

SUMMARY
Though most bacteria in nature are nutritionally limited and grow slowly, our understanding of core processes like transcription comes largely from studies in model organisms doubling rapidly. We previously identified a small protein of unknown function, SutA, in a screen of proteins synthesized in Pseudomonas aeruginosa during dormancy. SutA binds RNA polymerase (RNAP), causing widespread changes in gene expression, including upregulation of the ribosomal RNA genes. Here, using biochemical and structural methods, we examine how SutA interacts with RNAP and the functional consequences of these interactions. We show that SutA comprises a central α-helix with unstructured N- and C-terminal tails, and binds to the β1 domain of RNAP. It activates transcription from the rrn promoter by both the housekeeping sigma factor holoenzyme (Eσ70) and the stress sigma factor holoenzyme (EσS) in vitro, but has a greater impact on EσS. In both cases, SutA appears to affect intermediates in open complex formation, and its N-terminal tail is required for activation. The small magnitudes of in vitro effects are consistent with a role in maintaining activity required for homeostasis during dormancy. Our results add SutA to a growing list of transcription regulators that use their intrinsically disordered regions to remodel transcription complexes.
Keywords: Pseudomonas aeruginosa, Bacterial gene expression regulation, RNA polymerase beta subunit, Sigma factor, Intrinsically disordered proteins, Bacterial physiological phenomena
Abbreviated Summary
Mechanisms of bacterial transcription regulation that operate during slow growth and dormancy are not well understood, despite the fact that bacteria spend a lot of time in these states. We show here that a dormancy-specific regulator of P. aeruginosa, SutA, is an intrinsically disordered protein that binds to the β1 domain of RNA polymerase and directly enhances initiation at the ribosomal RNA promoter, likely by affecting the conformation of an intermediate in open complex formation.
Graphical Abstract
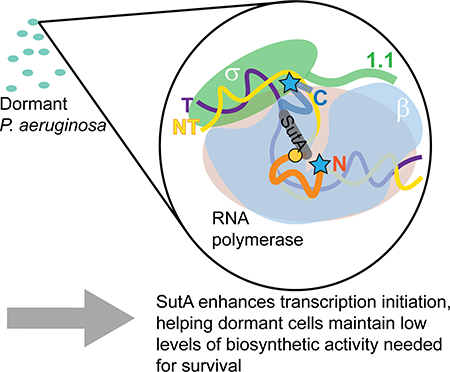
INTRODUCTION
Despite the fact that most natural environments do not allow bacteria to double every 20–30 minutes, our understanding of essential cellular processes—such as DNA replication, transcription, and translation—has been shaped by studies of a few model organisms growing exponentially at these rates, or responding to a rapid shift from exponential to slow growth. We do not know how the molecular machines responsible for transcription and translation (processes that are necessary to maintain homeostasis even when cell division is not occurring) adapt to long periods of reduced activity and low or uneven substrate availability (Bergkessel et al., 2016). P. aeruginosa and many other members of the Pseudomonadales order are notable opportunists, capable of using diverse substrates for rapid growth but also able to persist in dormancy for long periods (Udikovic-Kolic et al., 2014), making them attractive model organisms in which to explore the molecular strategies underpinning slow growth physiology. A better understanding of slow-growing or dormant states in P. aeruginosa also has clinical importance, as these states are thought to contribute to this organism’s antibiotic tolerance in chronic infections (Babin et al., 2017; Ciofu et al., 2015; Olivares et al., 2013).
Accordingly, in previous work, we used a proteomics-based screen to identify P. aeruginosa regulators that are preferentially expressed during hypoxia-induced growth arrest. We identified a non-essential RNAP-binding protein, SutA, that had broad impacts on gene expression and affected the ability of P. aeruginosa to form biofilms and produce virulence factors. Notably, SutA expression caused increased expression of the rRNA genes compared to what was seen in a ΔsutA strain under slow-growth conditions, and ChIP data showed both that SutA localized to rRNA promoters and that higher levels of RNAP localized to rRNA promoters when SutA was present. More broadly, RNA-Seq and ChIP-Seq data showed that expression of many housekeeping genes, including ribosomal protein genes, was enhanced when SutA was expressed during slow growth, and that SutA localized both at promoters and throughout gene bodies (Babin et al., 2016). The finding that a slow growth-specific regulator could act to enhance expression of these genes was initially counterintuitive, but recognizing that even cells experiencing a protracted period of nutrient limitation might benefit from the ability to transiently upregulate housekeeping genes for maintenance and repair, without fully re-entering exponential growth, helps resolve this paradox. As has been observed with other global regulators that directly bind RNAP but not DNA (Haugen et al., 2008; Hubin et al., 2017), SutA’s effects are broad but modest, potentially impacting the behavior of RNAP at every gene rather than enhancing or blocking the RNAP-promoter interaction at specific DNA sequences. In this study, we focus on how SutA might affect transcription initiation at the rrn promoters, as a model to better understand its impact more generally.
The regulation of the rrn promoters in E. coli is one of the best-studied examples of growth-rate-responsive control of bacterial gene expression. While they can drive extremely high levels of expression during exponential growth, they are rapidly and strongly down-regulated upon entry into stationary phase (Paul et al., 2004). This behavior depends on an extremely unstable open complex (OC) formed at rrn P1, which sensitizes initiation to conditions encountered during nutrient downshifts, such as decreased concentrations of the initiating nucleotides ([iNTPs]) (Murray et al., 2003). A second rrn promoter that drives low levels of expression and is less sensitive to regulatory inputs, P2, has been proposed as the mechanism by which some rRNA transcription can be maintained during stationary phase (Murray & Gourse, 2004), but this would imply that expression levels in E. coli are not actively modulated during protracted dormancy. The P. aeruginosa genome encodes four rrn operons whose upstream regions are very nearly identical throughout the putative promoter region. As in E. coli, there are multiple possible transcription start sites, but the growth phase-dependent regulation has not been described (Supporting Information, Fig. S1).
Expression of rrn is further modulated by diverse regulators acting at different stages of transcription initiation in different organisms. In many cases, the unstable OC is the target of additional regulation. For example, in E. coli, the signaling molecule (p)ppGpp and its co-regulator DksA bind to RNAP during early stationary phase and further destabilize the final rrn OC (Ross et al., 2016); the identities of the iNTPs (adenosine or guanosine) allow for direct coordination with the diminished energy stores available to drive translation ( Murray et al., 2003). Also, in many clades outside the Gammaproteobacteria, homologs of the global regulator CarD can enhance rRNA expression by directly stabilizing the OC (Bae et al., 2015; Hubin et al., 2017). By contrast, some factors that activate rrn P1 during rapid exponential growth in E. coli (e.g., Fis and DNA supercoiling) exert their effects before the final OC has formed, by helping to recruit RNAP or facilitating the initial opening of the double-stranded DNA (Leirmo & Gourse, 1991; Paul et al., 2004); in Mycobacteria, CarD, in conjunction with a second factor called RbpA, appears to stimulate OC formation in addition to its role in stabilizing the OC (Hubin et al., 2017). SutA lacks sequence or structural homology to any known transcription factor, raising the possibility that its mode of action is unique. Here, we report that SutA binds to a site on RNAP that is distinct from the binding sites of other regulators, that its activation of rrn transcription depends on its intrinsically disordered N- and C-terminal tails, and that its activity is modulated by the identity of the σ factor. Though our work focuses on a specific transcription factor and promoter in P. aeruginosa, the topic it tackles and the questions it raises are broadly relevant to understanding how bacteria survive periods of slow growth or dormancy in diverse environments.
RESULTS
SutA consists of a conserved α helix flanked by flexible tails
Because SutA is a small protein (105 amino acids) with no similarity to any known domains, we first explored its structural characteristics. We began by looking at structure predictions (using the Jpred4 algorithm for secondary structure and DISOPRED3 for intrinsic disorder) and sequence conservation (Buchan et al., 2013; Drozdetskiy et al., 2015; Jones & Cozzetto, 2015). SutA homologs are found in most organisms in the “Pseudomonadales-Oceanospirallales” clade of Gammaproteobacteria (Williams et al., 2010). Residues 56–76 are predicted to form an α-helix, followed by a β-strand comprising residues 81–84, but the rest of the protein has no predicted secondary structural elements, and residues 1–50 and 101–105 are predicted to be intrinsically disordered (Fig. 1A). While the central, potentially structured region is reasonably well conserved, some homologs completely lack the last 15–18 residues, and others lack most or all of the first 40 residues (Supporting Information, Fig. S2). This suggests that the N-and C-terminal tails (N-tail and C-tail) might function independently and that their removal might not affect folding/function of other regions.
Figure 1. NMR data confirm presence of α helix from residues 56–76 and flexible N- and C-terminal tails.

A. Primary sequence for SutA, with computational predictions indicated: underlining = intrinsic disorder; boxing = α-helix; gray shading = β-strand. B. Schematic of constructs used; wavy line = α-helix region; blue = C-tail; orange = N-tail. Schematics are aligned with residue numbers and NMR data in (C). C. Secondary chemical shift indices, residual dipolar coupling values, transverse relaxation rates, and peaks present in the positive amide NOE spectra following assignment of most backbone resonances for the full-length SutA. Secondary shifts were calculated using TALOS as part of the PINE automated assignment server. RDCs were measured by manual comparison of in-phase-anti-phase spectra between stretched gel and aqueous solution conditions. R2 values were calculated by fitting single exponential decay curves to peak integrals from spectra with increasing T2 delays. Positive NOE signal indicates that a peak was detected in the positive (1H-15N) NOE. The box indicates the location of the α-helix. D. One of many possible SutA structures modelled using the Robetta fragment server to incorporate chemical shift and RDC data, and PyRosetta. On the left, residues are colored by per-residue conservation score following alignment of 25 representative homologs (see Extended Experimental Procedures for details). On the right, residues are colored by charge.
For structural characterization by nuclear magnetic resonance (NMR), we purified 15N- and 13C-labeled full-length SutA and SutA 46–101, which lacks most of the predicted disordered residues. We also constructed two deletion mutants (Fig. 1B): SutA ΔN, retaining residues 41–105, and SutA ΔC, retaining residues 1–87.
