

SUMMARY
The G protein-coupled receptor (GPCR) CXCR4 is a co-receptor for HIV and is involved in cancers and autoimmune diseases. We characterized five purine or quinazoline core polyamine pharmacophores used for targeting CXCR4 dysregulation in diseases. All were neutral antagonists for wild-type (WT) CXCR4 and two were biased antagonists with effects on β-arrestin-2 only at high concentrations. These compounds displayed various activities for a constitutively active mutant (CAM). We use the IT1t-CXCR4 crystal structure and molecular dynamics (MD) simulations to develop two hypotheses for the activation of the N1193.35A CAM. The N1193.35A mutation facilitates increased coupling of TM helices III and VI. IT1t deactivates the CAM by disrupting the coupling between TM helices III and VI, mediated primarily by residue F872.53. Mutants of F872.53 in N1193.35A CXCR4 precluded constitutive signaling and prevented inverse agonism. This work characterizes CXCR4 ligands and provides a mechanism for N1193.35A constitutive activation.
Keywords: G protein-coupled receptor (GPCR), CXCR4, CXCL12, constitutively active mutant (CAM), molecular dynamics (MD)
Graphical Abstract
eTOC blurb:
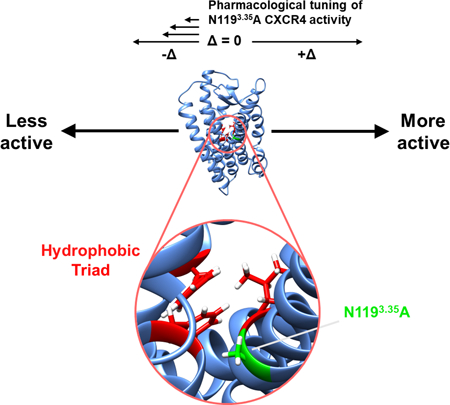
In this work, Rosenberg and co-authors characterize several potent small molecule CXCR4 antagonists. The authors discuss two different possible mechanisms for why the N1193.35A CXCR4 mutant exhibits constitutive signaling, providing biochemical evidence supporting the role of a “hydrophobic triad” observed only in the mutant.
INTRODUCTION
G protein-coupled receptors (GPCRs) are dynamic membrane proteins with multiple conformational states that give rise to orthosteric and allosteric binding for ligands, G proteins, and arrestins (Zhang et al., 2015). Historically, agonists and antagonists were thought to bind only to the orthosteric site but recent studies indicate allosteric ligands and their effects are a common occurrence for GPCRs (DeVree et al., 2016). Brought about by the intrinsic dynamic nature of GPCRs as well as various extracellular or intracellular binding molecules, distinct conformations of GPCRs result in differences in ligand binding kinetics, affinity, potency, protein dynamics, and biased signaling. Mutagenesis of GPCRs can also alter the basal signaling activity of receptors relative to the wild-type (WT) protein (Seifert and Wenzel-Seifert, 2002).
CXCR4 is a member of the class A GPCR family. It is essential for embryonic development, adult homeostasis, and chemotactic recruitment of B- and T-cells in response to a concentration gradient of its chemokine agonist CXCL12 (Chong and Mohan, 2009; Zou et al., 1998). CXCR4 also functions as a co-receptor for HIV (Feng et al., 1996). Mutants at the C-terminal region of the CXCR4 lead to Warts, Hypogammaglobulinemia, Immunodeficiency, and Myelokathexis (WHIM) syndrome through a gain of function mechanism (Hernandez et al., 2003). CXCR4 is involved in rheumatoid arthritis (Tamamura and Fujii, 2005) and more than 23 types of cancer (Truax et al., 2013). The small molecule antagonist AMD3100 was the first CXCR4 antagonist to enter clinical trials for HIV treatment (De Clercq, 2009), but was withdrawn within 30 days due to cardiotoxicity (Hendrix et al., 2004). However, AMD3100 received FDA approval for CD34+ hematopoietic stem cell mobilization necessary for autologous bone marrow transplants in non-Hodgkin lymphoma and multiple myeloma (Sanchez-Ortega et al., 2015). Cardiotoxicity in this clinical setting does not develop due to the lower doses and shorter timeframe necessary for this indication. Since CXCR4 is a therapeutic target for other diseases, there is a clear need for novel ligands with different functions, and an understanding of how these ligands achieve these functions.
To date, the three-dimensional structures of CXCR4 in complex with three ligands have been determined—the small molecule IT1t, the cyclic peptide CVX15, as well as the human herpesvirus-8 chemokine vMIP-II (Qin et al., 2015; Wu et al., 2010). While these structures provide insight into the inactive state conformations of the receptors, they fail to provide much information about the receptor conformations adopted upon agonist binding. To address this question, Wescott et al. conducted a thorough mutagenic profiling and modeling of CXCR4, identifying an intramolecular pathway through which CXCL12 binding causes intracellular G protein signaling (Wescott et al., 2016). Furthermore, additional complexes of GPCRs in other families provide insights into how activated receptors bind G proteins (Standfuss et al., 2011). Activated β2 adrenergic receptor in complex with Gαs reveals that the Gαs C-terminal helix inserts into the transmembrane region of the receptor, causing a large outward movement of the cytosolic end of TM helix VI, as well as a helical extension of the cytoplasmic end of TM V (Rasmussen et al., 2011). Other methods to study how GPCR ligands function and how receptors are activated involve studying constitutively active mutants (CAMs). For CXCR4, known CAMs arise from mutation of N1193.35 to either alanine or serine (Zhang et al., 2002). (The residue superscripts refer to the Ballesteros–Weinstein numbering scheme (Ballesteros and Weinstein, 1995)). Despite the aforementioned studies on agonist-induced G protein activation, the mechanism of activation of CAMs has received much less attention (Standfuss et al., 2011).
We studied the effects of purine and quinazoline analogues on WT CXCR4 and the N1193.35A CAM, two of which (B7 and CX0298) have never been characterized. These molecules include antagonists for CXCR4 developed as second-generation therapeutics for hematopoietic stem cell mobilization, first generation compounds for HIV-1 entry, and renoprotective treatment for acute kidney injury (Wu et al., 2012; Wu et al., 2015a; Wu et al., 2015b). We also studied the control antagonist N,N’-dicyclohexylcarbamimidothioic acid (5,6-dihydro-6,6-dimethylimidazo[2,1-b]thiazol-3-yl)methyl ester (IT1t) that was previously co-crystallized with CXCR4 (Wu et al., 2010) and is known to function as an inverse agonist on the CXCR4 N1193.35S CAM (Mona et al., 2016), as well as the antagonist AMD3100 which was previously identified as a partial agonist on the CXCR4 N1193.35A CAM (Zhang et al., 2002) but has yet to be co-crystallized with CXCR4. The purine and quinazoline CXCR4 antagonists cause varying effects on the CAM receptor, resulting in either neutral antagonism or inverse agonism.
To move towards an understanding of the structural features that contribute to CAM activity and facilitate inverse agonism by IT1t, we leveraged molecular dynamics to predict key features that contribute to CAM activity and performed experimental mutagenesis to test these predictions. These simulations suggested the formation of a hydrophobic triad composed of unique contacts between F872.53 and amino acids W2526.48 and L1203.36 in the apo CAM where the receptor should progress towards an active state. This triad facilitates allosteric signaling between TM helices III and VI that is not observed in all other systems associated with an inactive state of CXCR4. Experimental mutagenesis confirmed the importance of F872.53 to signaling in the CAM. Thus, we propose this hydrophobic triad of interactions mediated by F872.53 is critical for CXCR4 activation. Furthermore, we propose that inverse agonists such as IT1t and the previously uncharacterized compound B9 prevent the formation of this hydrophobic triad by altering the sidechain orientation of F872.53.
RESULTS
Functional activities of small molecule ligands with wild-type and N1193.35A CXCR4 in S. cerevisiae
We used a genetically modified strain of S. cerevisiae capable of expressing human Gαi2-coupled chemokine receptors to determine IC50 (and EC50) values of the compounds in this study. We previously used the CY12946 strain of yeast to measure receptor activation with a high signal-to-noise ratio via expression levels of β-galactosidase, leading to detectable enzymatic activity (Sachpatzidis et al., 2003). The excellent signal-to-noise ratio is key in allowing for the observation of very subtle differences in basal activity of either WT CXCR4 or mutants, as well as their signaling levels when dosed with various ligands. This strain also eliminates any interactions with other GPCRs (Liu et al., 2016), making it an indispensable tool for studying Gαi2 activity elicited by compounds and CXCR4 mutants.
The chemical structures of the compounds used in this study are shown in Fig. 1. Dose-response assays were performed for each compound with CXCL12-driven β-galactosidase activity to measure antagonist activity against WT CXCR4 (Fig. 2A). Each dose of the five compounds and the control antagonists (AMD3100 and IT1t) was performed in the presence of 2.0 μM CXCL12, a concentration that yields a high signal in this assay system (Fig. S1A). Standard logistic regressions were applied to extract IC50 values from each dose-response curve (Table 1). AMD3100 and IT1t had IC50 values of 49.2 nM and 0.198 nM, respectively. The remaining five compounds were antagonists with IC50 values that ranged between 55.1 pM to 7.75 nM. The two purine analogues B7 and B8 were extremely potent with IC50 values of 55.1 and 69.5 pM, respectively. These IC50 values are even lower than that of IT1t. The IC50 of another purine analogue CX0298 (0.167 nM) was not significantly different from that of the IT1t (0.198 nM) while the quinazoline analogue B9 showed a similar level of potency as the purine analogue CX344 with IC50 values of 7.18 and 7.75 nM, respectively.
FIGURE 1.

A. Structures of the FDA-approved drug AMD3100 and the small molecule antagonist IT1t. B. Structures of CXCR4 ligands based on the pharmacophores. Derivatives include B7, B8, B9, CX0298, and CX344. B8 and CX344 are compounds 18 and 25, respectively, in Wu et al (Wu et al., 2015b). B9 is compound 16 in Wu et al (Wu et al., 2015a).
FIGURE 2.

Effects of ligands on CXCR4 expressed in S. cerevisiae. Values represent the mean from at least two independent experiments, and error bars refer to the standard error of the mean (SEM). See also Table 1 for quantified results. A. Inhibition of CXCL12-induced CXCR4 activation by antagonist compounds. B. Ligand-induced behavior of CXCR4 mutant N1193.35A. Basal activity is set to 100% which corresponds to roughly 47% of the maximum activity of WT CXCR4 (see Fig. S1A). Although all compounds act as neutral antagonists on the wild-type CXCR4 receptor, they show different behaviors with a constitutively activate mutant. AMD3100 acts as a partial agonist, while IT1t shows inverse agonism. Compounds B8, B9, and CX344 also cause inverse agonism to a lesser degree than IT1t. Compounds B7 and CX0298 function as neutral antagonists.
Table 1.
