

SUMMARY
Hedgehog (Hh) signaling culminates in the conversion of the latent transcription factor Cubitus interruptus (Ci)/Gli into its activator form (CiA/GliA), but the underlying mechanism remains poorly understood. Here we demonstrate that Hh stimulates the phosphorylation of Ci by the Ser/Thr kinase Fused (Fu) and that Fu-mediated phosphorylation of Ci promotes its activation. We find that Fu directly phosphorylates Ci on Ser218 and Ser1230, which primes its further phosphorylation by CK1 on adjacent sties. These phosphorylation events alter Ci binding to the pathway inhibitor Suppressor of fused (Sufu) and facilitate the recruitment of Transportin and the transcriptional coactivator CBP. Furthermore, we provide evidence that Sonic hedgehog (Shh) activates Gli2 by stimulating its phosphorylation on conserved sites through the Fu family kinases ULK3 and mFu/STK36 in a manner depending on Gli2 ciliary localization. Hence, Fu family kinase-mediated phosphorylation of Ci/Gli serves as a conserved mechanism that activates the Hh pathway transcription factor.
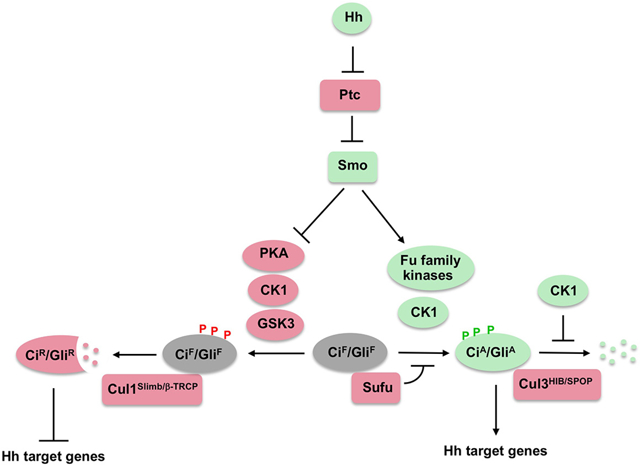
Graphical Abstract
eTOC Blurb
Hedgehog (Hh) signaling promotes phosphorylation that converts the latent transcription factor Cubitus interruptus (Ci)/Gli into its activator form, leading to activation of Hh target genes. Han et al. delineate a conserved phosphorylation cascade, involving Fused family kinases and CK1, that ultimately activates Ci/Gli.
INTRODUCTION
The Hedgehog (Hh) family of secreted proteins plays an essential role in embryonic development and adult tissue homeostasis (Briscoe and Therond, 2013; Jiang and Hui, 2008). Aberrant Hh pathway activity has been linked to a range of human diseases (Nieuwenhuis and Hui, 2005). Hh exerts it biological influence through a conserved signal transduction cascade that culminates in the conversion of the latent transcription factor Ci/Gli from a repressor (CiR/GliR) form into an activator form (CiA/GliA)(Aza-Blanc et al., 1997; Methot and Basler, 1999).
The core reception system for Hh consists of two multi-span transmembrane proteins: a twelve-span transmembrane protein Patched (Ptc) functions as the Hh receptor and a GPCR-family protein Smoothened (Smo) functions as an obligated signal transducer of the canonical Hh pathway (Briscoe and Therond, 2013; Jiang and Hui, 2008). In the absence of Hh ligand, Ptc inhibits Smo, allowing full-length Ci/Gli (CiF/GliF) to be phosphorylated by multiple kinases including protein kinase A (PKA), glycogen synthase kinase 3 (GSK3), and casein kinase 1 (CK1), which targets it for ubiquitination by SCFSlimb/β-TRCP, followed by proteasome-mediated partial degradation to generate CiR/GliR that actively repress a subset of Hh target genes (Chen and Jiang, 2013; Jiang and Struhl, 1995, 1998). Ci forms a complex with the kinesin-like protein Costal2 (Cos2) and the Ser/Thr kinase Fused (Fu), which prevents Ci nuclear localization and facilitates Ci phosphorylation and processing by recruiting PKA/CK1/GSK3 (Wang et al., 2000b; Wang and Jiang, 2004; Wang and Holmgren, 2000; Zhang et al., 2005).
In the presence of Hh ligand, binding of Hh to Ptc alleviates its inhibition of Smo, allowing Smo to be phosphorylated by multiple kinases including PKA (Drosophila only), CK1, and G protein-coupled receptor kinase 2 (GRK2) (Chen et al., 2010; Chen et al., 2011; Jia et al., 2004; Li et al., 2016; Li et al., 2014). Phosphorylation of Smo promotes its active conformation and accumulation on the cell surface (Drosophila) or in the primary cilium (vertebrate) (Chen et al., 2011; Zhao et al., 2007). Activated Smo blocks Ci/Gli processing into CiR/GliR by inhibiting Ci/Gli phosphorylation (Wang et al., 2000a; Zhang et al., 2005). However, blockage of Ci/Gli processing is insufficient for the conversion of CiF/GliF into CiA/GliA, a process that requires high levels of Hh (Strigini and Cohen, 1997).
In Drosophila, Fu converts CiF into a labile CiA by antagonizing Sufu, a conserved pathway inhibitor that binds Ci/Gli (Ohlmeyer and Kalderon, 1998). Loss of Sufu in Drosophila does not cause ectopic Hh pathway activation due to a dominant role of Cos2 in Ci inhibition (Preat et al., 1993). In mammals, however, Sufu plays an essential role in restricting Gli activation because Sufu knockout in mice resulted in phenotypes indicative of constitutive Hh pathway activation (Chen et al., 2009; Svard et al., 2006; Yin et al., 2019). The relevant Fu target(s) responsible for CiF-to-CiA conversion has remained elusive. Hh stimulated the phosphorylation of both Cos2 and Sufu in a manner depending on Fu (Lum et al., 2003; Ranieri et al., 2012); however, blocking Fu-mediated phosphorylation of either Cos2, Sufu, or both did not perturb Hh signaling under physiological conditions (Zadorozny et al., 2015; Zhou and Kalderon, 2011). Although it remains possible that unidentified phosphorylation events in Cos2 and Sufu may release their inhibition of Ci, Fu could phosphorylate other component(s) in the Hh pathway to activate Ci. Hh-stimulated phosphorylation of Gli2/3 has been implicated in the regulation of Gli activity (Humke et al., 2010; Niewiadomski et al., 2014), however, the precise Hh-induced phosphorylation events responsible for Gli activation and the relevant kinases involved have not been identified.
In this study, we found that Hh stimulated Ci phosphorylation by Fu on Ser218 and Ser1230, which primed its further phosphorylation by CK1 on adjacent sties. These phosphorylation events promoted Ci activation by reducing its binding to Sufu and consequently increasing its binding to Transportin (Trn) and CBP. Furthermore, we provided evidence that Shh activated Gli2 by stimulating its phosphorylation on conserved Ser residues though the Fu-family kinases ULK3 and STK36.
RESULTS
Fu-mediated phosphorylation of Sufu is not required for Ci activation
To determine whether Fu activates Ci by phosphorylating Sufu, we sought to systematically map the Fu-mediated phosphorylation sites on Sufu. Forced dimerization using a coiled-coil (CC) dimerization motif or activation loop phospho-mimetic mutations (EE: S151E and T154E) generated constitutively active forms of Fu (CC-Fu, FuEE and CC-FuEE) (Shi et al., 2011). In S2 cells, these active forms of Fu induced phosphorylation of coexpressed Sufu whereas a wild type or kinase dead form of Fu (CC-FuGV) failed to do so (Shi et al., 2011). Therefore, we coexpressed Flag-tagged Sufu (Flag-Sufu) with HA-tagged CC-FuEE in Sf9 cells and purified Flag-Sufu by immunoaffinity chromatography followed by LC-ESI-MS/MS analysis (Fig. S1A–B; see Methods). Eight phosphorylation sites in six locations were identified (Fig. S1C–D), including a cluster of sites (S321/S322/S324) that was implicated as Fu-mediated phosphorylation sites by a previous study (Zhou and Kalderon, 2011). To determine if any of these sites were essential for Fu-mediated activation of Ci, we mutated them to unphosphorylated Ala (A) individually or in combination (T39A, S98A, S161A, S290A, S321A/S234A, S460A, and SAall with all eight sites mutated). When coexpressed with FuEE, SufuT39A, SufuS98A, SufuS161A, S290A, and SufuS460A were phosphorylated similarly to SufuWT, as indicated by their similar mobility shift on the SDS-PAGE (Fig. S1E). By contrast, SufuS321A, SufuS321A/S234A and SufuSAall exhibited no mobility shift (Fig. S1E), suggesting that S321/S234 are the major Fu-mediated phosphorylation sites. In ptc-luc reporter assays, coexpression of SufuWT with a non-processed form of Ci (Ci−PKA), which has three PKA sites (S838, S856, and S892) mutated to Ala (Wang et al., 1999), suppressed the activity of Ci−PKA, and this inhibition was negated by CC-FuEE (Fig. S1F). Similar to SufuWT, all Sufu mutants including SufuSAall suppressed the activity of Ci−PKA and their inhibition was alleviated by CC-FuEE (Fig. S1F). In addition, mutating S321, S234 and three adjacent Ser residues in Sufu did not alter Hh pathway activity in wing imaginal discs (Zhou and Kalderon, 2011). Taken together, these observations suggest that Fu-mediated phosphorylation of Sufu is not essential for Ci activation.
Fu mediates Ci phosphorylation in response to Hh
In seeking for alternative Fu substrates, we noticed that Myc-Ci−PKA underwent a mobility shift on SDS PAGE when coexpressed with active forms of Fu in S2 cells, and that the mobility shift was abolished by λ phosphatase treatment (Fig. 1A), suggesting that activated forms of Fu can induce phosphorylation of Myc-Ci−PKA. Similarly, Hh-conditioned medium stimulated Myc-Ci−PKA phosphorylation mediated by FuWT but not by FuGV in S2 cells (Fig. 1B). In Cl8 cells, Hh induced phosphorylation of endogenous Ci after PKA was blocked by a pharmacological inhibitor H89 (Fig. 1C). Knockdown of either Smo or Fu by RNA interference (RNAi) abolished Hh-stimulated phosphorylation of Ci (Fig. 1D). These observations suggest that Hh signaling stimulates Ci phosphorylation through Smo/Fu on sites outside the previously identified PKA/GSK3/CK1 phosphorylation clusters. Hence, in addition to inhibiting PKA-mediated phosphorylation to block Ci processing, Hh also stimulates Ci phosphorylation by activating the Fu kinase.
