

Abstract
The effects of pituitary adenylate cyclase activating polypeptide (PACAP) on human lung cancer cell line NCI-1299 mitogen activated protein kinase (MAPK) tyrosine phosphorylation and vascular endothelial cell growth factor (VEGF) expression were investigated. PACAP-27 (100 nM) increased MAPK tyrosine phosphorylation 3-fold, 5 min after addition to NCI-H1299 cells. PACAP caused tyrosine phosphorylation in a concentration-dependent manner being half-maximal at 10 nM PACAP-27. PACAP-27 or PACAP-38 (100 nM) but not PACAP28–38 or VIP caused increased MAPK tyrosine phosphorylation using NCI-H1299 cells. Also, the increase in MAPK tyrosine phosphorylation caused by PACAP-27 was totally inhibited by 10 µM PACAP(6–38), a PAC1 receptor antagonist or 10 µM PD98059, a MAPKK inhibitor. These results suggest that PAC1 receptors regulate tyrosine phosphorylation of MAPK in a MAPKK-dependent manner. PACAP-27 (100 nM) caused increased VEGF mRNA in NCI-H1299 cells after 8 h. The increase in VEGF mRNA caused by PACAP-27 was partially inhibited by PACAP(6–38), PD98059 and H-89. Addition of VIP to NCI-H1299 cells caused increased VEGF mRNA, which was totally inhibited by H89, a PKA inhibitor. These results suggest that PAC1 and VPAC1 receptors regulate VEGF expression in lung cancer cells.
Keywords: PACAP, Lung cancer, Mitogen activated protein kinase, VEGF
1. Introduction
Pituitary adenylate cyclase activating polypeptide (PACAP) is a member of the vasoactive intestinal peptide (VIP) family of peptides [1]. Two forms of PACAP have been isolated, PACAP-27, which has 70% homology with VIP, and PACAP-38, which contains PACAP-27 plus a C-terminal extension of 11 amino acids [2,3]. Both PACAP-27 and PACAP-38 bind with high affinity to human lung cancer cells [4] and stimulate their growth. In addition, PACAP stimulates the proliferation of lovocolorectal and rat cerebellar cells [5,6] and is the most potent stimulant known to cause growth of gastric enterochromaffin cells [7].
PACAP and VIP elevate cAMP [8]. Both VPAC1 and PAC1 receptors interact with a stimulatory guanine nucleotide binding subunit (Gas) activating adenylate cyclase. In turn, the increase in intracellular cAMP may activate protein kinase (PK) A; H89 is a PKA enzyme inhibitor [9]. In contrast, PAC1 but not VPAC1 receptors interact with a guanine nucleotide binding subunit (Gαq/11) causing phosphatidylinositol (PI) turnover. The inositol-1,4,5-trisphosphate released may cause release of Ca2 + from the endoplasmic reticulum into the cytosol. PACAP-27 but not VIP elevates cytosolic Ca2 + using lung cancer cells [10]. The diacylglycerol released may activate PKC, which in turn phosphorylates substrates such as MAPKK which in turn phosphorylates MAPK; MAPKK is inhibited by PD98059 [11]. Here the effects of PACAP on MAPK tyrosine phosphorylation were investigated using lung cancer cells and NIH/3T3 cells transfected with PAC1 receptors. These results indicate that PAC1 receptors regulate MAPK activity as a result of PI turnover.
Phosphorylated MAPK may enter the nucleus and alter nuclear oncogene expression [12]. PACAP-27 transiently increases c-fos mRNA in lung cancer cells [13]. The effects of PACAP are reversed by the PAC1 receptor antagonist, PACAP(6–38) [14]. Also, PACAP-27 transiently increases c-jun mRNAs in lung cancer cells. After translation, the c-fos and c-jun proteins may form heterodimers and increase expression of growth factor genes. Recently, we showed that VIP and PGE2, which elevate intracellular cAMP in lung cancer cells, increased vascular endothelial cell growth factor (VEGF) mRNAs in lung cancer cell line NCI-H157 [15]. Here the effects of PACAP-27 were investigated on VEGF mRNA using lung cancer cell line NCI-H1299. Our results indicate that PACAP-27 increases MAPK tyrosine phosphorylation in a MAPKK-dependent manner. Also, PACAP-27 increased VEGF mRNAs and the increase in VEGF mRNAs was partially inhibited by PACAP(6–38), H89 and PD98059. In contrast, VIP increased VEGF was inhibited totally by H89 but not PACAP(6–38) or PD98059. These results indicate that both PAC1 and VPAC1 receptors regulate VEGF mRNA in lung cancer cells.
