

Abstract
Rubinstein–Taybi syndrome (RSTS) is an autosomal-dominant neurodevelopmental disease affecting 1:125,000 newborns characterized by intellectual disability, growth retardation, facial dysmorphisms and skeletal abnormalities. RSTS is caused by mutations in genes encoding for writers of the epigenetic machinery: CREBBP (~ 60%) or its homologous EP300 (~ 10%). No causative mutation is identified in up to 30% of patients. We performed whole-exome sequencing (WES) on eight RSTS-like individuals who had normal high-resolution array CGH testing and were CREBBP- and EP300-mutation -negative, to identify the molecular cause. In four cases, we identified putatively causal variants in three genes (ASXL1, KMT2D and KMT2A) encoding members of the epigenetic machinery known to be associated with the Bohring–Opitz, Kabuki and Wiedemann–Steiner syndromes. Each variant is novel, de novo, fulfills the ACMG criteria and is predicted to result in loss-of-function leading to haploinsufficiency of the epigene. In two of the remaining cases, homozygous/compound heterozygous variants in XYLT2 and PLCB4 genes, respectively, associated with spondyloocular and auriculocondylar 2 syndromes and in the latter an additional candidate variant in XRN2, a gene yet unrelated to any disease, were detected, but their pathogenicity remains uncertain. These results underscore the broad clinical spectrum of Mendelian disorders of the epigenetic apparatus and the high rate of WES disclosure of the genetic basis in cases which may pose a challenge for phenotype encompassing distinct syndromes. The overlapping features of distinct intellectual disability syndromes reflect common pathogenic molecular mechanisms affecting the complex regulation of balance between open and closed chromatin.
Introduction
Rubinstein–Taybi syndrome (RSTS, OMIM #180849, #613684) described in 1963 by Rubinstein, a pediatrician, and Taybi, a radiologist, is a rare neurodevelopmental multisystem malformation syndrome (Rubinstein and Taybi 1963) characterized by developmental delay and intellectual disability (DD/ID), growth retardation, skeletal anomalies including broad/short thumbs and/or big toes, and distinctive facial features (i.e. downslanting palpebral fissures, broad nasal bridge/convex nasal bridge, low hanging columella). A wide spectrum of other anomalies and malformations has also been reported in individuals with RSTS (Spena et al. 2015a, b). Phenotypic overlap between RSTS and other Mendelian conditions often makes the clinical diagnosis of RSTS difficult.
Variants in two genes, CREBBP (16p 13, OMIM #600140) and EP300 (22q13, OMIM #602700), underlie RSTS. CREBBP and EP300 encode two highly conserved, ubiquitously expressed and homologous lysine-acetyl-transferases (KAT) that act as “writers” of the epigenetic machinery, named CBP and p300 (Fahrner and Bjornsson 2014; Bjornsson 2015). Up to 60% of RSTS cases harbor de novo mutations in CREBBP, which is the “major” gene (http://www.lovd.nl/CREBBP) (Spena et al. 2015a, b), while approximately 10% of affected individuals have de novo alterations in EP300, the “minor” gene (http://chromium.liacs.nl/L0VD2/home.php?select_db=EP300) (Masuda et al. 2015; Wincent et al. 2015; Fergelot et al. 2016; Hamilton et al. 2016; López et al. 2016; Negri et al. 2016; Sellars et al. 2016). Point mutations (i.e. frameshift, nonsense, missense and splicing in order of prevalence) represent the majority of genetic mutations found in both CREBBP and EP300 patients followed by deletions (intragenic, whole gene or expanding to adjacent regions), translocations and inversions (Lacombe et al. 1992; Breuning et al. 1993; Masuda et al. 2015; Rusconi et al. 2015; Spena et al. 2015a, b; Negri et al. 2015, 2016). The genetic basis of ~ 30% of RSTS cases remains unknown.
Alterations in genes encoding proteins involved in the epigenetic regulation of chromatin dynamics, acting as “writers”, “erasers”, “readers” and “remodelers”, are associated with other Mendelian disorders of the epigenetic machinery showing phenotypic overlap with RSTS such as Bohring–Opitz, Wiedemann–Steiner and Kabuki syndrome (Fahrner and Bjornsson 2014; Bjornsson 2015) or to other rare conditions (i.e. Floating–Harbor and genitopatellar syndromes) whose the underlying genes encode proteins that interact directly with CBP and/or p300 (Spena et al. 2015a, b).
We performed exome sequencing on eight cases with RSTS diagnosis in which mutations in CREBBP and EP300 could not be identified. The rationale was that such novel genes would broaden our understanding of the epigenetic perturbations that lead to RSTS. We report the discovery of four novel de novo mutations within “epi-genes” associated with syndromic epigenetic disorders with clinical similarity to RSTS. We highlight the complex and unique phenotypes of these cases, accounting half of our WES-selected cohort, and comment on how epigenetic dynamics alterations could result in a wide spectrum of clinical features creating a continuum of overlapping syndromes. In two of the remaining four WES-processed cases, candidate variants were disclosed in genes associated with known syndromes not “tagged” as epigenetic disorders, but the pathogenicity of the variants remains unclear.
Materials and methods
Subjects
All individuals were assessed by a clinical geneticist. Written informed consent was obtained from all subjects and this study was approved by the local institution ethical committee and review board (http://www.unimi.it/ateneo/normativa/50486.htm) and performed according to the Declaration of Helsinki protocol. This study includes a subset of patients and their healthy parents selected according to the following criteria: (i) genetic test found negative to CREBBP/EP300 mutations, (ii) RSTS-like phenotype.
High-resolution array CGH
Array CGH experiments were performed with a commercially available 400 K 60-mer oligonucleotide microarray slide (Agilent Technologies Inc., Santa Clara, CA, USA) in accordance with the manufacturer’s instructions. Commercially available sex-matched control genomic DNA (Pro-mega) was used. Data were extracted and analyzed for copy number changes using Agilent CytoGenomics v.3.0.
Whole exome sequencing and data analysis
A total of 1 μg of genomic DNA was processed and enriched for exon sequences according to the protocol recommended for Roche/Nimblegen SeqCap EZ v3.0 (~ 62 MB target) (Roche, Basel, Switzerland). After quality check on the Agilent Bioanalyzer, libraries were sequenced on the Illumina HiSeq2000/2500 sequencers (Illumina, Applied Biosystems, Foster City, CA, USA). A standard bioinformatic pipeline was applied to analyze raw sequencing data, as previously described (Van der Auwera et al. 2013).
