

SUMMARY
Advances in cancer immunotherapies make it critical to identify genes that modulate antigen presentation and tumor-immune interactions. We report that DUX4, an early embryonic transcription factor that is normally silenced in somatic tissues, is re-expressed in diverse solid cancers. Both cis-acting inherited genetic variation and somatically acquired mutations in trans-acting repressors contribute to DUX4 re-expression in cancer. Although many DUX4 target genes encode self-antigens, DUX4-expressing cancers were paradoxically characterized by reduced markers of anti-tumor cytolytic activity and lower MHC Class I gene expression. We demonstrate that DUX4 expression blocks interferon-γ-mediated induction of MHC Class I, implicating suppressed antigen presentation in DUX4-mediated immune evasion. Clinical data in metastatic melanoma confirmed that DUX4 expression was associated with significantly reduced progression-free and overall survival in response to anti-CTLA-4. Our results demonstrate that cancers can escape immune surveillance by reactivating a normal developmental pathway and identify a therapeutically relevant mechanism of cell-intrinsic immune evasion.
Keywords: DUX4, cancer, immune evasion, immunotherapy
Graphical Abstract
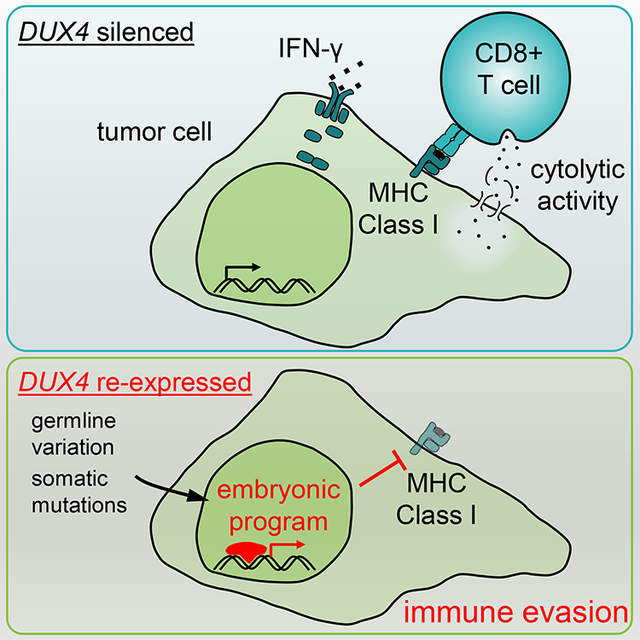
Chew and Campbell et al. report that DUX4, a preimplantation embryonic transcription factor that is normally silenced in somatic tissues, is re-expressed in many cancers. DUX4 suppresses anti-cancer immune activity by blocking interferon-γ-mediated induction of MHC Class I and is associated with reduced efficacy of immune checkpoint blockade therapy.
INTRODUCTION
Immune checkpoint blockade therapies, which act on T cell inhibitory receptors including CTLA-4 and PD-1, induce durable responses across diverse cancers. However, a majority of patients do not respond to these therapies, and initially responsive cancers may relapse (Ribas and Wolchok, 2018; Sharma et al., 2017; Topalian et al., 2015). Identifying molecular mechanisms that influence therapeutic response and relapse is critical in order to realize the full therapeutic potential of checkpoint blockade.
The efficacy of checkpoint blockade relies upon cytotoxic T cell recognition of antigens presented by MHC Class I on malignant cells. As a consequence, genetic lesions that suppress antigen presentation or blunt tumor-immune interactions can permit malignant cells to evade cytotoxic T cells. For example, loss-of-function mutations in B2M, JAK1, and JAK2, resulting in loss of MHC Class I expression (B2M) or response to interferon-γ (JAK1 and JAK2), have been identified in patients who relapsed following an initial response to checkpoint blockade (Sade-Feldman et al., 2017; Zaretsky et al., 2016). Copy number alterations affecting MHC Class I and interferon-γ response genes are likewise enriched in cancers that never respond to these therapies (Gao et al., 2016; Sade-Feldman et al., 2017). Tumors can also evade immune recognition by activating specific gene expression programs. For example, activation of WNT/β-catenin signaling promotes T cell exclusion from the melanoma microenvironment (Spranger et al., 2015), while depletion of LSD1 promotes anti-tumor immune activity (Sheng et al., 2018). Other modulators of tumor-immune interactions continue to be revealed by genetic screens and other methods (Manguso et al., 2017; Pan et al., 2018; Patel et al., 2017).
We performed a pan-cancer analysis of tumor transcriptomes in order to identify potential regulators of tumor-immune interactions. We sought to identify genes whose expression was restricted to cancers or immune-privileged sites such as the testes and early embryo. While such approaches have been historically used to identify cancer-testis (CT) antigens-proteins whose expression is normally restricted to the embryo and/or germ cells, but which can become re-expressed and antigenic in cancer (Caballero and Chen, 2009)-we hypothesized that such a search might also reveal regulators of antigen presentation and immune modulation. We therefore undertook an unbiased search for such genes using the transcriptomes of 9,759 samples from 33 distinct cancer types, 704 associated peritumoral normal samples, and 34 tissues from healthy individuals. Our analysis revealed that DUX4, an early embryonic transcription factor that is normally silenced in somatic tissues, is re-expressed in many solid cancer types. DUX4 re-expression in cancer results in suppression of MHC Class I-dependent antigen presentation, immune evasion, and resistance to immune checkpoint blockade.
RESULTS
Large-scale identification of genes with cancer-specific expression patterns
We sought to identify genes that were expressed in multiple cancer types, but not in corresponding peritumoral normal tissues or other somatic tissues isolated from healthy individuals. We compared the transcriptomes of 9,759 cancer samples from 33 distinct cancers (The Cancer Genome Atlas, TCGA) to the transcriptomes of 34 normal tissues, including peritumoral normal tissues (TCGA) and somatic tissues of healthy individuals (Illumina Human Body Map 2.0 and GTEx), as well as protein-level estimates from the Human Proteome Map (Kim et al., 2014; Kosti et al., 2016). We computed a quantitative measure of cancer-specific expression for each gene that was proportional to the numbers of cancer samples and types in which the gene was expressed and inversely proportional to the numbers of peritumoral normal samples and other healthy somatic tissues exhibiting detectable expression of the gene (Fig. 1A).
Figure 1. Identification of genes with cancer-specific expression.

(A) Overview of data sources and our strategy for identifying cancer-specific gene expression. We compared the expression of each gene in cancer samples (TCGA) to its corresponding expression in peritumoral samples (TCGA) and normal tissue from healthy individuals (Illumina Body Map 2.0, Human Proteome Map, and Genotype-Tissue Expression Project, GTEx). We defined the cancer specificity score for each gene as the logarithm of the fractions of cancer samples and types in which the gene was expressed divided by the fractions of peritumoral samples and normal tissues in which the gene was expressed.
(B) Ranked plot of cancer specificity scores of coding genes, restricted to genes that are not expressed in all tissue types. The double homeobox genes DUX4, DUXA, and DUXB are highlighted.
(C) Expression of cancer-specific genes across cancer types and samples. Each point corresponds to a gene highlighted in red in (B). y axis, number of cancer types (TCGA primary site) with at least one DUX4+ sample; x axis, total number of DUX4+ samples, irrespective of cancer type.
(D) DUX4 mRNA levels in DUX4+ cancer samples and during early embryogenesis (Hendrickson et al., 2017). TPM, transcripts per million.
See also Figure S1.
Our quantitative cancer-specific expression score allowed us to rank each gene according to its relative level of expression in malignant versus normal somatic tissue (Fig. 1B, S1A, Table S1). Our analysis highlighted many genes with known roles in tumorigenesis, including OCT4 pseudogenes (Hayashi et al., 2013) and genes that are recurrently translocated in cancer, such as TLX3 and members of the SSX gene family (Smith and McNeel, 2010). Many genes that exhibited the most cancer-specific expression patterns encoded known CT antigens, including members of the GAGE, MAGE, PAGE, PRAME, and SPANX gene families.
Given that CT antigens were strongly enriched among the most cancer-specific genes, we tested whether other classes of genes were preferentially expressed in cancers. We performed a Gene Ontology (GO)-based comparison of the 500 highest-scoring genes against a background set of non-ubiquitously expressed genes. We used non-ubiquitously expressed genes as a background set in order to avoid confounding our analysis with housekeeping genes. Genes involved in spermatogenesis comprised ~5% of the most cancer-specific genes (False Discovery Rate (FDR) < 10−3), consistent with our identification of many CT antigens. Unexpectedly, the most enriched biological pathway was transcriptional regulation (FDR < 10−5), which encompassed 19% of the highest-ranked genes. 28 of the 500 highest-ranked genes encode sequence-specific transcription factors, many of which are normally expressed only in germ cells or the embryo. Each of these factors could potentially influence tumor-immune interactions by modulating CT antigen expression, antigen presentation, or interferon signaling.
DUX4 is re-expressed in diverse cancers
Three genes (CGB5, SMC1B, and DUX4) exhibited the strongest pan-cancer signals. Expressed in testes but not in any queried somatic tissues, they each were also expressed in solid cancers arising from 26 distinct tissue types (Fig. 1B–C, S1B). SMC1B encodes a meiosis-specific subunit of cohesin (Revenkova et al., 2001); CGB5 encodes a subunit of chorionic gonadotropin; DUX4 encodes an early embryonic transcription factor (De Iaco et al., 2017; Hendrickson et al., 2017; Snider et al., 2010; Whiddon et al., 2017). Chorionic gonadotropin promotes maternal immunotolerance and is a biomarker of cancer (Kayisli et al., 2003; Stenman et al., 2004), suggesting that our ranking of cancer-specific genes enriched for potential mediators of tumor-immune interactions.
DUX4 was a particularly intriguing candidate for modulating tumor-immune interactions. DUX4 encodes a double homeobox transcription factor that acts as a pioneer factor demarcating the two-cell “cleavage” stage of early embryogenesis, after which it is epigenetically repressed (De Iaco et al., 2017; Hendrickson et al., 2017; Snider et al., 2010; Whiddon et al., 2017). DUX4 is expressed at low levels in the immune-privileged sites of the testis and thymus, but otherwise silenced in somatic tissues (Das and Chadwick, 2016; Snider et al., 2010). Several genes that are direct transcriptional targets of DUX4, such as PRAME genes, encode CT antigens that were first identified based on their cancer-specific expression and immunogenic potential (Chang et al., 2011; Ikeda et al., 1997). DUX4 was commonly expressed in cancers of the bladder, breast, cervix, endometrium, esophagus, lung, ovary, kidney, soft tissue, and stomach, and most commonly expressed in testicular germ cell cancers and thymomas (Fig. 1D).
DUX4 is expressed at physiological levels as a full-length mRNA
Since DUX4 is normally silenced in somatic cells, we first tested whether DUX4 was expressed at potentially physiologically relevant levels. We compared DUX4 mRNA levels in DUX4-expressing (DUX4+) cancer samples to DUX4 mRNA levels during early embryogenesis, including the cleavage stage, whose transcriptional program is driven by DUX4. DUX4 was typically expressed at levels ranging from ~2–10 transcripts per million (TPM) in DUX4+ cancer samples, comparable to its endogenous expression during embryogenesis (Fig. 1D).
