

Abstract
Alternative splicing of fibronectin increases expression of the EDA+ isoform of fibronectin (EDA+Fn), a damage-associated molecular pattern (DAMP) molecule, which promotes fibro-inflammatory disease through the activation of toll-like receptors (TLR). Our studies indicate that the fibronectin EDA domain drives two waves of gene expression in human dermal fibroblasts. The first wave seen at 2 hours consisted of inflammatory genes, VCAM1 and TNF. The second wave evaluated at 24 hours was composed of the fibrosis-associated cytokines, IL-10 and IL-13, and extracellular matrix genes fibronectin and osteopontin. Gene expression was coordinately regulated by the α4β1 integrin and the innate immune receptor, TLR4. Additionally, we found a significant TLR4/α4β1-dependent enrichment in the ratio of EDA+Fn to total fibronectin in response to EDA, consistent with EDA+Fn initiating further production of EDA+Fn. Our data also suggests that the EDA/α4β1 integrin interaction primes the cell for an enhanced response to TLR4 ligands. Our studies provide evidence that remodeling of the fibronectin matrix in injured or diseased tissue elicits an EDA-dependent fibro-inflammatory response in dermal fibroblasts. The data suggest a novel paradigm of DAMP based signaling whereby DAMP binding integrins cooperate with innate immune receptors to stimulate inflammation and fibrosis.
INTRODUCTION
Plasma fibronectin (pFn) is a soluble plasma protein which is synthesized by liver hepatocytes and assembled into the extracellular matrix of most tissues. Structurally, fibronectin is a modular protein whose secondary structure is organized into repeating, independently-folded domains (Type I, II, and III) based on amino acid sequence homology. Cellular fibronectin can also be synthesized locally by resident tissue fibroblasts and is increased in response to injury and pathological stimuli. Fibroblast synthesized fibronectin RNA undergoes alternative splicing which regulates the inclusion or exclusion of an additional Type III domain, Extra Domain A (EDA+Fn). EDA+Fn is selectively synthesized and polymerized into extracellular matrix by stromal fibroblasts. EDA+Fn is not detected in normal, resting tissue of the adult but is prominently expressed during development, wound healing, and in association with various disease processes (Schwarzbauer and DeSimone, 2011; White and Muro, 2011). The EDA domain of fibronectin is now widely recognized as a damage-associated molecular pattern (DAMP) capable of activating the innate immune receptor, toll-like receptor-4 (TLR4) on both immune and non-immune cells (Gondokaryono et al., 2007; Kelsh et al., 2014; McFadden et al., 2011; Okamura et al., 2001). That EDA+Fn contributes to prolonged inflammation and fibrosis has been documented in several mouse models of fibro-inflammatory disease (Arslan et al., 2011; Booth et al., 2012; Dhanesha et al., 2015; Julier et al., 2015; Khan et al., 2012; Kohan et al., 2011). Earlier research has highlighted a role for EDA+Fn and TLR4 in promoting cutaneous inflammation (McFadden et al., 2010; McFadden et al., 2011). More recently, keloid scars were found to have extensive amounts of EDA+Fn, which can potentially contribute to the increased inflammation and collagen production associated with this disease (Andrews et al., 2015; Kelsh et al., 2015). EDA+Fn dependent TLR4 signaling has been linked to the increased collagen production and myofibroblast differentiation seen in the skin of scleroderma patients. The TLR4 agonist activity of the EDA domain of fibronectin can be seen in the context of the intact molecule, proteolytic fragments and recombinant peptides (Julier et al., 2015; Bhattacharyya et al., 2014; Gondokaryono et al., 2007; Mogami et al., 2013; Doddapattar et al., 2015). However, the molecular mechanisms underlying EDA’s role in the initiation and persistence of fibrotic and inflammatory processes are not well understood.
Activation of TLR4 by its canonical ligand, lipopolysaccharide (LPS) requires the use of co-receptors or ancillary proteins, such as CD14 and MD2, to facilitate TLR4 activation (Park et al., 2009). The TLR4 co-receptors that control DAMP-initiated TLR4 activation and signaling are not well understood. Recently we reported that the interaction between the EDA domain and the α4β1 integrin receptor promotes a profibrotic contractile phenotype in dermal fibroblasts which is characterized by an increase in actin stress fibers, myosin light chain phosphorylation and fibronectin matrix (Shinde et al., 2015). These findings suggest that the α4β1 integrin may cooperate with innate immune receptors to control fibrotic outcomes in this cell type. In the current study we identify the EDA-binding integrin, α4β1, as essential to the TLR4-dependent induction of pro-fibrotic genes by the EDA domain. These data suggest that in dermal fibroblasts, the α4β1 integrin functions as a TLR4 co-receptor to initiate the fibro-inflammatory response to EDA+Fn.
