

Abstract
Introduction:
Signal transduction cascades drive cellular proliferation, apoptosis, immune, and survival pathways. Proteins have emerged as actionable drug targets because they are often dysregulated in cancer, due to underlying genetic mutations, or dysregulated signaling pathways. Cancer drug development relies on proteomic technologies to identify potential biomarkers, mechanisms-of-action, and to identify protein binding hot spots.
Areas covered:
Brief summaries of proteomic technologies for drug discovery include mass spectrometry, reverse phase protein arrays, chemoproteomics, and fragment based screening. Protein-protein interface mapping is presented as a promising method for peptide therapeutic development. The topic of biosimilar therapeutics is presented as an opportunity to apply proteomic technologies to this new class of cancer drug.
Expert opinion:
Proteomic technologies are indispensable for drug discovery. A suite of technologies including mass spectrometry, reverse phase protein arrays, and protein-protein interaction mapping provide complimentary information for drug development. These assays have matured into well controlled, robust technologies. Recent regulatory approval of biosimilar therapeutics provides another opportunity to decipher the molecular nuances of their unique mechanisms of action. The ability to identify previously hidden protein hot spots is expanding the gamut of potential drug targets. Proteomic profiling permits lead compound evaluation beyond the one drug, one target paradigm.
Keywords: biosimilar, drug discovery, hot spot, mass spectrometry, peptide, protein-protein interaction, reverse phase protein array, validation, cancer
1. Introduction
The technological tool box for drug development depends on proteomic methods. Drug development is no longer restricted to chemists using high throughput screening of large chemical libraries. Functional phenotypes are defining the paradigm for drug target identification. Signal transduction cascades drive cellular proliferation, death, immune, survival, and transcription and translation responses. These processes, and the proteins involved, have emerged as prime drug targets because they are often dysregulated in cancer and other diseases. The proteins may reside not only in the tumor cells, but in the stromal, or immune cells in the tumor microenvironment.
Precision medicine (or personalized medicine) incorporates genomic and/or proteomic signatures of a patient to develop treatment plans and ensure the highest chance of remission [1]. Precision medicine focuses on providing targeted therapies that selectively inhibit molecular drivers of a patient’s cancer. The goals are to reduce patient suffering from adverse effects of cytotoxic cancer agents and increase efficacy of cancer treatments. Over the period 1949-2014, a total of 150 anti-cancer drugs were approved by the FDA; of those, 61 were cytotoxic agents, while 89 were targeted therapies [2]. Targeted therapy continues to be a major focus of cancer drug development. Numerous antibody-based therapeutics have been developed and approved in the past 20 years [3,4]. However, in order to accelerate progress in treating disease, particularly against conditions such as cancer that frequently develop resistance to existing therapies, new targets for drug development must be discovered, as well as expanding our knowledge of the depth and breadth of protein interactions and epigenetic modifications caused by drug treatments.
Proteomics methodologies lend themselves to multiple stages of the drug discovery pipeline: a) to identify biomarkers or kinases implicated in the disease process, b) to validate the proteins to diagnosis or predict disease, and c) to validate the sensitivity, specificity, and predictive value. Proteomic assays employed in drug development span the gamut from immunohistochemistry for protein localization, western blotting and reverse phase protein arrays for quantifying protein/post-translational modifications, mass spectrometry for protein discovery and mass spectrometry multiple reaction monitoring for peptide quantification, to newer technologies such as click chemistry (chemoproteomics) and protein painting for deciphering protein-protein interaction hotspots [5-7]. These methodologies are often used in combination for drug target verification and validation. Therapeutic target validation requires three assessments of the drug target: 1) detection of the drug target in diseased tissue (cells), using a method that is sensitive and specific to quantify an increase/decrease in the drug target; 2) assays to prove that the target was inhibited/activated; and 3) correlation between drug target modulation and clinical outcome [8]. An integrated proteomics platform was used by Kuenzi et al [9] for repurposing ceritinib in ALK-negative lung cancer cell lines. Using this integrated approach, FAK1, RSK1/2 and IGF1R were identified as downstream signaling nodes modulated by ceritinib. Phosphorylated FAK1 Tyr397 was associated with a synergistic effect of ceritinib plus paclitaxel in RAS mutant cell lines. Emdal et al [10] used an integrative technology strategy for quantifying the Anaplastic Lymphoma Kinase (ALK) interactome, phosphotyrosine interactome, phosphoproteome, and proteome to produce a map of ALK actionable tyrosine kinase and adaptor protein signaling nodes in neuroblastoma cells. These two studies highlight the necessity for applying proteomic technologies to multiple areas of the drug discovery process.
Herein, we provide a brief summary of proteomic technologies for drug discovery, with a perspective on protein-protein interaction mapping as a promising method for small molecule/peptide therapeutic development. The US Food and Drug Administration (FDA) recently approved a number of biosimilar therapeutics, thus adding a new class of therapeutics to the protein inhibitor armamentarium [11-13]. The topic of biosimilar therapeutics is presented from the perspective of utilizing proteomic technologies to characterize potential differences between the predicate therapeutic and the biosimilar.
2. Proteomic technologies in drug development
2.1. Mass spectrometry
Mass spectrometry enables untargeted identification of thousands of peptides and proteins within solubilized specimens, revealing a multitude of macromolecules that may be relevant to therapeutic development. Mass spectrometry remains the standard method for performing bottom-up proteomics, global identification of proteins within complex sample matrices. Shotgun proteomics, generally performed by liquid chromatography paired with tandem mass spectrometry (LC-MS/MS) of protease digested proteins, facilitates cumulative identification of the functional proteome. In this method, detected ion fragments are compared to in-silico peptide sequences and ever-growing sequence databases (ProteinProspector, Expressionist, etc.) in order to identify peptides based on database matches [14].
The widespread adoption of selected reaction monitoring (SRM), also known as multiple reaction monitoring (MRM), has facilitated targeted, quantitative proteomic analysis. SRM/MRM quantitation of targeted proteins is performed by tandem MS/MS using triple quadrupole mass spectrometers (Figure 1). Selected precursors with one or more fragmented ions (aka transitions) of a specific mass are selected for detection. Parallel reaction monitoring (PRM) is a variation of SRM to facilitate quantifying specific precursors in a vast proteomic milieu [15]. PRM is based on MS/MS analysis of only the selected precursors. Cycling through selected precursors only for transition ions eliminates all other ions except the precursor ions for the specific fragment ions of interest, thus dramatically reducing the complexity of the data. PRM allows parallel detection of all target ion transitions, providing a method for simultaneously quantify multiple proteins/peptides [16,17]. SRM, MRM, and PRM techniques provide adequate sensitivity and selectivity for relative quantitation and can be combined with verified calibrators (stable isotopes) for absolute quantitation.
Figure 1. Multiple Reaction Monitoring for validating protein sequences.
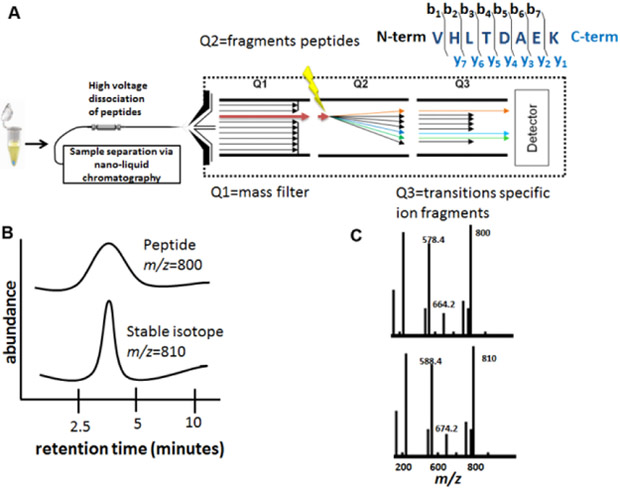
A) MRM workflow. Stable isotope standards are spiked into specimens prior to nano-liquid chromatography. Triple quadrupole mass spectrometers allow filtration of peptides by mass (quadrupole 1), fragmentation of peptides into b ions (from the protein N-terminus) and y ions (from the protein C-terminus) (quadrupole 2), and cycling (transition) of selected ions, prior to detection of unique protein fragments. B) The liquid chromatography retention time of the stable isotope standard is the same as its corresponding peptide. C) The mass/charge (m/z) ratio of the stable isotope standard is different from the peptide by a defined mass, allowing specific peptides to be quantified and validated. Reprinted/adapted by permission from Springer Nature: Book Publisher Springer, Molecular Profiling, 2nd edition, Chapter 20 Quantitative Mass Spectrometry by Isotope Dilution and Multiple Reaction Monitoring (MRM) by Paul Russo, Brian L. Hood, Nicholas W. Bateman, and Thomas P. Conrads. COPYRIGHT 2017.
Targeted proteomics permits hypothesis driven proteomics, wherein proteins of significance can be investigated even if they are of relatively low abundance in the sample matrix. Shi et al thoroughly described the evolution of targeted proteomics in their 2016 review [18]. The applicability of targeted proteomics by SRM extends to a multitude of research challenges. Mass spectrometry can be used to quantitate proteins that are poorly distinguished by commercially available antibodies, as seen in the recent use of LC-SRM to quantitate histone H2 variants [19]. SRM has also been employed to develop a breast cancer panel evaluating Estrogen Receptor (ER⍺), Progesterone Receptor (PR), and human epidermal growth factor receptor 2 (HER2) and post-translational modifications at nine different phosphorylation sites [20].Recently, a PRM method was developed for use in dogs with nonislet-cell tumor hypoglycemia using human QPrEST synthetic proteins, which are heavy isotope-labeled standards [21]. The PRM method was used to simultaneously quantify serum levels of insulin-like growth factors IGF-I, IGF-II, IGFBP-3, and big IGF-II, an incompletely processed form of IGF-II, in dogs [21]. The method allows evaluation of insulin-like growth factor signaling pathways before and after surgical removal of the tumor.
Post-translational modifications lend functional heterogeneity to gene products [22]. One gene can create a variety of functional proteins (proteoforms) through epigenetic modifications of the protein, including phosphorylation, acetylation, sumolyation, methylation, and glycosylation [23]. Many receptor tyrosine kinases, which are targets of Food and Drug Administration (FDA) approved therapies, modulate function via phosphorylation of serine, threonine, tyrosine residues [24,25]. Phosphoprotein enrichment strategies prior to mass spectrometry analysis include phospho-specific antibodies (e.g. anti-phosphotyrosine), titanium dioxide (TiO2) affinity chromatography [26], or immobilized metal ion affinity chromatography (IMAC) [27,28]. Antibody based enrichment strategies are limited by the antibody specificity for a single class of phosphorylation residues. For example, phospho-tyrosine antibody enrichment is specific for tyrosine phosphorylation, and fails to detect phosphorylation on serine or threonine residues. To detect 3 classes of phosphorylation, it is necessary to include phosphorylation-specific antibodies for serine and threonine residues [26]. IMAC or TiO2 methods allow simultaneous enrichment of all classes of phosphorylation, thus enabling broader molecular profiling of PTMs. IMAC enrichment of phosphorylated proteins has been used for biomarker discovery in high and low risk recurrence cohorts of human breast cancer tissues [28]. Differential phosphoprotein expression was shown for 117 phosphoproteins, with 19 selected and validated by SRM [28].
In the past year alone, mass spectrometry has facilitated drug discovery by identifying new biomarkers for acute myeloid leukemia [29], bladder cancer [30], and pancreatic cancer [31]. In addition, mass spectrometry methods for analyzing surface glycoproteins, an understudied proteomic family, have been created [32]. Quantitative proteomics by mass spectrometry, however, is still limited by method sensitivity and instrumentation, and often requires a priori protein enrichment by hydrogel nanoparticles [33,34], ligand library beads [35], FASP [36,37], SDS-PAGE, or immunoprecipitation [38]. Fortunately, mass spectrometry best practices are being implemented and endorsed by organizations, journals, and laboratories [19,33-35].