We next confirmed that cancers expressed a full-length transcript encoding the complete DUX4 transcription factor. Testing for full-length DUX4 mRNA expression was important for three reasons. First, alternative splicing generates multiple DUX4 isoforms, of which only the longest isoform includes the DUX4 C-terminal transcription activation domain (Geng et al., 2012; Snider et al., 2010). Second, DUX4 is recurrently translocated in round-cell sarcoma and B-cell acute lymphoblastic leukemia (B-ALL) to the CIC and IGH loci, generating fusion proteins containing N- and C-terminal truncations of DUX4, respectively (Kawamura-Saito et al., 2006; Lilljebjörn et al., 2016; Liu et al., 2016; Yasuda et al., 2016; Zhang et al., 2016). Because DUX4 requires both its N-terminal DNA-binding domains and its C-terminal activation domain to activate its target genes (Choi et al., 2016), neither fusion protein preserves endogenous DUX4 function. Third, DUX4 exhibits high sequence homology to its paralog DUX4C, such that errors in short read alignment could potentially confound estimates of DUX4 versus DUX4C mRNA levels.
We assessed DUX4 alternative splicing by testing whether the long or short isoform of DUX4 was preferentially expressed in cancers. We identified spliced reads that unambiguously distinguished between the two isoforms in approximately one-third of DUX4+ samples. Only four DUX4+ samples exhibited any evidence of short isoform expression, and in each case, we observed only one or two reads supporting the short isoform (data not shown). The vast majority of expressed DUX4 mRNA arose from the long isoform containing the complete open reading frame.
We next tested whether DUX4 was expressed in solid cancers as a full-length DUX4 mRNA, or instead as a truncated DUX4 mRNA consisting of the 5’ end of the DUX4 mRNA fused to another gene product (as occurs in B-ALL). We aligned reads from each DUX4+ solid cancer, preimplantation embryos, and B-ALL with DUX4 translocations to the full-length DUX4 mRNA sequence. Read coverage extended across the full-length DUX4 mRNA in preimplantation embryos, as expected, as well as in all DUX4+ solid cancers (Fig. 2A–C). In contrast, reads aligned only to the 5’-most half of the DUX4 mRNA in B-ALL, consistent with the known presence of DUX4 translocations in these leukemias (Fig. 2D). We did not observe read coverage patterns consistent with expression of a fusion gene encoding an N- or C-terminally truncated fragment of DUX4 in any DUX4+ solid cancer (Fig. S2A–B).
Figure 2. DUX4 is expressed as a full-length mRNA in diverse solid cancers.

(A) Schematic of DUX4 isoform that encodes the full-length transcription factor. Red, sequence encoding the DNA-binding homeodomains (HB1 and HB2); yellow, sequence encoding the C-terminal activation domain.
(B-D) Read coverage of the DUX4 isoform illustrated in (A) in embryos (Hendrickson et al., 2017), representative DUX4+ solid cancers, and B-ALL with DUX4-IGH translocations. Gray shaded box, open reading frame. Plot based on image from IGV.
(E) Numbers of RNA-seq reads containing DUX4’s polyadenylation site in its third exon (Exon 3 PAS), where each point corresponds to a single sample and is classified based on whether reads from that sample arose from a “permissive” 4qA161 allele or a “non-permissive” (10qA or 4qB) allele. The analyzed data are from poly(A)-selected libraries. ***, p < 0.001 by the one-sided Mann-Whitney U test.
(F) DUX4 mRNA levels (TPM) in samples from testicular germ cell tumors, segregated by DPPA2 and DPPA4 expression. <25th and >75th indicate the bottom and top quartiles. ***/****, p < 0.001/0.0001 by the one-sided Mann-Whitney U test. Error bars, standard deviations estimated by bootstrapping. TPM, transcripts per million.
(G) Association between predicted loss-of-function mutations affecting known or likely repressors of DUX4 expression with increased DUX4 mRNA levels. Colors indicate p-values computed with a one-sided Mann-Whitney U test. See also Fig. S2D.
(H) qRT-PCR measurement of PRPF8, DUX4, ZSCAN4, and TRIM43 mRNA levels following transfection of a pool of four PRPF8-targeting siRNAs or a control non-targeting siRNA into myoblasts isolated from a healthy (MB2401) or FSHD (MB073) individual. The FSHD myoblasts have a permissive genetic background that potentiates DUX4 expression. Error bars, standard deviation across biological replicates.
See also Figure S2.
Finally, we tested whether mis-alignment of reads from DUX4C to DUX4 was a confounding factor in our analysis. The DUX4C and DUX4 open reading frames are highly similar, with the exception of 32 residues at DUX4C’s C terminus. Identifying reads that mapped uniquely to the distinguishing regions of DUX4C and DUX4 revealed that while a few DUX4+ samples also exhibited detectable DUX4C expression, the vast majority of mapping reads aligned uniquely to DUX4 (Fig. S2C). Together with our analyses of DUX4 splicing and read coverage patterns, these data indicate that DUX4+ solid cancers express full-length DUX4 mRNA.
Genetic variation and somatic mutations contribute to DUX4 expression in cancer
Normally silenced in somatic cells, DUX4 becomes inappropriately re-expressed in the skeletal muscle of individuals with facioscapulohumeral muscular dystrophy (FSHD) due to cis- and/or trans-acting genetic variation that disrupts normal epigenetic repression of the DUX4 locus (Lemmers et al., 2012; 2010). We hypothesized that similar mechanisms might contribute to DUX4 expression in cancers. We first used reads mapping to the 3’ end of the DUX4 mRNA to assess whether the DUX4 mRNA was expressed from a “permissive” 4qA161 allele or a “non-permissive” (10qA or 4qB) allele, which respectively do or do not contain consensus polyadenylation sites that permit stable DUX4 mRNA expression (Lemmers et al., 2010; 2007). We observed significantly more reads arising from permissive alleles (Fig. 2E; p < 0.001), indicating that inherited genetic variation contributes to DUX4 expression in cancer as well as FSHD.
We next identified trans-acting regulators of DUX4 expression in cancer. Two recent studies reported that DPPA2 and DPPA4 activate expression of Dux, a murine double homeobox gene that is expressed during early embryogenesis like DUX4 (De Iaco et al., 2019; Eckersley-Maslin et al., 2019). Although mice lack DUX4 and the relationship between human DPPA2 and DPPA4 and DUX4 has not been tested, we wondered whether the same occurred in cancers. High DPPA2 and DPPA4 expression was strongly associated with DUX4 expression in testicular germ cell tumors but no other cancer types, suggesting that human DPPA2 and DPPA4 may activate DUX4 expression in some cell types (Fig. 2F). We next tested whether somatic mutations affecting known repressors of the DUX4 locus were associated with DUX4 expression. We identified all cancer samples with or without predicted loss-of-function mutations in 23 genes encoding validated or likely repressors of DUX4, including proteins encoded by Modifier of murine metastable epiallele (Momme) genes (Daxinger et al., 2013), components of the Nucleosome Remodeling Deacetylase and Chromatin Assembly Factor 1 complexes (Campbell et al., 2018), and other epigenetic factors (Haynes et al., 2018; Huichalaf et al., 2014; Ottaviani et al., 2009; Zeng et al., 2009) (Table S2). We tested whether samples with loss-of-function mutations in each gene exhibited elevated DUX4 expression relative to wild-type samples for each cancer type. Mutations in 12 tested genes were significantly associated with increased DUX4 expression in one or more cancer types (Fig. 2G, S2D), suggesting that loss of epigenetic repressors of the DUX4 locus contributes to DUX4 re-expression in cancer.
One of the strongest signals in our mutational analysis arose from PRPF8, which encodes a core spliceosomal protein. Although PRPF8 is not a known repressor of DUX4, we included PRPF8 in our analysis because we previously found that PRPF8 physically associates with the DUX4 locus (Campbell et al., 2018). We therefore experimentally tested whether PRPF8 inhibition induced DUX4 expression. We knocked down (KD) PRPF8 in myoblasts isolated from a healthy individual and an individual whose FSHD was caused by cis-acting genetic variation that potentiated DUX4 de-repression. PRPF8 KD resulted in increased DUX4 expression and up-regulation of the DUX4 targets ZSCAN4 and TRIM43, confirming the presence of transcriptionally active DUX4 protein following PRPF8 KD (Fig. 2H). The extent of up-regulation was much higher in FSHD cells, as expected. In addition to identifying PRPF8 as a repressor of DUX4 expression, our results demonstrate that both cis-acting genetic variation and somatic mutations in trans-acting repressors contribute to DUX4 expression in cancer.
DUX4 drives an early embryonic gene expression program in cancer
We next tested whether DUX4 mRNA was likely translated into a functional transcription factor in DUX4+ cancers. As a pioneer transcription factor, DUX4 induces a stereotyped cleavage- stage gene expression program, even when it is aberrantly expressed in somatic cells outside of its normal embryonic context (Fig. 3A). DUX4 also binds to and transcriptionally activates specific repetitive elements (Geng et al., 2012; Young et al., 2013), many of which characterize the cleavage-stage gene expression program (Hendrickson et al., 2017). We computed a high-confidence set of DUX4-induced target genes by intersecting the sets of genes associated with endogenous DUX4 expression during preimplantation embryogenesis as well as ectopic DUX4 expression in induced pluripotent stem cells (iPSCs) and myoblasts (Feng et al., 2015; Hendrickson et al., 2017). We additionally defined a compact set of DUX4-responsive repetitive elements that were induced irrespective of cell type (Table S3). We measured expression levels of each of these DUX4-induced genes and repetitive elements across each cancer cohort and compared their average expression in DUX4+ versus DUX4− cancers.
Figure 3. DUX4 drives an embryonic gene expression program in cancer.

(A) Differential gene expression induced by endogenous or ectopic DUX4. Each point represents mRNA levels for a single gene in samples with (y axis) or without (x axis) DUX4 expression. Plots illustrate comparisons between cleavage-stage embryos and zygotes, which have high and low DUX4 expression (left panel), iPSCs with or without DUX4 induction (center panel), and myoblasts with or without DUX4 induction (right panel) (Feng et al., 2015; Hendrickson et al., 2017). Red, high-confidence list of DUX4-induced genes identified by intersecting the sets of up-regulated genes in all three illustrated comparisons. TPM, transcripts per million.
(B) Expression of high-confidence DUX4 targets in DUX4+ cancers. Heat map illustrates the expression of each DUX4 target (rows) in each individual DUX4+ sample (columns) relative to the median expression across all DUX4− samples from that cancer type. The LSAU repetitive element corresponds to the beta satellite repeat, which is a multicopy genomic element like DUX4. Approximately 55% of LSAU’s 2,759 nt consensus sequence overlaps with part of DUX4’s consensus sequence, so its expression is invariably higher in DUX4+ samples when expression is quantified via short-read sequencing.
See also Figure S3.
DUX4 expression was strongly associated with increased expression of DUX4 targets, including coding genes, non-coding genes, and repetitive elements (Fig. 3B). Several trends were notable. First, while DUX4+ cancers exhibited increased expression of many DUX4-induced genes irrespective of cancer type, one or more specific target genes were particularly highly expressed in each cancer type. For example, almost all DUX4+ thymomas expressed high levels of the DUX4 target CCNA1, while few bladder or breast cancers did. Second, the quantitative level of DUX4 target induction was highly variable across DUX4+ cancer samples, and was only modestly correlated with DUX4 expression levels. Third, most DUX4 targets were strongly induced in almost all DUX4+ testicular germ cell cancers. Testicular germ cell cancers might constitute unusually permissive environments for DUX4 activity, perhaps because DUX4 is endogenously expressed in luminal cells in the testis, most likely in the germline (Snider et al., 2010). We conclude that DUX4 drives an early embryonic transcriptional program in diverse solid cancers.