We were able to assign resonances and determine backbone chemical shifts for about 85% of the residues of the full-length protein (Supporting Information, Table S1). Low sequence complexity and large regions of disorder caused a high degree of overlap in the spectra and made assignment difficult; spectra from the 46–101 variant were easier to assign, and served as a starting point for assignment of the full-length SutA. We focused on measuring secondary-structure chemical shift index values, R2 relaxation rates, and 1H-15N nuclear Overhauser effect (NOE) magnitude and sign to determine secondary-structure elements and degree of disorder for each residue that we could assign. We also embedded the protein in a stretched polyacrylamide gel to achieve weak alignment of the protein molecules, which allows for calculation of residual dipolar couplings (RDCs) by measuring differences in in-phase–antiphase spectra between the isotropic solution sample and the anisotropic stretched-gel sample (Fig. 1C). Dipolar couplings of the backbone N-H bonds can give restraints on the orientation vectors of the internuclear bonds; in an isotropic solution they sum to zero, but if the protein molecules are weakly aligned then residual dipolar couplings can be measured and provide useful restraints in ab initio structure prediction with only backbone assignments completed (Rohl & Baker, 2002). However, for SutA, the results of these analyses lend credence to the bioinformatics predictions and suggest that much of the protein outside a central α-helix is disordered. Residues 56–76 show the positive Cα and CO and negative Hα secondary chemical shifts associated with an α-helix structure (Wishart et al., 1991), and also show fast R2 relaxation rates and positive (1H-15N)NOE, suggesting that they are not disordered (Reddy & Rainey, 2010). RDCs for the helix region are also positive, as has been observed for α-helical regions of a partially denatured protein (Mohana-Borges et al., 2004). The short β-strand is less strongly supported, but secondary shifts for those residues are mostly of the appropriate sign for a β-strand (albeit of small magnitudes). In the N-tail, a small number of isolated residues have a positive NOE signal or secondary shifts that are not near zero, but in general, the residues of this region have the low R2, secondary shift, and RDC values that are characteristic of disorder. The C-tail has several residues that show somewhat higher R2 values and non-zero RDCs, suggestive of some degree of structure, but classic secondary structure elements are not apparent. We also compared 15N HSQC spectra for 15N-labeled ΔN and ΔC mutants to the full-length SutA (Supporting Information, Fig. S3–4). Deletion of either tail had minimal impact, affecting only the 2–4 residues adjacent to the newly created terminus.
The difficulty of making unambiguous assignments for all residues and the likelihood that much of the protein is intrinsically disordered precluded building a full NMR-based structural model of SutA. To model some of the conformations that might be adopted by SutA, we used the Robetta Server and PyRosetta to perform low-resolution Monte Carlo–based modeling, using the chemical shifts and RDC values from our NMR analysis to guide fragment library construction (Kim et al., 2004; Rohl & Baker, 2002). Figure 1D shows one resulting model. The most highly conserved residues are found in the α-helix, and the C-tail is also highly conserved among homologs that have it. The N-tail is less conserved and varies in length, but is generally quite acidic. The Supporting Information shows additional examples and modeling details (Supporting Information, Fig. S5 andExtended Experimental Procedures).
SutA binds to the β1 domain of RNAP
To investigate the binding interaction between SutA and RNAP, we used cross-linking and protein footprinting. We first asked whether SutA affects transcription by the closely related E. coli RNAP, for which extensive in vitro tools are available. Overexpressing SutA in E. coli did not lead to rrn upregulation in vivo as it did in P. aeruginosa (Supporting Information, Fig. S6), necessitating the use of the cognate P. aeruginosa proteins in all assays. We purified the core RNAP (E) natively from a ΔsutA strain using a protocol originally designed for E. coli RNAP and previously used to purify RNAP from P. aeruginosa (Burgess & Jendrisak, 1975; Kuznedelov et al., 2011). The P. aeruginosa homologs of σS, σ70, and DksA (as well as SutA) were heterologously expressed in E. coli with cleavable N-terminal hexahistidine tags and purified by metal affinity and size-exclusion chromatography (Supporting information, Fig. S7 and Extended Experimental Procedures). Because we found that SutA cross-linked most efficiently to E (Fig. 4 and Supporting Information, Fig. S8–9) and because our previous ChIP-Seq results suggested that SutA could interact with either initiating or elongating RNAP in vivo (Babin et al., 2016), we focused on interaction between SutA and E. First we used the homobifunctional reagent bis(sulfosuccinimidyl)suberate (BS3), which cross-links primary amines that are within about 25 Å of each other (Rappsilber, 2011). We added BS3 to complexes formed with purified E and SutA (Supporting Information, Figure S8). To avoid a possible scenario in which cross-links formed between lysine residues would inhibit the trypsin digestion that is typically used to generate peptides for LC-MS/MS, we instead used the peptidase Glu-C, which cuts following glutamate or aspartate residues, to digest cross-linked complexes, and subjected the resulting fragments to LC-MS/MS. Analysis with the software package Protein Prospector (Trnka et al., 2014) identified species that comprised one peptide from SutA and one peptide from RNAP (Supporting Information, Extended Experimental Procedures and Fig. S8, S10), which allowed mapping of cross-link sites.
Figure 4. Both σ factor and DNA compete with SutA for binding to RNAP.

A. Models based on E. coli σ70 and σS holoenzyme structures. The inset shows the perspective and extent of this view relative to the holoenzyme structure shown in (2B). The P. aeruginosa β sequence was threaded onto an E. coli crystal structure (PBD: 5UAG), and then the β subunit from this was docked into the Eσ70 cryoEM structure (left; PDB: 6CA0) or the EσS crystal structure (right; PDB:5IPN). Residues showing cross-link or cleavage reactivity with SutA (Fig. 2) that are visible in this view are colored magenta and numbered. Residues 168–212 of σ70, which are not visualized in the cryoEM structure, were modelled in as a flexible loop (AL). Two possible loop positions are shown (red and dark blue), one of which (red) could clash with both the DNA and SutA positions. In contrast, σS does not appear likely to directly contact SutA (right). B. Sequence and structure of template DNA surrounding transcription start site. C. Western blot showing cross-linking of L54BPA SutA to β, in the context of different σ factors and promoter DNA. D. Western blots showing β cleavage mediated by N-Fe or C-Fe SutA FeBABE conjugates. Sizes of cleavage products were estimated by comparison to β fragments of known sizes analyzed on large non-gradient gels (Supporting Information, Fig. S12 and S21); for some products (~), only approximate sizes can be determined. The blot for C-Fe was exposed for longer (4 minutes) than the blot for N-Fe (30 seconds). E. Cleavage of the DNA in the rrn promoter complexes formed by Eσ70 or EσS in the presence of N-Fe or C-Fe SutA, revealed by primer extension. Average log2-transformed enrichment in signal between the FeBABE reaction and a negative control reaction containing unmodified SutA, from triplicate measurements, is represented by color intensity for each base. FeBABE reactions and the negative control reactions to which they were normalized were run on the same gel. The DNA is depicted as an almost completely opened transcription bubble, because in this conformation the strongest DNA cleavages for the N- and C-tail FeBABE SutA would be in close proximity to the strongest protein cleavages for these reagents. However, the cross-linking data suggest that SutA may be displaced by the fully stabilized open complex.
We also used the photoreactive non-canonical amino acid p-benzoyl-L-phenylalanine (BPA), which, when irradiated with UV light, can form covalent bonds with a variety of moieties within 10 Å (Chin et al., 2002; Kauer et al., 1986). We introduced BPA at 9 different positions of SutA (residue 6, 11, 22, 54, 61, 74, 84, 89, or 100). We then formed complexes with purified E and each of the BPA-modified SutA proteins, irradiated them with UV light, and visualized cross-linked species following SDS-PAGE (Supporting Information, Fig. S9). For the most efficient cross-linkers (BPA at positions 54 and 84), we determined the sites of the cross-links on RNAP by identifying cross-linked peptides via StavroX (Götze et al., 2012) analysis of LC-MS/MS data after digestion of the complexes with trypsin (Supporting Information, Fig. S11).
Both cross-linking approaches identified interactions between the central region of SutA and the β1 domain or nearby regions of the β subunit of RNAP (Fig. 2A and B, green and orange). All SutA residues participating in the cross-links were within the α-helix (BS3) or just outside it (BPA). BPA cross-linking is sensitive to the orientations of the residues, so BPA residues within the helix that did not cross-link efficiently may not have been oriented optimally.
Figure 2. SutA interacts with the β subunit of RNAP.

A. A topology diagram of the contacts inferred by cross-linking (BS3, green lines; BPA, orange lines) and FeBABE-mediated cleavage (blue lines). Cross-linked residues were identified by LC-MS/MS and cleavage sites were determined by SDS-PAGE and Western blotting of the cleaved complexes, using a large-format gel system and Abcam antibody EPR18704, against the extreme C-terminus of the E. coli β. Cross-linking and cleavage reactions were performed using the core RNAP. See text and Extended Experimental Procedures for further details. B. Residues involved in cross-links or cleavages were mapped onto a structure generated by threading the P. aeruginosa sequence onto a crystal structure of the E. coli β subunit (PDB:5UAG). The relative position of the DNA in a crystal structure of the E. coli Eσ70 open complex is shown for reference. Inset shows the cryo-EM structure of E. coli Eσ70 (PDB: 6CA0), but with the P. aeruginosa sequence threaded model of the β subunit substituted in. Cross-linking and cleavage residues that are visible in this view (K45, K116, K481, M493, T586, and V721) are indicated in magenta for maximum contrast. The darker blue color indicates the positions of the β1 domain (the fragment purified for (C)) and pink indicates the βi9/β flap region that is shown in (A). C. 1H-15N HSQC spectra showing that chemical shifts for a handful of residues are perturbed when 15N-labeled SutA is mixed with unlabeled β1 domain (magenta) vs when it is analyzed alone (turquoise) or mixed with unlabeled σS (dark grey). A small number of background peaks show up only in the β1 domain mixture (magenta, lower right quadrant); these are most likely due to the fact that the protein concentration was lower in this sample.