RESULTS AND DISCUSSION
In order to assess alterations in fibrotic gene expression in response to EDA, we used real time RT-PCR microarray analysis and evaluated the changes in gene expression at early (2 hours) and late (24 hours) time points. To look at early gene induction, human dermal fibroblasts were seeded onto plasma fibronectin (pFn) alone or a mixture of pFn and the EDA domain for 2 hours. RNA was extracted and induced genes detected using both an extracellular matrix (ECM) and adhesion molecular array and a fibrosis array. Several genes were found to be upregulated >3.0 fold within 2 hours of seeding cells onto substrates coated with pFn and EDA compared with pFn alone (Figure 1a and b). These genes include tumor necrosis factor-alpha (TNF-α), interleukins 10 and 1A (IL-10, IL-1A), the chondroitin sulfate proteoglycan, Versican (VCAN) and the vascular cell adhesion molecule-1 (VCAM1) (Figure 1c and d). These genes have all been linked to the regulation of fibrosis and inflammation. At 2 hours, the most highly upregulated genes after seeding onto EDA were VCAM1 and TNF-α (Figure 1c and 1d). VCAM1 expression on stromal fibroblasts may have implications for the formation of tertiary lymph organs which are often seen in chronically inflamed tissue. It has been proposed that fibroblast VCAM1 by serving as the counter-receptor for the leukocyte α4β1 integrin promotes chronic inflammation by mediating lymphoid/stromal cell binding and preventing the emigration of leukocytes from sites of tissue damage (Buckley et al., 2015). These lymphoid/stromal cells can organize into structures, termed tertiary lymphoid organs, which promote chronic inflammation (Jones et al., 2016). TNF-α, a pro-inflammatory cytokine, has been shown to initiate a pathological cascade that begins with lingering inflammation ultimately leading to fibrotic disease in multiple organ systems (Duerrschmid et al., 2015; Matsui et al., 2014; Oikonomou et al., 2006). In addition to TNF-α and VCAM1, there was also an increase in chondroitin sulfate proteoglycan, Versican and the profibrotic cytokines IL-1 and IL-10. Of particular note, Versican has been identified as an activator of TLR2-dependent signaling to generate an inflammatory microenvironment (Kim et al., 2009), demonstrating its role as an ECM DAMP. Versican’s upregulation in response to EDA suggests a potential feed-forward loop in dermal fibroblasts, driven by ECM DAMPs. Collectively, the upregulation of these genes by EDA suggests an initial, robust pro-fibrotic inflammatory phenotype being evoked in these cells that can further lead to feed-forward fibro-inflammatory events.
Figure 1. Fibrosis gene expression profiling in response to EDA.

Human dermal fibroblasts were seeded onto wells coated with pFn (10 μg/ml) alone or a mixture of pFn (10 μg/ml) and EDA (40 μg/ml) in 0.1% BSA-DMEM for 2 hours (a–d) or 24 hours (e–h). Expression profiling of genes was performed with an Extracellular Matrix and Cell Adhesion Molecule array (a, e), as well as a Fibrosis array (b,f). Fold change lines indicate a 3.0-fold change in baseline. The genes whose expression was changed >3.0 fold are displayed in tables (c,d,g, and h).
Interestingly, we found that a different subset of genes became upregulated by EDA at 24 hours (Figure 1e and f). Several pro-fibrotic cytokines, TGF-β1, IL-10 and IL-13, were found to be upregulated by EDA (Figure 1g and h). TGF-β1 regulates fibrosis by promoting myofibroblast differentiation and the increased synthesis of collagen and EDA+fibronectin (Bochaton-Piallat et al., 2016). IL-10 and IL-13 are anti-inflammatory cytokines that have been implicated in Th2-dependent fibrosis (Kaviratne et al., 2004; Sziksz et al., 2015). The subsequent production of cytokines upon Th2 macrophage polarization can result in the generation of many different ECM-remodeling associated genes: LOX, MMP −2, −9, and procollagen –I and –III, demonstrating a critical link between the Th2 response and fibrogenesis (Kaminski et al., 2000; Decitre et al., 1998; Wang and Hirschberg, 2003; Underwood et al., 2000). Interestingly, IL-13 has been shown to indirectly activate latent TGF-β1 by regulating increased amounts of metalloproteases capable of releasing TGF-β1 from the ECM (Lanone et al., 2002; Lee et al., 2001). In our study, EDA resulted in increased TGF-β1 gene expression at 24 hours which, upon activation by IL-13, could lead to a feed-forward mechanism driving further production of EDA+Fn (White et al., 2010). Additionally, EDA induced the expression of the ECM proteins, osteopontin (SPP1) and fibronectin (FN1). This finding is in agreement with our recent finding that the α4β1 EDA interaction induces an increase in the accumulation of fibronectin matrix in human dermal fibroblasts (Shinde et al., 2015). Osteopontin is a multifunctional matricellular protein which has been implicated in organ fibrosis (Arriazu et al., 2017; Lancha et al., 2014; Lenga et al., 2008). Osteopontin has also been linked to myofibroblast differentiation in response to TGF-β, which in dermal and cardiac fibroblasts leads to a dramatic increase in EDA+Fn and alpha-smooth muscle actin (Lenga et al., 2008). The cell adhesion molecule, selectin (SELL) is a counter receptor for leukocyte selectins and like VCAM-1 could contribute to the formation of tertiary lymphoid tissue.