VarScan2 (http://www.dkoboldt.github.io/varscan) (Koboldt et al. 2012) and DeNovoGear (http://www.denovogear.weebly.com) (Ramu et al. 2013) algorithms were applied using standard parameters for de novo germ-line calling. To increase call specificity, only variants detected by both algorithms were considered as de novo candidates. Based on estimated disease prevalence of 1:125,000, we retained for further analysis de novo variants not reported in general population databases (ExAC: Exome Aggregation Consortium, http://exac.broadinstitute.org/ and gnomAD: Genome Aggregation Database, http://gnomad.broadinstitute.org/) and homozygous/compound heterozygous/hemizygous variants with allele frequency ≤ 0.3% without homozygous/ hemizygous individuals described in the above-mentioned databases. Only variants predicted to be likely protein altering (nonsynonymous substitutions, splice-site and insertions/deletions) were examined further and variants with CADD v1.2 Phred score < 15 were filtered out to discard most likely benign nonsynonymous variants. We excluded variants in genes associated with non-neurodevelopmental disorders. We classified the significance of candidate variants according to ACMG criteria (Richards et al. 2015) using Varsome tool (https://varsome.com/).
Variant validation and segregation analysis
Variants of interest were confirmed by Sanger sequencing following PCR amplification and segregation analysis was always performed in each trio. PCR reactions were performed following the protocol for GoTaq Flexi DNA Polymerase (Promega) with specific primer sets. The same pairs of primers were used for Sanger sequencing. Sequence variants were described according to HGVS nomenclature guidelines (http://varnomen.hgvs.org/).
In silico analyses
The deleterious potential of missense substitutions was assessed by PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml), SIFT (http://sift.jcvi.org/), SNPs&GO (http://snps-and-go.biocomp.unibo.it/snps-and-go/), Mut-Pred v.1.2 (http://mutpred.mutdb.org/), PMut (http://mmb.pcb.ub.es/PMut/analyses/new/), SNAP2 (https://rostlab.org/owiki/index.php/Snap2), Mutation Taster (http://www.mutationtaster.org/), Panther (http://www.ngrl.org.uk/Manchester/page/missense-prediction-tools), PhD SNP (http://snps.biofold.org/phd-snp/phd-snp.html), MetaSNP (http://snps.biofold.org/meta-snp/pages/methods.html) and IMutant2 (http://folding.biofold.org/cgi-bin/i-mutant2.0.cgi) programs.
Multiple sequence alignments were performed by submitting protein sequences derived from UniGene (http://www.ncbi.nlm.nih.gov/unigene) to Clustal program (http://www.ebi.ac.uk/Tools/msa/clustalo/).
Results
Variants detected by WES of CREBBP/EP300 mutation-negative patients with initial RSTS clinical diagnosis
DNA samples from eight probands with RSTS initial clinical diagnosis but no detected pathogenic variant in either CREBBP or EP300 by MLPA analysis, targeted sequencing and high-resolution array CGH were selected for WES. A first analysis considering only novel de novo variants with priority given to sequence changes in disease-associated genes belonging to the so-called epigenetic machinery allowed us to identify compelling candidate variants in four of eight patients (Table 1). Each variant was validated by Sanger sequencing and confirmed variants arose de novo. None of the identified variants is reported in ExAC and gnomAD databases.
Table 1.
Mutations detected in the four RSTS-like patients achieving molecular diagnosis
| Patient | Gene | cDNA | Position | Protein | Type | Associated Syndrome | Role in the epigenetic machinery |
ACMG classification (2015) |
|---|---|---|---|---|---|---|---|---|
| #80 | ASXL1 | c.3856C>T | Exon 12 | p.(Q1286*) | Stop-gain | Bohring–Opitz | Reader | Likely pathogenic |
| #173 | ASXL1 | c.4243C>T | Exon 12 | p.(R1415*) | Stop-gain | Bohring–Opitz | Reader | Pathogenic |
| #103 | KMT2A | c.5363+1delG | IVS 18 | Splicing | Wiedemann–Steiner | Writer | Pathogenic | |
| #95 | KMT2D | c.6040C>T | Exon 28 | p.(Q2014*) | Stop-gain | Kabuki | Writer | Pathogenic |
Position and type of the pathogenic variants, epigenetic role of the affected genes and associated syndromes are indicated
Two stop-gain variants, c.3856C>T, p.(Q1286*) and c.4243C>T, p.(R1415*), in exon 12 (Online Resource Fig. S1) of the Additional Sex Combs-Like 1 gene (ASXL1, OMIM *612990, NM_015338) were identified in cases #80 and #173, respectively. Heterozygous truncating variants in ASXL1, which encodes a protein belonging to the poly-comb group (PcG) and trithorax complex family, underlie Bohring–Opitz syndrome (BOPS, OMIM #605039), a rare congenital malformation disorder, first described in 1999 by Bohring (Bohring et al. 1999). Neither variant has been reported previously in BOPS families (Online Resource Table S1 and Fig. S1) (Arunachal et al. 2016). According to ACMG guidelines, the clinical significance of the two variants is likely pathogenic and pathogenic, respectively. Both variants meet the same ACMG criteria (PM1, PM2, PM4, PM6, PP2) with p.(R1415*) additionally fulfilling PS3 criterion, being recently uploaded in ClinVar database and classified as pathogenic.
Case #95 was found to have a novel stop-gain variant, c.6040C>T, p.(Q2014*) in exon 28 (Online Resource Fig. S1) of lysine (K)-specific methyltransferase 2D (KMT2D, formerly MLL2, OMIM #602113, NM_003482), encoding a methyltransferase responsible for histone 3 lysine 4 (H3K4) demethylation and trimethylation, an epigenetic mark of euchromatin and active transcription (Smith et al. 2011). KMT2D is the first identified and “major” gene for Kabuki syndrome (KS, OMIM #197420, #300867) a highly variable genetic condition characterized by growth deficiency, intellectual disability, minor skeletal anomalies and distinctive facial features (Niikawa et al. 1981). To date, more than 200 de novo mutations in KMT2D have been reported in about 56–75% of KS cases (Schott et al. 2016) (Online Resource Table S2 and Fig. S1). ACMG classification indicates the p.(Q2014*) variant as pathogenic fulfilling PVS1, PM2, PM4, PM6 and PP2 criteria.
In case #103, we found a c.5363+1delG variant at the donor site of intron 18 (Online Resource Fig. S1) of lysine-specific methyltransferase 2A (KMT2A, OMIM +159555, NM_001197104) encoding a writer of the epigenetic machinery. Truncating mutations of KMT2A underlie Wiedemann-Steiner syndrome (WDSTS, OMIM #605130), a heterogeneous disease described in 1989 by Wiedemann (Wiedemann et al. 1989), and characterized by hypertrichosis cubiti, intellectual disability and developmental delay together with a distinctive facial appearance and short stature (Aggarwal et al. 2017). To our knowledge, 26 mutations have been reported in clinically diagnosed WDSTS cases (Aggarwal et al. 2017) with the exception of one patient diagnosed with Cornelia de Lange syndrome (CdLS, OMIM #122470, #300590, #610759, #614701, #300882), a clinical and molecular heterogeneous plurimalformative syndrome with partial overlap to WDSTS (Yuan et al. 2015) (Online Resource Table S3 and Fig. S1). More recently, additional 29 KMT2A variants have been described in 33 patients selected as characterized by syndromic intellectual disability (Baer et al. 2018). The c.5363+1delG variant meets the PVS1, PM2 and PM6 ACMG criteria of pathogenicity, has a predicted null effect on a gene where loss-of-function is a known disease mechanism and is a previously unreported de novo variant.