2.2. Reverse phase protein arrays
Reverse phase protein arrays (RPPA) are a high throughput antibody-based approach for the quantitation of proteins and their post-translational modified forms [24,39-42]. Post-translational modifications dictate protein activity within the cell. RPPA technology has evolved into a key tool for translational proteomics, and is commonly employed to identify specific changes in the proteome and epigenome with maximum specificity and sensitivity for clinical trials and drug development [25,43-46]. RPPA is performed by using a robotic arrayer to print a protein-containing sample (bodily fluids, whole cell lysate, homogenized tissue, peripheral blood mononuclear cells, etc.) onto a slide coated with nitrocellulose or PVDF [47,48]. The slides are blocked and probed with validated antibodies, and chromogenic or fluorescent techniques are used to quantify the relative abundance of antigen within each array spot (Figure 2).
Figure 2. Reverse phase protein array technology.
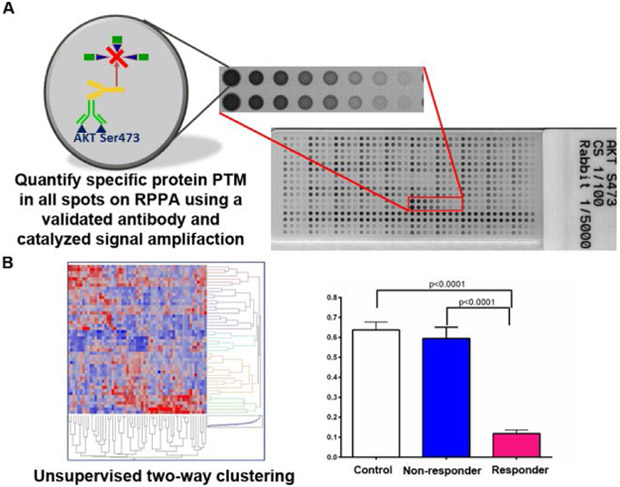
A) Reverse phase protein arrays are constructed by printing protein lysates onto a nitrocellulose slide, typically in replicate, in a two-fold dilution series. The arrays are printed with a robotic arrayer, ensuring highly reproducible spot morphology for specimens, controls, and calibrators. Each array is probed with a validated, single primary antibody that is specific for a protein or a post-translationally modified protein within the protein milieu of each spot. The protein of interest is quantified using spot analysis software. B) Multiple arrays, each probed with a different validated antibody, can be used to create functional protein network maps. Unsupervised two-way hierarchical clustering algorithms reveal potential biomarkers and cohorts of specimens with differentially expressed protein levels. Individual protein levels can be quantified and compared across treatment groups.
Hundreds of individual samples can be printed on a single slide and multiple slides are prepared for each sample set, providing an assay that simultaneously compares many samples for relative changes in protein abundance. This approach facilitates evaluation of treated versus control samples, primary versus metastatic cells, temporal studies, or any other comparative analyses. Other recent applications of RPPA include pinpointing protein off-target effects of therapeutics [49], deciphering mechanisms of actions for small molecule induction of protein synthesis [50], and identifying immune system defects associated with viral infection [51]. Mechanistic studies and phenotype screens at the post-translational level use RPPA for drug development. Pawlak et al recently described the attributes of RPPA phenotypic screening of 2D, 3D, and ex vivo models for drug development, lead compound optimization, and mechanism of action studies at the pathway level [45]. Epigenetic modifications are readily quantified using RPPA. Fingerprinting histone acetylation at various drug dosages and time frames was accomplished with RPPA in HCT116 colon cancer cell lines [46]. Sixty-two drug targets were evaluated by RPPA in a dose and time series to characterize an active analog of bisebromoamide, a compound derived from a marine cyanobacterium (Lyngbya species) [44]. These papers highlight the concept of quantifying multiple proteins that may be modulated by a drug, rather than limiting analysis to one drug/one target.
2.3. Chemoproteomics
Chemoproteomics is well-suited for identifying therapeutically relevant proteins that are unknown or understudied. A study of a drug’s off-target effects is limited by the breadth of the proteins that are screened. Screening a library of proteins known to be physiologically relevant to a disease may cover a few hundred proteins, and including relevant post-translationally modified versions of these proteins may increase that number two- to three-fold. However, this still only covers a minor percentage of proteins encoded by the human genome, the majority of which are never investigated for potential therapeutic benefits or consequences.
In chemoproteomics, bioactive small molecules, the chemical probes, are screened against cellular extracts or in vitro protein panels to identify high affinity binding partners. The probe and its associated protein are retrieved and proteins are usually identified by mass spectrometry. Affinity-based techniques affix the small molecule to a bead or similar moiety to facilitate extraction of the small molecule and its binding partners [52]. Activity-based techniques require co-incubation of the small molecule with an enzyme, such that it reacts with and covalently bonds to the enzyme’s active site. A linker segment connects the small molecule to a retrieval and/or detection moiety [53]. Click chemistry can be used to conjugate linkers to proteins or other macromolecules for retrieval [54]. This approach allows the original probe to be small and flexible in order to interact with the target enzyme, but facilitates retrieval after the substrate is covalently bound to the enzyme.
Chemoproteomics can be a powerful tool in drug discovery by providing a more complete picture of the potential protein interactions for any given therapeutic small molecule. This may reveal new mechanisms of action, and thereby, new targets for intervention. As many of the current therapeutic targets are enzymes such as kinases and proteases, activity based chemoproteomics provides a method for determining specificity for enzymes with similar catalytic domains or isoforms within the same enzyme family. The breadth of potential implications of chemoproteomics is highlighted by recent findings, such as the identification of a new therapeutic target for the treatment of lung cancer [55], the identification of a chemical class of inhibitors for an isoform of phospholipase A2 [56], the functional characterization of a member of the histone methyltransferase family [57], and the discovery of anti-proliferation effects resulting from off-targeting by a small molecule inhibitor not previously known to react with any proliferation related pathways [58].
2.4. Fragment screening
Small fragment screening can be used to identify the critical elements of protein binding within a small molecule. Fragment-based drug discovery (FBDD) screens proteins against the small fragments of chemical compounds, to identify the minimal motif needed to support ligand/protein interactions. The protein/ligand interaction is usually observed by Nuclear Magnetic Resonance spectroscopy (NMR), surface plasmon resonance (SPR), or fluorescence spectroscopy [59]. This technique allows a vast array of different basic chemical structures to be evaluated to reveal the key molecular interactions involved in protein binding. Once the chemical backbone needed for specific interaction is identified, the compounds can be further elaborated so that they achieve potency and efficacy in vitro and can be later modified for ideal performance in vivo. Thus, the fragment screening process provides fundamental structure- activity relationship information for a large library of molecular fragments against a given protein.
Fragment based drug discovery facilitates lead identification for designing any small molecule protein inhibitor. The increase in popularity of FBDD has driven new chemical processes for synthesizing fragment libraries and have promoted the expansion of the chemical space with novel and diverse families of small molecules that may serve as next generation protein inhibitors [59,60]. Recent successes of fragment screening include the development of new scaffolds for the design of metalloenzyme inhibitors [61], identification of selective inhibitors of Bruton’s Tyrosine Kinase with little off-target effects on structurally similar kinases [62], and the elucidation of inhibitors of kallikrein-5 [63], a protein implicated in Netherton syndrome.
2.5. Protein-protein interaction mapping
Protein painting is a method to determine specific protein-protein interaction hotspots and facilitate initial drug design [6,7]. In this method, small molecules act as molecular paints and bind to the solvent-accessible, exposed surfaces of proteins involved in protein-protein interactions and block protease cleavage sites. Regions of protein-protein interactions, however, are not exposed to the solvent, and protease cleavage sites in these positions are not blocked by bound molecular paints (Figure 3). Regions subjected to protease digestion are identified by mass spectrometry, and these fragments correlate to sequences involved in protein-protein interfaces [6]. The earliest example of protein painting was its application to identifying the interactions between interleukin 1-beta (IL1-β) and its receptor. Previously hidden points of interaction were revealed, and elucidation of the active portion of the protein/receptor interface facilitated the development of monoclonal antibodies against the target complex to limit signaling through the pathway [6]. Identifying the residues involved in native protein-protein interactions provides a potential new target for the development of inhibitors and antibody based therapeutics.
Figure 3. Hidden protein-protein interactions can be revealed using protein painting.
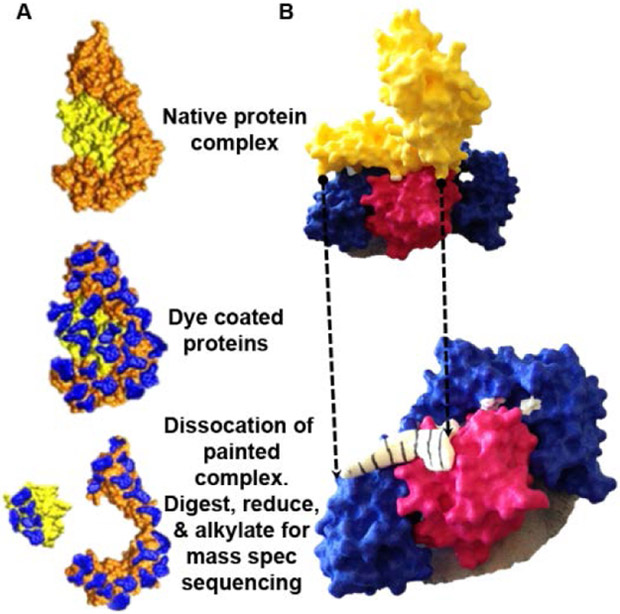
A) Overview of protein painting technology. Native protein complexes contain solvent inaccessible protein-protein interaction hot spots. Dyes coat only the protein surfaces of the native protein complex. Dissociation of the dye-coated proteins exposes sites of protein-protein interaction, which are identified by unpainted amino acids. The peptide sequence of the interaction hot spots is revealed by mass spectrometry sequencing of the dissociated protein complexes. B) 3-D printed model of interleukin-1β. Protein painting uncovers novel, hidden hot spots that can be exploited as potential small peptide inhibitor sites. The interaction site between the yellow domain and the blue and pink domains is hidden by conventional protein-protein interaction mapping. The white/black stripped area in the bottom panel represents a peptide inhibitor identified by protein painting of interleukin-1β [6].
The aforementioned proteomic technologies have been referenced extensively in the literature for therapeutic development. This perspective focuses on protein-protein interaction mapping for drug discovery.
3. Protein-protein interactions as targets for cancer drug development
Protein kinases are major molecular drug targets. Over the period of 1949-2014, a total of 150 anti-cancer drugs were approved by the FDA; of those, 61 were cytotoxic agents, while 89 were targeted therapies [2]. Currently, 37 FDA-approved kinase inhibitors are on the market, with an additional 150 kinase inhibitors in clinical trials [64]. The human kinome itself only contains 538 protein kinases [64], with a spectrum of relevancy to cancer therapy. In a search for new classes of molecular targets, protein-protein interactions may potentially provide a significant expansion in the number of new targets available for drug discovery. While determining the number of protein-protein interactions in humans is not straightforward, estimates based on experimental results cataloged in multiple databases indicate roughly 93,000 reliable protein-protein interactions, with estimates for the total number of protein-protein interactions in humans ranging from 130,000 to 650,000 [65]. The number of protein-protein interactions in the human body is at least 2-3 orders of magnitude greater than the number of kinases in the human body. Identifying and effectively targeting these protein-protein interactions could provide a plethora of new opportunities for drug development.
