (↓) and THRB (↑), by BDE-47 (Figs. 6B and 7). TH signaling is functional in human NPC models (Dach et al., 2017) and the divergent influence of PBDE exposure on expression of THRs has previously been identified in rat cerebellar granular neurons (Blanco et al., 2011), with corresponding functional consequences in human neurosphere assays (Schreiber et al., 2010). However, the specific roles of TRα and TRβ in this process have not been specifically elucidated to our knowledge. TRα overexpression has been linked to repression of NPC differentiation to a migratory neuroblast phenotype (Lopez-Juarez et al., 2012) and adult TRβ−/− mice exhibit increased number of proliferative and neuroblast cell populations in the subgranular zone (Kapoor et al., 2011). Thus, the divergent THR expression patterns induced by PBDEs may converge on a common neurotoxicological endpoint. However further studies would be necessary to address this.
TH-signaling is an important regulator of developmental and metabolic processes, including cholesterol homeostasis (O’Shea and Williams, 2002). Disruption in cholesterol/steroid homeostasis is linked with endocrine-disrupting chemicals (Casals-Casas and Desvergne, 2011) and dysregulation of these molecules is implicated in neurodegenerative risk (Liu et al., 2010). Here we observed upregulation of several cholesterol/steroid biosynthesis enzymes and transporters (Figure 6C), with common (eg, INSIG1) and novel (eg, NPC1 and LDLR) targets compared with our previous transcriptomic analyses of primary human cytotrophoblasts exposed to BDE-47 (Robinson et al., 2018). Our results suggest that the interplay between TH-signaling and molecules such as DHCR7 (Figure 7) in PBDE-neurotoxicity may warrant further investigation.
In diverse toxicological models, PBDEs generate oxidative stress and inflammation (Costa et al., 2015; He et al., 2008; Park et al., 2014; Peltier et al., 2012). Our transcriptomic results similarly indicate significant enrichment of pathways related to oxidative stress modulation (eg, response to oxygen) and upregulation of known molecules in the Nrf2-ARE (antioxidant response element) signaling pathway such as NFE2L2, NQ01, and HMOX1(Figure 6E) (Loboda et al., 2016). Previous in vitro studies using neuronal models have demonstrated BDE-47 induces oxidative stress (Costa et al., 2015; He et al., 2008; Tagliaferri et al., 2010), concurring with our transcriptomic data of relevant genes. Though responses to BDE-47 were generally observed at concentrations of > 1 µM, interestingly one of the few genes significantly dysregulated at 1 µM but not 10 µM was CYP2B6 (2-fold induction with 1 µM, Figure 6C). CYP2B6 is responsible for metabolizing environmental compounds and is the predominant isoform involved in the metabolism of PBDE (Feo et al., 2013). Our data may suggest that at lower concentrations, CYP2B6 is upregulated to mediate breakdown of PBDEs and at higher concentrations an inflammatory and prooxidant environment attenuates its transcription (Aitken et al., 2008; Aitken and Morgan, 2007), contributing to a toxic response.
Imbalances in reactive oxygen species (ROS)-mediated signaling can dramatically affect neuronal homeostasis and differentiation (as reviewed Vieira et al., 2011). Coinciding with an upregulation in stress response and Nrf2-ARE associated genes, we observed upregulation of apoptotic-related genes and pathways and downregulated expression of cell cycle factors, with the latter representing the most significantly enriched pathway in our dataset (Figure 6A). Among the cell cycle factors affected by BDE-47, we validated the downregulation of CDK1 (Figure 7), a critical promoter of cell survival and proliferation (Liu et al., 2008). The decreased expression of cell cycle factors may be involved in the reduced proliferation of NPCs observed with BDE-47 treatment (Figure 3), given corresponding changes in transcripts associated with mitotic spindle dynamics (eg, kinesins, ANLN, and DLGAP5) (Figure 6F). Additionally, BDE-47 significantly disrupted genes/pathways critical for neurogenesis (Figure 6D). PAX6, which is critical for regulating the balance of neural renewal and differentiation (Estivill-Torrus et al., 2002; Sansom et al., 2009), was significantly reduced in NPCs. A parallel upregulation in transcription for glutamate receptors (GRIA1) and neuronal adhesion molecules (NRCAM) was also observed (Figs. 6D and 7), indicating BDE-47 may alter the routes of differentiation in these cells. Collectively, these data suggest that PBDEs induce oxidative stress pathways in NPCs, leading to alterations in proliferation and potentially differentiation.