Additional de novo variants in genes not associated with epigenetic machinery syndromes have been also evaluated (Online Resource Table S4).
The variant c.560G>A, p.(G187E) in 5-prime,3-prime-exoribonuclease 2 (XRN2, OMIM *608851, NM_001317960) gene, not yet associated with a specific syndrome, was found in patient #169. The deleteriousness potential of the variant was assessed by performing in silico prediction analyses. Eight out of 11 programs predicted a probable disease causing role for the identified variant. Furthermore, multiple sequence alignments (Clustal program) evidenced the broad conservation of the involved amino acid residue from yeast to human. According to ACMG guidelines, this variant has uncertain significance.
Subsequently, variants analysis was also performed under AR-and X-linked model of Mendelian inheritance (Online Resource Table S4). Homozygous missense variant c.209G>A, p.(R70Q) in xylosyltransferase-2 gene (XYLT2, OMIM *608125) was found in patient #76 with a family record of consanguinity (the grandparents are cousins of second grade). Frameshift mutations in this gene are reportedly associated with spondyloocular syndrome (SOS, OMIM #605822).
Two variants in phospholipase C beta 4 gene (PLCB4 OMIM *600810), the paternal frameshift c.2607_2609dup, p.(Ser870dup) and the maternal missense c.2653G>A, p.(V885M), were detected in patient #169 found also carrier of the XRN2 variant. AR/AD mutations are reported in auriculocondylar syndrome 2 (ARCND2, OMIM #614669).
The clinical impact of all these variants was predicted as uncertain, following ACMG criteria.
The analysis of variants in the remaining cases #88 and #118 did not reveal any strong candidate variant.
Clinical phenotype of WES-processed cases with initial RSTS diagnosis
Case #80 (ASXL1)
Familial history is negative for genetic conditions and/or ID. Pregnancy was normal. The child was born at 41 weeks of gestational age by cesarean section; at birth, auxological parameters (not reported) were normal. He presented with sucking difficulties and hypotonia, and he developed a severe ID.
At the clinical evaluation (24 years), weight was 44 kg (< 3° centile), height was 166 cm (50°), and head circumference 57 cm (98°). He presented dolichocephaly, low hairline, low set and posteriorly rotated ears, synophrys, downslanting palpebral fissures, palpebral ptosis, hypertelorism, long eyelashes, high nasal bridge, low hanging columella, short philtrum, high and narrow palate, open mouth, malocclusion, thick vermilion of the lower lip (Fig. 1). Scoliosis and pectus excavatum were recognized, and low set and broad thumbs were evident. Corpus callosum hypoplasia, seizures and delayed bone age were also reported.
Fig. 1.
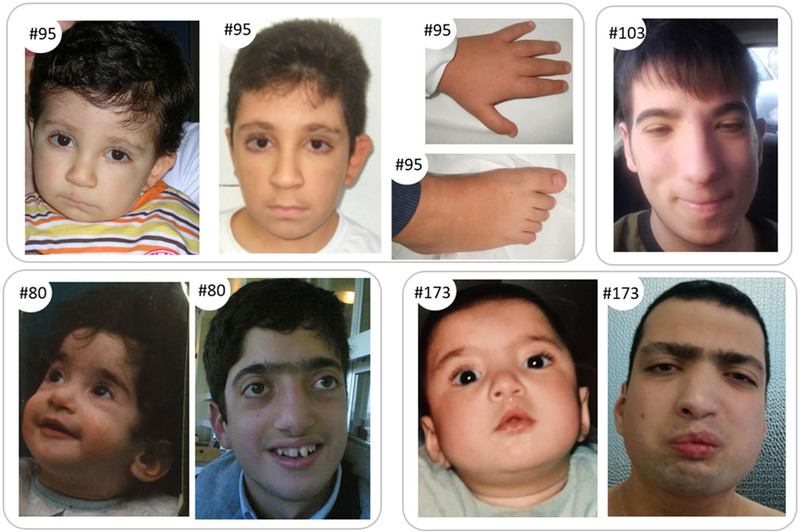
Features of patients #95, #80, #103 and #173. Top left: features of pt #95 found carrier of KMT2D mutation at 1 year (left) and 10 years (right) with particular of foot and hand. Top right: features of patient #103 at 16 years. Bottom: features of pt #80 and pt #173 found carriers of ASXL1 mutations. Faces are shown at 1 year (left) and in adulthood (right)
Case #173 (ASXL1)
Familial history is positive for macrocephaly and hydrocephalus in a cousin. Pregnancy was normal. The child was born at 37 weeks of gestational age by spontaneous delivery; at birth, weight was 2.400 kg (< 3°), length 45.5 cm (< 10°), OFC 31 cm (< 3°), and APGAR score was 6/8. Cleft lip was diagnosed.
He showed severe ID, autism spectrum disorder, increased levels of aggression and seizures.
At the clinical evaluation (22 years), weight was 60 kg (10° centile), height was 163 cm (3°), and head circumference 55.8 cm (50°). He presented low hairline, arched eyebrows, long eyelashes, downslanting palpebral fissures, convex nasal bridge, thick vermilion of the lower lip, micro-retrognathia, facial grimacing, hypotonic facies with full cheeks and low set ears with increased posterior angulation (Fig. 1); first toe was broad.
Additional medical problems and malformations were reported: corpus callosum hypoplasia, strabismus, hypermetropic astigmatism, left superior lateral incisor agenesis, gastroesophageal reflux, hip dislocation, patellar subluxation, hypothyroidism, constipation, frequent airways infections.
Case #103 (KMT2A)
Familial history is negative for genetic conditions and/or ID. Pregnancy was normal. The child was born at 39 weeks of gestational age by spontaneous delivery. Birth parameters were weight > 10°, head circumference 50° and length 15°. He presented with sucking difficulties and hypotonia, and a glabellar nevus flammeus that fades with age; a postnatal growth retardation was evident. He showed a moderate ID.
