

Abstract
Metal selectivity in P1B-type ATPase pumps appears to be determined by amino acid motifs on their transmembrane helices. We reveal the principles governing substrate promiscuity towards first-, second- and third- row transition metals in a transmembrane Zn2+/Cd2+/Hg2+/Pb2+ P-type ATPase (ZntA), by dissecting its coordination chemistry. Atomic resolution characterization in detergent micelles and lipid bilayers reveals a “plastic” transmembrane metal-binding site that selects substrates by unique and diverse, yet defined, coordination geometries and ligand-metal distances.
Graphical Abstract:
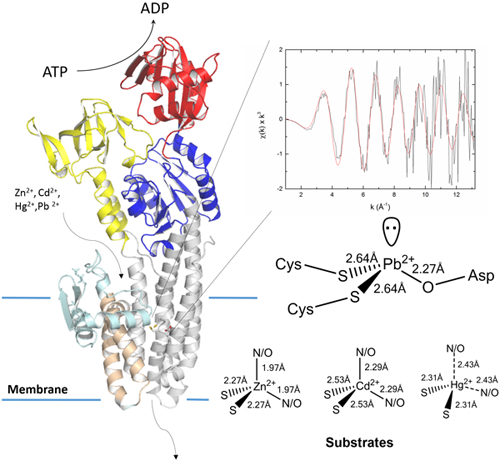
P1B-type ATPases are transmembrane primary-active pumps, conserved throughout all kingdoms of life, which utilize the energy generated by ATP hydrolysis to drive metal transport across biological membranes1. These transporters constitute an essential system for the selective translocation of transition metal ions to control the cellular concentrations of essential-but-toxic (e.g.: Cu+, Zn2+) and toxic (e.g.: Ag+, Cd2+, Pb2+) metals2.
The P1B-type ATPase transport cycle follows the Post-Albers scheme. P-type pumps alternate between two states, E1 and E2, allowing ion accessibility on opposite sides of the lipid bilayer1, coupled to conformational changes in cytoplasmic domains induced by ATP binding, hydrolysis, phosphorylation and dephosphorylation. Ion(s) bind to transmembrane site(s) (E1 state), are occluded within the membrane upon ATP hydrolysis and phosphorylation (E1P), and are then released from E2P to the opposite site, resulting in dephosphorylation to regenerate E1.
P1B-type ATPases possess 8 transmembrane helices (MA, -MB, and -M1–6) containing signature sequences for ion recognition2a, 3. Metal selectivity appears to be determined by conserved amino acid motifs in transmembrane helices TM4-5-6 providing ligands for coordination into transmembrane metal binding site(s) (TM-MBS). Based on these sequences, P1B-type ATPases are classified into subtypes: P1B-1 (Cu+/Ag+ exporters), P1B-2 (Zn2+/Cd2+/Pb2+), P1B-3 (Cu+/Cu2+), P1B-4 (Zn2+/Co2+), P1B-5 (probable Ni2+/Fe2+) and P1B-6/−7-types (unknown selectivity)2a, 3a.
The bioinorganic chemistry of pumps metal transport is unique. Selectivity and translocation are achieved through metal recognition with high thermodynamic stability and kinetic lability to guarantee selective binding and release through the transport cycle with a high turnover number. The P1B-2-type subclass evolved to control the concentrations of essential-but-toxic Zn2+ and provide resistance towards toxic Cd2+, Hg2+ and Pb2+ exposure. Thus, they represent an ideal system to investigate the structural coordination chemistry underlying a defined but promiscuous selectivity. The structural framework underlying Zn-pumps function was established through biochemical studies on P1B-2-type ZntA from E. coli and S. sonnei (99% identity) and from crystal structures in metal-free states3c, 45. Four key conserved residues in transmembrane signature motifs on the TM 4, 5 and 6 are critical for Zn2+ pumping activity. Cys392, Cys394 and Asp714 guarantee high-affinity substrate binding while Lys693 acts as a built-in counterion preventing proton countertransport3c. Atomic-level understanding of the coordination chemistry underlying metal binding and promiscuous selection at the transmembrane site remain poorly understood due to: i) difficulties in obtaining structural information on functional purified samples of metal-bound pumps; ii) the lack of comparative studies with transporters in their native lipid bilayer environment.