Figure 1. Characterization of Fu-mediated phosphorylation of Ci.

(A) Western blot analysis of Myc-Ci−PKA co-expressed with different forms of Fu in S2 cells treated with or without λ-protein phosphatase (λ-pp).
(B) Western blot analysis of Myc-Ci−PKA co-expressed with wild type (WT) or kinase dead (GV) Fu in S2 cells treated with Hh-conditioned medium.
(C-D) Western blot analysis of endogenous Ci from Cl8 cells treated with H89, the indicated dsRNAs, and Hh-conditioned medium.
(E) Schematic drawing of Ci protein. “ZF” and “CBP” indicate zinc finger domain and CBP binding domain, respectively. Red bars indicate the positions of potential Fu mediated Ci phosphorylation sites identified by MS. The primary sequences of two critical phosphorylation clusters (C-I and C-II) are shown below with phosphorylated S/T shown in red.
(F-G) ptc-luc reporter assay in S2 cells transfected with the indicated Ci−PKA constructs (50 ng/well) in the absence or presence of Sufu/CC-FuEE.
(H, K) ptc-luc reporter assay in S2 cells transfected with the indicated Ci constructs at low concentrations (5 ng/well) and treated with Hh-conditioned medium (+Hh) or control medium (−Hh).
(I-J) ptc-luc reporter assay in S2 cells transfected with increasing amount of the indicated Ci constructs (I), or fixed amount of the indicated Ci constructs (50 ng/well) and increasing amounts of Sufu (J).
Data in F, G, H, and K are mean ± SEM from three independent experiments. Data in I and J are mean ± SEM from two independent experiments. *p < 0.05, **p < 0.01 and ***p < 0.001 (Student’s t test). Also see Figure. S1, Figure. S2, and Table S1.
Mapping and characterizing Fu-mediated phosphorylation sites on Ci
To identify Fu-mediated phosphorylation sites on Ci, Flag-Ci was coexpressed with CC-FuEE, and phosphorylated Flag-Ci was purified and subjected to LC-ESI-MS/MS analysis (Fig. S2A–C; see Methods). A total of 19 phosphorylated Ser/Thr residues located in 9 regions (S1 to S9) were identified (Fig. S2D). S838 (S6) corresponds to a previously identified PKA site essential for Ci processing (Chen and Jiang, 2013) and will not be considered for further analysis.
We introduced S/T-to-A mutations into the identified sites in different combinations in the context of Ci−PKA (Supplementary Table 1) and analyzed the consequence of these mutations on Ci activation using the de-repression assay described in Fig. S1F. Because Ci−PKA is no longer processed into CiR and remains as full-length (Wang et al., 1999), the effect of mutating Fu phosphorylate sites in this background should reflect a change in CiA activity. We divided the phosphorylated sites into three group based on their locations: A (S1–4), B (S5, 7, 8), and C (S9) (Fig. S2E). Mutating group A or C alone slightly reduced whereas their combined mutations dramatically reduced Fu-mediated Ci activation (Fig. S2F). By contrast, mutating group B alone did not affect Fu-mediated Ci activation and mutating group B in conjunction with A/C did not have an additive effect (Fig. S2F). These observations suggest that groups A and C but not B may harbor phosphorylation sites important for Ci activation. Further mutagenesis analysis indicated that S218 and S220 are the important sites in group A (Fig. S2G–H) while phosphorylation residues in the cluster C have a redundant function (Fig.S2I). In the following, S218/S220 and S1229/S1230/T1232/S1233 were referred to as cluster I and cluster II, respectively (Fig. 1E). Hence, mutating either cluster I (SA-I) or cluster-II (SA-II) modestly reduced Fu-stimulated Ci activation while their combined mutations (AA) resulted in a more dramatic reduction (Fig 1F).
We then introduced phospho-mimetic mutation into either cluster I (SD-I), cluster II (SD-II), or both (DD). The phospho-mimetic Ci mutants exhibited consistently higher activity than the wild type Ci when expressed alone or together with Sufu/CC-FuEE (Fig. 1G). When the Ci constructs were expressed at low levels, Hh stimulated higher level of ptc-luc expression from Ci−PKA_DD but lower level from Ci−PKA_AA compared with Ci−PKA_WT (Fig. 1H). When expressed at low but increasing amounts in S2 cells, Ci−PKA_DD exhibited higher activity while Ci−PKA_AA exhibited slightly lower activity than Ci−PKA_WT (Fig. 1I). When a fixed amount of Ci was coexpressed with an increasing amount of Sufu, Ci−PKA_DD exhibited consistently higher activity than Ci−PKA_WT and Ci−PKA_AA before reaching a saturated amount of Sufu that inhibited all the Ci constructs (Fig. 1J), suggesting that Ci−PKA_DD is more resistant to Sufu inhibition. We also introduce AA and DD mutations in the wild type Ci (CiWT) background to generate CiAA and CiDD and found that CiDD exhibited higher whereas CiAA exhibited lower activity than CiWT in S2 cells upon Hh stimulation (Fig. 1K). Taken together, these results suggest that phosphorylation of C-I and C-II promoted Ci activation in S2 cells.
Phosphorylation of C-I and C-II promotes Ci activation in vivo
We next assessed whether phosphorylation of clusters I and II regulates Ci activity in vivo. To ensure equal expression of different Ci transgenes, UAS-Ci-PKA_WT, UAS-Ci-PKA_AA, UAS-Ci-PKA_DD, UAS-Ci WT, UAS-CiAA and, UAS-CiDD were introduced into flies using the phiC31 integrase system (Bischof et al., 2007). Because overexpression of Ci in wing imaginal discs resulted in constitutive Hh pathway activation that could mask the effect of mutating the Fu-mediated phosphorylation sites (Wang et al., 1999), a weak Gal4 driver, C765 (Ma et al., 2016), was used to express the Ci transgenes. We further modulated the expression of the Ci transgenes by growing larvae at 18 °C or 25 °C because Gal4 activity is temperature-sensitive (Duffy, 2002).
In wing imaginal discs, posterior (P) compartment cells express Hh but not Ci whereas anterior (A) compartment cells express Ci and are thus competent to respond to Hh. As a consequence, A-compartment cells near the A/P boundary accumulate CiF and express Hh target genes such as ptc (Fig. 2A–A”). Expression of UAS-Ci-PKA_WT with C765 (C765>Ci-PKA_WT) at 25°C resulted in ectopic ptc expression in P-compartment cells (Fig. 2B–B”). Expression of UAS-Ci-PKA_AA (C765>Ci-PKA_AA) resulted in lower levels while expression of UAS-Ci-PKA_DD (C765>Ci-PKA_DD) led to higher levels of ectopic ptc expression in P-compartment cells (Fig. 2C–C”, 2D–D”). C765>Ci-PKA_DD also induced ectopic ptc expression in A-compartment cells distant from the A/P boundary (arrowhead in Fig. 2D’), suggesting that the phospho-mimetic Ci exhibited constitutive activity. Consistent with this notion, C765>Ci-PKA_DD adult wings exhibited anterior overgrowth indictive of ectopic Hh signaling whereas adult wings expressing either C765>Ci-PKA_WT or C765>Ci-PKA_AA did not (Fig. 2A’”–D’”). The difference in Hh signaling activity among these Ci constructs was also obvious when larvae were grown at 18°C to further reduce transgenic expression (Fig. 2E–G’”). Similarly, C765>CiAA induced lower while C765>CiDD induced higher levels of ectopic ptc expression in P-compartment cells compared C765>CiWT (Fig. S3). Taken together, these results suggest that phosphorylation of C-I/C-II promotes Ci activation in wing imaginal discs.
Figure 2. Phosphorylation of C-I and C-II promotes Ci activity in vivo.

(A-D’”) Control (A-A”) wing disc or wing discs expressing UAS-Myc-Ci-PKA_WT (B-B”), UAS-Myc-Ci-PKA_AA (C-C”), or UAS-Myc-Ci-PKA_DD (D-D”) under the control of C765 Gal4 drive from larvae grown at 25 °C were immunostained to the expression of Ci (red) and Ptc (green). The corresponding adult wings were shown in A’”-D’”. (E-G’”) Wing discs expressing UAS-Myc-Ci-PKA_WT (E-E”‘), UAS-Myc-Ci-PKA_AA (F-F”‘), or UAS-Myc-Ci-PKA_DD (G-G”‘) under the control of C765 Gal4 drive from larvae grown at 18 °C were immunostained to the expression of Ci (red) and Ptc (green). Large magnification views of the posterior compartment of the wing discs are shown in E’”-G’”.
Scale bars are 50 μM for wing discs and 500 μM for adult wings. Also see Figure. S3.
Hh stimulates Ci phosphorylation at C-I and C-II through Fu
To further characterize Ci phosphorylation and its regulation by Hh, we generated phospho-specific antibodies that recognize Ci phosphorylated on T1232 (pT1232), doubly phosphorylated on S218 and S220 (pS218/220) or S1230 and S1233 (pS1230/1233) (Fig. 3A). Coexpression of FuEE but not FuGV with Myc-Ci−PKA induced Ci phosphorylation signals detected by pS218/220, pS1230/1233, and pT1232 antibodies (Fig. 3B). Mutating the corresponding S/T residues abolished the signals detected by these antibodies (Fig. 3C–D). Phosphorylation at C-I and C-II appeared to be independent of each other because mutating one cluster did not affect the phosphorylation of the other (Fig. 3C).
Figure 3. Hh induces Ci phosphorylation at C-I and C-II depending on Smo and Fu kinase activity.

(A) The antigen sequences for generating the three phospho-specific antibodies.
(B-D) Western blot analysis of Myc-Ci−PKA and its variants in S2 cells co-expressed with FuEE and FuGV with the indicated phospho-specific antibodies.
(E) Western blot analysis of Flag-Ci−PKA phosphorylation in S2 cells with or without Sufu coexpression and treated with Hh-conditioned medium or control medium.
(F) Western blot analysis of Flag-Ci−PKA phosphorylation in S2 cells treated with the indicated dsRNA and Hh-conditioned medium and cotransfected without or with the indicated Smo or Fu construct.
(G) Western blot analysis of HA-Fu and HA-FuIns from S2 cells expressing the indicated Fu constructs and treated with GFP or Fu dsRNA.
(H-I) Western blot analysis of endogenous Ci phosphorylation from Cl8 cells treated with or without Hh-conditioned medium and the indicated dsRNA.
(J) Western blot analysis of endogenous Ci phosphorylation (top panels) or ptc-luc reporter assay (bottom panel) in Cl8 cells treated with increasing concentrations of Hh-conditioned medium.