At the clinical evaluation (6 years), weight was 22 kg (50° centile), height 112 cm (10°), and head circumference 52 cm (50°); he presented with narrow forehead, low anterior hairline, synophrys, hypertelorism, arched and thick eyebrows, narrow and downslanting palpebral fissures, convex nasal bridge with low hanging columella, short philtrum, thin lips, facial grimacing, high palate, micrognathia, and posteriorly angulated ears (Fig. 1). Thumb and first toe were broad; pectus excavatum and hirsutism (not specifically localized on the elbow) were evident.
Other medical problems subsequently reported were kyphosis and obesity.
Case #95 (KMT2D)
Familial history is positive for hypothyroidism, seizures, autism; a brother of the proband was diagnosed with moderate ID and autism. The child was born at 37 weeks of gestational age with cesarean section for breech presentation and fetal distress. At birth, auxological parameters were: weight 25–50°, length 3–10°. During pregnancy, IUGR and weak fetal movements were referred; at birth, weight was 2.620 kg, length 44.6 cm, APGAR score was 9. In neonatal age, sucking difficulties are reported. Psychomotor retardation and hypotonia were evident.
At the age of 16 months, weight was 9 kg (3–10° centile), height 72 cm (< 3°), and head circumference 44 cm (3°).
At the clinical evaluation (9 years), weight was 36 kg (75° centile), height 127 cm (10°), and head circumference 53 cm (25°); the child had long eyelashes, low set ears, microstomia, thin lips, high palate, and micrognathia; thumb and first toe were broad, and brachydactyly was evident (Fig. 1).
Strabismus, myopia, lateral incisors agenesis, hypothyroidism, cryptorchidism (surgically treated), constipation and short stature (treated with growth hormone) were also reported.
Table 2 provides the detailed description of the four patients found carriers of de novo candidate variants with a definitive/strong role in disease. In particular, typical clinical signs of RSTS, KS, BOPS and WDSTS are detailed for comparison with features shown by patient #95 (carrier of KMT2D mutation), patients #80 and #173 (carriers of ASXL1 mutations) and patient #103 (carrier of KMT2A mutation).
Table 2.
Clinical signs of patients #95, #80, #173 and #103 compared to typical features of RSTS, KS, BOPS and WDSTS
| RSTS | #95 | KS | #80 | #173 | BOPS | #103 | WDSTS | |
|---|---|---|---|---|---|---|---|---|
| Gene with pathogenetic variant | KMT2D | ASXL1 | ASXL1 | KMT2A | ||||
| c.6040C>T, p.(Q2014*) | c.3856C>T, p.(Q1286*) | c.4243C>T, p.(R1415*) | c.5363 + ldelG | |||||
| Date of birth | 2006 | 1993 | 1994 | 2001 | ||||
| Time of clinical assessment | 2007 | 2006 | 2011 | 2008 | ||||
| Sex | male | male | male | male | ||||
| Dysmorphisms | ||||||||
| Low anterior hairline | + | − | − | − | + | + | − | − |
| Long eyelashes | + | + | − | + | + | − | + | + |
| Synophrys | +/− | − | − | + | + | − | + | + |
| Ptosis | + | + | + | − | − | − | − | − |
| Downslanting palpebral fissures | + | − | − | + | + | − | + | + |
| Upslanting palpebral fissures | − | − | − | − | − | + | − | − |
| Eversion of the lateral third of lower eyelid | − | + | + | − | − | − | − | − |
| Thick eyebrows | +/− | − | − | + | + | − | + | + |
| Narrow palpebral fissures | − | − | − | − | + | − | + | + |
| Hypertelorism | + | − | − | + | + | + | + | + |
| Prominent eyes | − | − | − | + | − | + | − | − |
| Low hanging columella | + | − | − | + | + | − | + | − |
| Broad nasal tip | − | + | − | + | + | − | + | − |
| Convex and wide nasal bridge | + | − | + | − | − | + | − | + |
| Anteverted nares | − | − | − | − | − | + | − | − |
| Thin lips | − | + | + | − | − | − | + | + |
| Facial grimacing | + | − | + | − | + | − | + | − |
| Microstomia | − | + | − | − | − | − | + | + |
| High-arched palate | + | − | + | + | na | − | + | + |
| Cleft lip/palate | − | − | + | − | + (cleft lip) | + | − | − |
| Micrognathia | + | + | + | + | + | + | + | + |
| Low set ears | + | + | — | + | +(with increased posterior angulation) | + | + | + |
| Strabismus | + | + | − | + | + | + | − | + |
| Flammeus nevus/angioma | +/− | − | − | − | − | + | + | − |
| Hypotonic facies with full cheeks | − | − | − | − | + | + | − | − |
| Growth failure | ||||||||
| IUGR | − | + | − | − | + | + | + | + |
| Birth parameters: weight–length–OFC (percentiles) | 25° to 3° to na | na(referred as normal) | <3° to <3° to 10° | <10° to 15° to 50° | ||||
| PNGR | + | + | + | − | + | + | + | + |
| PN (age) parameters: weight–height–OFC (percentiles) | 75°−10°−25° (9 y) | 25°−50°−98° (23 y) | 10°−3°−50°(21 y) | nk | ||||
| Intellectual disability | + | + (moderate) | + | + (severe) | + (severe) | + | + (moderate) | + |
| Speech delay/absence | +/− | + | +/− | + | + | + | − | + |
| Behavioral problems | +/− | − | +/− | +(hyperkinesis, agitation) | +(aggressivity, agitation) | + | − | + |
| Vision problems | ||||||||
| Myopia | + | + (and hypermetropic astigma-tism) | + (and hypermetropic astigma-tism) | + | — | |||
| Hearing loss | − | +/− | na | − | − | |||
| Teeth anomalies | + | +(absence of superior lateral incisors) | + | +(enamel hypoplasia, hypo-dontia) | +(absence of superior lateral incisors) | — | — | + |
| Musculoskeletal anomalies | ||||||||
| Broad thumbs | + | + | − | − | − | − | + | − |
| Angulated thumbs | +/− | − | − | − | − | − | − | − |
| Broad halluces | + | + | − | − | + | − | + | − |
| Clinodactyly | + | + | + | + | − | − | + | +/− |
| Brachydactyly | + | + | − | − | − | + | + | − |
| Camptodactyly | − | − | − | + | + | − | − | |
| Microcephaly | + | − | + | − | + | + | + | − |
| Trigonocephaly | − | − | − | − | − | + | − | + |
| Delayed bone age | + | − | − | + | na | − | − | + |
| Fetal fingertip pads | − | + | + | + | − | − | + | − |
| BOPS posture | − | − | − | − | − | + | − | − |
| Fixed contractures | − | − | − | + | − | + | − | − |
| Hypotonia | + | + | − | + | − | + | + | + |
| Organ anomalies | ||||||||
| Genitourinary anomalies | + | + (CO) | + | − | − | − | + (CO) | − |
| Heart defect | + | − | + | − | − | + | − | + |
| Brain anomalies | + | − | − | + (HCC) | + (ACC) | + | − | + |
| Seizures | + | + | − | + | + | + | + | − |
| Hypertrichosis | + | + | + | − | + | + | + | + |
| Keloids/naevi | + | − | − | − | − | +/− | − | − |
| Pilomatricoma | + | − | − | − | − | − | − | − |
| Frequent infections | + | − | + | − | − | + | − | − |
| Feeding problems | + | + | − | + | − | + | − | +/− |
| Gastroesophageal reflux | + | − | + | + | − | + | − | − |
| Others | Hypomobility, absence of superior lateral incisors, hypothyroidism, GH therapy for short stature | Buphthalmos; dolichocephaly; hyperreflexia; joint hyper-mobility, enamel hypolasia; abnormal tooth number | Acanthosis nigricans, striae distensae and erythema (chest); hepatomegaly, compulsive food research and eating; astigmatism, hypermetria | Kyphosis, pectus excavatum, obesity | ||||
Sign: + means present, sign − absent and sign +/− present in a few cases
ACC agenesis of corpus callosum, CO cryptorchidism, HCC hypoplasia of corpus callosum, na not assessed, nk not known
The typical RSTS heterogeneity in the number, type and severity of clinical findings is evident also in this cohort. Nevertheless, typical RSTS clinical signs, such as specific dysmorphic features (i.e. low hanging columella, etc.) or characteristic skeletal anomalies (i.e. broad thumb/hallux) are present in almost all patients (Table 2; Fig. 1).
