

Abstract
Non-alcoholic fatty liver disease (NAFLD) and its progressive form non-alcoholic steatohepatitis (NASH) affect 25% of the world population. NAFLD is predicted to soon become the main cause of liver morbidity and transplantation. The disease is characterized by a progressive increase of lipid accumulation in hepatocytes, which eventually induce fibrosis and inflammation, and can ultimately cause cirrhosis and hepatic carcinoma. Here, we created a patterned model of NAFLD on a chip using free fatty acid gradients to recapitulate a spectrum of disease conditions in a single continuous liver tissue. We established the NAFLD progression via quantification of intracellular lipid accumulation and transcriptional levels of fatty acid transporters and NAFLD pathogenesis markers. We then used this platform to create oxygen driven steatosis zonation mimicking the sinusoidal lipid distribution on a single continuous tissue and showed that this fat zonation disappears under progressed steatosis, in agreement with in vivo observations and recent computational studies. While we focus on free fatty acids and oxygen as the drivers of NAFLD, the microfluidic platform here is extensible to simultaneous use of other drivers.
Introduction
Non-alcoholic fatty liver disease (NAFLD)1 is a growing epidemic worldwide in part due to obesity and insulin resistance leading to liver accumulation of triglycerides and free fatty acids2; and is now the leading cause of chronic liver disease in the USA and Europe3, 4 and the second most common reason for liver transplantation2, 5. The prevalence of NAFLD stands at 20 – 30 % in Western countries and the overall world prevalence is estimated to be at 20 – 25 %3, 4, 6, 7. The prevalence amongst obese or diabetic patients is even higher, ~75 % for Western countries and 40 – 90 % worldwide4. Considering the rise of unhealthy diets and the growing patient population with obesity and diabetes, NAFLD is projected to become the leading cause of liver morbidity and mortality, and liver transplantation in the next 20 years2, 8. The current financial burden of NAFLD, with all incident and prevalent cases, is estimated at slightly over $100 billion in the United States3, 9. There are currently no FDA approved therapeutics for NAFLD treatment7, and the NASH drug market is expected to grow from $1 billion in 2015 to $25 billion till 202510. Accordingly, developing a progressive disease model and a high-throughput testing platform for NAFLD/NASH is crucial.
The past decade has resulted in numerous studies on the development of in vitro NAFLD models owing to the overwhelming increase in NAFLD cases. In general most of these in vitro models were limited due to: a) use of cell lines that do not properly recapitulate the lipid and carbohydrate metabolisms, b) use of supraphysiological levels of glucose and insulin along with other media components c) lack of perfusion (i.e. static models) resulting in incorrect chemical kinetics, d) models focusing only a specific phase of the disease, and e) low throughput11–17. Among these models, a few stand out despite their shortcomings. For example, Davidson et al. established a stable static long-term culture approach with primary hepatocyte and fibroblast co-cultures (a micropatterned co-culture system) where they studied effects of chronic hypo- and hyperglycemia; however, this study was limited by lack of flow11. A study which featured a continuous flow with physiologic glucose and insulin levels was conducted by Feaver et al., where multiple cell types were co-cultured in the HemoShear system18. This platform was used to create a lipotoxic model via the administration of a cocktail of free fatty acids; it was limited in its throughput allowing one experimental condition per device18. Since gut plays an important role in NAFLD pathogenesis, Lee et al. developed a gut-liver model studying the interactions among these systems, but was limited by its low throughput and use of a cell line instead of primary hepatocytes12.
Here we aimed to create an in vitro platform that can recapitulate different aspects of NAFLD progression and its drivers – such as exposure to free fatty acids and oxygen deprivation – where these drivers can be studied at multiple concentrations on a single microfluidic chip, a single continuous liver tissue under perfusion. This progressive fatty liver disease platform is based on our recent Metabolic Patterning on a Chip (MPOC) platform that takes advantage of enforced microfluidic gradients to enable gradual patterning of different metabolic activities for cells cultured across the length of a microfluidic device19. We have previously used this platform to recapitulate metabolic liver zonation in both rat and human primary hepatocytes using hormones, inducers and oxygen gradients19, 20. This approach can be used to study either a) the progression of NAFLD spectrum or b) the spatial physiological heterogeneity, i.e. zonal lipid accumulation21 and other heterogeneity22 observed during the disease, on a single integrated microfluidic chip. This approach results in reduced resource and pumping requirements compared to multiple well plate culture or multiple separate chip experiments.
NAFLD is a progressive disease in its pathogenesis and has multiple stages with different levels of fat accumulation, inflammation and cellular injury among others. Here, in the first part of our study, we captured the progressive nature of this disease in its fat accumulation aspect. We created patterned disease physiologies of different severity similar to the wide spectrum of cellular conditions in NAFLD. These in vitro physiologies that we create can simulate either the spatial heterogeneity and/or the progression of the disease. Specifically, we quantified the fat accumulation in primary rat hepatocytes upon exposure to a gradient of free fatty acids (FFA) and then confirmed the establishment of a progressive disease model via assessing the changes in expression levels of markers related to NAFLD and lipid metabolism.
The metabolic zonation of the liver - changes in metabolic activity across the liver sinusoid in response to dynamic concentration gradients and genetic drivers - is also found to be important in NAFLD pathogenesis. Specifically, triglyceride (TG) accumulation in the liver has predominantly been observed in hepatocytes in the pericentral zone for patients with NAFLD or NASH23, 24 and this has been primarily attributed to the gradual decrease in oxygen availability in the liver sinusoid25, 26. Accordingly, in the second part of this study, we investigated the effects of oxygen zonation on fat accumulation. While different in vitro oxygen zonation systems have been developed, none have established or measured its effect in intracellular lipid accumulation27, 28. Here, we established an oxygen gradient in our microfluidic platform and subjected primary rat hepatocytes to different fixed FFA concentrations with this oxygen gradient superimposed. Our results demonstrated that the largest effect of an oxygen gradient exposure on lipid accumulation is observed at the lower FFA concentrations, resulting in zonal fat accumulation, in line with predictions of a recent computational study25.