Hh stimulated phosphorylation of Ci−PKA at these two clusters whereas Sufu suppressed both basal and Hh-induced phosphorylation (Fig. 3E). Knockdown of Smo by a dsRNA targeting its 5’ UTR abolished Hh-induced phosphorylation of S218/220, S1230/1233 and T1232, which was rescued by the expression of exogenous Smo (Fig. 3F). Fu RNAi also abolished Hh-induced phosphorylation of Ci on these sites, which was rescued by transfection with a RNAi-insensitive wild type Fu (FuINS) but not its kinase dead (KR) form (Fig. 3F–G). Hh induced phosphorylation of endogenous Ci at C-I and C-II in Cl8 cells (Fig. 3H), which was abolished by either Smo or Fu RNAi (Fig. 3I). Interestingly, phosphorylation at these sites increased progressively in response to increasing levels of Hh, which correlated with increasing levels of Hh pathway activity (Fig. 3J), suggesting that Hh gradient was translated into a Ci phosphorylation and activity gradient.
Fu directly phosphorylates Ci on S218 and S1230
To determine whether Fu directly phosphorylates Ci, Flag-tagged CC-FuEE and CC-FuGV were purified by from Sf9 cells, incubated with GST fused with Ci fragments containing C-I site (GST-CiN) or C-II site (GST-CiC), and subjected to in vitro kinase assay using the pIMAGO system. As shown in Fig. 4A, CC-FuEE but not CC-FuGV induced phosphorylation of both GST-CiN and GST-CiC. Mutagenesis study of individual sites suggested that S218 and S1230 were directly phosphorylated by Fu (Fig. 4A).
Figure 4. Phosphorylation of Ci by Fu primes its further phosphorylation by CK1.

(A) In vitro kinase assay using CC-FuEE or CC-FuGV purified from Sf9 cells and GST fusion proteins containing wild type or mutant CiN/CiC fragments. Phosphorylation was detected by the pIMAGO kit. GST fusion proteins were visualized by coomassie blue gel staining (CB).
(B) Alignment Fu phosphorylation sites in Ci and Cos2.
(C-E) In vitro kinase assay using purified CC-FuEE or CC-FuGV and the indicated GST fusion proteins.
(F-H) In vitro kinase assay using purified CC-FuEE/recombinant CK1δ and the indicated GST fusion proteins. Phosphorylation was detected by western blot with the indicated antibodies.
(I) In vitro kinase assay using a recombinant CK1δ and the indicated GST-CiC fusion proteins. 4SA has S1229, S1230, T1232 and S1233 mutated to A.
A previous study identified S572 as the major Fu phosphorylation site on Cos2 (Nybakken et al., 2002). Alignment of the amino acid sequences surrounding CiS218. CiS1230, and CosS572 revealed that they all contain an acidic residue at the +6 position (Fig. 4B). Mutating this acidic residue to Ala (D224A, E1236A, and E578A) greatly reduced the phosphorylation of S218 and S1230 in Ci and abolished the phosphorylation of S572 in Cos2 (Fig. 4C–E), suggesting that an acidic residue at the +6 position is required for efficient phosphorylation of S/T at the 0 position by the Fu kinase.
Phosphorylation of Ci by Fu primes its further phosphorylation by CK1
CK1 phosphorylation sites contain an acidic residue or phosphorylated S/T at −2,−3, or −4 position with the following consensus: D/E/(p)S/(p)T[X1–3]S/T (underlined amino acid represents the CK1 phosphorylation site; X represents any amino acid) (Knippschild et al., 2005). Thus, phosphorylation of S218 by Fu could prime CK1 phosphorylation on S220 whereas phosphorylation of S1230 could prime CK1 phosphorylation on T1232 and S1233. In support of this, the phospho-specific antibody pS218/220 only recognized GST-CiN phosphorylated by both purified CC-FuEE and a recombinant CK1(Fig. 4F). Similarly, GST-CiC phosphorylated by both CC-FuEE and CK1 was recognized strongly by pS1230/1233 and pT1232 (Fig. 4G–H).
Of note, GST-CiC phosphorylated by CK1 alone was also weakly recognized by both pS1230/1233 and pT1232 antibodies (Fig. 4G–H), consistent with S1230/1233 and T1232 being predicted as CK1 sites. In vitro kinase assay using GST-CiC variants with different S/T mutated to A confirmed that pS1230, pT1232, and S1233 could be phosphorylated by the recombinant CK1 (Fig. 4I). Nevertheless, the observations that Hh-stimulated pS1230/1233 and pT1232 signals depended on the Fu kinase activity and that combination of CK1 with Fu induced much stronger phosphorylation at these sites than CK1 alone suggest that effective phosphorylation of C-II is achieved by a sequential phosphorylation event: Fu phosphorylates S1230, which primes CK1 to phosphorylate T1232 and S1233.
Fu-mediated phosphorylation of Ci alters its biding to Sufu
We next asked whether Fu-mediated phosphorylation of Ci affects its interaction with Sufu. In CoIP experiments, phospho-mimetic forms of Myc-Ci−PKA (SD-I, SD-II and DD) pulled down less HA-Sufu compared with Myc-Ci-PKA_WT (Fig. 5A). In addition, Flag-Ci−PKA bound to HA-Sufu exhibited reduced pS218/220 and pS1230/1233 signals compared with the whole population of Flag-Ci−PKA (Fig. 5B), suggesting that Sufu prefers to bind unphosphorylated Flag-Ci−PKA.
Figure 5. Phosphorylation of Ci reduces its binding to Sufu while increases its binding to Trn and dCBP.

(A-C) Western blot analysis (left panel) and quantitation (right panel) of co-immunoprecipitation experiments using lysates from S2 cells co-expressing the indicated Myc-Ci−PKA constructs and HA-Sufu (A, B) or Flag-MATH (C). TCL: total cell lysates.
(D-F) FRET efficiency between the indicated CFP-Ci constructs and YFP-Sufu in S2 cells with or without FuEE/FuKR co-expression.
(G, I) Western blot analysis of co-immunoprecipitation experiments using S2 cell lysates expressing Myc-Ci−PKA (WT, AA and DD) either alone or together with Sufu and with or without HA-FuEE/FuGV, and mixed with immunopurified Flag-Transportin (G) or Flag-dCBP (I) from Sf9 cells.
(H) Immunostaining (left panel) and quantitation (right panel) of S2 cells expressing Myc-Ci−PKA (WT and AA) without or with Flag-Sufu/HA-CC-FuEE. n=50 cells for each transfection. Scale bars, 10 μM.
Data in A-C are mean ± SD from two independent experiments. Data in D-F are mean ± SEM from two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t test).
CiA/GliA is labile due to its increased binding to HIB/SPOP, a Hh-induced MATH and BTB domain containing protein that targets CiA/GliA for ubiquitination and degradation, whereas binding of Sufu to Ci/Gli prevents its association with HIB/SPOP (Chen et al., 2009; Zhang et al., 2009; Zhang et al., 2006). Myc-Ci−PKA_DD exhibited increased binding to a Flag-tagged MATH domain of HIB (Flag-MATH) compared with Myc-Ci−PKA_WT (Fig. 5C), consistent with its reduced binding to Sufu.
To further characterize how Fu-mediated phosphorylation of Ci affect Sufu/Ci interaction, we carried out FRET analysis in S2 cells expressing an N-terminally CFP-tagged Ci−PKA (CFP-Ci−PKA) and a C-terminally YFP-tagged Sufu (YFP-Sufu) (Shi et al., 2011). In the absence of Fu cotransfection, FRET efficiency between CFP-Ci−PKA_WT and YFP-Sufu was >12% (Fig. 5D), suggesting that Sufu is in close contact with the N-terminus of Ci. Coexpression of HA-FuEE but not a kinase dead Fu (HA-FuKR) reduced the FRET efficiency blow 5% (Fig. 5D), confirming that Fu interferes with Sufu/Ci interaction through its kinase activity. We then coexpressed CFP-Ci−PKA_WT, CFP-Ci−PKA_DD, or CFP-Ci−PKA_AA with YFP-Sufu in the presence of HA-FuEE or HA-FuKR. As expected, CFP-Ci−PKA_DD/YFP-Sufu exhibited lower FRET efficiency than CFP-Ci−PKA_WT/Sufu-YFP in the presence of HA-FuKR but exhibited FRET efficiency similar to that of CFP-Ci−PKA_WT/YFP-Sufu in the presence of HA-FuEE (Fig. 5E). By contrast, CFP-Ci−PKA_AA/YFP-Sufu exhibited much higher FRET efficiency than CFP-Ci−PKA_WT/YFP-Sufu in the presence of HA-FuEE, suggesting that the association between Ci−PKA_AA/Sufu is resistant to the inhibition by Fu. However, the FRET efficiency from CFP-Ci−PKA_AA/YFP-Sufu or CFP-Ci−PKA_DD/YFP-Sufu was still downregulated by HA-FuEE, albeit to a lesser extent compared with that from CFP-Ci−PKA_WT/YFP-Sufu (Fig. 5E), implying that Fu can inhibit Sufu/Ci association through a mechanism other than phosphorylating Ci at C-I and C-II. Similar results were obtained with S2 cells transfected with CFP-CiWT, CFP-CiDD, or CFP-CiAA in conjunction with YFP-Sufu in the presence of FuEE or FuKR (Fig. 5F).
Fu-mediated phosphorylation of Ci facilitates its interaction with Trn and CBP
Sufu inhibits Ci nuclear translocation in part by blocking its interaction with Trn, which binds to an N-terminally situated nuclear localization signal in Ci (Shi et al., 2014a). To determine how Ci phosphorylation affect its interaction with Trn, Flag-Trn purified from SF9 cells was incubated with cell extracts derived from S2 cells transfected with different Ci constructs and with or without Sufu and Fu constructs, followed by immunoprecipitation and western blot analysis. As shown in Fig. 5G, Flag-Trn was coimmunoprecipitated with Myc-Ci−PKA_WT but this interaction was blocked by coexpression of Sufu (Fig. 5G; lanes 1–2). Coexpression of FuEE but not FuGV partially negated Sufu-mediated inhibition of Ci−PKA_WT/Trn association (Fig. 5E; lanes 3–4). Sufu also inhibited the association between Ci−PKA_AA/Trn (Fig. 5G; lanes 5–6) but this inhibition was released less efficiently by FuEE compared with Ci−PKA_WT (Fig. 5G; lane 7 compared with lane 3). Consistent with the reduced binding of Sufu to Myc-Ci−PKA_DD (Fig. 5A), Sufu inhibited the association between Myc-Ci−PKA_DD and Flag-Trn less effectively, and this inhibition was completely negated by FuEE (Fig. 5G; lanes 9–11). These results suggest that Fu-mediated Ci phosphorylation counteracts Sufu to promote Trn binding.