In particular, craniofacial anomalies such as the typical nose with prominent columella, downslanting of the palpebral fissures, and long eyelashes are present in 3/4 or 4/4 described patients.
Micrognathia is highly prevalent (4/4 patients), synophrys is evident in 3/4 patients, while grimacing smile is detected in two patients [#173 (ASXL1), #103 (KMT2A)].
Prenatal growth retardation is reported in two cases [pts #95 (KMT2D) and #173(ASXL1)] while postnatal growth retardation in 3/4 cases. Skeletal malformations, as expected, mainly concern thumbs and big toes, which are broad and short (3/4).
Intellectual disability (ranging from moderate to severe) is always present, and language is absent or very limited except for patient #103 (KMT2A).
Other RSTS-like signs recorded in our cases are dental anomalies [pts #80 (ASXL1), #95 (KMT2D), #173(ASXL1)], and behavioral problems [pts #80 (ASXL1), #95 (KMT2D), #173 (ASXL1)].
However, clinical features atypical for RSTS but described in the different syndromes associated with the identified pathogenic variants are also present: out of them buphthalmos [pt #80 (ASXL1)], contractures [pts #80 (ASXL1), #173 (ASXL1)], described in BOPS or eversion of the lateral third of the lower eyelids [pt #95 (KMT2D)], present in KS (Table 2; Fig. 1).
The clinical phenotype of the remaining patients #76, #88, #118, and #169 was also re-evaluated and supported the initial RSTS/RSTS-like diagnosis. Supplemental Table S5 summarizes the clinical features of these patients, compared to major typical signs of RSTS.
Discussion
We identified putatively causal variants in four patients with an initial RSTS diagnosis in genes known to underlie other multiple malformation syndromes with phenotypic features that overlap RSTS: the involved genes, namely ASXL1 (patients #80 and #173), KMT2D (patient #95) and KMT2A (patient #103), are known to be responsible for Bohring–Opitz, Kabuki and Wiedemann–Steiner syndromes, respectively.
The identified variants meet the pathogenicity criteria recommended by ACMG guidelines, including de novo occurrence, absence in general population and predicted LOF effect in genes (ASXL1, KMT2A and KMT2D) causing diseases through this proved pathogenetic mechanism. Type and gene localization of mutations are indiscernible from those described in BOPS, KS and WDSTS patients, respectively (Online Resource Tables S1–S3 and Fig. S1).
We also identified a novel likely pathogenic variant in XRN2 gene: the de novo missense mutation identified in patient #169 is predicted to cause a change of a highly evolutionary conserved aminoacid, p.(G187E) and is considered damaging by 8 out of 11 interrogated prediction softwares. XRN2 is not yet associated with a known human disease. It codes for a 5′ → 3′ exoribonuclease belonging to a large family of conserved enzymes in eukaryotes, ubiquitously expressed and acting in the nucleus. Studies performed in the yeast Schizosaccharomyces pombe (Tucker et al. 2016) demonstrate the activity of the protein in RNA silencing, suggesting a role in the epigenetic machinery, now expanded to include enzymes reversibly modifying mRNA (Koboldt et al. 2012; Van der Auwera et al. 2013). Further studies are needed to assess the potential pathogenic role of the XRN2 c.560G>A, p.(G187E) variant, although a different epigenetic mechanism involving RNA modification might be hypothesized to justify the phenotype of the carrier patient.
Finally, following the autosomal recessive inheritance model we found homozygous/composite heterozygous variants in XYLT2 (pt #76) and PLCB4 (pt #169) genes. XYLT2 and PLCB4 genes are associated with spondyloocular and auriculocondylar 2 syndromes, two diseases showing no clinical signs overlapping to RSTS. Clinical re-evaluation of patients #76 and #169 did not highlight any sign resembling those of spondyloocular and auriculocondylar 2 syndromes. Moreover, patient #169 is found carrier of both XRN2 and PLCB4 variants, making difficult to decipher the role of each gene in pathogenesis. In these cases, no clear pathogenetic role can be defined.
Provision of a definite molecular diagnosis to half of the WES-processed patients represents a main goal of this work combined to the disclosure of known genes implicated in epigenetic syndromes with partial phenotypic overlap with RSTS. Besides highlighting common molecular mechanisms underlying these syndromes, this result highlights the challenge of diagnosis of syndromic disorders spanning a broad phenotypic spectrum.
Genotype–phenotype correlations are very complex in the molecularly solved cases. All the eight patients were enrolled in this study as they received an initial diagnosis of Rubinstein–Taybi syndrome, supported by the presence of several typical signs (Table 2 and Table S5). In particular, broad thumb/hallux, typical supraorbital region features and low hanging columella are signs present in almost all the described patients that probably induced the clinician to formulate the RSTS or RSTS-like diagnosis. However, focusing on the four patients found carriers of likely patho-genetic variants, the patients also display additional signs, atypical in RSTS, as exemplified by the distinctive Kabuki sign of eversion of the lateral third of the lower eyelids in patient #95, who turned out to be carrier of a KMT2D mutation (Fig. 1).
