

Summary
Properly timed production of steroid hormones by endocrine tissues regulates juvenile-toadult transitions in both mammals (puberty) and holometabolous insects (metamorphosis). Nutritional conditions influence the temporal control of the transition, but the mechanisms responsible are ill-defined. Here we demonstrate that autophagy acts as an endocrine organ-specific, nutritionally-regulated, gating mechanism to help ensure productive metamorphosis in Drosophila. Autophagy in the endocrine organ is specifically stimulated by nutrient restriction at the early, but not the late third instar larva stage. The timing of autophagy induction correlates with the nutritional checkpoints which inhibit precocious metamorphosis during nutrient restriction in undersized larvae. Suppression of autophagy causes dysregulated pupariation of starved larvae which leads to pupal lethality, while forced autophagy induction results in developmental delay/arrest in well fed animals. Induction of autophagy disrupts production of the steroid hormone ecdysone at the time of pupariation not by destruction of hormone biosynthetic capacity, but rather by limiting the availability of the steroid hormone precursor cholesterol in the endocrine cells via a lipophagy mechanism. Interestingly, autophagy in the endocrine organ functions by interacting with endo/lysosome system, yet shows multiple features not fully consistent with a canonical autophagy process. Taken together, our findings demonstrate an autophagy mechanism in endocrine cells that helps shape the nutritional checkpoints and guarantee a successful juvenile-to-adult transition in animals confronting nutritional stress.
eTOC Blurb
Pan et al. identify a temporally-regulated, endocrine organ-specific autophagy process that prevents premature pupariation of Drosophila larva under nutritional stress. Autophagy limits cholesterol from entering the steroid hormone biosynthetic pathway and may be a general stress response mechanism controlling hormone production in other organisms.
Graphical Abstract
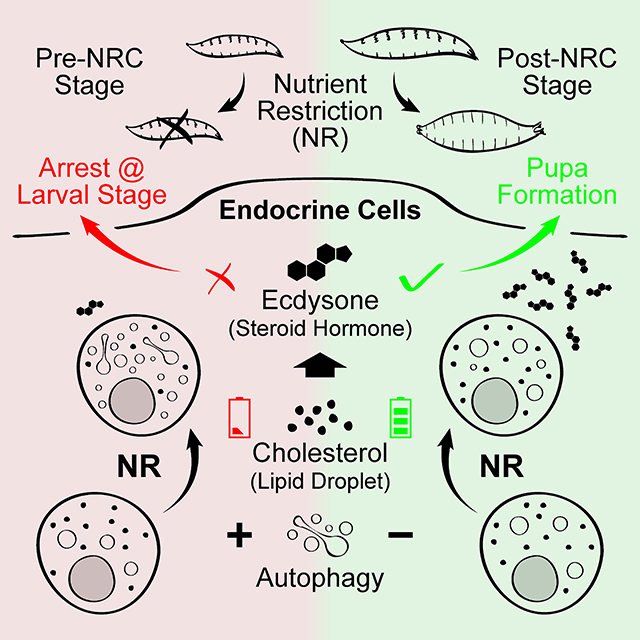
Introduction
During the juvenile stage, Drosophila larvae monitor internal and external cues to properly time the onset of metamorphosis. For example, among external cues, avoidance of light at the time of pupariation allows pupae to elude predation and diminish dehydration during this immobile phase of development [1]. Likewise, the monitoring of internal organ health and the ability to slow larval development in response to tissue insult is advantageous since it provides an opportunity to repair tissue damage before committing to irreversible differentiation during metamorphosis [2–6]. Together these monitoring systems help ensure a high probability of healthy adult eclosion.
Nutrition is another key factor that requires continuous assessment during the larval stages to ensure that sufficient nutrient stores are acquired before the cessation of feeding at the onset of metamorphosis. In Lepidoptera, two pre-metamorphosis nutritional checkpoints have been identified: critical weight is defined as the weight at which nutrient restriction (NR) no longer delays pupariation, while minimal viable weight is the minimal larval mass that is required for metamorphosis to occur [7]. In Drosophila, these two checkpoints occur at the same weight [7], but can be separated at certain circumstances [8, 9]. In this work, we refer to the two checkpoints collectively as the nutrient restriction checkpoints (NRCs), only distinguishing them from each other as required for additional clarity. By inhibiting pupariation of underweight larvae the NRCs help prevent a precocious transition that would otherwise lead to pupal lethality due to insufficient energy storage. After surpassing the checkpoints, the larvae actually slightly accelerate development during NR (Figure 1A) [7], perhaps providing an adaptive advantage when food sources become limiting.
Figure 1. Autophagy in the PG is temporally regulated during the L3 stage.

Schematic diagram showing the metamorphic timing response of larvae under NR stress. (B) Autophagy visualized by mCh-Atg8a in PG cells. Larvae were dissected and imaged immediately for fed groups, or after 4 hours starvation for NR groups. Magnified images of the indicated areas are also shown. Scale bar, 10 μm. (C and D) Quantification of the number (C) and the total area (D) of Atg8a-positive puncta per unit PG cell area (100 μm2) of fed and starved larvae. Mean ± SEM; p values by unpaired t-test (n=8; ns, not significant, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ##p<0.01, ####p<0.0001). (E and F) Autophagy visualized by mCh-Atg8a in pre-NRC (4 hours AL3E) phm>yw, phm>Atg1RNAi, phm>Atg9RNAi and phm>RhebAV4 PG tissues. (G) Quantification of the number of Atg8a-positive puncta per unit cell area (100 μm2) in the Atg gene knockdown and Rheb overexpression PGs. Mean ± SEM. (n=3–5).
Induction of macroautophagy (hereafter referred to as autophagy) is a common response in many tissues to NR which leads to degradation of cellular organelles and macromolecules into their constituent components, thereby helping satisfy nutrient and energy demands during NR [10]. This aspect of autophagy likely plays important roles during development. For example, autophagy aids in mobilization of nutrient stores to fuel the metamorphic remodeling process during the non-feeding pupal stage [11]. Given that determination of the NRCs involves starvation, the mechanistic output triggered by the checkpoints could involve weight- or temporally-regulated autophagy in the prothoracic gland (PG), the major steroid hormone-producing organ in larvae. In this model, we hypothesize that induction of autophagy prior to, but not after, the NRCs could limit hormone production and thereby slow development, allowing larvae more time to search for the additional nutrients needed to pass through the NRCs.
Here we confirm this model and show that autophagy is induced by NR in Drosophila PG cells specifically in the early, but not the late third instar (L3) stage, which suppresses ecdysone synthesis to prevent precocious pupariation during the pre-NRC stage. Furthermore, induction of autophagy disrupts ecdysone production by limiting the availability of the steroid hormone precursor cholesterol through interactions with endo/lysosome system. We also observed several non-conventional morphological features of the autophagy process in PG cells. These findings suggest that there may be a specialized autophagic machinery in steroidogenic cells that is specifically devoted to regulating cholesterol trafficking in response to nutritional stress. Given that several studies have suggested a role for autophagy in altering hormone synthesis in mammalian endocrine organs [12], our findings may be applicable in other species as a means of controlling hormone production.
Results
Autophagy in the PG is temporally regulated during the L3 stage
To test our hypothesis concerning temporally regulated autophagy induction, we monitored the NR induced formation of mCherry (mCh)-Atg8a labeled autophagosomes and autolysosomes in the PG at various time points pre- and post-NRC. In fed condition, a small number of Atg8a-positive vesicles were observed at all time points, suggesting an ongoing basal level of autophagy in the PG throughout the L3 stage (Figure 1B). Upon starvation both the number and total area of Atg8a-positive vesicles increased significantly in early L3 (6 hours after L3 ecdysis, AL3E) larvae (Figure 1B–1D). However, this effect of starvation on Atg8a-positive vesicles was significantly reduced at 12 hours AL3E and was further diminished as larvae grew into the mid L3 stage (24 hours AL3E) (Figure 1B–1D).