Environmental chemicals are implicated in epigenetic changes (Baccarelli and Bollati, 2009), with changes in methylation patterns potentially underlying adverse neurodevelopmental outcomes (Keil and Lein, 2016). PBDE exposures in utero are similarly associated with epigenetic changes (Woods et al., 2012) that could underlie transient or irreversible genomic changes, with the potential for transgenerational inheritance (Skinner and Guerrero-Bosagna, 2009). As epigenomic regulation is a pivotal process in neurogenesis, we also evaluated global methylation patterns in NPCs exposed to BDE-47. Our results indicate BDE-47 causes a modest, but consistent, global hypermethylation in the NPC methylome in a concentration-dependent manner (Figs. 8A–C), with diverse response when examined by chromosomal location (Figure 8D) and an overrepresentation of methylation at nonpromoter regions (Figure 8E). Interestingly, DM CpGs were almost exclusively found in variable (69.5%; β = 0.2‒0.8) or hypermethylated (29%, β > 0.8) CpG regions; only 1.5% of DM CpGs were hypomethylated (β < 0.2).The effects of DNA methylation outside the promoter region and first exon remains unclear, capable of both positively and negatively affecting transcription (Gagnon-Kugler et al., 2009; Jjingo et al., 2012). BDE-47 DM CpGs near the promoter region did not show significant enrichment in developmental processes such as cell differentiation, nervous system development, and synaptic signaling of genes, suggesting that NPC functional changes due to chemical exposure are driven by methylation at nonpromoter regions (Figure 8F). This would coincide with observations that nonpromoter methylation is a mediator of neuronal differentiation (Wu et al., 2010); however, additional studies are necessary for confirmation.
In conclusion, our study demonstrates the utilization of a hESC neural differentiation model to examine endpoints relevant for in vivo neurogenesis. Due to their (1) human relevance; (2) pluripotent capabilities to be differentiated into multiple cell types; and (3) representation of the continuum of neurodevelopment on cellular and molecular levels, hESCs can be utilized for many applications, including the screening of environmental chemicals for neurotoxicity. Using this multi-stage hESC differentiation model we assessed the effects of BDE-47 during windows of neurogenesis in vitro. Our results indicate that common PBDE congeners may be potent toxicants in neural progenitors, which induce functional changes and elicit transcriptomic alterations in pathways regulating neurodevelopment, hormone signaling, and environmental stress response. Our results concur with previous toxicological investigations in human in vitro and rodent model systems, and support growing epidemiological evidence tying exposure to these compounds to neurodevelopmental adverse outcomes. Future studies may examine the relationship between PBDEs, oxidative stress, and redox state in the context of NPC proliferation, differentiation, and epigenetic control. Proposed PBDE-gene interactions linked to neurotoxicity taken from our in vitro studies may also be investigated further in vivo. Our data support the utility of the hESC model system to better efforts towards screening environmental contaminants for potential neurotoxicity in humans.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
DECLARATION OF CONFLICTING INTERESTS
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Matthew Gormley and Katherine Ona for technical support; Dr Susan Fisher and Dr Tamara Zdravkovic for insightful conversations which led to the development of the hESC neural differentiation model system; and Dr Joe Costello and lab members for processing of DNA samples for methylation analyses.
FUNDING
This work was kindly supported by the National Institute of Child Health and Human Development (T32HD007263), the United States Environmental Protection Agency (RD883467801), and the National Institute of Environmental Health Sciences (K99ES023846, R00ES023846, P01ES022841).