To identify the positions of the N- and C-tails, we designed variants of SutA for affinity cleavage experiments. We introduced cysteine residues at SutA position 2, 32, or 98 and conjugated the chelated iron reagent, iron-(S)-1-[p-(bromoacetamido)benzyl]EDTA (FeBABE), to these cysteines. FeBABE catalyzes localized (estimated to occur within 12 Å of the FeBABE moiety) hydroxyl radical cleavage (Meares et al., 2003). We assembled complexes with the FeBABE-modified SutA variants and E, initiated the cleavage reactions, and analyzed the cleavage products by SDS-PAGE followed by Western blotting with a monoclonal antibody (Abcam EPR18704) against a peptide near the C-terminus of β. To map the cleavage sites, products were compared to C-terminal β fragments with known N-terminal endpoints (Supporting Information, Fig. S12). While the primary cleavage product of the N-terminal FeBABE (at residue 2; N-Fe) was in the cleft between the β1 and β2 (a.k.a. protrusion and lobe) domains, the strongest cleavage product of the C-terminal FeBABE (at residue 98; C-Fe) was in the long α-helix on the inside surface of β1 (designated α6 (Lane & Darst, 2010)), amongst BS3 and BPA cross-linking sites (Fig. 2A and B, blue). The FeBABE at residue 32 induced cleavage at both β positions, suggesting that the N-tail remains mobile to some degree even when bound to RNAP. All modified proteins used in the cross-linking and cleavage assays retained activity in in vitro transcription assays (Supporting Information, Fig. S13).
We also detected possible interactions with βi9, an insertion in the β flap domain (Opalka et al., 2010): BPA at residue 84 cross-linked to β967, and weak cleavage products were detected at β721 for the N-Fe variant and β1058 for the C-Fe variant (Fig. 2B). β967 and β484/493 residues that were cross-linked to BPA84 are too far apart to be reached from a single, stably bound position of SutA 84. However, we did not detect more than one shifted band after cross-linking with the 54 or 84 BPA variants (Supporting Information, Fig. S9), suggesting that two separate sites on β are not likely to be occupied by two SutA molecules at the same time. Instead, it may be that SutA’s inherent flexibility, combined with a binding interaction with the outside of the β1 domain that allows some rotation or translation of SutA, could allow for all of the observed cross-links and cleavages.
To corroborate SutA-β interaction without cross-linking or cleavage and assess which residues of SutA might directly participate, we conducted an NMR experiment. We were able to purify only a small amount of soluble β1 domain (dark blue in Fig. 2B), which we mixed with an approximately equimolar amount of 15N-labeled full-length SutA. As a control to rule out non-specific interactions, we mixed SutA with σS, which does not appear to bind SutA based on a microscale thermophoresis experiment (data not shown). We collected 15N HSQC spectra for these mixtures, as well as for SutA alone, using a Bruker 800 MHZ AV III HD spectrometer to help overcome difficulties introduced by the low concentration of β1 domain we could produce. While the peak positions for SutA alone and for the SutA+σS mixture were nearly identical, several SutA residues showed chemical shift perturbations in the β1 mixture, compared with the other two samples (Fig. 2C). Three of these residues, K95, D97 and K99, would be on the same side of an extended peptide chain in the C-tail, suggesting that this tail could directly interact with β1 in an extended conformation. However, the C-Fe SutA variant induced weaker cleavage than the N-Fe variant, suggesting that this interaction is probably not the only binding determinant. The other perturbed residues flank the α-helix, suggesting that the regions at the junctions with the flexible tails may change conformation upon binding to β.
SutA activates the rrn promoter in vitro
Next we investigated SutA effects on transcription. We focused on the rRNA promoter because our ChIP data suggested that SutA directly affects rrn initiation (Babin et al., 2016). Unlike its well-studied E. coli counterpart, the P. aeruginosa rrn initiation region has not been characterized. We used 5’-RACE to map the dominant rrn transcription start during stationary phase (when SutA is expressed) to a cytidine 8 bp downstream of a −10 consensus sequence. We produced a linear template of 150 bp containing the rrn promoter and 42 bp of transcribed sequence for use in single-turnover initiation experiments (Supporting Information, Fig. S14 and Extended Experimental Procedures).
Transcription initiation proceeds via a multi-step pathway consisting of: 1) formation of a closed complex between the double-stranded DNA and the RNAP holoenzyme; 2) initial DNA strand separation, followed by isomerization through several open intermediates into a final open complex (OC) in which the +1 position of the template DNA strand is loaded into the active site and the downstream DNA duplex is stably held by RNAP; 3 )initial abortive rounds of nucleotide addition; and 4) promoter clearance and transition into the elongation phase. Any of these steps could theoretically be affected by a regulator, and the details of this pathway differ at different promoters (Ruff, Record, et al., 2015). Much of the control of the E. coli P1 depends on the inherent instability of its OC (Paul et al., 2004), and the P. aeruginosa rrn promoter shares some of the features known to contribute to E. coli rrn P1 OC instability (Fig. 3A): suboptimal spacing (16 nt vs the optimal 17–18 nt) between near-consensus −35 and −10 hexamers, a GC-rich discriminator region, and a C residue 2 nt downstream of the −10 hexamer that cannot make productive contacts to σ70 (Haugen et al., 2006). However, the P. aeruginosa discriminator is 7 nt, one base shorter than that of E. coli, but still longer than the optimal 6 nt. Also, the initiating nucleotide is a cytidine rather than the adenosine or guanine iNTPs found in E. coli, indicating potentially different regulatory connections to cellular energy levels.
Figure 3. Effects of SutA variants on transcription initiation.

A. rrn promoter sequences of P. aeruginosa and E. coli (P1). −10 and −35 motifs are indicated in bold and boxed, transcription start sites are indicated by circles, and the discriminator region is noted (Disc.). B. Amount of transcript produced in the presence of varying concentrations of SutA or SutA variant protein, compared to the amount produced in the absence of SutA, expressed as a log2-transformed ratio. Single turnover initiation reactions were performed as schematized, and representative gel images are shown. Individual replicate values are plotted (n≥3), and lines connect the average of all replicates at each concentration. A representative full-length gel is shown in Supporting Information, Fig. S15. C. The heparin-resistant P. aeruginosa rrn OC is short-lived and its lifetime is not affected by SutA. The OC was formed with 20 nM Eσ70 and 15 nM promoter DNA and challenged with heparin. NTPs were added at the indicated times and transcription was allowed to proceed for 8 minutes before quenching the reaction and running on a 20% gel. Reactions were performed at least in duplicate. Representative primary data are shown in Supporting Information, Fig. S16. D. DksA and ppGpp, or low [iNTPs] repress initiation from the rrn promoter, and SutA does not overcome these effects. Reactions were generally performed as in (B), but in the left panel, 500 nM SutA and/or 250 nM DksA and 2.5 μM ppGpp were added as indicated, and on the right, varying concentrations of SutA and CTP+UTP, the first two nucleotides of the rrn transcript were used as indicated, in the absence of CpU dinucleotide. CTP and UTP were each present at the indicated concentration for [iNTPs]. RNAs were run on a 20% denaturing polyacrylamide gel and visualized by phosphorimaging. Symbols indicate the average value for the three replicates and lines represent the range of values observed in replicate experiments (n=3) (normalized such that the average signal for the 0 nM SutA condition for a given [iNTP] was the same across different gels). Representative primary data are shown in Supporting Information, Fig. S17 (DksA/ppGpp) and S18 ([iNTP]).
To directly measure the effects of SutA on transcription initiation, we performed single turnover initiation assays using the wild type (WT) SutA and the ΔN- and ΔC-tail variants described in Figure 1. Because EσS binds the rrn locus in vivo during stationary phase in E. coli (Raffaelle et al., 2005), we wanted to investigate whether SutA effects could be mediated through EσS or Eσ70, or both. We verified that both Eσ70 and EσS could drive specific initiation on our rrn promoter template, although EσS initiated much more weakly and showed some additional, non-specific products similar to those seen with E alone, and previously observed for EσS initiating on a linear template (Supporting Information, Fig. S15). We found that WT SutA increased transcription by both holoenzymes in vitro, but the magnitude of the effect was much larger for EσS (up to 6 times the amount of transcript produced in the absence of SutA) than for Eσ70 (up to 1.7 times) (Fig. 3B and Supporting Information, Fig. S13). In both cases, the effect saturated at concentrations of SutA between 125 and 500 nM, but transcription increased more than 2-fold for EσS even at 31 nM, the lowest concentration tested. The acidic N-tail is strictly required for activation, as the ΔN mutant inhibited transcription in a dose-dependent manner. The ΔC mutant was still able to enhance transcription, albeit with a shift in the concentration dependence evident with EσS. This shift may reflect C-tail interactions with EσS; as noted above, we observed that the chemical shifts of three residues in the C-tail were perturbed upon mixing with the β1 domain (Fig. 2C).
We next investigated whether SutA affects the rrn OC stability or interacts with other known regulatory inputs that impact OC stability. First, we directly measured the half-life of the heparin-resistant Eσ70 OC in a transcription-based assay. We measured an OC half-life that was very short, at about 45 seconds, but not quite as short as that seen for the E. coli rrnB P1 promoter under similar conditions (Barker et al., 2001). Addition of SutA at 125 or 500 nM had no significant effect (Fig. 3C, S16). We were unable to measure the OC stability for EσS on the rrn promoter because, especially in the absence of SutA, the signal was too weak at 0 s to accurately measure decay from that starting point.
Next, we measured effects of ppGpp/DksA and increasing [iNTPs], which repress and activate transcription from the E. coli rrn P1, respectively, in the presence or absence of SutA (Fig. 3D, S17–18). To assess the effects of [iNTPs], we replaced the CpU initiating dinucleotide used in Fig. 3B with CTP and UTP, and varied the concentration of both nucleotides together to ensure that transcription initiates at the same position. As observed in E. coli, rrn transcription was strongly diminished at low [iNTPs] and by DksA/ppGpp, but SutA did not significantly counter these effects. Taken together, these results suggest that while the P. aeruginosa rrn promoter forms an inherently unstable OC, which is sensitive to regulatory inputs that utilize its instability, SutA does not alter the stability of the Eσ70 OC.