To test whether the changes on Atg8a-positive vesicles result from autophagy induction or blockage of downstream autophagic flux, we tested autophagy mediated degradation by GFP-Ref(2)P [13]. Ref(2)P-positive protein aggregates were observed in fed larvae, which partially colocalized with Atg8a-positive autophagosomes (Figure S1A). Upon starvation, the number of Ref(2)P-positive structures did not change significantly but the size of these structures became markedly smaller (Figure S1A–S1C). These results show that protein degradation occurs normally during starvation and suggest that the changes on Atg8a-positive vesicles in PG cells represents an increased level of autophagy induction rather than blocked degradation.
The timing of the loss of autophagy induction by starvation correlates with the NRCs, i.e. starvation pre-NRCs strongly induces autophagy while starvation post-NRCs has a much weaker effect [7]. This observation is consistent with our hypothesis that a temporally-regulated autophagy switch in PG cells contributes to suppressing the ability of larvae to pupariate before the NRCs are surpassed.
Autophagy induction in the PG requires canonical autophagy pathway components
To determine whether autophagy induction in the PG requires the known components of autophagy machinery, we knocked down various Atg genes in the PG using available RNAi reagents (Figure 1E and S1D-S1H). We found that autophagy induction was significantly reduced by knocking down components of the autophagy initiation complex (Atg1, Atg17 and Atg101), the membrane expansion complex (Pi3K59F) and the membrane recycling system (Atg2, Atg9 and Atg18) [14] (Figure 1E, 1G and S1D-S1F). In addition, activation of the Target of Rapamycin (TOR) pathway by overexpressing Rheb (phm>Rheb) also effectively inhibited autophagy (Figure 1F and 1G). These findings indicate that the autophagy in the PG employs similar machinery and upstream control mechanisms to those found in canonical autophagy in other tissues [15, 16].
Autophagy inhibition in the PG suppresses the NR checkpoints
To determine the effect of autophagy induction on pupariation control, we first tested pupariation activity of larvae when autophagy was suppressed in the PG. In fed animals, autophagy suppression by phm>Atg1RNAi, phm>Atg9RNAi or phm>Rheb did not significantly affect developmental timing nor larval growth rate. (Figure S2A and S2B). Early L3 starvation (2–8 hours AL3E, pre-NRC stage) caused delayed pupariation and decreased pupariation rate in control larvae (Figure 2A, 2E and S2C-S2F), which is consistent with previous reports [17, 18]. Phm>Atg1RNAi and phm>Atg9RNAi larvae showed higher rates of pupariation (although delayed) during early L3 starvation (Figure 2B, 2C and 2E), indicating a shift of the minimal viable weight checkpoint in these larvae. Similarly, the majority of phm>Rheb larvae pupariated without delay regardless of the starting point of starvation (Figure 2D and 2E), showing that both nutritional checkpoints are altered. In contrast to pupariation, the eclosion rate under starvation was not affected by autophagy suppression (Figure 2F and S2G). Taken together, these data demonstrate that autophagy induction blocks pupariation activity of larvae during early stage NR. Upon autophagy suppression many undersized larvae formed pupa, but the pupae did not produce viable adults (Figure 2F). So this mechanism likely improves the chance of survival during NR stress by enabling underweight larva to search for new food sources instead of initiating an ill-fated attempt at metamorphosis.
Figure 2. Autophagy suppression in the PG causes inappropriate pupariation during early stage NR.

(A-D) Relationship between start time of starvation and the time from L2/L3 molting to pupariation in phm>yw (A), phm>Atg1RNAi (B), phm>Atg9RNAi (C) and phm>RhebAV4 (D) larvae (n > 30 for each time point). (E and F) Percentage of larvae that manage to pupariate (E) and that eclose from pupa (F) during starvation treatment starting at different time points.
See also Figure S2.
Forced autophagy induction in the PG causes developmental delay/arrest in fed larvae
To further corroborate our finding that autophagy controls pupariation, we next examined whether ectopically activating autophagy is sufficient to block larval pupariation in the absence of NR stress. Constant autophagy induction in the PG (phm>Atg1 or phm>TSC½) caused L1/L2 developmental arrest (data not shown), so we employed inducible phmGeneSwitch (phmGS)-Gal4 (Figure S3A and S3B) to activate autophagy specifically after L2/L3 molting [19]. Without RU486 induction, neither phmGS>Atg1 nor phmGS>TSC½ induced autophagy or changed developmental timing (Figure S3C and S3D). However, RU486 administration markedly stimulated autophagy (Figure S3C) and caused developmental delay/arrest in both larvae (Figure 3A and 3B). Autophagy induction by Atg1 overexpression may cause cell apoptosis [20]. We detected apoptotic PG cells using antibody targeting cleaved caspase-3 [21] but found no cleaved caspase-3 positive cells in RU486 treated phmGS>Atg1 and phmGS>TSC½ larvae (Figure S3E). Therefore, we conclude that autophagy induction does not causes apoptosis and that RU486 feeding likely causes developmental defects by inducing inappropriate levels of autophagy in the PG.
Figure 3. Forced autophagy induction in the PG causes developmental delay/arrest in fed larvae.

(A and B) Pupariation timing curves of phmGS>yw, phmGS>Atg1 and phmGS>TSC½ larvae fed with 0.5 μg/ml (A) or 1.5 μg/ml (B) RU486 at 4 hours (pre-NRC) or 12 hours AL3E (post-NRC) time points. Mean ± SEM, n=3.
See also Figure S3.
Intriguingly, the extent of the autophagy-induced developmental defects correlated with the length and the strength of autophagy induction. Autophagy induction at either pre-NRC (4 hours AL3E) or post-NRC (12 hours AL3E) time points (Figure S3F) caused developmental delay/arrest. However, early induction (4 hours AL3E) resulted in a longer delay and a higher rate of developmental arrest than late induction (12 hours AL3E). Similarly, feeding higher concentrations of RU486 also resulted in stronger developmental defects (Figure 3A and 3B). These data demonstrate that strong induction of autophagy in the PG is capable of blocking pupariation regardless of larva body weight and nutritional status and functions in a “dose” dependent manner. We propose that as wildtype larvae grow and surpass the NRCs, NR is no longer able to induce autophagy to the threshold level required to block pupariation, thereby enabling larvae to initiate metamorphosis even when nutrient access is limited.
Autophagy blocks ecdysone synthesis by limiting cholesterol availability in PG cells
We then wished to determine how autophagy disrupts developmental timing and pupariation control. Since pupariation activity is directly regulated by ecdysone [22], we measured the level of pupariation triggering ecdysone pulse at larval wandering stage. In autophagy suppression larvae, the wandering stage ecdysone peak was comparable to the control without starvation. However, pre-NRC (4 hours AL3E) starvation prevented the rise of ecdysone titer at wandering stage in control, but not phm>Rheb larvae (Figure 4A), which is consistent with the increased pupariation of phm>Rheb larvae (Figure 2D and 2E). Phm>Atg1RNAi larvae showed a modest but insignificant increase in the ecdysone titer (Figure 4A), which may be attributed to the delayed and unsynchronized pupariation of these larvae under pre-NRC starvation (Figure 2B). In a complementary experiment, we saw very limited ecdysone production at larval wandering stage in autophagy induction larvae (Figure 4B). Thus, we conclude that autophagy blocks larva pupariation by disrupting ecdysone production in PG cells.
Figure 4. Autophagy blocks developmental transition by disrupting ecdysone synthesis.

(A) Quantification of ecdysone/20-hydroxyecdysone titers in fed and starved phm>yw, phm>Atg1RNAi and phm>RhebAV4 larvae at different timing stages. (B) Quantification of ecdysone/20-hydroxyecdysone levels in phmGS>yw, phmGS>Atg1 and phmGS>TSC½ larvae at different stages with or without 1.5 μg/ml RU486 feeding. (A and B) Mean ± SEM; p values by unpaired t-test (n=4; ns, not significant, ****p<0.0001).
See also Figure S4.