Despite the promising opportunity, targeting protein-protein interactions has proven to be extraordinarily difficult. Fundamentally, protein-protein interactions are structurally different than classical protein-ligand interactions; protein-protein interfaces differ in terms of the size and topology of interface as well as chemical residues found at the interface. For a generic protein-protein interface, an average surface area of interaction is ~1000 Å2 and involves ~28 amino acid residues [65]. This area, significantly larger than a generic ligand-receptor binding interface, is generally devoid of classical small molecule binding pockets that can be exploited as targets for small molecule therapeutics. Thus, protein-protein interfaces are often flat and relatively featureless [65]. The interface region itself can be segregated into a “core” region which is solvent-inaccessible and defines the location of protein-protein interaction hotspots, and a “rim” region of the interaction that is solvent accessible [65]. Protein-protein interaction hotspots are residues that contribute at least 2 kcal mol−1 of total binding energy to the complex, thus representing the most compelling targets for disruption of a protein-protein interface as these hotspot residues drive binding affinity between the interacting proteins [65]. Alanine scanning can be used to determine hotspot residues [44], but this technique is time consuming; crystallography can be used to define whole interfaces, but does not selectively distinguish hotspot residues. While computational prediction of hotspot residues in protein-protein interactions is improving, most computational methods still rely on structural information first to determine hotspots, which becomes challenging as many therapeutically relevant targets lack detailed structural information [66]. Analysis of known protein-protein interaction hotspot residues has shown a strong overrepresentation of tyrosine and arginine as hotspot residues, while isoleucine, valine, leucine, and alanine are strongly underrepresented [67], suggesting certain patterns of primary sequences may likely be drivers of binding affinity in protein-protein interactions. Successful identification of protein-protein interaction hotspot residues remains a key to developing modulators of protein-protein interactions.
3.1. Successful protein-protein interaction inhibitors as cancer therapeutics
It is worth noting that while hormonal therapies and kinase inhibitors are two large classes of targeted cancer treatments that disrupt downstream protein signaling, neither are considered true protein-protein interaction inhibitors. Both therapeutic classes are primarily designed to fit in small molecule binding pockets, i.e. steroid binding pockets or ATP-binding pockets. For this reason, compounds from these two classes are not considered protein-protein interaction inhibitors in this article despite the protein-protein interactions of their respective targets.
Despite the difficulty in designing protein-protein interaction inhibitors, several compounds that disrupt relevant protein-protein interactions are either FDA approved or in clinical trials as cancer therapies. Examples are described below.
3.1.1. Bcl-2 family inhibitors
Apoptosis is governed by a complex protein signaling network, including members of the B-cell lymphoma-2 (Bcl-2) protein family. Bcl-2 family proteins consist of both pro-apoptotic members and pro-survival members, and the balance of these two protein functionalities help maintain appropriate balance between cell survival and cell death [68]. Bcl-2, the first member discovered in the mid-1980s and the namesake for the family, was discovered in B-cell lymphoma where its overexpression contributes to increased lymphomagenesis [68]. The pro-survival activity of Bcl-2 is inhibited when a helical BH3-domain of one of the pro-apoptotic members of Bcl-2 family bind to a deep binding cleft in Bcl-2 [69]. Inhibitors disrupting the Bcl-2/BH-3 domain protein-protein interaction could restore appropriate apoptotic function to cancer cells overexpressing Bcl-2.
Development of small molecules targeting Bcl-2 or other pro-survival members of the Bcl-2 family, including Bcl-XL or Bcl-w, began soon after Bcl-2 was discovered. Abbot reported the first-in-class Bcl-XL inhibitor ABT-737 in 2005 as the optimized product of a fragment screening campaign [70]. Poor bioavailability of ABT-737 prompted further development of Bcl-2 family inhibitors, yielding a second generation inhibitor ABT-263 that potently inhibits Bcl-XL as well as Bcl-2 [71]. ABT-263 advanced to the clinic in 2006, but was ultimately implicated in an increased risk of thrombocytopenia due to Bcl-XL inhibition causing apoptosis of blood platelets [72]. Additional optimization of ABT-263 produced the Bcl-2 selective agent vABT-199, eliminating the concomitant thrombocytopenia [73]. ABT-199 was approved by the FDA in 2016 for chronic lymphocytic leukemia (CLL) patients with chromosome 17 deletions who had been treated with at least one other therapy [74].
3.1.2. PD-1/PD-L1 inhibitors
Programmed cell death protein 1 (PD-1) is a T-cell receptor discovered in 1992 whose expression is induced on T-cell receptor (TCR) activation and maintained in the presence of consistent immune stimulation [75]. In order to reduce autoimmunity in the presence of consistently activated T-cells, antigen presenting cells express programmed cell death ligand 1 (PD-L1), a transmembrane protein with an extracellular region that binds to PD-1 and inhibits T-cell mediated immune response [76]. Constitutive expression of PD-1 can lead to reduced immune response to tumors through T-cell exhaustion [77], while overexpression of PD-L1 by tumors can inactivate T-cells and shield tumor cells from immune response [76]. In this way, inhibition the PD-1/PD-L1 complex represents an attractive target for cancer therapies. Due to the function of the PD-1/PD-L1 complex as a “check” on the immune system, therapies targeting PD-1 and PD-L1 fall into a class of immuno-oncology treatments known as “checkpoint inhibitors” [78]. The co-crystal structure of the extracellular domains of human PD-1 and PD-L1 was solved in 2015; it revealed a large, relatively flat interaction domain that may prove difficult to target with small molecules [79].
Given this flat interaction domain, it is unsurprising that all FDA approved therapies targeting the PD-1/PD-L1 complex are antibodies. The first approved antibodies targeted PD-1, including pembrolizumab (approved 9/2014) and nivolumab (approved 12/2014) [80], have been shown to bind near the site of interaction with PD-L1 [81], although pembrolizumab’s binding site on PD-1 overlaps more significantly with the PD-L1 binding site than does nivolumab. Following the approval of anti-PD-1 antibodies, three anti-PD-L1 antibodies were approved, including atezolizumab (approved 5/2016), avelumab (approved 3/2017), and durvalumab (approved 05/2017) [80]. Crystal structures indicate that the binding sites of all three anti-PD-L1 antibodies overlap at least partially with the PD-1 binding interface [82]. A third anti-PD-1 antibody, cemiplimab (approved 9/2018) is now also available, although its binding site is not yet reported [83]. Pembrolizumab was first approved in 2014 for melanoma, and since then PD-1/PD-L1 checkpoint inhibitors have been approved for 10 different cancer indications, including the most recent indication of advanced cutaneous squamous cell carcinoma (CSCC) with cemiplimab [80,83]. Given the immense patient and press response to PD-1/PD-L1 checkpoint inhibitors based on a number of high profile clinical trials and personal stories, including the use of pembrolizumab in 2015 to cure President Jimmy Carter’s melanoma that had spread to his brain [84], it is likely that additional generations of inhibitors will be developed. While small molecule inhibitors of the PD-1/PD-L1 complex have been reported, only two small molecules have advanced to a Phase I clinical trial [85]. These compounds include CA-170, whose structure is not yet available, and BMS-986189, which is described as a macrocyclic peptide [85].
3.1.3. BET bromodomain/acetylated histone inhibitors
Chromatin remodeling is an essential process in which tightly wound DNA segments are made accessible to transcriptional machinery by a variety of regulatory mechanisms, such as DNA methylation, remodeling via SWI/SNF complexes, or histone acetylation. Histone acetyltransferases (HATs) transfer acetyl groups from acetyl-CoA to lysine residues on the N-terminal tails of histones, reducing the net positive charge of the histone and thus reducing the electrostatic interactions between the histones and the negatively-charged DNA [86]. Following acetylation, proteins recognizing the polyacetylated histones can bind and recruit downstream transcriptional machinery and regulators to the newly-accessible DNA segment. The most well-characterized protein domain recognizing poly-acetylated histone tails is the bromodomain, a left-handed four-helix bundle that forms a hydrophobic pocket for acetyl-lysine binding [87]. The mammalian bromodomain and extra-terminal domain-containing (BET) protein family is therefore an attractive target for cancer therapeutics, as many cancer cells are particularly vulnerable to disruption of transcriptional regulation due to their relaxed control of gene expression [87].
Currently no FDA-approved BET inhibitors are on the market for cancer therapies, but as of 2017 there were 16 BET inhibitors in clinical trials, primarily in Phase I and primarily for cancer indications [87]. One notable exception is the small molecule BET inhibitor RVX-208, a somewhat selective agent for the second bromodomain in BET family proteins which is currently in Phase III clinical trials in for reduction in cardiovascular events in Type-2 diabetes patients with coronary artery disease [87,88]. While small-molecule bromodomain inhibitors have shown activity in a wide variety of solid-tumor models, including NUT-midline carcinoma and tumors of the breast, prostate, lung, liver, pancreas, intestine, and brain, some concerns over healthy cell toxicity and biological process governed by BET proteins remain [87,89]. As the many BET inhibitors progress through clinical trials, more of the basic biology of BET proteins and potential side effects of their inhibition will hopefully be elucidated.
3.1.4. MDM2/p53 inhibitors
Tumor suppressor protein p53 is a well-recognized pro-apoptotic transcription factor in which loss of function mutations contribute to over 50% of human cancers [90]. In addition to loss of function mutations in the p53 gene, p53 can also be inactivated through N-terminal binding to the murine double mutant 2 (MDM2) oncogene, which inhibits p53 though a variety of mechanisms [91]. MDM2 binding to p53 can initiate degradation of p53 though ubiquitination and recruitment of the 26S proteasome as a E3 ubiquitin ligase, promote nuclear export of p53, and directly prevent association with DNA, eliminating p53’s role as a transcription factor [91]. Structural studies of the p53-MDM2 complex have indicated that the MDM2-bound alpha-helical p53 peptide binds in a defined hydrophobic cleft of MDM2, where binding is mediated by primarily through p53 residues Phe19, Trp23, and Leu26 [92]. This well-defined cleft has proved amenable to small molecule inhibition.
As of 2017, there were nine MDM2/p53 small molecule inhibitory compounds in clinical development, although there are no FDA approved MDM2/p53 inhibitors [93]. Many of these small molecules were originally designed to mimic binding of the three key p53 residues, although further medicinal chemistry optimization has resulted in small molecule inhibitors with >1000x the affinity for MDM2 than the p53 peptide [92]. Structural studies have indicated that these high-affinity compounds take advantage of additional π-π stacking and electrostatic interactions in the binding cavity not seen in the native p53-MDM2 complex [92]. Early clinical results indicate that some MDM2 inhibitors cause thrombocytopenia, which is thought to be a target-based side effect rather than an off-target effect. These side effects lead to dose-limiting toxicity for this class of compounds [92]. Further clinical studies are needed to determine appropriate dosing windows to ensure robust anti-tumor activity with acceptably low on-target toxicity.
3.1.5. SMAC mimetics
In addition to Bcl-2 inhibitors discussed above, there have been additional efforts made to disrupt cancer cell’s resistance to apoptosis through dysregulation of the BCL-2 pathway. The intrinsic apoptotic pathway in cells is reliant on the pore-forming properties of BCL-2 family proteins BAK and BAX, which allow the formation of pores in the outer mitochondrial wall that releases cytochrome c and second mitochondrial-derived activator of caspases (SMAC) into the cytosol [94]. The release of cytochrome c initiates apoptosis through downstream signaling leading to the formation of an apoptosome that activates multiple caspases, while the release of SMAC allows for SMAC binding of a number of diverse inhibitor of apoptosis (IAP) proteins, preventing further inhibition of the intrinsic apoptotic pathway [94]. As anticipated, upregulation of IAP proteins or mutation in BAK and BAX proteins allow cancer cells to resist apoptosis. SMAC itself is an alpha helical protein that dimerizes and binds to IAP proteins though their baculovirus IAP repeat (BIR) domains; crystal structures and biochemical data of SMAC interactions with the BIR3 domains of IAP protein XIAP show a conserved 4-amino acid sequence in SMAC (Ala-Val-Pro-Ile), which constitutes the binding domain. Inhibitor development has proceeded via small molecule mimetics designed around the AVPI peptide [94].
No FDA-approved SMAC mimetics are on the market, although there are six SMAC mimetics currently in clinical trials [94]. The most advanced on these compounds, LC161 and birinapant, have reached Phase II trials [94]. Interestingly, in a mouse model of glioblastoma, both SMAC inhibitors LC161 and birinapant were found to synergize with anti-PD-1 therapy, leading to a possibility of a combination of SMAC inhibitors with immune checkpoint inhibitors as an effective cancer treatment [95]. Toxicity studies have indicated dose-limiting toxicities in some patients, but in general SMAC mimetics appear to be well tolerated and can be combined with conventional chemotherapies [96].