2. Methods
2.1. Cell culture
NCI-H1299 and H157 cells were cultured in RPMI-1640 containing 10% heat-inactivated fetal bovine serum (FBS, Life Technologies) [16]. The cells were split weekly with trypsin-EDTA (Life Technologies). The cells were mycoplasma-free and were used when they were in exponential growth phase after incubation at 37 °C in 5% CO2/95% air. NIH/3T3 cells were cultured in DMEM containing 10% calf serum. The cells were transfected with PAC1 receptor splice variants (SVs) using the pCKL-SRα/NEO expression vector at the EcoR1 restriction site. Following linearization of the vector with restriction enzyme AatII, 20 µg of recombinant plasmid was transfected into NIH/3T3 cells by electroporation (2 × 107 cells, 500 mFd, 025 kV). The cells were cultured in DMEM containing 10% calf serum and 250 µg/ml geneticin [17].
2.2. MAPK tyrosine phosphorylation
The ability of PACAP-like peptides to phosphorylate MAPK was investigated using Western blot techniques [18]. NCI-H1299 or NIH/3T3 cells were cultured in 15 cm dishes. When a monolayer of cells formed, they were placed in SIT media containing 0.5% FBS overnight. Three hours before treatment, cells were placed in fresh ST media. Cells were treated with 100 nM PACAP-27 for 5 min in the presence or absence of inhibitors. The cells were washed twice with PBS and lysed in buffer containing 50 mM Tris/HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, 1% deoxycholate, 1% NaN3, 1 mM EGTA, 0.4 mM EDTA, 1.5 µ/ml aprotinin, 1.5 µg/ml leupeptin, 1 mM PMSF and 0.2 mM Na2VO4 (Sigma) and sonicated for 5 s at 4 °C. The resulting lysate was centrifuged at 10000 × g for 15 min. Extract protein concentration was measured using Bio-Rad protein assay reagent (Pierce Chemical), and 150 µg/ml of protein was incubated with 4 µg of anti-MAPK (Upstate Biotechnologies) antibody, 4 µg of goat anti-rabbit IgG and 30 µl of protein A-agarose (Sigma) overnight at 4 °C. The immunoprecipitates were washed three times with PBS and analyzed by SDS/PAGE and Western blotting. Immunoprecipitates were fractionated using 10% polyacrylamide gels. The resulting nitrocellulose membranes were blocked overnight at 4 °C using blotto (5% nonfat dried milk in solution containing 50 mM Tris/HCl (pH 8.0), 2 mM CaCl2, 80 mM NaCl, 0.05% Tween 20 and 0.02% NaN3 (Sigma). Then the nitrocellulose membranes were treated with anti-phospho MAP kinase Ab (Upstate Biotechnologies) and incubated for 2 h at 25 °C with anti-mouse IgG-horseradish peroxidase conjugate. The membrane was washed for 10 min with blotto and twice for 10 min with washing solution (50 mM Tris/HCl (pH 8.0), 2 mM CaCl2, 80 mM NaCl, 0.05% Tween 20 and 0.02% NaN3; Sigma). The blot was incubated with enhanced chemiluminescence detection reagent for 5 min and exposed to Hyperfilm ECL (Amersham). The intensity of the bands was determined using a densitometer.
2.3. VEGF mRNA
The ability of PACAP-like peptides to stimulate VEGF expression was investigated. For the VEGF experiments, NCI-H1299 cells were cultured with SIT medium containing 0.5% fetal bovine serum. After 4 h, the cells were treated with 100 nM PACAP-27 for 8 h in the presence or absence of inhibitors. Total RNA was isolated using guanidinium isothiocyanate (Fluka). Ten micrograms of denatured RNA was separated in a 0.66 M formaldehyde 1% agarose gel. The RNA was fractionated using Northern blot techniques. The gel was treated with ethidium bromide to assess RNA integrity. The RNA was blotted onto a nytran membrane overnight and the membrane hybridized with DNA probes labeled with 32P-dCTP using a Bethesda Research Laboratories random priming kit. The membrane was apposed to Kodak XAR-2 film at −80 °C for 1 day and the autoradiogram developed. The autoradiograms were analyzed using a Molecular Dynamics densitometer.
3. Results
3.1. PACAP-like peptides cause MAPK tyrosine phosphorylation
The ability of PACAP-27 to cause MAPK tyrosine phosphorylation was investigated by Western blot. Fig. 1 shows that 1 nM PACAP-27 weakly, but 10 to 100 nM PACAP-27 strongly caused MAPK tyrosine phosphorylation after addition to NCI-H1299 cells. MAPK tyrosine phosphorylation was maximal (2.8-fold increase) 5 min after addition of PACAP-27 to NCI-H1299 cells (data not shown). These results indicate that PACAP-27 causes MAPK tyrosine phosphorylation in a time- and concentration-dependent manner after addition to NCI-H1299 cells. Similar results were observed using NCI-H157 cells (data not shown).
Fig. 1.