Ecdysone is synthesized from cholesterol by a series of redox reactions that involve a number of enzymes encoded by the “Halloween” genes [23]. Considering that autophagy commonly exerts degradative functions, we sought to determine whether autophagy disrupts ecdysone production by degrading the Halloween enzymes. Examination of Phantom (Phm), Disembodied (Dib) and Spookier (Spok) protein levels in the PG using immunohistochemical (IHC) method indicates that autophagy induction does not substantially deplete any of these enzymes (Figure S4A–S4C), suggesting that autophagy likely does not function through ecdysone biosynthetic protein degradation.
Since previous studies demonstrate that autophagy can mediate cholesterol transportation [24, 25], we next examined whether autophagy affects cholesterol homeostasis in the PG. As a steroid hormone producing organ, the PG maintains a pool of cholesterol storage in lipid droplets. We observed a number of lipid droplets in PG cells of pre-NRC fed larvae, consistent with previous findings [25, 26], while the number of lipid droplets significantly decreased after NR treatment. In contrast, we observed an increased number of lipid droplets in autophagy suppression larvae compared with control, while the number did not significantly change during NR (Figure 5A and 5B). Additionally, autophagy induction caused a decrease in lipid droplet number in PG cells (Figure 5C and 5D). Together, these data show that autophagy induces clearance of lipid droplets in PG cells.
Figure 5. Autophagy constrains cholesterol availability in PG cells by interacting with cholesterol trafficking pathway.

(A) Lipid droplets visualized by Nile Red staining in phm>yw, phm>Atg1RNAi and phm>RhebAV4 PG cells before and after NR. Scale bar, 10 μm. (B) Quantification of number of lipid droplet per unit cell area (100 μm2) in the PGs tested in (A). (C) Lipid droplets visualized by Nile Red staining in phmGS>yw, phmGS>Atg1 and phmGS>TSC½ larvae with or without RU486 feeding. Scale bar, 10 μm. (D) Quantification of number of lipid droplet per unit cell area (100 μm2) in the PGs tested in (C). (B and D) Mean ± SEM; p values by unpaired t-test (n=5; ns, not significant, *p<0.05, **p<0.01, ****p<0.0001). (E) Phm>mCh-Atg8a PGs were incubated ex vivo with 22-NBD-cholesterol and then imaged by fluorescence microscopy. Arrows indicate overlaps of mCherry and NBD signals and the numbered images show zoom-in views of the corresponding areas indicated by the arrows. Scale bar, 10 μm. (F) Images of PGs expressing mCh-Atg8a and EYFP-NPC1a under fed and NR condition. Arrows indicate overlaps between mCherry and EYFP signals. Scale bar, 10 μm. (G) Lipophagy structures in PG cells visualized under TEM. Pre-NRC (4 hours AL3E) larvae were starved for 6 hours before sample preparation. Upper left, a double-membrane bound lipophagic vesicle; lower left, a doublemembrane structure that occupies part of a lipid droplet; upper middle and right, an early stage, autophagic isolation membrane that is wrapping up a lipid droplet; lower middle and right, a lipid droplet processed by a late-stage autolysosome. Black arrows indicate double membrane structures. Scale bars, 0.5 μm. (H-J) Pupariation timing curve of phmGS>yw (H), phmGS>Atg1 (I) and phmGS>TSC½ (J) larvae with indicated treatments starting at 12 hours AL3E. EtOH, ethanol; Chol, 40 μg/ml cholesterol; RU486, 0.5 μg/ml RU486; RU486+Chol, 40 μg/ml cholesterol plus 0.5 μg/ml RU486.
See also Figure S4.
To confirm the interaction between autophagy and cholesterol trafficking, we cultured PG tissue ex vivo with NBD-cholesterol. Many Atg8a-positive vesicles overlapped with the NBD signal, suggesting that these autophagic vesicles contain cholesterol (Figure 5E). mCh-Atg8a also showed colocalization with EYFP-NPC1a (Figure 5F), a key transmembrane protein mediating cholesterol trafficking [27], consistent with the notion that autophagy functions to transport cholesterol. We then visualized the ultrastructure of autophagy in PG cells using transmission electron microscopy (TEM). In the PG of starved pre-NRC animals, we observed double-membrane bound vesicles containing homogenous content of light density (Figure 5G), typical of structures found in cells undergoing lipid-processing autophagy (lipophagy) [28]. In addition, we observed lipophagy related structures which exhibited a cluster of small, single or double membrane bound vesicles that contain similar light-density content (Figure S4D) as well as double membrane bound autophagic vesicles containing non-lipid cargoes (Figure S4E). These structures were seldomly found in phm>Atg1RNAi or phm>Rheb PG cells, confirming their autophagic nature (Figure S4F–S4I). Together, the data from both fluorescence and electron microscopy confirm that autophagy participates in cholesterol trafficking in PG cells.
To examine whether reduction in cholesterol availability to the hormone biosynthetic enzymes is responsible for the autophagy-induced developmental defects, we fed cholesterol to the autophagy induction larvae. Cholesterol feeding fully rescued the developmental delay/arrest in these larvae (Figure 5H–5J), demonstrating that cholesterol insufficiency, rather than induction of PG cell death or disruption of the hormone biosynthetic capacity, is the likely underlying cause of the autophagy-induced ecdysone deficiency and developmental defects.
Autophagy affects cholesterol trafficking by interacting with late endosome/lysosomes
Previous studies have shown that autophagy delivers cholesterol esters to late endosomes and lysosomes for hydrolysis [24]. Thus we examined whether similar autophagic flux occurs in the PG using specific vesicle markers. Firstly, we observed a high level of colocalization (revealed by Mander’s Colocalization Coefficient, MCC) between mCh-Atg8a and GFP-Vamp7 (Figure 6A and 6B), a marker targeting both late endosomes and lysosomes [29, 30], confirming the interaction between autophagy and the endolysosome system. However, we saw a different colocalization profile when labeling late endosomes and lysosomes separately. mCh-Atg8a exhibited frequent colocalization with late endosome marker EYFP-Rab7 (Figure 6C and 6D), but much lower level of overlap with lysosome marker GFP-Lamp1 (Figure 6E and 6F). These data are inconsistent with what we and others observed in fat body tissues (Figure S5A) where mCh-Atg8a overlaps frequently with GFP-Lamp1 [31]. A caveat for using the GFP-Lamp1 marker is that its GFP tag faces the intravesicular space and thus may be quenched by the acidity of lysosomes. To overcome this limitation, we employed a Spinster (Spin) marker with cytoplasm-facing GFP tag [32] which largely overlaps with Lamp1 in lysosomes [33, 34], but still observed a comparatively low level of colocalization with mCh-Atg8a (Figure 6G and 6H). These results suggest that the autophagosomes either interact more frequently with late endosomes than lysosomes or persist in autophago-endosome stage for a longer time before transforming into autolysosome stage. Nevertheless, it is important to recognize that the endocytic pathway markers such as Rab7 and Lamp1 do not exclusively localize on the compartments they are proposed to label [35]. In the tests with lysosome markers the MGreen value (percentage of green-positive pixels that are also red-positive) increased upon starvation (Figure 6F and 6H), showing that a fraction of autophagosomes did fuse with lysosomes during NR. Under TEM we also observed autolysosome structures (Figure S4E), supporting the existence of autophago-lysosome communication. Taken together, we conclude that the autophagy in PG cells communicates with endolysosome system, but with an altered dynamics.
Figure 6. Autophagy affects cholesterol trafficking by interacting with endo/lysosomes.

(A, C, E and G) Images of PGs expressing mCh-Atg8a and GFP-Vamp7 (A), EYFP-Rab7 (C), GFP-Lamp1 (E) or GFP-Spin (G). Arrows indicate overlaps of mCh-Atg8a and EYFP/GFP signals. Scale bars, 10 μm. (B, D, F and H) Mander’s colocalization analyses of corresponding images in (A, C, E and G). Mean ± SEM; p values by unpaired t-test (n=5–7; ns, not significant, **p<0.01, ***p<0.001). (I to L) Autophagy visualized by tandem tagged Atg8a in phm>dicer (I), phm>SyxRNAi,dicer (J), phm>Snap29RNAi,dicer (K) and phm>Vamp7RNAi,dicer (L) PGs. Scale bars, 10 μm. (M) Pupariation activity of SNARE proteins and Rab8 knockdown larvae during early L3 (4 hours AL3E) starvation. The scattered dots indicate the time of pupariation of each individual larva. The underscored numbers indicate the percentage rate of pupariation. For each group n > 50 larvae.