3.2. Addressing developmental challenges for protein-protein interaction inhibitors
Based on the five case studies presented in the previous section, clearly protein-protein interactions are viable targets for cancer therapeutics, but much work remains to be done in accelerating the process from discovery of potential protein-protein interaction targets, to development of small molecule or biologic drugs. In our perspective, there are three key areas in which substantial scientific progress would accelerate the development of protein-protein interaction inhibitors: 1) improving methods and techniques to more quickly identify hotspot residues of protein-protein interactions, 2) increasing development of small molecule moieties suited to selectively target protein-protein interactions, and 3) improving bioavailability and stability of peptides and peptidomimetic drugs. Medicinal chemistry and drug discovery have a long history of developing drugs that target small molecule binding pockets. However, a bias remains in pharmaceutical screening decks and programs towards small, Rule-of-5 compliant molecules that are often ill-suited to targeting protein-protein interactions [97]. Successful protein-protein interactions inhibitors are often larger (>400 Da), more lipophilic (AlogP > 4 (molecular hydrophobicity), and contain more hydrogen bonds than traditional small-molecule drugs [97]. Additionally, while it might seem natural to pursue peptide-based therapies for protein-protein interactions, significant optimization must be done to peptide-based drugs to render them orally stable, conformationally constrained, and if necessary, cell-penetrable.
While improvements in small molecule and peptide-based therapies for protein-protein interactions remain the purview of medicinal chemistry, improved methods to identify protein-protein interaction hotspots is within the proteomic realm. To that end, we offer a perspective on the role of proteomics in developing new techniques to detect protein-protein interaction hotspots, specifically discussing protein painting, and how these techniques can be improved to lead directly from hotspot identification to drug discovery. Furthermore, we discuss how advancements in medicinal chemistry related to protein-protein interaction inhibitors have a direct role in transforming proteomics from primarily diagnostics to the drug discovery.
3.3. Protein painting as a gateway into cancer drug discovery
3.3.1. Identification of protein-protein interaction hotspots via protein painting
While methods such as immunoprecipitation, pull-down assays, or far western blotting can be used to identify interacting protein partners, these methods do not give structural information about protein-protein interactions that can be used to develop interaction inhibitors. To identify locations of protein-protein interactions, the gold standard is obtaining co-crystal structures of the interacting proteins. Crystallography is unsurpassed by any other method for resolution of protein-protein interfaces, yet obtaining co-crystal structures of proteins of interest can be incredibly difficult. Other structure-based techniques, such as NMR or cryogenic electron microscopy (cryoEM), both have great promise in expanding the number of proteins that can be structurally characterized, but at present can only solve structures of either relatively small proteins (NMR) or relatively large proteins (cryoEM). We look with great excitement toward developments in cryo-electron crystallography (or micro electron diffraction, microED), which may ease the burden in protein crystallography by making structure determination possible with protein crystals smaller than conventionally allowed with X-ray diffraction [98]. However, this technique has not become commonplace. Furthermore, for a large, relatively featureless interface identified via crystallography (XRD or microED), only a few amino acids may be driving the majority of the binding affinity. Alanine scanning can reveal drivers of binding affinity, but this technique is quite laborious [44]. As previously noted, computational predictions of hotspot residues are helpful, but often rely on protein structures.
Methods for identifying protein interfaces in the absence of crystallographic data have mainly focused on mass spectrometry techniques, including hydrogen deuterium exchange (HDX), hydroxy radical foot printing, crosslinking mass spectrometry, or protein painting [6,7]. Each technique has the advantage in that additional structural information is not needed. Protein amino acid sequence is sufficient to identify regions subject to structural change. Protein mass spectrometry is a mainstay technique in proteomics laboratories, and mass-spectrometry based structural proteomics is a key area in which proteomics expertise segues into drug discovery. Protein painting answers both structural and drug discovery questions: Where do two proteins interact? and Can a small peptide inhibitor be developed to the interaction domain? Protein painting can be easily adopted by other proteomics laboratories, allowing protein-protein interaction mapping a more prominent place in cancer drug discovery than previously thought possible.
Protein painting was designed to alleviate a few of the concerns associated with comparable structural mass spectrometry techniques, such as non-native conditions (low pH, etc.), high false positive rates, or laborious protocols requiring specialized software (Figure 3) [7]. The technique is simple to perform, and requires no specialized expertise beyond a background in protein mass spectrometry. First, protein painting begins with preforming the complex of interest from isolated, purified protein binding partners. The proteins may be recombinant, or isolated protein complexes from cells through immunoprecipitation. Once the complex is preformed, either through 1 hr. incubation of recombinant proteins or isolation of complexes from cells, it is rapidly pulsed (~5 minutes) with small molecule dyes. These dyes bind promiscuously to protein surfaces, rapidly covering solvent accessible regions of the protein. Small molecule dyes with various binding chemistries can be applied simultaneously to ensure the highest possible coverage of the solvent-accessible surface of the protein complex. Dye pulsing experiments can be conducted in any basic biological buffer in which the protein is stable, with the caveat that if detergents are used, they must be removed prior to mass spectrometry using detergent removal columns. Once the protein complex is covered, any unbound dye can be removed via gel filtration, and the protein complex prepared for mass spectrometry via denaturation, alkylation, and trypsinization (Figure 4). The small molecule dyes remain bound through mass spectrometry preparation and block trypsin cleavage sites [7]. Blocked trypsin sites leave solvent accessible protein surfaces undigested, and thus undetected in mass spectrometry. By comparing the sequences of proteins painted in isolation to a painted protein complex, those peptides that appear differentially in the complexed sample are hit peptides from the protein-protein interface. The output of protein painting is solvent-accessible peptide sequences derived from the protein-protein interface. These peptide sequences represent good targets for developing protein-protein interaction inhibitors.
Figure 4. Protein painting workflow for deciphering protein binding hotspots.

Protein complexes are pre-formed in vitro, followed by coating with organic dye to ‘paint’ the protein surfaces. The inaccessible binding pockets exclude the dye molecules. Unbound dye is removed by gel filtration. The painted protein complex is denatured, digested, and the amino acid sequences are identified by liquid chromatography mass spectrometry. Only those proteins that we not painted by the dye molecules are susceptible to trypsin digestion, and thus can be readily identified by mass spectrometry.
3.3.2. Protein painting to identify drug targets for small molecule or biologic inhibitor development
The output of protein painting experiments is a series of tryptic peptides known to be present at a protein-protein interface where the tryptic lysine or arginine residue is inaccessible to solvent. Accessible lysine and arginine residues are dye bound, and uncleaved by trypsin. Any residues that are not dye covered, such as internal residues that are uncovered during denaturation, are subtracted from the data set by comparison to each protein painted in isolation. These tryptic peptides can be biologically validated via pull down assays to ensure that the identified peptide disrupts complex formation and thus is found at the protein-protein interface. Once validated, these peptides for can be characterized via NMR or circular dichroism spectroscopy (CD) to determine structural features. Even in the absence of other structural information, these validated peptides provide a starting target for small molecule development or biologic inhibitor development. Luchini et al described developing an antibody against a protein painting hit as a successful strategy toward for a specific protein-protein interaction site [6]. In their study of the ternary complex of interleukin 1 β (IL1β), interleukin 1 receptor accessory protein (IL1RAcP), and interleukin 1 receptor 1 (IL1R1), Luchini et al generated an antibody specifically against the protein painting hit on IL1RAcP (Arg286 peptide) that corresponded to the three-way hotspot of the ternary complex based on comparison to the determined crystal structure [6]. This antibody eliminated ternary complex formation [6].
3.3.3. Protein painting as a method to develop lead compound peptides
Protein painting offers a method for the generation of initial peptides for further optimization as peptide or peptidomimetic therapeutics. Once a tryptic peptide identified via protein painting experiments has been validated for complex disruption by pull-down assays, it can serve as a starting place for further medicinal chemistry. While the statement that a peptide is a good starting place for inhibitor generation might strain credibility for a medicinal chemist even as recently as 10 years ago, there have been a series of recent developments in the field of peptide therapeutics that are starting to change the perspective towards development of peptide therapeutics (reviewed in [99].
3.3.4. Cell penetrating peptides
One obvious objection to the development of peptide therapeutics is the difficulty of inhibiting intracellular targets. However, the identification of several classes of cell penetrating peptides for conjugation to a molecular cargo has allowed for the development of inhibitors that by themselves have less than optimal cell penetrating properties due to size or hydrophobicity. Classes of cell penetrating peptides, often initially identified from pathogenic molecules, include cationic peptides, amphipathic peptides, and hydrophobic peptides [100]. Some peptides tags, even as short as six amino acids, can help overcome one of the first hurdles towards the development of peptide therapeutics, and can also assist in intestinal absorption. As of 2017, there were at least 8 different compounds bearing a cell-penetrating-peptide domain in clinical trials.
3.3.5. Stapled macrocyclic peptides
A second concern regarding peptide therapeutics is the conformational flexibility inherent in polypeptide structures. This conformational flexibility results in a significant entropic cost of binding to the target as compared a relatively constrained small molecule inhibitor. In order to constrain peptide therapeutics, much work has been done in recent years in optimizing cyclization strategies, including development of monocycles, bicycles, and stapling strategies in which linkers are added between amino acid side chains or side chains are coupled together directly to reduce conformational flexibility [101]. Examples of such stapling strategies include lactam scanning, in which side chains of lysine and glutamic acid are coupled, or ring closing metathesis, in which hydrocarbon side chains may be stapled [99]. Together, these cyclization and stapling strategies have primarily been pursued by small biotech companies, although the use of these technologies is now reaching larger pharmaceutical companies. A Japanese company, for example, has almost 100 distinct drug discovery programs and has partnered with 18 different pharmaceutical companies, including licensing their technology to 5 large pharma companies [101]. The majority of companies in the constrained peptide space were founded after 2005, at which time the first constrained peptide products began to enter Phase I/II clinical trials [102]. We are now beginning to see significant numbers of constrained peptides entering clinical trials, with roughly 50 non-insulin peptide drugs approved and marketed worldwide [99]. ALRN-6924, for example, is a constrained peptide entering Phase II targeting the MDM2-p53 complex [102] after showing an acceptable safety profile and anti-tumor activity in a Phase I trial published in 2017 [103].
3.3.6. Orally bioavailable peptides
A final significant hurdle for peptide therapeutics to overcome is poor pharmacokinetic properties. While peptide therapeutics can be incredibly specific for their target, poor absorption via the intestinal mucosa or subsequent proteolytic cleave in the serum can significantly reduce the amount of peptide available for target engagement [104]. In addition to reducing conformational flexibility, cyclization of peptides is one key way to improve oral bioavailability by eliminating cleavable N-and C-termini and protecting peptide bonds from proteolytic enzymes [105]. Additional methods, such as chemical modification or substitution of amino acids, are often also necessary to reduce proteolytic cleavage of the peptide therapeutic. Common strategies include substitution of L-amino acids with either D-analogs, α-methylated or N-methylated analogs, β-amino acid analogs, or aza analogs [99]. Strategies such as replacing beta-turn amino acids with small molecule mimics [106] or replacing carbonyl groups in peptide bonds with oxetanyl rings [107] are additional peptide modifications that resist proteolytic cleavage. Beyond modification of the peptide itself, formulation approaches such as peptide PEGylation, attaching poly-ethylene glycol substituents to amino acid side chains, can further improve peptide pharmacokinetics [108]. Delivery mechanisms such as liposome or chitosan nanoparticles have also been proposed as formulations to address oral bioavailability problems [109].
Peptide therapeutics will continue to grow as a viable alternative to small molecule and antibody-based drugs. Small molecules have difficulty effectively targeting protein-protein interfaces and antibodies are restricted to extracellular target. Protein-protein interactions are a prime case study demonstrating that peptide therapeutics may prove to be the most efficacious drug discovery platform.