Summary of compound properties
| Compound name | S. cerevisiae IC50, WT CXCR4 (nM)a | S. cerevisiae IC50 [EC50 for AMD3100], N1193.35A CXCR4 (nM)a | CXCL12 binding IC50, WT CXCR4 (nM)b | HIV-1 IC50 (nM)b | β-arrestin-2 IC50, WT CXCR4 (nM)a | Cytotoxicity CC50 (μM) | |
|---|---|---|---|---|---|---|---|
| CEM cells | TZM-bl cells | ||||||
| AMD3100 | 49.2 (39.1, 61.3) | [47.2 (19.7, 107)] | 12.0 ± 1.1d | 4 – 10g | 29 (16, 54) | NTj | NTj |
| B7 | 0.0551 (0.0386, 0.0785) | NRc | 28.0 ± 2.7 | 2.4 ± 0.4 | 90 (61, 130) | >50 | >50 |
| B8 | 0.0695 (0.0507, 0.0946) | 83.0 (1.87, 2430) | 18.1 ± 2.8e | 2.0 ± 0.1e | 170 (83, 590) | >50e | >50e |
| B9 | 7.18 (6.39, 8.06) | 21.0 (6.21, 75.3) | 39.7 ± 5.1f | 25.2 ± 24.9 | 700 (210, 2200000) | >50 | >50 |
| CX0298 | 0.167 (0.130, 0.213) | NRc | 27.8 ± 1.9 | 4.2 ± 0.3 | 1600 (370, NRi) | >50 | >50 |
| CX344 | 7.75 (6.87, 8.73) | 98.9 (29.6, 392) | 4.2 ± 0.4e | 0.61 ± 0.27e | 55 (31, 120) | >50e | >50e |
| IT1t | 0.198 (0.163, 0.240) | 13.7 (6.08, 32.5) | 2.1± 0.4d | 40h | NTj | NTj | NTj |
95% confidence intervals are provided in parentheses
Error is presented as standard deviation of the mean (SD)
NR = not reported. These compounds function as neutral antagonists against the CAM
As reported by Van Hout et al., 2017 (Van Hout et al., 2017), in which a fluorescently-labeled CXCL12 was used
As reported by Wu et al., 2015 (Wu et al., 2015b)
As reported by Wu et al., 2015 (Wu et al., 2015a)
As reported by Schols et al., 1997 (Schols et al., 1997)
As reported by Thoma et al., 2008 (Thoma et al., 2008)
NR = not reported. The upper confidence interval is not defined
NT = not tested
Previous work using the N1193.35A/S CAMs demonstrated that AMD3100 functioned as a partial agonist and also indicated that T140—a polyphemusin analogue similar to the CXCR4 co-crystallized antagonist CVX15 (Wu et al., 2010)—functioned as an inverse agonist (Zhang et al., 2002). A more recent study identified that IT1t also functions as an inverse agonist on the N1193.35S CAM (Mona et al., 2016). We reproduced the partial agonism induced by AMD3100 and the inverse agonism elicited by IT1t when performing dose-response studies using the N1193.35A CAM (Fig. 2B). Basal CAM activity is roughly 47% of the maximum WT response activated by CXCL12 (Fig. S1A). Thus, AMD3100 is able to enhance the signal to ~83% maximum of the WT signaling levels in this assay system, while IT1t is able to reduce the signal to ~25%. Against the CAM, the four purine and single quinazoline core polyamine pharmacophores functioned as either neutral antagonists that did not change the active-inactive equilibrium (B7 and CX0298) or inverse agonists with lower efficacies than that of IT1t (Fig. 2B). Classification of the compounds’ functionalities at 1 μM was achieved by performing one-way ANOVA followed by Dunnett’s multiple comparison test with DMSO-treated samples set as the control group— B7 and CX0298 were not significantly different from controls (classifying them as neutral antagonists) whereas B8, B9, and CX344 were (p < 0.05, classifying them as inverse agonists). IC50 values were typically at least an order of magnitude higher against the N1193.35A CAM than WT CXCR4 (Table 1). AMD3100 had a similar EC50 value against the CAM as its corresponding IC50 value against the WT receptor, with values of 47.2 and 49.2 nM, respectively. Initial dose-response studies on the CXCR4 CAM in the presence of 2.0 μM CXCL12 showed that all of the compounds function as antagonists, suggesting that the compounds are able to prevent CXCL12-mediated signaling and reduce the CAM’s activity (Fig. S1B).
Compound-mediated CXCL12 and HIV-1 inhibition and toxicity in human cell lines
The effects of two of the purine core-derived polyamines on competitive binding experiments with 125I-CXCL12 and IC50 for anti-HIV activities were previously reported (Wu et al., 2015a; Wu et al., 2015b), but two purine core-derived polyamines have not been previously characterized. The quinazoline analogue was only tested for competitive binding against 125I-CXCL12 (Wu et al., 2015a). We used mammalian cells to characterize B7 and CX0298 (as well as the anti-HIV activity of B9).
HEK-293 cells were stably transfected with human CXCR4 to perform 125I-CXCL12 radioligand binding experiments, while TZM-bl cells (a HeLa-derived cell line) were used to measure anti-HIV-1 activity (Wu et al., 2015a; Wu et al., 2015b). The IC50 values for the competitive binding assays against 125I-CXCL12 for B7, B9, and CX0298 were all very similar—approximately 30 nM—whereas the IC50 values for anti-HIV activity were 2.3, 25.2, and 3.6 nM, respectively. The IC50 values determined from experiments using S. cerevisiae were typically lower than those obtained using mammalian cells with a notable exception being CX344, which exhibited similar IC50 values. None of the compounds exhibited toxicity against either TZM-bl or CEM (T-lymphoblast) mammalian cell lines (Table 1).
Effects of compounds on CXCR4-mediated β-arrestin-2 signaling in mammalian cells
In addition to G-protein signaling, CXCR4 signals through the cytosolic adapter protein β-arrestin-2 to activate ERK1/2 in mammalian cells, but yeast do not have homologous G protein-coupled receptor kinases (GRKs) and arrestin proteins to test this effect. β-arrestin-2-mediated signaling downstream of CXCR4 and other GPCRs promotes scaffolding of ERK1/2 on β-arrestin-2, activating ERK1/2 as a cytosolic target (Tohgo et al., 2002). We investigated the effects of the compounds on CXCR4 signaling through this pathway using click beetle luciferase complementation assays for association of CXCR4 with β-arrestin-2 upon CXCL12 stimulation (Fig. 3). Dose-response studies with compound concentrations ranging from 10 nM to 10 μM allowed us to determine IC50 values (Table 1). AMD3100 was the most potent inhibitor of β-arrestin-2 signaling with an IC50 value of 29 nM. Compounds B7, B8, and CX344 were the next most potent with values of 90, 170, and 55 nM, respectively. B9 and CX0298 exhibited low potency, with values of approximately 700 and 1600 nM, respectively. We note that all of the compounds were able to completely inhibit CXCL12-driven β-arrestin-2 coupling to CXCR4 at concentrations of 10 μM.
FIGURE 3.

Inhibition of CXCL12-induced recruitment of β-arrestin-2 to WT CXCR4 expressed on mammalian cells. Values represent the mean from at least two independent experiments, and error bars refer to the standard error of the mean (SEM). A. The kinetic traces for each drug treatment are shown. B. Dose-response curves generated from the kinetic data shown in Fig. 3A at time = 20 minutes. While the antagonists displayed various potencies in preventing β-arrestin-2 signaling, they displayed similar levels of efficacy, with all being able to completely inhibit signaling at concentration of 10 μM. Luminescence is proportional to association of the labeled proteins. All graphs normalized to untreated controls for each individual trial. See also Table 1 for quantified results.
Mechanism of CXCR4 N1193.35A constitutive activity
A comparison of two active structures of GPCRs—a rhodopsin CAM complexed to a Gα C-terminal peptide (Standfuss et al., 2011) and an agonist-β2 adrenergic receptor-Gαβγ complex (Rasmussen et al., 2011)—indicates their mechanism of activation is different, with the former suggesting that agonist binding causes an initial rotation of TM VI, and the latter suggesting instead an initial inward movement of TM V (Tehan et al., 2014). These observations raise the possibility that that individual receptors may exhibit unique mechanisms of activation. We sought to utilize structural information, the observed inverse agonism of IT1t and B9, and molecular dynamics simulations to understand how the N1193.35A mutant exerts its constitutive activity.
To analyze the structural basis of the constitutive activity of the N1193.35A mutant, we examined the interactions of N1193.35 in the structure of CXCR4 bound to IT1t. The structure, representing an inactive state of CXCR4, shows that N1193.35 is in the middle of TM helix III. The N1193.35 side chain atoms form a network of hydrogen bonds with residues found on two other TM helices. This type of network is not observed anywhere else in the transmembrane region. The N1193.35 side chain oxygen atom forms a hydrogen bond with Nε2 of H2947.45 while the N1193.35 side chain amide nitrogen and H2947.45 Nε2 form hydrogen bonds with a carboxylate oxygen of D842.50 (Fig. S2A). (It is also worth noting that one of the CXCR4 monomers in this structure displays a second rotamer of D842.50 in which the side chain is not directed towards N1193.35). We hypothesized that this network of hydrogen bonds involving three TM helices in a region with a low dielectric constant provides substantial free energy for stabilizing the inactive state of CXCR4. Indeed, these same three residues—as well as an additional residue, S1233.39—have been postulated to function in CXCR4 as an allosteric binding site for sodium ions (Taddese et al., 2018). Sodium allostery in class A GPCRs seems to function as a general regulator of receptor activation by stabilizing the inactive state conformation and in many cases the binding pocket residues are highly conserved (Katritch et al., 2014).
We rationalized that the N1193.35A/S mutants impart constitutive signaling through loss of sodium binding to the allosteric pocket of CXCR4, possibly in conjunction with the upward movement of TM III due to a loss of hydrogen bonding that “anchors” the helix in place. As TM III is known to move upward during GPCR activation (Tehan et al., 2014), the loss of this stabilizing hydrogen bond network offers a rational explanation for why the N1193.35A mutation exhibits constitutive activity. It is interesting to note that CXCR4 has the DRY motif at the C-terminal end of TM III (consisting of residues D1333.49, R1343.50, and Y1353.51) but there is no aspartate or glutamate at TM VI position 6.30 to make an ionic lock that stabilizes the inactive state (Schneider et al., 2010), suggesting the interactions at N1193.35 may substitute for the ionic lock.
We attempted to mimic the constitutive activity of the N1193.35A/S mutants by targeting the two residues to which hydrogen bonds are formed (and which are also part of the putative sodium binding site), D842.50 and H2947.45. In total, we generated four single mutants—D842.50A, and H2947.45A/F/I—and three double mutants with the same His mutations coupled with the D842.50A mutation. Intriguingly, we found that none of the seven mutants exhibited constitutive activity similar to the N1193.35A mutant, but instead signaled at levels comparable to WT CXCR4 (Fig. S2B). The results from the double mutants are particularly convincing, as they would disrupt any hydrogen bond “anchors” that stabilize the N1193.35 position as seen in the crystal structure and would theoretically destabilize binding of sodium to the allosteric binding site. These results indicate that N1193.35 must somehow play another, yet uncharacterized, role in stabilizing the inactive form of CXCR4.
We next performed molecular dynamics simulations of inactive and active systems for CXCR4 using both conventional and accelerated molecular dynamics algorithms. Starting from the crystal structure of CXCR4 bound to IT1t, we mutated N1193.35 to alanine. Then two additional systems were generated by removing IT1t from the complex. Due to IT1t’s inverse agonist effect on N1193.35A, the holo structures of the WT and CAM receptors and the apo form of the WT receptor represent inactive systems of CXCR4, while the apo form of the N1193.35A mutant represents an active system.