In the following, we show that we were able to create an in vitro progressive NAFLD model on a single microfluidic chip, where varying levels of fatty liver conditions were achieved. We also used this platform to create an oxygen gradient and exposed primary hepatocytes to different FFA concentrations in this system to investigate the role of oxygen on lipid accumulation and NAFLD progression. This progressive disease platform can allow for dissection of NAFLD progression and most importantly can be built upon for high throughput drug testing. Similarly, while we have limited ourselves to free fatty acid and oxygen concentrations, other effectors such as inflammatory cues can also be superimposed onto the existing conditions to recapitulate an even wider spectrum of conditions in NAFLD and its further progression.
Results
Primary hepatocyte cultures perfused with a free fatty acid (FFA) gradient.
The majority of in vitro NAFLD platforms utilize free fatty acid (FFA) supplemented media to induce hepatic steatosis13–15, 17, 18. Here, we treated rat primary hepatocytes with linoleic acid (LA) to induce lipid accumulation. As demonstrated in Figure 1, an FFA gradient of 0 – 2 mM was generated by supplying media with 0 mM and 2 mM LA to the inlet on the left and on the right of the gradient generator, respectively. We decided on the FFA concentrations based on data from clinical studies, which showed that test subjects with healthy liver had an approximate FFA concentration of 0.7 mM ± 0.4 mM whereas NAFLD patients had an approximate FFA concentration of 1.1 mM ± 0.6 mM29, and in some cases as high as 3 mM30. Following the exposure of primary hepatocytes to 0 – 2 mM LA gradient for 24 hours, the cells were either stained for quantification of viability and fat storage or lysed to assess the changes in the gene expression levels.
Figure 1. Establishment of the progressive NAFLD model.
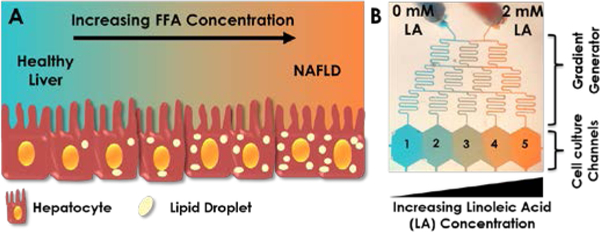
(A) Cartoon demonstration of lipid accumulation in hepatocytes during NAFLD progression, which can be achieved in vitro by an increasing supply of free fatty acid (FFA) concentration. (B) Visual demonstration of gradient generation in the microfluidic device. Experimental gradient was created by supplying one inlet with regular media (0 mM linoleic acid, LA) and the other with 2 mM LA. The inlets were connected to two syringes controlled by a single pump. These media were mixed in the gradient generator and were fed to each channel in the cell culture chambers labelled 1–5. Artificial food colouring was added to the media in the two syringes in this image to demonstrate formation of gradients.
Cell viability and lipid accumulation following FFA gradient exposure.
We measured the amount of stored lipids in the hepatocytes in each channel of the microfluidic device following the FFA exposure. Representative channel pictures are shown in Figure 2A, where we observed an increase in the Oil-Red-O stain along the FFA gradient. Bright-field images of channels also indicated the formation of lipid vesicles inside the cell cytoplasm, especially in channels 4 and 5. We performed image quantification of lipid staining by assigning the absolute values of channel 1 and channel 5 as 0 % and 100 %, respectively; and by normalizing the absolute values of the other channels accordingly. As shown in Figure 2B, the lipid accumulation in hepatocytes in channels 2, 3 and 4 was 19 %, 48 % and 85 %, respectively. Cell viability of hepatocytes in each channel was determined via a live/dead cell assay kit. The viability did not change significantly across the FFA gradient and was within the range of 84 % - 87 % (Fig. 2C), indicating that the explored FFA range was not toxic to hepatocytes. Representative channel pictures of live/dead staining (bright-field, Hoechst 33342 and ethidium homodimer-1 images) are shown in Figure S1.
Figure 2. Lipid accumulation and cell viability upon linoleic acid (LA) gradient exposure.
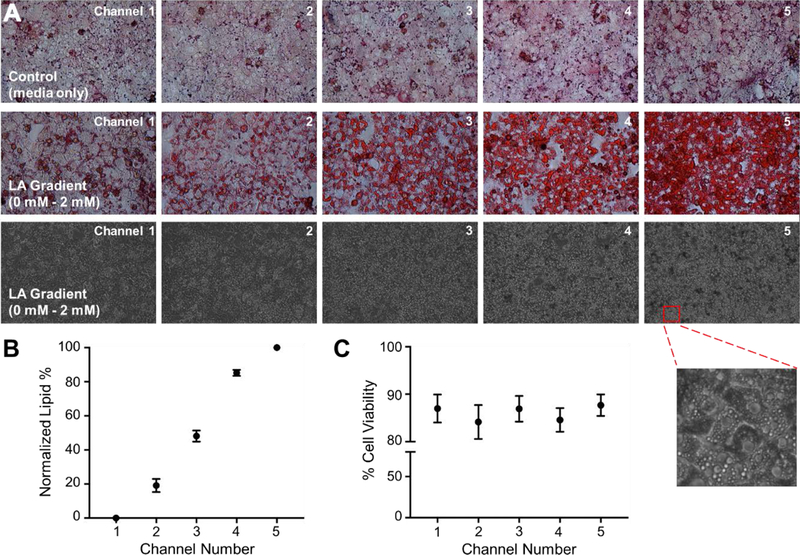
(A) Representative images of channels stained with oil-red-o (top panel: control group with no LA treatment; middle panel: hepatocytes subjected to 0 – 2 mM LA). Bright-field images of the 0 – 2 mM LA device are shown in the bottom panel where lipid droplets can be seen in the magnified image of hepatocytes in channel 5. (B) Quantified lipid accumulation in the 0 – 2 mM LA device. (C) Percentage cell viability in each channel was assessed via live-dead kit following the 24 hr perfusion with 0 – 2 mM LA gradient. All data are presented as mean ± SEM.
Gene expression profile of NAFLD/lipid metabolism markers upon FFA gradient exposure.