See also Figure S5.
We then examined whether the autophago-endolysosome interaction is required for regulation of cholesterol trafficking by autophagy. In this test we introduced a tandem fluorescent-tagged GFP-mCh-Atg8a (tfAtg8a) marker [36], whose GFP tag can be rapidly quenched by acidity upon autophago-endolysosome fusion [37]. In PG cells, the GFP of the majority of tfAtg8a marker was quenched (Figure 6I), indicating that most autophagic vesicles, even under fed conditions, are acidic. Depletion of v-ATPase subunits Vha26 and Vha55 effectively rescued the GFP signal of the markers (Figure S5B and S5C), further verifying this finding [31].
Three SNARE family proteins, Syx17, Snap29 and Vamp7, are known to mediate the autophago-endolysosome fusion [38, 39]. Knockdown of Syx17 and Snap29 caused an accumulation of small, cloudy looking Atg8a-positive structures in PG cells during starvation (Figure 6J and 6K), consistent with findings in fat body cells [39]. Knockdown of Vamp7 resulted in accumulation of Atg8a vesicles of larger size (Figure 6L, Figure S5D and S5E), similar to the effects of the vesicle fusion inhibitor Bafilomycin A1 in fat body cells [31]. These data indicate that depletion of the SNAREs disrupts autophagic flux in PG cells. All three groups of SNARE knockdown larvae pupariated precociously during pre-NRC starvation (Figure 6M), phenocopying the autophagy suppression larvae (Figure 2B–2D). These results show that the autophagosome-endolysosome interaction plays an essential role in the function of autophagy on pupariation control likely through alterations in cholesterol trafficking.
Intriguingly, disruption of autophagic flux by depleting Syx17, Snap29 or Vamp7 did not rescue the GFP signal of tfAtg8a (Figure 6J–6L), in contrast to the requirement of these SNAREs for autophagosome acidification in other cell types [39, 40]. This data indicates that the acidification of autophagosomes in PG cells may not depend on fusion with endolysosomes, further suggesting an autophagic flux in PG cells not fully consistent with that in other well-studied cell/tissue types.
Autophagy in the PG is highly dynamic and exhibits additional non-conventional morphological features
The specific function and altered autophagic flux inspire us to look into the dynamics of autophagy in the PG. When measuring autophagy induction (Figure 1B–1D), we also examined the size distribution of Atg8a-positive vesicles. In early L3 (6 hours AL3E) PG cells, the frequency of large-sized (> 1.0 μm2) vesicles significantly decreased during NR, while that of the small-sized (< 1.0 μm2) vesicles increased. In contrast, this effect of NR on vesicle size was reduced as larvae progressed to the mid L3 stage (24 hours AL3E) (Figure S6A and S6B). The simultaneous increase in vesicle number coupled with reduced vesicle size indicate a highly dynamic process which has not been reported in NR-induced autophagy in other tissues such as the fat body [41]. To examine this possibility, we observed PG cells maintained in ex vivo culture using time-lapse live imaging. In fed early L3 larvae, the Atg8a-positive vesicles maintained their size and shape and exhibited limited mobility (Video S1). Strikingly, after starvation in early, but not mid L3 stage larvae, Atg8a vesicles showed highly dynamic movements (Video S1) and formation of tubule-like structures (Figure S6C–S6E and Video S2–S4). We also observed shrinkage and fragmentation of large vesicles into small ones (Figure S6F and Video S5) and ring-like structures budding out of existing large-size vesicles (Figure S6G, S6H and Video S6). These observations explain the changes on vesicle size distribution during NR and suggest dynamic changes during autophagy induction in the PG.
We also noticed that most of the tubular and ring-like structures were sensitive to fixative since they were rarely seen in fixed tissues (Figure S6I). Therefore, the apparent formation of small-size vesicles in fixed tissue likely represents fragmentation of the tubular network upon fixation. Further, all Atg8a-positive structures in the PG are extremely sensitive to detergents even after fixation (Figure S6I), which is in stark contrast to the autophagic structures in fat body and other vesicles such as lysosomes in the PG (Figure S6I). Although not fully understood, the dynamics and the non-conventional morphological features of the autophagy in the PG suggest that it performs a function distinct from canonical NR-induced autophagy.
Discussion
Temporally-regulated autophagy in the PG controls the NRCs
The Drosophila NRCs are characterized by an abrupt switch in the response to starvation, suggesting that the determinants of the checkpoints should be regulated (1) by nutritional signals and (2) in a developmental stage dependent fashion. In this study, we identify autophagy as a NR induced, developmental stage dependent mechanism that is the functional output of the checkpoints, i.e. it is the mechanism responsible for prevention of pupariation before larvae have enough stored nutrients to develop into viable adults.
In the PG, most characterized NR-responsive pathways, such as insulin/insulin-like and TOR signaling, are essential for development and are repressed under NR [18, 42, 43]. In contrast, autophagy is induced by NR and suppression of autophagy did not disrupt development of well-fed larvae (Figure S2A), although a basal level of Atg8a positive vesicles are always present in PG cells (see below). We propose that autophagy has evolved as a specific stress responsive “STOP” pathway, which works together with essential “ON” pathways, to exert precise developmental timing control under nutritional stress.
Similar to other factors that influence the NRCs [43, 44], autophagy induction is temporally regulated. Measured by the number of Atg8a positive puncta, the regulation of autophagy inducibility is progressive (Figure 1C) and does not exhibit an “all or none” switch that precisely correlates with the NRCs. However, when measuring the area of Atg8a puncta there is a very strong correlation with the time of the NRCs (Figure 1D). We also found that autophagy suppresses pupariation in an “dose” dependent manner, in which stronger autophagy induction causes more severe developmental defects (Figure 3). This explains the findings that strong post-NRC TOR suppression (Figure 3), but not suppression using a weaker P0206-Gal4 driver, delays development [42]. We reason that the NRCs correlates with a certain threshold of autophagy induction, which can no longer be reached when the checkpoints are surpassed.
Autophagy is inducible through genetic manipulations during the post-NRC stage (Figure S3C), indicating that the silencing of autophagy in this stage is likely due to changes in upstream signaling. A compensatory pathway may function to antagonize the suppression of insulin/insulin-like and TOR pathway during NR, which resembles what happens during the organ sparing process in some imaginal tissues in post-NRC larvae [45]. It is also possible that some pathway that can regulate TOR activity, such as Hippo pathway [46], may function differently before and after the NRCs. In any case, the mechanism that controls the developmental stage dependent autophagy suppression requires further study.
Autophagy disrupts hormone synthesis by targeting cholesterol trafficking in PG cells
In previous studies, most known pathways exert control of ecdysone synthesis by targeting hormone biosynthetic enzymes [8, 18, 42, 44]. However, we found that autophagy induction disrupts ecdysone synthesis likely by depleting cholesterol storage in the PG, which represents a new dimension of regulation of the ecdysone producing pathway. This mechanism is efficient from a design principle standpoint since it allows a more rapid means to ramp up hormone production when conditions turn favorable than does destruction of biosynthetic capacity which would require time for re-synthesis of ecdysone production components. Furthermore, insects are strict cholesterol auxotrophs [47], which potentially makes them particularly sensitive to this type of regulation. Nevertheless, some studies indicate that autophagy in mammalian endocrine organs can also affect hormone synthesis [48, 49], indicating that this mechanism may be universally employed as a means of controlling hormone production in response to nutritional stress.