AUTHOR CONTRIBUTIONS
J.R. designed and directed all aspects of the project; J.R., H.S., N.L., I.K., C.J.F., and N.A. performed the experiments; J.R. drafted all figures and conducted the statistical analyses; J.R. and Y.K. conducted the methylation data analyses; J.R. and H.C. interpreted the experimental results and wrote the article.
REFERENCES
- Aitken A. E., Lee C. M., Morgan E. T. (2008). Roles of nitric oxide in inflammatory downregulation of human cytochromes p450. Free Radic. Biol. Med. 44, 1161–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken A. E., Morgan E. T. (2007). Gene-specific effects of inflammatory cytokines on cytochrome p4502c, 2b6 and 3a4 mRNA levels in human hepatocytes. Drug Metab. Dispos. 35, 1687–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccarelli A., Bollati V. (2009). Epigenetics and environmental chemicals. Curr. Opin. Pediatr. 21, 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal-Price A. K., Coecke S., Costa L., Crofton K. M., Fritsche E., Goldberg A., Grandjean P., Lein P. J., Li A., Lucchini R. (2012). Advancing the science of developmental neurotoxicity (DNT): Testing for better safety evaluation. ALTEX 29, 202–215. [DOI] [PubMed] [Google Scholar]
- Blanco J., Mulero M., Lopez M., Domingo J. L., Sanchez D. J. (2011). BDE-99 deregulates BDNF, Bcl-2 and the mRNA expression of thyroid receptor isoforms in rat cerebellar granular neurons. Toxicology 290, 305–311. [DOI] [PubMed] [Google Scholar]
- Borenfreund E., Puerner J. A. (1985). Toxicity determined in vitro by morphological alterations and neutral red absorption. Toxicol. Lett. 24, 119–124. [DOI] [PubMed] [Google Scholar]
- Bradner J. M., Suragh T. A., Wilson W. W., Lazo C. R., Stout K. A., Kim H. M., Wang M. Z., Walker D. I., Pennell K. D., Richardson J. R., et al. (2013). Exposure to the polybrominated diphenyl ether mixture DE-71 damages the nigrostriatal dopamine system: Role of dopamine handling in neurotoxicity. Exp. Neurol. 241, 138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casals-Casas C., Desvergne B. (2011). Endocrine disruptors: From endocrine to metabolic disruption. Annu. Rev. Physiol. 73, 135–162. [DOI] [PubMed] [Google Scholar]
- Chen A., Yolton K., Rauch S. A., Webster G. M., Hornung R., Sjodin A., Dietrich K. N., Lanphear B. P. (2014). Prenatal polybrominated diphenyl ether exposures and neurodevelopment in U.S. Children through 5 years of age: The home study. Environ. Health Perspect. 122, 856–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy B., Darlington R. B., Finlay B. L. (2001). Translating developmental time across mammalian species. Neuroscience 105, 7–17. [DOI] [PubMed] [Google Scholar]
- Colleoni S., Galli C., Gaspar J. A., Meganathan K., Jagtap S., Hescheler J., Sachinidis A., Lazzari G. (2011). Development of a neural teratogenicity test based on human embryonic stem cells: Response to retinoic acid exposure. Toxicol. Sci. 124, 370–377. [DOI] [PubMed] [Google Scholar]
- Costa L. G., de Laat R., Tagliaferri S., Pellacani C. (2014). A mechanistic view of polybrominated diphenyl ether (PBDE) developmental neurotoxicity. Toxicol. Lett. 230, 282–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa L. G., Giordano G. (2007). Developmental neurotoxicity of polybrominated diphenyl ether (PBDE) flame retardants. Neurotoxicology 28, 1047–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa L. G., Pellacani C., Dao K., Kavanagh T. J., Roque P. J. (2015). The brominated flame retardant BDE-47 causes oxidative stress and apoptotic cell death in vitro and in vivo in mice. Neurotoxicology 48, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne E., Pierce K. (2005). Brain overgrowth in autism during a critical time in development: Implications for frontal pyramidal neuron and interneuron development and connectivity. Int. J. Dev. Neurosci. 23, 153–170. [DOI] [PubMed] [Google Scholar]