A disordered acidic loop in σ70 modulates SutA binding
We wondered what difference between σ70 and σS could explain the difference in SutA’s impact on rrn initiation by Eσ70 compared to EσS. Domains 2, 3, and 4 are highly similar, and both σ70 and σS have flexible acidic regions, referred to as 1.1, near their N-termini (Gowrishankar et al., 2003). However, σ70 contains a large (~245 amino acids) insertion, termed the “non-conserved region” or NCR, which is not present in σS (Fig. 4A). Crystal and cryoEM structures show that most of the NCR is situated relatively far from the β1 binding site of SutA, contacting the β’ subunit on the opposite side of the main channel of RNAP, but an acidic stretch of ~40 residues within the NCR is too flexible to be resolved in these structures (herein AL for Acidic Loop) (Liu et al., 2016; Narayanan et al., 2018).
To investigate possible interactions between the AL of σ70 and SutA, we threaded the P. aeruginosa sequence onto the β subunit of an E. coli RNAP crystal structure (Molodtsov et al., 2017), docked that model into the recent cryoEM structure of the E. coli Eσ70 OC (Narayanan et al., 2018), and modeled the missing AL (using the E. coli sequence for both the structured and flexible regions of σ70) using the MODELLER software suite (Yang et al., 2012). The highly flexible AL could occupy a wide range of positions, some of which would stay well above the DNA in the main channel (position 1) and some of which would clash with the DNA (position 2), and also reach the β1 residues that participate in SutA cross-links. In contrast, σS has no corresponding flexible region and remains far from the SutA cross-links (Fig. 4A).
To determine whether the AL might contribute to the observed differences between Eσ70 and EσS activation by SutA, we constructed and purified a P. aeruginosa σ70 mutant lacking residues 171–214 (ΔAL), which correspond to the region missing in the E. coli structure, verified that it still had activity in in vitro (Supporting Information, Fig. S19) and repeated our cross-linking and cleavage assays using Eσ70, EσS, or Eσ70ΔAL holoenzymes instead of just E. In the absence of DNA, SutA L54BPA cross-linked to Eσ70 less efficiently than to E or EσS. Interestingly, Eσ70ΔAL largely restored the cross-linking to the levels seen with E or EσS (Fig. 4C, lanes 1–4), suggesting that the σ70 AL modulates the SutA interaction with Eσ70. The difference in cross-linking efficiency between Eσ70 and Eσ70ΔAL decreased at higher SutA concentrations, as might be expected if SutA and AL are competing to occupy a similar space.
SutA competes with DNA in the final open complex
Our cross-linking and cleavage results suggested that SutA’s position on RNAP might allow it to compete with the promoter DNA. To explore this possibility, we added to our cross-linking assay either a double-stranded (ds) rrn promoter DNA or a bubble template in which the region of the DNA that forms the transcription bubble in the OC was non-complementary (Fig. 4B). The dsDNA requires σ to melt the DNA strands, and will support a native population of promoter complex intermediates. By contrast, the bubble template obviates the need for σ and would be expected to stabilize an OC formed with the holoenzyme, but this complex may not represent the dominant native complex, as the E. coli rrn P1 does not form a stable final OC (Haugen et al., 2006; Ruff, Record, et al., 2015). The addition of the bubble DNA had a large negative effect on SutA binding that was synergistic with the presence of σ (Fig. 4C, lanes 5–8). Cross-linking could still be readily detected in the absence of σ, and to a lesser extent when σS was present, but not with either σ70 or σ70ΔAL; longer exposures revealed that cross-linking did occur at low efficiency (Supporting Information, Fig. S20). Addition of dsDNA allowed more SutA binding, but still less than in the absence of DNA (Fig. 4C, lanes 9–11).
The flexible SutA tails approach the transcription bubble
The BPA cross-linking reports on the interactions established by the central region of SutA, but gives no information on the position of its flexible tails, and a decrease in cross-linking could be due to a loss of binding or a change of SutA conformation. We used FeBABE cleavage assays with the N-Fe and C-Fe SutA variants (Fig. 4D and Supporting Information, Fig. S21) to address these questions. We found that the addition of σ70 had a large negative effect on cleavage induced by C-Fe, but little effect on cleavage induced by N-Fe. The σ70ΔAL mutant partially restored C-Fe cleavage levels to those observed with E or EσS. Together with the 54BPA cross-linking results (Fig. 4C), this suggests that the σ70AL does not fully displace SutA, but instead interferes with a binding interaction of the C-tail, consistent with our observation that the ΔC mutant required higher concentrations for maximal activity on EσS but not on Eσ70 (Fig. 3B).
By contrast, the addition of template DNA had a large negative effect for both C-Fe and N-Fe cleavage reactions, as well as for BPA cross-linking (Fig. 4C and D). This suggested that DNA might induce SutA dissociation, rather than its repositioning, prompting us to investigate whether SutA and DNA could form a ternary complex with RNAP holoenzyme. We measured FeBABE SutA-dependent cleavage of the template and non-template DNA strands using primer extension. In order to account for background cleavage or primer extension stops, we normalized the signal of each primer extension band in the FeBABE SutA-containing samples to the signal in the corresponding band from a control reaction (run on the same gel) that contained WT SutA, which should not be able to induce localized radical cleavage. We saw stronger cleavage with EσS than with Eσ70, but in both cases the signal was relatively weak, as might be expected for a factor that does not directly bind DNA (Fig. 4E and Supporting Information, Fig. S22). In the EσS complex, C-Fe induced cleavage of both strands between residues −8 and −12, suggesting that it remains near the upstream fork junction of the transcription bubble. N-Fe cleaved the template strand near the upstream junction but also cleaved both strands further downstream. For Eσ70, the cleavage was weaker overall and showed a different pattern; for C-Fe in particular, more non-template strand cleavage occurred in the downstream half of the transcription bubble, and less in the upstream half. This difference could reflect the AL-mediated repositioning of the C-tail, or differences in the OC intermediates formed by EσS and Eσ70.
DISCUSSION
As part of their response to fluctuating environmental conditions, bacteria produce regulators that directly bind RNAP and modify its behavior, eliciting global changes in gene expression patterns, in addition to producing different DNA-binding transcription factors that help recruit RNAP to specific genes (reviewed in Haugen et al., 2008). We previously identified SutA as a global regulator that binds RNAP and contributes to a broad response to protracted growth arrest, enhancing ongoing, low-level expression of housekeeping genes (Babin et al., 2016). SutA directly affects initiation at the rrn promoter, prompting comparison to other regulators that affect rRNA expression by directly binding RNAP, such as DksA, ppGpp, CarD, and RbpA. DksA and ppGpp, which appear to operate similarly in P. aeruginosa and E. coli, broadly destabilize OCs, leading to down-regulation of rrn P1 and activation of amino acid biosynthesis genes in response to nutrient downshifts (Ross et al., 2016). In contrast, CarD and RbpA, constitutively expressed in Mycobacteria, together enhance OC formation, and CarD stabilizes the final OC, modestly increasing rrn initiation in vitro (Boyaci et al., 2019; Hubin et al., 2017). SutA is distinct from these examples: though it acts on a P. aeruginosa rrn promoter that also forms an unstable OC with Eσ70, its activity does not affect the stability of this complex. Instead, SutA appears to affect intermediates in open complex formation, and may be displaced from the final OC, as we discuss below.
A model for SutA interactions with promoter complexes
Unlike DksA and CarD, which have well-defined structures (Boyaci et al., 2019; Perederina et al., 2004), SutA is largely intrinsically disordered, with its flexible tails playing key functional roles. Cross-linking and cleavage patterns suggest a SutA binding site that is close to but distinct from that of CarD (Boyaci et al., 2019), and far from the sites occupied by RbpA and DksA/ppGpp (Boyaci et al., 2019; Hubin et al., 2017; Ross et al., 2016). Although the extreme flexibility of SutA and the relatively large distances over which our cross-linking and cleavage reagents could act (10–25 Å) preclude precise docking of SutA onto RNAP, a binding site on the outside of the β1 domain is consistent with our data. SutA failed to activate rrn transcription in E. coli in vivo and failed to bind the E. coli Eσ70 in vitro (Supporting Information, Fig. S6, S20–21), indicating that its binding site is in a region that is different between the two polymerases. Most of the β residues are identical (72%) or similar (87%) between E. coli and P. aeruginosa, but two β1 loops that contain residues involved in BS3 cross-linking (K45 and K116) are among a small number of regions with reduced similarity (Supporting Information, Fig. S23). In this study we were limited in our ability to directly test whether any of these differences individually could result in loss of SutA binding, due to the fact that we were natively purifying the chromosomally encoded RNAP from P. aeruginosa, but future studies would benefit from an overexpression system for purifying this RNAP that would allow generating and testing mutant enzymes in vitro. From the proposed binding site for the SutA helix, its flexible tails could reach into the main channel of RNAP through the cleft between β1 and β2 (Fig. 5).
Figure 5. Model for SutA interaction with RNAP.

The model is based on patterns of cross-linking and FeBABE cleavage shown in Figures 2 and 4; the N-tail of SutA is shown in orange, its alpha helix in gray, and its C-tail in dark blue; blue stars represent positions of FeBABE modifications at SutA residues 2, 32, and 98; orange circles represent positions of BPA modifications at SutA residues 54 and 84; green hexagons represent positions of lysines involved in BS3 cross-links at residues 60, 62, and 69. A. In the absence of DNA, the N-tail, which is critical for activity (Fig. 3B), is located in or near the cleft between β1 and β2. The C-tail is located near the top of β1 in E and EσS but is displaced by the AL in Eσ70 (Fig. 2, 4D), as represented by the more transparent C-tail. NMR chemical shift perturbations suggested a direct interaction between SutA and β1 (Fig. 2C); removal of the C-tail affected the concentration dependence of the transcriptional impact of SutA on EσS but not Eσ70 (Fig. 3B); and the β cleavage by C-Fe was stronger for EσS than for Eσ70 (Fig. 4D). B. For both holoenzymes (represented by generic σ) in the presence of rrn promoter DNA, L54BPA cross-linking and β cleavage by C-Fe was dramatically reduced, whereas more N-Fe cleavage was retained (Fig. 4C and D, lanes 9–11). At the same time, DNA cleavage (Fig. 4E) suggests that SutA may be bound to some promoter complex intermediates. DNA cleavage signal for C-Fe occurred near the upstream junction of the bubble, consistent with positioning of the C-tail near the top of β1, while N-Fe cleaved the template strand upstream of the start site and the non-template strand at the start site, potentially consistent with positioning in the β1/β2 cleft. The OC intermediates on which SutA is proposed to act are likely to be dynamic, and the lack of cross-linking or cleavage when promoter complexes are formed with a bubble template that artifically stabilizes the OC suggests that SutA is displaced in the final OC. C. Very weak cleavages and one cross-link of I84BPA suggest an additional possible position for SutA at the base of β1 that could be a transient intermediate in binding, release, or other dynamics, but it does not seem possible for both sites to be occupied at the same time.