DUXB is not essential for the core DUX4 transcriptional program in cancer
A recent study reported that Duxbl was recurrently amplified in a murine model of rhabdomyosarcoma and that its human ortholog DUXB was expressed in many human cancers (Preussner et al., 2018). Although we also observed frequent DUXB expression in cancers, DUXB did not rank highly on our cancer specificity index because it is expressed in many healthy tissues (Fig. 1B). Nonetheless, as DUXB expression is promoted by DUX4 (Fig. 3A–B), we wondered whether DUXB might contribute to the DUX4-induced gene expression program. We established a myoblast cell line with a doxycycline-inducible DUXB transgene and performed RNA-seq on cells with or without doxycycline treatment. Although DUXB lacks DUX4’s C-terminal transcriptional activation domain, DUXB induction resulted in statistically significant up-regulation of a small set of genes. However, DUXB had no effects on our high-confidence set of DUX4 targets (Fig. S3B–C, Table S4), suggesting that it is not essential for the DUX4-driven embryonic expression program in cancer.
DUX4 is associated with reduced anti-tumor immune activity
As many DUX4 targets encode CT antigens (Chang et al., 2011; Ikeda et al., 1997), we wondered whether DUX4 re-expression might promote anti-tumor immune activity. We therefore tested whether DUX4-expressing cancers exhibited gene expression signatures of high immune infiltration. To our surprise, multiple lines of evidence indicated that DUX4 expression was associated with decreased, rather than increased, anti-tumor immune activity. First, we identified genes that were consistently differentially expressed in multiple cancer types in DUX4+ versus DUX4− samples and performed a Gene Ontology (GO)-based analysis of enriched functional categories (Fig. S3A). Almost all enriched GO terms belonged to immune-related categories. However, contrary to our hypothesis that DUX4 expression might trigger immune surveillance, the immune-related GO terms were uniformly associated with decreased gene expression in DUX4+ versus DUX4− cancers (Fig. 4A). Second, we noticed that many immune cell-specific genes were downregulated in DUX4+ cancers, suggesting that reduced immune cell infiltration might underlie this GO enrichment signature (Table S5). We therefore estimated infiltration of different immune cell types in DUX4+ and DUX4− cancers with the TIMER algorithm (Li et al., 2017). DUX4 expression was associated with reduced infiltration of diverse immune cells, most notably cytotoxic CD8+ T cells, in many cancers (Fig. 4B, S4A–B). Natural killer (NK) cell-specific markers exhibited similarly reduced expression in many DUX4+ cancer types (Fig. S4C). Third, we estimated anti-tumor immune activity by measuring the expression of GZMA and PRF1, which encode two key cytolytic factors expressed by cytotoxic T cells and NK cells (Rooney et al., 2015), to find that cytolytic activity was markedly lower in most DUX4+ versus DUX4− cancers (Fig. 4C, S4D).
Figure 4. DUX4 is associated with cancer immune evasion.

(A) Gene Ontology (GO) terms that were enriched for genes that were differentially expressed in DUX4+ versus DUX4− samples across multiple cancer types. Plot is restricted to the child-most GO terms with a False Discovery Rate (FDR) ≤ 0.01 after Benjamini–Hochberg correction. Circle areas are proportional to −log10 (FDR).
(B) Estimated CD8+ T cell infiltration in DUX4− and DUX4+ cancers, where infiltration was estimated with the TIMER method (Li et al., 2017). */**, p < 0.05/0.01 by the one-sided Mann-Whitney U test.
(C) Mean estimated cytolytic activity in DUX4− and DUX4+ cancers. Cytolytic activity was estimated as the geometric mean of GZMA and PRF1 gene expression (Rooney et al., 2015). Error bars, standard deviations estimated by bootstrapping.
(D) Mean expression of canonical MHC Class I genes, where the mean was computed over all DUX4− and DUX4+ cancers for each type. Error bars, standard deviations estimated by bootstrapping. TPM, transcripts per million.
(E) As (D), but for the illustrated datasets. Feng et al. 2015 introduced DUX4 via lentivirus into 54–1 and MB135 myoblasts (Feng et al., 2015), Rickard et al. 2015 sorted DUX4− and DUX4+ myoblasts following induction of differentiation for myoblasts that spontaneously express DUX4 (Rickard et al., 2015), and Eidahl et al. 2016 transfected DUX4-expressing plasmids into WS236 myoblasts (Eidahl et al., 2016). n, number of replicates.
See also Figure S4.
As immunosuppressive regulatory T cells (Tregs) play important roles in tolerance of self-antigens (Sakaguchi et al., 2008), we wondered whether DUX4 might create an immunosuppressive environment by altering Treg recruitment. The Treg marker gene FOXP3 was significantly down-regulated in DUX4+ samples in a few, although not most, cancer types (Fig. S4E). However, this association did not persist when we estimated Treg infiltration with the more sophisticated CIBERSORT algorithm (Newman et al., 2015; Thorsson et al., 2018) (Fig. S4F), suggesting that DUX4-mediated immunosuppression is likely not explained by Treg recruitment.
DUX4 suppresses MHC Class I expression
As DUX4 promotes CT antigen expression yet DUX4+ cancers exhibited low anti-tumor immune activity, we wondered whether antigen presentation might be suppressed in DUX4-expressing cells. Reduced expression or loss of the HLA (Human Leukocyte Antigen) or B2M (β2 microglobulin) genes, which encode MHC Class I molecules that display peptides for immune recognition, is a common mechanism by which cancers evade immune surveillance (Shukla et al., 2015). We therefore compared the expression levels of MHC Class I genes in DUX4+ and DUX4− cancers to find that DUX4 expression was associated with reduced expression of B2M, HLA-A, HLA-B, and HLA-C in most cancer types (Fig. 4D).
Decreased expression of MHC Class I genes could be a direct consequence of DUX4 expression, or alternatively might be an independent event that enhances the survival of DUX4-expressing cancers. For example, cancers that evade immune surveillance might be under reduced immune selection, enabling them to subsequently express DUX4 and its antigenic targets without deleterious consequences. To distinguish between those possibilities, we studied acute DUX4 expression in cell culture. We re-analyzed two RNA-seq datasets in which DUX4 was ectopically expressed in DUX4− cells (Eidahl et al., 2016; Feng et al., 2015) and one dataset in which differentiating myoblasts that spontaneously expressed DUX4 were flow-sorted into DUX4+ and DUX4− pools (Rickard et al., 2015). We found that DUX4-expressing cells exhibited reduced levels of MHC Class I genes relative to DUX4− control cells in all three datasets (Fig. 4E). As immunoediting is not a potential confounding factor in these cell culture experiments, we conclude that DUX4 is a cell-intrinsic suppressor of MHC Class I.
DUX4 suppresses interferon-γ-mediated induction of MHC Class I-dependent antigen presentation
Tumor-infiltrating immune cells induce antigen presentation on malignant cells by secreting interferon-γ, resulting in up-regulation of MHC Class I genes via JAK1, JAK2, and STAT1-dependent signal transduction (Friedman et al., 1984; Schroder et al., 2004). We noted that DUX4 expression was associated with reduced expression of JAK1, JAK2, and/or STAT1 in many cancer types (Fig. 5A). JAK1, JAK2, and STAT1 exhibited similarly reduced expression following acute DUX4 expression in cultured myoblasts (Fig. 5B) as well as after the onset of endogenous DUX4 expression in preimplantation embryos (Fig. 5C). Finally, we noted that JAK1, JAK2, and STAT1 each exhibited multiple peaks of DUX4 binding in our published ChIP-seq dataset of acute DUX4 expression in cultured myoblasts (Fig. S5A) (Geng et al., 2012).
Figure 5. DUX4 blocks interferon-γ-mediated up-regulation of MHC Class I-dependent antigen presentation.

(A) Mean expression of genes encoding the illustrated components of the interferon-g signaling pathway, where the mean was computed over all DUX4− and DUX4+ cancers for each type. Error bars, standard deviations estimated by bootstrapping. TPM, transcripts per million.
(B) As (A), but for the illustrated datasets.
(C) As (A), but illustrating gene expression during preimplantation embryonic development.
(D-I) Immunoblots probing MHC Class I, DUX4, and GAPDH protein following treatment of the indicated cell lines, each of which was engineered to contain a doxycycline-inducible DUX4 expression construct, with interferon-γ (IFN-γ) and/or doxycycline (Dox) to induce DUX4. a-MHC I, pan-MHC Class I probe.
(J-K) Levels of MHC Class I on the cell surface following treatment of the indicated cell lines with interferon-γ (IFN-γ) and/or doxycycline (Dox) to induce DUX4. Cell surface levels of MHC Class I were probed with a pan-MHC Class I antibody.
See also Figure S5.
We therefore tested whether DUX4 could block interferon-g-mediated induction of MHC Class I. We initially used MB135iDUX4 cells, which permit doxycycline-inducible DUX4 expression in a myoblast cell line. MB135iDUX4 cells are a validated model of the biological consequences of DUX4 expression that recapitulate the cleavage-stage transcriptional program (Hendrickson et al., 2017; Jagannathan et al., 2016; Whiddon et al., 2017). Treatment with interferon-γ resulted in robust up-regulation of MHC Class I protein, as expected, while inducing DUX4 by adding doxycycline effectively blocked this up-regulation of MHC Class I (Fig. 5D). DUX4 induction drove expression of the DUX4-activated embryonic gene ZSCAN4 independent of interferon-g treatment while suppressing interferon-γ-mediated up-regulation of B2M, HLA-A, HLA-B, and HLA-C mRNA (Fig. S5B–C). Adding doxycycline in the absence of the inducible DUX4 construct had no effect on interferon-γ-mediated induction of MHC Class I (Fig. S5D).
We next tested whether DUX4 blocked interferon-γ-mediated induction of MHC Class I protein in cancer cells. We introduced a doxycycline-inducible DUX4 construct into six cell lines derived from five cancer types: breast adenocarcinoma (MCF-7), cervical cancer (HeLa), melanoma (Mel375 and Mel526), rhabdomyosarcoma (A204), and testicular teratocarcinoma (SuSa). Treatment with interferon-γ induced higher levels of MHC Class I protein in all tested cell lines, and DUX4 suppressed this induction (Fig. 5E–I). Treating the parental cell lines with doxycycline had no effect on interferon-γ-mediated induction of MHC Class I (Fig. S5D).
Finally, we confirmed that DUX4 blocked interferon-γ-mediated induction of MHC Class I-dependent antigen presentation. As peptide binding is required for MHC Class I stability and cell surface localization (Townsend et al., 1989), we used flow cytometry to measure how DUX4 affected levels of MHC Class I on the cell surfaces of representative untransformed (MB135) and cancer (HeLa) cell lines. Interferon-γ treatment drove robust up-regulation of MHC Class I on the cell surface, as expected, while DUX4 abrogated this induction in both cell types (Fig. 5JK, S5E–F). DUX4-mediated suppression of MHC Class I was particularly notable in HeLa cells, where DUX4 suppressed cell surface levels of MHC Class I beyond the basal state even in the presence of interferon-γ. Together, these data demonstrate that DUX4 blocks interferon-γ-mediated induction of MHC Class I and antigen presentation in untransformed and cancer cells.