4. Biosimilars
The growth of the biosimilar market-space is anticipated to lower treatment cost and improve public access to biological therapeutics. As patents end on first generation biologics, production of “generic” versions is sought to maximize profits for the manufacturer and to promote availability to end-users. The approval pipeline for biosimilars, however, differs substantially from the methodology for small molecule approval. The burgeoning growth of biologicals as clinical treatments emphasizes the importance of proteomics in identifying new targets and new entities regulating those targets. However, the development of biosimilars requires critical scrutiny into how proteins are synthesized and modified, and how seemingly minor differences in producing a mature protein can have significant outcomes in efficacy and toxicity. Development of a biosimilar requires intensive characterization and understanding of the contributions of protein complexity to protein activity.
Developing a biologic from lead identification through clinical trials differs from the small molecule pipeline. Emphasis is placed on mechanism of action, efficacy, tolerability, and a comparison of clinical benefit to toxicity and risk. However, biological manufacturing is subject to differences in the producing organisms’ biology, and thus, the synthesized macromolecule has added levels of complexity that reach beyond a primary sequence. Complete details regarding the methods of biosimilar production are generally unavailable, the higher-level details of the biosimilars structure and function remain unknown [110]. Thus, the re-creation of a biological as a biosimilar presents new challenges that are absent from generic small molecule manufacturing [12,13].
Biosimilar protein production typically begins with a genetically engineered cell line that synthesizes the desired protein. The protein is then extracted, purified, and formulated into a final drug product [13]. The final protein is, therefore, a result of its synthesis conditions, including organism or cell line used, culture conditions, cell cycle, cell age, and all related biological factors that affect protein synthesis and post-translational modifications. As a result, a protein intended to serve as a potential biosimilar may differ significantly from the bio-original with respect to glycosylation, phosphorylation, methylation, oxidation, deamidation, and other post-translational modifications that contribute directly to its half-life, aggregation ability, bioavailability, target-affinity, off-targeting effects, and activation of complement and immune response [11-13]. These complications create a challenge to biosimilar development that requires thorough characterization of the mature protein and a critical investigation into whether differences between the bio-original structure and the biosimilar are clinically meaningful and create a difference in efficacy or toxicity.
4.1. The stepwise pathway to proving biosimilarity requires a complete proteomics analysis
The Food and Drug Administration released guidance for demonstrating biosimilarity to a reference in 2015 [111]. The guideline highlights that a biosimilar must be “highly similar” to the reference product with the exception of “minor differences in clinically inactive components”, and emphasizes that no significant difference exists in “safety, purity, and potency of the product” [111].
The stepwise approach begins with a qualitative and quantitative structural and functional comparison of the candidate biosimilar to the reference biologic. The structural comparison of candidate to reference should encompass all reviewable characteristics, including primary structures, higher order structures and aggregation, post translational modifications, naturally occurring chemical modifications, and intentional chemical modifications. Functional evaluations are expected to evaluate pharmacologic activity of the candidate via biological assays, binding assays, and enzyme kinetics [111]. Herein, the goal is to demonstrate that minimal differences exist between the two entities, and that differences between the two are not clinically meaningful. Understanding of the reference biological’s mechanism of action and safety risks may help rationalize how minor structural differences in the biosimilar may not contribute any functional change in comparison to the reference biologic [111]. Proving that no clinically meaningful difference exists is the most essential component of biosimilar development, and determining which deviations have clinical consequences provides an opportunity to re-optimize formulation methods to alleviate these discrepancies. Thus, biosimilar development reverts to fundamental analysis of protein structure-function relationships.
Generally, methods for determining protein structure, function, and formulation purity are well established. Primary structure is often determined by mass spectrometry-based peptide mapping, intact mass analysis, sequence coverage analysis, and N-terminal or C-terminal sequencing [112,113]. Higher order structure can be evaluated by Fourier transform infrared spectroscopy, differential scanning calorimetry, circular dichroism spectroscopy, free thiol analysis, and disulfide bond analysis [103,104]. Charge is evaluated by capillary zone electrophoresis, isoelectric focusing, and ion exchange chromatography. Aggregation is analyzed via gel and liquid based chromatography. Affinities are determined via ELISA and SPR [112,113].
Upon completion of structural and functional comparisons, the candidate should progress to animal studies for evaluation of safety, toxicity, immunogenicity, and to provide insight into pharmacodynamics (PD) and pharmacokinetics (PK) [111]. The comprehensiveness of animal data will likely reflect the results of the structural/functional analysis, as candidates that are near-equivalent to the bio-originals will require less supplemental animal data to conclude biosimilarity. The concept of comprehensiveness extends to clinical studies as well. Comparative human PK and PD studies and an immunogenicity assessment are expected, but more clinical data will be required in proportion to the amount of uncertainty into structural differences between the candidate and the bio-original [111].
4.2. Requirements for biosimilarity promote better understanding of protein glycosylation and its effects
The clinical immunogenicity assessment is outlined in detail in the guideline, highlighting its significance in the development of biosimilars [111]. Immunogenicity can greatly reduce efficacy of a biologic and/or may induce an allergic reaction [13,114]. The guideline directs that predicting immunogenicity based on structural and functional data is usually not feasible, and therefore, a clinical study to compare the immunogenicity of the candidate to the reference is expected [111]. This reality challenges the proteomic community to evolve its understanding of immunogenicity and the impacts of more poorly characterized post-translational modifications, namely glycosylation, on binding and recognition.
Glycosylation, a commonly occurring post-translational modification seen in recombinant proteins, impacts the activity, stability, function, and immunogenicity of the protein [13,114]. Antibody formulations are highly affected, as glycosylation plays a role in antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity [13,114]. Hajba et al thoroughly detail the relationship between specific glycans and the elicited response [114]. Glycosylation is a challenge in the formulation of biosimilars, as the specific means of bio-original synthesis, purification, and formulation are not known or cannot be identically replicated. When developing a biosimilar, manufacturers are often at the mercy of the host organism and culture conditions, and these features will directly regulate the glycosylation of the protein, with little to no regulation offered to the manufacturer. The glycosylation patterns of the biosimilar candidate are likely to differ from the original, but many of the added or lost post-translational modifications will have an unknown effect on activity or immunogenicity until they are tested in many patients.
Mass spectrometry techniques provide a means to characterize the location and identity of the added oligosaccharides and glycan derivatives, but challenges still exist in directly correlating the structural characteristics to functional differences. As such, manufacturers developing biosimilars may not identify clinically meaningful differences between candidates and bio-originals until they perform immunogenicity studies as part of clinical trials. This reality greatly increases the cost of biosimilar development in comparison to small molecular therapeutics and highlights the demand for better elucidation of the “sugar code” and the specificity of glycosylation identity in protein recognition.
The current state of the art for characterizing N-glycosylation involves three levels of analysis: the intact protein, glycopeptide, and released glycans [114]. Intact glycoproteins are analyzed by MS. For IgG antibodies, this provides information into the symmetry or asymmetry of glycans between chains [112-114]. Second order analysis of the glycopeptide by tandem MS/MS is performed following digestion and reduction of the intact glycoprotein and reveals the location and variation of glycan structures at sites along the protein. Finally, glycans are cleaved from the protein backbone and exoglycosidase-based sequencing can be performed to determine the identity of the glycan components [112-114]. This stepwise approach provides a fingerprint of the type and location of glycosylations on the candidate protein and the bio-original.
Glycosylation remains an understudied and underappreciated aspect of protein post-translational modification. However, as biosimilars continue to be developed, the demand for glycosylation characterization increases. Differences that arise in immunogenicity studies will be traced back to glycosylation fingerprints obtained through these characterization workflows. These structure/function analyses will expand knowledge of the effects of different glycan profiles in patients with differing genomes, phenotypes, and immune states. Biosimilar manufacturers will likely correlate features of the host cell organisms and cell culture conditions responsible for generating their protein to the final glycosylation profiles they observe. We can expect biosimilar development to expand our working knowledge of protein glycosylation structure/function relationships and to drive research into how the sugar code is written and delivered to proteins in vivo.
5. Perspectives
Future methodological improvements to protein painting
While protein painting holds promise as a technique to identify lead peptides for drug development, there are a couple key areas of improvement that are needed. The first and most obvious weakness in the technique as it stands now is that resolution is limited by the density of tryptic cleavage sites in the protein partners of interest. This issue is of course common to multiple mass spectrometry techniques; it will be essential going forward to adapt protein painting to work with other proteases such as chymotrypsin or AspN to ensure cleavage site presence in the protein-protein interaction sites to be mapped. While trypsin’s high specificity has made it the protease of choice in mass spectrometry, there are other well-validated proteases that are suitable to such kinds of analysis and whose digestions can be run in parallel with trypsin [115]. Validating new cohorts of molecular dyes to be used in alternative digestions will aid in the goal of developing a single dye mixture that effectively bocks all protease cleavage sites of many diverse proteins. The expansion in resolution of this technique will allow for analysis of different types of proteins; as of now, protein painting is most effective on soluble, intracellular proteins with abundant lysine or arginine residues at their surfaces. One can imagine that validation of this technique with proteases such as chymotrypsin may allow for effective interrogation of binding site on membrane bound proteins or other signaling molecules; chymotrypsin’s specificity is thought most orthogonal to trypsin [116], and would represent a likely starting point for further protein painting optimization.
Additionally, the number of proteins and complexes analyzed via protein painting is relatively small. As this number increases, one can image being able to generate more detailed statistical models comparing known hotspots to mass spectrometry output. Specifically, rather than relying on complete solvent inaccessibility to determine hotspots, it may be able to model the percentage reduction in peptide spectral matches for a given peptide in a painted versus complexed sample to better determine “rim” regions around protein hotspots. These regions may have limited solvent accessibility and only some fraction of the total protein may bind to the molecular dyes, leading to apparent inconsistency in mass spectrometry detection. With a sufficiency large sample set of proteins with known interaction regions, protein painting may be able to provide a more nuanced structural analysis of protein interaction regions beyond simple “totally solvent inaccessible” verses “solvent accessible” as is currently possible.
Finally, widespread adoption of protein painting technique requires validation of the technique in multiple laboratories. Standardized parameters, such as which molecular dyes to use and the dye concentration for specific proteins should be standardized. Developing an “automated” approach, with a published list of various control protein complexes to run with each standard dye cocktail, would allow comparative data analysis from different labs.
Many proteomics groups already spend significant experimental time identifying and investigating protein signaling pathways and interrogating protein interactions that transmit biological information; therefore it seems natural to translate institutional knowledge of ‘which proteins interacts’ to a more structural knowledge of ‘where within the proteins do the interactions take place’. We would be remiss not to mention the many groups in the field of computational proteomics/computational structural biology who also contribute significant insights into protein-protein interactions. As the data sets for interacting proteins sites grow through various experimental techniques, modelling these interactions will only become more accurate. The goals of protein-protein interaction mapping are to: a) increase experimental knowledge of structure-function based techniques, b) allow better prediction of interaction sites in protein complexes, c) provide confirmation of recombinant and/or biosimilar protein binding sites, and d) provide structural information necessary for creating bio-functional therapeutics.
6. Conclusion
Proteomics informs both drug discovery and diagnostics. Proteomics techniques include biomarker discovery and quantification by mass spectrometry, and RPPA for phenotypic characterization, patient stratification, and elucidating drug mechanisms-of-action. Proteomics has a profound role in drug development due to key advancements being made in structural mass spectrometry and medicinal chemistry of peptide therapeutics. Drug discovery is perhaps the most exciting role of proteomics, given the importance of protein signaling networks and protein-protein interactions in cancer pathology.
While immune-oncology drugs hold promise as cancer treatments, not every cancer will be susceptible to anti-immune related treatments, and not every patient will respond to them without adverse side effects. Maintaining a diverse set of cancer treatment options ensures the greatest chance of success for treating multiple patients. The future of cancer drug development depends on consistent improvements in our understanding of cancer biology, yielding new or multiple protein targets for drug development, and adoption of new proteomic technologies for deciphering protein-protein interactions.