When expressed in S2 cells, Ci−PKA_WT and Ci−PKA_AA were largely localized in the nucleus after the cells were treated with the nuclear export inhibitor LMB (Fig. 5H). Overexpression of Sufu blocked the nuclear localization of both Ci−PKA_WT and Ci−PKA_AA while coexpression of CC-FuEE with Sufu restore the nuclear localization of Myc-Ci−PKA_WT but not Myc-Ci−PKA_AA (Fig. 5H), suggesting that Fu-mediated phosphorylation of Ci is essential to overcome Sufu-mediated inhibition of Ci nuclear localization.
In addition to blocking Ci nuclear transport, Sufu also inhibits the activity of nuclear Ci by blocking Drosophila CBP (dCBP) recruitment (Han et al., 2015). As shown in Fig. 5I, Sufu blocked the association between dCBP and Myc-Ci−PKA_WT or Myc-Ci−PKA_AA but to a lesser extent the association between dCBP and Myc-Ci−PKA_DD (lanes 1–2, 5–6, 9–10). Sufu-mediated inhibition of Myc-Ci−PKA_WT/dCBP or Myc-Ci−PKA_DD/dCBP interaction was largely alleviated by FuEE but not by FuGV (Fig. 5I; lanes 3, 11); however, Sufu-mediated blockage of Myc-Ci−PKA_AA/dCBP association was only slightly negated by FuEE (Fig. 5I, lane 7). Hence, Fu-mediated phosphorylation of Ci promotes the recruitment of dCBP by antagonizing Sufu.
Shh stimulates Gli2 activity by phosphorylating it on S230/S232
Sequence alignment of the Ci/Gli family of proteins indicated that C-I is conserved among the three Gli proteins, which corresponds to S230/S232 in mouse Gli2 (Fig. 6A). Because Gli2 is a major contributor of GliA, we focused on Gli2 to analyze the function of this conserved phosphorylation cluster. We generated NIH3T3 cells in which the endogenous Gli2 was depleted by a shRNA targeting its 3’ UTR and replaced by Myc-tagged wild type mouse Gli2 (Gli2WT), a phosphorylation deficient form with S230/S232 mutated to A (Gli2AA; Fig. 6A), or a phospho-mimetic form with S230/S232 mutated to D (Gli2DD; Fig. 6A) through lentiviral transduction. Western blot analysis indicated that the levels of exogenously expressed Gli2 proteins were comparable to one another but slightly higher than that of the endogenous Gli2 (Fig. 6B). Consistent with our previous finding (Han et al., 2017), Gli2 RNAi diminished the expression of Shh target gene Gli1 induced by the Smo agonist SAG, which was completely rescued by Myc-Gli2WT (Fig. 6C). In response to SAG, Myc-Gli2DD activated Gli1 more efficiently than Myc-Gli2WT whereas Myc-Gli2AA exhibited reduced activity (Fig. 6C). In addition, Myc-Gli2DD exhibited higher basal activity and was more resistant to Sufu-mediated inhibition than Myc-Gli2WT and Myc-Gli2AA (Fig. 6C–D). In CoIP experiments, Myc-Gli2DD pulled down less Flag-Sufu than Myc-Gli2WT (Fig. 6E), suggesting that phosphorylation of Gli2 on S230/232 attenuated its binding to Sufu.
Figure 6. Shh signaling activates Gli2 by inducing its phosphorylation.

(A) Alignment of amino acid sequences surrounding the N-terminal phosphorylation sites in Ci, mouse (m) and human (h) Gli proteins.
(B) Western blot analysis to show Gli2 protein levels from NIH3T3 cells expressing the indicated shRNA and Myc-Gli2 lentiviral constructs.
(C) Relative Gli1 mRNA levels measured by RT-qPCR in NIH3T3 cells expressing the indicated shRNA and Myc-Gli2 lentiviral constructs with or without SAG. Data are mean ± SEM from two independent experiments. **p < 0.01 and ***p < 0.001 (Student’s t test).
(D) Gli-luc reporter activity in NIH3T3 cells expressing the indicated Myc-Gli2 constructs and increasing amounts of Flag-Sufu. Data are mean ± SEM from two independent experiments.
(E) Western blot analysis (left) and quantification (right) of Flag-Sufu coimmunoprecipitated with Myc-Gli2WT or Myc-Gli2DD. Data are mean ± SD from two independent experiments. **p < 0.01 (Student’s t test).
(F) Western blot analysis of Myc-Gli2 phosphorylation in NIH3T3 cells transduced with lentivirus expressing shRNA targeting Gli2 3’ UTR and lentivirus expressing either Myc-Gli2WT or Myc-Gli2AA in the absence or presence of SAG. The antigen sequence for generating the pS230/232 antibody is shown on the top.
(G) Western blot analysis of Myc-Gli2 phosphorylation (top) or Gli-luc reporter activity (bottom) in NIH3T3 cells transduced with lentivirus expressing shRNA targeting the 3’ UTR of Gli2 and lentivirus expressing Myc-Gli2WT (NIH3T3Gli2-shRNA/Myc-Gli2) in the presence of increasing amounts of Shh-N.
(H) Western blot analysis of SAG-induced Myc-Gli2 phosphorylation in NIH3T3Gli2-shRNA/Myc-Gli2 cells in the absence or presence of DN-Kif3b coexpression.
(I) Western blot analysis of SAG-induced Myc-Gli2 phosphorylation (top) or images of SAG-induced Myc-Gli2 ciliary localization (bottom) in NIH3T3Gli2-shRNA/Myc-Gli2 cells transduced with lentiviruses expressing shRNA targeting either GFP (control) or Kapβ2.
Ac-Tub: acetylated tubulin. Scale bars, 2.5 μM. Also see Figure S4.
We developed a phospho-specific antibody that recognized a Gli2 epitope with both S230 and S232 phosphorylated (pS230/232) (Fig. 6F). Using this antibody, we demonstrated that SAG stimulated Gli2 phosphorylation on S230/S232 (Fig. 6F). Furthermore, increasing levels of Shh-N progressively increased the phosphorylation of S230/S232, which correlated with the increasing levels of pathway activity measured by the Gli-luc reporter assay (Fig. 6G). Gli2 phosphorylation depended on the presence of primary cilia because expression of a dominant negative Kif3b (DN-Kif3b), which blocked primary cilium formation (Chen et al., 2009), abolished SAG-induced phosphorylation signal detected by the pS230/232 antibody (Fig. 6H).
In response to Hh, Gli proteins accumulate at the primary cilium (Chen et al., 2009; Haycraft et al., 2005). Ciliary localization of Gli depends on karyopherin-β2 (Kapβ2) that binds to a PY-NLS located in the N-terminal region of Gli proteins (Han et al., 2017). Kapβ2 depletion by shRNA diminished SAG-induced Gli2 ciliary localization and consequently abolished S230/S232 phosphorylation (Fig. 6I), suggesting that Hh signaling stimulates Gli2 phosphorylation depending on its ciliary localization.
Because the Gli2 epitope used for generating the pS230/S232 antibody is highly conserved among the three Gli proteins (Gli1, Gli2 and Gli3) (Fig. 6A), pS230/232 is likely to recognize Gli1 and Gli3 phosphorylated on the corresponding sites. Indeed, using the pS230/232 antibody, we found that SAG stimulated phosphorylation of Myc-Gli1 and Myc-Gli3 expressed in NIH-3T3 cells (Fig. S4A–B). To determine whether Shh stimulated the phosphorylation of endogenous Gli proteins on these sites, we took advantage of a recently developed, protein A- and HA-tagged Gli3 knock-in mice (Gli3TAP) and derived MEF cells from E15.5 embryos (Gli3TAP-MEF) from these mice (Zhang et al., 2019). Treating Gli3TAP-MEF cells with Shh-N induced phosphorylation of Gli3TAP detected by the pS230/232 antibody (Fig. S4C), suggesting that Shh induced phosphorylation of endogenous Gli3 on these conserved sites.
ULK3 and STK36 mediate Gli2 phosphorylation in response to Shh
The closest homolog of Fu in mammals is STK36 (also called mFu) (Fig. 7A) (Maloverjan and Piirsoo, 2012). STK36/mFu synergized with Gli1 and Gli2 to activate the Hh pathway when coexpressed in cultured cells (Murone et al., 2000), and rescued Hh signaling defects in zebrafish caused by the loss of zebrafish Fu homolog (Wilson et al., 2009). However, mice with STK36/mFu knocked out did not show any discernible Hh signaling defects during embryonic development (Chen et al., 2005; Merchant et al., 2005), suggesting that other kinases may substitute STK36/mFu to regulate Gli proteins during development. Indeed, overexpression of another Fu-related kinase ULK3 could also increase Gli activity in cultured cells (Maloverjan et al., 2010). We found that HA-tagged wild type ULK3 (HA-ULK3WT) but not its kinase dead form (HA-ULK3KR) phosphorylated coexpressed Myc-Gli2 on S230/S232 in NIH3T3 cells (Fig. 7B–C). Similar to Drosophila Fu, forced dimerization of ULK3 (HA-CC-ULK3) stimulated its kinase activity toward phosphorylating Myc-Gli2 (Fig. 7C). In addition, ULK3 could phosphorylate coexpressed Myc-Gli1 and Myc-Gli3 on the conserved sites in NIH3T3 cells (Fig. S4D–E). Immunopurified Flag-ULK3WT but not Flag-ULK3KR phosphorylated a GST-Gli2 fusion protein in in vitro kinase assay (Fig. S5A). Mutating S230 but not S232 to A abolished this phosphorylation, suggesting that ULK3 directly phosphorylates S230 (Fig. S5A). Interestingly, mutating D236 to A in GST-Gli2 also diminished phosphorylation of S230 by ULK3 (Fig. S5B), suggesting that ULK3, like Drosophila Fu, also prefers an acidic residue at +6 position for its target site. S232 conforms a CK1 site after S230 is phosphorylated by Ulk3. Indeed, the pS230/232 antibody only recognized GST-Gli2 after it was phosphorylated by both ULK3 and CK1 (Fig. S5B). These results suggest that ULK3 directly phosphorylates S230, which primes Ci phosphorylation on S232 by CK1.
Figure 7. ULK3/STK36 mediates Gli2 phosphorylation and activation.

(A) Family tree of Fu-related kinases.
(B-C) Western blot analysis of Myc-Gli2 phosphorylation in NIH3T3 cells transfected with the indicated Gli2 and ULK3 constructs.