DUX4 promotes resistance to immune checkpoint blockade
As immune checkpoint blockade relies upon antigen presentation, resistance to these therapies is strongly associated with loss of antigen presentation as well as loss of interferon-γ signal transduction (Gao et al., 2016; Sade-Feldman et al., 2017; Zaretsky et al., 2016). We therefore hypothesized that DUX4-mediated suppression of antigen presentation might promote resistance to checkpoint blockade. We analyzed RNA-seq data from pretreatment biopsies across two cohorts in which patients with metastatic melanoma were treated with anti-CTLA-4 (Van Allen et al., 2015) or anti-PD-1 (Hugo et al., 2016). Biopsies from patients whose disease was clinically classified as non-responsive to anti-CTLA-4 exhibited significantly higher levels of DUX4 transcriptional activity relative to biopsies from responsive patients (Fig. 6A). Stratifying patients according to DUX4 transcriptional activity revealed dramatic and statistically significant differences in both progression-free and overall survival following anti-CTLA-4 therapy (Fig. 6B–C). Increased DUX4 transcriptional activity was similarly associated with failure to respond to anti-PD-1 therapy (Fig. 6A) and decreased overall survival following anti-PD-1 treatment (Fig. 6D), although the differences were not statistically significant, perhaps because of the smaller size of the anti-PD-1 cohort (27 versus 41 patients) or a more predictive role for MHC
Figure 6. DUX4 expression is associated with resistance to immune checkpoint blockade.

(A) Fractions of DUX4 target genes (Table S3) that are expressed in pre-treatment biopsies taken from metastatic melanoma patients who received anti-CTLA-4 (Van Allen et al., 2015) or anti-PD-1 (Hugo et al., 2016) therapy. Clinical responses were classified in the original studies according to RECIST (anti-CTLA-4) (Eisenhauer et al., 2009) or irRECIST (anti-PD-1) (Wolchok et al., 2009). p-value computed with the Wilcoxon rank-sum test.
(B) Progression-free survival for patients treated with anti-CTLA-4 whose pre-treatment biopsies fell within the top or bottom terciles of DUX4 target gene expression. p-value computed with the log-rank test.
(C) As (B), but for overall survival.
(D) As (B), but for the cohort of patients treated with anti-PD-1.
Class I expression in response to anti-CTLA-4 than anti-PD-1 therapy (Rodig et al., 2018). We conclude that DUX4-mediated suppression of MHC Class I-dependent antigen presentation is a clinically relevant biomarker for response to immune checkpoint blockade.
DISCUSSION
Our finding of DUX4 expression in diverse solid cancers was notable for several reasons. First, as DUX4 acts as a pioneer transcription factor that contributes to zygotic genome activation, DUX4 re-expression in cancer provides a functional link between early embryogenesis and cancer (De Iaco et al., 2017; Hendrickson et al., 2017; Whiddon et al., 2017). Second, prior to the recent discovery of DUX4’s embryonic role, DUX4 was best known for causing FSHD when aberrantly expressed in skeletal muscle due to genetic variation that causes loss of its normal epigenetic repression in somatic tissues (Lemmers et al., 2010; 2012). Third, while DUX4’s normal transcription factor activity has not been previously reported to play a role in cancer, the presence of recurrent translocations involving the DUX4 locus in round-cell sarcoma and B-ALL strongly suggests that DUX4 has pro-oncogenic capacity, at least when expressed as part of the CIC-DUX4 or DUX4-IGH fusion proteins (Kawamura-Saito et al., 2006; Lilljebjörn et al., 2016; Liu et al., 2016; Yasuda et al., 2016; Zhang et al., 2016). Finally, our observation that DUX4 activity is significantly associated with failure to respond to immune checkpoint blockade (Fig. 6) provides a clinical motivation for determining when, where, and how DUX4 becomes re-expressed in cancers. Future prospective studies of larger cohorts are essential to confirm our results and test the clinical utility of using DUX4 activity as a predictive biomarker of response to checkpoint blockade.
DUX4 is aberrantly expressed in both FSHD muscle and cancers, but the physiological consequences of DUX4 expression in these two disease states are quite different. Sustained expression of DUX4 in skeletal muscle causes apoptosis (Eidahl et al., 2016; Kowaljow et al., 2007), in contrast to DUX4’s importance during early embryogenesis and apparent compatibility with many malignancies. Determining why early embryos and cancer cells can tolerate DUX4 expression, while muscle cells cannot, may give insight into possible mechanisms for treating FSHD. Another notable difference is the frequent presence of inflammation and lymphocytic infiltration in FSHD muscle (Arahata et al., 1995) versus reduced immune infiltration in DUX4+ cancers. As DUX4 suppresses MHC Class I in both untransformed muscle cells and cancer cells (Fig. 5D–I, S5B–D), further work is required to determine why DUX4 expression results in immune attack in FSHD muscle but immune evasion in cancers.
Our finding of full-length DUX4 expression and transcriptional activity in diverse solid cancers is mechanistically distinct from the prior identification of DUX4 translocations in other cancer types. All DUX4-IGH fusion proteins lack DUX4’s C-terminal activation domain and do not activate DUX4 target gene expression (Lilljebjörn et al., 2016; Liu et al., 2016; Tanaka et al., 2018; Yasuda et al., 2016; Zhang et al., 2016). The CIC-DUX4 fusion protein combines CIC’s high mobility group-box DNA-binding domain with the transcriptional activation domain of DUX4, and so dysregulates CIC target genes but should not activate DUX4 target gene expression (Specht et al., 2014). While the DUX4-IGH and CIC-DUX4 fusion proteins likely possess oncogenic capacities, neither activates the gene expression program that is characteristic of totipotent embryonic cells expressing full-length DUX4.
Since DUX4 re-expression is common in many cancers, why has it not been previously detected? First, DUX4 is a multicopy gene that lies within the D4Z4 macrosatellite repeat array in the subtelomeric region of chromosome 4q (Gabriëls et al., 1999; Lee et al., 1995). The repetitive nature of DUX4’s genomic locus and its highly variable copy number in the human population (Wijmenga et al., 1993) render it difficult to study, possibly hindering its prior identification as a cancer-specific gene. The continued development of experimental and computational techniques for querying repetitive genomic loci may facilitate the identification of additional genes like DUX4 that play unexpected roles in cancer. Second, the DUX4 mRNA is rapidly turned over by nonsense-mediated decay (Feng et al., 2015) and is present at relatively low abundance in its normal developmental context as well as in cancers. Third, because the cleavage-stage embryonic transcriptome that DUX4 activates was only recently characterized (De Iaco et al., 2017; Hendrickson et al., 2017; Whiddon et al., 2017), it would not have been revealed by Gene Ontology-like enrichment analyses of cancer-expressed genes. For all of these reasons, quantifying expression of the genes that we identified as robust DUX4 targets in both embryos and cancers (Table S3) may prove to be an efficient method for identifying DUX4-expressing cancers.
Our data implicate DUX4 in MHC Class I-dependent antigen presentation, but do not exclude the possibility that DUX4 regulates tumor-immune interactions via other mechanisms as well. For example, DUX4 could influence T cell exclusion from the tumor microenvironment. We noted that many DUX4+ cancers exhibited reduced levels of the chemoattractants CXCL9 and CXCL10 (Fig. S5G), which promote lymphocyte recruitment to tumors (Homey et al., 2002). We experimentally confirmed that DUX4 prevented interferon-g-stimulated increases in CXCL9 and CXCL10 mRNA in myoblasts (Fig. S5H), suggesting that DUX4 may influence chemokine signaling in addition to altering antigen presentation. Further work is required to confirm that DUX4 influences CXCL9 and CXCL10 levels in cancer cells and identify all of the potentially diverse means by which DUX4 contributes to immune evasion.
In addition to facilitating immune evasion, DUX4 might promote tumorigenesis through additional mechanisms that regulate normal early embryonic development. For example, the DUX4 target ZSCAN4 is required for telomere maintenance and extension in embryonic stem cells (Zalzman et al., 2010). ZSCAN4 is activated in most DUX4+ solid cancers, where it may similarly contribute to the replicative potential of these cancers. Another example is the DUX4 target CCNA1, which encodes an A-type cyclin that is essential for male meiosis (Liu et al., 1998) and aberrantly expressed in many myeloid malignancies (Krämer et al., 1998). Ectopic expression of CCNA1 in the murine hematopoietic lineage caused abnormal myelopoiesis and sporadic progression to acute myeloid leukemia (Liao et al., 2001). These are just two examples illustrating how DUX4 targets’ normal roles in the totipotent cleavage-stage embryo and germ cells may contribute to tumorigenesis. While the functional roles of many DUX4 target genes (Table S3) are undefined, we hypothesize that other DUX4 targets may directly contribute to cancer initiation and progression.
As others have long noted, preimplantation embryos bear many qualitative similarities to malignant cells, including de-/re-programming of cell state, infinite replicative potential, and the capacity to effectively invade other tissues (Ben-Porath et al., 2008; Hanahan and Weinberg, 2000; Monk and Holding, 2001; Pierce, 1983). As DUX4 activates the transcriptional program defining the cleavage stage of early embryogenesis, DUX4 targets and downstream factors presumably enable preimplantation embryos to acquire these cancer-like characteristics. DUX4’s ability to suppress MHC Class I-dependent antigen presentation provides a mechanistic connection between preimplantation development and immune evasion, which is now widely recognized as a hallmark of cancer (Hanahan and Weinberg, 2011). Our discovery that diverse cancers express transcriptionally active DUX4 suggests a model whereby re-expression of an embryonic transcription factor activates an early developmental program characteristic of totipotent cells, resulting in the transformation of somatic cells into malignancies that can evade immune destruction.
STAR★Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents and resources may be directed to and will be fulfilled by the lead contact, Dr. Robert K. Bradley, at Fred Hutchinson Cancer Research Center (rbradley@fredhutch.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines.
A204 cells were purchased from ATCC (Cat# HTB-82) and have been cultured in the Tapscott laboratory since 2012. HeLa cells were purchased from ATCC (Cat# CCL-2) and have been cultured long-term in the Tapscott laboratory. All myoblast lines were obtained as de-identified primary cells from the Fields Center for FSHD and Neuromuscular Research at the University of Rochester Medical Center (https://www.urmc.rochester.edu/neurology/fields-center.aspx) and immortalized by retroviral transduction of CDK4 and hTERT (Stadler et al., 2011) in the Tapscott laboratory. MB135iDUX4 cells have also been described previously (Jagannathan et al., 2016). MCF-7 cells were purchased from ATCC (Cat# HTB-22) and have been cultured in the Tapscott laboratory since 2017. Mel375 and Mel526 cells were generously provided by Dr. Seth Pollack (Pollack et al., 2012). SuSa cells were purchased from DSMZ (Cat# ACC 747) and have been cultured in the Tapscott laboratory since 2014.
Cell culture.