We observed a number of interhelical contacts, residue pairs with a minimum distance between groups of atoms of 3 Å or less, that best distinguish between active and inactive forms of CXCR4. Specifically, these involved contacts between TM helix pairs I-II, II-IV, III-IV, I-II, II-VI, II-III, and III-VI, in order of decreasing contribution to the variance explained by the first principal component from our time series of pairwise distances between amino acid side chains (Fig. S3). While the relative positions of TM helices were generally preserved, we observed bending of the extracellular end of TM helix II towards TM helices I and VII and of the intracellular end of TM helix V towards TM helix VI (Fig. S4) in the active system. These bending motions were not observed in any inactive system to the same extent as the active system. We measured the average angle between the conformation of the extracellular end of TM helix II in the initial conformation of holo WT and the conformations of the extracellular end of TM helix II in the last 10 ns of each simulation trajectory and found mean bending magnitudes of 28.6° in apo CAM, 11.0° in apo WT, 14.8° in holo CAM, and 11.4° in holo WT (Fig. S4C). We performed an analogous measurement for the bending of the intracellular end of TM helix V and found bending magnitudes of 20.1° in apo CAM, 6.4° in apo WT, 6.7° in holo CAM, and 4.4° in holo WT (Fig. S4D). The intracellular movement of TM helix V towards TM helix VI is one known conformational change observed in activation of GPCRs (Rasmussen et al., 2011; Tehan et al., 2014).
We also observed differences in allosteric coupling between active and inactive simulation systems based on a pairwise residue correlation matrix that describes the coupling of changes in side-chain contacts for all residue pairs. From the residue correlation matrix we extracted maximum correlations between helices by finding the maximum correlated residue pair that occurs across each helix pair. To identify those changes that correspond to differences between active and inactive states of CXCR4, we calculated the mean and standard deviation for differences in maximum helix pair correlation between every inactive system and the active system. As a result, we found TM helix pairs I-VI and III-VI exhibited significantly higher (α=0.05, two-sided) maximum correlations in the active state than in the inactive state by differences of 0.35 and 0.47, respectively. TM helix pairs II-IV, II-V, and IV-VII exhibited significantly higher (α=0.05, two-sided) maximum correlations in the inactive state than in the active state by differences of 0.29, 0.22, and 0.15, respectively (Fig. 4). In terms of root-mean-square fluctuations (RMSF) of backbone Cα from the average conformation, we observed a mean increase of 0.14 nm for residues T902.56 to A1002.66 from TM helix II in the active apo CAM when compared to holo WT CXCR4. The bending in helix II (previously described, Figure S4C) likely contributes to the larger fluctuations in this region (Fig. S5A). IT1t binding to the apo CAM was observed to decrease backbone RMSF in this region by an average of 0.13 nm for residues V962.62 to A1002.66 (Fig. S5C). Additionally, an average increase of 0.17 nm RMSF for backbone Cα was observed in the N-terminal unstructured region prior to TM helix I in apo CAM compared to holo CAM due to removal of steric constraints imposed by IT1t binding (Fig. S5C).
FIGURE 4.

Differences between active and inactive systems in maximum allosteric signals between TM helices (ignoring signals within the same helices) in a heatmap representation. Allosteric signals represent coupling of dynamics between TM helices, and were determined with the TimeScapes software using a neighbor contact exclusion of 3 residues and a distance cutoff of 6 Å. Mean differences in maximum correlations between TM helices within active or inactive simulations systems are colored on a scale from red to blue in the heatmap. Red suggests that allosteric coupling is higher in the inactive systems, while blue suggests that the allosteric coupling is higher in the active system. To account for variation in the comparison, those differences that are significant (as determined by a t-test, α = 0.05) are marked with a magenta star. See also Figures S3, S4, and S5.
At the level of individual amino acids, we observed unique coupling between TM helices III and VI through F872.53 in TM helix II as a result of forming a hydrophobic triad in the apo CAM (Figs. 4, 5B, 5C, and 6A). These interactions involved contacts between residue pairs F872.53-L1203.36 and F872.53-W2526.48 (Fig. 5B and C). Contacts are defined as residue pairs with a minimum distance of 3 Å or less between two groups of atoms, each group corresponding to a distinct residue. Binding of IT1t was observed to disrupt this coupling by F872.53 preferring to interact with W942.59, a residue proximal to the binding site that is repositioned as a result of ligand binding (Figs. 5A, 6A). In the apo WT system, the hydrophobic triad between TM helices II and VI was not observed, likely due to the presence of a number of other polar and aromatic interactions among residues such as Y451.39, F872.53, Y1163.32, N1193.35 and H2947.45 in TM helices I, II, III, VI and VII, respectively (Fig. S6). This may be partially explained by F872.53 interacting with H2947.45 (Fig. 5D). Examples of other interactions in apo WT include W942.59 interacting with Y451.39 instead of F872.53 or Y1163.32 (Fig. 5E and F). These observed interactions will indirectly affect the behavior of F872.53 and other residues involved in signal propagation such as W2526.48 and F2927.43. For example, we observed formation of contacts between T902.56 and F2927.43 only in apo forms of CXCR4 (Fig. 5G). Additionally, we observed that apo and holo forms of CXCR4 differ in the probability of maintaining a contact between W2526.48 and F2927.43 (Fig. 5H).
FIGURE 5.

Minimum distances between atom groups corresponding to selected residue pairs throughout molecular dynamics trajectories for apo CAM (blue), apo WT (orange), holo CAM (green), and holo WT (red). Note that the only active system is apo CAM. Each panel from (A) to (H) displays the distance-time profile for a different residue pair, as indicated by panel titles. Distances are shown in nm, and time index is shown in ns. The first 100 ns represent data from conventional molecular dynamics (MD) simulations, while the last 100 ns represent data from accelerated molecular dynamics simulations. When changing from conventional to accelerated MD algorithms, velocities and positions from the end of the prior simulation are used to restart the simulation resulting in a continuous trajectory. See also Figure S6.
FIGURE 6.

A. Networks of van der Waals contacts in N1193.35A, WT, IT1t-N1193.35A, and IT1t-WT in representative conformations from accelerated molecular dynamics trajectories. Residues of CXCR4 are colored by helix according to the inset key with non-carbon atoms colored according to the CPK convention. IT1t is colored purple with non-carbon atoms colored according to the CPK convention. Van der Waals contacts are denoted by orange lines between atom pairs. Note that the hydrophobic triad is only formed in the active system, N1193.35A, with the side-chain of F872.53 flipping towards the intracellular side of CXCR4. B. Effect of F872.53 single and double mutants (i.e., coupled with the N1193.35A mutation) on CXCR4 signaling in S. cerevisiae. While the F872.53 single mutants signal at only slightly higher levels than WT, the F872.53 double mutants almost completely reduce N1193.35A-induced CXCR4 signaling back to WT levels (DMSO-treated cells). Cells expressing WT CXCR4 and treated with DMSO do not signal at levels significantly different than any of the F872.53 mutants, but N1193.35A treated with DMSO signals significantly higher than any other receptor variant tested (**** = p < 0.0001, as determined by two-way ANOVA followed by Tukey’s multiple comparisons test). Treatment of the cells with 1 μM inverse agonist IT1t or B9 significantly reduces N1193.35A activity (p < 0.0001) as expected, but does not exert any effects on the F872.53 single or double mutants. Values represent the mean from three independent experiments, and error bars refer to the standard error of the mean (SEM). See also Figure S7.
To evaluate the importance of the hydrophobic triad across class A GPCRs, we retrieved all sequences of human proteins that match hidden Markov model profiles from human CXCR4 including the profiles for the CXCR4 chemokine receptor N-terminal domain (Pfam: PF12109) and for class A GPCRs (Pfam: PF00001). After filtering these sequences for uniqueness, we observed a phenylalanine at the position of F872.53 in 79% of sequences. Though at the position of L1203.36 we only observed a leucine in 48% of sequences, we observed a phenylalanine at this same position in 48% of sequences resulting in conservation of hydrophobicity. Finally, we observed a tryptophan at the position of W2526.48 in 85% of sequences. Thus, this hydrophobic triad appears to be conserved in human GPCRs that match the class A GPCR profile; however, it is possible that substitution of L1203.36 for phenylalanine may affect the balance of inactive and active conformers for the associated sequences.
We next sought to biochemically test via mutagenesis whether disrupting the F872.53-mediated coupling of TM helices III and VI in the CAM could prevent constitutive signaling. We reasoned that this residue was most likely responsible for the differences in maximum helix pair correlation observed between helix pair III-VI (Fig. 4). Furthermore, we desired to see whether disruption of this coupling could eliminate any inverse agonist properties of both IT1t and B9 on the CAM, as the former was observed in molecular dynamics (MD) simulations to disrupt F872.53-mediated coupling through binding and promoting interactions between F872.53 and W942.59 (Fig. 6A). With the coupling already disrupted via mutagenesis, we reasoned that IT1t—and possibly the other studied inverse agonist B9—would lose their inverse agonist properties when administered to the CAM.
Generation of two F872.53 single mutants—to an alanine and to a threonine—resulted in a modest (~twofold) increase in signaling compared to WT CXCR4. Interestingly, these same mutations coupled with the N1193.35A mutation almost entirely eliminated the constitutive signaling elicited by the N1193.35A mutation, with the mutants signaling at levels less than threefold higher than WT CXCR4. While WT did not signal significantly different than any of the F872.53 mutants, N1193.35A on its own signaled significantly higher than any of the receptor variants, with p < 0.0001 as determined by two-way ANOVA followed by Tukey’s multiple comparisons test (Fig. 6B, DMSO-treated samples). Importantly, we observed that all of the CXCR4 receptor variants were expressed at similar levels suggesting that differences in signaling is not attributable to differences in expression. (Fig. S7). Treatment of N1193.35A with either 1 μM of IT1t or B9 significantly reduced signaling levels when compared to N1193.35A treated with DMSO (p < 0.0001, determined using the same means as above), but the compounds had no significant effect on signaling for any of the other strains (Fig. 6B). We also note that when treated with IT1t or B9, N1193.35A signaling levels were still significantly higher than any other IT1t- or B9-treated strain (p ≤ 0.02 for IT1t-treated strains and p < 0.0001 for B9-treated strains, respectively). Altogether, this data suggests that the compounds’ inverse agonism involves disruption of F872.53-mediated coupling between helices III and VI.
DISCUSSION
Activation of GPCRs is based on the equilibrium between active and inactive states (Schneider and Seifert, 2010). Mutations that lead to a significantly lowered energy barrier from the inactive to active state result in constitutive activity (Tsukamoto and Farrens, 2013). CAMs allow ligands that bind to a receptor’s orthosteric site to be classified based on the propensity of the compound to alter receptor signaling, something that is not possible with many WT receptors with low basal activity as an external agonist would be required to elicit a signal. In these cases, competition between compound and agonist for binding to the orthosteric site would confound interpretation of how a compound acts on its own. Neutral antagonists against a WT receptor can demonstrate partial agonism in CAMs because of the decreased energy needed to get to the active state. More frequently, CAMs can identify neutral antagonists on the WT receptor as compounds that have intrinsic inverse agonist properties that shift the equilibrium of a CAM towards an inactive state. Neutral ligands of CAMs are defined as not having any effect on receptor equilibrium in the absence of other ligands. In the absence of an external agonist, the equilibrium for CXCR4 lies strongly towards the inactive state. CXCL12 and CAMs such as the N1193.35A mutant shift the equilibrium towards the active state.