To reveal the coordination chemistry defining metal selection and examine the structural features underlying promiscuity we characterized biochemically and by X-ray absorption spectroscopy (XAS) the coordination properties of a Zn-pump from P. aeruginosa (PaZntA) bound to all its metal substrates.
PaZntA possess a ferredoxin-like N-terminal MBD carrying at least a metal–binding site centered on a MDCxxE motif corresponding to the CxxC motif characterized in other ZntAs, flanked by a His/Cys-rich sequence potentially involved in additional metal binding5. However, regulatory cytoplasmic metal binding domains present in P1B-2-type ATPases (MBDs) are not essential to confer metal selectivity and transport.3c, 6 Thus, the N-terminal MBD was truncated to exclusively preserve the transmembrane metal binding site(s). PaZntA shares high homology with Cu(I) ATPases and Zn(II) ATPases, possessing 40% identical and 30% similar amino acids outside the N-MBD with S. sonnei ZntA and ~70% identical positions in TM4-5-6, allowing the generation of reliable structural homology models (Fig. 1A–B).
Figure 1:

(A) PaZntA51–740 3D homology model based on the A. Fulgidus CopA cryo-EM model (PDB 3J09, which include the N-terminal MBD) and (B) close-up view of the expected high-affinity TM-MBS. Cys391, Cys393 and Asp721 (conserved in ZntAs) are responsible for transmembrane substrate binding, while Lys700 on M6 acts as a built-in counterion. A similar model was also obtained based on the structure of S. Sonnei ZntA (PDB 3J09). (C) Relative ATPase activity of PaΔZntA121–740 in proteoliposomes as a function of different metals (40 µM, see Material and Methods; Zn(II)-ATPase activity ∼ 0.9 nmol/(mg·min) ) and (D) the influence of P-type ATPases inhibitors (VO43-, AlF4-, ouabain) and non-hydrolysable nucleotide analogues (AMPPCP).
We established protocols for wtPaZntA and PaΔZntA121–740 expression, purification in detergent micelles and reconstitution in artificial lipid bilayers (Fig. S1). Detergent-solubilized wtPaZntA showed a Zn2+-dependent ATPase activity with KM of 32 ± 11 μM, and Vmax of 1.7 ± 0.4 nmol/(mg·min). For PaΔZntA121–740 the Vmax was reduced to approx. 30 % (0.6 ± 0.1 nmol/(mg·min)) with KM of 13.8 ± 7 μM, in agreement with the ATPase regulatory role by N-MBD (Fig. S2). Whether the N-MBD acts as a metal sensor and/or directly regulate phosphorylation/dephosphorylation rates as a function of metal bound to its MDCxxE motif remains to be established. To verify the PaΔZntA121–740 functional reconstitution in proteoliposomes, the ATP hydrolysis and metal selectivity were determined by metal-dependent ATPase activity. Full-length wtPaZntA and PaΔZntA121–740 proteoliposomes showed a Michaelis-Menten Zn(II)-dependent ATPase activity with KM of 26 ± 4 and 26 ± 7 μM, and Vmax of 1.9 ± 0.2 and 1.4 ± 0.2 nmol/(mg·min), respectively (Fig. S3). Substrate selectivity indicates that activity in PaΔZntA121–740 proteoliposomes depends on group 12 transition metals (Zn2+/Cd2+/Hg2+) and Pb2+ (Fig. 1C). The selectivity profile follows the same order as detergent-solubilized wtPaZntA, confirming that selectivity stems from metal binding to the high-affinity transmembrane site and is not imparted by N-MBD (Fig. S4). Inhibitors of P1B-type ATPases (AlF4− and VO4−) abolished the metal-dependent activation of ATPase activity, but not inhibitors of other P-type ATPases (ouabain, Na/K ATPase). Furthermore, the absence of activity with the non-hydrolysable ATP analogue AMPPCP confirmed functional reconstitution of PaZntA in lipid bilayers (Fig. 1D), despite PaZntA appears to possess a lower specific ATPase activity than other Zn2+ P-type ATPases4b, 7.