A204 and A204iDUX4 cells were maintained in RPMI 1640 Medium (Gibco) supplemented with 10% HyClone Fetal Bovine Serum (GE Healthcare Life Sciences), 100 U/100 μg penicillin/streptomycin (Gibco), and, for A204iDUX4, 1.5 μg/ml puromycin (Sigma-Aldrich). HeLa and HeLaiDUX4 cells were maintained in Dulbecco’s Modified Eagle Medium (Gibco) supplemented with 10% HyClone Fetal Bovine Serum, 100 U/100 μg penicillin/streptomycin, and, for HeLaiDUX4, 1.5 μg/ml puromycin. MB2401, MB073, MB135, MB135iDUX4, and MB135iDUXB myoblasts were maintained in Ham’s F-10 Nutrient Mix (Gibco) supplemented with 20% HyClone Fetal Bovine Serum, 100 U/100 μg penicillin/streptomycin (Gibco), 10 ng/ml recombinant human basic fibroblast growth factor (Promega Corporation), 1 μM dexamethasone (Sigma-Aldrich), and, for MB135iDUX4 and MB135iDUXB, 3 μg/ml or 2 μg/ml puromycin (Sigma-Aldrich), respectively. MCF-7 and MCF-7iDUX4 cells were maintained in Eagle’s Minimal Essential Medium (Quality Biological) supplemented with 10% HyClone Fetal Bovine Serum, 100 U/100 μg penicillin/streptomycin, 10 μg/ml insulin (Sigma-Aldrich), and, for MCF7iDUX4, 1.5 μg/ml puromycin. Mel375, Mel375iDUX4, Mel526, and Mel526iDUX4 cells were maintained in RPMI 1640 Medium containing HEPES (Gibco) supplemented with 10% HyClone Fetal Bovine Serum, 100 U/100 μg penicillin/streptomycin, 2 mM L-Glutamine (Gibco), 1% MEM Non-Essential Amino Acids Solution (Gibco), 1 mM Sodium Pyruvate (Gibco), and, for Mel375iDUX4 and Mel526iDUX4,2 μg/ml or 1 μg/ml puromycin, respectively. SuSa and SuSaiDUX4 cells were maintained in RPMI 1640 Medium supplemented with 10% HyClone Fetal Bovine Serum, 100 U/100 μg penicillin/streptomycin, and, for SuSaiDUX4, 1 μg/ml puromycin. A204iDUX4, HeLaiDUX4, MCF-7iDUX4, Mel375iDUX4, Mel526iDUX4, SuSaiDUX4, and MB135iDUXB cell lines were generated as previously described for MB135iDUX4 cells (Jagannathan et al., 2016).
METHOD DETAILS
Data sources.
RNA-seq reads were downloaded from CGHub (TCGA), the Gene Expression Omnibus (accession numbers GSE85632, GSE85935, GSE78220) (Eidahl et al., 2016; Hendrickson et al., 2017; Hugo et al., 2016), the NCBI sequence read archive (SRA) database (accession number SRP058319) (Rickard et al., 2015), dbGaP (accession number phs000452.v2.p1) (Van Allen et al., 2015), the Japanese Genotype-Phenotype Archive (accession number JGAS00000000047) (Yasuda et al., 2016), and the European Genomephenome Archive (accession number EGAD00001002112) (Lilljebjörn et al., 2016). Sample annotations and gene expression data were downloaded from the GTEx portal (www.gtexportal.org). ChIP-seq reads were downloaded from the Gene Expression Omnibus (accession number GSE33838) (Geng et al., 2012).
RNA-seq library preparation.
Total RNA integrity was checked using a 4200 TapeStation System (Agilent Technologies) and quantified using a Trinean DropSense 96 UV-Vis spectrophotometer (Caliper Life Sciences). The RNA-seq libraries were prepared from total RNA using the TruSeq RNA Sample Prep Kit v2 (Illumina). Library size distribution was validated using a 4200 TapeStation System. Additional library quality control, blending of pooled indexed libraries, and cluster optimization was performed using a Qubit 2.0 Fluorometer (Life Technologies-Invitrogen). RNA-seq libraries were pooled (16-plex) and clustered onto 2 flow cell lanes. Sequencing was performed using an Illumina HiSeq 2500 in high-output mode employing a single-end, 100 base read length sequencing strategy. All work was carried out by the Fred Hutch Genomics Shared Resource.
Genome annotation, RNA-seq and ChIP-seq read mapping, and gene expression estimation.
A genome annotation was created by merging the UCSC knownGene (Meyer et al., 2013), Ensembl 71 (Flicek et al., 2013), and MISO v2.0 (Katz et al., 2010) annotations. RNA-seq reads were mapped to this annotation as previously described (Dvinge et al., 2014). In brief, RSEM v1.2.4 (Li and Dewey, 2011) was modified to call Bowtie v1.0.0 (Langmead et al., 2009) with the option ‘-v 2’ and then used to map all reads to the merged genome annotation. Remaining unaligned reads were then mapped to the genome (GRCh37/hg19 assembly) and a database of potential splice junctions with TopHat v2.0.8b (Trapnell et al., 2009). All gene expression estimates were normalized using the trimmed mean of M values (TMM) method (Robinson and Oshlack, 2010). For GTEx data, per-tissue gene expression estimates were obtained by computing the median expression over all samples for a given tissue following TMM normalization. Read alignments to the full-length DUX4 cDNA were performed with Kallisto v0.43.0’s pseudoalignment function (Bray et al., 2016). Subsequent visualization of the resulting read coverage was performed with IGV (Thorvaldsdottir et al., 2013). ChIP-seq reads were mapped to the genome with Bowtie v1.0.0 (Langmead et al., 2009) with the arguments ‘-v 2 -k 1 -m 1 --best --strata’. ChIP-seq peaks were called with MACS v 2.1.1.20160309 (Zhang et al., 2008) with the arguments ‘callpeak -g hs’.
Cancer-specific expression score.
The cancer-specific expression score of each gene was defined as the logarithm of the fractions of cancer samples and types in which the gene was expressed divided by the fractions of peritumoral samples and normal tissues in which the gene was expressed. We used the following thresholds to define a gene as expressed or not expression in a given sample. Following TMM normalization, each gene within each sample was classified as expressed (>1.5 TPM), not expressed (<0.5 TPM in normal tissue), or uncertain (<1.5 TPM, but >0.5 TPM). For peritumoral samples, we relaxed the threshold for defining genes as not expressed to <1.0 TPM in order to allow for potential cancer field effects, sample contamination, or other confounding factors. We defined DUX4+ and DUX4− cancer samples as those with DUX4 expression >1.5 TPM or <0.5 TPM.
DUX4 polyadenylation site usage.
The alleles from which DUX4 mRNAs were expressed in a sample were determined from diagnostic polymorphisms in exon 2 of DUX4 (Snider et al., 2010). This information was inferred from the nucleotide sequences at genomic positions chrUn_gl000228:114,025–114,056 in mapped RNA-seq reads, corresponding to the sequences shown in Table 4 of Snider et al., 2010. Mapped RNA-seq reads containing the DUX4 exon 3 polyadenylation sites were defined as reads that overlapped the ATATATTAAA sequence at chrUn_gl000228:114,642–114,651 with no mismatches.
Somatic mutation analysis.
TCGA somatic mutation calls from the Mutect pipeline (Cibulskis et al., 2013), together with their phenotypic impact as predicted by PolyPhen (Adzhubei et al., 2010) were obtained using the GDCquery_Maf function from TCGAbiolinks (Colaprico et al., 2016). Mutations were segregated as deleterious (frameshift, nonsense, and predicted deleterious mutations), low impact (silent and predicted tolerated) or other. In each cancer cohort, for each gene tested, samples with deleterious mutations were compared to samples with low impact or no mutations.
DUX4 and DUX4C comparison.
RNA-seq reads that mapped uniquely to full-length DUX4 mRNA or DUX4C mRNA were extracted using samtools view (Li et al., 2009) with the following coordinates in the GRCh37/hg19 assembly: DUX4, chrUn_gl000228:113631–113879; DUX4C, chr4:190942696–190942795 and chrUn_gl000228:26525–26624 (those two loci have identical genomic sequences). The density of reads was calculated as the total number of reads mapping to the region, normalized by the lengths of the regions queried.
DUX4 and DUX4-s isoform comparison.
Uniquely identifying splice junctions for the long and short isoforms of DUX4 mRNA were identified by their 5’ splice sites at chrUn_gl000228:113,887 and chrUn_gl000228:113,081, respectively.
DUXB differential gene expression analysis.
DUXB target genes were defined as those genes that were expressed at >5 TPM following DUXB induction, exhibited a fold-change >2 relative to uninduced samples, and had an associated Bayes factor of >10 as computed with Wagenmakers’s framework (Wagenmakers et al., 2010) in both experimental replicates.
DUX4 differential gene expression analysis.
A high-confidence set of DUX4 target genes were defined as expressed at >5 TPM in DUX4+ samples, with a greater than 4-fold change over DUX4− samples, and with a Bayes factor of >10 as computed with Wagenmakers’s framework (Wagenmakers et al., 2010) in all experimental replicates, across samples from pre-implantation embryos (Hendrickson et al., 2017), myoblasts (Feng et al., 2015) and iPSCs (Hendrickson et al., 2017).
Differential gene expression and Gene Ontology analyses.
Genes that were differentially expressed in DUX4+ versus DUX4− cancer samples were determined with a two-sided Mann-Whitney test, as implemented in wilcox.test in R, with a p-value threshold of 0.01 and a fold-change threshold of 2.0. Gene Ontology (GO) terms that were enriched amongst genes that exhibited increased or decreased expression in DUX4+ versus DUX4− samples were identified with the GOseq method (Young et al., 2010), with a false-discovery rate threshold of 0.01. The intersection between the resulting significant GO terms that were identified for each cancer type was then computed, and only the child-most terms of the resulting intersection were analyzed further and reported.
Quantification of immune cell infiltration.
Estimates of immune cell infiltration of TCGA primary tumors were downloaded from the TIMER web server (Li et al., 2017) and cross-referenced to our classification of each sample as DUX4+ or DUX4-. A one-sided Mann-Whitney U test was used to test for a statistically significant difference in immune cell infiltration between DUX4− and DUX4+ cancer samples.
Cloning.
The DUXB gene was codon altered, synthesized by IDT custom gene synthesis, and subcloned by restriction enzyme digest into the NheI and SalI sites of the pCW57.1 vector, a gift from David Root (Addgene plasmid #41393).
siRNA transfection.
FlexiTube GeneSolution siRNAs targeting PRPF8 (Cat. no. GS10594) and a non-targeting Negative Control siRNA (Cat. no. 1022076) were obtained from Qiagen. Transfection of siRNAs into myoblasts was carried out as previously described (Campbell et al., 2018).
MHC Class I immunoblotting and induction by interferon-γ.
Cells were treated with 1 μg/ml doxycycline hyclate (Sigma-Aldrich) for four hours and then stimulated with 100–200 ng/ml interferon-g (R&D Systems) for 14–17 hours. Whole-cell protein extracts were obtained by lysing cells directly in 4X SDS sample buffer (500 mM Tris-HCl pH 6.8, 8% SDS, 20% 2-mercaptoethanol, 0.004% bromophenol blue, 30% glycerol) or RIPA buffer (150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 25 mM Tris-HCl pH 7.4), followed by sonication. Protein extracts were run on NuPAGE 4–12% precast polyacrylamide gels (Invitrogen) and transferred to nitrocellulose membrane (Invitrogen). Membranes were blocked in PBS containing 0.1% Tween-20 and 5% non-fat dry milk before overnight incubation at 4 °C with primary antibodies. Membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies in block solution for 1 hour at room temperature and chemiluminescent substrate (Thermo Fisher Scientific) was used for detection on film or with a ChemiDox MP Imaging System (Bio-Rad). Membranes were stripped with Restore Western Blot Stripping Buffer (Pierce) before being re-probed.
Real-time qPCR.
Total RNA was extracted from whole cells using the NucleoSpin RNA kit (Machery-Nagel) according to the manufacturer’s instructions. Isolated RNA was treated with DNase I (Thermo Fisher Scientific), heat inactivated, and reverse transcribed into cDNA using SuperScript III (Thermo Fisher Scientific) and oligo(dT) primers (Invitrogen) following the manufacturer’s protocol. Quantitative PCR was carried out on a QuantStudio 7 Flex (Applied Biosystems) using primers specific for each mRNA and iTaq SYBR Green Supermix (Bio-Rad Laboratories, Inc.). The relative expression levels of target genes were normalized to that of reference housekeeping genes by using the delta-delta-Ct method (Livak and Schmittgen, 2001) after confirming equivalent amplification efficiencies of reference and target molecules.