Compounds B8 and CX344 have been examined for anti-HIV activity (Wu et al., 2015b). We sought to fully characterize B8 and CX344, as well as two previously uncharacterized CXCR4 antagonists—B7 and CX0298—alongside the partially-characterized compound B9 (Fig. 1). To this end, we utilized both S. cerevisiae strain CY12946 expressing functional CXCR4 and mammalian cells to determine IC50 values for each of the compounds with both WT CXCR4 and the constitutively active N1193.35A mutant. We also determined the anti-HIV potential of these compounds, as well as their CXCR4/β-arrestin-2 signaling profiles. Finally, we examined the N1193.35A mutant itself and attempted to determine both how it functions as a CAM and how the inverse agonists IT1t and B9 are able to shift receptor equilibrium back towards an inactive state.
Use of the S. cerevisiae strain CY12946 expressing functional CXCR4 eliminates any interactions with other GPCRs, allowing for specific signaling responses to be easily observed, something that is more difficult to achieve in mammalian cells (Liu et al., 2016). The outputs of assays with this yeast strain also exhibit an incredibly high signal-to-noise, making it an ideal reporter system by which to study CXCR4 mutants and their subtle differences in response to different ligands. The end-point signal generated from these assays is a result of four main factors: 1. receptor expression; 2. receptor signaling activity through Gαi2 (either basal or elicited by ligands); 3. integration of downstream signals; and 4. subsequent transcription, translation, and enzymatic activity of β-galactosidase. Barring drastic changes in expression of variant receptors, interpretation of the assay outputs can mainly be attributed to the second factor, i.e., receptor signaling activity. Enzymatic activity of expressed β-galactosidase from these assays can be easily measured.
Yeast expressing WT CXCR4 and stimulated with CXCL12 demonstrated that all of the tested compounds have stronger antagonist effects than the clinically used AMD3100 (Fig. 2A). Further, studies of compounds in mammalian cell inhibition assays demonstrated that all of the compounds were potent at inhibiting HIV-1 and CXCL12-mediated CXCR4 binding. In all cases, the compounds exhibited lower IC50 values in yeast than mammalian cells (with the exception of CX344, which had lower IC50 values in mammalian cells than in S. cerevisiae). Furthermore, there was a difference in trends for the four purine and the single quinazoline polyamines that had the lowest IC50 values between cell types with B7, B8, and CX0298 having the lowest IC50 values in S. cerevisiae, and yet in assays with mammalian cells CX344 consistently displayed the lowest IC50 values. This finding suggests that differences in growth media, the components of the cell membrane, and post-translational modifications of CXCR4, which differ in mammalian and yeast cells, may affect compound potency. For example, many chemokine receptors expressed in mammalian cells undergo tyrosine sulfation, which increases their affinities for their cognate ligands (Ludeman and Stone, 2014), but the same process does not occur in yeast (Kanan et al., 2012). As such, sulfation of tyrosine residues in mammalian cells likely makes it more difficult for the compounds to displace CXCL12 than in yeast and could also explain the difference in trends. These results may also be explained by the presence of serum albumin in mammalian assays, a protein that has the tendency to bind to small molecule ligands with different affinities that affect CXCR4 binding (Zhang et al., 2016).
We also tested if CXCR4 antagonists blocking only Gαi2 signaling in S. cerevisiae might function as biased ligands affecting recruitment of the cytosolic adapter protein β-arrestin-2 in mammalian cells (Fig. 3). Using a luciferase complementation assay for association of WT CXCR4 with β-arrestin-2, we were able to perform dose-response studies for each compound to determine inhibition of CXCR4 association with β-arrestin-2 upon CXCL12 stimulation. We determined that each of the compounds was able to completely inhibit β-arrestin-2 association with CXCR4 at concentrations of 10 μM, but that the potencies ranged quite dramatically. B9 and CX0298 exhibited very low activity, with IC50 values of ~700 and ~1600 nM, characterizing them as biased antagonists that affect β-arrestin-2 interactions with CXCR4 only at high concentrations. The differences in potencies may be useful in future studies aimed at biasing CXCR4 to signal through β-arrestin-2 rather than the canonical G protein pathways.
Dose-response assays with each of the ligands against the N1193.35A CAM in S. cerevisiae revealed various functionalities not discerned when tested against the WT receptor co-treated with CXCL12 (Fig. 2B). AMD3100 functioned as a partial agonist in the CAM, as previously reported (Zhang et al., 2002). Compounds B7 and CX0298 functioned as neutral antagonists, while the remaining compounds B8, B9, CX344, and IT1t all functioned as inverse agonists. IT1t was previously reported to exhibit inverse agonist properties on N1193.35S (Mona et al., 2016), and was the most efficacious of the four inverse agonists. The results with AMD3100 and IT1t are especially intriguing as IT1t was co-crystallized with a construct of CXCR4 in the inactive state (Wu et al., 2010), yet the structure of the human drug AMD3100 in complex with CXCR4 has not been solved, particularly due to use of the inactive CXCR4 constructs used to solve the structures of CXCR4 complexed to IT1t, CVX15, or vMIP-II (Qin et al., 2015; Wu et al., 2010). CVX15 is chemical analog of cyclin peptide T140, which was also identified as an inverse agonist with the CXCR4 CAM (Zhang et al., 2002). Inverse agonism of vMIP-II has not been tested. The observation that IT1t and CVX15 function as inverse agonists against a CXCR4 CAM lends credence to the hypothesis that ligands with intrinsic inverse agonism have a higher likelihood of crystallizing with GPCR constructs in a stabilized inactive state. CAMs could be used to differentiate inverse agonists from neutral antagonists against WT receptors with low basal activity to expedite the process of crystal formation with structurally uncharacterized GPCRs stabilized in the inactive state.
The N1193.35A CXCR4 CAM was identified before the structure of CXCR4 was determined (Wu et al., 2010; Zhang et al., 2002), but its mechanism for constitutive signaling was never investigated. To that end, we identified a hydrogen bond network formed among residues N1193.35, D842.50, and H2947.45 in the crystal structure of CXCR4 (Wu et al., 2010). These three residues, in conjunction with S1233.39, also serve as a putative allosteric site for sodium ion binding (Taddese et al., 2018). We rationalized that this hydrogen bond network located in the extremely hydrophobic transmembrane environment served to stabilize the inactive state of the receptor, potentially by sodium ion binding—a known GPCR inactivation mechanism (Katritch et al., 2014)—or by anchoring TM III down and preventing it from moving upward, a movement observed as a general mechanism for GPCR activation (Tehan et al., 2014). Four single mutants and three double mutants of CXCR4 were generated to test this hypothesis. The seven mutants signaled at levels similar to that of WT CXCR4, thereby ruling out any significant stabilization of the receptor’s inactive state through this hydrogen bond network. These experimental results are in disagreement with recent MD simulations that propose that the N1193.35A CAM functions by preventing spontaneous sodium binding to the allosteric site (Cong and Golebiowski, 2018), suggesting that the CAM must function through yet another mechanism.
MD simulations of CXCR4 in separate active and inactive systems were employed to study the mechanism of the CAM. From these simulations, a number of dynamic features exhibited only in the active system—the CAM in an apo state—were observed. For example, F872.53 in TM helix II seemed to increase allosteric communication between TM helices III and VI only for the active system (Fig. 4). This change may be less likely in inactive systems as suggested by the decreased RMSF of amino acid side-chains in TM helix II. Notably, the side chain of F872.53 only forms a hydrophobic triad of contacts in the active system. This triad involves the formation of contacts between F872.53, L1203.36, and W2526.48, residues strongly conserved across human class A GPCRs (Fig. 6A). In WT CXCR4, the proximity of polar N1193.35 may be sufficient to prevent formation of the hydrophobic triad as these contacts were not observed. In both apo forms of CXCR4, W942.59 exhibits different behavior. Specifically, a contact between Y451.39 and W942.59 is unformed only in the apo CAM (Fig. 5E), while a contact between W942.59 and Y1163.32 is unformed only in apo WT (Fig. 5F). In a prior mutagenesis study, Y451.39H, W942.59R, and Y1163.32S were shown to decrease activation of CXCR4 when stimulated with CXCL12 (Wescott et al., 2016). Our F872.53 mutants (discussed below) were able to almost completely eliminate N1193.35A signaling. Together, these mutations indicate the importance of TM helix II in activation of CXCR4. Prior studies have also shown that F2927.43 and W2526.48 are in closer proximity during an inactive state than during an active state; furthermore, both F2927.43 and W2526.48 have been shown to be key residues involved in signal propagation through the transmembrane region of CXCR4 (Wescott et al., 2016). Similarly, we observed that the apo CAM is most likely to un-form a contact between W2526.48 and F2927.43 followed by apo WT, holo CAM, and holo WT in order of decreasing probability (Fig. 5H).
When IT1t is bound to CXCR4, F872.53 interacts primarily with W942.59 (Fig. 5A), a key residue involved in initiation of a signaling response (Wescott et al., 2016). Our hypothesis is that IT1t inhibits activation of CXCR4 by preventing the formation of the conserved, hydrophobic triad (Fig. 6A) and therefore disrupting TM helix II-mediated coupling of TM helices III and VI. In our simulations, this coupling—also responsible for N1193.35A’s constitutive signaling—seems to be mediated by F872.53. Subsequent coupling of the N1193.35A mutation with mutations of F872.53 resulted in signaling levels not significantly different to WT CXCR4, confirming its critical role in CAM activity (Fig. 6B). Furthermore, we observed that perturbing F872.53-mediated coupling of helices III and VI via mutation resulted in a complete loss of inverse agonism elicited by either IT1t or B9, as treatment of N1193.35A/F872.53A and N1193.35A/F872.53T double mutants with inverse agonists did not further reduce activity of CXCR4 (Fig. 6B). Although the mutants trend to higher activities than WT (but are not statistically significant), there is no downward trend upon compound treatment. IT1t’s loss of inverse agonist activity on the F872.53 mutants is to be expected as the helix III-VI coupling has already been perturbed, but it was interesting to see that B9 functioned in a similar manner. While the complex of B9 interacting with CXCR4 has not been solved, our results suggest that B9 may disrupt F872.53-mediated coupling of helices III and VI in a similar manner to that of IT1t.
Activation of CXCR4 requires a more compact arrangement of intracellular ends of TM helices III, V, and VII around TM helix VI (Wescott et al., 2016). Our simulations likely capture a portion of these dynamical changes that occur during the initial phases of activation as only the active system exhibits increased coupling of motions between TM helices III and VI. Additionally, simulations were able to observe bending of the extracellular end of TM helix II towards the binding pocket of IT1t and bending of the intracellular end of TM helix V towards TM helix VI in the active system. The intracellular end of TM helix V is especially interesting as key residues involved in switches (Y2195.58) for receptor activation and in G protein coupling (L2265.65) are located within this region (Wescott et al., 2016).