To reconcile the initial clinical to the final molecular diagnosis, one need to recall that the RSTS diagnosis has been formulated during infancy of our patients and phenotypic changes manifested during growth were only monitored at clinical re-evaluation upon WES results. In addition, clinical features of syndromes such as BOPS have been defined only in the last years, hampering the referring clinician to raise this diagnostic hypothesis. Indeed, a specific alternative diagnosis (i.e. BOPS for patients #80 and #173, KS for #95 and WDSTS for #103) has been taken into account only after the molecular findings. However, the phenotype of patient #103, found carrier of KMT2A mutation associated to WDSTS, remains RSTS-matching also in his adolescence. Moreover, he does not show the generalized or localized (i.e. hypertricosis cubiti) hypertrichosis which is a typical sign of WDSTS (Aggarwal et al. 2017) and conversely maintains the distinctive RSTS facies (Fig. 1). Similarly, patients found carriers of ASXL1 mutations (#80 and #173) display an atypical RSTS phenotype, but their clinical signs are only in part reminiscent of BOPS features and in a milder form (e.g. exophthalmos in one out of two patients, camp-todactyly, mild growth delay). In the literature, the BOPS patients show a severe overall clinical presentation that may compromise the survival in the childhood; despite the mutations identified in these patients are similar to those of BOPS patients in term of type and gene localization, our patients show very limited BOPS features (Table 2; Fig. 1).
Furthermore, we do not exclude that mutations in the same gene give rise to different or composite phenotypes which may be attributed to stochastic progression during development, to differences in the genetic background between patients and to different strength mutations. Several CREBBP and EP300 mutations were identified in the last years by NGS approaches in patients who had not, or only in a very limited way, the RSTS features (Woods et al. 2014; Masuda et al. 2015; Dauwerse et al. 2016; Menke et al. 2016, 2018; Sellars et al. 2016). Conversely, the same applies to BOPS, KS and WDSTS syndromes and patients featuring with WDSTS were shown mutated in genes underlying the main form of Cornelia de Lange syndrome (Yuan et al. 2015).
Clinical genomics is increasingly revealing molecular pathogenesis and promises to foster remarkable advances in the molecular diagnosis of epigenetic machinery syndromes, as already shown for chromatin-related disorders. Examples from the recent literature are mutations of the gene for ankyrin repeat domain-containing protein 11 (ANKRD11, OMIM *611192) which are usually associated with KBG syndrome which is characterized by macrodontia of the upper central incisors, distinctive craniofacial features, short stature, skeletal anomalies and intellectual disability (Sirmaci et al. 2011). Recently, few patients with features consistent with Cornelia de Lange syndrome (partially overlapping with KBG) sorted out from a large CdLS cohort were found carriers of ANKRD11 mutations (Parenti et al. 2016).
Despite the wide presentation of our patients’ phenotype, which may be defined peculiar or unique, we can grasp a slight overlapping feature between the patients, attested by their initial RSTS diagnosis. Though the identified genetic causes are distinct, all the involved genes belong to the epigenetic apparatus: KMT2A and KMT2D are known writers (just like CBP and p300), ASXL1 is classified as reader. Alteration in one of the component of the epigenetic machinery can perturb the equilibrium of opening/closing chromatin, but also the crosstalk between the different players of this vast and complicated interconnected network may be imbalanced (Allis and Jenuwein 2016). Information flow is bi-directional forming a feedback loop among individual epigenetic machinery. Nevertheless, disruption of histone marks leads to disruption of DNA methylation levels and vice versa (Jin et al. 2008; Sobreira et al. 2017) but also one histone mark alteration can influence the global histone modification. In this view, different overlapping disorders can develop from similar shared epigenetic modification and the specific cell population sensitive to loss of epigenetic machinery component at specific time can induce the mild differences underlying the different features of epigenetic syndromes.
These findings highlight then on one hand the known overlap between RSTS and other conditions and on the other underline the expansion of the phenotypic spectrum of variants in genes encoding epigenetic machinery.
In summary, mutations in genes other than CREBBP or EP300 such as ASXL1, KMT2A, and KMT2D, known to be causative of the epigenetic syndromes BOPS, WDSTS and KS, respectively, are found associated with a specific, different and composite phenotype which seems to be more RSTS-typical in early childhood.
The patients herein described might be the top of a platform of cases that are overlooked by conventional workflow and could be easily detected by NGS targeted to multigene panel. Results from this work support the view that different molecular causes, all belonging to the same epigenetic interplay, underpin different/unique overlapping phenotypes: these multiple causes should be considered in order to perform the appropriate diagnosis and management of patients with epigenetic syndromes.
Supplementary Material
Acknowledgements
We thank the patients’ families for participating in this study. CG thanks the Italian Association of Rubinstein–Taybi patients “RTS Una Vita Speciale ONLUS” for its support and Dr. Giordano, Dr. Ficcadenti, Dr. Cavaliere, Dr. Vitiello for providing clinical data of patients #76, #88, #118 and #169, respectively. This work was supported by University of Milan young researcher grant to CG (Dotazione d’Ateneo linea 2 del piano di sostegno alla ricerca), by Associazione “RTS Una Vita Speciale ONLUS” (project #DigiRare) to CG and by a Ministry of Health grant to Istituto Auxologico Italiano IRCCS (08C623_2016) to PF.
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s00439–019-01985-y) contains supplementary material, which is available to authorized users.