The regulation of cholesterol storage by autophagy in the PG is confirmed by multiple lines of evidences. We observed cholesterol-containing autophagosomes under fluorescence microscopy (Figure 5E) and lipophagy related structures under TEM (Figure 5G), strongly suggesting that autophagy interacts with a cholesterol trafficking pathway. Previous studies show that the late endosome rather than the lysosome is the key compartment that mediates cholesterol trafficking [50], while cholesterol can egress from late endosomes to further destinations before reaching the lysosome [51, 52]. We also find a stronger correlation of the autophagosome marker with late endosomes than with lysosomes (Figure 6A–6H), suggesting a more important role of the interaction between autophagosome and late endosome. Further, the cholesterol trafficking mediator NPC1a strongly localizes to autophagic vesicles in the PG (Figure 5F), while its mammalian homolog NPC1 is also primarily a late endosomal protein that only transiently associates with lysosomes [53]. Taken together, these observations demonstrate that autophagy interacts with cholesterol trafficking pathway, possibly removing or sequestering cholesterol from progressing into the ecdysone biosynthetic pathway.
How autophagy depletes cholesterol storage in the PG remains an unresolved question. Unlike proteins or triglycerides, cholesterol is typically not catabolized into small molecules. It is known that excessive cholesterol can be cleared from a cell via a specific recycling process involving cholesterol efflux [54]. Autophagy facilitated cholesterol efflux has been reported in macrophages in mammalian system [24]. It is possible that autophagy works in a similar way to facilitate cholesterol efflux in the PG. Interestingly, Rab8 is required for both secretory autophagy [55] and cholesterol efflux pathway [56–58]. We find that knockdown of Rab8 in the PG leads to impairment of the NRCs similar to blocking the autophagoe-endolysosome interaction (Fig. 6M), providing an indirect evidence that support this hypothesis.
Non-conventional morphological features of the autophagy in PG cells
Although the autophagy process in the PG requires most of the canonical autophagy machinery (Figure 1E–1G and Figure S1), it exhibits some morphological features that are not commonly observed. We found dynamic Atg8a vesicle movements in PG cells from starved pre-NRC animals, during which vesicle compartmentation, tubule- and ring-like structure formation were observed (Figure S6C–S6H). Autophagy-relevant tubular structures have been reported in previous studies [59, 60], but the structures we observed are likely different. The tubule structures formed during lysosomal reformation [59] were visualized by the lysosome marker (Lamp1), which does not show clear colocalization with Atg8a in PG cells (Figure 6E and 6F). The tubular network found in muscle cells was detected by both Atg8a and Spinster marker [60], but the two markers also overlap well in muscles, unlike what we found in PG cells. Moreover, the formation of the muscle tubular network is not stimulated by starvation [60], also suggesting that it is distinct from the autophagy in PG cells.
Intriguingly, during cholesterol trafficking it has been shown that NPC1-positive vesicles can form tubular and ring-like structures [61]. In PG cells, NPC1a localizes on some autophagic vesicles (Figure 5F), thus the Atg8a-positive tubular and ring-like structures may be NPC1a-positive and represent the structures mediating cholesterol trafficking. In addition, the clustered lipophagy-like vesicles we observed under TEM (Figure S4D) may represent the autophagosomes undergoing fragmentation, which links the vesicle fragmentation process with the cholesterol trafficking pathway. We hypothesize that the dynamic movement of Atg8a-positive structures facilitates cholesterol removal from the ecdysone biosynthetic enzymes either by an efflux mechanism or sequestering it to a novel membrane compartment. In either case, the removal of the cholesterol attenuates hormone synthesis and thereby prevents nonproductive pupariation of larvae during nutritional stress.
Lastly, it has recently been reported that autophagy is also required to produce ecdysone [46], which seems at odds with our findings. However, as we describe above there are always Atg8a positive vesicles in the PG even under well fed condition and these vesicles tend to be large (Figure 1B). Starvation pre-NRC leads to induction of a highly dynamic movement and a size reduction of the autophagic structures, but post-NRC the dynamics and vesicle size change are reduced (Figure S6). Thus we propose that there are two types of autophagic processes occurring in the PG: a constitutive level in well fed larva that is not so dynamic and is responsible for aiding flux of cholesterol into the biosynthetic pathway and a second highly inducible process that only occurs pre-NRC which is responsible for shunting cholesterol away from the biosynthetic pathway. Future studies aimed at elucidating the mechanism responsible for switching off the inducibility and the highly dynamic nature of the autophagic process postNRC may aid in understanding how cholesterol flux can be directed into and away from the hormone biosynthetic pathway.
STAR Methods
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael B. O’Connor (moconnor@umn.edu). This study did not generate new unique reagents.
Experimental Model and Subject Details
Flies
Unless noted, all flies were reared on standard agar-cornmeal food supplemented with yeast at 25 °C. Flies were cultured in 12:12 light-dark cycles, however, all experiments were carried out under constant light to avoid the potential impact of circadian cycle on developmental timing. Both male and female flies were used in all experiments. Phm-Gal4 and phmGeneSwitch-Gal4 were generated by O’Connor Lab. UAS-mCh-Atg8a, UAS-GFP-mCh-Atg8a, UAS-GFP-Lamp1, UAS-GFP-Ref(2)P and UAS-TSC½ were obtained from T. Neufeld. UAS-GFP-Spin was a kind kind gift from G. Davis. UAS-GFP-Vamp7 was a kind gift from A. Kiger. UAS-Vha26RNAi and UAS-Vha55RNAi were obtained from Vienna Drosophila Resource Center (VDRC). All the other lines were obtained from Bloomington Drosophila Stock Center (BDSC).
Method details
Developmental staging and starvation assay
Before egg collection, flies were transferred to constant light environment for at least 2 days and all subsequent treatments were carried out under constant light. Eggs were collected on apple juice plates with yeast paste. Twenty-four hours after egg collection, early L1 larvae were transferred to standard lab fly food with yeast paste. One and half days later, larvae were carefully staged every 2 hours and newly-molted L3 larvae were picked out and transferred to new fly food without yeast paste. Then the L3 larvae were cultured for a period as needed before carrying out starvation assay or other treatments. For starvation assay larvae were transferred to 1% agar. Drops of water were added occasionally on top of the agar to keep moisture. Larvae were then monitored every 2 hours until they pupariated or died.
Fluorescence microscopy
Larvae were dissected in PBS and fixed using 3.7% formalderhyde for 15 mins at room temperature. For samples that express fluorescent-tagged proteins, tissues were washed in PBS (3 times, 10 mins each) and mounted in 70% glycerol/PBS for imaging. For samples treated with staining dyes or immunohistochemical assay, the methods are described in the following sections. Following sample preparation, all images were captured using Zeiss LSM710 confocal microscope. To be noted, detergent was strictly avoided in all imaging process involving visualization of autophagy in the PG.
Time-lapse imaging
Larvae were firstly dissected carefully in M3 culture medium to isolate the brain-ring gland complexes. The complexes were then transferred to a glass-bottom culture dish (MatTek) with a drop of M3 medium, after which the brain tissues were quickly separated from the ring glands and removed from the dish. A coverslip was then positioned on top of the ring glands to prevent sample movements, and more M3 medium was added to the dish before imaging. The samples were imaged with a Zeiss LSM 710 confocal microscope using the time-lapse configuration.
Immunohistochemistry
Larvae were dissected in PBS and fixed using 3.7% formalderhyde for 15 mins at room temperature. Tissues were washed in PBS containing 0.1% Triton X-100 (PBT) for 3 times (10 mins each) and then permeablized and blocked simultaneously using PBT containing 5% normal goat serum (NGS) for 1 hour. Tissues were then incubated with primary antibody (anti-Phm, 1:1000, anti-Dib, 1:1000, anti-Spok, 1:500, anti-cleaved caspase-3, 1:200) in PBT containing 10% NGS overnight at 4 degrees, followed by PBT washes (5 times, 10 mins each) and then post-secondary incubation for 2 hours at room temperature. DAPI staining occurred for 5 minutes at the pen-ultimate washing step after secondary antibody incubation. Finally, tissues were transferred to 70% glycerol/PBS mounting medium and then mounted on glass slide for imaging. Images were captured using a Zeiss LSM 710 confocal microscope.