We further characterized the kinetics of expression of several of these genes in dermal fibroblasts by real time RT-PCR over a 24-hour time frame. Interestingly, we found two distinct waves of gene expression: an initial wave of inflammatory genes (i.e., VCAM1, TNF) seen at 2 hours (Figure 2a) followed by a late-phase wave of fibrotic genes at 24 hours that included the ECM proteins, osteopontin and fibronectin as well as the fibrosis-associated cytokines, IL-10, IL-13 (Figure 2b). Of particular interest was the upregulation of fibronectin expression. Inflamed and fibrotic skin is characterized by the increased deposition of the alternatively spliced EDA+Fn isoform into the extracellular matrix (Andrews et al., 2015; Bhattacharyya et al., 2014; Fullard and O’Reilly, 2016; McFadden et al., 2010). To assess whether EDA could also influence the isoform of fibronectin being synthesized, we utilized a primer specific to the EDA region of fibronectin or a primer that recognizes all isoforms of fibronectin and performed RT-PCR. We found that although there was no change in the overall expression of fibronectin at 2 hours, there was a change in the relative levels of the EDA+ isoform of fibronectin. At 2 hours, cells seeded onto EDA substrates exhibited an increase in the proportion of fibronectin mRNA containing the EDA domain compared to cells seeded to a control Type III (III-10n) module of fibronectin (Figure 2c). A quantitation of three separate experiments demonstrated a significant increase in the proportion of EDA+Fn being synthesized, suggesting that seeding cells onto EDA resulted in a rapid change in the alternative splicing of the Fn transcript (Figure 2d). To evaluate whether the changes in Fn splicing was accompanied by increased matrix deposition of the EDA+ isoform of Fn, cell layers were solubilized and analyzed by Western blot for expression of the EDA+ isoform of Fn using the IST-9 antibody directed at the EDA domain. As shown in Fig. 2e, EDA+Fn was markedly enhanced in cells seeded onto substrates coated with pFn+EDA.
Figure 2. EDA induces two distinct waves of gene expression.

Fibroblasts were seeded onto wells coated with a mixture of pFn (10 μg/ml) and EDA or the control module III-10n (40 μg/ml). RNA was extracted and gene expression evaluated by real time RT-PCR. (a,b) Expression of VCAM1, TNF, IL-13, IL-10, SPP1 and fibronectin mRNA was assessed at 2, 4 or 24 hours. Fold change values are expressed relative to cells plated on control well (pFn + III-10n). Data are representative of one experiment performed three times. Error bars indicate standard error for quadruplicate samples for 1 of 3 representative experiments. Data at 2 and 24 hours was analyzed by ANOVA. *P<0.001. (c) RNA was extracted and expression of EDA+Fn, total Fn and β-Actin was analyzed by RT-PCR at 2 hours. (d) Graph shows quantitation of data in (c). Ratio of EDA+Fn:Total Fn was calculated using a molecular mass correction factor and levels were normalized to β-Actin. Error bars indicate standard error of triplicate samples. Statistical analysis to determine significance was performed using Student’s t-test, *P<0.05. (e) Fibroblasts were seeded onto wells coated with pFn (10 µg/ml) or a mixture of pFn and the EDA module (40 µg/ml). Cell monolayers were solubilized after 5 hours and lysates analyzed by Western blot for the expression of the EDA+ isoform of fibronectin. Blots were stripped and reprobed for FAK which served as loading control.
EDA is a known ligand for the α4β1 integrin receptor and an established agonist for TLR4. To assess the contribution of each of these receptors to EDA-dependent gene induction, cells were pre-treated with blocking antibodies to either TLR4 or the α4 subunit of the integrin. We found that the upregulation of both TNF and VCAM1 in response to seeding cells onto EDA was significantly inhibited by blocking antibodies to either TLR4 or α4 (Figure 3a and b). To further assess the role of α4β1 integrin, the α4 subunit was knocked down using siRNA. Western blot analysis indicated that α4 protein levels were unaffected by control, non-targeting (NT) siRNA, while the α4 siRNA markedly decreased α4 protein levels even 24 hours after lifting and replating cells (Figure 3c). As shown in Figure 3d, control cells treated with NT siRNA exhibited increased levels of the inflammatory mediators, VCAM1 and TNF, in response to EDA which was significantly inhibited under conditions of α4 knockdown. Similarly, α4 knockdown completely prevented the EDA-mediated induction of IL-10 and IL-13 (Figure 3e). Furthermore, pretreatment of cells with inhibitors to TLR4 (TAK242) and/or NF-κB (Bay 11–782) completely inhibited expression of all EDA-induced genes. Taken together, these data suggest that both the α4 integrin and TLR4 on fibroblasts cooperatively regulate the NF-κB dependent induction of fibro-inflammatory gene expression in response to EDA.