Proteomics groups are poised to supply novel protein-protein interaction data as potential targets for cancer therapeutics, and also to provide small interfering peptides identified via mass spectrometry techniques as lead compounds for optimization. Peptide therapeutics may represent a better approach than antibodies or small molecules as protein-protein interaction inhibitors in cancer. Key challenges to overcome include conformational flexibility and protease degradation, but our perspective posits that peptides that interfere with protein-protein interactions may be the next wave of therapeutics to change clinical cancer treatment. Proteomic laboratories, already leaders in personalized medicine, are poised to adapt to these new advancements by incorporating existing institutional knowledge in mass spectrometry with protein structural techniques, thus providing new peptide lead molecules for cancer therapy and further development of biosimilar therapeutics.
7. Expert Opinion
Proteomic technologies are indispensable for drug discovery. Proteomics allows biomarker identification, quantification, and proteome wide comparisons between different cell types, treatment conditions, or patients. Functional drug discovery enhances direct patient care. Reverse phase protein arrays are used for patient stratification for drug treatment, quantifying post-translationally modified proteins and epigenetic modifications, developing phenotypic fingerprints, and characterizing drug mechanisms of action. Protein-protein interaction assays, fragment analysis, and chemoproteomics reveal previously unknown mechanisms of action, and thereby, new drug targets. Proteomics delivers the actionable, functional information required for drug development and precision medicine, thus impacting treatment decisions, drug utilization, and economic factors for patients and pharmaceutical companies.
Standardized processes and procedures are required for any assay that will be used for pre-clinical drug development, to develop a companion diagnostic assay, to be used as a laboratory developed test, or to gain FDA approval. Mass spectrometry protein identification and RPPA technology have matured into standardized well-controlled, robust technologies, and have been adopted world-wide. Protein-protein interaction assays need to meet these same standards. The greatest hurdle to overcome for designing small peptide inhibitors is determining the affinity and specificity of the small peptide with the 20,230 known proteins in the human proteome [117]. This hurdle may be overcome through in silico analysis, and/or by developing the assays into massively parallel, high-throughput systems. Despite these limitations, our ability to identify previously hidden protein hot spots is expanding our gamut of potential drug targets and therapeutic compounds. Imagine the ability to design small peptide inhibitors or activators that target vulnerable protein-interaction hot spots within an individual patient’s tumor. Designer small peptides may be the next generation of cancer therapeutics.
The main limitation to implementing protein painting stem from its recent introduction and limited adoption beyond a single laboratory. Rapid commercialization of new technologies in general will facilitate widespread adoption. The clinical and scientific community should not be complacent regarding the long timeframe from invention to commercialization to clinical acceptance. The catch-22 dilemma in technology development is the need for financial investment to commercialize a product/drug/device; however the investors demand demonstrated clinical utility to ensure a return on their investment, which of course requires funding. Research funding for technology development must exist well-beyond the initial idea stage, and support studies through clinical implementation. Without investment in technology development, potentially paradigm shifting technologies will be forgotten or shelved. A technical limitation to advancing protein painting to multiple laboratories is lot-to-lot variability of the dyes used to coat the proteins during protein painting. Impurities in the dye formulation may impart desirable or undesirable binding characteristics. Availability of specific dye lots should be considered before commencing multi-center studies.
A mindset transformation needs to occur in drug discovery for drug developers, scientists, investors, and clinicians. We need to discard the concepts of ‘one hit wonder drugs’ and ‘one drug/one target’. These concepts are naïve assumptions and fail to account for biological variability between individuals as well as similarities between protein motifs and ligand binding domains. Alternative splicing of mRNA creates various protein isoforms, which are not routinely profiled in clinical specimens. The biological redundancy in protein motifs and ligand binding domains potentially creates specificity issues for drugs and small peptides. Characterizing functional phenotypes of individual cancer specimens, utilizing the full spectrum of proteomics technologies, will eventually permit true designer drug for individualized cancer treatments.
Biosimilar therapeutics may gain broader, more rapid acceptance if the proteomic community can deliver a biosimilar product profile relating to protein-protein interactions and post-translational modifications, both of which can potentially modulate on and off-target drug outcomes. Proteomic technologies will be critical for assessing the true clinical effectiveness of biosimilar therapeutics. Molecular nuances of a biosimilar therapeutics unique mechanism of action will only be gleaned through extensive pre-clinical proteomic assessments.
The next five years will hopefully produce advances in three key areas that have the potential to transform drug discovery and treatment: 1) characterizing the dark proteome, 2) identifying mechanisms of acquired drug resistance, and 3) elucidation of the synergy and/or antagonism of commonly prescribed medications with other cancer drugs. Our knowledge of the structure, function and protein expression of the “dark proteome” will propel future drug discovery [118,119]. The dark proteome consists of intrinsically disordered proteins that are mis-folded or fail to follow a well-defined folding structure and thus are missed by traditional homology modeling, structure determination, and in silico methods [120]. Imagine the myriad of potential drug targets that could have functional significance if we simply knew they existed and were able to precisely characterize their structure and/or interactions [118,119,121].
Within 5 years, proteomic technology will aid in deciphering acquired drug resistance. Even though we currently have the tools to identify actionable drug targets, and develop appropriately matched molecular targeted inhibitors, cancer cells frequently develop drug resistance. Proteomics is well-suited to quantifying proteins, identifying changes in signal transduction pathways, and deciphering changes in protein-protein interactions – all of which may be implicated in drug resistance. The hurdle to overcome is access to biospecimens from patients prior to therapy and after acquiring drug resistance.
A completely understudied area is the proteomic effects of commonly prescribed medications with cancer drugs and the association/correlation with treatment outcomes. Pharmacogenomics has identified many drug metabolism pathways for all classes of drugs, such as the cytochrome oxidase enzymes pathways [122]. However, concurrent administration of neuromodulators, anti-hypertensive, anti-inflammatory, and diabetes medications exert molecular effects that could either enhance or diminish treatment outcomes due to deletion of metabolites, as is the case with glutathione depletion during acetaminophen metabolism [123]. Are concomitant medications responsible for the subsets of patients that respond, or do not respond, to treatment? How does an individual patient’s pharmacogenomic profile contribute to treatment success rates? Are there individual patient differences in the dark proteome that influence drug response? Do clinical trials need to be redesigned to assess efficacy based on pharmacogenomic profiles and concomitant medications? Can protein-protein interaction mapping assist in deciphering these multiple drug-protein interactions? The answers to these questions may ultimately yield higher treatment success rates and spare patients unnecessary toxicity.
As proteomics evolves into standardized research protocols, routine clinical assays, and companion diagnostic kits, the filed will face an “optimization versus standardization” dilemma. We are at risk of losing creativity and optimization that stem from protocol modifications that can enhance new discoveries. On one hand, assay standardization is required by regulatory agencies, professional laboratory standards, and good laboratory practice. While standardized assays ensure rigor and reproducibility, we must continue to teach and understand the underlying biological and chemical principles of the assays. We need to actively avoid the ‘kit’ mentality in which highly complex assays are followed like recipes without understanding the mechanism of action of each reagent, the expected biological/chemical interactions, and the principles of instrument operation. We must actively encourage creativity, critical thinking, and the willingness to take risks, when and where appropriate, to propel scientific advances.
Article highlights.
Proteomic profiling and quantification by mass spectrometry and reverse phase protein arrays yield mechanism-of-action information and phenotypic hits to advance lead compound evaluation beyond the one drug, one target paradigm. Chemoproteomics provides a method for determining specificity for enzymes with similar catalytic domains or isoforms within the same enzyme family.
Protein-protein interaction mapping via chemoproteomics, fragment based screening, and protein painting are essential for identifying small peptide inhibitors/activators that may be the next generation of cancer therapeutics.
The development of protein-protein interaction inhibitors could be advanced by a) improving methods to more rapidly identify hotspot residues of protein-protein interactions in native protein complexes, b) increasing development of small molecule moieties suited to selectively target protein-protein interactions, and c) improving the bioavailability and stability of peptides and peptidomimetic drugs for intracellular delivery.
Therapeutic target validation requires three assessments of the drug target: 1) detection of the drug target in diseased tissue (cells), using a method that is sensitive and specific to quantify an increase/decrease in the drug target; 2) assays to prove that the target was inhibited/activated; and 3) correlation between drug target modulation and clinical outcome.
Characterizing biosimilar therapeutics should entail a critical investigation into whether differences between the bio-original structure and a biosimilar are clinically meaningful and create differences in efficacy or toxicity.
The goals of protein-protein interaction mapping are to: a) increase experimental knowledge of structure-function based techniques, b) allow better prediction of interaction sites in protein complexes, c) provide confirmation of recombinant and/or biosimilar protein binding sites, and d) provide structural information necessary for creating bio-functional therapeutics.
Acknowledgements
The authors would like to thank Lance Liotta for editorial advice.
Funding
This work was supported in part by George Mason University, and the National Institutes of Health Innovative Molecular Analysis Technologies (IMAT) program grant 1R33CA20693701, “Protein painting identifies therapeutic targets at protein-protein interfaces” and the National Institute of Arthritis and Musculoskeletal and Skin Diseases grant R01AR068436 “Probes to Target the 3-way Hotspot of IL1RacP to Abolish Aberrant Interleukin Inflammation” (to Lance Liotta).
Footnotes
Declaration of interest
A.Haymond, J.Davis, and V. Espina receive salary support from NIH Innovative Molecular Analysis Technologies (IMAT ) grant 1R33CA20693701.
A. Haymond receives salary support from National Institute of Arthritis and Musculoskeletal and Skin Diseases grant R01AR068436-03 and Center for Innovative Technologies (CIT) CRCF Award MF18-007-LS. A. Haymond has stock ownership and is a consultant for Monet Pharmaceuticals.
V. Espina is an inventor of the protein painting technology and, as a university employee, is entitled to receive patent royalties per university policies. V. Espina is a consultant for Avant Diagnostics.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as:
* of interest
** of considerable interest
- [1].Precision Medicine [Internet]. Natl. Cancer Inst 2015. [cited 2018 Dec 7]. Available from: https://www.cancer.gov/about-cancer/treatment/types/precision-medicine.
- [2].Sun J, Wei Q, Zhou Y, et al. A systematic analysis of FDA-approved anticancer drugs. BMC Syst. Biol. [Internet]. 2017. [cited 2018 Dec 7];11 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5629554/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the “high-hanging fruit.” Nat. Rev. Drug Discov. 2018;17:197–223.* Inclusive review and list of antibody-based drugs.
- [4].Marsden CJ, Eckersley S, Hebditch M, et al. The Use of Antibodies in Small-Molecule Drug Discovery. J. Biomol. Screen. 2014;19:829–838.* Discusses applications of antibody and protein affinity reagents in drug discovery.
- [5].Tyler DS, Vappiani J, Cañeque T, et al. Click chemistry enables preclinical evaluation of targeted epigenetic therapies. Science. 2017;356:1397–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Luchini A, Espina V, Liotta LA. Protein painting reveals solvent-excluded drug targets hidden within native protein–protein interfaces. Nat. Commun. 2014;5:4413.**Protein painting original paper, provides detailed methods.
- [7].Dailing A, Luchini A, Liotta L. Unlocking the secrets to protein-protein interface drug targets using structural mass spectrometry techniques. Expert Rev. Proteomics 2015;12:457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lee J, Han JJ, Altwerger G, et al. Proteomics and biomarkers in clinical trials for drug development. J. Proteomics. 2011;74:2632–2641.*Provides a detailed discussion of target validation strategies in drug discovery.
- [9].Kuenzi BM, Remsing Rix LL, Stewart PA, et al. Polypharmacology-based ceritinib repurposing using integrated functional proteomics. Nat. Chem. Biol. 2017;13:1222–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Emdal KB, Pedersen A-K, Bekker-Jensen DB, et al. Integrated proximal proteomics reveals IRS2 as a determinant of cell survival in ALK-driven neuroblastoma. Sci. Signal 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bhatt V Current Market and Regulatory Landscape of Biosimilars. Am J Manag Care. 2018;24:S0.*Up-to-date overview of the biosimilar regulatory environment.