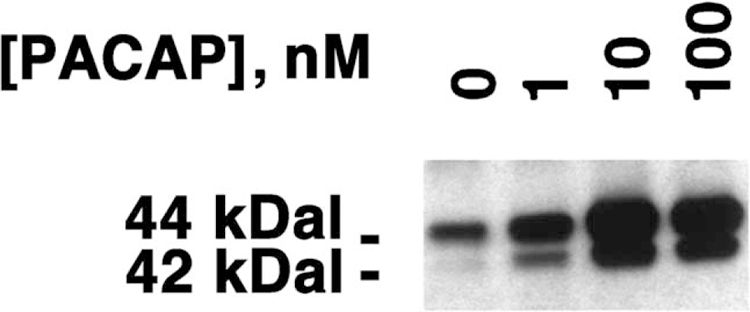
NCI-H1299 MAPK tyrosine phosphorylation. PACAP-27 at varying concentrations was added to NCI-H1299 cells, and after 5 min, the MAPK tyrosine phosphorylation was determined by Western blot. This experiment is representative of three others.
The specificity of MAPK tyrosine phosphorylation was investigated. Table 1 shows that 100 nM PACAP-27 or PACAP-38 strongly increased tyrosine phosphorylation of MAPK 2.8-fold relative to the control 5 min after addition to NCI-H1299 cells. In contrast, the inactive PACAP fragment PACAP(28–38) had little effect on MAPK tyrosine phosphorylation as did PACAP(6–38), a receptor antagonist, or VIP, a VPAC1 and VPAC2 receptor agonist. Table 1 shows that 10 µM PACAP(6–38) inhibited the increase in tyrosine phosphorylation of MAPK caused by 100 nM PACAP-27. These results indicate that PACAP(6–38) is a PAC1 receptor antagonist using NCI-H1299 cells.
Table 1.
MAPK tyrosine phosphorylation in NCI-H1299 cells
| Addition | Percentage MAPK tyrosine phosphorylation |
|---|---|
| None | 100±6 |
| PACAP-27, 100 nM | 290±26 |
| PACAP-38, 100 nM | 275±38 |
| PACAP(28 – 38), 100 nM | 110±12 |
| PACAP(6– 38), 10 µM | 98±9 |
| PACAP(6– 38) + PACAP-27 | 108±11 |
| VIP, 100 nM | 106±8 |
The mean value ± S.D. of four determinations are indicated.
PACAP-27 increased the tyrosine phosphorylation of the 44 and 42-kDa bands of MAPK 5 min after addition to NCI-H1299 cells. The relative density of the tyrosine phosphorylation of the 44 relative to the 42 kDa band was, however, 2:1. Because MAPK is phosphorylated by MAPKK, the effects of PD98059, an MAPKK inhibitor, were investigated. Fig. 2 shows that PD98059 (1 µM) had little effect on MAPK tyrosine phosphorylation; however, 10 µM PD98059 strongly inhibited the ability of 100 nM PACAP-27 to cause tyrosine phosphorylation of MAPK. These results indicate that PD98059 inhibits in a concentration-dependent manner the ability of PACAP-27 to cause tyrosine phosphorylation of MAPK using NCI-H1299 cells.
Fig. 2.
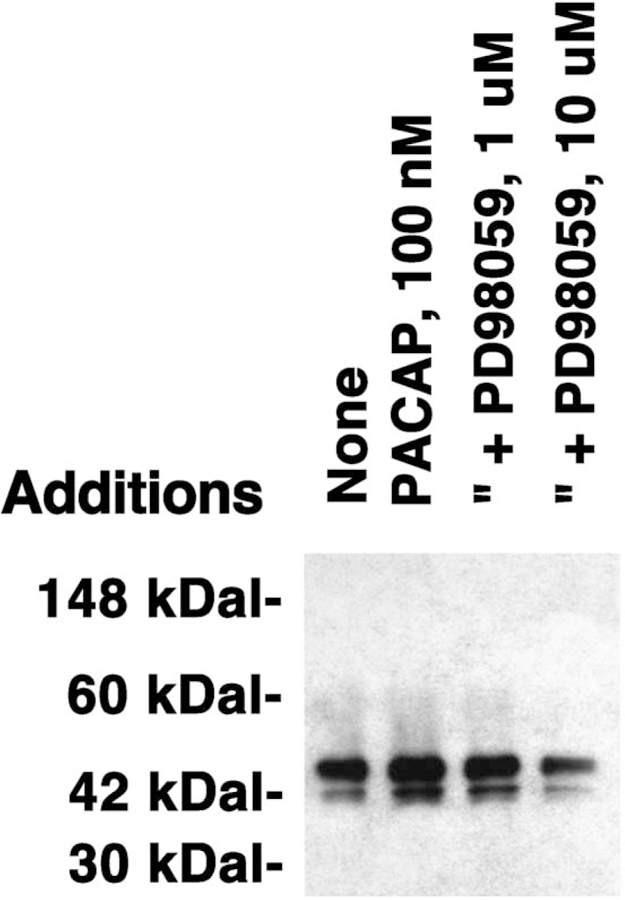
MAPK tyrosine phosphorylation and PD98059. The MAPK tyrosine phosphorylation was determined 5 min after no additions, and the addition of 100 nM PACAP-27, 100 nM PACAP-27 plus 1 µM PD98059 and 100 nM PACAP-27 plus 10 µM PD98059. This experiment is representative of two others.