(D, F, H) Relative Gli1 mRNA levels measured by RT-qPCR in clonal NIH3T3 cells transduced with lentiviruses expressing Cas9 and the indicated sgRNA with or without the indicated rescue constructs and Shh-N.
(E, G, I) Western blot analysis of Myc-Gli2 phosphorylation in clonal NIH3T3 cells transduced with lentiviruses expressing Cas9 and the indicated sgRNA with or without the indicated rescue constructs and Shh-N treatment.
(J) Western blot analysis of Gli2 phosphorylation in in vitro cultured SmoM2-driven medulloblastoma cells transduced with lentiviruses expressing Myc-Gli2WT or Myc-Gli2AA and treated with the indicated siRNA.
(K) Relative growth of in vitro cultured SmoM2-driven medulloblastoma cells treated with the indicated siRNAs and with or without lentiviral transduction of the indicated Gli2 constructs. Data in D, F, H, K are mean ± SEM from two independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001 (Student’s t test). Also see Figures S4–S7.
We next asked whether ULK3 is required for Shh-induced phosphorylation of S230/S232. We applied the CRISPR/Cas9 system to inactivate ULK3 using two independent guide RNAs: sgRNA_ULK3–1 and sgRNA_ULK3–2. Clonal NIH3T3 cell lines containing either sgRNA_ULK3–1 or sgRNA_ULK3–2 exhibited a reduction in Shh-induced Gli1/Ptch1 expression and S230/S232 phosphorylation (Fig. 7D–E; Fig. S6A), which was rescued by lentiviral expression of ULK3WT but not ULK3KR (Fig. 7D–E; Fig. S6A).
The incomplete blockage of Gli2 phosphorylation by ULK3 depletion suggests that other kinases such as STK36 might act in parallel to promote Gli2 phosphorylation in NIH3T3 cells. Indeed, gene editing of STK36 by the CRISPR/Cas9 system using two independents guided RNAs, sgRNA_S1 and sgRNA_S2, also reduced Shh-induced Gli1/Ptch1 expression and S230/S232 phosphorylation, which was rescued by expression of wild type STK36 (STK36WT) but not its kinase dead form (STK36KR) (Fig. 7F–G; Fig. S6B). Simultaneous inactivation of ULK3 and STK36 resulted in a more dramatic reduction in Shh-stimulated Gli1/Ptch1 expression and S230/S232 phosphorylation, which was rescued by coexpression of exogenous ULK3 and STK36 (Fig. 7H–I; Fig. S6C). Exogenous expression of either ULK3 or STK36 alone in ULK3/STK36 doubly depleted cells only partially rescued Shh-stimulated Gli1/Ptch1 expression and S230/S232 phosphorylation (Fig. 7H–I; Fig. S6C), suggesting that ULK3 and STK36 have overlapping but not identical function in promoting Gli activation in NIH3T3 cells.
We noticed that Shh still induced Gli1/Ptch1 expression in ULK3/STK36 double-depleted cells, albeit at much lower levels compared with wild type cells (Fig. 7H; Fig. S6C). This residual pathway activity could be due to the inhibition of GliR by Shh. Indeed, Shh could block the processing of both Gli2 and Gli3 in ULK3/STK36 doubly depleted cells (Fig. S6D–E), suggesting that the Fu-family kinases are not required for the regulation of Gli processing.
We also used siRNA to knock down ULK3 and STK36 either individually or in combination in NIH3T3 cells and obtained results similar to those obtained in experiments using the CRISPR/Cas9-mediated gene editing system (Fig. S7A–C). In addition, knockdown of ULK3/STK36 diminished Shh-induced phosphorylation of endogenous Gli3 in Gli3TAP-MEF cells (Fig. S4F).
ULK3 and STK36 are required for Gli activation in medulloblastoma cells
We next determined whether ULK3/STK36 regulates Gli phosphorylation and activation in medulloblastoma driven by an oncogenic form of Smo (SmoM2)(Shi et al., 2014c; Xie et al., 1998). Primary medulloblastoma cells derived from SmoM2, CAG-CreER mouse cerebella were cultured in vitro and treated with siRNA for ULK3 and/or STK36, followed by infection with lentiviruses expressing Myc-Gli2WT, Myc-Gli2AA, or Myc-Gli2DD. Knockdown of either ULK3 or STK36 reduced Myc-Gli2 phosphorylation (Fig. 7J), Shh target gene expression (Fig. S7D), and medulloblastoma cell growth (Fig. 7K) with ULK3 knockdown exhibiting stronger defects than STK36 knockdown. Combined knockdown of ULK3 and STK36 resulted in more dramatic defects (Fig. 7J–K; Fig. S7D). Overexpression of Gli2 transgenes rescued Shh target gene expression and medulloblastoma cell growth with Gli2DD acting more whereas Gli2AA less potently than Gli2WT (Fig. 7J–K; Fig. S7D). Taken together, these results suggest ULK3/STK36 promotes Gli2 phosphorylation and activation in SmoM2-induced medulloblastoma cells.
DISCUSSION
How Hh signaling converts Ci/Gli into its activator form has remained poorly understood. Here we reveal a conserved mechanism by which Hh stimulates Ci/Gli activation by inducing its phosphorylation through the Fu family kinases (Fig. 8). Furthermore, we show that phosphorylation of Ci/Gli contributes to the formation of CiA/GliA by attenuating Sufu binding, thus promoting Ci/Gli nuclear localization and coactivator binding.
Fig. 8. Gli phosphorylation code.

Ci and Gli2/3 are phosphorylated on distinct set of sites by different combination of kinases in the absence or presence of Hh, which generate CiR/GliR and CiA/GliA, respectively. Phosphorylation sites are color-coded to reflect their positive (green) or negative (red) influence on Hh pathway activity. See text for details.
Fu-mediated Ci phosphorylation contributes to CiA formation
Although Fu has been implicated as a critical mediator of CiA formation, the relevant phosphorylation event(s) that contributes to Ci activation has remained an enigma. Through MS mapping coupled with mutagenesis and functional analysis, we found that Fu phosphorylated Ci on two Ser/Thr clusters that are required for optimal Hh pathway activation. Using phospho-specific antibodies, we demonstrated that phosphorylation of these sites was induced by Hh. Furthermore, we demonstrated that Fu directly phosphorylated S218 and S1230, priming its further phosphorylation on S220 and T1232/S1233 by CK1. In addition, we found that an acidic residue at the +6 position is critical for Fu/ULK3 to efficiently phosphorylate their substrates. The identification of a phosphorylation consensus site for the Fu family kinases may facilitate the identification of physiologically relevant phosphorylation events mediated by these kinases.
We noticed that both Hh and constitutively active forms of Fu could further induce the activity of Ci−PKA_AA and Ci−PKA_DD, suggesting that Ci can be activated through additional mechanism(s) in parallel to C-I/C-II phosphorylation. There are several non-mutually exclusive possibilities. 1) Hh signaling could inhibit phosphorylation of Ci on PKA sites 4 and 5, as phosphorylation of these sites attenuates Ci activity independent of Ci processing (Price and Kalderon, 1999; Wang et al., 1999). 2) Hh signaling could induce Ci phosphorylation by CK1 on multiple HIB/SPOP degrons to protect CiA from premature degradation by Cul3HIB/SPOP (Shi et al., 2014b; Zhang et al., 2006). 3) Fu could activate Ci by phosphorylating additional sites on Ci that could have been missed by our MS analysis. 4) Fu could allosterically regulate Ci/Sufu interaction, e.g., by interacting with Sufu and/or Ci to change their conformations. Future work is needed to explore these possibilities.
ULK3/STK36-mediated phosphorylation and activation of Gli
How Hh converts GliF into GliA in mammals has remained a mystery given that STK36 knockout in mice did not cause any discernible Hh signaling defects during embryonic development. Here we found that Hh signaling stimulated phosphorylation of Gli proteins on a conserved Ser cluster, which promoted Gli2 activity by attenuating Sufu binding. Hh stimulated Gli2 phosphorylation on these sites depending on its ciliary localization and the Fu family kinases ULK3/STK36 in cultured cells. The involvement of ULK3 and perhaps other kinases could explain the lack of developmental defects associated with STK36 knockout. It is possible that the relative contribution of ULK3 and STK36 to Gli activation may vary depending on the cell types and developmental contexts. Double knockout of ULK3 and STK36 is needed to address their precise roles in mammalian Hh signaling during development and tissue homeostasis.
Ci/Gli phosphorylation code
Previous studies identified a set of phosphorylation sites in Ci/Gli that is essential for Ci/Gli processing to generate CiR/GliR (Chen and Jiang, 2013). Here, we identified a distinct set of phosphorylation sites in Ci/Gli essential for the formation of CiA/GliA. Hence, Ci/Gli undergoes two distinct sets of phosphorylation events that influence Ci/Gli activity in opposite directions depending the Hh signaling states (Fig. 8). In the “Hh signaling off” state, Ci/Gli undergoes extensive phosphorylation mediated by PKA, CK1 and GSK3, which targets it for Cul1Slimb/βTRCP-mediated ubiquitination, followed by proteasome-mediated proteolysis to generate CiR/GliR. In the “Hh signaling on” state, Ci/Gli undergoes phosphorylation mediated by Fu/ULK3/STK36 and CK1, which promotes Ci/Gli activation by antagonizing Sufu. Interestingly, we found that phosphorylation of Ci at C-I/C-II and Gli2 at S230/S232 increased progressively in response to increasing amounts of Hh, which correlated with progressively increased pathway activity, suggesting that the Hh signal gradient may generate a Ci/Gli phosphorylation gradient, which in turn contributes to the formation of a Ci/Gli activity gradient.
It is likely that ULK3/STK36 could phosphorylate additional sites, e.g., in the C-terminal region of Gli proteins to promote Gli activation. A recent study identified a set of Ser/Thr residues (Pc, Pc, Pe, Pf, and Pg) whose collected mutations attenuated Gli2 activation (Niewiadomski et al., 2014). We noticed that Pf corresponds to S230, which could explain why collective mutations of Pc-Pg reduced Gli2 activity. It remains to be determined whether phosphorylation of other sites in Pc-Pg, especially Pg/S248, whose phosphorylation appeared to be induced by SAG, also contributes to Gli2 activation.