Figure 3. EDA induces VCAM1 and TNF expression through Integrin α4β1 and TLR4.

Fibroblasts were pre-treated with blocking antibodies (10 µg/ml) to the α4β1 integrin (MAB16983) or TLR4 for 30 minutes. Cells were then plated on mixtures of pFn (10 μg/ml) and EDA (40 μg/ml) or the control module (C) III-10n (40 μg/ml) in 0.1% BSA-DMEM for 2 hours. RNA was extracted and VCAM1 (a) and TNF (b) mRNA levels were assessed by real time RT-PCR. Fibroblasts were transiently transfected with 10 nM α4 siRNA or 10 nM non-targeting (NT) siRNA for 4 days. (c) Transfected cells were assessed for α4 knockdown by western blot both before and after replating onto Fn substrates. Cells were pre-treated with 1 μM TLR4 inhibitor (TAK 242), 1 μM NF-kB inhibitor (Bay 11–0872) or DMSO prior to being seeded onto mixtures of pFn (10 μg/ml) and EDA or III-10n (40 μg/ml) in 0.1% BSA-DMEM for 2 hours. RNA was extracted and (d) VCAM1 and TNF or (e) IL-10 and IL13 mRNA levels were assessed by real time RT-PCR. Fold change values are expressed relative to non-targeting (NT) siRNA treated cells plated onto pFn and III-10n. Error bars indicate standard error of quadruplicate samples for one of three representative experiments. Statistical analysis was performed using a two-way ANOVA with Tukey post-hoc test. In VCAM and IL-10 experiments, *P<0.05, **P<0.01 for α4 knockdown cells, TAK and Bay treated cells plated to EDA. In TNF and IL-13 experiments, #P<0.05, ##P<0.01 for α4 knockdown cells and TAK treated cells compared with NT siRNA treated cells plated onto pFn and III-10n.
The role of α4β1 and TLR4 in the synthesis of the EDA+ isoform of fibronectin was also investigated. Fibroblasts were pretreated with a control mouse IgG or blocking antibodies to TLR4 and α4β1, either alone or in combination prior to seeding onto substrates coated with pFn and EDA or pFn and the control III-10n. After 2 hours, there was little change in total fibronectin mRNA levels, consistent with the data shown in Figure 2; however, there was a 5-fold increase in EDA+Fn mRNA levels (Figure 4a). This increase was inhibited in the presence of α4β1 and TLR4 blocking antibodies. When used in combination, antibodies to both receptors completely abrogated induction of EDA+Fn mRNA in response to EDA (Figure 4a). Knock-down of the α4 integrin using siRNA as well as inhibition of TLR4 signaling with the TAK inhibitor each prevented the increase in EDA+Fn mRNA in response to EDA (Figure 4b). These data indicate that both the α4β1 integrin and TLR4 regulate alternative splicing of fibronectin in response to EDA.
Figure 4. α4β1 and TLR4 coordinately regulate the ratio of EDA+Fn:total Fn levels in response to EDA.

(a) Prior to seeding cells onto substrates, cells were incubated with blocking antibody (10 μg/ml) to the α4β1 integrin (α4) or TLR4. Mouse IgG (IgG) was used as a control. Real time RT-PCR analysis of EDA+Fn, Total Fn, as well as β-Actin mRNA was assessed. (b) Fibroblasts were transiently transfected with 10 nM α4 siRNA or 10 nM non-targeting (NT) siRNA for 4 days. Transfected cells were pre-treated with 1 μM TLR4 inhibitor (TAK 242) or DMSO prior to being seeded to mixtures of pFn (10 μg/ml) and EDA or the control module III-10n (40 μg/ml) in 0.1% BSA-DMEM for 2 hours. RNA was extracted and EDA+Fn and total Fn mRNA levels were assessed by real time RT-PCR analysis. Fold change values are expressed relative to NT siRNA treated cells plated to control wells. Error bars indicate standard error of quadruplicate samples for one of three representative experiments. Statistical analysis was performed using a two-way ANOVA with Tukey post-hoc test. **P<0.01, ***P<0.001 compared to IgG control or siRNA control.
Our data highlight a previously unreported link between TLR4 and integrin α4β1 in facilitating fibrotic outcomes in response to EDA. Earlier reports have suggested cooperativity between innate immune signaling and integrins. In immune cells, integrins have been shown to both positively and negatively regulate the TLR-dependent production of inflammatory mediators in response to pathogens (Acharya et al., 2016; Gianni et al., 2012; Han et al., 2010; Marre et al., 2010; Yee and Hamerman, 2013). Our studies suggest a new role for integrins in DAMP-based signaling as ligand binding TLR co-receptors required for TLR activation and downstream signaling in non-immune cells. The data further suggest that it may be possible to selectively target DAMP-mediated fibro-inflammatory responses without compromising host defense against pathogens.