The ability of PAC1 receptor splice variants to tyrosine phosphorylate MAPK was investigated. Table 2 shows that using NIH/3T3 cells transfected with the PAC1 hop receptor SV, 100 nM PACAP-27 increased MAPK tyrosine phosphorylation 5-fold. Using NIH/3T3 cells transfected with the PAC1 basic, hip or hip-hop receptor SV, PACAP stimulated MAPK 3-, 2.5- and 2-fold, respectively. There was little tyrosine phosphorylation of MAPK in NIH/3T3 cells containing the vector control. These results indicate that the PAC1 hop receptor SV strongly increases MAPK tyrosine phosphorylation.
Table 2.
Tyrosine phosphorylation of NIH/3T3 MAPK
| PAC1 receptor SV | Percentage MAPK tyrosine phosphorylation |
|---|---|
| Basic | 260±35 |
| None | 108±6 |
| Hip | 320±30 |
| Hop | 510±60 |
| Hip-hop | 210±25 |
The percentage MAPK tyrosine phosphorylation caused by 100 nM PACAP-27 is indicated. The mean value ± S.D. of four determinations are indicated.
3.2. PACAP-like peptides increase VEGF mRNA
The effects of PACAP-like peptides on VEGF mRNA were investigated by Northern blot. Fig. 3 shows that PACAP-27 and VIP increased VEGF mRNA 8 h after addition to NCI-H1299 cells. Fig. 4 shows that the effects of 100 nM PACAP-27 are time-dependent with some stimulation occurring after a 4-h incubation with NCI-H1299 cells and maximal stimulation occurring after 8 or 16 h. After a 24 h incubation of PACAP-27 with NCI-H1299 cells, the amount of VEGF mRNA started to decline. These results indicate that PACAP-27 transiently increases VEGF mRNA after addition to NCI-H1299 cells.
Fig. 3.
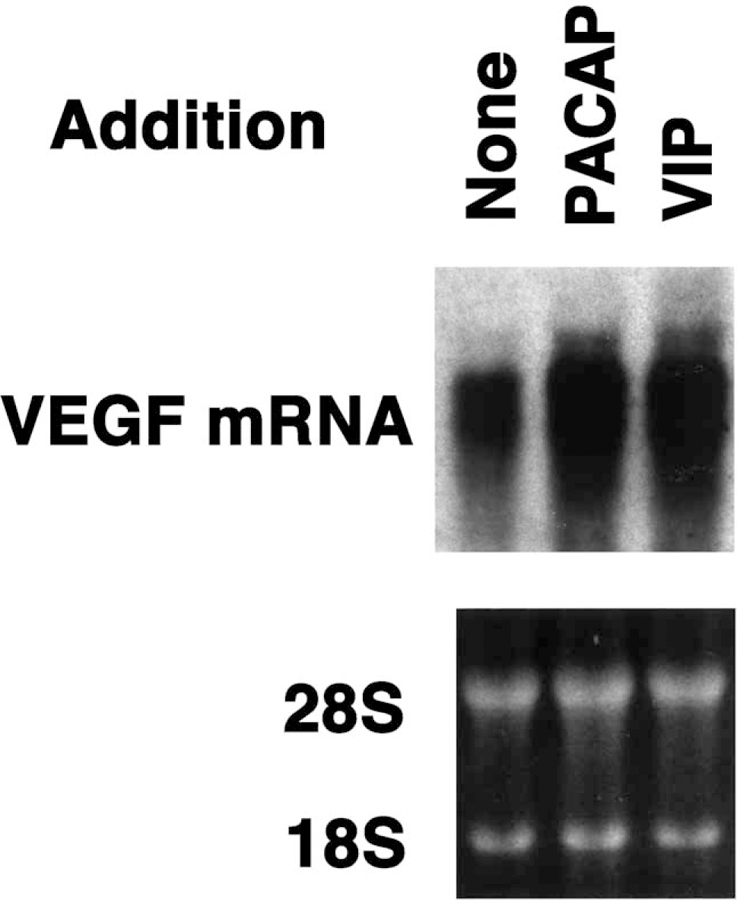
VEGF mRNA. (Top) VEGF mRNA was determined by Northern blot 8 h after no additions, and the addition of 100 nM PACAP-27 and 100 nM VIP to NCI-H1299 cells. (Bottom) Equal amounts of RNA were loaded onto the gels based on ethidium bromide staining of the 28S and 18S rRNA. This experiment is representative of three others.