The presence of a single transmembrane metal binding site was investigated by PaΔZntA121–740 titration with all its metal substrates followed by metal quantification by Inductively-coupled plasma mass spectrometry (ICP-MS) revealing metal-to-protein ratios of: 0.99 ± 0.04 mol/mol for Zn2+, 1.36 ± 0.23 mol/mol for Cd2+, 0.95 ± 0.24 mol/mol for Hg2+, and 0.82 ± 0.18 mol/mol for Pb2+. Cd2+ binding was confirmed by absorption spectroscopy (Fig. S5). The metal-induced absorption intensity at 252nm plotted as function of Cd-to-protein ratios showed a breakpoint at ~1 Cd2+ equiv./mol, confirming the presence of a single high-affinity transmembrane binding site8.
To obtain details of the structure and coordination of the metal centers, we generated metal-bound forms of PaΔZntA121–740 (1 eq. M2+/PaΔZntA121–740, M2+= Zn2+, Cd2+, Hg2+, Pb2+) in Cymal-7 micelles. Zn and Cd K-edge, and Hg and Pb L3-edge extended X-ray absorption fine structure (EXAFS) spectra were collected. The data are presented in Fig. 2A–H, with the corresponding best fits and Fourier transforms. To prevent interference from potential binding to the His6 tag, thrombin cleavage was optimized to obtain a tag-free protein (Fig S6).
Figure 2:

XAS analysis of PaΔZntA121–740–M2+. K-edge experimental EXAFS data (black line) and best fits (red line) with the corresponding Fourier transforms for ΔZntA121–740–Zn2+ (A,B), ΔZntA121–740–Cd2+ (C,D), and L3-edge EXAFS data for ΔZntA121–740–Pb2+ (E,F) and ΔZntA121–740–Hg2+ (G,H; see Materials and Methods). The parameters for the best fits are listed in Table 1. (I) Models of the metal binding sites in PaΔZntA121–740–Zn2+, PaΔZntA121–740–Cd2+, PaΔZntA121–740–Hg2+ and PaΔZntA121–740–Pb2+ with the coordination geometries and metal-ligand bond distances.
In PaΔZntA-Zn2+, the best EXAFS fit was obtained with two N/O ligands at 1.97 Å, and two S ligands at 2.27 Å (Fig. 2A–B and Table 1). Fitting with 1S3N/O coordination and penta-coordinated geometry including an additional independent ligand (either S or N/O) resulted in worse fits. The bond distances are consistent with those of protein sites where Zn is bound in a tetrahedral or distorted tetrahedral coordination9 by mixed S and N/O ligands, and with that of E. coli ZntA10 (Fig. 2I). Based on functional data for ZntA homologues and the key residues conservation among Zn2+ P-type pumps, the proposed ligands are two thiolate sulfurs (Cys391 and Cys393) and two oxygen ligands from Asp721 in bidentate fashion, likely resulting in distortion from ideal tetrahedral geometry due to constraints imposed by the Asp O-C-O angle.3c, 4d, 4f. We generated mutants in all the proposed coordinating residues (C391A, C393A, C391A, C393A, D721A and D721N) and determined the effect on the Zn2+-dependent ATPase activity (Fig.3). In agreement with the proposed model the ATPase activity was dramatically affected in all mutants. In addition, single mutation in either coordinating Cys (C391 or C393) abolished the activity to the same extent as the double Cys mutant (C391 or C393) confirming the nature of the 2 S scatters in EXAFS analysis and excluding possible Cl− coordination (similar scattering properties in EXAFS). Moreover, mutation of D721 to either Asn or Ala resulted in similar abolishment of pump activity (Fig. 3), supporting bidentate coordination by the Asp side chain rather than the possible presence of a H2O molecule in the coordination shell.
Table 1.
EXAFS best curve-fitting parameters for ΔZntA121–740–M2+.