Flow cytometry.
Cells were treated with 1 ug/ml doxycycline hyclate (Sigma-Aldrich) for four hours and then stimulated with 200 ng/ml interferon-γ (R&D Systems) for 16 hours. Cells were harvested using trypsin, washed with FACS Buffer (1X DPBS, 2% FBS), and then stained with a 1:50 dilution of FITC-conjugated MHC Class I antibody (Biolegend) for 30 minutes at 4°C. Following staining, cells were washed again and resuspended in FACS buffer for analysis by flow cytometry using BD LSRFortessa X-50 with BD FACSDiva software (BD Biosciences). Data were analyzed using FlowJo v10.5.3.
Antibodies.
The following antibodies were used: GAPDH (6C5) (GeneTex, Inc. GTX28245; Lot #23184, Lot #821705388, or Lot #821803139), MHC Class I (F-3) (Santa Cruz Biotechnology sc-55582; Lot #C0817 or Lot #L1118), FITC anti-human HLA-A,B,C (W6/32) (BioLegend 311404; Lot #B223038), ZSCAN4 (Invitrogen PA5-32106; Lot #SK2479411A), Peroxidase AffiniPure Goat Anti-Mouse IgG (H+L) (Jackson ImmunoResearch 115-035-146), and Peroxidase AffiniPure Goat Anti-Rabbit (H+L) (Jackson ImmunoResearch 111-035-144), and a previously described rabbit monoclonal antibody against DUX4 (E14-3) that was produced in collaboration with Epitomics (Geng et al., 2011).
Primers.
The following primers were used:
B2M F: ACTGAATTCACCCCCACTGA (Zhang et al., 2005)
B2M R: CCTCCATGATGCTGCTTACA
CXCL9 F: TCTTTTCCTCTTGGGCATCA (Zeisel et al., 2013)
CXCL9 R: TAGTCCCTTGGTTGGTGCTG
CXCL10 F: GTGGCATTCAAGGAGTACCTC (Wang et al., 2012)
CXCL10 R: TGATGGCCTTCGATTCTGGATT
DUX4 F: CGGAGAACTGCCATTCTTTC (Shadle et al., 2017)
DUX4 R: CAGCCAGAATTTCACGGAAG
HLA-A F: CGACGCCGCGAGCCAGA (Kruse et al., 2015)
HLA-A R: GCGATGTAATCCTTGCCGTCGTAG
HLA-B F: CTACCCTGCGGAGATCA (Meissner et al., 2010)
HLA-B R: ACAGCCAGGCCAGCAACA
HLA-C F: GGAGACACAGAAGTACAAGCG (Kruse et al., 2015)
HLA-C R: CGTCGTAGGCGTACTGGTCATA
PRPF8 F: ACCCAATCTCCCATAGGCAC (Zeisel et al., 2013)
PRPF8 R: AGGAAGGGCTCCACAAACTC
RPL13A F: AACCTCCTCCTTTTCCAAGC (Geng et al., 2012)
RPL13A R: GCAGTACCTGTTTAGCCACGA
RPL27 F: GCAAGAAGAAGATCGCCAAG (Shadle et al., 2017)
RPL27 R: TCCAAGGGGATATCCACAGA
TRIM43 F: ACCCATCACTGGACTGGTGT (Geng et al., 2012)
TRIM43 R: CACATCCTCAAAGAGCCTGA
ZSCAN4 F: TGGAAATCAAGTGGCAAAAA (Geng et al., 2012)
ZSCAN4 R: CTGCATGTGGACGTGGAC
siRNAs.
The following siRNAs were used:
siCTRL: AATTCTCCGAACGTGTCACGT
siPRPF8–1: ACGGGCATGTATCGATACAAA
siPRPF8–2: ATGGCTTGTCATCCTGAATAA
siPRPF8–3: CAACGTCGTCATCAACTATAA
siPRPF8–4: CTCATCGTGGACCACAACATA
Survival analyses.
Survival analyses and corresponding statistical tests were performed with the Kaplan-Meier estimator and log-rank test as implemented in the R package survival (Therneau and Grambsch, 2000).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analysis and visualization.
Data analysis was performed in the R programming environment and relied on Bioconductor (Huber et al., 2015), dplyr (Wickham and Francois), and ggplot2 (Wickham, 2009). The RT-qPCR panels were generated using GraphPad Prism Software (version 7.0, www.graphpad.com). Flow cytometry data was analyzed with FloJo (version 10.5.3, www.flowjo.com/solutions/flowjo).
DATA AND SOFTWARE AVAILABILITY
Experimental data and plasmids.
Relevant plasmids are available through Addgene (https://www.addgene.org/Stephen_Tapscott/).
Accession codes.
FASTQ files from the DUXB RNA-seq experiment have been deposited in NCBI’s GEO database (accession number GSE128917).
Supplementary Material
Table S1 – related to Figure 1.
Table of cancer-specific expression scores for all genes illustrated in Fig. 1B.
Table S2 – related to Figure 2.
Table of genes tested for association of loss-of-function mutations with increased DUX4 expression in TCGA cancers (Fig. 2G, S2D).
Table S3 – related to Figure 3.
Table of high-confidence DUX4 targets (DUX4-induced genes and repetitive elements illustrated in Fig. 3B).
Table S4 – related to Figure 3 and STAR Methods.
Table of high-confidence DUXB targets (DUXB-induced genes illustrated in Fig. S3C).
Table S5 – related to Figure 4.
Table of data illustrated graphically in Fig. 4A. Each row corresponds to an enriched GO term, and includes ancestral terms not shown in Fig. 4A. Columns named by cancer type indicate −log10 (FDR for enrichment of GO term) for that cancer type. Columns “Genes in multiple cancers”/“Genes in any cancer” indicate differentially associated genes that were differentially expressed in more than one/one or more cancer types in association with DUX4 reactivation.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GAPDH (6C5) | GeneTex, Inc. | GTX28245 RRID:AB_370675 |
| MHC Class I (F-3) | Santa Cruz Biotechnology | sc-55582 RRID:AB_831547 |
| FITC anti-human HLA-A,B,C (W6/32) | BioLegend | 311404 RRID:AB_314873 |
| ZSCAN4 | Invitrogen | PA5–32106 RRID:AB_2549579 |
| DUX4 (14–3) | Geng et al., 2011 | N/A |
| Peroxidase AffiniPure Goat Anti-Mouse IgG (H+L) | Jackson ImmunoResearch | 115–035-146 RRID:AB_2307392 |
| Peroxidase AffiniPure Goat Anti-Rabbit (H+L) | Jackson ImmunoResearch | 111–035-144 RRID:AB_2307391 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Insulin | Sigma-Aldrich | I1882 |
| Recombinant human basic fibroblast growth factor | Promega | G5071 |
| Dexamethasone | Sigma-Aldrich | D4902 |
| Puromycin dihydrochloride | Sigma-Aldrich | P8833 |
| Doxycycline hyclate | Sigma-Aldrich | D9891 |
| Recombinant human IFN-gamma | R&D Systems | 285IF100 |
| Polybrene | Sigma-Aldrich | 107689 |
| Penicillin/streptomycin | Gibco/Thermo Fisher | 15140122 |
| Sodium Pyruvate | Gibco/Thermo Fisher | 11360070 |
| DNase I | Gibco/Thermo Fisher | 18068015 |
| MEM Non-Essential Amino Acids Solution | Gibco/Thermo Fisher | 11140050 |
| Critical Commercial Assays | ||
| Lipofectamine RNAiMAX | Life Technologies | 13778150 |
| SuperScriptIII First-Strand System | Life Technologies | 18080051 |
| iTaq SYBR Green Supermix | Bio-Rad | 1725124 |
| TruSeq RNA Library Preparation Kit | Illumina | RS-122–2001 |
| NucleoSpin RNA kit | Machery-Nagel | 740955 |
| Key Instruments | ||
| QuantStudio 7 Flex | Applied Biosystems | N/A |
| 4200 TapeStation System | Agilent Technologies | N/A |
| Trinean DropSense 96 UV-Vis spectrophotometer | Caliper Life Sciences | N/A |
| Qubit 2.0 Fluorometer | Life Technologies | N/A |
| HiSeq 2500 | Illumina | N/A |
| LSRFortessa X-50 | BD Biosciences | N/A |
| Source Data | ||
| The Cancer Genome Atlas RNA-seq data | CGHub RRID:SCR_002657 | N/A |
| RNA-seq data for Eidahl et al., 2016; Hendrickson et al., 2017; Hugo et al., 2016 | NCBI Gene Expression Omnibus RRID:SCR_005012 | GSE85632, GSE85935, GSE78220 |
| RNA-seq data for Rickard et al., 2015 | NCBI Sequence Read Archive RRID:SCR_004891 | SRP058319 |
| RNA-seq data for Van Allen et al., 2015 | dbGaP RRID:SCR_002709 | phs000452.v2.p1 |
| RNA-seq data for Yasuda et al., 2016 | Japanese Genotype-Phenotype Archive RRID:SCR_003118 | JGAS00000000047 |
| RNA-seq data for Lilljebjörn et al., 2016 | European Genome-phenome Archive RRID:SCR_004944 | EGAD00001002112 |
| GTEx Sample Annotations and Gene Expression | GTEx portal RRID:SCR_013042 | www.gtexportal.org |
| ChIP-seq data for Geng et al., 2012 | NCBI Gene Expression Omnibus RRID:SCR_005012 | GSE33838 |
| Deposited Data | ||
| RNA-seq data for this study | NCBI Gene Expression Omnibus RRID:SCR_005012 | GSE128917 |
| Experimental Models: Cell Lines | ||
| MB135 cells (female) | Snider et al., 2010 | N/A |
| MB2401 cells (male) | Campbell et al., 2018 | N/A |
| MB073 cells (male) | Campbell et al., 2018 | N/A |
| A204 cells (female) | ATCC | HTB-82 RRID:CVCL_1058 |
| HeLa cells (female) | ATCC | CCL-2 RRID:CVCL_0030 |
| MCF-7 cells (female) | ATCC | HTB-22 RRID:CVCL_0031 |
| Mel375 cells (female) and Mel526 cells (unknown) | Dr. Seth Pollack (Pollack et al., 2012) | N/A |
| SuSa cells (male) | DSMZ | ACC 747 RRID:CVCL_L280 |
| MB135iDUX4 cells (female) | Jagannathan et al., 2016 | N/A |
| MB135iDUXB cells (female) | This study | N/A |
| A204iDUX4 cells (female) | This study | N/A |
| HeLaiDUX4 cells (female) | This study | N/A |
| MCF-7iDUX4 cells (female) | This study | N/A |
| Mel375iDUX4 cells (female) | This study | N/A |
| Mel526iDUX4 cells (unknown) | This study | N/A |
| SuSaiDUX4 cells (male) | This study | N/A |
| Oligonucleotides | ||
| Primers for qPCR and cloning See STAR★Methods for sequences |
This study | N/A |
| FlexiTube GeneSolution siRNAs targeting PRPF8 See STAR★Methods for sequences | Qiagen | GS10594 |
| Non-targeting Negative Control siRNA See STAR★Methods for sequence |
Qiagen | 1022076 |
| Recombinant DNA | ||
| pCW57.1 | Addgene | 41397 RRID:Addgene_41397 |
| pCW57.1-DUX4-CA | Addgene | 99281 RRID:Addgene_99281 |
| pCW57.1-DUXB-CA | This study, available on Addgene | 125027 RRID:Addgene_125027 |
| Software and Algorithms | ||
| Bowtie v1.0.0 |
Langmead et al, 2009 RRID:SCR_005476 |
github.com/BenLangmead/bowtie/ |
| RSEM v.1.2.4 | Li and Dewey, 2011 RRID:SCR_013027 | deweylab.github.io/RSEM/ |
| TopHat v2.0.8b | Trapnell et al, 2009 RRID:SCR_013035 | ccb.jhu.edu/software/tophat/index.shtml |
| MISO v2.0 |
Katz et al., 2010 RRID:SCR_003124 |
genes.mit.edu/burgelab/miso/ |
| Samtools v1.3.1 |
Li et al., 2009 RRID:SCR_002105 |
www.htslib.org |
| Kallisto v0.43.0 | Bray et al., 2016 RRID:SCR_016582 | pachterlab.github.io/kallisto/ |
| MACS v2.1.1.20160309 | Zhang et al., 2008 RRID:SCR_013291 | github.com/taoliu/MACS/ |
| IGV v2.3.90 | Thorvaldsdottir et al., 2013 RRID:SCR_011793 | software.broadinstitute.org/software/igv/ |
| Bioconductor |
Huber et al., 2015 RRID:SCR_006442 |
www.bioconductor.org |
| GOseq |
Young et al., 2010 RRID:SCR_017052 |
bioconductor.org/packages/release/bioc/html/goseq.html |
| dplyr | Wickham and Francois RRID:SCR_016708 |
cran.r-project.org/web/packages/dplyr/index.html |
| ggplot2 | Wickham et al., 2009 RRID:SCR_014601 | ggplot2.org |
| GraphPad Prism v7.0 | RRID:SCR_002798 | www.graphpad.com |
| FlowJo v10.5.3 | RRID:SCR_008520 | www.flowjo.com/solutions/flowjo |
| BD FACSDiva | RRID:SCR_001456 | www.bdbiosciences.com/instruments/software/facsdiva/index.jsp |
Highlights.