- Cowell W. J., Lederman S. A., Sjodin A., Jones R., Wang S., Perera F. P., Wang R., Rauh V. A., Herbstman J. B. (2015). Prenatal exposure to polybrominated diphenyl ethers and child attention problems at 3-7 years. Neurotoxicol. Teratol. 52, 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dach K., Bendt F., Huebenthal U., Giersiefer S., Lein P. J., Heuer H., Fritsche E. (2017). BDE-99 impairs differentiation of human and mouse NPCs into the oligodendroglial lineage by species-specific modes of action. Sci. Rep. 7, 44861.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingemans M. M., Ramakers G. M., Gardoni F., van Kleef R. G., Bergman A., Di Luca M., van den Berg M., Westerink R. H., Vijverberg H. P. (2007). Neonatal exposure to brominated flame retardant BDE-47 reduces long-term potentiation and postsynaptic protein levels in mouse hippocampus. Environ. Health Perspect. 115, 865–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingemans M. M., van den Berg M., Westerink R. H. (2011). Neurotoxicity of brominated flame retardants: (In)direct effects of parent and hydroxylated polybrominated diphenyl ethers on the (developing) nervous system. Environ. Health Perspect. 119, 900–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du P., Zhang X., Huang C. C., Jafari N., Kibbe W. A., Hou L., Lin S. M. (2010). Comparison of beta-value and m-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 11, 587.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskenazi B., Chevrier J., Rauch S. A., Kogut K., Harley K. G., Johnson C., Trujillo C., Sjodin A., Bradman A. (2013). In utero and childhood polybrominated diphenyl ether (PBDE) exposures and neurodevelopment in the chamacos study. Environ. Health Perspect. 121, 257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estivill-Torrus G., Pearson H., van Heyningen V., Price D. J., Rashbass P. (2002). Pax6 is required to regulate the cell cycle and the rate of progression from symmetrical to asymmetrical division in mammalian cortical progenitors. Development 129, 455–466. [DOI] [PubMed] [Google Scholar]
- Evans M. J., Kaufman M. H. (1981). Establishment in culture of pluripotential cells from mouse embryos. Nature 292, 154–156. [DOI] [PubMed] [Google Scholar]
- Fathi A., Hatami M., Hajihosseini V., Fattahi F., Kiani S., Baharvand H., Salekdeh G. H. (2011). Comprehensive gene expression analysis of human embryonic stem cells during differentiation into neural cells. PLoS One 6, e22856-e22856.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feo M. L., Gross M. S., McGarrigle B. P., Eljarrat E., Barcelo D., Aga D. S., Olson J. R. (2013). Biotransformation of BDE-47 to potentially toxic metabolites is predominantly mediated by human CYP2B6. Environ. Health Perspect. 121, 440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederiksen M., Vorkamp K., Thomsen M., Knudsen L. E. (2009). Human internal and external exposure to PBDES - A review of levels and sources. Int. J. Hyg. Environ. Health 212, 109–134. [DOI] [PubMed] [Google Scholar]
- Fritsche E., Barenys M., Klose J., Masjosthusmann S., Nimtz L., Schmuck M., Wuttke S., Tigges J. (2018). Development of the concept for stem cell-based developmental neurotoxicity evaluation. Toxicol. Sci. 165, 14–20. [DOI] [PubMed] [Google Scholar]
- Gagnon-Kugler T., Langlois F., Stefanovsky V., Lessard F., Moss T. (2009). Loss of human ribosomal gene CpG methylation enhances cryptic RNA polymerase ii transcription and disrupts ribosomal RNA processing. Mol. Cell 35, 414–425. [DOI] [PubMed] [Google Scholar]