In order to explore the progression of NAFLD at the transcriptional level, we investigated the gene expression levels of several NAFLD markers and genes associated with lipid metabolism in primary hepatocytes upon exposure to an FFA gradient of 0 – 2 mM. As demonstrated in Figure 3, the expression levels of several markers gradually increased along the FFA gradient. The expression levels of lipogenesis markers Acetyl-CoA carboxylase α (ACACA) and fatty acid synthase (FASN) increased by up to 2.3 and 2.8-fold, respectively. Acyl-CoA synthetase long chain family member 4 (ACSL4) expression increased by up to 2.2-fold (in channel 5) whereas the elongation of long-chain fatty acids family member 6 (ELOVL6) expression increased by up to 2.6-fold. For all these targets, the changes in fold expression in channels 4 and 5 were statistically significant compared to channel 1 (Figs. 3, S2).
Figure 3. Fold gene expression changes of NAFLD pathogenesis and lipid metabolism markers.
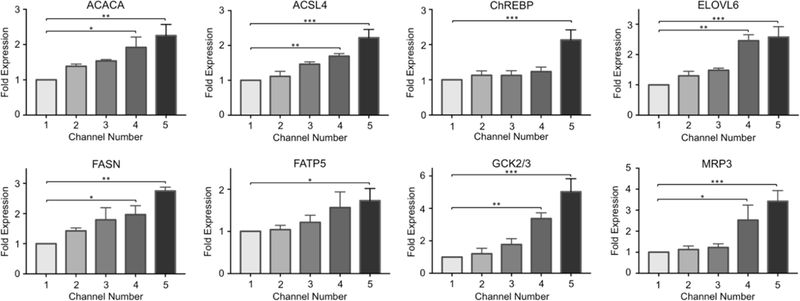
The changes in the mRNA levels of the following markers were quantified via quantitative real-time PCR: acetyl-CoA carboxylase α (ACACA), acyl-CoA synthetase long chain family member 4 (ACSL4), carbohydrate responsive element-binding protein (ChREBP), elongation of long-chain fatty acids family member 6 (ELOVL6), fatty acid synthase (FASN), fatty acid transporter protein 5 (FATP5), glucokinase (GCK) and efflux transport multidrug resistance-associated protein 3 (MRP3). The data are presented as mean ± SEM. N ≥ 3, * = p < 0.05, ** = 0.010 < p < 0.050, *** = 0.001 < p < 0.010.
The expression levels of fatty acid transporter protein 5 (FATP5), responsible for FFA uptake, increased with higher linoleic acid supplementation as well, although a statistically significant change in fold expression was only observed in cells of channel 5. Glucokinase (GCK) expression was increased by up to 5-fold, which is associated with triglyceride formation and is an activator of hepatic lipogenesis; and the efflux transport multidrug resistance-associated protein 3 (MRP3) expression increased by up to 3.4-fold upon FFA treatment. For both targets, the changes in expression levels were significant in channels 4 and 5, compared to the first channel of devices. All expression data were normalized to the first channel. Lastly, the expression of carbohydrate responsive element-binding protein (ChREBP), a transcriptional factor contributing to the regulation of triglyceride synthesis, was found to be significantly upregulated by 2.1-fold in channel 5.
Effect of oxygen deprivation on lipid accumulation.
Following the establishment of a lipid accumulation gradient in our platform accompanied by an increase in several NAFLD markers, we used this device to investigate the effect of oxygen deprivation on fat storage. A spatially heterogenous lipid accumulation is observed in the liver due to metabolic zonation; and insufficient oxygen supply is usually associated with NAFLD and steatosis development26. Accordingly, we created a chemically induced oxygen gradient via oxygen quenching by using culture media supplemented with 0.13 % sulfite and 13 μM cobalt in one inlet and regular culture media with no sulfite or cobalt in the other inlet as demonstrated in Figure 4A. Consequently, we generated an oxygen gradient of 11.2 ± 0.8% - 6.9 ± 0.7% inside the device channels as measured by the VisiSens oxygen detection system (Fig. 4B). This established oxygen concentration range is closer to the physiological oxygen gradient of 3.9% - 8.6% along the liver sinusoid31, 32, compared to the conventional open-air cell cultures at the atmospheric oxygen pressure and concentration of 20%.
Figure 4. Oxygen gradient generation in the microfluidic device.
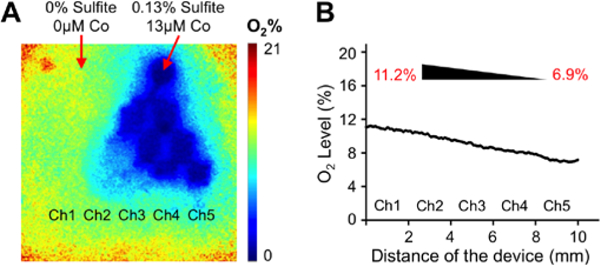
(A) Representative oxygen gradient profile measured by the Visisens oxygen detection system. Inlet 1: 0% sulfite/0 μM cobalt and inlet 2: 0.13% sulfite/13 μM cobalt. (B) The descending oxygen levels along the culture chamber with increasing sulfite/cobalt concentrations. An oxygen gradient of 11.2% – 6.9% was generated.
We supplemented this media with different FFA concentrations (0.25 mM, 0.5 mM, and 1 mM LA) to investigate the effect of oxygen deprivation on lipid accumulation at different levels of fatty acid availability (Figs. 5A, B). Following the perfusion of devices for 24 hours, we quantified the lipid accumulation via Oil-Red-O staining. Representative channel pictures of hepatocytes subjected to the oxygen gradient are shown in Figure 5A. Additional Oil-Red-O images are also given in Figure S3, where we subjected hepatocytes to different FFA and sulfite/cobalt concentrations in well plates. During image quantification of device experiments, we normalized the absolute values of each channel to 0 mM - 2 mM LA in the control device, of which channels 1 and 5 were assigned to 0 % and 100 %, respectively. The obtained values were fit via linear regression as shown in Figure 5C. The resulting slopes, which indicates the relative effect of oxygen on zonal lipid accumulation, were 7.8, 6.4 and 3.7 for oxygen gradient devices supplemented with 0.25 mM, 0.5 mM, and 1 mM linoleic acid, respectively.