Ecdysteroid titer measurement
The ecdysteroid titers of larvae were measured using the 20-hydroxyecdysone Enzyme Immunoassay (EIA) kit, (Cayman Chemicals) which detects both ecdysone and 20-hydroxyecdysone. Briefly, frozen larvae were homogenized in methanol and ecdysteroids were extracted as described previously [71]. The extracts were evaporated in a Speed Vac and the residue resuspended in EIA buffer (Cayman Chemical) and analyzed following the manufacturer’s protocol. A standard curve was determined using a dilution series containing a known amount of purified 20-hydroxyecdysone solution provided by the kit. Absorbance at 415 nm was detected using a benchtop microplate reader (Bio-Rad).
Ex vivo incubation assay
The assay was adapted from previous studies [72, 73]. Briefly, wandering larvae were dissected in M3 culture medium and the brain-ring gland complexes were carefully isolated. The complexes were then incubated in M3 medium containing 10% fetal bovine serum and 0.5% dilution of 10 μM NBD-cholesterol for 6 hours at room temperature. The samples were subsequently fixed in 3.7% formaldehyde for 15 mins and then imaged using a Zeiss LSM 710 confocal microscope.
Nile Red staining for lipid droplets
Larvae were dissected in PBS and fixed using 3.7% formalderhyde for 15 mins at room temperature. After washed in PBS (3 times, 10 mins each), tissues were incubated in 1 μg/ml Nile Red solution in PBS for 30 mins. Then samples were washed in PBS (3 times, 10 mins each) again and mounted in 70% glycerol/PBS for imaging. Images were captured using Zeiss LSM 710 confocal microscope.
Electron microscopy
Larvae were dissected in cacodylate buffer and fixed in fixative containing 2% paraformaldehyde and 2.5% glutaraldehyde in cacodylate buffer. Then samples were post-fixed in 1% osmium tetroxide for 1 hour, dehydrated progressively in acetone, and embedded in Poly/Bed 812 epoxy resin. Following heat polymerization, resin blocks were sectioned using Leica UC7 microtome toward 70 nm ultra-thin sections. Finally, the section grids were post-stained by uranyl acetate and lead citrate and then imaged using FEI Tecnai G2 F30 transmission electron microscope.
Quantification and Statistical Analysis
Quantification of vesicles
The number and area of vesicles were quantified using imageJ software. Briefly, the vesicles were selected using the “threshold” function. Then the number and total area of the vesicles were calculated automatically using the “analyze particles” function in the software.
Quantification of colocalization
Colocalization analysis was carried out in ImageJ software using the JACoP plugin [74]. Briefly, the particles in each channel were selected manually in the plugin in a similar way to the “threshold” function. Then Mander’s Colocalization Coefficient was automatically calculated by the plugin.
Statistics
GraphPad Prism software was used to carry out statistical analyses. All experiments were repeated at least three times and Student’s t-test (unpaired t-test with Welch’s correction) was used to determine statistical significance. p<0.05 was considered significant in all analyses. No specific method was used to determine whether the data met assumptions of the statistical approach. Details of statistical parameters of each test are indicated in the corresponding figure legends.
Data and Code Availability
This study did not generate/analyze any dataset/code.
Supplementary Material
Video S1. Autophagy dynamics in the PG visualized by time-lapse live imaging. Related to Figure 1 and Figure S6.
Phm>mCh-Atg8a PG tissues were cultured ex vivo and observed under fluorescence microscope. Samples from fed pre-NRC (A), starved pre-NRC (B), fed post-NRC (C) and starved post-NRC (D) larvae were shown. For fed groups, PG tissues were imaged immediately upon dissection. For starvation groups larvae were starved for 6 hours before dissection and imaging process.
Video S2. Tubular network formation during autophagy induction. Related to Figure 1 and Figure S6.
Pre-NRC larvae were starved for 6 hours and then dissected for imaging. The video shows a representative event of tubular structure movement within a tubule network. Representative frames from this movie are shown in Figure. S6C.
Video S3. Tubule expansion and retraction during autophagy induction. Related to Figure 1 and Figure S6.
Representative frames from this movie are shown in Figure. S6D.
Video S4. A tubular structure fuses with neighboring vesicle during autophagy induction. Related to Figure 1 and Figure S6.
The arrow shows the site where the fusion occurs. Representative frames from this movie are shown in Figure. S6E.
Video S5. Fragmentation of autophagic structure during autophagy induction. Related to Figure 1 and Figure S6.
The video shows a large-sized autophagic structures fragmenting into multiple smaller vesicles. Representative frames from this movie are shown in Figure. S6F.
Video S6. Formation of ring-like structure during autophagy induction. Related to Figure 1 and Figure S6.
The arrow shows the formation site of the ring-like structure. Representative frames from this movie are shown in Figure. S6G.
Highlights.
Autophagy in the PG is temporally regulated in response to nutrition.
PG autophagy prevents precocious pupariation of undersized larva.
PG autophagy alters cholesterol trafficking to block hormone production.
PG autophagy is dynamic and exhibits novel features.
Acknowledgements
We thank J. Simon and members of O’Connor laboratory for critical review of the manuscript. We are grateful to A. Neisch and members of Neufeld laboratory for many helpful discussions and to G. Marqués for providing technical assistance with live imaging methods. We thank F. Zhou, H. Lee, B. Zhi and C. Frethem for carrying out sectioning and imaging works and providing expertized assistance on the TEM experiments. We also thank the Vienna Drosophila RNAi Center, Drosophila Genetics Resource Center, and Bloomington Drosophila Stock Center for fly stocks. Parts of this work were carried out in the Characterization Facility, University of Minnesota, a member of the NSF-funded Materials Research Facilities Network (www.mrfn.org) via the MRSEC program. This work was supported by grant 1R35GM118029 from NIGMS to M.B.O.