| M2+ eq. | Scattering paths [b] | N [a] | R (Å) | σ2 (Å2) | F | |
|---|---|---|---|---|---|---|
| ΔZnta-Zn2+ | 1 | Zn-S | 2 | 2.268(5) | 0.0069(4) | 0.412 |
| Zn-N/O | 2 | 1.969(4) | 0.0030(3) | |||
| ΔZnta-Cd2+ | 1 | Cd-S | 2 | 2.529(8) | 0.0025(4) | 0.472 |
| Cd-N/O | 2 | 2.29(1) | 0.002(1) | |||
| ΔZnta-Pb2+ | 1 | Pb-S | 2 | 2.642(6) | 0.0061(3) | 0.584 |
| Pb-N/O | 1 | 2.27(1) | 0.006(1) | |||
| ΔZnta-Hg2+ | 1 | Hg-S | 2 | 2.312(7) | 0.007(1) | 0.392 |
| Hg-N/O | (2) | 2.433(5) | 0.0017(7) |
Coordination numbers (N), interatomic distances (R), Debye-Waller factors σ2 (mean-square deviations in interatomic distance). The fit-error function F is: , X(k): EXAFS oscillation; k: photo-electron wave number). In parentheses are the standard deviations for best-fit parameters (± values on last digit)
Figure 3:

ATPase activity of PaΔZntA121–740 mutants (C391A, C393A, C391A/C393A, D721N and D721A) relative to wtPaΔZntA121–740 in detergent micelles, in the presence of the corresponding metal substrates (40 µM).
In PaΔZntA-Cd2+, the best fit was obtained with two N/O ligands at 2.29 Å, and two S ligands at 2.52 Å (Fig. 2C–D and Table 1, extended k-range fitting up to 14 Å−1 is reported in Fig. S7). These bond-lengths are consistent with other protein Cd2+ sites in tetrahedral/distorted tetrahedral coordination (Fig. 2I)9. Because of the larger Cd2+ ionic radius, the bond lengths significantly increased by 0.3 Å indicating plasticity in the selection site to accommodate first- and second- row transition metal substrates. The capability of uptake and binding of Zn2+ and Cd2+ at the plastic TM-MBS is compatible with the presence of a conserved electronegative funnel connecting the cytoplasmic membrane interface to the intramembranous high-affinity site3c. The funnel and the absence of a selectivity filter allow specific uptake of cellular Zn2+ or Cd2+. In agreement with the proposed Zn2+ model, mutation of coordinating C391, C393 and D721 residues dramatically affected the Cd2+ dependent ATPase activation.
To date, the understanding of Pb2+ transporters chemistry underlying detoxification is limited. By generating PaΔZntA121–740-Pb2+ samples, we have characterized for the first time the binding site and bonding geometry in a Pb2+ transporter. The EXAFS data revealed the existence of two ligand shells. The best EXAFS fit in PaΔZntA-Pb2+ was obtained with one N/O ligand at 2.27 Å, and two S ligands at 2.64 Å, (Fig. 2E–F and Table 1). The bond distances are consistent with trigonal pyramidal Pb2+ sites in which one tetrahedron position accommodates the Pb2+ lone pair (hybridization) (Fig. 2I). Based on the abolished Pb2+-dependent ATPase activity in all our PaΔZntA121–740 mutants (Fig. 3) and corresponding mutagenesis studies in E. coli4d, 4f and S. sonnei3c ZntA, the Pb2+ site is formed by Cys391, Cys393 and Asp7213c, 4d, 4f.
This novel PbS2O/N site indicates that the binding environment in ZntAs differs from the frequently observed favorable [Pb(II)(SR)3]− in Pb-substituted zinc fingers11 and de novo designed three helical bundles12. Pb2+ can be coordinated by S/O/N/P-containing ligands with coordination numbers from 2 to 9. Nevertheless, the favorable geometry in a thiol-rich environment is trigonal pyramidal with a lone pair occupying the apical position (hemidirected)13. Analysis of PDB for Pb2+-bound proteins reveals that thiolate ligands are indeed typically present in trigonal pyramidal [Pb(II)(SR)3]− geometries9. Because of the soft nature of Pb2+ expected from Pearson’s theory and the high enthalpy of Pb-S bond formation14, Pb(II) is thermodynamically more tightly bound by thiolates than carboxylate ligands. Thus, the observed 2S1N/O binding indicates a reduced Pb2+ affinity compared to 3S sites. This and the intrinsic kinetic lability of CysS-Pb(II) bonds could favor dissociation from the site in the Post-Albers cycle. Indeed, Pb(II) activation results in the highest ATPase turnover rate among all substrates (Fig. 1C).