Figure 5. Effect of oxygen deprivation on lipid accumulation.
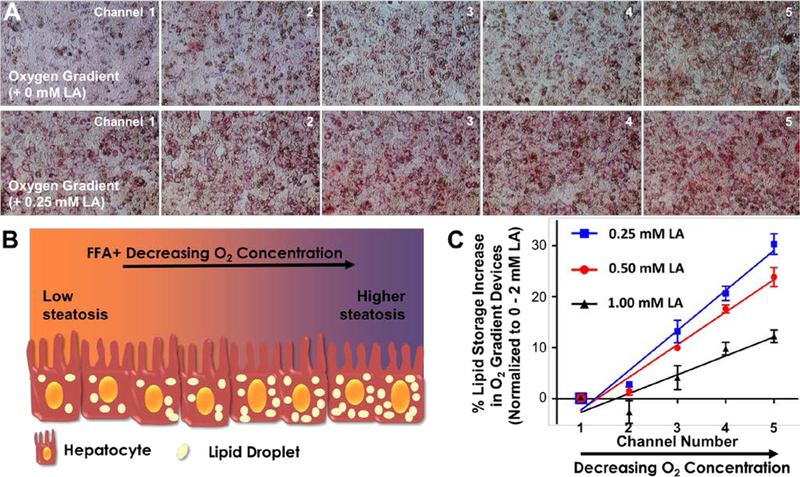
(A) Representative channel pictures of hepatocytes subjected to an oxygen gradient of 11.2% – 6.9%, top panel: no additional linoleic acid (LA) supplementation, bottom panel: in the presence of 0.25 mM LA supplementation. (B) Cartoon representation of the hypothesized effect of oxygen deprivation on lipid accumulation (represented by beige ellipses). (C) Quantification of percent lipid accumulation in oxygen gradient devices supplemented with varying levels of LA. The absolute values of each channel were normalized to the 0 mM - 2 mM LA control device, of which channels 1 and 5 were assigned to 0 % and 100 %, respectively. The solid lines for all three groups represent the linear regression analyses which are used to determine the slope for each group and thus the magnitude of the effects the oxygen gradient. The data are presented as mean ± SEM.
This data suggested that the difference in the lipid content of hepatocytes in channels 1 and 5 was more drastic for the device supplemented with the lowest FFA. Thus, the oxygen gradient had the highest effect on fat storage at the lowest FFA supplementation, which is in good agreement with the results of a recent computational work25, where the authors concluded that the ratio of accumulated TG concentration in the pericentral region (low oxygen) over the periportal region (high oxygen) is higher at lower FFA concentrations in blood.
Discussion
NAFLD is a progressive disease, both in pathogenesis and with respect to the native zonal heterogeneity across the liver tissue. As such, it is important for an in vitro disease model to capture these characteristics to reliably investigate both the disease itself and effective treatment options. Here, we established a progressive NAFLD on a chip platform by perfusing rat primary hepatocytes with a free fatty acid gradient of 0 – 2 mM to reflect the progressive nature of the disease (Fig. 1).
Despite the generation of a linear linoleic acid gradient, the quantification of fat storage in hepatocytes in different channels indicated a slightly non-linear response showing a saturation towards channels 4 and 5, as demonstrated in Figure 2B. There has been a debate on the exact mechanism of FFA uptake, where two mechanisms are considered: simple diffusion of FFAs across the cell membrane and transport-mediated uptake of FFAs33. For the latter, FFA transport follows Michaelis-Menten kinetics, where its uptake approaches a steady state as the FFA concentration increases outside the cell34. Our results did not indicate a clear linear lipid accumulation, which would have been expected in the case of simple diffusion. The shape of the second-half of the lipid accumulation graph (channels 3–5, Fig. 2B) resembles the beginning of a Michaelis-Menten type of saturation curve, characteristic of a transporter-mediated mechanism. As such, higher LA concentrations should result in a saturation of the FFA uptake. This could imply that FFA intake was transporter-mediated in a non-linear fashion, rather than through simple diffusion, for higher LA concentrations (> 1 mM). We studied the regulation of a long chain FFA transporter, FATP5, for further insight into the mechanism of LA uptake. The expression level of this transporter was significantly upregulated in channel 5, suggesting its involvement in the hepatic uptake of LA and upholds the idea of a transporter mediated uptake mechanism. It is important to note that this does not rule out the possibility of simple diffusion occurring simultaneously, as suggested by others35.
In addition to lipid accumulation, we also investigated the changes in expression levels of several NAFLD markers and lipid metabolism influencers upon exposure to the established FFA gradient (Fig. 3). ACSL family members catalyze the initial step of FFA activation and ACSL4 has a high affinity for polyunsaturated fatty acids such as linoleic acid36. A strong correlation has been shown between NAFLD progression and ACSL4 overexpression. A 2.8-fold increase in its mRNA levels was observed in the liver of NAFLD patients and the expression levels of ACSL4 were also increased in hepatocellular carcinoma37, 38. In agreement with these findings, we observed an up to 2–2-fold increase in our gradient model. FATP5 is a FATP family member only expressed in the liver and is responsible for FFA transport across the cell membrane as discussed above39. It is commonly used as a NAFLD marker and FATP5 knockdown in mice resulted in decreased lipid accumulation40. We observed an up to 1.9-fold increase in the expression levels of this transporter protein. Member 6 of the ELOVL family regulates FFA composition and was shown to play a role in insulin resistance and liver fibrosis in NAFLD patients41. A 3-fold increase in its mRNA levels was also observed in mice fed a high fat diet42. Similarly, we observed a gradual increase to up to 2.6-fold in our NAFLD platform.