Extended author information available on the last page of the article
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Aggarwal A, Rodriguez-Buritica DF, Northrup H (2017) Wiedemann–Steiner syndrome: novel pathogenic variant and review of literature. Eur J Med Genet 60:285–288. 10.1016/j.ejmg.2017.03.006 [DOI] [PubMed] [Google Scholar]
- Allis CD, Jenuwein T (2016) The molecular hallmarks of epigenetic control. Nat Rev Genet 17:487–500. 10.1038/nrg.2016.59 [DOI] [PubMed] [Google Scholar]
- Arunachal G, Danda S, Omprakash S, Kumar S (2016) A novel denovo frameshift mutation of the ASXL1 gene in a classic case of Bohring–Opitz syndrome. Clin Dysmorphol 25:101–105. 10.1097/MCD.0000000000000126 [DOI] [PubMed] [Google Scholar]
- Baer S, Afenjar A, Smol T, Piton A, Gérard B, Alembik Y, Bienvenu T, Boursier G, Boute O, Colson C, Cordier MP, Cormier-Daire V, Delobel B, Doco-Fenzy M, Duban-Bedu B, Fradin M, Geneviève D, Goldenberg A, Grelet M, Haye D, Heron D, Isidor B, Keren B, Lacombe D, Lèbre AS, Lesca G, Masurel A, Mathieu-Dramard M, Nava C, Pasquier L, Petit A, Philip N, Piard J, Rondeau S, Saugier- Veber P, Sukno S, Thevenon J, Van-Gils J, Vincent-Delorme C, Willems M, Schaefer E, Morin G (2018) Wiedemann-Steiner syndrome as a major cause of syndromic intellectual disability: a study of 33 French cases. Clin Genet 94:141–152. 10.1111/cge.13254 [DOI] [PubMed] [Google Scholar]
- Bjornsson HT (2015) The Mendelian disorders of the epigenetic machinery. Genome Res 25:1473–1481. 10.1101/gr.190629.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohring A, Silengo M, Lerone M, Superneau DW, Spaich C, Braddock SR, Poss A, Opitz JM (1999) Severe end of Opitz trigonocephaly (C) syndrome or new syndrome? Am J Med Genet 85:438–446. 10.1002/(SICI)1096-8628(19990827)85:5<438::AID-AJMG2>3.0.C0;2-A [DOI] [PubMed] [Google Scholar]
- Breuning MH, Dauwerse HG, Fugazza G, Saris JJ, Spruit L, Wijnen H, Tommerup N, van der Hagen CB, Imaizumi K, Kuroki Y, van den Boogaard MJ, de Pater JM, Mariman EC, Hamel BC, Himmelbauer H, Frischauf AM, Stallings R, Beverstock GC, van Ommen GJ, Hennekam RC (1993) Rubinstein-Taybi syndrome caused by submicroscopic deletions within 16p13.3. Am J Hum Genet 52:249–254 [PMC free article] [PubMed] [Google Scholar]
- Dauwerse JG, van Belzen M, van Haeringen A, van Santen G, van de Lans C, Rahikkala E, Garavelli L, Breuning M, Hennekam R, Peters D (2016) Analysis of mutations within the intron20 splice donor site of CREBBP in patients with and without classical RSTS. Eur J Hum Genet 24:1639–1643. 10.1038/ejhg.2016.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrner JA, Bjornsson HT (2014) The Mendelian disorders of the epigenetic machinery. Mendelian disorders of the epigenetic machinery: tipping the balance of chromatin states. Annu Rev Genom Hum Genet 15:269–293. 10.1146/annurev-genom-090613-094245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fergelot P, Van Belzen M, Van Gils J, Afenjar A, Armour CM, Arveiler B, Beets L, Burglen L, Busa T, Collet M, Deforges J, de Vries BB, Dominguez Garrido E, Dorison N, Dupont J, Francannet C, Gar-ciá-Minaúr S, Gabau Vila E, Gebre-Medhin S, Gener Querol B, Geneviève D, Gérard M, Gervasini CG, Goldenberg A, Josifova D, Lachlan K, Maas S, Maranda B, Moilanen JS, Nordgren A, Parent P, Rankin J, Reardon W, Rio M, Roume J, Shaw A, Smigiel R, Sojo A, Solomon B, Stembalska A, Stumpel C, Suarez F, Terhal P, Thomas S, Touraine R, Verloes A, Vincent-Delorme C, Wincent J, Peters DJ, Bartsch O, Larizza L, Lacombe D, Hennekam RC (2016) Phenotype and genotype in 52 patients with Rubinstein- Taybi syndrome caused by EP300 mutations. Am J Med Genet A 170:3069–3082. 10.1002/ajmg.a.37940 [DOI] [PubMed] [Google Scholar]
- Hamilton MJ, Newbury-Ecob R, Holder-Espinasse M, Yau S, Lillis S, Hurst JA, Clement E, Reardon W, Joss S, Hobson E, Blyth M, Al-Shehhi M, Lynch SA, Suri M, DDD Study (2016) Rubinstein-Taybi syndrome type 2: report of nine new cases that extend the phenotypic and genotypic spectrum. Clin Dysmorphol 25:135–145. 10.1097/MCD.0000000000000143 [DOI] [PubMed] [Google Scholar]
- Jin B, Tao Q, Peng J, Soo HM, Wu W, Ying J, Fields CR, Delmas AL, Liu X, Qiu J, Robertson KD (2008) DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum Mol Genet 17:690–709. 10.1093/hmg/ddm341 [DOI] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK (2012) VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22:568–576. 10.1101/gr.129684.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacombe D, Saura R, Taine L, Battin J (1992) Confirmation of assignment of a locus for Rubinstein-Taybi syndrome gene to 16p13.3. Am J Med Genet 44:126–128. 10.1002/ajmg.1320440134 [DOI] [PubMed] [Google Scholar]
- López M, Seidel V, Santiáñnez P, Cervera-Acedo C, Castro-de Castro P, Domínguez-Garrido E (2016) First case report of inherited Rubinstein–Taybi syndrome associated with a novel EP300 variant. BMC Med Genet 17:97 10.1186/s12881-016-0361-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda K, Akiyama K, Arakawa M, Nishi E, Kitazawa N, Higuchi T, Katou Y, Shirahige K, Izumi K (2015) Exome sequencing identification of EP300 mutation in a proband with coloboma and imperforate anus: possible expansion of the phenotypic spectrum of Rubinstein-Taybi syndrome. Mol Syndromol 6:99–103. 10.1159/000375542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke LA, van Belzen MJ, Alders M, Cristofolik F, DDD Study, Ehmke N, Fergelot P, Foster A, Gerkes EH, Hoffer MJ, Horn D, Kant SG, Lacombe D, Leon E, Maas SM, Melis D, Muto V, Park SM, Peeters H, Peters DJ, Pfundt R, van Ravenswaaij–Arts CM, Tartaglia M, Hennekam RC(2016) CREBBP mutations in individuals without Rubinstein–Taybi syndrome phenotype. Am J Med Genet A 170:2681–2693. 10.1002/ajmg.a.37800 [DOI] [PubMed] [Google Scholar]
- Menke LA, DDD study, Gardeitchik T, Hammond P, Heimdal KR, Houge G, Hufnagel SB, Ji J, Johansson S, Kant SG, Kinning E, Leon EL, Newbury-Ecob R, Paolacci S, Pfundt R, Ragge NK, Rinne T, Ruivenkamp C, Saitta SC, Sun Y, Tartaglia M, Terhal PA, van Essen AJ, Vigeland MD, Xiao B, Hennekam RC(2018) Further delineation of an entity caused by CREBBP and EP300 mutations but not resembling Rubinstein-Taybi syndrome. Am J Med Genet A 176:862–876. 10.1002/ajmg.a.38626 [DOI] [PubMed] [Google Scholar]