- [12].Rugo HS, Rifkin RM, Declerck P, et al. Demystifying biosimilars: development, regulation and clinical use. Future Oncol. [Internet]. 2018; Available from: https://www.futuremedicine.com/doi/10.2217/fon-2018-0680. [DOI] [PubMed] [Google Scholar]
- [13].Isaacs J, Gonçalves J, Strohal R, et al. The biosimilar approval process: how different is it? Consid. Med. 2017;1:3–6.*Provides details regarding the biosimilar approval process.
- [14].Iwamoto N, Shimada T. Recent advances in mass spectrometry-based approaches for proteomics and biologics: Great contribution for developing therapeutic antibodies. Pharmacol. Ther. 2018;185:147–154. [DOI] [PubMed] [Google Scholar]
- [15].Russo P, Hood BL, Bateman NW, et al. Quantitative Mass Spectrometry by Isotope Dilution and Multiple Reaction Monitoring (MRM). Methods Mol. Biol. Clifton NJ. 2017;1606:313–332. [DOI] [PubMed] [Google Scholar]
- [16].Bourmaud A, Gallien S, Domon B. Parallel reaction monitoring using quadrupole-Orbitrap mass spectrometer: Principle and applications. Proteomics. 2016;16:2146–2159.**Extensive description of parallel reaction monitoring.
- [17].Peterson AC, Russell JD, Bailey DJ, et al. Parallel Reaction Monitoring for High Resolution and High Mass Accuracy Quantitative, Targeted Proteomics. Mol. Cell. Proteomics MCP. 2012;11:1475–1488.*Initial paper describing parallel reaction monitoring with comparison to SRM.
- [18].Shi T, Song E, Nie S, et al. Advances in targeted proteomics and applications to biomedical research. Proteomics. 2016;16:2160–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].El Kennani S, Adrait A, Permiakova O, et al. Systematic quantitative analysis of H2A and H2B variants by targeted proteomics. Epigenetics Chromatin. 2018;11:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chen Y, Britton D, Wood ER, et al. Quantitative proteomics of breast tumors: Tissue quality assessment to clinical biomarkers. Proteomics. 2017;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Valdés A, Lewitt M, Wiss E, et al. Development of a Parallel Reaction Monitoring-MS Method To Quantify IGF Proteins in Dogs and a Case of Nonislet Cell Tumor Hypoglycemia. J. Proteome Res. 2019;18:18–29.**A detailed description of parallel reaction monitoring test development across species.
- [22].Schlüter H, Apweiler R, Holzhütter H-G, et al. Finding one’s way in proteomics: a protein species nomenclature. Chem. Cent. J. 2009;3:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Aebersold R, Agar JN, Amster IJ, et al. How many human proteoforms are there? Nat. Chem. Biol. 2018;14:206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mueller C, deCarvalho AC, Mikkelsen T, et al. Glioblastoma Cell Enrichment Is Critical for Analysis of Phosphorylated Drug Targets and Proteomic–Genomic Correlations. Cancer Res. 2014;74:818–828. [DOI] [PubMed] [Google Scholar]
- [25].Wolf DM, Yau C, Sanil A, et al. DNA repair deficiency biomarkers and the 70-gene ultra-high risk signature as predictors of veliparib/carboplatin response in the I-SPY 2 breast cancer trial. NPJ Breast Cancer. 2017;3:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Possemato AP, Paulo JA, Mulhern D, et al. Multiplexed Phosphoproteomic Profiling Using Titanium Dioxide and Immunoaffinity Enrichments Reveals Complementary Phosphorylation Events. J. Proteome Res. 2017;16:1506–1514.*Good description of phosphoprotein enrichment strategies.
- [27].Arshad OA, Danna V, Petyuk VA, et al. An integrative analysis of tumor proteomic and phosphoproteomic profiles to examine the relationships between kinase activity and phosphorylation. Mol. Cell. Proteomics MCP. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Narumi R, Murakami T, Kuga T, et al. A strategy for large-scale phosphoproteomics and SRM-based validation of human breast cancer tissue samples. J. Proteome Res. 2012;11:5311–5322. [DOI] [PubMed] [Google Scholar]
- [29].Wang D, Tan G, Wang H, et al. Identification of novel serum biomarker for the detection of acute myeloid leukemia based on liquid chromatography-mass spectrometry. J. Pharm. Biomed. Anal. 2019;166:357–363. [DOI] [PubMed] [Google Scholar]
- [30].Geijsen AJMR, Brezina S, Keski-Rahkonen P, et al. Plasma metabolites associated with colorectal cancer: A discovery-replication strategy. Int. J. Cancer. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li H, Mao Y, Xiong Y, et al. A Comprehensive Proteome Analysis of Peripheral Blood Mononuclear Cells (PBMCs) to Identify Candidate Biomarkers of Pancreatic Cancer. Cancer Genomics Proteomics. 2019;16:81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sun F, Suttapitugsakul S, Wu R. Enzymatic Tagging of Glycoproteins on the Cell Surface for Their Global and Site-Specific Analysis with Mass Spectrometry. Anal. Chem. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fredolini C, Meani F, Reeder KA, et al. Concentration and Preservation of Very Low Abundance Biomarkers in Urine, such as Human Growth Hormone (hGH), by Cibacron Blue F3G-A Loaded Hydrogel Particles. Nano Res 2008;1:502–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Magni R, Espina BH, Liotta LA, et al. Hydrogel nanoparticle harvesting of plasma or urine for detecting low abundance proteins. J. Vis. Exp. JoVE. 2014;e51789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Castagna A, Cecconi D, Sennels L, et al. Exploring the hidden human urinary proteome via ligand library beads. J. Proteome Res. 2005;4:1917–1930. [DOI] [PubMed] [Google Scholar]
- [36].Tubaon RM, Haddad PR, Quirino JP. Sample Clean-up Strategies for ESI Mass Spectrometry Applications in Bottom-up Proteomics: Trends from 2012 to 2016. Proteomics. 2017;17. [DOI] [PubMed] [Google Scholar]
- [37].León IR, Schwämmle V, Jensen ON, et al. Quantitative assessment of in-solution digestion efficiency identifies optimal protocols for unbiased protein analysis. Mol. Cell. Proteomics MCP. 2013;12:2992–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Huang BX, Kim H-Y. Effective identification of Akt interacting proteins by two-step chemical crosslinking, co-immunoprecipitation and mass spectrometry. PloS One. 2013;8:e61430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Paweletz CP, Charboneau L, Bichsel VE, et al. Reverse phase protein microarrays which capture disease progression show activation of pro-survival pathways at the cancer invasion front. Oncogene. 2001;20:1981–1989.*Original description of reverse phase protein arrays.
- [40].Akbani R, Ng PKS, Werner HMJ, et al. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat. Commun. 2014;5:3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Akbani R, Becker K-F, Carragher N, et al. Realizing the promise of reverse phase protein arrays for clinical, translational, and basic research: a workshop report: the RPPA (Reverse Phase Protein Array) society. Mol. Cell. Proteomics MCP. 2014;13:1625–1643.*Summary of reverse phase protein array technology and clinical utility.
- [42].Mueller C, Liotta LA, Espina V. Reverse phase protein microarrays advance to use in clinical trials. Mol. Oncol. 2010;4:461–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hayashi N, Manyam GC, Gonzalez-Angulo AM, et al. Reverse-phase protein array for prediction of patients at low risk of developing bone metastasis from breast cancer. The Oncologist. 2014;19:909–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Johnston HJ, Boys SK, Makda A, et al. Naturally Inspired Peptide Leads: Alanine Scanning Reveals an Actin-Targeting Thiazole Analogue of Bisebromoamide. ChemBioChem. 2016;17:1621–1627.*Application of alanine screening and reverse phase protein arrays for drug discovery.
- [45].Pawlak M, Carragher NO. Reverse Phase Protein Arrays elucidate mechanisms-of-action and phenotypic response in 2D and 3D models. Drug Discov. Today Technol 2017;23:7–16.**This paper describes RPPA technology for phenotypic hits in drug discovery.
- [46].Partolina M, Thoms HC, MacLeod KG, et al. Global histone modification fingerprinting in human cells using epigenetic reverse phase protein array. Cell Death Discov. 2017;3:16077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gallagher RI, Espina V. Reverse phase protein arrays: mapping the path towards personalized medicine. Mol. Diagn. Ther. 2014;18:619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Liotta LA, Espina V, Mehta AI, et al. Protein microarrays: meeting analytical challenges for clinical applications. Cancer Cell. 2003;3:317–325.* Detailed description of the theory and technology supporting reverse phase protein arrays.
- [49].C O’Farrell A, Miller IS, Evans R, et al. Implementing Reverse Phase Protein Array profiling as a sensitive method for the early pre-clinical detection of off-target toxicities associated with Sunitinib Malate. Proteomics Clin. Appl. 2019;e1800159. [DOI] [PubMed] [Google Scholar]
- [50].Davis JB, Calvert V, Roberts S, et al. Induction of nerve growth factor by phorbol 12-myristate 13-acetate is dependent upon the mitogen activated protein kinase pathway. Heliyon. 2018;4:e00617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].He S, Fu Y, Guo J, et al. Cofilin hyperactivation in HIV infection and targeting the cofilin pathway using an anti-α4β7 integrin antibody. Sci. Adv. 2019;5:eaat7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Saxena C Affinity-based chemoproteomics with small molecule-peptide conjugates. Methods Mol. Biol. Clifton NJ. 2012;803:39–54. [DOI] [PubMed] [Google Scholar]
- [53].Drewes G, Knapp S. Chemoproteomics and Chemical Probes for Target Discovery. Trends Biotechnol. 2018;36:1275–1286. [DOI] [PubMed] [Google Scholar]
- [54].Chen Y-C, Zhang C. A chemoproteomic method for identifying cellular targets of covalent kinase inhibitors. Genes Cancer. 2016;7:148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Counihan JL, Wiggenhorn AL, Anderson KE, et al. Chemoproteomics-Enabled Covalent Ligand Screening Reveals ALDH3A1 as a Lung Cancer Therapy Target. ACS Chem. Biol. 2018;13:1970–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhou J, Mock ED, Martella A, et al. Activity-Based Protein Profiling Identifies α-Ketoamides as Inhibitors for Phospholipase A2 Group XVI. ACS Chem. Biol. 2019;14:164–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Scheer S, Ackloo S, Medina TS, et al. A chemical biology toolbox to study protein methyltransferases and epigenetic signaling. Nat. Commun. 2019;10:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Li W, Zhou Y, Tang G, et al. Chemoproteomics Reveals the Antiproliferative Potential of Parkinson’s Disease Kinase Inhibitor LRRK2-IN-1 by Targeting PCNA Protein. Mol. Pharm. 2018;15:3252–3259. [DOI] [PubMed] [Google Scholar]
- [59].Dalvit C NMR methods in fragment screening: theory and a comparison with other biophysical techniques. Drug Discov. Today. 2009;14:1051–1057. [DOI] [PubMed] [Google Scholar]
- [60].Luise N, Wyatt PG. Diversity-oriented synthesis of bicyclic fragments containing privileged azines. Bioorg. Med. Chem. Lett. 2019;29:248–251. [DOI] [PubMed] [Google Scholar]
- [61].Dick BL, Cohen SM. Metal-Binding Isosteres as New Scaffolds for Metalloenzyme Inhibitors. Inorg. Chem. 2018;57:9538–9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Qiu H, Liu-Bujalski L, Caldwell RD, et al. Discovery of potent, highly selective covalent irreversible BTK inhibitors from a fragment hit. Bioorg. Med. Chem. Lett. 2018;28:2939–2944. [DOI] [PubMed] [Google Scholar]
- [63].White GV, Edgar EV, Holmes DS, et al. Kallikrein 5 inhibitors identified through structure based drug design in search for a treatment for Netherton Syndrome. Bioorg. Med. Chem. Lett. 2019;29:821–825. [DOI] [PubMed] [Google Scholar]
- [64].Bhullar KS, Lagarón NO, McGowan EM, et al. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol. Cancer [Internet]. 2018. [cited 2018 Dec 7];17 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5817855/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Rosell M, Fernández-Recio J. Hot-spot analysis for drug discovery targeting protein-protein interactions. Expert Opin. Drug Discov. 2018;13:327–338. [DOI] [PubMed] [Google Scholar]
- [66].Jiang J, Wang N, Chen P, et al. Prediction of Protein Hotspots from Whole Protein Sequences by a Random Projection Ensemble System. Int. J. Mol. Sci [Internet]. 2017. [cited 2018 Dec 7];18 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5536031/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ofran Y, Rost B. Protein–Protein Interaction Hotspots Carved into Sequences. PLoS Comput. Biol [Internet]. 2007. [cited 2018 Dec 7];3 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1914369/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kelly PN, Strasser A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ. 2011;18:1414–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ku B, Liang C, Jung JU, et al. Evidence that inhibition of BAX activation by BCL-2 involves its tight and preferential interaction with the BH3 domain of BAX. Cell Res. 2011;21:627–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. [DOI] [PubMed] [Google Scholar]
- [71].Garland W, Benezra R, Chaudhary J. Chapter Fifteen - Targeting Protein–Protein Interactions to Treat Cancer—Recent Progress and Future Directions In: Desai MC, editor. Annu. Rep. Med. Chem. [Internet]. Academic Press; 2013. [cited 2018 Dec 10]. p. 227–245. Available from: http://www.sciencedirect.com/science/article/pii/B9780124171503000156. [Google Scholar]
- [72].Mullard A Pioneering apoptosis-targeted cancer drug poised for FDA approval. Nat. Rev. Drug Discov. 2016;15:147–149. [DOI] [PubMed] [Google Scholar]
- [73].Souers AJ, Leverson JD, Boghaert ER, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013;19:202–208. [DOI] [PubMed] [Google Scholar]
- [74].Press Announcements - FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality [Internet]. 2018. [cited 2018 Dec 10]. Available from: https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm495253.htm. [Google Scholar]
- [75].Simon S, Labarriere N. PD-1 expression on tumor-specific T cells: Friend or foe for immunotherapy? Oncoimmunology [Internet]. 2017. [cited 2018 Dec 10];7 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5739549/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wang X, Teng F, Kong L, et al. PD-L1 expression in human cancers and its association with clinical outcomes. OncoTargets Ther. 2016;9:5023–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015;6:e1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].NCI Dictionary of Cancer Terms. Immune Checkpoint Inhibitor. [Internet]. Natl. Cancer Inst 2011. [cited 2018 Dec 10]. Available from: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/immune-checkpoint-inhibitor.