In summary, we report the effects of five antagonists on WT and N1193.35A CXCR4 signaling, the inhibition of HIV-1 and CXCL12 binding, and the identification of two biased antagonists. We use the structure of CXCR4-IT1t to develop a model for the mechanism of activation in the N1193.35A CAM. While disrupting the unique hydrogen bond network involving three TM helices is not involved in constitutive activity, MD simulations (a) predict increased coupling of helices III and VI through F872.53 in the CAM, (b) model the structural changes associated with activation of N1193.35A in greater detail, and (c) are consistent with experimental observations describing receptor activation of WT CXCR4 (Wescott et al., 2016). Mutants of F872.53 confirm the importance of the phenylalanine in constitutive activity and also eliminate the inverse agonism of IT1t and B9.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Elias Lolis (elias.lolis@yale.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cells Lines
The S. cerevisiae strain used to express a functional human CXCR4 is CY12946 (MATα FUS1p-HIS3 GPA1Gαi2(5) can1 far1∆1442 his3 leu2 sst2∆2 ste14::trp1::LYS2 ste3∆1156 tbt1–1 trp1 ura3), which we have described previously (Sachpatzidis et al., 2003). Human CXCR4 is expressed from the constitutive phosphoglycerate kinase (PGK1) promoter under the selectable marker LEU2 in the plasmid Cp4181. When the pheromone pathway is activated by CXCR4, plasmid Cp1584 expresses the β-galactosidase under the control of a FUS1 promoter. This plasmid contains the TRP1 selectable marker.
Human CXCR4 cDNA was subcloned into the pIRES2-EGFP vector, transfected into HEK-293 cells (female), and selected by 1 mg/mL G418 sulfate and EGFP (through high content analysis). The selected clone was maintained in DMEM supplemented with 10% fetal bovine serum (FBS) and 0.5 mg/mL G418 sulfate. CEM and TZM-bl cells (both female) were cultured as described previously (Paintsil et al., 2007). MDA-MB-231 cells (female) were cultured in DMEM with 10% FBS with 1% glutamine/penicillin/streptomycin. All cells were cultured at 37°C in a humidified incubator with 5% CO2. Cell identities were verified by short tandem repeat profiling performed by the University of Michigan DNA sequencing core.
METHOD DETAILS
Chemical synthesis of B7 and CX0298
Linkers I and II for CX0298 and B7, respectively, are shown in Scheme S1A and were synthesized by following a similar synthetic sequence described in a manuscript (Wu et al., 2015a) or in a patent (Yen et al., 2006). The core molecules for CX0298 and B7 were synthesized by following a similar synthetic sequence reported previously (Wu et al., 2015b). Scheme S1B outlines the synthesis steps used to combine linkers and core molecules in order to generate compounds B7 and CX0298. The chemical structure of both compounds were authenticated by both 1H and 13C NMR and mass spectrometry. All test compounds displayed more than 95% purity, as determined by an Agilent 1100 series HPLC system using a C18 column as previously described (Wu et al., 2015b).
NMR profiling of B7 and CX0298
Unless otherwise stated, all materials used were commercially available and used as supplied. Reactions requiring anhydrous conditions were performed in flame-dried glassware and cooled under an argon or nitrogen atmosphere. Reactions were carried out under argon or nitrogen and monitored by analytical thin layer chromatography performed on glass-backed plates (5 × 10 cm) pre-coated with silica gel 60 F254 as supplied by Merck. Visualization of the resulting chromatograms was performed by looking under an ultraviolet lamp (λ = 254 nm). Flash chromatography was used routinely for purification and separation of product mixtures using silica gel 60 of 230−400 mesh size as supplied by Merck. Eluent systems are given in volume/volume concentrations. 1H and 13C NMR spectra were recorded on a Varian Mercury-300 (300 MHz) and a Varian Mercury-400 (400 MHz). Chloroform-d, methanol-d4 or deuterium oxide-d2 was used as the solvent and TMS (δ 0.00 ppm) as an internal standard. Chemical shift values are reported in ppm relative to the TMS in delta (δ) units. Multiplicities are recorded as s (singlet), br s (broad singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), dt (doublet of triplets), and m (multiplet). Coupling constants (J) are expressed in Hertz. Electrospray mass spectra (ESMS) were recorded as m/z values using an Agilent 1100 MSD mass spectrometer. IUPAC nomenclature of compounds was determined with ACD/Name Pro software.
The NMR profiles of N-Cyclohexyl-N’-{4-[(2-piperazin-1-yl-9H-purin-6-ylamino)-methyl]-cyclohexylmethyl}-propane-1,3-diamine hydrochloride salt (B7) were as follows: 1H NMR (400 MHz, D2O) δ 8.34 (s, 1H), 4.12 (t, J=4. 8 Hz, 4H), 3.54 (d, J=6.0 Hz, 2H), 3.44 (t, J=4.8 Hz, 4H), 3.21−3.12 (m, 4H), 2.98 (d, J=6.8 Hz, 2H), 2.18−2.02 (m, 4H), 1.96−1.80 (m, 6H), 1.78−1.62 (m, 3H), 1.42−1.30 (m, 5H), 1.23−1.05 (m, 5H); 13C NMR (100 MHz, D2O) δ 153.54, 151.92, 146.44, 139.36, 105.49, 57.46, 53.55, 46.71, 44.93, 42.67, 41.85, 41.28, 36.86, 34.45, 29.21, 29.17, 28.77, 24.40, 23.82, 22.71; ESMS m/z: 484.3 (M+1).
The NMR profiles of N-Cyclohexyl-N’-(3-{4-[(2-piperazin-1-yl-9H-purin-6-ylamino)-methyl]-[1,2,3]triazol-1-yl}-propyl)-propane-1,3-diamine hydrochloride salt (CX0298) were as follows: 1H NMR (400 MHz, D2O) δ 8.48(s, 1H), 8.11 (s, 1H), 4.92 (s, 2H), 4.58 (t, J=6.8 Hz, 2H), 4.07 (t, J=5.2 Hz, 4H), 3.36 (t, J=5.2 Hz, 4H), 3.21−3.08 (m, 6H), 2.34 (m, 2H), 2.16−2.02 (m, 4H), 1.81 (m, 2H), 1.63 (m, 1H), 1.40−1.14 (m, 6H); 13C NMR (100 MHz, D2O) δ 157.41, 154.13, 150.34, 146.94, 141.64, 126.94, 107.79, 60.02, 50.16, 47.33, 47.19, 45.24, 44.26, 43.76, 38.33, 31.34, 28.63, 26.97, 26.39, 25.39; ESMS m/z: 497.3 (M+1).
S. cerevisiae luciferase assays
CY12946 yeast were transformed with Cp4181 plasmids encoding either WT or mutant CXCR4, as well as plasmid Cp1584 containing β-galactosidase on an inducible FUS1 promoter. Individual clones were grown up in yeast synthetic Leu− Trp− (L−W−) dropout media and used to make glycerol stocks as well as for sub-culturing. All media lacking leucine and tryptophan for growth of the target yeast strain were obtained and prepared as described previously (Ausube et al., 1993). Liquid cultures were sub-cultured by inoculating into fresh media at a ratio of 1:100 (v/v). All cultures were grown in a shaker incubator at 30°C and 200 rpm. Cultures were sub-cultured at least once and allowed to grow to saturation (OD600 of >2) before being used for signaling experiments. For dose-response experiments, compounds were dissolved in either water (AMD3100) or DMSO (all other compounds) at a stock concentration of 50 mM.
Compounds were serially diluted in the assays. For experiments in which CXCL12 was co-administered to the yeast, the dilutions were prepared in 96-well plates with the initial dilution at a concentration of 10−4 M. Each subsequent well received an aliquot from the previous well to create a higher dilution by one-half log unit. Wells were covered with aluminum seals and stored at 4°C until use. CXCL12 itself was prepared by diluting from a 1.75 mM stock to a 40 μM working stock in dH2O. For experiments in which CXCL12 was not co-administered, the compounds were initially diluted to 2 mM, with subsequent tenfold dilutions to allow us to test concentrations one log unit apart.
For signaling experiments, subcultured yeast cultures at an OD600 of >2 were diluted into fresh L− W− media. For all experiments the final OD600 was 0.1. Note that the final OD600 is the number obtained after addition of the compounds, dH2O or DMSO, and CXCL12 (if added). The diluted cells were added to a clear, flat-bottom, white, 96-well plate with each well having a final volume of 100 μL (including compounds, dH2O or DMSO, and CXCL12, if present). Compounds accounted for 5% of the well volume and CXCL12 accounted for another 5% of well volume when present. The final concentration of CXCL12, when present, was 2 μM. Assays were performed in triplicate (for dose-response assays without CXCL12 and to test F872.53 CXCR4 mutants), quadruplicate (for dose-response assays with CXCL12), or with six replicates (to test the basal activities of all other CXCR4 mutants).
Once the assay plates were prepared, they were subsequently coated with Breathe-Easy sealing membranes and placed into shaker incubators without the lids, allowing for gas exchange throughout the assay. The yeast cells were allowed to grow at 200 rpm for 2 – 4 hours at 30°C. β-galactosidase expression was quantified using the Beta-Glo assay system. Plates were first allowed to cool to room temperature before a sample of each well was transferred to a new plate containing an equal volume of Beta-Glo reagent. Typically, 80 μL of each well solution was added to 80 μL of the Beta-Glo reagent with vigorous mixing. The plates were then covered with aluminum seals and shaken at 800 rpm for 40 minutes on a plate shaker. The aluminum seal was removed and luminescence signals were measured using an Infinite M200 plate reader (TECAN) with i-control software. Data were plotted by deducting blanks (i.e., wells without cells but containing media) and then performing nonlinear regression analyses using GraphPad software. Results represent data from ≥ two independent experiments (with compounds or CXCL12 alone) or three independent experiments (with CXCR4 mutants).
Radioligand Binding Assays
These procedures were based on a previously published experiment (Wu et al., 2012). Briefly, a stable HEK-293 cell line expressing human CXCR4 was grown with DMEM supplemented with 10% fetal bovine serum and 0.5 mg/mL G418 sulfate in a humidified incubator with 5% CO2 at 37°C. For membrane purification, cells were homogenized in ice-cold buffer A (50 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 2.5 mM EDTA, 10% sucrose) with freshly prepared 1 mM PMSF. The homogenate was centrifuged at 3500 g for 15 minutes at 4° C. The supernatant was removed and re-centrifuged at 43000 g for 0.5 h at 4°C. The pellet was resuspended in buffer A and stored at −80°C until use. 2 – 4 μg of purified membrane was incubated with 0.16 nM 125I−CXCL12 and compounds of interest in the incubation buffer (50 mM HEPES-NaOH, pH 7.4, 100 mM NaCl, 5 mM MgCl2, 1 mM CaCl2, 0.5% BSA). Nonspecific binding was defined in the presence of 50 μM AMD3100. The reaction mixtures were incubated for 1.5 h at 30°C, transferred to a 96-well GF/B filter plate (Millipore Corp., Billerica, MA), terminated by manifold filtration, and washed with ice-cold wash buffer (50 mM HEPES-NaOH, pH 7.4, 100 mM NaCl) four times. The radioactivity bound to the filter was measured by a Topcount detector (PerkinElmer Inc., Waltham, MA). IC50 values from three independent experiments were determined by the concentrations of compounds required to inhibit 50% of the specific binding of 125I-CXCL12 and were calculated by nonlinear regression using GraphPad software.