Finally, we analyzed the EXAFS data of PaΔZntA121–740-Hg2+ (Fig. 2G–H and Table 1). Frequently, in metalloproteins Hg2+ is bound in digonal or trigonal 2S/3S coordination. We initially obtained a best fit with 2S ligands at 2.32 Å. However, the introduction of additional 1 or 2 N/O ligands resulted in comparable F values, preventing definitive assignment of the coordination environment from EXAFS. However, analysis of the X-ray absorption near edge structure (XANES) features in PaΔZntA121–740-Hg2+ (Fig. S8), revealed the absence of a pronounced 2p3/2 → 6s/5d transition at 12280 eV, the absence of a shoulder at 12295 eV and an absorption maximum below 12330 eV (in linear bis-L-cysteinate Hg(Cys)2 is above 12330 eV)15, consistent with a non-linear Hg2+ complex. The conservation and proximity of the Asp721 and the high frequency of Hg2+ structures in the PDB adopting irregular 3-/4- coordinated geometries, suggest linear coordination distortion and Hg2+ binding in irregular trigonal/tetrahedral geometries with possible weak N/O interactions (Fig. 2I). To address this, the effect of D721A or D721N mutations on the Hg2+-dependent ATPase activation profiles was investigated. D721A and D721N PaΔZntA121–740 mutants failed to be activated by Hg2+ (Fig. 3), strongly supporting the involvement of Asp721 side chain in Hg2+ coordination. Indeed, linear 2S Hg2+ coordination resulting in high affinity and reduced kinetic lability could prevent metal release or dramatically reduce turnover numbers.
To address whether the transmembrane site plasticity is preserved in the lipid bilayer, where structural constrains are imposed by the anisotropic nature of the protein-membrane interaction, we characterized the metal-bound forms of the pump reconstituted in lipid bilayers. PaΔZntA121–740ZntA was successfully reconstituted in proteoliposomes and its metal-bound forms generated for comparative XANES in PaΔZntA121–740-Zn2+/Cd2+/Hg2+/Pb2+ (Fig. S8) and for comparative EXAFS in the Zn2+/Cd2+/Hg2+ bound forms (Fig. S9). The close correspondence of all XANES features for PaΔZntA121–740-M2+ in detergent-micelles with those in lipid bilayers (with minor differences only in PaΔZntA121–740-Zn2+) and the similar ligand-to-metal distances from EXAFS fits, revealed sites with almost identical coordination (Table S1).
The results reveal “coordination plasticity” in metal recognition to guarantee substrate promiscuity in metal pumps. In contrast to ion channels, where size-selectivity filters select substrates in gated or non-gated conducting pores16, selectivity in zinc pumps is governed primarily by coordination chemistry and geometry resulting in metal binding with different coordination numbers and bond distances, while preserving the apparent coordination properties. Moreover, coordination divergence form ideal ligands sets and geometries typically present in high affinity binding sites could result in inefficient metal release required by the catalytic cycle. Thus, divergence from these ideal templates appears required for substrate promiscuity and efficient metal translocation.
Supplementary Material
Acknowledgments
The work was supported by the Robert A. Welch Foundation (AT-1935–20170325 to G.M), by the National Institute Of General Medical Sciences of the National Institutes of Health (R35GM128704 to G.M.) and by a Marie Curie Fellowship (European Commission, No. 252961 to G.M.). This work was also supported by P30 GM103335 to L. Z. through the Nebraska Redox Biology Center. G.W.I. is supported by an NSERC postdoctoral fellowship (PDF; Natural Sciences and Engineering Research Council of Canada). We thank the staff at Beamline 7–3/9–3, Stanford Synchrotron Radiation Lightsource (SSRL) for support in data collection. SSRL is operated for the DOE and supported by OBER and NIH. We thank O. Lewinson and A. T. Lee for the cDNA of PaZntA, and Claudia Andreini for support with MetalPDB analysis (http://metalweb.cerm.unifi.it/). We thank Prof. D. C. Rees (California Institute of Technology) and Prof. P. Nissen (Aarhus University) for invaluable help in project development and discussions.