ChREBP is a transcription factor promoting transcription of lipogenic genes, including ACACA and FASN, and contributes to TG synthesis by stimulating pyruvate kinase expression43, 44. We observed a 2.1-fold increase in its mRNA levels in hepatocytes in channel 5, which were subjected to 2 mM FFA. More recently, NAFLD has been associated with increased expression of other genes involved in de novo lipogenesis, such as ACACA and FASN, which are considered to be new targets for NAFLD treatment and diagnostic markers for NAFLD45. In our microfluidic platform, we observed a gradual increase in the expression levels of ACACA and FASN (up to 2.3 and 2.8-fold, respectively), suggesting a change in the lipogenesis metabolism of hepatocytes subjected to the FFA gradient.
Liver biopsies from patients showed that mRNA expression of GCK, another activator of hepatic lipogenesis, was positively correlated with accumulated TG content in the liver46. Similar to this in vivo observation, we measured a gradual increase in GCK mRNA levels upon administration of a 0 – 2 mM LA gradient. In channel 5, where hepatocytes had the highest lipid accumulation, we detected a 5-fold upregulation. Another pathogenesis marker in NAFLD is the dysregulation of bile acid synthesis and metabolism. MRP3 contributes to the transport of biliary excretion and was shown to get upregulated in both expression and activity in NAFLD patients as well as animal models47, 48. In mice with hepatic steatosis, the mRNA levels of MRP3 increased by 3.3-fold, compared to wild-type animals49. Comparable to these results, we observed an increase of up-to 3.4-fold in MRP3 expression levels in rat primary hepatocytes.
We note that some of the studied genes respond to the FFA gradients in a linear fashion while the others do not. This supports the notion that FFAs activate different transcription factors that then modulate the expression of the downstream genes through different pathways50. Gene regulation can further be modulated by other factors such as the specific promoter region of each gene and their location in the genome. We then posit that our gradient platform is suitable to study such dose and time response relationships between the FFAs and these downstream genes. A higher throughput model with more conditions than 5 chambers can be used to capture the complete range of the expected sigmoidal response curve.
We also investigated the effect of oxygen on lipid accumulation at different fixed concentrations of FFA supplementation to demonstrate the prowess of our platform and to confirm recent in vivo and computational studies with intriguing insights. The in vivo studies on livers of mice fed with a steatosis-inducing diet revealed that lipid accumulation differs along different zones of the liver51. Histology analysis indicated that hepatocytes in the pericentral zone accumulate more lipid droplets compared to those in the periportal zone, paralleling the oxygen gradients (higher and lower oxygen levels in the periportal and pericentral zones, respectively) across the sinusoid1. A recent computational study investigated the influence of the oxygen gradient and plasma FFA gradient on the rate of fat accumulation in hepatocytes along the liver sinusoid25. The authors modeled the lipid metabolism under a high-fat diet and analyzed the FFA oxidation rate and TG accumulation. They highlighted oxygen as a major contributor to zonal fat accumulation due to its critical role in mitochondrial FA oxidation. In addition, their results showed that the pericentral steatosis dominance decreases with increasing FFA supplementation (over a range of 0 – 2000 μmol/min FFA in blood). This indicates that the oxygen gradient driven difference in TG accumulation - between the periportal and pericentral compartments - disappears with increasing FFA concentrations. This hypothesis was further supported by an in vivo study which showed a gradual loss of fat zonation in overfed mice21, but was yet to be tested in vitro.
We have previously performed hypoxia studies, where we generated chemically induced oxygen gradients of 6.9% - 0.3%20. Here, we generated a more physiologically relevant oxygen gradient of 11.2% - 6.9% in a similar fashion (Fig. 4). After subjecting rat hepatocytes to this established gradient for 24 hr, we quantified the fat storage and observed higher lipid accumulation in the 5th channel, successfully reproducing lipid zonation in vitro for the very first time. We then performed experiments in the presence of 0.25, 0.5 and 1 mM LA supplementation to assess the lipid zonation at different levels of FFA consumption. Our results showed that the difference of lipid accumulation in channels 1 and 5 decreased with increasing FFA concentrations (Fig. 5), in agreement with the computational25 and in vivo studies21. This loss of zonation is attributed to the saturation in lipid accumulation at higher FFA exposure, whereby lipid accumulation increases in all channels (zones) attenuating the differences in oxygen concentration.
Experimental
Microfluidic device design and fabrication.
The microfluidic devices (Fig. 1B) are fabricated via soft lithography and based on the Metabolic Patterning on-a-Chip (MPOC) design19. Briefly, the system consists of a gradient generator connected to a cell culture chamber area of 17 mm2 (10 mm x 1.7 mm) and a height of 200 μm. The Polydimethylsiloxane (PDMS, Sylgard 184, Dow Corning, Midland, MI) polymer solution was prepared via mixing the elastomer and the curing agent at a 10:1 ratio, which was then poured over a SU-8 negative pattern with the microfluidic design on a silicon wafer. The bubbles were removed using a vacuum chamber for 30 min, and then the mold was cured at 65 °C for 24 hrs. Subsequently, the PDMS was peeled-off from the wafer, the individual device molds were cut off, and the inlet and outlet ports were punched out with a 1.5 mm biopsy puncher. The PDMS molds were bonded to glass microscope slides (75 × 25 mm, Thermo Fisher Scientific, Grand Island, NY, USA) via oxygen plasma treatment aligning the cell culture outlets to the edge of the glass slide to allow media outflow. For gene expression assays, an alternative design was used, which contains four physical barriers dividing the cell culture chamber into five distinct channels. This allows for sectional cell lysis and collection within the microfluidic gradient.
Rat primary hepatocyte culture.