- [79].Zak KM, Kitel R, Przetocka S, et al. Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1. Structure. 2015;23:2341–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Gong J, Chehrazi-Raffle A, Reddi S, et al. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J. Immunother. Cancer [Internet]. 2018. [cited 2018 Dec 10];6 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5778665/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lee JY, Lee HT, Shin W, et al. Structural basis of checkpoint blockade by monoclonal antibodies in cancer immunotherapy. Nat. Commun. [Internet]. 2016. [cited 2018 Dec 10];7 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5095608/.* Comprehensive overview of immune-oncology using checkpoint inhibitors.
- [82].Lee HT, Lee JY, Lim H, et al. Molecular mechanism of PD-1/PD-L1 blockade via anti-PD-L1 antibodies atezolizumab and durvalumab. Sci. Rep [Internet]. 2017. [cited 2018 Dec 10];7 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5514103/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Research C for DE and. FDA approves cemiplimab-rwlc for metastatic or locally advanced cutaneous squamous cell carcinoma. FDA [Internet]. 2019. [cited 2019 May 19]; Available from: /drugs/drug-approvals-and-databases/fda-approves-cemiplimab-rwlc-metastatic-or-locally-advanced-cutaneous-squamous-cell-carcinoma.
- [84].“I want what Jimmy Carter had”: Patients clamor for Keytruda [Internet]. STAT. 2015. [cited 2018 Sep 20]. Available from: https://www.statnews.com/2015/12/18/jimmy-carter-cancer-drug-keytruda/. [Google Scholar]
- [85].Wang T, Wu X, Guo C, et al. Development of Inhibitors of the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Signaling Pathway. J. Med. Chem. [Internet]. 2018. [cited 2018 Dec 10]; Available from: 10.1021/acs.jmedchem.8b00990. [DOI] [Google Scholar]
- [86].Verdone L, Caserta M, Di Mauro E. Role of histone acetylation in the control of gene expression. Biochem. Cell Biol. Biochim. Biol. Cell 2005;83:344–353. [DOI] [PubMed] [Google Scholar]
- [87].Xu Y, Vakoc CR. Targeting cancer cells with BET bromodomain inhibitors. Cold Spring Harb. Perspect. Med. [Internet]. 2017. [cited 2018 Dec 11];7 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5495050/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Wang Q, Li Y, Xu J, et al. Selective inhibition mechanism of RVX-208 to the second bromodomain of bromo and extraterminal proteins: insight from microsecond molecular dynamics simulations. Sci. Rep. 2017;7:8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Andrieu G, Belkina AC, Denis GV. Clinical trials for BET inhibitors run ahead of the science. Drug Discov. Today Technol. 2016;19:45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Ozaki T, Nakagawara A. Role of p53 in Cell Death and Human Cancers. Cancers. 2011;3:994–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Liao G, Yang D, Ma L, et al. The development of piperidinones as potent MDM2-P53 protein-protein interaction inhibitors for cancer therapy. Eur. J. Med. Chem. 2018;159:1–9. [DOI] [PubMed] [Google Scholar]
- [92].Zhao Y, Aguilar A, Bernard D, et al. Small-Molecule Inhibitors of the MDM2–p53 Protein–Protein Interaction (MDM2 Inhibitors) in Clinical Trials for Cancer Treatment. J. Med. Chem. 2015;58:1038–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Tisato V, Voltan R, Gonelli A, et al. MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J. Hematol. OncolJ Hematol Oncol. 2017;10:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Bai L, Smith DC, Wang S. Small-Molecule SMAC Mimetics as New Cancer Therapeutics. Pharmacol. Ther. 2014;144:82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Beug ST, Beauregard CE, Healy C, et al. Smac mimetics synergize with immune checkpoint inhibitors to promote tumour immunity against glioblastoma. Nat. Commun. [Internet]. 2017. [cited 2018 Dec 11];8 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5330852/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Fulda S Promises and Challenges of Smac Mimetics as Cancer Therapeutics. Clin. Cancer Res. 2015;21:5030–5036. [DOI] [PubMed] [Google Scholar]
- [97].Bakail M, Ochsenbein F. Targeting protein–protein interactions, a wide open field for drug design. Comptes Rendus Chim. 2016;19:19–27. [Google Scholar]
- [98].Shi D, Nannenga BL, Iadanza MG, et al. Three-dimensional electron crystallography of protein microcrystals. eLife [Internet]. 2013. [cited 2018 Dec 11];2 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3831942/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Henninot A, Collins JC, Nuss JM. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018;61:1382–1414.**Excellent review of peptide drug discovery.
- [100].Guidotti G, Brambilla L, Rossi D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017;38:406–424. [DOI] [PubMed] [Google Scholar]
- [101].Morrison C Constrained peptides’ time to shine? Nat. Rev. Drug Discov. 2018;17:531–533. [DOI] [PubMed] [Google Scholar]
- [102].Cary DR, Ohuchi M, Reid PC, et al. Constrained Peptides in Drug Discovery and Development. J. Synth. Org. Chem. Jpn. 2017;75:1171–1178. [Google Scholar]
- [103].Meric-Bernstam F, Saleh MN, Infante JR, et al. Phase I trial of a novel stapled peptide ALRN-6924 disrupting MDMX- and MDM2-mediated inhibition of WT p53 in patients with solid tumors and lymphomas. J. Clin. Oncol. 2017;35:2505–2505. [Google Scholar]
- [104].Otvos L, Wade JD. Current challenges in peptide-based drug discovery. Front. Chem. [Internet]. 2014. [cited 2018 Dec 12];2 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4126357/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Nielsen DS, Shepherd NE, Xu W, et al. Orally Absorbed Cyclic Peptides. Chem. Rev. 2017;117:8094–8128. [DOI] [PubMed] [Google Scholar]
- [106].Eckhardt B, Grosse W, Essen L-O, et al. Structural characterization of a β-turn mimic within a protein–protein interface. Proc. Natl. Acad. Sci. 2010;107:18336–18341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Möller GP, Müller S, Wolfstädter BT, et al. Oxetanyl Amino Acids for Peptidomimetics. Org. Lett. 2017;19:2510–2513. [DOI] [PubMed] [Google Scholar]
- [108].Hamley IW. PEG–Peptide Conjugates. Biomacromolecules. 2014;15:1543–1559. [DOI] [PubMed] [Google Scholar]
- [109].Muheem A, Shakeel F, Jahangir MA, et al. A review on the strategies for oral delivery of proteins and peptides and their clinical perspectives. Saudi Pharm. J. 2016;24:413–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Sharma A, Reddy P, Kuppermann BD, et al. Biosimilars in ophthalmology: “Is there a big change on the horizon?” Clin. Ophthalmol. Auckl. NZ 2018;12:2137–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].U.S Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, et al. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product: Guidance for Industry [Internet]. 2015. Available from: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf.
- [112].Lee KH, Lee J, Bae JS, et al. Analytical similarity assessment of rituximab biosimilar CT-P10 to reference medicinal product. mAbs. 2018;10:380–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Thennati R, Singh SK, Nage N, et al. Analytical characterization of recombinant hCG and comparative studies with reference product. Biol. Targets Ther. 2018;12:23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Hajba L, Szekrényes Á, Borza B, et al. On the glycosylation aspects of biosimilarity. Drug Discov. Today. 2018;23:616–625. [DOI] [PubMed] [Google Scholar]
- [115].Giansanti P, Tsiatsiani L, Low TY, et al. Six alternative proteases for mass spectrometry–based proteomics beyond trypsin. Nat. Protoc. 2016;11:993–1006. [DOI] [PubMed] [Google Scholar]
- [116].Tsiatsiani L, Heck AJR. Proteomics beyond trypsin. FEBS J. 2015;282:2612–2626. [DOI] [PubMed] [Google Scholar]
- [117].Paik Y-K, Lane L, Kawamura T, et al. Launching the C-HPP neXt-CP50 Pilot Project for Functional Characterization of Identified Proteins with No Known Function. J. Proteome Res. 2018;17:4042–4050.* Provides a reference for the number of human proteins and the fraction of proteins classified within the dark proteome.
- [118].Schafferhans A, O’Donoghue SI, Heinzinger M, et al. Dark Proteins Important for Cellular Function. PROTEOMICS. 2018;18:1800227.*Provides a discussion of cellular functions modulated by dark proteins.
- [119].Perdigão N, Heinrich J, Stolte C, et al. Unexpected features of the dark proteome. Proc. Natl. Acad. Sci. U. S. A. 2015;112:15898–15903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Bhowmick A, Brookes DH, Yost SR, et al. Finding Our Way in the Dark Proteome. J. Am. Chem. Soc. 2016;138:9730–9742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Perdigão N, Rosa A. Dark Proteome Database: Studies on Dark Proteins. High-Throughput. 2019;8:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Issa NT, Wathieu H, Ojo A, et al. Drug Metabolism in Preclinical Drug Development: A Survey of the Discovery Process, Toxicology, and Computational Tools. Curr. Drug Metab 2017;18:556–565.*Comprehensive review of pharmacogenomics and computational tools to assist in drug development.
- [123].Agrawal S, Khazaeni B. Acetaminophen Toxicity StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019. [cited 2019 May 18]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK441917/. [PubMed] [Google Scholar]