The β1/β2 cleft has been shown to accommodate the non-template strand during the early rounds of nucleotide addition, when it must scrunch to allow additional bases of the downstream DNA to enter the enzyme before the upstream contacts are released (Winkelman et al., 2015); on E. coli rrnB P1, scrunching occurs even before initiation (Winkelman et al., 2016). Interestingly, the β1/β2 cleft is also the site of several point mutations that suppress the auxotrophy phenotype of a ΔdksA mutant in E. coli, consistent with a model where modulating its interaction with the DNA could be functionally important in growth-phase-dependent gene expression regulation by DksA (Rutherford et al., 2009). Our results show that the fully melted promoter DNA inhibits SutA binding, as cleavage by both the N-tail and C-tail FeBABE are diminished, as is L54BPA cross-linking (Fig. 4C). This suggests that DNA and SutA may compete for similar contacts with RNAP on β1 or near the β1/β2 cleft. The DNA must undergo large conformational changes during open complex formation; several studies have suggested that multiple intermediates are involved, and that the importance and characteristics of these intermediates differ based on the promoter sequence (Hubin et al., 2017; Ruff, Drennan, et al., 2015; Rutherford et al., 2009). We hypothesize that the flexible, acidic N-tail of SutA influences the conformation of the DNA in early promoter complex intermediates and then causes SutA to be displaced from the final OC, in which the transcription bubble has been stabilized by contacts between the DNA and RNAP (Fig. 5). The fact that the N-terminal truncation mutant actually inhibits initiation in a concentration-dependent manner (Fig. 3B) is consistent with this hypothesis: lacking its acidic N-tail, SutA not only fails to helpfully influence an OC intermediate, it also fails to be displaced to allow initiation to proceed.
Proximity of the SutA tails to the DNA in at least some promoter complexes, consistent with the potential to influence DNA conformation, is indicated by FeBABE-mediated cleavage of the DNA (Fig. 4E). Cleavage was much stronger for EσS than for Eσ70, especially with C-Fe, possibly reflecting that Eσ70 favors more advanced OC intermediates, since SutA binding is strongly disfavored by the bubble template which locks the final OC (Fig. 4D), or that the decreased association of the SutA C-tail with Eσ70 in the absence of DNA allows SutA to be more easily displaced during OC formation. This is analogous to the regulatory mechanism of σ70 1.1, an acidic flexible region that binds in the main RNAP channel in early promoter complexes and must be ejected to accommodate the promoter DNA that binds to the same site in the final OC (Hook-Barnard & Hinton, 2009; Mekler et al., 2002; Murakami, 2013; Vuthoori et al., 2001; Wilson & Dombroski, 1997). Like SutA, σ70 1.1 stimulates initiation at some promoters but does not affect the stability of their OCs (Vuthoori et al., 2001).
The roles of acidic disordered regions in SutA regulation and beyond
In addition to the critical role of SutA’s unstructured acidic N-tail in mediating its enhancement of rrn transcription, we also found that an unstructured, acidic region of σ70, the AL, directly modulates SutA’s activity, possibly by interfering with the ability of the C-tail to bind β1. The AL is part of the NCR region that is unique to σ70 (Feklistov et al., 2014), so this interaction may contribute to the difference in the SutA’s effects on initiation by Eσ70 versus EσS. The NCR makes contacts with the upstream DNA duplex (Narayanan et al., 2018), and our modeling suggests that the AL could be positioned near the upstream junction of the transcription bubble. Like region 1.1 and the SutA N-tail, the σ70 AL is a highly dynamic element that could modulate the DNA trajectory in early intermediates in open complex formation, before the bubble is locked in place in the final OC, and we found that in addition to affecting the RNAP-SutA interaction, σ70 ΔAL also has mild defects in initiation at the rrn promoter on its own (Supporting Information, Fig. S19). Because the OC formation pathways and the relative occupancies of the intermediates vary among promoters (Ruff, Record, et al., 2015), we expect the effects of these unstructured elements to be varied too.
Dynamic interactions of intrinsically disordered (and often highly acidic) modules play key roles in eukaryotic transcriptional regulators, bacteriophage proteins, and σ factors. Flexible regions can gain access to and remodel dynamic regions of transcription complexes, as in the case of the phage proteins Gp2 and Nun, leading to inhibition of RNA synthesis (Bae et al., 2013; Kang et al., 2017). They also can bind or mimic flexible nucleic acid sequences, as in the case of the λN protein (Krupp et al., 2019) or σ1.1 (Murakami, 2013), and activate transcription. Although their precise mechanisms of action have not been determined, the δ subunit of RNAP in B. subtilis and other Gram-positive bacteria and the recently described RNAP-binding factor AtfA from Acinetobacter baylyi both contain substantial acidic, unstructured domains. Both proteins can bind to core RNAP, compete with nucleic acids for interaction with RNAP, and broadly affect transcription (Lopez de Saro et al., 1995; Prajapati et al., 2016; Weiss et al., 2014; Withers et al., 2014). These proteins are not direct SutA homologs, but further exploration of this group of small, acidic, and largely unstructured factors may allow for a more general understanding of their roles in modulating bacterial transcription. In eukaryotic systems, important roles for flexible acidic domains of transcriptional activators such as Gcn4 or Ino2 are widely appreciated: they can serve as flexible protein-protein interaction domains, capable of mediating interactions whose structural constraints vary depending on nuances of nucleic acid sequences, chromatin states, and other aspects of the intracellular environment (Pacheco et al., 2018; Staller et al., 2018; Tuttle et al., 2018). While their disordered nature has made many of these domains difficult to study using traditional structural and biochemical approaches, it is becoming increasingly clear that they play critical roles in many aspects of transcriptional regulation in all domains of life, and SutA adds to this growing body of evidence.
Implications for the in vivo role of SutA during slow growth
SutA is unique as a global regulator of transcription in that it appears to be specifically expressed under conditions of resource limitation, when overall rates of transcription are very low, and may function just to help maintain low levels of ongoing gene expression. Our previous in vivo ChIP data suggested that SutA may associate with RNAP both during initiation, especially at the rrn promoters, and during elongation, but here we have chosen to focus our efforts on understanding details of SutA’s interactions with RNAP at the rrn promoters (Babin et al., 2016). While the magnitudes of the SutA effects in vitro on Eσ70 were modest (just under 2-fold) and the total amount of transcription increase driven by SutA on EσS was small (a 3–6-fold increase, but from a very low baseline level of transcription), these results match our previous in vivo observation that expression of SutA during slow growth causes approximately a 2-fold increase in nascent rrn transcript (Babin et al., 2016). In contrast to well-studied activators that drive dramatic increases in transcription for a small subset of genes during an active stress response, by binding DNA and recruiting RNAP, SutA binds directly to RNAP, likely altering kinetic parameters that describe its interactions with the DNA during open complex formation. Factors that act in this way, including DksA/ppGpp, CarD, and RbpA as well as SutA, potentially affect initiation at every promoter in a positive or negative way, depending on the characteristics of the promoter (Galburt, 2018; Jensen et al., 2019). The stimulatory effects of these factors on transcription initiation are never very large: from less than 2-fold to 8-fold for DksA/ppGpp on amino acid biosynthesis and stress response gene promoters (Blankschien et al., 2009; Girard et al., 2017; Paul et al., 2005); less than 2-fold for RbpA on an rrn promoter; and 3–4-fold for RbpA plus CarD on an rrn promoter(Hubin et al., 2017). Changing the general behavior of RNAP enough to drive larger effects than this on specific promoters would likely have very detrimental effects on its ability to transcribe from other promoters, and detrimental effects on the cell as a whole. Additionally, SutA is expressed, and has presumably evolved to function, under conditions where overall rates of transcription are very low. Its key contribution to cellular fitness in these conditions may be to adapt RNAP to the challenges associated with very low overall metabolic activity, such as decreased negative supercoiling, changes in solute concentrations, and changes in the complement of nucleoid associated proteins. In this context, a very small stimulation of transcription initiation may be physiologically significant, if it is the difference between being able to produce a small number of new ribosomes to replace damaged ones, or not being able to make any ribosomes at all. A deep understanding of regulation during non-growing states will require measuring small effects, both in vivo and in vitro, but the pervasiveness and importance of these states in natural environments makes rising to this challenge worthwhile.