Footnotes
Declaration of Interests
The authors declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yamanaka N, Romero NM, Martin FA, Rewitz KF, Sun M, O’Connor MB, and Leopold P (2013). Neuroendocrine control of Drosophila larval light preference. Science 341, 1113–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colombani J, Andersen DS, and Leopold P (2012). Secreted peptide Dilp8 coordinates Drosophila tissue growth with developmental timing. Science 336, 582–585. [DOI] [PubMed] [Google Scholar]
- 3.Garelli A, Gontijo AM, Miguela V, Caparros E, and Dominguez M (2012). Imaginal discs secrete insulin-like peptide 8 to mediate plasticity of growth and maturation. Science 336, 579–582. [DOI] [PubMed] [Google Scholar]
- 4.Garelli A, Heredia F, Casimiro AP, Macedo A, Nunes C, Garcez M, Dias AR, Volonte YA, Uhlmann T, Caparros E, et al. (2015). Dilp8 requires the neuronal relaxin receptor Lgr3 to couple growth to developmental timing. Nat. Commun 6, 8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colombani J, Andersen DS, Boulan L, Boone E, Romero N, Virolle V, Texada M, and Leopold P (2015). Drosophila Lgr3 Couples Organ Growth with Maturation and Ensures Developmental Stability. Curr. Biol 25, 2723–2729. [DOI] [PubMed] [Google Scholar]
- 6.Boone E, Colombani J, Andersen DS, and Leopold P (2016). The Hippo signalling pathway coordinates organ growth and limits developmental variability by controlling dilp8 expression. Nat. Commun 7, 13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mirth CK, and Riddiford LM (2007). Size assessment and growth control: how adult size is determined in insects. Bioessays 29, 344–355. [DOI] [PubMed] [Google Scholar]
- 8.Shimell M, Pan X, Martin FA, Ghosh AC, Leopold P, O’Connor MB, and Romero NM (2018). Prothoracicotropic hormone modulates environmental adaptive plasticity through the control of developmental timing. Development 145, dev159699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stieper BC, Kupershtok M, Driscoll MV, and Shingleton AW (2008). Imaginal discs regulate developmental timing in Drosophila melanogaster. Dev. Biol 321, 18–26. [DOI] [PubMed] [Google Scholar]
- 10.Russell RC, Yuan HX, and Guan KL (2014). Autophagy regulation by nutrient signaling. Cell Res. 24, 42–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McPhee CK, and Baehrecke EH (2009). Autophagy in Drosophila melanogaster. Biochim. Biophys. Acta 1793, 1452–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao F, Li G, Liu C, Gao H, Wang H, Liu W, Chen M, Shang Y, Wang L, Shi J, et al. (2018). Autophagy regulates testosterone synthesis by facilitating cholesterol uptake in Leydig cells. J. Cell Biol 217, 2103–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeVorkin L, and Gorski SM (2014). Monitoring autophagic flux using Ref(2)P, the Drosophila p62 ortholog. Cold Spring Harb. Protoc 2014, 959–966. [DOI] [PubMed] [Google Scholar]
- 14.Feng Y, He D, Yao Z, and Klionsky DJ (2014). The machinery of macroautophagy. Cell Res. 24, 24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diaz-Troya S, Perez-Perez ME, Florencio FJ, and Crespo JL (2008). The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy 4, 851–865. [DOI] [PubMed] [Google Scholar]
- 16.Galluzzi L, Pietrocola F, Levine B, and Kroemer G (2014). Metabolic control of autophagy. Cell 159, 1263–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beadle GW, Tatum EL, and Clancy CW (1938). Food Level in Relation to Rate of Development and Eye Pigmentation in Drosophila Melanogaster. Biol. Bull 75, 447–462. [Google Scholar]
- 18.Mirth C, Truman JW, and Riddiford LM (2005). The role of the prothoracic gland in determining critical weight for metamorphosis in Drosophila melanogaster. Curr. Biol 15, 1796–1807. [DOI] [PubMed] [Google Scholar]
- 19.Roman G, Endo K, Zong L, and Davis RL (2001). P[Switch], a system for spatial and temporal control of gene expression in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 98, 12602–12607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scott RC, Juhasz G, and Neufeld TP (2007). Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr. Biol 17, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fan Y, and Bergmann A (2010). The cleaved-Caspase-3 antibody is a marker of Caspase-9-like DRONC activity in Drosophila. Cell Death Differ. 17, 534–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamanaka N, Rewitz KF, and O’Connor MB (2013). Ecdysone control of developmental transitions: lessons from Drosophila research. Annu. Rev. Entomol 58, 497–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilbert LI (2004). Halloween genes encode P450 enzymes that mediate steroid hormone biosynthesis in Drosophila melanogaster. Mol. Cell. Endocrinol 215, 1–10. [DOI] [PubMed] [Google Scholar]
- 24.Ouimet M, Franklin V, Mak E, Liao X, Tabas I, and Marcel YL (2011). Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 13, 655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Danielsen ET, Moeller ME, Yamanaka N, Ou Q, Laursen JM, Soenderholm C, Zhuo R, Phelps B, Tang K, Zeng J, et al. (2016). A Drosophila Genome-Wide Screen Identifies Regulators of Steroid Hormone Production and Developmental Timing. Dev. Cell 37, 558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Talamillo A, Herboso L, Pirone L, Perez C, Gonzalez M, Sanchez J, Mayor U, Lopitz-Otsoa F, Rodriguez MS, Sutherland JD, et al. (2013). Scavenger receptors mediate the role of SUMO and Ftz-f1 in Drosophila steroidogenesis. PLoS Genet. 9, e1003473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang X, Suyama K, Buchanan J, Zhu AJ, and Scott MP (2005). A Drosophila model of the Niemann-Pick type C lysosome storage disease: dnpc1a is required for molting and sterol homeostasis. Development 132, 5115–5124. [DOI] [PubMed] [Google Scholar]
- 28.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, and Czaja MJ (2009). Autophagy regulates lipid metabolism. Nature 458, 1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Advani RJ, Yang B, Prekeris R, Lee KC, Klumperman J, and Scheller RH (1999). VAMP-7 mediates vesicular transport from endosomes to lysosomes. J. Cell Biol 146, 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jean S, Cox S, Nassari S, and Kiger AA (2015). Starvation-induced MTMR13 and RAB21 activity regulates VAMP8 to promote autophagosome-lysosome fusion. EMBO Rep. 16, 297–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mauvezin C, Nagy P, Juhasz G, and Neufeld TP (2015). Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat. Commun 6, 7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dermaut B, Norga KK, Kania A, Verstreken P, Pan H, Zhou Y, Callaerts P, and Bellen HJ (2005). Aberrant lysosomal carbohydrate storage accompanies endocytic defects and neurodegeneration in Drosophila benchwarmer. J. Cell Biol 170, 127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rong Y, McPhee CK, Deng S, Huang L, Chen L, Liu M, Tracy K, Baehrecke EH, Yu L, and Lenardo MJ (2011). Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc. Natl. Acad. Sci. USA 108, 7826–7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sweeney ST, and Davis GW (2002). Unrestricted synaptic growth in spinster-a late endosomal protein implicated in TGF-beta-mediated synaptic growth regulation. Neuron 36, 403–416. [DOI] [PubMed] [Google Scholar]
- 35.Humphries W.H.t., Szymanski CJ, and Payne CK (2011). Endo-lysosomal vesicles positive for Rab7 and LAMP1 are terminal vesicles for the transport of dextran. PLoS One 6, e26626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mauvezin C, Ayala C, Braden CR, Kim J, and Neufeld TP (2014). Assays to monitor autophagy in Drosophila. Methods 68, 134–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Itakura E, Kishi-Itakura C, and Mizushima N (2012). The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151, 1256–1269. [DOI] [PubMed] [Google Scholar]
- 39.Takats S, Nagy P, Varga A, Pircs K, Karpati M, Varga K, Kovacs AL, Hegedus K, and Juhasz G (2013). Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J. Cell Biol 201, 531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Furuta N, Fujita N, Noda T, Yoshimori T, and Amano A (2010). Combinational soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins VAMP8 and Vti1b mediate fusion of antimicrobial and canonical autophagosomes with lysosomes. Mol. Biol. Cell 21, 1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scott RC, Schuldiner O, and Neufeld TP (2004). Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev. Cell 7, 167–178. [DOI] [PubMed] [Google Scholar]
- 42.Layalle S, Arquier N, and Leopold P (2008). The TOR pathway couples nutrition and developmental timing in Drosophila. Dev. Cell 15, 568–577. [DOI] [PubMed] [Google Scholar]