Rat primary hepatocytes were isolated from female Lewis rats by the Cell Resource Core at the Massachusetts General Hospital (according to the protocol #2011N000111 approved by the institutional Animal Care and Use Committee). The cell culture chambers of the microfluidic devices were coated with a 50 μg/mL fibronectin (Sigma-Aldrich) solution in Phosphate Buffered Saline (PBS). Approximately 5 × 104 hepatocytes (with 90–95% viability) were injected into the cell chamber, incubated for 30 min at 37 °C at 10% CO2 and then washed with fresh media. The cells were seeded and maintained for the first 24 hr in Dulbecco’s modified eagle’s medium (DMEM, Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich), 0.5 U/mL insulin, 7 ng/mL glucagon, 20 ng/mL epidermal growth factor, 7.5 μg/mL hydrocortisone, 200 U/mL penicillin, 200 μg/mL streptomycin, and 50 μg/mL gentamycin. After 24 hrs, the cells were treated with the corresponding experimental gradient for 24 hrs as described in the following “Generation of the fatty acid gradient” and “Generation of the oxygen gradient” sections. The basal experimental media consisted of William’s medium E supplemented with 200 U/mL penicillin, 200 μg/mL streptomycin, and 5% Bovine Serum Albumin (BSA). For well plate experiments, approximately 6 × 105 hepatocytes were seeded on collagen coated 12 well plates (Corning, New York, NY, USA). The cells were incubated for 30 min at 37 °C and then washed with fresh DMEM media.
Generation of the fatty acid gradient.
The cells were exposed to a gradient of 0 – 2 mM linoleic acid (Sigma-Aldrich) in basal experimental media. The linoleic acid (LA) was dissolved in Dimethyl sulfoxide (DMSO) at 1 g/mL and a 30 mM stock solution was prepared in the basal experimental media. This solution was used to prepare the 2 mM treatment solution. After 20–24 hrs of culture stabilization following cell seeding, the corresponding minimum (0 mM LA) and maximum (2 mM LA) media were loaded into two syringes and were connected to the two inlets of the gradient generator, which exposes the cell chamber to the established gradient. The syringes were attached to a syringe pump (Harvard Apparatus, Holliston, MA, USA), which perfused the media at 120 μL/hr for 24 hr inside an incubator at 37 °C in 10% CO2.
Generation of the oxygen gradient.
An oxygen gradient was generated using sodium sulfite and cobalt nitrate, a commonly employed approach for oxygen removal and depletion20, 52. The oxygen scavenger solution was prepared with 0.13% (w/v) sodium sulfite and 13 μM cobalt nitrate in basal experimental medium. 20–24 hours after cell seeding, media with and without oxygen scavengers were loaded into 3 mL syringes individually and connected to the the gradient generator of the microfluidic device. The devices were perfused at 120 μL/hr for 24 hr at 37 °C in 10% CO2.
For oxygen measurement in the microfluidic device, the VisiSens system with a VisiSens 2D sensor film (PreSens/TD system GmbH, Germany) was used according to the manufacturer’s instructions. The VisiSens sensor film was adhered to the glass microscope slide via super glue. The device was attached on the VisiSens sensor film after oxygen plasma treatment. The oxygen level in the microfluidic device was detected via the VisiSens system with a modular detector unit for 2D read-out of fluorescent oxygen sensor foils. The images were visualized by VisiSens AnalytiCal 1 software, followed by the quantification of the oxygen profile through ImageJ software (NIH, Bethesda, MD, USA).
Oil-Red-O Staining.
Following experimental treatment, cell culture media was removed, and the cells were washed with PBS and fixed with 4% paraformaldehyde (Santa Cruz Biotechnology, Dallas, TX, USA) in PBS for 15 min. For the microfluidic devices, the PDMS chip was removed following the fixation, leaving only the fixed cells on the glass slide. The cells were washed with diH2O three times and then stained with a solution of 60% Oil Red O (ScienCell, Carlsbad, CA, USA) in diH2O for 15 min. The cells were washed three more times with diH2O. Colored bright field images were taken using a Nikon Diaphot TMD inverted microscope. Ten consecutive pictures were taken for each microfluidic device, spanning the entire cell culture chamber. For well plate experiments, 3–5 representative pictures were taken in each well. All image quantification was processed with ImageJ (NIH, Bethesda, MD, USA). The pictures were converted to grey-scale (using the split channel function in ImageJ (NIH, Bethesda, MD, USA)) and the average intensities of each picture was obtained. The values were normalized to the minimum and maximum of the 0 – 2 mM LA device to re-scale the values into a percentage scale.
Cell viability assay.
The microfluidic device was washed with PBS to remove cell culture media. The cells were stained with 1:1000 Hoechst 33342 nuclear dye (Thermo Fisher Scientific) and ethidium homodimer-1 (Thermo Fisher Scientific) in PBS for 15 min at 37 °C according to the manufacturer’s instructions. The cells were washed with PBS and fluorescent images were obtained using the EVOS fluorescence microscope (Thermo Fisher Scientific), spanning the whole length of the cell culture chamber. Using ImageJ, the pictures were converted to grey-scale. The threshold and particle analysis built-in tools were used to count the total number of nuclei from the Hoechst 33342 fluorescence and the dead nuclei from the ethidium homodimer-1 fluorescence. The viability was represented as the ratio of dead nuclei to the total number of nuclei present in each microfluidic channel.
RNA isolation and RT-PCR analysis.
After inducing progressive steatosis with a gradient of linoleic acid, the sectioned microfluidic devices were perfused with lysis buffer (Zymo Research, Irvine, CA, USA) and the lysates from each section were collected in separate tubes. RNA was extracted and purified with the quick-RNA microprep kit (Zymo Research) according to the manufacturer’s instructions. A total of 10–20 ng of RNA was reverse transcribed with cDNA synthesis kit (iScript, Bio-rad, Portland, ME, USA) according to the manufacturer’s instructions. Quantitative PCR was performed in a ViiA 7 Real-time PCR system (Thermo Fisher Scientific) using power SYBR Green PCR master mix kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. The PCR primers for each target are given in SI Table 1. The relative mRNA expression was quantified using the comparative Ct (ΔΔCt) method on the ViiA 7 Real-time PCR system.
Statistical analysis.
All quantitative data are presented as the mean ± standard error of the mean (SEM) from at least three devices seeded with primary rat hepatocytes from different isolations. GraphPad Prism software (Graphpad Software Inc., San Diego, CA) was used for statistical analysis of qPCR results. Statistical significance of the results was assessed using one-way ANOVA where channels 2–5 were compared to channel 1. P-values less than 0.05 were considered statistically significant.