Our results clearly show that SutA can interact with both Eσ70 and EσS. σ70 and σS are closely related σ factors with partially overlapping promoter specificities (Feklistov et al., 2014; Liu et al., 2016; Schulz et al., 2015). Our previous RNA-Seq results imply that SutA interactions with both holoenzymes are likely to be functionally relevant, as some affected genes are bona fide σS regulon members (Schulz et al., 2015), but overall the affected genes are biased toward classic targets of σ70 (Babin et al., 2016). In the context of severe limitation in which SutA is expressed, its ability to interact more strongly with EσS and potentially stimulate the activity of this holoenzyme toward promoters that are classic Eσ70 targets, as well as its own regulon, could be significant. In both E. coli and P. aeruginosa, the activities and relative abundances of EσS and Eσ70 change throughout different growth phases, with σS upregulated at the transition to stationary phase (Battesti et al., 2015; Schuster et al., 2004), and much of Eσ70 sequestered by the 6S RNA late in stationary phase (Wassarman & Storz, 2000). In addition, EσS and Eσ70 appear to be differentially sensitive to changes in cellular conditions that occur during stationary phase.Moreover, in specific cases that have been examined in vitro, EσS initiation efficiency increases under the stationary phase-associated condition, while Eσ70 initiation efficiency decreases (Bordes et al., 2003; Meyer & Grainger, 2013). These characteristics of EσS may in part explain why σS ChIP signal at the rrn promoters increases in stationary phase in E. coli and σ70 ChIP signal decreases (Raffaelle et al., 2005). The ability of SutA to enhance initiation by EσS and Eσ70 differentially could allow greater flexibility during different stages of growth arrest. For example, SutA may enable baseline levels of housekeeping gene expression regardless of which holoenzyme is most available and active. Our results also suggest that SutA can efficiently associate with the core enzyme E, and its possible role in influencing transcription elongation, independent of interactions with σ factors, will be an exciting question to explore in the future. More work will certainly be required to fully understand how SutA contributes to the regulatory architecture that allows P. aeruginosa to thrive during dormancy, but this study provides important mechanistic insight into the function of this global regulator, laying the foundation for such efforts.
EXPERIMENTAL PROCEDURES
See Extended Experimental Procedures in SI for additional details about all experiments, for strain construction details, and for tables of strains and primers used.
Protein purification
P. aeruginosa core RNAP was purified as previously described (Burgess & Jendrisak, 1975; Hager et al., 1990; Kuznedelov et al., 2011). N-terminal 6xHis-tagged SutA, SutA variants, DksA, σS, σ70, σ70ΔAL, and β1 were heterologously expressed in E. coli and purified by metal affinity chromatography followed by cleavage of the 6xHis tag with TEV protease and size exclusion chromatography. For NMR experiments, cells were grown in minimal media prepared with 15NH4Cl or 13C glucose or both. For BPA cross-linking, amber stop codons were introduced at positions of interest and BPA was incorporated via amber suppression following co-transformation of the SutA plasmid with pEVOL-pBpF (Chin et al., 2002). For preparation of FeBABE variants, cysteine residues were introduced at positions of interest and following purification of the protein, the FeBABE moeity was conjugated to the cysteine (Meares et al., 2003). Conjugation efficiencies were estimated to be 57%, 38%, and 76% respectively for the residue 2, 32, and 98 variants.
NMR experiments
Data were collected from SutA proteins at concentrations of 300 μM. For the 46–101 variant, the following spectra were acquired on a Varian Inova 600 MHz NMR with a triple resonance inverse probe running VnmrJ 4.2A: 15N HSQC, 13C HSQC, HNCO, HNCA, HNCACB, CBCACONH, HNCOCA, HNCACO, CCONH, and 15N HSQC experiments modified for measurement of T2 and of 15N-1H NOE. For the full-length protein, 15N HSQC, 13C HSQC, HNCACB, and CBCACONH spectra were acquired at 7 °C on a Bruker AV III 700 MHz spectrometer with a TCI cryoprobe running Topspin 3.2, but 15N HSQC experiments modified for measurement of T2 and of 15N-1H NOE were collected on the Varian Inova 600 MHz NMR, as were 15N HSQC spectra for the SutA ΔN and SutA ΔC SutA proteins. 15N13C-labeled full-length SutA was embedded in a stretched polyacrylamide gel for measurement of residual dipolar couplings as previously described (Mohana-Borges et al., 2004), using the Varian Inova 600 MHz NMR. To assess SutA binding to β1 by NMR, 15N-labeled SutA and β1 fragment were mixed together and the resulting complex subjected to size exclusion chromatography, resulting in a final concentration of complex of approximately 25 μM. In addition, 15N-labeled SutA was mixed with σS at 50 μM each, and 13C15N-labeled SutA at 50 μM was measured alone. These 15N HSQC spectra were acquired on a Bruker 800 MHZ AV III HD spectrometer with a TCI cryoprobe at 25 °C. Peak assignments and analysis were done using the PINE Server, CcpNmr Analysis Suite, and MestreNova software.
Cross-linking experiments and data analysis
BS3 cross-linking of E and SutA was carried out as described (Rappsilber, 2011), with modifications. Cross-linked complexes were subjected to in-solution digestion by the Glu-C peptidase, and the resulting fragments were analyzed by LC-MS/MS on an Orbitrap Elite Hybrid Ion Trap MS. Cross-linked pepties were identified as described (Trnka et al., 2014), with modifications. BPA cross-linking was achieved by irradiatiation with UV light, complexes were digested in solution with trypsin, and analyzed by LC-MS/MS on a Q Exactive HF Orbitrap MS. Cross-linked peptides were identified using the StavroX software package (Götze et al., 2012).
FeBABE experiments and analysis
Cleavage reactions of complexes were initiated by the addition of ascorbate and hydrogen peroxide to final concentrations of 5 mM each (Meares et al., 2003). For measuring protein cleavage, reactions were quenched by the addition of SDS loading buffer and were evaluated by SDS-PAGE followed by Western blotting, using a monoclonal antibody raised against a peptide from the extreme C-terminus of E. coli β (EPR18704 from Abcam). To generate standards for size comparison, several different C-terminal fragments of RpoB with endpoints ranging from residue 355 to 1062 were overexpressed in E. coli and crude lysates from these strains were subjected to SDS-PAGE and Western blotting alongside the FeBABE cleavage products. For measuring DNA cleavage, reactions were quenched with thiourea and treated with proteinase K. The DNA was precipitated, and subjected to primer extension using Cy3- or Cy5-labeled primers complementary to the non-template or template strand respectively. Products were separated on denaturing 12% polyacrylamide gels and imaged by laser scanner.
In vitro transcription experiments were carried out as previously described with minor modifications (Artsimovitch & Henkin, 2009). Briefly, reactions containing 15 nM template, 20 nM RNAP holoenzyme, and other factors as indicated (SutA or DksA/ppGpp) were assembled in TGA buffer (20 mM Tris-acetate pH 8.0, 2 mM Na-acetate, 2 mM Mg-acetate, 4 % glycerol, 0.1 mM DTT, 0.1 mM EDTA) and open complexes were allowed to form at 37°C for 6 minutes. For single turnover reactions, 5x nucleotide mix (unless otherwise indicated: 375 μM initiating dinucleotide, 250 μM each NTP not carrying 32P label (ATP, UTP, and either CTP or GTP), 100 μM cold NTP of the same type as that carrying the label (either CTP or GTP), 0.75 μCi α32P GTP or CTP (3000 Ci mmol−1, 10 mCi ml−1, Perkin Elmer, Waltham MA), and 100 μg ml−1 heparin) was added and transcription was allowed to continue for 8 minutes before reactions were quenched with STOP buffer (8 M urea, 10 mM EDTA, 0.8x TBE, 2 mg ml−1 bromophenol blue, 2 mg ml−1 xylene cyanol FF, 2 mg ml−1 amaranth). For open complex stability assays, heparin was added to a master mix of open complex to a final concentration of 20 μg ml−1 first, and then aliquots were withdrawn and added to 5x nucleotide mix at the indicated time points, and transcription allowed to proceed for 8 minutes before quenching with STOP buffer. Quenched reactions (2 μl) were run on a denaturing 20% polyacrylamide TBE gel and the gel was exposed to a phosphorimaging screen without drying. Phosphorimager screens were scanned on a Typhoon FLA 9000 gel imaging system and images were quantified using the FIJI analysis suite (RRID:SCR_002285)(Schindelin et al., 2012).
Supplementary Material
ACKNOWLEDGMENTS
We thank Ben Ramirez (University of Illinois at Chicago) for helping us with preliminary NMR studies of SutA, Jacqueline Barton (Caltech) for giving us access to her lab to perform experiments involving radioactivity, Nate Glasser for help with HPLC measurements to quantify SutA, Hsiau-Wei (Jack) Lee and Aimee Marceau (University of California, Santa Cruz) for help with the NMR binding experiment using the Bruker AVIII HD 800 MHz NMR, Weidong Hu (City of Hope) for help with NMR experiments using the Bruker AV III 700 MHz spectrometer, and Julia Kardon and Niels Bradshaw (Brandeis University) and members of the Newman lab for feedback on the project at different stages. MB was supported by a post-doctoral fellowship from the Cystic Fibrosis Foundation (BERGKE16F0). Grants from the NIH (GM067153) to IA and grants from the HHMI and NIH (R01HL117328) to DKN supported this work. The Proteome Exploration Laboratory is supported by the Beckman Institute and NIH 1S10OD02001301. This work was also supported by the Institute for Collaborative Biotechnologies through grant W911NF-09–0001 from the U.S. Army Research Office. The content of the information does not necessarily reflect the position or the policy of the Government, and no official endorsement should be inferred. The authors have no conflicts of interest to disclose.
Footnotes
Data availability statement: Additional raw data that support the findings of this study are openly available in figshare at 10.6084/m9.figshare.8259017.