- Negri G, Milani D, Colapietro P, Forzano F, Della Monica M, Rusconi D. Consonni L, Caffi LG, Finelli P, Scarano G, Magnani C, Selicorni A, Spena S, Larizza L, Gervasini C (2015) Clinical and molecular characterization of Rubinstein–Taybi syndrome patients carrying distinct novel mutations of the EP300 gene. Clin Genet 87:148–154. 10.1111/cge.12348 [DOI] [PubMed] [Google Scholar]
- Negri G, Magini P, Milani D, Colapietro P, Rusconi D, Scarano E, Bonati MT, Priolo M, Crippa M, Mazzanti L, Wischmeijer A, Tamburrino F, Pippucci T, Finelli P, Larizza L, Gervasini C (2016) From whole gene deletion to point mutations of EP300-positive Rubinstein–Taybi Patients: new insights into the mutational spectrum and peculiar clinical hallmarks. Hum Mutat 37:175–183. 10.1002/humu.22922 [DOI] [PubMed] [Google Scholar]
- Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajii T (1981) Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J Pediatr 99:565–569 [DOI] [PubMed] [Google Scholar]
- Parenti I, Gervasini C, Pozojevic J, Graul-Neumann L, Azzollini J, Braunholz D, Watrin E, Wendt KS, Cereda A, Cittaro D, Gillessen-Kaesbach G, Lazarevic D, Mariani M, Russo S, Werner R, Krawitz P, Larizza L, Selicorni A, Kaiser FJ (2016) Broadening of cohesinopathies: exome sequencing identifies mutations in ANKRD11 in two patients with Cornelia de Lange-overlapping phenotype. Clin Genet 89:74–81. 10.1111/cge.12564 [DOI] [PubMed] [Google Scholar]
- Ramu A, Noordam MJ, Schwartz RS, Wuster A, Hurles ME, Cartwright RA, Conrad DF (2013) DeNovoGear: de novo indel and point mutation discovery and phasing. Nat Methods 10:985–987. 10.1038/nmeth.2611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein JH, Taybi H (1963) Broad thumbs and toes and facial abnormalities. A possible mental retardation syndrome. Am J Dis Child 105:588–608. 10.1001/archpedi.1963.02080040590010 [DOI] [PubMed] [Google Scholar]
- Rusconi D, Negri G, Colapietro P, Picinelli C, Milani D, Spena S, Magnani C, Silengo MC, Sorasio L, Curtisova V, Cavaliere ML, Prontera P, Stangoni G, Ferrero GB, Biamino E, Fischetto R, Piccione M, Gasparini P, Salviati L, Selicorni A, Finelli P, Larizza L, Gervasini C (2015) Characterization of 14 novel deletions underlying Rubinstein–Taybi syndrome: an update of the CREBBP deletion repertoire. Hum Genet 134:613–626. 10.1007/s00439-015-1542-9 [DOI] [PubMed] [Google Scholar]
- Schott DA, Blok MJ, Gerver WJ, Devriendt K, Zimmermann LJ, Stumpel CT (2016) Growth pattern in Kabuki syndrome with a KMT2D mutation. Am J Med Genet A 170:3172–3179. 10.1002/ajmg.a.37930 [DOI] [PubMed] [Google Scholar]
- Sellars EA, Sullivan BR, Schaefer GB (2016) Whole exome sequencing reveals EP300 mutation in mildly affected female: expansion of the spectrum. Clin Case Rep 4:696–698. 10.1002/ccr3.598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirmaci A, Spiliopoulos M, Brancati F, Powell E, Duman D, Abrams A, Bademci G, Agolini E, Guo S, Konuk B, Kavaz A, Blanton S, Digilio MC, Dallapiccola B, Young J, Zuchner S, Tekin M (2011) Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am J Hum Genet 89:289–294. 10.1016/jjajhg.2011.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E, Lin C, Shilatifard A (2011) The super elongation complex (SEC) and MLL in development and disease. Genes Dev 25:661–672. 10.1101/gad.2015411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira N, Brucato M, Zhang L, Ladd-Acosta C, Ongaco C, Romm J, Doheny KF, Mingroni-Netto RC, Bertola D, Kim CA, Perez AB, Melaragno MI, Valle D, Meloni VA, Bjornsson HT (2017) Patients with a Kabuki syndrome phenotype demonstrate DNA methylation abnormalities. Eur J Hum Genet 25:1335–1344. 10.1038/s41431-017-0023-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spena S, Gervasini C, Milani D (2015a) Ultra-rare syndromes: the example of Rubinstein-Taybi syndrome. J Pediatr Genet 4:177–186. 10.1055/s-0035-1564571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spena S, Milani D, Rusconi D, Negri G, Colapietro P, Elcioglu N, Bedeschi F, Pilotta A, Spaccini L, Ficcadenti A, Magnani C, Scarano G, Selicorni A, Larizza L, Gervasini C (2015b) Insights into genotype-phenotype correlations from CREBBP point mutation screening in a cohort of 46 Rubinstein-Taybi syndrome patients. Clin Genet 88:431–440. 10.1111/cge.12537 [DOI] [PubMed] [Google Scholar]
- Tucker JF, Ohle C, Schermann G, Bendrin K, Zhang W, Fischer T, Zhang K (2016) A novel epigenetic silencing pathway involving the highly conserved 5′–3′ exoribonuclease Dhp1/Rat1/Xrn2 in Schizosaccharomyces pombe. PLoS Genet 12:e1005873. 10.1371/journal.pgen.1005873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA (2013) From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinform 43:11.10.1–33. 10.1002/0471250953.bi1110s43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann HR, Kunze J, Grosse FR, Dibbern H (1989) A syndrome of abnormal facies, short stature, and psychomotor retardation In: Atlas of clinical syndromes: a visual aid to diagnosis for clinicians and practicing physicians, 2nd edn Wolfe Publishing Ltd, London, pp 198–199 [Google Scholar]
- Wincent J, Luthman A, van Belzen M, van der Lans C, Albert J, Nordgren A, Anderlid BM (2015) CREBBP and EP300 mutational spectrum and clinical presentations in a cohort of Swedish patients with Rubinstein–Taybi syndrome. Mol Genet Genom Med 4:39–45. 10.1002/mgg3.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods SA, Robinson HB, Kohler LJ, Agamanolis D, Sterbenz G, Khalifa M (2014) Exome sequencing identifies a novel EP300 frame shift mutation in a patient with features that overlap Cornelia de Lange syndrome. Am J Med Genet A 164A:251–258. 10.1002/ajmg.a.36237 [DOI] [PubMed] [Google Scholar]
- Yuan B, Pehlivan D, Karaca E, Patel N, Charng WL, Gambin T, Gonzaga-Jauregui C, Sutton VR, Yesil G, Bozdogan ST, Tos T, Koparir A, Koparir E, Beck CR, Gu S, Aslan H, Yuregir OO, Al Rubeaan K, Alnaqeb D, Alshammari MJ, Bayram Y, Atik MM, Aydin H, Geckinli BB, Seven M, Ulucan H, Fenercioglu E, Ozen M, Jhangiani S, Muzny DM, Boerwinkle E, Tuysuz B, Alkuraya FS, Gibbs RA, Lupski JR (2015) Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes. J Clin Invest 125:636–651. 10.1172/JCI77435 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.