The early embryonic transcription factor DUX4 is active in many human cancers
DUX4-expressing cancers are characterized by low anti-tumor immune activity
DUX4 blocks interferon-g-mediated induction of MHC Class I and antigen presentation
DUX4 is significantly associated with failure to respond to anti-CTLA-4 therapy
ACKNOWLEDGEMENTS
We thank Heidi Dvinge for TCGA transcriptome analyses, Sean Bennett and S. Jessica Kumar for technical assistance, and Sujatha Jagannathan and Shirleen Soh for manuscript feedback. This research was supported in part by NIH/NINDS P01 NS069539 (RKB, SJT, NAS), NIH/NIAMS R01 AR045203 (SJT, SCS, AEC), Friends of FSH Research (AEC, SJT), and the Chris Carrino Foundation for FSHD (SJT, AEC). RKB is a Scholar of The Leukemia & Lymphoma Society (1344–18). GLC is a Mahan Fellow. The results shown here are in part based upon data generated by the TCGA Research Network: https://cancergenome.nih.gov/. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS.
Footnotes
DECLARATION OF INTERESTS
GLC, AEC, SJT, and RKB are inventors on a provisional patent application submitted by Fred Hutchinson Cancer Research Center that covers DUX4 expression in cancers and response to immunotherapy.
REFERENCES
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, and Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nat Methods 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arahata K, Ishihara T, Fukunaga H, Orimo S, Lee JH, Goto K, and Nonaka I (1995). Inflammatory response in facioscapulohumeral muscular dystrophy (FSHD): immunocytochemical and genetic analyses. Muscle Nerve Suppl 2, S56–S66. [PubMed] [Google Scholar]
- Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, and Weinberg RA (2008). An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 40, 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Caballero OL, and Chen Y-T (2009). Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci 100, 2014–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell AE, Shadle SC, Jagannathan S, Lim J-W, Resnick R, Tawil R, van der Maarel SM, and Tapscott SJ (2018). NuRD and CAF-1-mediated silencing of the D4Z4 array is modulated by DUX4-induced MBD3L proteins. Elife 7, e31023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang T-C, Yang Y, Yasue H, Bharti AK, Retzel EF, and Liu W-S (2011). The expansion of the PRAME gene family in Eutheria. PLoS ONE 6, e16867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S-H, Gearhart MD, Cui Z, Bosnakovski D, Kim M, Schennum N, and Kyba M (2016). DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I, et al. (2016). TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res 44, e71–e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, and Chadwick BP (2016). Influence of Repressive Histone and DNA Methylation upon D4Z4 Transcription in Non-Myogenic Cells. PLoS ONE 11, e0160022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daxinger L, Harten SK, Oey H, Epp T, Isbel L, Huang E, Whitelaw N, Apedaile A, Sorolla A, Yong J, et al. (2013). An ENU mutagenesis screen identifies novel and known genes involved in epigenetic processes in the mouse. Genome Biol 14, R96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Iaco A, Coudray A, Duc J, and Trono D (2019). DPPA2 and DPPA4 are necessary to establish a 2C-like state in mouse embryonic stem cells. EMBO Rep e47382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Iaco A, Planet E, Coluccio A, Verp S, Duc J, and Trono D (2017). DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat Genet [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H, Ries RE, Ilagan JO, Stirewalt DL, Meshinchi S, and Bradley RK (2014). Sample processing obscures cancer-specific alterations in leukemic transcriptomes. Proceedings of the National Academy of Sciences 111, 16802–16807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckersley-Maslin M, Alda-Catalinas C, Blotenburg M, Kreibich E, Krueger C, and Reik W (2019). Dppa2 and Dppa4 directly regulate the Dux-driven zygotic transcriptional program. Genes Dev [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eidahl JO, Giesige CR, Domire JS, Wallace LM, Fowler AM, Guckes SM, Garwick-Coppens SE, Labhart P, and Harper SQ (2016). Mouse Dux is myotoxic and shares partial functional homology with its human paralog DUX4. Hum Mol Genet 25, 4577–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, et al. (2009). New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). European Journal of Cancer 45, 228–247. [DOI] [PubMed] [Google Scholar]
- Feng Q, Snider L, Jagannathan S, Tawil R, van der Maarel SM, Tapscott SJ, and Bradley RK (2015). A feedback loop between nonsense-mediated decay and the retrogene DUX4 in facioscapulohumeral muscular dystrophy. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, et al. (2013). Ensembl 2013. Nucleic Acids Res 41, D48–D55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RL, Manly SP, McMahon M, Kerr IM, and Stark GR (1984). Transcriptional and posttranscriptional regulation of interferon-induced gene expression in human cells. Cell 38, 745–755. [DOI] [PubMed] [Google Scholar]
- Gabriëls J, Beckers MC, Ding H, De Vriese A, Plaisance S, van der Maarel SM, Padberg GW, Frants RR, Hewitt JE, Collen D, et al. (1999). Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene 236, 25–32. [DOI] [PubMed] [Google Scholar]
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. (2016). Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 167, 397–404.e399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng LN, Tyler AE, and Tapscott SJ (2011). Immunodetection of human double homeobox 4. Hybridoma (Larchmt) 30, 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng LN, Yao Z, Snider L, Fong AP, Cech JN, Young JM, van der Maarel SM, Ruzzo WL, Gentleman RC, Tawil R, et al. (2012). DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev Cell 22, 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2000). The hallmarks of cancer. Cell 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Arao T, Togashi Y, Kato H, Fujita Y, De Velasco MA, Kimura H, Matsumoto K, Tanaka K, Okamoto I, et al. (2013). The OCT4 pseudogene POU5F1B is amplified and promotes an aggressive phenotype in gastric cancer. Oncogene 34, 199–208. [DOI] [PubMed] [Google Scholar]
- Haynes P, Bomsztyk K, and Miller DG (2018). Sporadic DUX4 expression in FSHD myocytes is associated with incomplete repression by the PRC2 complex and gain of H3K9 acetylation on the contracted D4Z4 allele. Epigenetics Chromatin 11, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson PG, Doráis JA, Grow EJ, Whiddon JL, Lim J-W, Wike CL, Weaver BD, Pflueger C, Emery BR, Wilcox AL, et al. (2017). Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat Genet [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homey B, Müller A, and Zlotnik A (2002). Chemokines: agents for the immunotherapy of cancer? Nat. Rev. Immunol 2, 175–184. [DOI] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. (2016). Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 165, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huichalaf C, Micheloni S, Ferri G, Caccia R, and Gabellini D (2014). DNA methylation analysis of the macrosatellite repeat associated with FSHD muscular dystrophy at single nucleotide level. PLoS ONE 9, e115278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda H, Lethé B, Lehmann F, van Baren N, Baurain JF, de Smet C, Chambost H, Vitale M, Moretta A, Boon T, et al. (1997). Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity 6, 199–208. [DOI] [PubMed] [Google Scholar]
- Jagannathan S, Shadle SC, Resnick R, Snider L, Tawil RN, van der Maarel SM, Bradley RK, and Tapscott SJ (2016). Model systems of DUX4 expression recapitulate the transcriptional profile of FSHD cells. Hum Mol Genet [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, and Burge CB (2010). Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods 7, 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura-Saito M, Yamazaki Y, Kaneko K, Kawaguchi N, Kanda H, Mukai H, Gotoh T, Motoi T, Fukayama M, Aburatani H, et al. (2006). Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet 15, 2125–2137. [DOI] [PubMed] [Google Scholar]
- Kayisli UA, Selam B, Guzeloglu-Kayisli O, Demir R, and Arici A (2003). Human chorionic gonadotropin contributes to maternal immunotolerance and endometrial apoptosis by regulating Fas-Fas ligand system. J. Immunol 171, 2305–2313. [DOI] [PubMed] [Google Scholar]
- Kim M-S, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, et al. (2014). A draft map of the human proteome. Nature 509, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosti I, Jain N, Aran D, Butte AJ, and Sirota M (2016). Cross-tissue Analysis of Gene and Protein Expression in Normal and Cancer Tissues. Sci Rep 6, 24799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowaljow V, Marcowycz A, Ansseau E, Conde CB, Sauvage S, Mattéotti C, Arias C, Corona ED, Nuñez NG, Leo O, et al. (2007). The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul. Disord 17, 611–623. [DOI] [PubMed] [Google Scholar]
- Krämer A, Hochhaus A, Saussele S, Reichert A, Willer A, and Hehlmann R (1998). Cyclin A1 is predominantly expressed in hematological malignancies with myeloid differentiation. Leukemia 12, 893–898. [DOI] [PubMed] [Google Scholar]
- Kruse V, Hamann C, Monecke S, Cyganek L, Elsner L, Hübscher D, Walter L, Streckfuss-Bömeke K, Guan K, and Dressel R (2015). Human Induced Pluripotent Stem Cells Are Targets for Allogeneic and Autologous Natural Killer (NK) Cells and Killing Is Partly Mediated by the Activating NK Receptor DNAM-1. PLoS ONE 10, e0125544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Goto K, Matsuda C, and Arahata K (1995). Characterization of a tandemly repeated 3.3-kbKpnl unit in the facioscapulohumeral muscular dystrophy (FSHD) gene region on chromosome 4q35. Muscle & Nerve 18, S6–S13. [PubMed] [Google Scholar]
- Lemmers RJLF, Tawil R, Petek LM, Balog J, Block GJ, Santen GWE, Amell AM, van der Vliet PJ, Almomani R, Straasheijm KR, et al. (2012). Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmers RJLF, van der Vliet PJ, Klooster R, Sacconi S, Camaño P, Dauwerse JG, Snider L, Straasheijm KR, van Ommen GJ, Padberg GW, et al. (2010). A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 329, 1650–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmers RJLF, Wohlgemuth M, van der Gaag KJ, van der Vliet PJ, van Teijlingen CMM, de Knijff P, Padberg GW, Frants RR, and van der Maarel SM (2007). Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am J Hum Genet 81, 884–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS, Li B, and Liu XS (2017). TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res 77, e108–e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao C, Wang XY, Wei HQ, Li SQ, Merghoub T, Pandolfi PP, and Wolgemuth DJ (2001). Altered myelopoiesis and the development of acute myeloid leukemia in transgenic mice overexpressing cyclin A1. Proc Natl Acad Sci USA 98, 6853–6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilljebjörn H, Henningsson R, Hyrenius-Wittsten A, Olsson L, Orsmark-Pietras C, Palffy von S, Askmyr M, Rissler M, Schrappe M, Cario G, et al. (2016). Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun 7, 11790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Matzuk MM, Sung WK, Guo Q, Wang P, and Wolgemuth DJ (1998). Cyclin A1 is required for meiosis in the male mouse. Nat Genet 20, 377–380. [DOI] [PubMed] [Google Scholar]