REFERENCES
- Artsimovitch I, & Henkin TM (2009). In vitro approaches to analysis of transcription termination. Methods, 47(1), 37–43. doi: 10.1016/j.ymeth.2008.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babin BM, Atangcho L, van Eldijk MB, Sweredoski MJ, Moradian A, Hess S, . . . Tirrell DA (2017). Selective Proteomic Analysis of Antibiotic-Tolerant Cellular Subpopulations in Pseudomonas aeruginosa Biofilm. MBio, 8(5), 16. doi: 10.1128/mBio [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babin BM, Bergkessel M, Sweredoski MJ, Moradian A, Hess S, Newman DK, & Tirrell DA (2016). SutA is a bacterial transcription factor expressed during slow growth in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A, 113(5), E597–605. doi: 10.1073/pnas.1514412113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae B, Chen J, Davis E, Leon K, Darst SA, & Campbell EA (2015). CarD uses a minor groove wedge mechanism to stabilize the RNA polymerase open promoter complex. Elife, 4. doi: 10.7554/eLife.08505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae B, Davis E, Brown D, Campbell EA, Wigneshweraraj S, & Darst SA (2013). Phage T7 Gp2 inhibition of Escherichia coli RNA polymerase involves misappropriation of σ70 domain 1.1. Proc Natl Acad Sci U S A, 110(49), 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker MM, Gaal T Fau - Josaitis CA, Josaitis Ca Fau - Gourse RL, & Gourse RL (2001). Mechanism of regulation of transcription initiation by ppGpp. I. Effects of ppGpp on transcription initiation in vivo and in vitro. J Mol Biol, 305(4), 16. [DOI] [PubMed] [Google Scholar]
- Battesti A, Majdalani N, & Gottesman S (2015). Stress sigma factor RpoS degradation and translation are sensitive to the state of central metabolism. Proc Natl Acad Sci U S A, 112(16), 5159–5164. doi: 10.1073/pnas.1504639112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergkessel M, Basta DW, & Newman DK (2016). The physiology of growth arrest: uniting molecular and environmental microbiology. Nat Rev Microbiol, 14(9), 549–562. doi: 10.1038/nrmicro.2016.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankschien MD, Lee JH, Grace ED, Lennon CW, Halliday JA, Ross W, . . . Herman C (2009). Super DksAs: substitutions in DksA enhancing its effects on transcription initiation. EMBO J, 28(12), 1720–1731. doi: 10.1038/emboj.2009.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordes P, Conter A, Morales V, Bouvier J, Kolb A, & Gutierrez C (2003). DNA supercoiling contributes to disconnect σS accumulation from σS-dependent transcription in Escherichia coli. Mol Microbiol, 48(2), 11. [DOI] [PubMed] [Google Scholar]
- Boyaci H, Chen J, Jansen R, Darst SA, & Campbell EA (2019). Structures of an RNA polymerase promoter melting intermediate elucidate DNA unwinding. Nature, 565(7739), 382–385. doi: 10.1038/s41586-018-0840-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan DW, Minneci F, Nugent TC, Bryson K, & Jones DT (2013). Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res, 41(Web Server issue), W349–357. doi: 10.1093/nar/gkt381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RR, & Jendrisak JJ (1975). A procedure for the rapid, large-scale purification of Escherichia coli DNA-dependent RNA polymerase involving Polymin P precipitation and DNA-cellulose chromatography. Biochemistry, 14(21), 4634–4638. [DOI] [PubMed] [Google Scholar]
- Chin JW, Martin AB, King DS, Wang L, & Schultz PG (2002). Addition of a photocrosslinking amino acid to the genetic code of Escherichia coli. Proc Natl Acad Sci U S A, 99(17), 11020–11024. doi: 10.1073/pnas.172226299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofu O, Tolker-Nielsen T, Jensen PO, Wang H, & Hoiby N (2015). Antimicrobial resistance, respiratory tract infections and role of biofilms in lung infections in cystic fibrosis patients. Adv Drug Deliv Rev, 85, 7–23. doi: 10.1016/j.addr.2014.11.017 [DOI] [PubMed] [Google Scholar]
- Drozdetskiy A, Cole C, Procter J, & Barton GJ (2015). JPred4: a protein secondary structure prediction server. Nucleic Acids Res, 43(W1), W389–394. doi: 10.1093/nar/gkv332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feklistov A, Sharon BD, Darst SA, & Gross CA (2014). Bacterial sigma factors: a historical, structural, and genomic perspective. Annu Rev Microbiol, 68, 357–376. doi: 10.1146/annurev-micro-092412-155737 [DOI] [PubMed] [Google Scholar]
- Galburt EA (2018). The calculation of transcript flux ratios reveals single regulatory mechanisms capable of activation and repression. Proc Natl Acad Sci U S A, 115(50), 10. doi: 10.1073/pnas.1809454115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard ME, Gopalkrishnan S, Grace ED, Halliday JA, Gourse RL, & Herman C (2017). DksA and ppGpp Regulate the sigmaS Stress Response by Activating Promoters for the Small RNA DsrA and the Anti-Adapter Protein IraP. J Bacteriol, 200(2), 12. doi: 10.1128/jb.00463-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götze M, Pettelkau J, Schaks S, Bosse K, Ihling CH, Krauth F, . . . Sinz A (2012). StavroX—A Software for Analyzing Crosslinked Products in Protein Interaction Studies. J Am Soc Mass Spectrom, 23, 12. doi: 10.1007/s13361-011-0261-2) [DOI] [PubMed] [Google Scholar]
- Gowrishankar J, Yamamoto K, Subbarayan PR, & Ishihama A (2003). In Vitro Properties of RpoS (σS) Mutants of Escherichia coli with Postulated N-Terminal Subregion 1.1 or C-Terminal Region 4 Deleted. J Bacteriol, 185(8), 2673–2679. doi: 10.1128/jb.185.8.2673-2679.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager DA, Jin DJ, & Burgess RR (1990). Use of Mono Q high-resolution ion-exchange chromatography to obtain highly pure and active Escherichia coli RNA polymerase. Biochemistry, 29(34), 7890–7894. [DOI] [PubMed] [Google Scholar]
- Haugen SP, Berkmen MB, Ross W, Gaal T, Ward C, & Gourse RL (2006). rRNA promoter regulation by nonoptimal binding of sigma region 1.2: an additional recognition element for RNA polymerase. Cell, 125(6), 1069–1082. doi: 10.1016/j.cell.2006.04.034 [DOI] [PubMed] [Google Scholar]
- Haugen SP, Ross W, & Gourse RL (2008). Advances in bacterial promoter recognition and its control by factors that do not bind DNA. Nat Rev Microbiol, 6(7), 507–519. doi: 10.1038/nrmicro1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook-Barnard IG, & Hinton DM (2009). The promoter spacer influences transcription initiation via sigma70 region 1.1 of Escherichia coli RNA polymerase. Proc Natl Acad Sci U S A, 106(3), 737–742. doi: 10.1073/pnas.0808133106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubin EA, Fay A, Xu C, Bean JM, Saecker RM, Glickman MS, . . . Campbell EA (2017). Structure and function of the mycobacterial transcription initiation complex with the essential regulator RbpA. Elife, 6. doi: 10.7554/eLife.22520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen D, Manzano AR, Rammohan J, Stallings CL, & Galburt EA (2019). CarD and RbpA modify the kinetics of initial transcription and slow promoter escape of the Mycobacterium tuberculosis RNA polymerase. Nucleic Acids Res(1362–4962 (Electronic)), 14. doi: 10.1093/nar/gkz449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, & Cozzetto D (2015). DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics, 31(6), 857–863. doi: 10.1093/bioinformatics/btu744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JY, Olinares PD, Chen J, Campbell EA, Mustaev A, Chait BT, . . . Darst SA (2017). Structural basis of transcription arrest by coliphage HK022 Nun in an Escherichia coli RNA polymerase elongation complex. Elife, 6. doi: 10.7554/eLife.25478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer JC, Erickson-Viitanen S, Wolfe HR, & DeGrado WF (1986). p-Benzoyl-L-phenylalanine, A New Photoreactive Amino Acid. J Biol Chem, 261(23), 6. [PubMed] [Google Scholar]
- Kim DE, Chivian D, & Baker D (2004). Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res, 32(Web Server issue), W526–531. doi: 10.1093/nar/gkh468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupp F, Said N, Huang YH, Loll B, Burger J, Mielke T, . . . Wahl MC (2019). Structural Basis for the Action of an All-Purpose Transcription Anti-termination Factor. Mol Cell. doi: 10.1016/j.molcel.2019.01.016 [DOI] [PubMed] [Google Scholar]
- Kuznedelov K, Semenova E, Knappe TA, Mukhamedyarov D, Srivastava A, Chatterjee S, . . . Severinov K (2011). The antibacterial threaded-lasso peptide capistruin inhibits bacterial RNA polymerase. J Mol Biol, 412(5), 842–848. doi: 10.1016/j.jmb.2011.02.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane WJ, & Darst SA (2010). Molecular evolution of multisubunit RNA polymerases: sequence analysis. J Mol Biol, 395(4), 671–685. doi: 10.1016/j.jmb.2009.10.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leirmo S, & Gourse RL (1991). Factor-independent activation of Escherichia coli rRNA transcription. I. Kinetic analysis of the roles of the upstream activator region and supercoiling on transcription of the rrnB P1 promoter in vitro. J Mol Biol, 220(3), 555–568. [DOI] [PubMed] [Google Scholar]
- Liu B, Zuo Y, & Steitz TA (2016). Structures of E. coli sigmaS-transcription initiation complexes provide new insights into polymerase mechanism. Proc Natl Acad Sci U S A, 113(15), 4051–4056. doi: 10.1073/pnas.1520555113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Saro FJ, Woody AY, & Helmann JD (1995). Structural Analysis of the Bacillus subtilis δFactor: A Protein Polyanion which Displaces RNA from RNA Polymerase. J Mol Biol, 252, 14. [DOI] [PubMed] [Google Scholar]
- Meares CF, Datwyler SA, Schmidt BD, Owens J, & Ishihama A (2003). Principles and Methods of Affinity Cleavage in Studying Transcription. Methods Enzymol, 371, 25. [DOI] [PubMed] [Google Scholar]
- Mekler V, Kortkhonjia E, Mukhopadhyay J, Knight J, Revyakin A, Kapanidis AN, . . . Ebright RH (2002). Structural Organization of Bacterial RNA Polymerase Holoenzyme and the RNA Polymerase-Promoter Open Complex. Cell, 108, 16. [DOI] [PubMed] [Google Scholar]