Fig. 4.
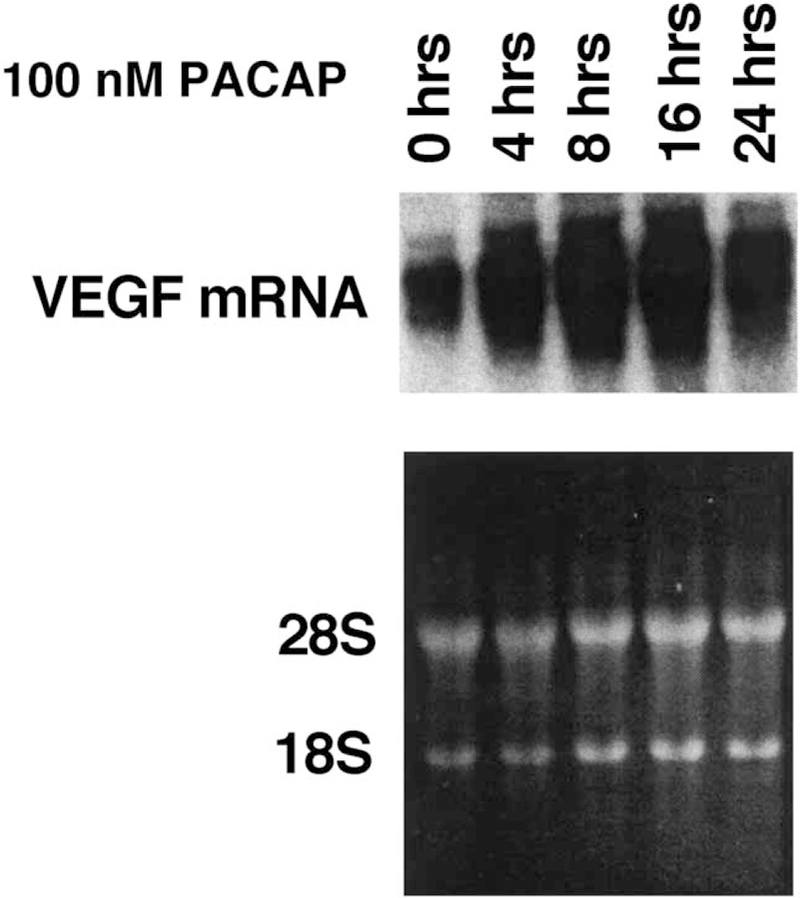
PACAP-27 and VEGF mRNA. (Top) VEGF mRNA was determined by Northern blot as a function of time after the addition of 100 nM PACAP-27 to NCI-H1299 cells. (Bottom) Equal amounts of RNA were loaded onto the gels based on ethidium bromide staining of the 28S and 18S rRNA. This experiment is representative of two others.
Table 3 shows that the increase in NCI-H1299 VEGF mRNA caused addition of PACAP-27 (1.9-fold) was partially inhibited by PACAP(6–38), PD98059 and H89. In contrast, the increase in NCI-H1299 VEGF mRNA caused by addition of VIP (1.5-fold) was not inhibited by PACAP(6–38) or PD98059, but H89 inhibited totally the increase in VEGF mRNA caused by VIP. These results indicate that the mechanisms by which VIP and PACAP-27 increase VEGF mRNAs are different.
Table 3.
VEGF mRNA
| Addition | Percentage VEGF mRNA |
|---|---|
| None | 100±8 |
| PACAP-27, 100 nM | 194±13 ** |
| VIP, 100 nM | 152±11 * |
| PACAP-27 + PACAP(6–38) | 135±14 * |
| VIP + PACAP(6–38) | 157±15 * |
| PACAP+ 50 µM H89 | 144±15 * |
| VIP + H89 | 112±9 |
| PACAP-27 + 10 µM PD98059 | 141±12 * |
| VIP + PD98059 | 144±9 * |
Agents were added to NCI-H1299 cells at the indicated concentrations and the RNA extracted after 8 h. The relative percentage VEGF mRNA (mean ± S.D. of four determinations) is indicated; using Student’s t-test.
p < 0.05.
p < 0.01.
4. Discussion
Previously, we found that PACAP-27 bound with high affinity to lung cancer cells, causing elevated cAMP and phosphatidylinositol turnover [4]. Also, PACAP-27 increased c-fos and c-jun mRNAs [13] and increased the clonal growth of lung cancer cells [4]. In contrast, VIP bound with high affinity causing elevated cAMP but had no effect on PI turnover [8]. Here the mechanisms by which PACAP-27 altered MAPK tyrosine phosphorylation and VEGF mRNAs were investigated.