- 43.Ohhara Y, Kobayashi S, and Yamanaka N (2017). Nutrient-Dependent Endocycling in Steroidogenic Tissue Dictates Timing of Metamorphosis in Drosophila melanogaster. PLoS Genet. 13, e1006583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koyama T, Rodrigues MA, Athanasiadis A, Shingleton AW, and Mirth CK (2014). Nutritional control of body size through FoxO-Ultraspiracle mediated ecdysone biosynthesis. Elife 3, e03091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng LY, Bailey AP, Leevers SJ, Ragan TJ, Driscoll PC, and Gould AP (2011). Anaplastic lymphoma kinase spares organ growth during nutrient restriction in Drosophila. Cell 146, 435–447. [DOI] [PubMed] [Google Scholar]
- 46.Texada MJ, Malita A, Christensen CF, Dall KB, Faergeman NJ, Nagy S, Halberg KA, and Rewitz K (2019). Autophagy-Mediated Cholesterol Trafficking Controls Steroid Production. Dev. Cell 48, 659–671. [DOI] [PubMed] [Google Scholar]
- 47.Clayton RB (1964). The Utilization of Sterols by Insects. J. Lipid Res 5, 3–19. [PubMed] [Google Scholar]
- 48.Gawriluk TR, Ko C, Hong X, Christenson LK, and Rucker EB 3rd (2014). Beclin-1 deficiency in the murine ovary results in the reduction of progesterone production to promote preterm labor. Proc. Natl. Acad. Sci. USA 111, E4194–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshii SR, Kuma A, Akashi T, Hara T, Yamamoto A, Kurikawa Y, Itakura E, Tsukamoto S, Shitara H, Eishi Y, et al. (2016). Systemic Analysis of Atg5-Null Mice Rescued from Neonatal Lethality by Transgenic ATG5 Expression in Neurons. Dev. Cell 39, 116–130. [DOI] [PubMed] [Google Scholar]
- 50.Kobayashi T, Beuchat MH, Lindsay M, Frias S, Palmiter RD, Sakuraba H, Parton RG, and Gruenberg J (1999). Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat. Cell Biol 1, 113–118. [DOI] [PubMed] [Google Scholar]
- 51.Mobius W, van Donselaar E, Ohno-Iwashita Y, Shimada Y, Heijnen HF, Slot JW, and Geuze HJ (2003). Recycling compartments and the internal vesicles of multivesicular bodies harbor most of the cholesterol found in the endocytic pathway. Traffic 4, 222–231. [DOI] [PubMed] [Google Scholar]
- 52.Wojtanik KM, and Liscum L (2003). The transport of low density lipoprotein-derived cholesterol to the plasma membrane is defective in NPC1 cells. J. Biol. Chem. 278, 14850–14856. [DOI] [PubMed] [Google Scholar]
- 53.Higgins ME, Davies JP, Chen FW, and Ioannou YA (1999). Niemann-Pick C1 is a late endosome-resident protein that transiently associates with lysosomes and the trans-Golgi network. Mol. Genet. Metab 68, 1–13. [DOI] [PubMed] [Google Scholar]
- 54.Ikonen E (2008). Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol 9, 125–138. [DOI] [PubMed] [Google Scholar]
- 55.Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, and Deretic V (2011). Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 30, 4701–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kanerva K, Uronen RL, Blom T, Li S, Bittman R, Lappalainen P, Peranen J, Raposo G, and Ikonen E (2013). LDL cholesterol recycles to the plasma membrane via a Rab8a-Myosin5b-actin-dependent membrane transport route. Dev. Cell 27, 249–262. [DOI] [PubMed] [Google Scholar]
- 57.Linder MD, Mayranpaa MI, Peranen J, Pietila TE, Pietiainen VM, Uronen RL, Olkkonen VM, Kovanen PT, and Ikonen E (2009). Rab8 regulates ABCA1 cell surface expression and facilitates cholesterol efflux in primary human macrophages. Arterioscler. Thromb. Vasc. Biol 29, 883–888. [DOI] [PubMed] [Google Scholar]
- 58.Linder MD, Uronen RL, Holtta-Vuori M, van der Sluijs P, Peranen J, and Ikonen E (2007). Rab8-dependent recycling promotes endosomal cholesterol removal in normal and sphingolipidosis cells. Mol. Biol. Cell 18, 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al. (2010). Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465, 942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnson AE, Shu H, Hauswirth AG, Tong A, and Davis GW (2015). VCP-dependent muscle degeneration is linked to defects in a dynamic tubular lysosomal network in vivo. Elife 4, e07366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ko DC, Gordon MD, Jin JY, and Scott MP (2001). Dynamic movements of organelles containing Niemann-Pick C1 protein: NPC1 involvement in late endocytic events. Mol. Biol. Cell 12, 601–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Warren JT, Petryk A, Marques G, Parvy JP, Shinoda T, Itoyama K, Kobayashi J, Jarcho M, Li Y, O’Connor MB, et al. (2004). Phantom encodes the 25-hydroxylase of Drosophila melanogaster and Bombyx mori: a P450 enzyme critical in ecdysone biosynthesis. Insect Biochem. Mol. Biol 34, 991–1010. [DOI] [PubMed] [Google Scholar]
- 63.Parvy JP, Blais C, Bernard F, Warren JT, Petryk A, Gilbert LI, O’Connor MB, and Dauphin-Villemant C (2005). A role for betaFTZ-F1 in regulating ecdysteroid titers during post-embryonic development in Drosophila melanogaster. Dev. Biol 282, 84–94. [DOI] [PubMed] [Google Scholar]
- 64.Gibbens YY, Warren JT, Gilbert LI, and O’Connor MB (2011). Neuroendocrine regulation of Drosophila metamorphosis requires TGFbeta/Activin signaling. Development 138, 2693–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ono H, Rewitz KF, Shinoda T, Itoyama K, Petryk A, Rybczynski R, Jarcho M, Warren JT, Marques G, Shimell MJ, et al. (2006). Spook and Spookier code for stage-specific components of the ecdysone biosynthetic pathway in Diptera. Dev. Biol. 298, 555–570. [DOI] [PubMed] [Google Scholar]
- 66.Kaieda Y, Masuda R, Nishida R, Shimell M, O’Connor MB, and Ono H (2017). Glue protein production can be triggered by steroid hormone signaling independent of the developmental program in Drosophila melanogaster. Dev. Biol 430, 166–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chang YY, and Neufeld TP (2009). An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol. Biol. Cell 20, 2004–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nezis IP, Shravage BV, Sagona AP, Lamark T, Bjorkoy G, Johansen T, Rusten TE, Brech A, Baehrecke EH, and Stenmark H (2010). Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. J. Cell Biol 190, 523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pulipparacharuvil S, Akbar MA, Ray S, Sevrioukov EA, Haberman AS, Rohrer J, and Kramer H (2005). Drosophila Vps16A is required for trafficking to lysosomes and biogenesis of pigment granules. J. Cell Sci 118, 3663–3673. [DOI] [PubMed] [Google Scholar]
- 70.Tapon N, Ito N, Dickson BJ, Treisman JE, and Hariharan IK (2001). The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell 105, 345–355. [DOI] [PubMed] [Google Scholar]
- 71.Warren JT, Yerushalmi Y, Shimell MJ, O’Connor MB, Restifo LL, and Gilbert LI (2006). Discrete pulses of molting hormone, 20-hydroxyecdysone, during late larval development of Drosophila melanogaster: correlations with changes in gene activity. Dev. Dyn 235, 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim J, and Neufeld TP (2015). Dietary sugar promotes systemic TOR activation in Drosophila through AKH-dependent selective secretion of Dilp3. Nat. Commun 6, 6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Enya S, Ameku T, Igarashi F, Iga M, Kataoka H, Shinoda T, and Niwa R (2014). A Halloween gene noppera-bo encodes a glutathione S-transferase essential for ecdysteroid biosynthesis via regulating the behaviour of cholesterol in Drosophila. Sci. Rep 4, 6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bolte S, and Cordelieres FP (2006). A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc 224, 213–232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Autophagy dynamics in the PG visualized by time-lapse live imaging. Related to Figure 1 and Figure S6.
Phm>mCh-Atg8a PG tissues were cultured ex vivo and observed under fluorescence microscope. Samples from fed pre-NRC (A), starved pre-NRC (B), fed post-NRC (C) and starved post-NRC (D) larvae were shown. For fed groups, PG tissues were imaged immediately upon dissection. For starvation groups larvae were starved for 6 hours before dissection and imaging process.
Video S2. Tubular network formation during autophagy induction. Related to Figure 1 and Figure S6.
Pre-NRC larvae were starved for 6 hours and then dissected for imaging. The video shows a representative event of tubular structure movement within a tubule network. Representative frames from this movie are shown in Figure. S6C.
Video S3. Tubule expansion and retraction during autophagy induction. Related to Figure 1 and Figure S6.
Representative frames from this movie are shown in Figure. S6D.
Video S4. A tubular structure fuses with neighboring vesicle during autophagy induction. Related to Figure 1 and Figure S6.
The arrow shows the site where the fusion occurs. Representative frames from this movie are shown in Figure. S6E.
Video S5. Fragmentation of autophagic structure during autophagy induction. Related to Figure 1 and Figure S6.
The video shows a large-sized autophagic structures fragmenting into multiple smaller vesicles. Representative frames from this movie are shown in Figure. S6F.
Video S6. Formation of ring-like structure during autophagy induction. Related to Figure 1 and Figure S6.
The arrow shows the formation site of the ring-like structure. Representative frames from this movie are shown in Figure. S6G.
Data Availability Statement
This study did not generate/analyze any dataset/code.