- Liu Y-F, Wang B-Y, Zhang W-N, Huang J-Y, Li B-S, Zhang M, Jiang L, Li J-F, Wang M-J, Dai Y-J, et al. (2016). Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine 8, 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, Collins NB, Bi K, LaFleur MW, Juneja VR, et al. (2017). In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner TB, Li A, Biswas A, Lee K-H, Liu Y-J, Bayir E, Iliopoulos D, van den Elsen PJ, and Kobayashi KS (2010). NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proceedings of the National Academy of Sciences 107, 13794–13799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, et al. (2013). The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res 41, D64–D69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk M, and Holding C (2001). Human embryonic genes re-expressed in cancer cells. Oncogene 20, 8085–8091. [DOI] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA (2015). Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottaviani A, Rival-Gervier S, Boussouar A, Foerster AM, Rondier D, Sacconi S, Desnuelle C, Gilson E, and Magdinier F (2009). The D4Z4 macrosatellite repeat acts as a CTCF and A-type lamins-dependent insulator in facio-scapulo-humeral dystrophy. PLoS Genet 5, e1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, Tsoucas D, Qiu X, Lim K, Rao P, et al. (2018). A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, Gartner JJ, Jia L, Steinberg SM, Yamamoto TN, et al. (2017). Identification of essential genes for cancer immunotherapy. Nature 548, 537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce GB (1983). The cancer cell and its control by the embryo. Rous-Whipple Award lecture. Am. J. Pathol 113, 117–124. [PMC free article] [PubMed] [Google Scholar]
- Pollack SM, Li Y, Blaisdell MJ, Farrar EA, Chou J, Hoch BL, Loggers ET, Rodler E, Eary JF, Conrad EU, et al. (2012). NYESO-1/LAGE-1s and PRAME are targets for antigen specific T cells in chondrosarcoma following treatment with 5-Aza-2-deoxycitabine. PLoS ONE 7, e32165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preussner J, Zhong J, Sreenivasan K, Günther S, Engleitner T, Künne C, Glatzel M, Rad R, Looso M, Braun T, et al. (2018). Oncogenic Amplification of Zygotic Dux Factors in Regenerating p53-Deficient Muscle Stem Cells Defines a Molecular Cancer Subtype. Cell Stem Cell [DOI] [PubMed] [Google Scholar]
- Revenkova E, Eijpe M, Heyting C, Gross B, and Jessberger R (2001). Novel meiosis-specific isoform of mammalian SMC1. Mol Cell Biol 21, 6984–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickard AM, Petek LM, and Miller DG (2015). Endogenous DUX4 Expression in FSHD Myotubes is Sufficient to Cause Cell Death and Disrupts RNA Splicing and Cell Migration Pathways. Hum Mol Genet [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, and Oshlack A (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodig SJ, Gusenleitner D, Jackson DG, Gjini E, Giobbie-Hurder A, Jin C, Chang H, Lovitch SB, Horak C, Weber JS, et al. (2018). MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci Transl Med 10, eaar3342. [DOI] [PubMed] [Google Scholar]
- Rooney MS, Shukla SA, Wu CJ, Getz G, and Hacohen N (2015). Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane J-P, Bjorgaard SL, Hammond MR, Vitzthum H, Blackmon SM, et al. (2017). Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun 8, 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Yamaguchi T, Nomura T, and Ono M (2008). Regulatory T cells and immune tolerance. Cell 133, 775–787. [DOI] [PubMed] [Google Scholar]
- Schroder K, Hertzog PJ, Ravasi T, and Hume DA (2004). Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75, 163–189. [DOI] [PubMed] [Google Scholar]
- Shadle SC, Zhong JW, Campbell AE, Conerly ML, Jagannathan S, Wong C-J, Morello TD, van der Maarel SM, and Tapscott SJ (2017). DUX4-induced dsRNA and MYC mRNA stabilization activate apoptotic pathways in human cell models of facioscapulohumeral dystrophy. PLoS Genet 13, e1006658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA, and Ribas A (2017). Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 168, 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, Li Y, Chen H, Yang H, Hsu P-H, et al. (2018). LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, Stevens J, Lane WJ, Dellagatta JL, Steelman S, et al. (2015). Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HA, and McNeel DG (2010). The SSX family of cancer-testis antigens as target proteins for tumor therapy. Clin. Dev. Immunol 2010, 150591–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider L, Geng LN, Lemmers RJLF, Kyba M, Ware CB, Nelson AM, Tawil R, Filippova GN, van der Maarel SM, Tapscott SJ, et al. (2010). Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet 6, e1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht K, Sung Y-S, Zhang L, Richter GHS, Fletcher CD, and Antonescu CR (2014). Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged Ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosomes Cancer 53, 622–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Bao R, and Gajewski TF (2015). Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235. [DOI] [PubMed] [Google Scholar]
- Stadler G, Chen JC, Wagner K, Robin JD, Shay JW, Emerson CP, and Wright WE (2011). Establishment of clonal myogenic cell lines from severely affected dystrophic muscles - CDK4 maintains the myogenic population. Skelet Muscle 1, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenman U-H, Alfthan H, and Hotakainen K (2004). Human chorionic gonadotropin in cancer. Clin. Biochem 37, 549–561. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Kawazu M, Yasuda T, Tamura M, Hayakawa F, Kojima S, Ueno T, Kiyoi H, Naoe T, and Mano H (2018). Transcriptional activities of DUX4 fusions in B-cell acute lymphoblastic leukemia. Haematologica haematol 2017.183152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therneau TM, and Grambsch PM (2000). Modeling Survival Data: Extending the Cox Model (Springer, New York: ). [Google Scholar]
- Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang T-H, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, et al. (2018). The Immune Landscape of Cancer. Immunity 48, 812–830.e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, and Mesirov JP (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinformatics 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Drake CG, and Pardoll DM (2015). Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend A, Ohlén C, Bastin J, Ljunggren HG, Foster L, and Kärre K (1989). Association of class I major histocompatibility heavy and light chains induced by viral peptides. Nature 340, 443–448. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, et al. (2015). Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenmakers E-J, Lodewyckx T, Kuriyal H, and Grasman R (2010). Bayesian hypothesis testing for psychologists: a tutorial on the Savage-Dickey method. Cogn Psychol 60, 158–189. [DOI] [PubMed] [Google Scholar]
- Wang X, Spandidos A, Wang H, and Seed B (2012). PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res 40, D1144–D1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiddon JL, Langford AT, Wong C-J, Zhong JW, and Tapscott SJ (2017). Conservation and innovation in the DUX4-family gene network. Nat Genet [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2009). ggplot2: elegant graphics for data analysis (Springer; New York: ). [Google Scholar]
- Wickham H, and Francois R dplyr: A Grammar of Data Manipulation.
- Wijmenga C, Frants RR, Hewitt JE, van Deutekom JC, van Geel M, Wright TJ, Padberg GW, Hofker MH, and van Ommen GJ (1993). Molecular genetics of facioscapulohumeral muscular dystrophy. Neuromuscul. Disord 3, 487–491. [DOI] [PubMed] [Google Scholar]
- Wolchok JD, Hoos A, O’Day S, Weber JS, Hamid O, Lebbe C, Maio M, Binder M, Bohnsack O, Nichol G, et al. (2009). Guidelines for the Evaluation of Immune Therapy Activity in Solid Tumors: Immune-Related Response Criteria. Clin. Cancer Res 15, 7412–7420. [DOI] [PubMed] [Google Scholar]
- Yasuda T, Tsuzuki S, Kawazu M, Hayakawa F, Kojima S, Ueno T, Imoto N, Kohsaka S, Kunita A, Doi K, et al. (2016). Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat Genet [DOI] [PubMed] [Google Scholar]
- Young JM, Whiddon JL, Yao Z, Kasinathan B, Snider L, Geng LN, Balog J, Tawil R, van der Maarel SM, and Tapscott SJ (2013). DUX4 binding to retroelements creates promoters that are active in FSHD muscle and testis. PLoS Genet 9, e1003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MD, Wakefield MJ, Smyth GK, and Oshlack A (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 11, R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalzman M, Falco G, Sharova LV, Nishiyama A, Thomas M, Lee S-L, Stagg CA, Hoang HG, Yang H-T, Indig FE, et al. (2010). Zscan4 regulates telomere elongation and genomic stability in ES cells. Nature 464, 858–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. (2016). Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med 375, 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Yitzhaky A, Bossel Ben-Moshe N, and Domany E (2013). An accessible database for mouse and human whole transcriptome qPCR primers. Bioinformatics 29, 1355–1356. [DOI] [PubMed] [Google Scholar]
- Zeng W, de Greef JC, Chen Y-Y, Chien R, Kong X, Gregson HC, Winokur ST, Pyle A, Robertson KD, Schmiesing JA, et al. (2009). Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet 5, e1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, McCastlain K, Yoshihara H, Xu B, Chang Y, Churchman ML, Wu G, Li Y, Wei L, Iacobucci I, et al. (2016). Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ding L, and Sandford AJ (2005). Selection of reference genes for gene expression studies in human neutrophils by real-time PCR. BMC Mol Biol 6, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 – related to Figure 1.
Table of cancer-specific expression scores for all genes illustrated in Fig. 1B.
Table S2 – related to Figure 2.
Table of genes tested for association of loss-of-function mutations with increased DUX4 expression in TCGA cancers (Fig. 2G, S2D).
Table S3 – related to Figure 3.
Table of high-confidence DUX4 targets (DUX4-induced genes and repetitive elements illustrated in Fig. 3B).
Table S4 – related to Figure 3 and STAR Methods.
Table of high-confidence DUXB targets (DUXB-induced genes illustrated in Fig. S3C).
Table S5 – related to Figure 4.
Table of data illustrated graphically in Fig. 4A. Each row corresponds to an enriched GO term, and includes ancestral terms not shown in Fig. 4A. Columns named by cancer type indicate −log10 (FDR for enrichment of GO term) for that cancer type. Columns “Genes in multiple cancers”/“Genes in any cancer” indicate differentially associated genes that were differentially expressed in more than one/one or more cancer types in association with DUX4 reactivation.
Data Availability Statement
Experimental data and plasmids.
Relevant plasmids are available through Addgene (https://www.addgene.org/Stephen_Tapscott/).
Accession codes.
FASTQ files from the DUXB RNA-seq experiment have been deposited in NCBI’s GEO database (accession number GSE128917).