- Meyer AS, & Grainger DC (2013). The Escherichia coli Nucleoid in Stationary Phase. Adv Appl Microbiol, 83, 69–86. doi: 10.1016/B978-0-12-407678-5.00002-7 [DOI] [PubMed] [Google Scholar]
- Mohana-Borges R, Goto NK, Kroon GJ, Dyson HJ, & Wright PE (2004). Structural characterization of unfolded states of apomyoglobin using residual dipolar couplings. J Mol Biol, 340(5), 1131–1142. doi: 10.1016/j.jmb.2004.05.022 [DOI] [PubMed] [Google Scholar]
- Molodtsov V, Scharf NT, Stefan MA, Garcia GA, & Murakami KS (2017). Structural basis for rifamycin resistance of bacterial RNA polymerase by the three most clinically important RpoB mutations found in Mycobacterium tuberculosis. Mol Microbiol, 103(6), 1034–1045. doi: 10.1111/mmi.13606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami KS (2013). X-ray crystal structure of Escherichia coli RNA polymerase sigma70 holoenzyme. J Biol Chem, 288(13), 9126–9134. doi: 10.1074/jbc.M112.430900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray HD, & Gourse RL (2004). Unique roles of the rrn P2 rRNA promoters in Escherichia coli. Mol Microbiol, 52(5), 1375–1387. doi: 10.1111/j.1365-2958.2004.04060.x [DOI] [PubMed] [Google Scholar]
- Murray HD, Schneider DA, & Gourse RL (2003). Control of rRNA Expression by Small Molecules Is Dynamic and Nonredundant. Mol Cell, 12(1), 125–134. doi: 10.1016/s1097-2765(03)00266-1 [DOI] [PubMed] [Google Scholar]
- Narayanan A, Vago FS, Li K, Qayyum MZ, Yernool D, Jiang W, & Murakami KS (2018). Cryo-EM structure of Escherichia coli sigma(70) RNA polymerase and promoter DNA complex revealed a role of sigma non-conserved region during the open complex formation. J Biol Chem, 293(19), 7367–7375. doi: 10.1074/jbc.RA118.002161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares J, Bernardini A, Garcia-Leon G, Corona F, M BS, & Martinez JL (2013). The intrinsic resistome of bacterial pathogens. Front Microbiol, 4, 103. doi: 10.3389/fmicb.2013.00103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opalka N, Brown J, Lane WJ, Twist KA, Landick R, Asturias FJ, & Darst SA (2010). Complete structural model of Escherichia coli RNA polymerase from a hybrid approach. PLoS Biol, 8(9). doi: 10.1371/journal.pbio.1000483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco D, Warfield L, Brajcich M, Robbins H, Luo J, Ranish J, & Hahn S (2018). Transcription Activation Domains of the Yeast Factors Met4 and Ino2: Tandem Activation Domains with Properties Similar to the Yeast Gcn4 Activator. Mol Cell Biol, 38(10). doi: 10.1128/MCB.00038-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BJ, Berkmen MB, & Gourse RL (2005). DksA potentiates direct activation of amino acid promoters by ppGpp. Proc Natl Acad Sci U S A, 102(22), 7823–7828. doi: 10.1073/pnas.0501170102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BJ, Ross W, Gaal T, & Gourse RL (2004). rRNA transcription in Escherichia coli. Annu Rev Genet, 38, 749–770. doi: 10.1146/annurev.genet.38.072902.091347 [DOI] [PubMed] [Google Scholar]
- Perederina A, Svetlov V, Vassylyeva MN, Tahirov TH, Yokoyama S, Artsimovitch I, & Vassylyev DG (2004). Regulation through the secondary channel--structural framework for ppGpp-DksA synergism during transcription. Cell, 118(3), 297–309. doi: 10.1016/j.cell.2004.06.030 [DOI] [PubMed] [Google Scholar]
- Prajapati RK, Sengupta S, Rudra P, & Mukhopadhyay J (2016). Bacillus subtilis delta Factor Functions as a Transcriptional Regulator by Facilitating the Open Complex Formation. J Biol Chem, 291(3), 1064–1075. doi: 10.1074/jbc.M115.686170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaelle M, Kanin EI, Vogt J, Burgess RR, & Ansari AZ (2005). Holoenzyme switching and stochastic release of sigma factors from RNA polymerase in vivo. Mol Cell, 20(3), 357–366. doi: 10.1016/j.molcel.2005.10.011 [DOI] [PubMed] [Google Scholar]
- Rappsilber J (2011). The beginning of a beautiful friendship: cross-linking/mass spectrometry and modelling of proteins and multi-protein complexes. J Struct Biol, 173(3), 530–540. doi: 10.1016/j.jsb.2010.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy T, & Rainey JK (2010). Interpretation of biomolecular NMR spin relaxation parameters. Biochem Cell Biol, 88(2), 131–142. doi: 10.1139/o09-152 [DOI] [PubMed] [Google Scholar]
- Rohl CA, & Baker D (2002). De novo determination of protein backbone structure from residual dipolar couplings using Rosetta. J Am Chem Soc, 124(11), 2723–2729. [DOI] [PubMed] [Google Scholar]
- Ross W, Sanchez-Vazquez P, Chen AY, Lee JH, Burgos HL, & Gourse RL (2016). ppGpp Binding to a Site at the RNAP-DksA Interface Accounts for Its Dramatic Effects on Transcription Initiation during the Stringent Response. Mol Cell, 62(6), 811–823. doi: 10.1016/j.molcel.2016.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff EF, Drennan AC, Capp MW, Poulos MA, Artsimovitch I, & Record MT Jr. (2015). E. coli RNA Polymerase Determinants of Open Complex Lifetime and Structure. J Mol Biol, 427(15), 2435–2450. doi: 10.1016/j.jmb.2015.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff EF, Record MT Jr., & Artsimovitch I (2015). Initial events in bacterial transcription initiation. Biomolecules, 5(2), 1035–1062. doi: 10.3390/biom5021035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford ST, Villers CL, Lee JH, Ross W, & Gourse RL (2009). Allosteric control of Escherichia coli rRNA promoter complexes by DksA. Genes Dev, 23(2), 236–248. doi: 10.1101/gad.1745409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, . . . Cardona A (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods, 9(7), 676–682. doi: 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz S, Eckweiler D, Bielecka A, Nicolai T, Franke R, Dotsch A, . . . Haussler S (2015). Elucidation of sigma factor-associated networks in Pseudomonas aeruginosa reveals a modular architecture with limited and function-specific crosstalk. PLoS Pathog, 11(3), e1004744. doi: 10.1371/journal.ppat.1004744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster M, Hawkins AC, Harwood CS, & Greenberg EP (2004). The Pseudomonas aeruginosa RpoS regulon and its relationship to quorum sensing. Mol Microbiol, 51(4), 973–985. doi: 10.1046/j.1365-2958.2003.03886.x [DOI] [PubMed] [Google Scholar]
- Staller MV, Holehouse AS, Swain-Lenz D, Das RK, Pappu RV, & Cohen BA (2018). A High-Throughput Mutational Scan of an Intrinsically Disordered Acidic Transcriptional Activation Domain. Cell Syst, 6(4), 444–455 e446. doi: 10.1016/j.cels.2018.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trnka MJ, Baker PR, Robinson PJ, Burlingame AL, & Chalkley RJ (2014). Matching cross-linked peptide spectra: only as good as the worse identification. Mol Cell Proteomics, 13(2), 420–434. doi: 10.1074/mcp.M113.034009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle LM, Pacheco D, Warfield L, Luo J, Ranish J, Hahn S, & Klevit RE (2018). Gcn4-Mediator Specificity Is Mediated by a Large and Dynamic Fuzzy Protein-Protein Complex. Cell Rep, 22(12), 3251–3264. doi: 10.1016/j.celrep.2018.02.097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udikovic-Kolic N, Wichmann F, Broderick NA, & Handelsman J (2014). Bloom of resident antibiotic-resistant bacteria in soil following manure fertilization. Proc Natl Acad Sci U S A, 111(42), 15202–15207. doi: 10.1073/pnas.1409836111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuthoori S, Bowers CW, McCracken A, Dombroski AJ, & Hinton DM (2001). Domain 1.1 of the sigma(70) subunit of Escherichia coli RNA polymerase modulates the formation of stable polymerase/promoter complexes. J Mol Biol, 309(3), 561–572. doi: 10.1006/jmbi.2001.4690 [DOI] [PubMed] [Google Scholar]
- Wassarman KM, & Storz G (2000). 6S RNA Regulates E. coli RNA Polymerase Activity. Cell, 101, 11. [DOI] [PubMed] [Google Scholar]
- Weiss A, Ibarra JA, Paoletti J, Carroll RK, & Shaw LN (2014). The delta subunit of RNA polymerase guides promoter selectivity and virulence in Staphylococcus aureus. Infect Immun, 82(4), 1424–1435. doi: 10.1128/IAI.01508-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KP, Gillespie JJ, Sobral BW, Nordberg EK, Snyder EE, Shallom JM, & Dickerman AW (2010). Phylogeny of gammaproteobacteria. J Bacteriol, 192(9), 2305–2314. doi: 10.1128/JB.01480-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C, & Dombroski AJ (1997). Region 1 of sigma70 is required for efficient isomerization and initiation of transcription by Escherichia coli RNA polymerase. J Mol Biol, 267(1), 60–74. doi: 10.1006/jmbi.1997.0875 [DOI] [PubMed] [Google Scholar]
- Winkelman JT, Chandrangsu P, Ross W, & Gourse RL (2016). Open complex scrunching before nucleotide addition accounts for the unusual transcription start site of E. coli ribosomal RNA promoters. Proc Natl Acad Sci U S A, 113(13), E1787–1795. doi: 10.1073/pnas.1522159113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelman JT, Winkelman BT, Boyce J, Maloney MF, Chen AY, Ross W, & Gourse RL (2015). Crosslink Mapping at Amino Acid-Base Resolution Reveals the Path of Scrunched DNA in Initial Transcribing Complexes. Mol Cell, 59(5), 768–780. doi: 10.1016/j.molcel.2015.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Sykes BD, & Richards FM (1991). Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J Mol Biol, 222(2), 311–333. [DOI] [PubMed] [Google Scholar]
- Withers R, Doherty GP, Jordan M, Yang X, Dixon NE, & Lewis PJ (2014). AtfA, a new factor in global regulation of transcription in Acinetobacter spp. Mol Microbiol, 93(6), 1130–1143. doi: 10.1111/mmi.12723 [DOI] [PubMed] [Google Scholar]
- Yang Z, Lasker K, Schneidman-Duhovny D, Webb B, Huang CC, Pettersen EF, . . . Ferrin TE (2012). UCSF Chimera, MODELLER, and IMP: an integrated modeling system. J Struct Biol, 179(3), 269–278. doi: 10.1016/j.jsb.2011.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.