IC50 determination for HIV-1 inhibition
The IC50 for HIV-1 inhibition was based on luciferase activity using TZM-bl cells (a HeLa-derived cell line) that assesses the efficacy of the CXCR4 antagonists to prevent HIV-1 infectivity (Wu et al., 2015b). TZM-bl cells express high levels of CD4, CCR5 and CXCR4 receptors, and contain HIV-1 long terminal repeat-driven β-galactosidase and luciferase reporter cassettes that are activated by HIV-1 Tat expression (Paintsil et al., 2007). For the luciferase reporter experiments, 5 × 103 cells/well were cultured in 96-well plates for 1 – 2 days in a 37°C incubator with 5% CO2. The cells were treated with serial dilution of each compound for 30 minutes prior to infecting with HIV-1 IIIB at a multiplicity of infection (MOI) of one. β-galactosidase activity from each well were recorded with a FARCyte machine. The standard deviation was based on ≥ three independent experiments.
Cytotoxicity Assays
For cytotoxicity assays both the acute lymphoblastic lymphoma CEM cells and TZM-bl cells were used. Cells (1 × 104/well) were cultured in a 24-plate overnight before treating with the CXCR4 antagonists for 72 hours. 0.5% methylene blue in 50% ethanol was used to fix and stain the cells for two hours at room temperature. Excess dye was removed by washing with water. Dried plates were re-suspended in 1% sarkosyl for 3 hours at room temperature. Methylene blue was oxidized by living cells to a colorless product, while dead cells remained blue colored. Cell growth was quantitated based on the amount of methylene blue adsorbed into cells as measured by a spectrophotometer at 595 nm. All experiments were performed in triplicate wells and repeated at least three times to get the standard deviation.
Arrestin signaling pathway determination
We previously described MDA-MB-231 cells stably expressing a click beetle green luciferase complementation reporter for interaction of CXCR4 with β-arrestin-2 (Coggins et al., 2014). We performed complementation assays with these cells as reported (Coggins et al., 2014). Briefly, cells were pre-treated with various concentrations of compounds for one hour and then administered 200 ng/mL CXCL12-α. Cells were imaged live at intervals in order to track the course of the signaling in real time (n = 4 replicates per condition; two independent experiments). IC50 values were determined at the point of maximal CXCL12-induced signal (i.e., 20 minutes post-CXCL12 stimulation).
Analysis of the N119A CXCR4 CAM
The structure of the CXCR4-T4 lysozyme fusion protein complexed with IT1t (PDB 3ODU) was obtained from the Protein Databank (PDB) and displayed in PyMOL. Coordinates of T4 lysozyme and water molecules were removed. Interactions involving N119 were evaluated, and were used as the basis for designing additional mutants to understand the mechanism of the constitutively active mutants N1993.35A/S.
Generation of additional CXCR4 mutants
Site-directed mutagenesis was used to mutate CXCR4 within the Cp4181 plasmid. Following mutagenesis using the PfuTurbo polymerase and our mutagenic oligos, the methylated plasmid DNA strands encoding WT CXCR4 were degraded with DpnI for 1 – 2 hours at 37°C. DNA was then transformed using E. coli XL10-Gold Ultracompetent cells and plated on LB/ampicillin plates overnight to select for modified plasmids. Individual clones were grown in liquid LB/ampicillin media and DNA was subsequently purified using commercial miniprep kits (Qiagen) for sequencing. Plasmids with verified mutants were used to transform yeast cells via the lithium acetate method (Agatep et al., 1998).
Molecular Dynamics simulations
The crystal structure of CXCR4 in complex with IT1t was retrieved from the PDB with identifier 3ODU. Non-canonical residues were removed (lysozyme), and the resulting gap was remodeled with a loop refinement protocol from Modeller (Fiser et al., 2000). Hydrogens were then added according to the most probable protonation state at pH 7.0 as predicted by PROPKA (Olsson et al., 2011). For all subsequently described simulations, the CHARMM36 forcefield (Best et al., 2012) was used to parameterize protein, lipids, and solvent, and the CGENFF webserver (Vanommeslaeghe et al., 2009) was used to generate CHARMM-compatible parameters for the small-molecule IT1t. All simulations were performed in Nanoscale Molecular Dynamics (Phillips et al., 2005) with Langevin temperature and pressure control, particle mesh Ewald electrostatics, and bond constraints imposed by the SHAKE algorithm.
To prepare for simulations, the CXCR4(N119)-IT1t complex was aligned along the z-axis, solvated in an aqueous sodium chloride solution at 150 mM with a net system charge of zero, and placed in a phosphatidylcholine (POPC) lipid bilayer in the x-y plane using Visual Molecular Dynamics, VMD (Humphrey et al., 1996). The membrane was melted by restraining all atoms not within the POPC tails, minimizing the energy for 2 ps, and simulating the system dynamics for 500 ps. Next, harmonic constraints of were imposed on all CXCR4 atoms, and the system was once again minimized for 2 ps and equilibrated for 500 ps while preventing hydration of the membrane-protein interface. Finally, the protein was released and the entire system was equilibrated for 500 ps.
After equilibrating the WT-IT1t simulation system, we generated three additional simulation systems: N1193.35A-IT1t, WT (apo), and N1193.35A (apo). The N1193.35A mutation was performed in VMD (Humphrey et al., 1996) with the Mutator plugin, and removal of IT1t was performed by deletion of the molecule in the structural files. Each of the four simulation environments were then subject to a 100 ns molecular dynamics simulation.
In order to sample conformational space at an improved rate, we used each of the prior trajectories from standard molecular dynamics to parameterize a corresponding accelerated simulation for each of the four simulation systems. These simulations were performed for 100 ns with a boost potential applied to dihedral angles. The energy threshold, E, and acceleration factor, α, for application of the boost potential were calculated according to
where 〈Vdihedral〉 represents the average dihedral energy from the initial 100 ns molecular dynamics trajectories. These parameters are consistent with prior simulations of GPCRs (Gaieb et al., 2016; Miao et al., 2013).
Analysis of all molecular dynamics trajectories was performed with custom Python (Foundation) scripts leveraging the MDTraj library (McGibbon et al., 2015). Allosteric signals were quantified with the signals method from the TimeScapes software (Wriggers et al., 2009). Briefly, this method determined all pairwise residue distances where contacts are considered to occur. For this approach, we consider a contact to occur if for a simulation the distance value for one standard deviation below the mean is below a cutoff distance of 6 Å. Furthermore, we excluded all contacts between residue pairs separated by less than 3 amino acids. TimeScapes then calculated all pairwise correlations for distance time-series and projected the results into a residue correlation matrix. For each residue pair in the correlation matrix, the projection is achieved by summing all individual pairs of distance correlations that involve at least one residue from the residue pair.
Sequence similarity analyses
The full sequence for human CXCR4 (Uniprot: P61073) was uploaded to the PHMMER method on the HMMER webserver (Potter et al., 2018) with default parameters. Results were filtered to only those sequences from Homo sapiens, and all redundant sequences (based on accession identifiers) were removed. Amino acid frequencies at positions 87, 120, and 252 were calculated using custom Python scripts leveraging the Biopython (Cock et al., 2009) library.
S. cerevisiae receptor expression analysis
Yeast strains containing CXCR4 were grown up in Leu− Trp− synthetic dropout media as described above. A negative control strain lacking plasmid cp4181 was grown in Trp− synthetic dropout media. Saturated yeast cells were lysed in pre-chilled solution containing 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1mM EDTA, 1% Triton X-100, and a protease inhibitor cocktail. Glass beads were used to aid in cell lysis. The resulting lysate was spun down for 30 minutes at 4°C in a microcentrifuge and the supernatant was collected. BCA assays were performed against a BSA standard in order to normalize protein levels, and the normalized samples were then mixed 1:1 with 2x LDS sample buffer containing 100 mM DTT. The samples were incubated at room temperature for five minutes and were then run on 4 – 12% bis-tris polyacrylamide gels, 15 μg protein per lane. One gel was used to measure total protein levels for each sample with a Coomassie-based dye, while the other gel was used to transfer proteins to a nitrocellulose membrane. The membrane was promptly blocked for one hour at room temperature with 5% non-fat dry milk dissolved in 1x TBST. After blocking, the membrane was transferred to a solution containing rabbit anti-CXCR4 primary antibody in 3% milk dissolved in 1x TBST, and was incubated overnight at 4°C. The membrane was then washed three times with 1x TBST (five minutes each) and then incubated for one hour at room temperature in a solution containing anti-rabbit-IRDye800 in 3% milk dissolved in 1x TBST. The membrane was washed again as above and was imaged on an Odyssey CLx scanner. Image Studio Lite was used to analyze expression levels.