PACAP-27, but not VIP, caused MAPK tyrosine phosphorylation in a time- and concentration-dependent manner using NCI-H157 or H1299 cells. PACAP-27 (10 nM) half maximally increased MAPK tyrosine phosphorylation. MAPK tyrosine phosphorylation caused by 100 nM PACAP-27 was totally inhibited by PACAP(6–38), a PAC1 receptor antagonist, or PD98059, a MAPKK inhibitor. Similarly in primary rat brain cerebellar cultures, nanomolar concentrations of PACAP-27 or PACAP-38 but not VIP, caused tyrosine phosphorylation of MAPK [18]. Also, PACAP enhanced survival of the cerebellar cultures exposed to HK medium which lacked serum for 48 h. In the HK medium, approximately 80% of the cells underwent apoptosis; however, if PACAP-27 or PACAP-38 was added to the medium, only 40% of the cells died [18]. These results indicate that PAC1 receptor activation results in increased survival of cerebellar cells. In lung cancer cells, PACAP-27 stimulated, whereas PACAP(6–38) inhibited clonal proliferation [4].
The PAC1 receptor contains seven transmembrane domains and four SVs [19,20]. The null or “basic” receptor has 467 amino acids whereas SV-1 or the “hip” receptor has a 28 amino acid insert in the third cytosolic domain. The 495 amino acid SV-2 or the “hop” PAC1 receptor has a different 28 amino acid insert in the third cytosolic domain. The 523 amino acid SV-3 or “hip-hop” receptor has both 28 amino acid inserts into the third cytosolic domain. All of the PAC1 receptor SVs strongly elevate cAMP [18]. In contrast, the PAC1 receptor SVs have altered ability to cause PI turnover and increase c-fos mRNA with the order of PAC1 receptor SV efficacy being hop>basic = hip-hop>hip. Similarly, the ability of PACAP-27 to cause MAPK tyrosine phosphorylation is greatest if NIH/3T3 cells contain the PAC1 hop receptor. Preliminary data (T. Moody, unpublished) indicate that lung cancer cells have predominantly PAC1 receptor hop mRNA. Also, cerebellar cells have PAC1-hop receptor mRNA [18].
MAPKK may be a key upstream enzyme in the MAPK tyrosine phosphorylation process. The ability of PACAP-27 to cause MAPK tyrosine phosphorylation in lung cancer cells is totally inhibited by PD98059. It remains to be determined if ras and/or raf are additional upstream regulators. Because VIP has no effect on MAPK tyrosine phosphorylation, PKA activation does not contribute to MAPK tyrosine phosphorylation in human lung cancer cells. In cerebellar cells, however, MAPK tyrosine phosphorylation was inhibited by PD98059 and H89 [18]. Similarly, using GH4C1 pituitary cells, PD98059, H89 and GF109203X, a PKC inhibitor, inhibited MAPK tyrosine phosphorylation caused by PACAP-27 [21]. Preliminary data (T. Moody, unpublished) indicate that H7, which inhibits PKC, inhibits the ability of PACAP-27 to cause tyrosine phosphorylation of MAPK in lung cancer cells. Phosphorylated MAP kinase may phosphorylate sis-inducing factor (SIS) causing nuclear translocation and interaction with the sis-inducing element (SIE) resulting in increased nuclear oncogene expression [12]. Subsequently c-fos and c-jun may form heterodimers and activated AP-1 sites in growth factor genes [22].
Previously, we found that VIP caused increased VEGF mRNA [15]. The increase in VEGF mRNA caused by addition of 100 nM VIP to NCI-H1299 cells was totally inhibited by H89 but not PD98059 or PACAP(6–38). Thus, VPAC1 receptors may regulate the increase in VEGF mRNA in a PKA-dependent manner. H89 partially inhibited the increase in VEGF mRNA caused by addition of 100 nM PACAP-27 to NCI-H1299 cells. The increase in VEGF mRNA caused by PACAP-27 was partially inhibited by PACAP(6–38) and PD98059. These results suggest that PAC1 receptors may regulate the increase in VEGF mRNA induced by PACAP-27 in a PKC-dependent manner. Also, VPAC1 receptors may regulate the increase in VEGF mRNA caused by PACAP-27 in a PKA-dependent manner. Preliminary data (T. Moody, unpublished) indicate that the increase in VEGF mRNA caused by addition of PACAP-27 to NCI-H1299 cells was totally inhibited by addition of PD98059 and H89.
Previously, we showed that VEGF mRNA for the 121, 165 and 189 but not 205 amino acid forms were present in lung cancer cells [15]. VIP increased the immunoreactive VEGF secretion from lung cancer cells [15]. Preliminary data (T. Moody, unpublished) indicate that PACAP significantly increased immunoreactive VEGF secretion from lung cancer cells. The secreted VEGF may diffuse to endothelial cells and activate KDR/Flk-1 receptors facilitating the proliferation of endothelial cells. Angiogenesis is an essential step in both the growth of primary cancer tumors and metastasis of the primary tumors to secondary sites [23,24].