- Gene Ontology Consortium. (2015). Gene ontology consortium: Going forward. Nucleic Acids Res. 43(Database issue), D1049–D1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He P., He W. H., Wang A. G., Xia T., Xu B. Y., Zhang M., Chen X. M. (2008). Pbde-47-induced oxidative stress, DNA damage and apoptosis in primary cultured rat hippocampal neurons. Neurotoxicology 29, 124–129. [DOI] [PubMed] [Google Scholar]
- Herbstman J. B., Mall J. K. (2014). Developmental exposure to polybrominated diphenyl ethers and neurodevelopment. Curr. Environ. Health Rep. 1, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbstman J. B., Sjodin A., Apelberg B. J., Witter F. R., Halden R. U., Patterson D. G., Panny S. R., Needham L. L., Goldman L. R. (2008). Birth delivery mode modifies the associations between prenatal polychlorinated biphenyl (PCB) and polybrominated diphenyl ether (PBDE) and neonatal thyroid hormone levels. Environ. Health Perspect. 116, 1376–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbstman J. B., Sjodin A., Kurzon M., Lederman S. A., Jones R. S., Rauh V., Needham L. L., Tang D., Niedzwiecki M., Wang R. Y., et al. (2010). Prenatal exposure to PBDEs and neurodevelopment. Environ. Health Perspect. 118, 712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hites R. A. (2004). Polybrominated diphenyl ethers in the environment and in people: A meta-analysis of concentrations. Environ. Sci. Technol. 38, 945–956. [DOI] [PubMed] [Google Scholar]
- Huang D. W., Sherman B. T., Tan Q., Collins J. R., Alvord W. G., Roayaei J., Stephens R., Baseler M. W., Lane H. C., Lempicki R. A. (2007). The DAVID gene functional classification tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 8, R183.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jjingo D., Conley A. B., Yi S. V., Lunyak V. V., Jordan I. K. (2012). On the presence and role of human gene-body DNA methylation. Oncotarget 3, 462–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor R., Ghosh H., Nordstrom K., Vennstrom B., Vaidya V. A. (2011). Loss of thyroid hormone receptor beta is associated with increased progenitor proliferation and neurod positive cell number in the adult hippocampus. Neurosci. Lett. 487, 199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keil K. P., Lein P. J. (2016). DNA methylation: A mechanism linking environmental chemical exposures to risk of autism spectrum disorders? Environ. Epigenet. 2, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstreuer N. C., Smith A. M., West P. R., Conard K. R., Fontaine B. R., Weir-Hauptman A. M., Palmer J. A., Knudsen T. B., Dix D. J., Donley E. L., et al. (2011). Identifying developmental toxicity pathways for a subset of ToxCast chemicals using human embryonic stem cells and metabolomics. Toxicol. Appl. Pharmacol. 257, 111–121. [DOI] [PubMed] [Google Scholar]
- Kojima H., Takeuchi S., Uramaru N., Sugihara K., Yoshida T., Kitamura S. (2009). Nuclear hormone receptor activity of polybrominated diphenyl ethers and their hydroxylated and methoxylated metabolites in transactivation assays using Chinese hamster ovary cells. Environ. Health Perspect. 117, 1210–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krtolica A., Ilic D., Genbacev O., Miller R. K. (2009). Human embryonic stem cells as a model for embryotoxicity screening. Regen. Med. 4, 449–459. [DOI] [PubMed] [Google Scholar]