Conclusions
In this study, we established a microfluidic progressive NAFLD platform to capture the wide spectrum of NAFLD on a single integrated chip. We subjected rat primary hepatocytes to a linoleic acid gradient of 0 – 2 mM and observed an increasing lipid accumulation across the length of the cell culture chamber, while the cell viability did not vary significantly across the five channels of the device. We also investigated the change in expression levels of several hepatic markers of lipid metabolism including fatty acid transporter FATP5, which showed a significant increase in channel 5 only. The expression levels of other NAFLD markers, such as ACSL4, were also significantly upregulated, mostly in channels 4 and 5, where the hepatocytes were treated with > 1 mM FFA. Finally, we investigated the role of oxygen deprivation in lipid accumulation and found that the difference in lipid accumulation among hepatocytes subjected to an oxygen gradient of 11.2% – 6.9% decreased gradually upon an increase in FFA supplementation, in agreement with the results of recently published computational and in vivo studies. To our knowledge, this is the first in vitro demonstration of lipid zonation.
The developed model can either simulate the progression of the disease spectrum or aid in dissecting the effects of spatial heterogeneity during any stage of the disease. This progressive high-throughput disease model can be used for hypothesis testing about NAFLD progression and its heterogeneity, including inflammatory cues, as well as for compound screening and efficacy testing of drugs against progressively severe NAFLD conditions. While we have limited the throughput of the device in this study to 5 chambers, the extension to higher throughput models with higher number of chambers is possible. We aim to conduct such studies of efficacy and other therapies, and higher throughput implementation of the platform in the near future.
Supplementary Material
Acknowledgements
This research was supported by grants from the National Institutes of Health (NIH# 5R01EB023812 and NIH# 5P41EB002503) and a Massachusetts General Hospital (MGH), Executive Committee on Research (ECOR) Interim Support fund. B.B. was supported by the NIH Ruth L. Kirschtein National Research Service Award F32 (NIH/NIBIB 1F32EB026916) and a Shriners Hospitals for Children Postdoctoral Fellowship Grant (award no: 84311). S.M. was supported by Shriners Hospitals for Children Postdoctoral Fellowship Grant (award no: 84001). The authors would also like to acknowledge the MGH Cell Resource Core; Proteomics & Genomics Core Shared Facility, Morphology and Imaging Shared Facility, Nano-Micro Core Special Shared Facility, and Translational Regenerative Medicine Shared Facility provided at the Shriners Hospital for Children.
Footnotes
Electronic Supplementary Information (ESI) available: [Representative channel pictures of live/dead staining; Statistical significance comparison of fold gene expression changes of hepatocytes in device channels; Oil-red-o staining of primary rat hepatocytes cultured in well-plates; Sequences of qPCR primers].
Conflicts of interest
There are no conflicts to declare.
References
- 1.Angulo P and Lindor KD, J Gastroenterol Hepatol, 2002, 17, S186–S190. [DOI] [PubMed] [Google Scholar]
- 2.Benedict M and Zhang X, World J Hepatol, 2017, 9, 715–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J and Bugianesi E, Nat Rev Gastroenterol Hepatol, 2018, 15, 11–20. [DOI] [PubMed] [Google Scholar]
- 4.LaBrecque DR, Abbas Z, Anania F, Ferenci P, Khan AG, Goh KL, Hamid SS, Isakov V, Lizarzabal M, Penaranda MM, Ramos JF, Sarin S, Stimac D, Thomson AB, Umar M, Krabshuis J and LeMair A, J Clin Gastroenterol, 2014, 48, 467–473. [DOI] [PubMed] [Google Scholar]
- 5.Patel V, Sanyal AJ and Sterling R, Clin Liver Dis, 2016, 20, 277–292. [DOI] [PubMed] [Google Scholar]
- 6.Rinella M and Charlton M, Hepatology, 2016, 64, 19–22. [DOI] [PubMed] [Google Scholar]
- 7.Diehl AM and Day C, N Engl J Med, 2017, 377, 2063–2072. [DOI] [PubMed] [Google Scholar]
- 8.Calzadilla Bertot L and Adams LA, Int J Mol Sci, 2016, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Younossi ZM, Blissett D, Blissett R, Henry L, Stepanova M, Younossi Y, Racila A, Hunt S and Beckerman R, Hepatology, 2016, 64, 1577–1586. [DOI] [PubMed] [Google Scholar]
- 10.Cassidy S and Syed BA, Nat Rev Drug Discov, 2016, 15, 745–746. [DOI] [PubMed] [Google Scholar]
- 11.Davidson MD, Ballinger KR and Khetani SR, Sci Rep, 2016, 6, 28178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee SY and Sung JH, Biotechnol Bioeng, 2018, DOI: 10.1002/bit.26793. [DOI] [PubMed] [Google Scholar]
- 13.Mashek DG and Grummer RR, J Dairy Sci, 2003, 86, 2390–2396. [DOI] [PubMed] [Google Scholar]
- 14.Ricchi M, Odoardi MR, Carulli L, Anzivino C, Ballestri S, Pinetti A, Fantoni LI, Marra F, Bertolotti M, Banni S, Lonardo A, Carulli N and Loria P, J Gastroenterol Hepatol, 2009, 24, 830–840. [DOI] [PubMed] [Google Scholar]
- 15.Janorkar AV, King KR, Megeed Z and Yarmush ML, Biotechnol Bioeng, 2009, 102, 1466–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao F, Xie P, Jiang J, Zhang L, An W and Zhan Y, Int J Mol Sci, 2014, 15, 4019–4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gori M, Simonelli MC, Giannitelli SM, Businaro L, Trombetta M and Rainer A, PLoS One, 2016, 11, e0159729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feaver RE, Cole BK, Lawson MJ, Hoang SA, Marukian S, Blackman BR, Figler RA, Sanyal AJ, Wamhoff BR and Dash A, JCI Insight, 2016, 1, e90954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang YBA, Eo J, Mert S, Yarmush ML and Usta OB, Sci Rep, 2018, 8, 8951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang YBA, Eo J, Bai H, Bulutoglu B, Yarmush ML and Usta OB, submitted.