As tumors grow, they co-opt existing blood vessels for their nutrient blood supply [25]. VEGF may develop new vasculature within tumors where endothelial cells migrate, proliferate and invade the basement membrane. Shortly thereafter, the existing vasculature becomes destabilized through release of Ang-2 by endothelial cells. This loss of vascular integrity can lead to hypoxia, which induces hypoxia-inducible factor (HIF)-1, leading to VEGF up-regulation [26]. It remains to be determined if PACAP-27 causes PI-3 kinase/Akt activation leading to stabilization of HIF-1, and increased MAPK tyrosine phosphorylation leading to VEGF gene expression.
While there are numerous factors that stimulate angiogenesis such as VEGF, b-fibroblast growth factor, and hepatocyte growth factor/scatter factor, VEGF appears to be of primary importance. Anti-VEGF monoclonal antibodies (mAbs) and the KDR/flk-1 receptor tyrosine kinase inhibitor decreased hepatic tumor burden, vessel count and tumor proliferative index in mouse models of colon cancer [27,28]. Preliminary data (T. Moody, unpublished) indicate that VIPhyb, which inhibits xenograft proliferation in nude mice, causes smaller and reduced numbers of blood vessels in tumors.
In summary, PACAP-27 stimulates MAPK tyrosine phosphorylation and VEGF mRNA in lung cancer cells lines. In remains to be determined if PACAP stimulates angiogenesis of lung cancer tumors.
Acknowledgements
The authors thank Drs. D. Brenneman, I. Gozes and S. Wank for helpful discussions.
Abbreviations:
- PACAP
pituitary adenylate cyclase activating polypeptide
- VIP
vasoactive intestinal peptide
- MAPK
mitogen activated protein kinase
- VEGF
vascular endothelial cell growth factor
- PI
phosphatidylinositol
- PK
protein kinase
References
- [1].Said SI, Mutt V. Polypeptide with broad biological activity: isolation from the small intestine. Science 1970;69:1217–8. [DOI] [PubMed] [Google Scholar]
- [2].Miyata A, Arimura A, Dahl RR, Uehara A, Jiang L, Culler MD, et al. Isolation of a novel 38 residue-hypothalamic polypeptide which stimulates adenylate cyclase in pituitary cells. Biochem Biophys Res Commun 1989;164:567–74. [DOI] [PubMed] [Google Scholar]
- [3].Miyata A, Jiang L, Dahl RR, Kitada C, Kubo K, Fujino M, et al. Isolation of a neuropeptide corresponding to the N-terminal 27 residues of the pituitary adenylate cyclase activating polypeptide with 38 residues (PACAP38). Biochem Biophys Res Commun 1990;170: 643–8. [DOI] [PubMed] [Google Scholar]
- [4].Moody TW, Zia F, Bitar K, Coy DH. PACAP(6–38) is a PACAP type I receptor antagonist on small cell lung cancer cells. In: Rosselin G, editor. International symposium on vasoactive intestinal peptide, pituitary adenylate cyclase activating polypeptide and related regulatory peptides Singapore: World Scientific; 1994. p. 537–44. [Google Scholar]
- [5].Chastre E, DiGioia Y, Djelloul S, Empereur S, Louvet C, Couldray AM, et al. Pharmacological characterization of VIP/PACAP Type II receptors in normal and immortalized rat fetal intestinal epithelial cells. In: Rosselin G, editor. International symposium on vasoactive intestinal peptide, pituitary adenylate cyclase activating polypeptide and related regulatory peptides Singapore: World Scientific; 1994. p. 568–74. [Google Scholar]
- [6].DiCicco-Bloom E, Lu N. VIP/PACAP regulate peripheral and central neurogenesis and survival. In: Rosselin G, editor. International symposium on vasoactive intestinal peptide, pituitary adenylate cyclase activating polypeptide and related regulatory peptides Singapore: World Scientific; 1994. p. 593–4. [Google Scholar]
- [7].Lauffer JM, Tang LH, Zhang T, Hinoue T, Rabbar S, Odo M, et al. PACAP mediates the neural proliferation pathway of Mastomys Enterochromaffin-like cell transformation. Regulatory Pept 2001;102: 157–64. [DOI] [PubMed] [Google Scholar]
- [8].Lee M, Jensen RT, Huang SC, Bepler G, Korman L, Moody TW. Vasoactive intestinal polypeptide binds with high affinity to non-small cell lung cancer cells and elevates cAMP levels. Peptides 1990;11: 1205–9. [DOI] [PubMed] [Google Scholar]
- [9].Hidaka H, Kobayashi R. Pharmacology of protein kinase inhibitors. Annu Rev Pharmacol Toxicol 1992;32:377–97. [DOI] [PubMed] [Google Scholar]