- Lam J., Lanphear B. P., Bellinger D., Axelrad D. A., McPartland J., Sutton P., Davidson L., Daniels N., Sen S., Woodruff T. J. (2017). Developmental PBDE exposure and IQ/ADHD in childhood: A systematic review and meta-analysis. Environ. Health Perspect. 125, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F., Xie Q., Li X., Li N., Chi P., Chen J., Wang Z., Hao C. (2010). Hormone activity of hydroxylated polybrominated diphenyl ethers on human thyroid receptor-beta: In vitro and in silico investigations. Environ Health Perspect. 118, 602–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T., Wang W. B., Pan Y. W., Xu L. H., Xia Z. G. (2013). A hydroxylated metabolite of flame-retardant PBDE-47 decreases the survival, proliferation, and neuronal differentiation of primary cultured adult neural stem cells and interferes with signaling of ERK5 MAP kinase and neurotrophin 3. Toxicol. Sci. 134, 111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C. K., Kim S. K., Ko D. S., Cho J. W., Jun J. H., An S. Y., Han J. H., Kim J. H., Yoon Y. D. (2009). Differential cytotoxic effects of mono-(2-ethylhexyl) phthalate on blastomere-derived embryonic stem cells and differentiating neurons. Toxicology 264, 145–154. [DOI] [PubMed] [Google Scholar]
- Liu J. P., Tang Y., Zhou S., Toh B. H., McLean C., Li H. (2010). Cholesterol involvement in the pathogenesis of neurodegenerative diseases. Mol. Cell. Neurosci. 43, 33–42. [DOI] [PubMed] [Google Scholar]
- Liu P., Kao T. P., Huang H. (2008). CDK1 promotes cell proliferation and survival via phosphorylation and inhibition of FOXO1 transcription factor. Oncogene 27, 4733–4744. [DOI] [PubMed] [Google Scholar]
- Loboda A., Damulewicz M., Pyza E., Jozkowicz A., Dulak J. (2016). Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 73, 3221–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Juarez A., Remaud S., Hassani Z., Jolivet P., Simons J. P., Sontag T., Yoshikawa K., Price J., Morvan-Dubois G., Demeneix B. A. (2012). Thyroid hormone signaling acts as a neurogenic switch by repressing Sox2 in the adult neural stem cell niche. Cell Stem Cell 10, 531–543. [DOI] [PubMed] [Google Scholar]
- Main K. M., Kiviranta H., Virtanen H. E., Sundqvist E., Tuomisto J. T., Tuomisto J., Vartiainen T., Skakkebaek N. E., Toppari J. (2007). Flame retardants in placenta and breast milk and cryptorchidism in newborn boys. Environ. Health Perspect. 115, 1519–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazdai A., Dodder N. G., Abernathy M. P., Hites R. A., Bigsby R. M. (2003). Polybrominated diphenyl ethers in maternal and fetal blood samples. Environ. Health Perspect. 111, 1249–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald T. A. (2002). A perspective on the potential health risks of PBDEs. Chemosphere 46, 745–755. [DOI] [PubMed] [Google Scholar]
- Meeker J. D. (2012). Exposure to environmental endocrine disruptors and child development. Arch. Pediatr. Adolesc. Med. 166, 952–958. [PubMed] [Google Scholar]
- Meerts I., van Z. J. J., Luijks E. A. C., van L., Bol I., Marsh G., Jakobsson E., Bergman A., Brouwer A. (2000). Potent competitive interactions of some brominated flame retardants and related compounds with human transthyretin in vitro. Toxicol. Sci. 56, 95–104. [DOI] [PubMed] [Google Scholar]
- Mundy W. R., Freudenrich T. M., Crofton K. M., DeVito M. J. (2004). Accumulation of PBDE-47 in primary cultures of rat neocortical cells. Toxicol. Sci. 82, 164–169. [DOI] [PubMed] [Google Scholar]
- O’Shea P. J., Williams G. R. (2002). Insight into the physiological actions of thyroid hormone receptors from genetically modified mice. J. Endocrinol. 175, 553–570. [DOI] [PubMed] [Google Scholar]
- Pal R., Mamidi M. K., Das A. K., Bhonde R. (2011). Human embryonic stem cell proliferation and differentiation as parameters to evaluate developmental toxicity. J. Cell Physiol. 226, 1583–1595. [DOI] [PubMed] [Google Scholar]