QUANTIFICATION AND STATISTICAL ANALYSIS
The numbers or replicates used for each assay and the software used to analyze the assay results are reported in Method Details and in figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| IRDye 800CW Donkey anti-Rabbit IgG (H + L) Antibody | LI-COR Biosciences | Cat# 925–32213, RRID:AB_2715510 |
| Monoclonal anti-CXCR4 produced in rabbit (clone UMB2) | Abcam | Cat# ab124824, RRID:AB_10975635 |
| Bacterial and Virus Strains | ||
| Escherichia coli XL10-Gold Ultracompetent cells | Stratagene | Cat# 200314 |
| HIV-1 IIIB | NIH AIDS Reagent Program | Cat# 398 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 1-pentanol | Alfa Aesar | Cat# A13093 |
| 125I-CXCL12 | PerkinElmer | Cat# NEX346 |
| 1,4-Dithiothreitol | Sigma-Aldrich | Cat# 10708984001 |
| 2,6-Dichloropurine | AK Scientific | Cat# 865279 |
| 37% HCl (aq) | Fluka | Cat# 10190802 |
| Agar | Fisher Scientific | Cat# DF0140 |
| AMD3100 | Sigma-Aldrich | Cat# A5602 |
| Argon | Sinda Gas | Cat# 90111 |
| B7 | This paper | Custom |
| B8 (Wu et al., 2015b) | Synthesized according to published literature | Custom |
| B9 (Wu et al., 2015a) | Synthesized according to published literature | Custom |
| BD Difco™ Yeast Nitrogen Base without Amino Acids | Fisher Scientific | Cat# DF0919 |
| Bovine Serum Albumin Fraction V | Roche | Cat# 10735094001 |
| Calcium chloride | Sigma-Aldrich | Cat# C5080 |
| Carbowax PEG 3350 | Electron Microscopy Services | Cat# 50–249-15 |
| Chloroform-d | Sigma-Aldrich | Cat# 151823 |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich | Cat# 11836170001 |
| CX0298 | This paper | Custom |
| CX344 (Wu et al., 2015b) | Synthesized according to published literature | Custom |
| Deuterium oxide-d | Sigma-Aldrich | Cat# 151882 |
| Dichloromethane | Sigma-Aldrich | Cat# 270997 |
| DMEM | Thermo-Fisher | Cat# 11965092 |
| DMSO | AmericanBio | Cat# AB00435 |
| dNTP Mix (10 mM ea) | Thermo-Fisher | Cat# 18427013 |
| DpnI | New England Biolabs | Cat# R0176S |
| EDTA, 0.5M solution, pH 8.0 | AmericanBio | Cat# AB00502 |
| Ethyl acetate | Macron | Cat# UN1173 |
| Fetal Bovine Serum | Thermo-Fisher | Cat# 10437036 |
| G418 | Sigma-Aldrich | Cat# A1720 |
| Glucose | Alfa Aesar | Cat# A16828 |
| HEPES | Sigma-Aldrich | Cat# H3375 |
| Histidine | Sigma-Aldrich | Cat# H0750000 |
| IT1t | Tocris | Cat# 4596 |
| Leucine | Sigma-Aldrich | Cat# L0375000 |
| Lithium acetate | Sigma-Aldrich | Cat# L6883 |
| Luria Broth Base | Thermo-Fisher | Cat# 12795027 |
| Magnesium chloride | Sigma-Aldrich | Cat# 2444–01 |
| Methanol-d | Sigma-Aldrich | Cat# 151947 |
| Methylene Blue solution | Sigma-Aldrich | Cat# 1808 |
| N-(3-Aminopropyl)cyclohexylamine | Alfa Aesar | Cat# L11250 |
| N-Lauroylsarcosine sodium salt | Sigma-Aldrich | Cat# L9150 |
| NuPAGE LDS Sample Buffer (4X) | Thermo-Fisher | Cat# NP0008 |
| NuPAGE MES SDS Running Buffer (20X) | Thermo-Fisher | Cat# NP0002 |
| Penicillin-Streptomycin-Glutamine | Thermo-Fisher | Cat# 10378016 |
| PfuTurbo DNA Polymerase | Agilent | Cat# 600250 |
| Piperazine | Acros | Cat# 203–808-3 |
| Poly(ethyleneimine) solution | Fluka Analytical | Cat# P3143 |
| Phenylmethylsulfonyl fluoride | Sigma-Aldrich | Cat# P7626 |
| Propargylamine | Alfa Aesar | Cat# H53495 |
| Recombinant CXCL12 (Murphy et al., 2007) | Synthesized according to published literature | Custom |
| Recombinant CXCL12-α | R&D Systems | Cat# 350-NS |
| Salmon Sperm DNA | Thermo-Fisher | Cat# AM9680 |
| SeeBlue Plus2 Pre-stained Protein Standard | Thermo-Fisher | Cat# LC5925 |
| SimplyBlue SafeStain | Thermo-Fisher | Cat# LC6060 |
| Sodium chloride | Sigma-Aldrich | Cat# S9888 |
| Sucrose | JT Baker | Cat# 4072 |
| TBST Buffer, 10X solution, pH 7.4 | AmericanBio | Cat# AB14330 |
| Tetramethylsilane | Sigma-Aldrich | Cat# 87921 |
| Trans-4-(aminomethyl)cyclohexane carboxylic acid | HY Biocare | Cat# TA5528 |
| Triton X-100 | Sigma-Aldrich | Cat# X100 |
| TsOH.H2O | Sigma-Aldrich | Cat# 402885 |
| Uracil | Sigma-Aldrich | Cat# U0750 |
| Yeast Synthetic Drop-out Medium Supplements without histidine, leucine, tryptophan and uracil | Sigma-Aldrich | Cat# Y2001 |
| Critical Commercial Assays | ||
| Beta-Glo Assay System | Promega | Cat# E4780 |
| Pierce BCA Protein Assay Kit | Thermo-Fisher | Cat# 23225 |
| Deposited Data | ||
| RCSB Protein Data Bank | (Wu et al., 2010) | PDB: 3ODU |
| Experimental Models: Cell Lines | ||
| CEM | NIH AIDS Reagent Program | Cat# 117 |
| HEK-293 | ATCC | Cat# CRL-1573 |
| MDA-MB-231 | ATCC | Cat# HTB-26 |
| TZM-bl | NIH AIDS Reagent Program | Cat# 8129 |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae strain CY12946 | Gift from John Manfredi (Cadus Pharmaceutical Corporation) | N/A |
| Oligonucleotides | ||
| Primer F87A F: GTGGCCGACCTCCTCGCTGTCATCACGCTTCCC | This paper | N/A |
| Primer F87A R: GGGAAGCGTGATGACAGCGAGGAGGTCGGCCAC | This paper | N/A |
| Primer F87T F: GTGGCCGACCTCCTCACTGTCATCACGCTTCCC | This paper | N/A |
| Primer F87T R: GGGAAGCGTGATGACAGTGAGGAGGTCGGCCAC | This paper | N/A |
| Primer N119A F: GTCATCTACACAGTCGCACTCTACAGCAGTGTC | This paper | N/A |
| Primer N119A R: GACACTGCTGTAGAGTGCGACTGTGTAGATGAC | This paper | N/A |
| Recombinant DNA | ||
| pIRES2-EGFP | Clontech Laboratories | Cat# 6064–1 |
| Plasmid cp1584 encoding β-galactosidase | Gift from John Manfredi (Cadus Pharmaceutical Corporation) | N/A |
| Plasmid cp4181 encoding CXCR4 | Gift from John Manfredi (Cadus Pharmaceutical Corporation) | N/A |
| Software and Algorithms | ||
| ACD/Name pro software | ChemDraw | https://www.acdlabs.com/products/draw_nom/nom/name/ |
| CGenFF | University of Maryland | https://cgenff.umaryland.edu/ |
| GraphPad Prism | GraphPad Software Inc. | https://www.graphpad.com/scientificsoftware/prism/ |
| i-control | TECAN | https://lifesciences.tecan.com/software-overview |
| Image Studio Lite | LI-COR | https://www.licor.com/bio/products/software/image_studio_lite/ |
| MDTraj | Stanford University | https://github.com/mdtraj/mdtraj |
| Modeller | University of California, San Francisco | https://salilab.org/modeller/ |
| Nanoscale Molecular Dynamics | University of Illinois at Urbana-Champaign | http://www.ks.uiuc.edu/Research/namd/ |
| PHMMER | Howard Hughes Medical Institute and Harvard University | https://www.ebi.ac.uk/Tools/hmmer/search/phmmer |
| PROPKA | University of Copenhagen | https://github.com/jensengroup/propka-3.1 |
| PyMOL | Schrödinger | https://pymol.org/2/ |
| Python3 | Python Software Foundation | https://www.python.org/ |
| TimeScapes | Biomachina.org | http://timescapes.biomachina.org/ |
| UCSF Chimera | University of California, San Francisco | https://www.cgl.ucsf.edu/chimera/ |
| Visual Molecular Dynamics | University of Illinois at Urbana-Champaign | http://www.ks.uiuc.edu/Research/vmd/ |
| Other | ||
| 300 MHz Nuclear magnetic resonance spectroscopy | Varian Inc. | Cat# 103843 |
| 400 MHz Nuclear magnetic resonance spectroscopy | Varian Inc. | Cat# 239228 |
| 96-well Flat Clear Bottom White Polystyrene TC-treated Microplates | Corning | Cat# 3610 |
| 96-well GF/B filter plate | Millipore | Cat# MAHFB1H |
| AlumaSeal II Sealing Film | Dot Scientific | Cat# T489 |
| Breathe-Easy film | USA Scientific | Cat# 9123–6100 |
| Fluorescence scanner | LI-COR | Model# Odyssey CLx |
| Glass backed plates precoated silica gel 60 F254 | Merck | Cat# 1057150001 |
| Glass beads, acid-washed 425–600 μm | Sigma-Aldrich | Cat# G8772 |
| High-performance liquid chromatography system | Agilent | Model# 1100 series |
| iBlot Gel Transfer Device | Thermo-Fisher | Cat# IB1001 |
| iBlot Transfer Stack, nitrocellulose | Thermo-Fisher | Cat# IB301032 |
| Mass spectrometer | Agilent | Model# 1100 series |
| Microplate reader | TECAN | Model# Infinite M200 |
| Microplate reader | TECAN | Model# FARCyte |
| NuPAGE 4–12% Bis-Tris Protein Gels, 1.0 mm, 10-well | Thermo-Fisher | Cat# NP0321 |
| QIAprep Spin Miniprep Kit | Qiagen | Cat# 27106 |
| Silica gel 60 F254 (230–400 mesh) | Merck | Cat# 1093859025 |
| TopCount NXT | PerkinElmer | Cat# C991201 |
SIGNIFICANCE.
CXCR4 is a GPCR involved in homeostasis, HIV infection, and cancer. Herein we characterize ligands that function as neutral antagonists for WT CXCR4 but have different functions on the CXCR4 constitutively active mutant N1193.35A. We tested two previously uncharacterized ligands for their ability to inhibit HIV-1 infection and CXCL12 binding. We also tested all ligands for G protein coupling and β-arrestin-2 signaling. To understand the mechanism of the N1193.35A CAM, we generated mutants of CXCR4 based on structural data and molecular dynamics simulations. We identified that F872.53 is responsible for constitutive activity through formation of a hydrophobic triad (comprised of residues F872.53, L1203.36, and W2526.48) and mediates inverse agonism of two of the tested ligands.
Highlights:
Antagonists of wild-type CXCR4 exhibit differential effects on the N1193.35A mutant
The mutant’s constitutive activity stems from residues forming a hydrophobic triad
Perturbation of the hydrophobic triad via mutation ablates constitutive activity
ACKNOWLEDGEMENTS
We would like to thank Will Hungerford (Yale Center for Molecular Discovery) for performing liquid chromatography/mass spectrometry and NMR analyses on the five compounds at Yale. We would also like to thank Keith Weise and Joel Sexton (Yale Pharmacology) for their expertise in S. cerevisiae. This work was supported by the Yale Cancer Center Pilot Funds and NIH grant R01AI082295 (EL). ER was supported by F31CA220854. GDL acknowledges grant support from U01CA210152, R01CA196018, R01CA195655, and R37CA222563. KEL was supported by R50CA221807. We are grateful to the National Health Research Institutes and Ministry of Science and Technology of the Republic of China (MOST 101-2325-B-400-016) for financial support (KSS). RESH received funding from the University of California President’s Dissertation-Year Fellowship. Computations were performed using the computer clusters and data storage resources of the UCR’s High-Performance Computing Center (http://hpcc.ucr.edu), which were funded by grants from NSF (MRI-1429826) and NIH (1S10OD016290-01A1), and using GPU workstations in the Biomolecular Modeling and Design Lab (https://biomodel.engr.ucr.edu). We also gratefully acknowledge the support of NVIDIA Corporation with the donation of the Tesla K40 GPU used for parts of this research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
KEL and GDL receive research funding from Polyphor.
DATA AND SOFTWARE AVAILABILITY
All software used in this study is reported in Method Details and indicated in the Key Resources Table.