Although we could detect a rapid (2 hour) change in the level of TNFα RNA when cells were seeded onto pFn and EDA (Figure 2), TNF-α protein was not detected in the medium after 5 hours (data not shown). To explore this further, we took advantage of our previous finding that EDA could synergize with a second fibronectin DAMP, III-1c, to enhance cytokine expression in fibroblasts (Kelsh et al., 2014). III-1c represents a stable intermediate structure in fibronectin which is predicted to form during force induced unfolding of fibronectin’s III-1 domain (Gao et al., 2003). Like EDA, III-1c works through TLR4 to activate the NFκB dependent release of cytokines (TNF-α, IL-8) from human fibroblasts (You et al., 2010; Kelsh et al., 2014). Therefore, we asked whether cells adherent to EDA were more responsive to III-1c and whether α4β1 was regulating this response. To address this question, III-1c was added to cells adherent to either pFn or pFn and EDA and TNF-α released into the medium over a 5 hour period was measured by Enzyme-linked immunosorbent assay (ELISA). Figure 5 shows that when III-1c is added to cells adherent to pFn and EDA, there is a 4-fold increase in TNF-α when compared to cells seeded onto pFn alone. The EDA dependent increase in TNF-α release in response to III-1c was significantly inhibited under conditions of α4 knockdown. In contrast, α4 knockdown had no effect on III-1c stimulated synthesis of TNF-α by cells adherent to pFn alone. These results suggest that the α4β1 integrin specifically regulates the EDA dependent synthesis of cytokines by human fibroblasts and further suggests that cells adherent to EDA+Fn may be “primed” for a more robust inflammatory response to additional TLR4 ligands.
Figure 5. The α4β1 integrin regulates an enhanced response to TLR4 ligands.
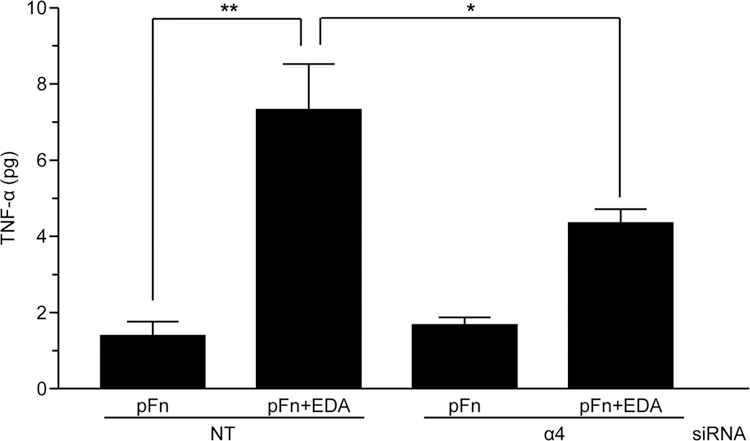
Human dermal fibroblasts were transiently transfected with 10 nM α4 siRNA or non-targeting (NT) siRNA and seeded onto wells coated with pFn (10 µg/ml) or a mixture of pFn (10 µg/ml) and EDA (40 µg/ml). Cells were then treated with III-1c for 5 hours and conditioned medium analyzed for TNF-α protein by ELISA. The data are expressed as picograms (pg) of TNF-α synthesized by 106 cells over a 5 hour period. Data were analyzed by ANOVA. **P<0.001; *P<0.05.
MATERIALS AND METHODS
Knockdown of the integrin subunit α4 using small interfering RNA
To suppress the expression of the α4 integrin subunit using siRNA, fibroblasts were cultured at 30–40% confluence in complete medium then transfected for 4 days with 25 nM ON-TARGET plussiRNA targeting α4 or a non-targeting (NT) control siRNA (Dharmacon, Lafayette, CO). α4 knockdown was confirmed by immunoblotting using a rabbit monoclonal antibody to α4 (D2E1). Antibodies to α4 integrin and GAPDH were obtained from Cell Signaling (Danvers, MA).
Gene Profiling
Human dermal fibroblasts were grown overnight in complete medium as previously described (You et al., 2010). Cells were trypsinized and placed in serum-free medium prior to plating onto different fibronectin substrates (pFn alone or pFn mixed with EDA) (Shinde et al., 2015). Cells appeared equally adherent and well spread on both substrates. Incubation times and doses of inhibitors are listed in the respective figure legends. The TLR4 (TAK242) and NFκB (BAY11–7082) inhibitors were purchased from EMD Millipore (Billerica, MA). Blocking antibody to α4 (MAB16983) was from Millipore (Temecula, CA). Blocking antibody to human TLR4 was obtained from R&D Systems (Minneapolis, MN). Subsequently, total RNA was isolated from fibroblasts using RNeasy extraction kit (Qiagen, Valencia, CA). An RT2 First Strand kit (Qiagen) was used to convert 1.5 μg of RNA into cDNA. The cDNA was applied to either an Extracellular Matrix and Cell Adhesion Molecule Array or a Fibrosis Array (Qiagen). A MyiQ cycler system (Bio-Rad Laboratories, Hercules, CA) was utilized for real-time PCR detection. Gene expression profiling was analyzed using an Excel-based PCR array data analysis template provided by the manufacturer. Relative gene expression was calculated as the difference in fold-change upon treatment and normalized against 5 housekeeping genes using the ΔΔCt method.