- Park H. R., Kamau P. W., Loch-Caruso R. (2014). Involvement of reactive oxygen species in brominated diphenyl ether-47-induced inflammatory cytokine release from human extravillous trophoblasts in vitro. Toxicol. Appl. Pharmacol. 274, 283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual A., Aranda A. (2013.) Thyroid hormone receptors, cell growth and differentiation. Biochimica et Biophysica Acta 1830, 3908–3916. [DOI] [PubMed] [Google Scholar]
- Patel J., Landers K., Li H., Mortimer R. H., Richard K. (2011). Thyroid hormones and fetal neurological development. J. Endocrinol. 209, 1–8. [DOI] [PubMed] [Google Scholar]
- Peltier M. R., Klimova N. G., Arita Y., Gurzenda E. M., Murthy A., Chawala K., Lerner V., Richardson J., Hanna N. (2012). Polybrominated diphenyl ethers enhance the production of proinflammatory cytokines by the placenta. Placenta 33, 745–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieters T., van Roy F. (2014). Role of cell–cell adhesion complexes in embryonic stem cell biology. J. Cell Sci. 127, 2603.. [DOI] [PubMed] [Google Scholar]
- Repetto G., del Peso A., Zurita J. L. (2008). Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 3, 1125–1131. [DOI] [PubMed] [Google Scholar]
- Rice D., Barone S. Jr (2000). Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environ. Health Perspect. 108(Suppl. 3), 511–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson V. M., Staskal D. F., Ross D. G., Diliberto J. J., DeVito M. J., Birnbaum L. S. (2008). Possible mechanisms of thyroid hormone disruption in mice by BDE 47, a major polybrominated diphenyl ether congener. Toxicol. Appl. Pharmacol. 226, 244–250. [DOI] [PubMed] [Google Scholar]
- Robinson J. F., Gormley M. J., Fisher S. J. (2016). A genomics-based framework for identifying biomarkers of human neurodevelopmental toxicity. Reprod. Toxicol. 60, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, J. F., Kapidzic, M., Hamilton, E. G., Chen, H., Puckett, K.W., Zhou, Y., Ona, K., Parry, E., Wang, Y., Park, J. S., et al. (2018). Genomic profiling of bde-47 effects on human placental cytotrophoblasts. Toxicol. Sci 167, 211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed A. I., Bhagabati N. K., Braisted J. C., Liang W., Sharov V., Howe E. A., Li J., Thiagarajan M., White J. A., Quackenbush J. (2006). TM4 microarray software suite. Methods Enzymol. 411, 134–193. [DOI] [PubMed] [Google Scholar]
- Sansom S. N., Griffiths D. S., Faedo A., Kleinjan D. J., Ruan Y., Smith J., van Heyningen V., Rubenstein J. L., Livesey F. J. (2009). The level of the transcription factor Pax6 is essential for controlling the balance between neural stem cell self-renewal and neurogenesis. PLoS Genet. 5, e1000511.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schecter A., Johnson-Welch S., Tung K. C., Harris T. R., Papke O., Rosen R. (2007). Polybrominated diphenyl ether (PBDE) levels in livers of U.S. Human fetuses and newborns. J .Toxicol. Environ. Health A 70, 1–6. [DOI] [PubMed] [Google Scholar]
- Schreiber T., Gassmann K., Gotz C., Hubenthal U., Moors M., Krause G., Merk H. F., Nguyen N. H., Scanlan T. S., Abel J., et al. (2010). Polybrominated diphenyl ethers induce developmental neurotoxicity in a human in vitro model: Evidence for endocrine disruption. Environ. Health Perspect. 118, 572–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senut M. C., Sen A., Cingolani P., Shaik A., Land S. J., Ruden D. M. (2014). Lead exposure disrupts global DNA methylation in human embryonic stem cells and alters their neuronal differentiation. Toxicol. Sci. 139, 142–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R., Lam A., Li M. C., Ngan M., Menenzes S., Zhao Y. (2007). Analysis of gene expression data using BRB-arraytools. Cancer Inform. 3, 11–17. [PMC free article] [PubMed] [Google Scholar]