Footnotes
Electronic Supplementary Information (ESI) available: [Experimental methods, materials and supporting results]. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Bublitz M, Morth JP and Nissen P, J. Cell. Sci, 2011, 124, 2515. [DOI] [PubMed] [Google Scholar]
- 2.(a) Arguello JM, Eren E and Gonzalez-Guerrero M, BioMetals, 2007, 20, 233; [DOI] [PubMed] [Google Scholar]; (b) Ma Z, Jacobsen FE and Giedroc DP, Chem. Rev, 2009, 109, 4644; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Boal AK and Rosenzweig AC, Chem Rev, 2009, 109, 4760; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Waldron KJ and Robinson NJ, Nature reviews, 2009, 7, 25. [DOI] [PubMed] [Google Scholar]
- 3.(a) Smith AT, Smith KP and Rosenzweig AC, J. Biol. Inorg. Chem, 2014, 19, 947; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sitsel O, Gronberg C, Autzen HE, Wang K, Meloni G, Nissen P and Gourdon P, Biochemistry, 2015, 54, 5673; [DOI] [PubMed] [Google Scholar]; (c) Wang K, Sitsel O, Meloni G, Autzen HE, Andersson M, Klymchuk T, Nielsen AM, Rees DC, Nissen P and Gourdon P, Nature, 2014, 514, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Rensing C, Mitra B and Rosen BP, Proc. Natl. Acad. Sci. U.S.A, 1997, 94, 14326; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sharma R, Rensing C, Rosen BP and Mitra B, J Biol Chem, 2000, 275, 3873; [DOI] [PubMed] [Google Scholar]; (c) Hou Z and Mitra B, J. Biol. Chem, 2003, 278, 28455; [DOI] [PubMed] [Google Scholar]; (d) Dutta SJ, Liu J, Hou Z and Mitra B, Biochemistry, 2006, 45, 5923; [DOI] [PubMed] [Google Scholar]; (e) Liu J, Dutta SJ, Stemmler AJ and Mitra B, Biochemistry, 2006, 45, 763; [DOI] [PubMed] [Google Scholar]; (f) Dutta SJ, Liu J, Stemmler AJ and Mitra B, Biochemistry, 2007, 46, 3692. [DOI] [PubMed] [Google Scholar]
- 5.(a) Liu J, Stemmler AJ, Fatima J and Mitra B, Biochemistry, 2005, 44, 5159; [DOI] [PubMed] [Google Scholar]; (b) Banci L, Bertini I, Ciofi-Baffoni S, Su XC, Miras R, Bal N, Mintz E, Catty P, Shokes JE and Scott RA, J. Mol. Biol, 2006, 356, 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitra B and Sharma R, Biochemistry, 2001, 40, 7694. [DOI] [PubMed] [Google Scholar]
- 7.Eren E and Arguello JM, Plant. Physiol, 2004, 136, 3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vašák M, Kägi JHR and Hill HA, Biochemistry, 1981, 20, 2852. [DOI] [PubMed] [Google Scholar]
- 9.Putignano V, Rosato A, Banci L and Andreini C, Nucleic Acids Res, 2018, 46, D459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raimunda D, Subramanian P, Stemmler T and Arguello JM, Biochim Biophys Acta, 2012, 1818, 1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Payne JC, ter Horst MA and Godwin HA, J. Am. Chem. Soc, 1999, 121, 6850. [Google Scholar]
- 12.Neupane KP and Pecoraro VL, Angew. Chem. Int. Ed. Engl, 2010, 49, 8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Cangelosi V, Ruckthong L and Pecoraro VL, Met. Ions Life Sci, 2017, 17; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Magyar JS, Weng TC, Stern CM, Dye DF, Rous BW, Payne JC, Bridgewater BM, Mijovilovich A, Parkin G, Zaleski JM, Penner-Hahn JE and Godwin HA, J. Am. Chem. Soc, 2005, 127, 9495. [DOI] [PubMed] [Google Scholar]
- 14.Andersen RJ, diTargiani RC, Hancock RD, Stern CL, Goldberg DP and Godwin HA, Inorg. Chem, 2006, 45, 6574. [DOI] [PubMed] [Google Scholar]
- 15.(a) Jalilehvand F, Leung BO, Izadifard M and Damian E, Inorg. Chem, 2006, 45, 66; [DOI] [PubMed] [Google Scholar]; (b) Manceau A, Lemouchi C, Rovezzi M, Lanson M, Glatzel P, Nagy KL, Gautier-Luneau I, Joly Y and Enescu M, Inorg. Chem, 2015, 54, 11776. [DOI] [PubMed] [Google Scholar]
- 16.Gouaux E and Mackinnon R, Science, 2005, 310, 1461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.