Aberrant Hh pathway activation has been implicated in many types of human cancer and Smo inhibitors have entered the clinic to treat “Hh-driven” cancers such as advanced BCC; however, clinical trials on other types of cancer haven been disappointing (Wu et al., 2017). A commonly used biomarker for Hh pathway activation in cancer samples is Gli1 expression; however, Gli1 expression could also be activated by noncanonical mechanisms (Lauth and Toftgard, 2007). Gli phosphorylation detected by the pS230/232 antibody may reflect the canonical Hh pathway activation more faithfully than Gli1 expression and could be explored as a biomarker for selecting cancer patients who may benefit from “anti-Smo” therapies. In addition, our finding that inhibition of ULK3/STK36 blocks SmoM2-driven medulloblastoma cell growth suggests that the Fu-family kinases are potential drug targets for Hh-driven cancers.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Author, Dr. Jin Jiang (Jin.Jiang@UTSouthwestern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
S2 cells (male origin) were cultured in Drosophila SFM (Thermo Fisher Scientific) supplemented with 10% FBS (Millipore Sigma F4135) at 24 °C. Clone8 cells were cultured in Shields and Sang M3 Medium (Millipore Sigma) with 2.5% of FBS, 2.5% fly extract and 0.5mg/ml insulin (Millipore Sigma). Sf9 cells (female origin) were cultured in SF900III medium containing 10% FBS at 27 °C. NIH 3T3 cells were cultured in DMEM (Millipore Sigma) containing 10% BCS (ATCC). Gli3TAP MEF cells were isolated from P15.5 embryo of Gli3TAP mice. HEK293T and Gli3TAP MEF cells were cultured in DMEM containing 10% FBS (Millipore Sigma). For primary medulloblastoma cell culture, tumor cells from SmoM2, CAG-creER mice (Mao et al., 2006) with spontaneous occurring medulloblastoma were dissociated and cultured in DMEM/F12 medium containing 25mM KCl, N2 supplement (Invitrogen) and 10%FBS (Sigma Aldrich). Hh-conditioned medium was prepared by culturing S2 cells stably expressing Hh-N with 0.7mM CuSO4 induction for 1 day. The resultant medium was filtered and harvested. Hh-conditioned medium was used at 6:4 ratios with regular medium, which was defined as 100% in Figure 3.
Drosophila strains and husbandry
All Drosophila stocks were cross maintained and crossed at room temperature (~20 °C). Their progenies were grown at 18 or 25 °C as described in the text. Transgenic flies expressing Ci constructs were generated using the phiC31 integration system with the transgenes inserted into the 75A10 (for UAS-Myc-CiAA, UAS-Myc-CiAA, and UAS-Myc-CiDD) or 75B1 (for UAS-Myc-Ci-PKA_WT, UAS-Myc-Ci-PKA_AA, and UAS-Myc-Ci-PKA_DD) attP locus (Bischof et al., 2007; Jia et al., 2009). UAS-Myc-Ci-PKA was described previously (Han et al., 2015). Gal4 driver C765 (BL#36523) was used to drive the expression UAS transgenes. Virgin females of C765 were crossed with males expressing Ci constructs. Wing imaginal discs described in Fig. 2 and Fig. S3 were derived from both female and male late third instar larvae. Adult wings described in Fig. 2 were derived from females.
METHOD DETAILS
Plasmid Generation and Cell Transfection
Myc-Ci−PKA variants with point mutations in different phosphorylation sites (see supplemental Table 1) were generated using PCR-based site directed mutagenesis. N-terminally Flag-tagged wild type Sufu and its mutants, and Ci−PKA with three copies of Flag tag (Flag-Ci−PKA) were subcloned into the pUAST vector using NotI and KpnI sites. All baculoviral constructs were made with pFastBac1 as the backbone using EcoRI and Kpn I sites. All GST-fusion proteins were subcloned into the pGEX 4T-1 vector using EcoRI and XhoI sites. N-terminally 6×Myc-tagged mouse Gli2WT (Myc-Gli2WT), Gli2AA (Myc-Gli2AA), and Gli2DD (Myc-Gli2DD), along with N-terminally 3×HA-tagged mouse Ulk3WT (HA-ULK3WT), Ulk3KR (HA-ULK3KR), CC-Ulk3WT(HA-CC-ULK3WT), human Stk36WT (HA-Stk36WT) and Stk36KR (HA-Stk36KR) were subcloned into pcDNA3.1(+) vector using EcoRI and XbaI sites or into the lentiviral vector, FUXW, using BamHI and XbaI sites. sgRNAs targeting Ulk3 and Stk36 were cloned into the Lentiguide-Puro plasmid at the BsmBI site. FuINS constructs were made by replacing the coding sequence targeted by Fu dsRNA with a synthesized DNA fragment with alternative codon for the same primary sequence (Genescript, NJ). Please see Supplemental Table 3 for the full list of plasmids used in this study. Transfection of S2 cells was performed using Calcium Phosphate Transfection Kit (Specialty Media) following the instructions of the manufacturer. NIH 3T3 and HEK293T cells were transfected using PolyJet in vitro DNA transfection kit (SignaGen). For Baculovirus production, sf9 cells were transfected with Cellfetin II Reagent (Thermo Fisher Scientific).
Protein Expression, Purification and Mass Spec
Baculovirus was prepared following manufacturer’s instruction (Bac to Bac Baculovirus Expression System, Thermo Fisher Scientific). Sf9 cells were infected with corresponding baculovirus at the density of 1–2×106/ml, and cells were collected 2 days after the infection. In order to purify Flag-Ci, resultant cell lysate were incubated with Flag M2 affinity agarose (Millipore Sigma) and bound protein was eluted by 2× SDS loading buffer. The elute was further applied onto Superdex 200 10/300 GL column and Ci containing fractions were collected, lyophilized, re-dissolved, and separated on SDS-PAGE gel. After coomassie blue staining, Ci band was isolated for post-translational modification analysis. In case of Sufu purification, only one-step Flag affinity purification was performed. For mass spec analysis, standard tryptic digestion was conducted and the resultant samples was run on Orbitrap Fusion Lumos mass-spectrometry platform, using a short reverse-phase LC-MS/MS method. The putative post-translational modification was identified using Proteome Discoverer 2.2.
Flag-CC-FuEE and FuGV expressed in Sf9 cells using the baculovirus system were purified by Flag M2 affinity agarose and eluted with 200ug/ml 3×Flag peptide. The purity of Sf9-derived Fu kinase was confirmed by SDS-PAGE and coomassie blue staining. Only one major band was detected and at least 95% purity was achieved. HA-mUlk3WT and HA-mUlk3KR were expressed in HEK293T cells by transient transfection, purified with Pierce anti-HA agarose (Thermo Fisher Scientific), and eluted with 250ug/ml HA peptide. GST and GST-fusion proteins were expressed in BL21 with IPTG (0.1mM) induction at 30°C for 3–4 hours, purified with Glutathione Sepharose 4B (GE Healthcare), and eluted with 10mM reduced Glutathione.
Phospho Antibodies Generation
Phospho-Ci antibodies were made by Abmart (Shanghai, China) using the following phospho-peptides as antigens: RKRALS(p)SS(p)PYSDS for pS218/220, and QIIDSS(p)MTS(p)LPEL and QIIDSSMT(p)SLPEL for pS1230/1233 and PT1232, respectively. Phospho-mGli2 antibody was made by Genemed Synthesis (San Antonio, Tx) using RKRALS(p)IS(p)PLSDA as the antigen. The phospho-specific antibodies were purified by positive and negative selection using affinity column conjugated with the corresponding phosphorylated and non-phosphorylated peptides sequentially.
In vitro Kinase Assay
In vitro kinase assay was conducted by incubating reaction mixture (25uL) containing 150mM Tris-HCl pH7.5, 0.2mM Mg2+/ATP, 1ug substrate (GST/GST fusion proteins) and appropriate amount of protein kinase at 30 °C for 30 mins. In the case of radioactive kinase assay, 1uL of γ−32P ATP (Perkin Elmer) was added. The reaction was terminated by adding 2× SDS loading buffer. The resultant samples were load on SDS-PAGE and subject to further analysis by autoradiography, immunoblots with phospho-specific antibodies, or pIMAGO with fluor-680 detection kit and LI-COR Odyssey platform for western blot analyses of phosphorylated proteins (Millipore Sigma). In the case of sequential in vitro kinase assay, glutathione beads pre-absorbed with GST or GST-fusion proteins were used; after the first round of kinase assay, glutathione beads were centrifuged and thoroughly washed by 1× kinase assay buffer 3 times. And these beads were used as substrates for the second of kinase assay.
Dual Luciferase Assay
S2 or Cl8 cells were seeded in 12-well plate at the concentration of 1×106/ml and the next day each well was transfected with 0.5 μg ptc-Luc reporter vector, 25ng RL-PolIII internal control vector as well as 0.5 μg of each other construct. Cells were collected 48 hours after the transfection. For Gli-luc reporter assay, NIH 3T3 cells were seeded in 12-well plate and transfected with 0.4 μg 8×Gli-Luc reporter vector and 0.1 μg PRL-SV40 Renilla vector, together with 0.5 μg each other construct were transfected. The luciferase activity was determined using dual-luciferase reporter assay system (Promega) and FLUOstar OPTIMA (BMG Labtech).
RNA Isolation and RT-qPCR
For RT-qPCR with cell samples, total RNA was extracted from cells using RNeasy Plus Mini Kit (Qiagen), cDNA was synthesized with High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and qPCR was performed using Fast SYBR Green Master Mix (Applied Biosystems) and a Bio-rad CFX96 real-time PCR system. GADPH expression level was used as a normalization control. Please see Supplemental Table 2 for primer sequences.
FRET Analysis
CFP/YFP-tagged constructs were transfected into S2 cells together with an ub-Gal4 expression vector. Cells were washed with PBS, fixed with 4% formaldehyde for 20 minutes and mounted on slides in 80% glycerol. CFP signals were acquired with 100× objective of Zeiss LSM710 confocal microscope before (BP) and after (AP) photobleaching YFP. Each data set was calculated using 10–20 individual cells. YFP was photobleached with full power of the 514 nM laser line for 1–2 min at the top half of the cells, leaving the bottom half as an internal control. The intensities of CFP signals were quantified by ImageJ software. The FRET efficiency was calculated using the formula: FRET%=[(CFPAP-CFPBP)/CFPAP] x100.
Lentivirus Production
HEK293T/17 (ATCC) cells were seeded and transfected with psPAX2 and pM2.G, along with pLKO.1-puro (for shRNA lentivirus) or FUXW (for protein expressing lentivirus). After 48 hours, virus containing culture media was collected, filtered and centrifuged at 20,000g for 2 hours; the resultant precipitant was re-suspended in small volume of culture medium and stored at −80°C for future use. All shRNA lentiviral vectors were purchased from Millipore Sigma.