REFERENCES
- Agatep R, Kirkpatrick R, Parchaliuk D, Woods R, and Gietz R (1998). Transformation of Saccharomyces cerevisiae by the lithium acetate/single-stranded carrier DNA/polyethylene glycol protocol. Tech Tips Online
- Ausube F, Brent R, Kingston R, Moore D, Siedman J, Smith J, and Struhl K, eds. (1993). Current Protocols in Molecular Biology (John Wiley and Sons; ). [Google Scholar]
- Ballesteros JA, and Weinstein H (1995). Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods in Neurosci 25, 366–428. [Google Scholar]
- Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, and MacKerell AD (2012). Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 Dihedral Angles. J Chem Theory Comput 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong BF, and Mohan C (2009). Targeting the CXCR4/CXCL12 axis in systemic lupus erythematosus. Expert Opin Ther Targets 13, 1147–1153. [DOI] [PubMed] [Google Scholar]
- Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, et al. (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggins NL, Trakimas D, Chang SL, Ehrlich A, Ray P, Luker KE, Linderman JJ, and Luker GD (2014). CXCR7 controls competition for recruitment of beta-arrestin 2 in cells expressing both CXCR4 and CXCR7. PLoS One 9, e98328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong X, and Golebiowski J (2018). Allosteric Na(+)-binding site modulates CXCR4 activation. Phys Chem Chem Phys 20, 24915–24920. [DOI] [PubMed] [Google Scholar]
- De Clercq E (2009). The AMD3100 story: the path to the discovery of a stem cell mobilizer (Mozobil). Biochem Pharmacol 77, 1655–1664. [DOI] [PubMed] [Google Scholar]
- DeVree BT, Mahoney JP, Velez-Ruiz GA, Rasmussen SG, Kuszak AJ, Edwald E, Fung JJ, Manglik A, Masureel M, Du Y, et al. (2016). Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature 535, 182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Broder CC, Kennedy PE, and Berger EA (1996). HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272, 872–877. [DOI] [PubMed] [Google Scholar]
- Fiser A, Kinh Gian Do R, and Sali (2000). Modeling Loops in Protein Structures. Protein Sci 9, 1753–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foundation, P.S. Python Language Reference, version 3.5.
- Gaieb Z, Lo DD, and Morikis D (2016). Molecular mechanism of biased ligand conformational changes in CC chemokine receptor 7. J Chem Inf Model 56, 1808–1822. [DOI] [PubMed] [Google Scholar]
- Hendrix CW, Collier AC, Lederman MM, Schols D, Pollard RB, Brown S, Jackson JB, Coombs RW, Glesby MJ, Flexner CW, et al. (2004). Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. J Acquir Immune Defic Syndr 37, 1253–1262. [DOI] [PubMed] [Google Scholar]
- Hernandez PA, Gorlin RJ, Lukens JN, Taniuchi S, Bohinjec J, Francois F, Klotman ME, and Diaz GA (2003). Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nature Genet 34, 70–74. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K (1996). VMD: Visual molecular dynamics. J Mol Graph 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Kanan Y, Hamilton RA, Sherry DM, and Al-Ubaidi MR (2012). Sulfotyrosine. Exp Eye Res 105, 85–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V, and Stevens RC (2014). Allosteric sodium in class A GPCR signaling. Trends Biochem Sci 39, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Wong W, and AP IJ (2016). Human G protein-coupled receptor studies in Saccharomyces cerevisiae. Biochem Pharmacol 114, 103–115. [DOI] [PubMed] [Google Scholar]
- Ludeman JP, and Stone MJ (2014). The structural role of receptor tyrosine sulfation in chemokine recognition. Br J Pharmacol 171, 1167–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGibbon RT, Beauchamp KA, Harrigan MP, Klein C, Swails JM, Hernández CX, Schwantes CR, Wang LP, Lane TJ, and Pande VS (2015). MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys J 109, 1528–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y, Nichols S, Gasper P, Metger V, and McCammon J (2013). Activation and dynamic network of the M2 muscarinic receptor. Proc Natl Acad Sci USA 110, 10982–10987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mona CE, Besserer-Offroy E, Cabana J, Leduc R, Lavigne P, Heveker N, Marsault E, and Escher E (2016). Design, synthesis, and biological evaluation of CXCR4 ligands. Org Biomol Chem 14, 10298–10311. [DOI] [PubMed] [Google Scholar]
- Murphy JW, Cho Y, Sachpatzidis A, Fan C, Hodsdon ME, and Lolis E (2007). Structural and functional basis of CXCL12 (stromal cell-derived factor-1 alpha) binding to heparin. J Biol Chem 282, 10018–10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson MHM, Søndergaard CR, Rostkowski M, and Jensen JH (2011). PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J Chem Theory Comput 7, 525–537. [DOI] [PubMed] [Google Scholar]
- Paintsil E, Dutschman GE, Hu R, Grill SP, Lam W, Baba M, Tanaka H, and Cheng YC (2007). Intracellular metabolism and persistence of the anti-human immunodeficiency virus activity of 2’,3’-didehydro-3’-deoxy-4’-ethynylthymidine, a novel thymidine analog. Antimicrob Agents Chemother 51, 3870–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, and Schulten K (2005). Scalable molecular dynamics with NAMD. J Comput Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter SC, Luciani A, Eddy SR, Park Y, Lopez R, and Finn RD (2018). HMMER web server: 2018 update. Nucleic Acids Res 46, W200–w204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Kufareva I, Holden LG, Wang C, Zheng Y, Zhao C, Fenalti G, Wu H, Han GW, Cherezov V, et al. (2015). Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 347, 1117–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. (2011). Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachpatzidis A, Benton BK, Manfredi JP, Wang H, Hamilton A, Dohlman HG, and Lolis E (2003). Identification of allosteric peptide agonists of CXCR4. J Biol Chem 278, 896–907. [DOI] [PubMed] [Google Scholar]
- Sanchez-Ortega I, Querol S, Encuentra M, Ortega S, Serra A, Sanchez-Villegas JM, Grifols JR, Pujol-Balaguer MM, Pujol-Bosch M, Marti JM, et al. (2015). Plerixafor in patients with lymphoma and multiple myeloma: effectiveness in cases with very low circulating CD34+ cell levels and preemptive intervention vs remobilization. Bone Marrow Transplant 50, 34–39. [DOI] [PubMed] [Google Scholar]
- Schneider EH, Schnell D, Strasser A, Dove S, and Seifert R (2010). Impact of the DRY motif and the missing “ionic lock” on constitutive activity and G-protein coupling of the human histamine H4 receptor. J Pharmacol Exp Ther 333, 382–392. [DOI] [PubMed] [Google Scholar]
- Schneider EH, and Seifert R (2010). Sf9 cells: a versatile model system to investigate the pharmacological properties of G protein-coupled receptors. Pharmacol Ther 128, 387–418. [DOI] [PubMed] [Google Scholar]
- Schols D, Struyf S, Damme JV, Esté JA, Henson G, and Clercq ED (1997). Inhibition of T-tropic HIV Strains by Selective Antagonization of the Chemokine Receptor CXCR4. J Exp Med 186, 1383–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert R, and Wenzel-Seifert K (2002). Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch Pharmacol 366, 381–416. [DOI] [PubMed] [Google Scholar]
- Standfuss J, Edwards PC, D’Antona A, Fransen M, Xie G, Oprian DD, and Schertler GF (2011). The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature 471, 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddese B, Deniaud M, Garnier A, Tiss A, Guissouma H, Abdi H, Henrion D, and Chabbert M (2018). Evolution of chemokine receptors is driven by mutations in the sodium binding site. PLoS Comput Biol 14, e1006209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamura H, and Fujii N (2005). The therapeutic potential of CXCR4 antagonists in the treatment of HIV infection, cancer metastasis and rheumatoid arthritis. Expert Opin Ther Targets 9, 1267–1282. [DOI] [PubMed] [Google Scholar]
- Tehan BG, Bortolato A, Blaney FE, Weir MP, and Mason JS (2014). Unifying Family A GPCR Theories of Activation. Pharmacol Ther 143, 51–60. [DOI] [PubMed] [Google Scholar]
- Thoma G, Streiff MB, Kovarik J, Glickman F, Wagner T, Beerli C, and Zerwes HG (2008). Orally bioavailable isothioureas block function of the chemokine receptor CXCR4 in vitro and in vivo. J Med Chem 51, 7915–7920. [DOI] [PubMed] [Google Scholar]
- Tohgo A, Pierce KL, Choy EW, Lefkowitz RJ, and Luttrell LM (2002). beta-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J Biol Chem 277, 9429–9436. [DOI] [PubMed] [Google Scholar]
- Truax VM, Zhao H, Katzman BM, Prosser AR, Alcaraz AA, Saindane MT, Howard RB, Culver D, Arrendale RF, Gruddanti PR, et al. (2013). Discovery of tetrahydroisoquinoline-based CXCR4 antagonists. ACS Med Chem Lett 4, 1025–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto H, and Farrens DL (2013). A constitutively activating mutation alters the dynamics and energetics of a key conformational change in a ligand-free G protein-coupled receptor. J Biol Chem 288, 28207–28216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hout A, D’Huys T, Oeyen M, Schols D, and Van Loy T (2017). Comparison of cell-based assays for the identification and evaluation of competitive CXCR4 inhibitors. PLoS One 12, e0176057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, et al. (2009). CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wescott MP, Kufareva I, Paes C, Goodman JR, Thaker Y, Puffer BA, Berdougo E, Rucker JB, Handel TM, and Doranz BJ (2016). Signal transmission through the CXC chemokine receptor 4 (CXCR4) transmembrane helices. Proc Natl Acad Sci U S A 113, 9928–9933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wriggers W, Stafford K.a., Shan Y, Piana-Agostinetti S, Maragakis P, Lindorff-Larsen K, Miller PJ, Eastwood MP, Dror RO, and Shaw DE (2009). Automated Event Detection and Activity Monitoring in Long Time-Scale Molecular Dynamics. J Chem Theory Comput 5, 2595–2605. [DOI] [PubMed] [Google Scholar]
- Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, et al. (2010). Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CH, Chang CP, Song JS, Jan JJ, Chou MC, Wu SH, Yeh KC, Wong YC, Hsieh CJ, Chen CT, et al. (2012). Discovery of novel stem cell mobilizers that target the CXCR4 receptor. ChemMedChem 7, 209–212. [DOI] [PubMed] [Google Scholar]
- Wu CH, Song JS, Chang KH, Jan JJ, Chen CT, Chou MC, Yeh KC, Wong YC, Tseng CT, Wu SH, et al. (2015a). Stem cell mobilizers targeting chemokine receptor CXCR4: renoprotective application in acute kidney injury. J Med Chem 58, 2315–2325. [DOI] [PubMed] [Google Scholar]
- Wu CH, Wang CJ, Chang CP, Cheng YC, Song JS, Jan JJ, Chou MC, Ke YY, Ma J, Wong YC, et al. (2015b). Function-Oriented Development of CXCR4 Antagonists as Selective Human Immunodeficiency Virus (HIV)-1 Entry Inhibitors. J Med Chem 58, 1452–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen CF, Hu CK, Chou MC, Tseng CT, Wu CH, Huang YH, Chen SJ, and King CHR (2006). Pyrimidine compounds (Google Patents)
- Zhang D, Zhao Q, and Wu B (2015). Structural Studies of G Protein-Coupled Receptors. Mol Cells 38, 836–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Cai QY, Cai ZX, Fang Y, Zheng CS, Wang LL, Lin S, Chen DX, and Peng J (2016). Interactions of Bovine Serum Albumin with Anti-Cancer Compounds Using a ProteOn XPR36 Array Biosensor and Molecular Docking. Molecules 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WB, Navenot JM, Haribabu B, Tamamura H, Hiramatu K, Omagari A, Pei G, Manfredi JP, Fujii N, Broach JR, et al. (2002). A point mutation that confers constitutive activity to CXCR4 reveals that T140 is an inverse agonist and that AMD3100 and ALX40–4C are weak partial agonists. J Biol Chem 277, 24515–24521. [DOI] [PubMed] [Google Scholar]
- Zou YR, Kottmann AH, Kuroda M, Taniuchi I, and Littman DR (1998). Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 393, 595–599. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.