Conventional and real time RT-PCR
Total RNA was isolated from adherent cells and assayed for expression of EDA+Fn, total Fn, IL-10, IL-13, SPP1, or β-actin by reverse transcription PCR (RT-PCR). PCR primers for amplification of EDA+Fn were designed and synthesized based on primers from (Bhattacharyya et al., 2014). Fn, TNF, VCAM1, SPP1, IL-10, IL-13 and β-actin primers were purchased from SABiosciences (Frederick, MD). Real time RT-PCR was performed and data analyzed as previously described (You et al., 2010). Conventional RT-PCR for EDA+Fn, Fn and β-actin was carried out under the conditions: 94°C for 30 seconds, 60°C for 1 minute, and 72°C for 30 seconds, with 23 amplification cycles. Products were electrophoresed on a 1.2% Tris/Borate/EDTA gel and signals were quantified using a BioRad ChemiDoc™ Imager.
Preparation of recombinant fibronectin modules
The cDNA for the fibronectin EDA domain and the III-1c domain were inserted into bacterial expression vector pQE-30 in-frame with an N-terminal 6x His Tag (Qiagen, Inc., Valencia, CA), transformed and recombinantly expressed and purified as previously described (Kelsh et al., 2014). The His-tagged recombinant fibronectin Type III module, FnIII-10n, was prepared as previously described (Klein et al., 2003) and served as a control.
Immunoblot Analysis
Cell layer lysates were prepared and immunoblotted as previously described (Ambesi and McKeown-Longo, 2014). Antibodies to EDA+Fn (IST-9; sc-59826) and FAK (A-17; sc-557) were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Human TNF-α ELISA
Human dermal fibroblasts were cultured and α4 integrin subunit expression suppressed using siRNA as indicated above. Cells were lifted, rinsed once in serum-free media, and seeded onto dishes coated with pFn (10 µg/ml) or a mixture of pFn (10 µg/ml) and EDA (40 µg/ml) in the presence of 10 µM III-1c for 5 hours. Conditioned medium was concentrated using an Amicon Ultra-4 10K centrifugal filter and analyzed for TNF-α protein expression using a human TNF-α ELISA kit (BD Biosciences, San Diego, CA) as directed by the manufacturer.
ACKNOWLEDGMENTS
This study was supported by grants CA058626 and AR067956 from the National Institutes of Health to PJ McK-L.
Abbreviations:
- TLR4
toll-like receptor 4
- EDA+Fn
extra domain A-fibronectin
- Fn
fibronectin
- DAMP
damage-associated molecular pattern
- pFn
plasma fibronectin
- RNA
ribonucleic acid
- TNF
tumor necrosis factor
- VCAM1
vascular cell adhesion molecule-1
- ECM
extracellular matrix
- NT
non-targeting siRNA
- FAK
focal adhesion kinase
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
REFERENCES
- Acharya M, Sokolovska A, Tam JM, Conway KL, Stefani C, Raso F, et al. αv Integrins combine with LC3 and atg5 to regulate Toll-like receptor signalling in B cells. Nature Commun 2016; 7:10917-DOI: 10.1038/ncomms10917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambesi A, McKeown-Longo PJ. Conformational remodeling of the fibronectin matrix selectively regulates VEGF signaling. J Cell Sci 2014; 127:3805–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews JP, Marttala J, Macarak E, Rosenbloom J, Uitto J. Keloid pathogenesis: potential role of cellular fibronectin with the EDA domain. J Invest Dermatol 2015; 135:1921–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriazu E, Ge X, Leung TM, Magdaleno F, Lopategi A, Lu Y, et al. Signalling via the osteopontin and high mobility group box-1 axis drives the fibrogenic response to liver injury. Gut 2017; 66:1123–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arslan F, Smeets MB, Riem Vis PW, Karper JC, Quax PH, Bongartz LG, et al. Lack of fibronectin-EDA promotes survival and prevents adverse remodeling and heart function deterioration after myocardial infarction. Circ Res 2011; 108:582–92. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Tamaki Z, Wang W, Hinchcliff M, Hoover P, Getsios S, et al. FibronectinEDA promotes chronic cutaneous fibrosis through toll-like receptor signaling. Science Translational Med 2014; 6:232ra50-DOI: 10.1126/scitranslmed.3008264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochaton-Piallat ML, Gabbiani G, Hinz B. The myofibroblast in wound healing and fibrosis: answered and unanswered questions. F1000Res 2016; April 25; 5:pii: F1000 Faculty Rev-752-doi: 10.12688/f1000research.8190.1.ecollection 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth AJ, Wood SC, Cornett AM, Dreffs AA, Lu G, Muro AF, et al. Recipient-derived EDA fibronectin promotes cardiac allograft fibrosis. J Pathol 2012; 226:609–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley CD, Barone F, Nayar S, Benezech C, Caamano J. Stromal cells in chronic inflammation and tertiary lymphoid organ formation. Annu Rev Immunol 2015; 33:715–45. [DOI] [PubMed] [Google Scholar]