CRISPR Clonal Cell Generation
NIH 3T3 cells were first transduced by lentivirus expressing a Flag-tagged Cas9 (LentiCas9-Blast) and selected with 10ug/ml blasticidin (Millipore Sigma). The expression of Cas9 was further confirmed by immunoblot analysis of the resultant cell lines. For single gene manipulation, the above Cas9-expressing cells were infected by lentivirus expressing sgRNAs (Lentiguide-Puro) targeting Ulk3 or Stk36 (see Supplemental Table 2 for sgRNA sequences). After selection with 5ug/ml Puromycin for 1–2 weeks, emerging cell colonies were picked and screened for SAG-mediated Gli expression by RT-qPCR. Individual cell lines exhibiting the worst response to SAG treatment were frozen for further studies. For double gene manipulation, cell lines edited by Ulk3 sgRNAs were further infected by lentivirus expressing sgRNAs targeting Stk36.
Double-Stranded RNA (dsRNA)
The double-stranded RNAs (dsRNAs) were generated using the MEGAscript High Yield Transcription Kit (Ambion). Please see Supplemental Table 2 for primer sequences. For transfection, cells (S2 or Cl8) were seeded at the density of 1×106/ml with serum-free medium; 10–20ug of dsRNA was added for each 10cm plate and the cells were further incubated for 2–3 hours before switching to regular growth medium.
Small Interfering RNA (siRNA)
All siRNAs are purchased from Millipore Sigma and transfected using Lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific) at the concentration of 20nM.
Please see Supplemental Table 2 for siRNA sequences.
Western Blot Quantification
Western blot quantification was performed by using appropriate fluorogenic secondary antibody and the blot was scanned and quantified with an Odyssey CLx imaging system (LI-COR).
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments were carried out independently two to three times as described in figure legends. All quantification data were analyzed by two-tailed Student’s t-test. P-values are described in figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse Anti-Flag antibody | Millipore Sigma | Clone ID: M2; Cat#1804; RRID: AB_439685 |
| Mouse Anti-HA (F-7) antibody | Santa Cruz Biotechnology | Cat#sc-7392; RRID: AB_627809 |
| Mouse Anti-Myc antibody | Santa Cruz Biotechnology | Clone ID: 9E10; Cat#sc-40; RRID: AB_627268 |
| Rabbit Anti-Myc antibody | Abcam | Cat#ab9106; RRID: AB_307014 |
| Rat Anti-Ci antibody | DSHB | Cat#2A1; RRID: AB_2109711 |
| Mouse Anti-Fu antibody | DSHB | Cat#22F10; RRID: AB_528258 |
| Goat Anti-PKC ζ antibody | Santa Cruz Biotechnology | Clone ID: c-20; Cat#sc-216; RRID: AB_2300359 |
| Mouse Anti-α-tubulin antibody | Millipore Sigma | Clone ID: DM1A; Cat#T9026; RRID: AB_477593 |
| Mouse Anti-GFP antibody | Takara Biol | Cat#632380; RRID: AB_10013427 |
| Mouse Anti-acetylated Tubulin antibody | Millipore Sigma | Clone ID:6-11b1; Cat#T7451; RRID: AB_609894 |
| Goat Anti-Gli2 antibody | R&D Systems | Cat#AF3635; RRID: AB_2111902 |
| Goat Anti-Gli3 antibody | R&D Systems | Cat#AF3690; RRID: AB_2232499 |
| Bacterial and Virus Strains | ||
| NEB5-Alpha Competent E. Coli | New England Biolabs | Cat#C29871 |
| BL21 Competent E.Coli | New England Biolabs | Cat#C2530H |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Leptomycin B | Millipore Sigma | Cat#L2913 |
| Acti-Stain 670 phalloidin | Cytoskeleton | Cat#PHDN1-A |
| Bluo-Gal | Thermo Fisher Scientific | Cat#15519028 |
| L-Glutathione Reduced | Millipore Sigma | Cat#G4251 |
| SAG | Millipore Sigma | Cat#566660 |
| DAPI | Millipore Sigma | Cat#D9542 |
| ATP [γ −32P] | Perkin Elmer | Cat#BLU002A |
| Insulin | Millipore Sigma | Cat#I6634 |
| Fetal Bovine Serum | Millipore Sigma | Cat#F4135 |
| Fetal Bovine Serum | Millipore Sigma | Cat#F0926 |
| Calf Bovine Serum Iron Fortified | ATCC | Cat#ATCC30-2030 |
| 3× Flag Peptide | Millipore Sigma | Cat#F4799 |
| HA Synthetic Peptide | Thermo Fisher Scientific | Cat#26184 |
| Casein Kinase I | New England Biolabs | Cat#P6030 |
| Recombinant Human Sonic Hedgehog Protein (Shh-N) | R&D Systems | Cat#8908-HSH/CF |
| Lambda Protein Phosphatase | New England Biolabs | Cat#P0753 |
| Critical Commercial Assays | ||
| Dual Luciferase Reporter Assay System | Promega | Cat#E1910 |
| MEGAscript T7 High Yield Transcription Kit | Thermo Fisher Scientific | Cat#AM1334 |
| pIMAGO-biotin Phosphoprotein Detection Kit | Millipore Sigma | Cat#67221 |
| Anti-Flag M2 Affinity Gel | Millipore Sigma | Cat#A2220 |
| Pierce Anti-HA Argarose | Thermo Fisher Scientific | Cat#26181 |
| Glutathione Sepharose 4B | GE Healthcare | Cat#17075601 |
| SimplyBlue SafeStain | Thermo Fisher Scientific | Cat#LC6060 |
| RNeasy Plus Mini Kit | Qiagen | Cat#74134 |
| High Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368814 |
| Fast SYBR Green Master Mix | Applied Biosystems | Cat#4385612 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat#G7572 |
| Experimental Models: Cell Lines | ||
| Mouse: NIH/3T3 | ATCC | Cat#ATCC CRL-1658 |
| Human: HEK293T/17 | ATCC | Cat#ATCC CRL-11268 |
| D. melanogaster. S2 | Thermo Fisher Scientific | Cat#R69007 |
| D. melanogaster. S2R+ | DGRC | Cat#150 |
| D. melanogaster. Clone 8 | DGRC | Cat#151 |
| S. frugiperda: Sf9 | Thermo Fisher Scientific | Cat#11496015 |
| Experimental Models: Organisms/Strains | ||
| Mouse: CAGGS-CreER; R26-SmoM2 | Mao et al., 2006 | N/A |
| Mouse: Gli3TAP Knock-In | Zhang et al., 2019 | N/A |
| D. melanogaster. UAS-Cr-PKA-WT | Han et al., 2015 | N/A |
| D. melanogaster. UAS-C-PKA-AA | This Paper | N/A |
| D. melanogaster. UAS-C-PKA-DD | This Paper | N/A |
| D. melanogaster. UAS-CiWT | This Paper | N/A |
| D. melanogaster. UAS-CiAA | This Paper | N/A |
| D. melanogaster. UAS-CiDD | This Paper | N/A |
|
D. melanogaster. C765-Gla4 y1 w*; P{GawB}C-765 |
Bloomington Drosophila Stock Center | FBti0002765 |
| Oligonucleotides | ||
| Please see Supplemental Table 2 for oligo/primer sequences | N/A | N/A |
| Recombinant DNA | ||
| Please see Supplemental Table 3 for the list of plasmids used in this study | N/A | N/A |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
Highlights.
Hh activates Ci by stimulating its phosphorylation through Fu
Fu directly phosphorylates Ci and primes its further phosphorylation by CK1
Fu-mediated phosphorylation of Ci recruits Trn and CBP by antagonizing Sufu
Shh activates Gli2 by inducing its phosphorylation via ULK3 and STK36
ACKNOWLEDGEMENT
We thank Dr. Xiaochun Li for Shh-N and DSHB for antibodies. This work was supported by grants from NIGMS (GM118063) and Welch Foundation (I-1603) to Jin Jiang (J.J.). J.J. is a Eugene McDermott Endowed Scholar in Biomedical Science at UTSW.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCE
- Aza-Blanc P, Ramirez-Weber F, Laget M, Schwartz C, and Kornberg T (1997). Proteolysis that is inhibited by Hedgehog targets Cubitus interruptus protein to the nucleus and converts it to a repressor. Cell 89, 1043–1053. [DOI] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, and Basler K (2007). An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci U S A 104, 3312–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe J, and Therond PP (2013). The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol 14, 418–431. [DOI] [PubMed] [Google Scholar]
- Chen CH, von Kessler DP, Park W, Wang B, Ma Y, and Beachy PA (1999). Nuclear trafficking of Cubitus interruptus in the transcriptional regulation of Hedgehog target gene expression. Cell 98, 305–316. [DOI] [PubMed] [Google Scholar]
- Chen MH, Gao N, Kawakami T, and Chuang PT (2005). Mice deficient in the fused homolog do not exhibit phenotypes indicative of perturbed hedgehog signaling during embryonic development. Mol Cell Biol 25, 7042–7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MH, Wilson CW, Li YJ, Law KK, Lu CS, Gacayan R, Zhang X, Hui CC, and Chuang PT (2009). Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev 23, 1910–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, and Jiang J (2013). Decoding the phosphorylation code in Hedgehog signal transduction. Cell Res 23, 186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Li S, Tong C, Zhao Y, Wang B, Liu Y, Jia J, and Jiang J (2010). G protein-coupled receptor kinase 2 promotes high-level Hedgehog signaling by regulating the active state of Smo through kinase-dependent and kinase-independent mechanisms in Drosophila. Genes Dev 24, 2054–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Sasai N, Ma G, Yue T, Jia J, Briscoe J, and Jiang J (2011). Sonic Hedgehog dependent phosphorylation by CK1alpha and GRK2 is required for ciliary accumulation and activation of smoothened. PLoS Biol 9, e1001083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy JB (2002). GAL4 system in Drosophila: a fly geneticist’s Swiss army knife. Genesis 34, 1–15. [DOI] [PubMed] [Google Scholar]
- Han Y, Shi Q, and Jiang J (2015). Multisite interaction with Sufu regulates Ci/Gli activity through distinct mechanisms in Hh signal transduction. Proc Natl Acad Sci U S A 112, 6383–6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Xiong Y, Shi X, Wu J, Zhao Y, and Jiang J (2017). Regulation of Gli ciliary localization and Hedgehog signaling by the PY-NLS/karyopherin-beta2 nuclear import system. PLoS Biol 15, e2002063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, and Yoder BK (2005). Gli2 and gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet 1, e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humke EW, Dorn KV, Milenkovic L, Scott MP, and Rohatgi R (2010). The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev 24, 670–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H, Liu Y, Yan W, and Jia J (2009). PP4 and PP2A regulate Hedgehog signaling by controlling Smo and Ci phosphorylation. Development 136, 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Tong C, Wang B, Luo L, and Jiang J (2004). Hedgehog signalling activity of Smoothened requires phosphorylation by protein kinase A and casein kinase I. Nature 432, 1045–1050. [DOI] [PubMed] [Google Scholar]