- 21.Hall Z, Bond NJ, Ashmore T, Sanders F, Ament Z, Wang X, Murray AJ, Bellafante E, Virtue S, Vidal-Puig A, Allison M, Davies SE, Koulman A, Vacca M and Griffin JL, Hepatology, 2017, 65, 1165–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scupakova K, Soons Z, Ertaylan G, Pierzchalski KA, Eijkel GB, Ellis SR, Greve JW, Driessen A, Verheij J, De Kok TM, Olde Damink SWM, Rensen SS and Heeren RMA, Anal Chem, 2018, 90, 5130–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chalasani N, Wilson L, Kleiner DE, Cummings OW, Brunt EM and Ünalp A, J Hepatol, 2008, 48, 829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeh MM and Brunt EM, Gastroenterology, 2014, 147, 754–764. [DOI] [PubMed] [Google Scholar]
- 25.Schleicher J, Dahmen U, Guthke R and Schuster S, J R Soc Interface, 2017, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kietzmann T, Redox Biol, 2017, 11, 622–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allen JW and Bhatia SN, Biotechnol Bioeng, 2003, 82, 253–262. [DOI] [PubMed] [Google Scholar]
- 28.Sato A, Kadokura K, Uchida H and Tsukada K, Biochemical and Biophysical Research Communications, 2014, 453, 767–771. [DOI] [PubMed] [Google Scholar]
- 29.Gambino R, Bugianesi E, Rosso C, Mezzabotta L, Pinach S, Alemanno N, Saba F and Cassader M, Int J Mol Sci, 2016, 17, 479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang J, Zhao Y, Xu C, Hong Y, Lu H, Wu J and Chen Y, Sci Rep, 2014, 4, 5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolfle D, Schmidt H and Jungermann K, Eur J Biochem, 1983, 135, 405–412. [DOI] [PubMed] [Google Scholar]
- 32.Jungermann K and Kietzmann T, Hepatology, 2000, 31, 255–260. [DOI] [PubMed] [Google Scholar]
- 33.Glatz JF, Luiken JJ and Bonen A, Physiol Rev, 2010, 90, 367–417. [DOI] [PubMed] [Google Scholar]
- 34.Stremmel W and Theilmann L, Biochim Biophys Acta, 1986, 877, 191–197. [DOI] [PubMed] [Google Scholar]
- 35.Bradbury MW, Am J Physiol Gastrointest Liver Physiol, 2006, 290, G194–198. [DOI] [PubMed] [Google Scholar]
- 36.Kang MJ, Fujino T, Sasano H, Minekura H, Yabuki N, Nagura H, Iijima H and Yamamoto TT, Proceedings of the National Academy of Sciences, 1997, 94, 2880–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang YC, Wu CH, Chu JS, Wang CK, Hung LF, Wang YJ, Ho YS, Chang JG and Lin SY, World J Gastoenterol, 2005, 11, 2557–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Westerbacka J, Kolak M, Kiviluoto T, Arkkila P, Siren J, Hamsten A, Fisher RM and Yki-Jarvinen H, Diabetes, 2007, 56, 2759–2765. [DOI] [PubMed] [Google Scholar]
- 39.Musso G, Gambino R and Cassader M, Obes Rev, 2010, 11, 430–445. [DOI] [PubMed] [Google Scholar]
- 40.Doege H, Grimm D, Falcon A, Tsang B, Storm TA, Xu H, Ortegon AM, Kazantzis M, Kay MA and Stahl A, J Biol Chem, 2008, 283, 22186–22192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamada K, Mizukoshi E, Sunagozaka H, Arai K, Yamashita T, Takeshita Y, Misu H, Takamura T, Kitamura S, Zen Y, Nakanuma Y, Honda M and Kaneko S, Liver Int, 2015, 35, 582–590. [DOI] [PubMed] [Google Scholar]
- 42.Matsuzaka T, Shimano H, Yahagi N, Kato T, Atsumi A, Yamamoto T, Inoue N, Ishikawa M, Okada S, Ishigaki N, Iwasaki H, Iwasaki Y, Karasawa T, Kumadaki S, Matsui T, Sekiya M, Ohashi K, Hasty AH, Nakagawa Y, Takahashi A, Suzuki H, Yatoh S, Sone H, Toyoshima H, Osuga J and Yamada N, Nat Med, 2007, 13, 1193–1202. [DOI] [PubMed] [Google Scholar]
- 43.Cohen JC, Horton JD and Hobbs HH, Science, 2011, 332, 1519–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uyeda K and Repa JJ, Cell Metab, 2006, 4, 107–110. [DOI] [PubMed] [Google Scholar]
- 45.Mitsuyoshi H, Yasui K, Harano Y, Endo M, Tsuji K, Minami M, Itoh Y, Okanoue T and Yoshikawa T, Hepatol Res, 2009, 39, 366–373. [DOI] [PubMed] [Google Scholar]
- 46.Peter A, Stefan N, Cegan A, Walenta M, Wagner S, Konigsrainer A, Konigsrainer I, Machicao F, Schick F, Haring HU and Schleicher E, J Clin Endocrinol Metab, 2011, 96, E1126–1130. [DOI] [PubMed] [Google Scholar]
- 47.Cobbina E and Akhlaghi F, Drug Metab Rev, 2017, 49, 197–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X and Deng R, IntechOpen, 2018, 1–33. [Google Scholar]
- 49.Martin IV, Schmitt J, Minkenberg A, Mertens JC, Stieger B, Mullhaupt B and Geier A, Biol Chem, 2010, 391, 1441–1449. [DOI] [PubMed] [Google Scholar]
- 50.Jump DB, Y BD. Xu Wang, J., Christian B and Demeure O, The Journal of Nutrition, 2005, 135, 2503–2506. [DOI] [PubMed] [Google Scholar]
- 51.Schleicher J, Guthke R, Dahmen U, Dirsch O, Holzhuetter HG and Schuster S, Biochim Biophys Acta, 2014, 1841, 62–69. [DOI] [PubMed] [Google Scholar]
- 52.Shimizu S, Eguchi E, Kamiike W, Waguri S, Uchiyama Y, Matsuda H and Tsuhimoto Y, Oncogene, 1996, 12, 2045–2050. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.