- [10].Moody TW, Zia F, Makheja A. PACAP elevates cytosolic calcium in small cell lung cancer cell lines. Peptides 1993;14:241–6. [DOI] [PubMed] [Google Scholar]
- [11].Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD98059 is a specific inhibitor of the activation of mitogen-activated protein kinase in vitro and in vivo. J Biol Chem 1995;270:27489–94. [DOI] [PubMed] [Google Scholar]
- [12].Whitmarsh AJ, Davies RJ. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J Mol Med 1996;74:589–607. [DOI] [PubMed] [Google Scholar]
- [13].Draoui M, Hida T, Jakowlew S, Birrer M, Zia F, Moody TW. PACAP stimulates c-fos mRNAs in small cell lung cancer cells. Life Sci 1996; 59:307–13. [DOI] [PubMed] [Google Scholar]
- [14].Leyton J, Coelho T, Hida T, Jakowlew S, Birrer M, Fridkin M, et al. PACAP hybrid antagonizes PACAP receptor splice variants. Life Sci 1997;61:631–9. [DOI] [PubMed] [Google Scholar]
- [15].Casibang M, Purdom S, Zia F, Jakowlew S, Neckers L, Ben-Av P, et al. Prostaglandin E2 and VIP increase VEGF mRNAs in lung cancer cells. Lung Cancer 2001;31:203–12. [DOI] [PubMed] [Google Scholar]
- [16].Carney DN, Gazdar AF, Bepler G, Guccion JG, Marangos PJ, Moody TW, et al. Establishment and identification of small cell lung cancer cell lines having classic and variant features. Cancer Res 1995;45: 2913–23. [PubMed] [Google Scholar]
- [17].Pisegna JR, Wand SA. Cloning and characterization of the signal transduction of four splice variants of the human pituitary adenylate cyclase activating polypeptide receptor. Evidence of dual coupling to adenylate cyclase and phospholipase C. J Biol Chem 1996;271: 17267–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Villalba M, Bockaert J, Journot L. Pituitary adenylate cyclase-activating polypeptide (PACAP-38) protects cerebellar granule neurons from apoptosis by activating the mitogen-activated protein kinase (MAP kinase) pathway. J Neurosci 1997;170:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pisegna JR, Wank SA. Molecular cloning and functional expression of the pituitary adenylate cyclase activating polypeptide type 1 receptor. Proc Natl Acad Sci U S A 1993;90:6345–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Spengler D, Waeber C, Pantaloni C, Holsboer F, Bockaert J, Seeburg PH, et al. Differential signal transduction by five splice variants of the PACAP receptor. Nature 1993;365:170–5. [DOI] [PubMed] [Google Scholar]
- [21].LePechon-Vallee C, Magalon K, Rasolonjanaary R, Enjalbert A, Gerard C. Vasoactive intestinal polypeptide and pituitary adenylate cyclase-activating polypeptides stimulate mitogen-activated protein kinase in the pituitary cell line GH4C1 by a 3′,5′-cyclic adenosine monophosphate pathway. Endocrinology 2000;72:46–56. [DOI] [PubMed] [Google Scholar]
- [22].Moody T, Walters J, Casibang M, Zi F, Gozes Y. VPAC1 receptors and lung cancer. Ann N Y Acad Sci 2000;921:26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Folkman J What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst 1989;82:4–6. [DOI] [PubMed] [Google Scholar]
- [24].Fidler IJ, Ellis LM. The implications of angiogenesis to the biology and therapy of cancer metastasis. Cell 1994;79:185–8. [DOI] [PubMed] [Google Scholar]
- [25].Holash J, Maisonpierre PC, Compton D. Vessel cooption, regression and growth in tumors mediated by angiopoietins and VEGF. Science 1999;284:1994–8. [DOI] [PubMed] [Google Scholar]
- [26].Ellis LM, Liu WB, Fan F, Reinmuth N, Shaheen RM, Jung YD, et al. Role of angiogenesis inhibitors in cancer treatment. Oncology 2001; 15:39–46. [PubMed] [Google Scholar]
- [27].Warren RS, Yuan H, Matli MR. Regulation by vascular endothelial growth factor of human colon cancer tumorigenesis in a mouse model of experimental liver metastasis. J Clin Invest 1995;95:1789–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shaheen RM, Davis DW, Liu W. Antiangiogenic therapy targeting the tyrosine kinase receptor for vascular endothelial growth factor receptor inhibits the growth of colon cancer liver metastasis and induces tumor and endothelial cell apoptosis. Cancer Res 1999;59:5412–6. [PubMed] [Google Scholar]