- Jiang J, and Hui CC (2008). Hedgehog signaling in development and cancer. Dev Cell 15, 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, and Struhl G (1995). Protein kinase A and hedgehog signaling in Drosophila limb development. Cell 80, 563–572. [DOI] [PubMed] [Google Scholar]
- Jiang J, and Struhl G (1998). Regulation of the Hedgehog and Wingless signalling pathways by the F-box/WD40-repeat protein Slimb. Nature 391, 493–496. [DOI] [PubMed] [Google Scholar]
- Knippschild U, Gocht A, Wolff S, Huber N, Lohler J, and Stoter M (2005). The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell Signal 17, 675–689. [DOI] [PubMed] [Google Scholar]
- Lauth M, and Toftgard R (2007). Non-canonical activation of GLI transcription factors: implications for targeted anti-cancer therapy. Cell cycle 6, 2458–2463. [DOI] [PubMed] [Google Scholar]
- Li S, Li S, Han Y, Tong C, Wang B, Chen Y, and Jiang J (2016). Regulation of Smoothened Phosphorylation and High-Level Hedgehog Signaling Activity by a Plasma Membrane Associated Kinase. PLoS Biol 14, e1002481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Ma G, Wang B, and Jiang J (2014). Hedgehog induces formation of PKA-Smoothened complexes to promote Smoothened phosphorylation and pathway activation. Sci Signal 7, ra62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum L, Zhang C, Oh S, Mann RK, von Kessler DP, Taipale J, Weis-Garcia F, Gong R, Wang B, and Beachy PA (2003). Hedgehog signal transduction via Smoothened association with a cytoplasmic complex scaffolded by the atypical kinesin, Costal-2. Mol Cell 12, 1261–1274. [DOI] [PubMed] [Google Scholar]
- Ma G, Li S, Han Y, Li S, Yue T, Wang B, and Jiang J (2016). Regulation of Smoothened Trafficking and Hedgehog Signaling by the SUMO Pathway. Dev Cell 39, 438–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloverjan A, and Piirsoo M (2012). Mammalian homologues of Drosophila fused kinase. Vitam Horm 88, 91–113. [DOI] [PubMed] [Google Scholar]
- Maloverjan A, Piirsoo M, Michelson P, Kogerman P, and Osterlund T (2010). Identification of a novel serine/threonine kinase ULK3 as a positive regulator of Hedgehog pathway. Exp Cell Res 316, 627–637. [DOI] [PubMed] [Google Scholar]
- Mao J, Ligon KL, Rakhlin EY, Thayer SP, Bronson RT, Rowitch D, and McMahon AP (2006). A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res 66, 10171–10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant M, Evangelista M, Luoh SM, Frantz GD, Chalasani S, Carano RA, van Hoy M, Ramirez J, Ogasawara AK, McFarland LM, et al. (2005). Loss of the serine/threonine kinase fused results in postnatal growth defects and lethality due to progressive hydrocephalus. Mol Cell Biol 25, 7054–7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot N, and Basler K (1999). Hedgehog controls limb development by regulating the activities of distinct transcriptional activator and repressor forms of Cubitus interruptus. Cell 96, 819–831. [DOI] [PubMed] [Google Scholar]
- Murone M, Luoh SM, Stone D, Li W, Gurney A, Armanini M, Grey C, Rosenthal A, and de Sauvage FJ (2000). Gli regulation by the opposing activities of fused and suppressor of fused. Nat Cell Biol 2, 310–312. [DOI] [PubMed] [Google Scholar]
- Nieuwenhuis E, and Hui CC (2005). Hedgehog signaling and congenital malformations. Clin Genet 67, 193–208. [DOI] [PubMed] [Google Scholar]
- Niewiadomski P, Kong JH, Ahrends R, Ma Y, Humke EW, Khan S, Teruel MN, Novitch BG, and Rohatgi R (2014). Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep 6, 168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nybakken K, Vokes SA, Lin TY, McMahon AP, and Perrimon N (2005). A genome-wide RNA interference screen in Drosophila melanogaster cells for new components of the Hh signaling pathway. Nat Genet 37, 1323–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nybakken KE, Turck CW, Robbins DJ, and Bishop JM (2002). Hedgehog-stimulated phosphorylation of the kinesin-related protein Costal2 is mediated by the serine/threonine kinase fused. J Biol Chem 277, 24638–24647. [DOI] [PubMed] [Google Scholar]
- Ohlmeyer JT, and Kalderon D (1998). Hedgehog stimulates maturation of Cubitus interruptus into a labile transcriptional activator. Nature 396, 749–753. [DOI] [PubMed] [Google Scholar]
- Preat T, Therond P, Limbourg-Bouchon B, Pham A, Tricoire H, Busson D, and Lamour-Isnard C (1993). Segmental polarity in Drosophila melanogaster: genetic dissection of fused in a Suppressor of fused background reveals interaction with costal-2. Genetics 135, 1047–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MA, and Kalderon D (1999). Proteolysis of cubitus interruptus in Drosophila requires phosphorylation by protein kinase A. Development 126, 4331–4339. [DOI] [PubMed] [Google Scholar]
- Ranieri N, Ruel L, Gallet A, Raisin S, and Therond PP (2012). Distinct phosphorylations on kinesin costal-2 mediate differential hedgehog signaling strength. Developmental cell 22, 279–294. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H, Hui C, Nakafuku M, and Kondoh H (1997). A binding site for Gli proteins is essential for HNF-3beta floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development 124, 1313–1322. [DOI] [PubMed] [Google Scholar]
- Shi Q, Han Y, and Jiang J (2014a). Suppressor of fused impedes Ci/Gli nuclear import by opposing Trn/Kapbeta2 in Hedgehog signaling. J Cell Sci 127, 1092–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Li S, Jia J, and Jiang J (2011). The Hedgehog-induced Smoothened conformational switch assembles a signaling complex that activates Fused by promoting its dimerization and phosphorylation. Development 138, 4219–4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Li S, Li S, Jiang A, Chen Y, and Jiang J (2014b). Hedgehog-induced phosphorylation by CK1 sustains the activity of Ci/Gli activator. Proc Natl Acad Sci U S A 111, E5651–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Zhang Z, Zhan X, Cao M, Satoh T, Akira S, Shpargel K, Magnuson T, Li Q, Wang R, et al. (2014c). An epigenetic switch induced by Shh signalling regulates gene activation during development and medulloblastoma growth. Nat Commun 5, 5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strigini M, and Cohen SM (1997). A Hedgehog activity gradient contributes to AP axial patterning of the Drosophila wing. Development 124, 4697–4705. [DOI] [PubMed] [Google Scholar]
- Svard J, Heby-Henricson K, Persson-Lek M, Rozell B, Lauth M, Bergstrom A, Ericson J, Toftgard R, and Teglund S (2006). Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev Cell 10, 187–197. [DOI] [PubMed] [Google Scholar]
- Wang B, Fallon JF, and Beachy PA (2000a). Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100, 423–434. [DOI] [PubMed] [Google Scholar]
- Wang G, Amanai K, Wang B, and Jiang J (2000b). Interactions with Costal2 and suppressor of fused regulate nuclear translocation and activity of cubitus interruptus. Genes Dev 14, 2893–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, and Jiang J (2004). Multiple Cos2/Ci interactions regulate Ci subcellular localization through microtubule dependent and independent mechanisms. Dev Biol 268, 493–505. [DOI] [PubMed] [Google Scholar]
- Wang G, Wang B, and Jiang J (1999). Protein kinase A antagonizes Hedgehog signaling by regulating both the activator and repressor forms of Cubitus interruptus. Genes Dev 13, 2828–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QT, and Holmgren RA (2000). Nuclear import of cubitus interruptus is regulated by hedgehog via a mechanism distinct from Ci stabilization and Ci activation. Development 127, 3131–3139. [DOI] [PubMed] [Google Scholar]
- Wilson CW, Nguyen CT, Chen MH, Yang JH, Gacayan R, Huang J, Chen JN, and Chuang PT (2009). Fused has evolved divergent roles in vertebrate Hedgehog signalling and motile ciliogenesis. Nature 459, 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Zhang Y, Sun B, McMahon AP, and Wang Y (2017). Hedgehog Signaling: From Basic Biology to Cancer Therapy. Cell Chem Biol 24, 252–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Murone M, Luoh S-M, Ryan A, Gu Q, Zhang C, Bonifas JM, Lam C-W, Hynes M, Goddard A, et al. (1998). Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 391, 90–92. [DOI] [PubMed] [Google Scholar]
- Yin WC, Satkunendran T, Mo R, Morrissy S, Zhang X, Huang ES, Uuskula-Reimand L, Hou H, Son JE, Liu W, et al. (2019). Dual Regulatory Functions of SUFU and Targetome of GLI2 in SHH Subgroup Medulloblastoma. Dev Cell 48, 167–183e165. [DOI] [PubMed] [Google Scholar]
- Zadorozny EV, Little JC, and Kalderon D (2015). Contributions of Costal 2-Fused interactions to Hedgehog signaling in Drosophila. Development 142, 931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Shi Q, Chen Y, Yue T, Li S, Wang B, and Jiang J (2009). Multiple Ser/Thr-rich degrons mediate the degradation of Ci/Gli by the Cul3-HIB/SPOP E3 ubiquitin ligase. Proceedings of the National Academy of Sciences of the United States of America 106, 21191–21196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Zhang L, Wang B, Ou CY, Chien CT, and Jiang J (2006). A hedgehog-induced BTB protein modulates hedgehog signaling by degrading Ci/Gli transcription factor. Dev Cell 10, 719–729. [DOI] [PubMed] [Google Scholar]
- Zhang W, Zhao Y, Tong C, Wang G, Wang B, Jia J, and Jiang J (2005). Hedgehog-regulated costal2-kinase complexes control phosphorylation and proteolytic processing of cubitus interruptus. Dev Cell 8, 267–278. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Zhan X, Kim B, and Wu J (2019). A proteomic approach identifies SAFB-like transcription modulator (SLTM) as a bidirectional regulator of GLI family zinc finger transcription factors. J Biol Chem 294, 5549–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Tong C, and Jiang J (2007). Hedgehog regulates smoothened activity by inducing a conformational switch. Nature 450, 252–258. [DOI] [PubMed] [Google Scholar]
- Zhou Q, and Kalderon D (2011). Hedgehog Activates Fused through Phosphorylation to Elicit a Full Spectrum of Pathway Responses. Dev Cell 20, 802–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.