- Decitre M, Gleyzal C, Raccurt M, Peyrol S, Aubert-Foucher E, Csiszar K, et al. Lysyl oxidase-like protein localizes to sites of de novo fibrinogenesis in fibrosis and in the early stromal reaction of ductal breast carcinomas. Lab Invest 1998; 78:143–51. [PubMed] [Google Scholar]
- Dhanesha N, Ahmad A, Prakash P, Diddapattar P, Lentz SR, Chauhan AK. Genetic ablation of extra domain A of fibronectin in hypercholesteroiemic mice improves stroke outcome by reducing thrombo-inflammation. Ciruclation 2015; 132:2237–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doddapattar P, Gandhi C, Prakash P, Dhanesha N, Grumbach IM, Dailey ME, et al. Fibronectin splicing variants containing extra domain A promote atherosclerosis in mice through Toll-like Receptor 4. Arterioscler Thromb Vasc Biol 2015; 35:2391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerrschmid C, Trial J, Wang Y, Entman ML, Haudek SB. Tumor necrosis factor: a mechanistic link between angiogensin-II-induced cardiac inflammation and fibrosis. Circ Heart Fail 2015; 8:352–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullard N, O’Reilly S. Role of innate immune system in systemic sclerosis. Semin Immunopathol 2016; 37:511–7. [DOI] [PubMed] [Google Scholar]
- Gao M, Craig D, Lequin O, Campbell ID, Vogel V, Schulten K. Structure and functional significance of mechanically unfolded fibronectin type III1 intermediates. Proc Natl Acad Sci 2003; 100:14784–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni T, Leoni V, Chesnokova LS, Hutt-Fletcher LM, Campadelli-Fiume G. αvβ3-integrin is a major sensor and activator of innate immunity to herpes simplex virus-1. Proc Natl Acad Sci USA 2012; 109:19792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondokaryono SP, Ushio H, Niyonsaba F, Hara M, Takenaka H, Jayawardana ST, et al. The extra domain A of fibronectin stimulates murine mast cells via toll-like receptor 4. J Leukocyte Biol 2007; 82:657–65. [DOI] [PubMed] [Google Scholar]
- Han C, Jin J, Xu S, Liu H, Li N, Cao X. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol 2010; 11:734–42. [DOI] [PubMed] [Google Scholar]
- Jones GW, Hill DG, Jones SA. Understanding immune cells in tertiary lymphoid organ development: It is all starting to come together. Front Immunol 2016; 7:401-eCollection, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julier Z, Martino MM, de Titta A, Jeanbart L, Hubbelll JA. The TLR4 agonist fibronectin extra domain A is cryptic, exposed by elastase-2; use in a fibrin matrix cancer vaccine. Sci Rep 2015; 5:8569-doi: 10.1038/srep08569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski N, Allard JD, Pittet JF, Zuo F, Griffiths MJ, Morris D, et al. Global analysis of gene expression in pulmonary fibrosis reveals distinct programs regulating lung inflammation and fibrosis. Proc Natl Acad Sci USA 2000; 97:1778–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaviratne M, Hesse M, Leusink M, Cheever AW, Davies SJ, McKerrow JH, et al. IL-13 activates a mechanism of tissue fibrosis that is completely TGF-β independent. J Immunol 2004; 173:4020–9. [DOI] [PubMed] [Google Scholar]
- Kelsh R, You R, Horzempa C, Zheng M, McKeown-Longo PJ. Regulation of the innate immune response by fibronectin: Synergism between the III-1 and EDA domains. PLoS ONE 2014; 9:e102974-doi: 10.1371/journal.pone.0102974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsh RM, McKeown-Longo PJ, Clark RAF. EDA fibronectin in keloids create a vicious cycle of fibrotic tumor formation. J Invest Dermatol 2015; 135:1714–8. [DOI] [PubMed] [Google Scholar]
- Khan MM, Gandhi C, Chauhan N, Stevens JW, Motto DG, Lentz SR, et al. Alternatively-spliced extra domain A of fibronectin promotes acute inflammation and brain injury after cerebral ischemia in mice. Stroke 2012; 43:1376–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009; 457:102–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RM, Zheng M, Ambesi A, van de Water L, McKeown-Longo PJ. Stimulation of extracellular matrix remodeling by the first type III repeat in fibronectin. J Cell Sci 2003; 116:4663–74. [DOI] [PubMed] [Google Scholar]
- Kohan M, Muro AF, Bader R, Berkman N. The extra domain A of fibronectin is essential for allergen-induced airway fibrosis and hyperresponsiveness in mice. J Allergy Clin Immunol 2011; 127:439–46. [DOI] [PubMed] [Google Scholar]