- Skinner M. K., Guerrero-Bosagna C. (2009). Environmental signals and transgenerational epigenetics. Epigenomics 1, 111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenzel D., Huttner W. B. (2013). Role of maternal thyroid hormones in the developing neocortex and during human evolution. Front. Neuroanat. 7, 19.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stummann T. C., Hareng L., Bremer S. (2009). Hazard assessment of methylmercury toxicity to neuronal induction in embryogenesis using human embryonic stem cells. Toxicology 257, 117–126. [DOI] [PubMed] [Google Scholar]
- Tagliaferri S., Caglieri A., Goldoni M., Pinelli S., Alinovi R., Poli D., Pellacani C., Giordano G., Mutti A., Costa L. G. (2010). Low concentrations of the brominated flame retardants BDE-47 and BDE-99 induce synergistic oxidative stress-mediated neurotoxicity in human neuroblastoma cells. Toxicol. In Vitro 24, 116–122. [DOI] [PubMed] [Google Scholar]
- Theunissen P. T., Pennings J. L., van Dartel D. A., Robinson J. F., Kleinjans J. C., Piersma A. H. (2013). Complementary detection of embryotoxic properties of substances in the neural and cardiac embryonic stem cell tests. Toxicol. Sci. 132, 118–130. [DOI] [PubMed] [Google Scholar]
- Viberg H., Fredriksson A., Eriksson P. (2003). Neonatal exposure to polybrominated diphenyl ether (PBDE 153) disrupts spontaneous behaviour, impairs learning and memory, and decreases hippocampal cholinergic receptors in adult mice. Toxicol. Appl. Pharmacol. 192, 95–106. [DOI] [PubMed] [Google Scholar]
- Vieira H. L. A., Alves P. M., Vercelli A. (2011). Modulation of neuronal stem cell differentiation by hypoxia and reactive oxygen species. Prog. Neurobiol. 93, 444–455. [DOI] [PubMed] [Google Scholar]
- Watson R. E., DeSesso J. M., Hurtt M. E., Cappon G. D. (2006). Postnatal growth and morphological development of the brain: A species comparison. Birth Defects Res B Dev. Reprod. Toxicol .77, 471–484. [DOI] [PubMed] [Google Scholar]
- Wegiel J., Kuchna I., Nowicki K., Imaki H., Wegiel J., Marchi E., Ma S. Y., Chauhan A., Chauhan V., Bobrowicz T. W., et al. (2010). The neuropathology of autism: Defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 119, 755–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn V. D., Haimov-Kochman R., Paquet A. C., Yang Y. J., Madhusudhan M. S., Gormley M., Feng K. T., Bernlohr D. A., McDonagh S., Pereira L., et al. (2007). Gene expression profiling of the human maternal-fetal interface reveals dramatic changes between midgestation and term. Endocrinology 148, 1059–1079. [DOI] [PubMed] [Google Scholar]
- Woodruff T. J., Zota A. R., Schwartz J. M. (2011). Environmental chemicals in pregnant women in the United States: NHANES 2003-2004. Environ. Health Perspect. 119, 878–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods R., Vallero R. O., Golub M. S., Suarez J. K., Ta T. A., Yasui D. H., Chi L.-H., Kostyniak P. J., Pessah I. N., Berman R. F., et al. (2012). Long-lived epigenetic interactions between perinatal PBDE exposure and Mecp2308 mutation. Hum. Mol. Genet. 21, 2399–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Coskun V., Tao J. F., Xie W., Ge W. H., Yoshikawa K., Li E., Zhang Y., Sun Y. E. (2010). Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science 329, 444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zota A. R., Mitro S. D., Robinson J. F., Hamilton E. G., Park J. S., Parry E., Zoeller R. T., Woodruff T. J. (2018). Polybrominated diphenyl ethers (PBDES) and hydroxylated PBDE metabolites (OH-PBDES) in maternal and fetal tissues, and associations with fetal cytochrome P450 gene expression. Environ. Int. 112, 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.