- Lancha A, Rodríquez A, Catalán V, Becerril S, Sáinz N, Ramírez B, et al. Osteopontin deletion prevents the developmenty of obesity and hepatic steatosis via impaired adipose tissue matrix remodeling and reduced inflammation and fibrosis in adipose tissue and liver in mice. PLoS ONE 2014; 9:e98398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanone S, Zheng T, Zhu Z, Liu W, Lee CG, Ma B, et al. Overlapping and enzyme-specific contributions of matrix metalloproteinases-9 and −12 in IL-13 induced inflammation and remodeling. J Clin Invest 2002; 110:463–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor β1. J Exp Med 2001; 194:809–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenga Y, Koh A, Perera AS, McCulloch CA, Sodek J, Zohar R. Osteopontin expression is required for myofibroblast differentiation. Circ Res 2008; 102:319–27. [DOI] [PubMed] [Google Scholar]
- Marre ML, Petnicki-Ocwieja T, DeFrancesco AS, Darcy CT, Hu LT. Human integrin α3β1 regulates TLR2 recognition of lipopeptides from endosomal compartments. PLoS ONE 2010; 5:e12871-doi: 10.1371/journal.pone.0012871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui Y, Tomaru U, Miyoshi A, Ito T, Fukaya S, Miyoshi H, et al. Overexpression of TNF-α converting enzyme promotes adipose tissue inflammation and fibrosis induced by high fat diet. Exp Mol Pathol 2014; 97:354–8. [DOI] [PubMed] [Google Scholar]
- McFadden JP, Baker BS, Powles AV, Fry L. Psoriasis and extra domain A fibronectin loops. Brit J Dermatol 2010; 163:5–11. [DOI] [PubMed] [Google Scholar]
- McFadden JP, Basketter DA, Dearman RJ, Kimber IR. Extra domain A-positive fibronectin-positive feedback loops and their association with cutaneous inflammatory disease. Clin Dermatol 2011; 29:257–65. [DOI] [PubMed] [Google Scholar]
- Mogami H, Kishore AH, Shi H, Keller PW, Akgul Y, Word RA. Fetal fibronectin signaling induces matrix metalloproteases and cyclooxygenase-2 (COX-2) in amnion cells and preterm birth in mice. J Biol Chem 2013; 288:1953–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikonomou N, Harokopos V, Zalevsky J, Valavanis C, Kotanidou A, Szymkowski DE, et al. Soluble TNF mediates the transition from pulmonary inflammation to fibrosis. PLoS ONE 2006; 1:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, et al. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem 2001; 276:10229–33. [DOI] [PubMed] [Google Scholar]
- Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopoly-saccharaide recognition by the TLR4-MD-2 complex. Nature 2009; 458:1191–5. [DOI] [PubMed] [Google Scholar]
- Schwarzbauer JE, DeSimone DW. Fibronectins, their fibrillogenesis, and in vivo functions. Cold Spring Harbor Perspectives in Biology 2011; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde AV, Kelsh R, Peters JH, Sekiguchi K, Van De Water L, McKeown-Longo PJ. The α4β1 integrin and the EDA domain of fibronectin regulate a profibrotic phenotype in dermal fibroblasts. Matrix Biol 2015; 41:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sziksz E, Pap D, Lippai R, Béres NJ, Fekete A, Szabó AJ, et al. Fibrosis related inflammatory mediators: Role of the IL-10 cytokine family. Mediators of Inflammation 2015; 2015:15 pages-Article ID 764641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood DC, Osborn RR, Bochnowicz S, Webb EF, Rieman DJ, Lee JC, et al. SB 239063, a p38 MAPK inhibitor, reduces neutrophilia, inflammatory cytokines, MMP-9, and fibrosis in lung. Am J Physiol Lung Cell Mol Physiol 2000; 279:L895–L902. [DOI] [PubMed] [Google Scholar]
- Wang S, Hirschberg R. BMP7 antagonizes TGF-β-dependent fibrogenesis in mesangial cells. Am J Physiol Renal Physiol 2003; 284:F1006–F1013. [DOI] [PubMed] [Google Scholar]
- White ES, Muro AF. Fibronectin splice variants: understanding their multiple roles in health and disease using engineered mouse models. IUBMB Life 2011; 63:538–46. [DOI] [PubMed] [Google Scholar]
- White ES, Sagana RL, Booth AJ, Yan M, Cornett AM, Bloomheart CA, et al. Control of fibroblast fibronectin expression and alternative splicing via the PI3K/Akt/mTOR pathway. Exp Cell Res 2010; 316:2644–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee NY, Hamerman JA. β2 integrins inhibit TLR responses by regulating NF-κB pathway and p38 MAPK activation. Eur J Immunol 2013; 43:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You R, Zheng M, McKeown-Longo PJ. The first type III repeat in fibronectin activates an inflammatory pathway in dermal fibroblasts. J Biol Chem 2010; 285:36255–9. [DOI] [PMC free article] [PubMed] [Google Scholar]