

Abstract
Background
Dent disease type 1 (DD1) is a rare X-linked disorder caused mainly by CLCN5 mutations. Patients may present with nephrotic range proteinuria leading to erroneous diagnosis of focal segmental glomerulosclerosis (FSGS) and unnecessary immunosuppressive treatments.
Methods
The following cohorts were screened for CLCN5 mutations: Chronic Kidney Disease in Children (CKiD; n=112); Multicenter FSGS-Clinical Trial (FSGS-CT) (n=96) and Novel therapies for resistant FSGS Trial (FONT) (n=30). Urinary α1-microglobulin (α1-M), albumin (A), total protein (TP) and creatinine (Cr) were assessed from CKiD subjects (n=104); DD1 patients (n=14) and DD1 carriers (DC; n=8). TP/Cr, α1M/Cr, α1M/TP and A/TP from the CKiD cohort were compared with DD1 and DC.
Results
No CLCN5 mutations were detected. TP/Cr was lower in DC and CKiD with tubulointerstitial disease than DD1 and CKiD with glomerular disease (P < 0.002). α1M/Cr was higher in DD1 than CKiD and DC (P < 0.001). A/TP was lower in DD1, DC and CKiD with tubulointerstitial disease, higher in CKiD with glomerular disease (P < 0.001). Thresholds for A/TP of ≤ 0.21 and α1-M/Cr ≥ 120 mg/g (> 13.6 mg/mmol) creatinine were good screens for Dent disease.
Conclusions
CLCN5 mutations were not seen in screened CKiD/FSGS cohorts. In our study, a cut off of TP/Cr > 600 mg/g (> 68 mg/mmol) and A/TP < 0.3 had a high sensitivity and specificity to distinguish DD1 from both CKiD glomerular and tubulointerstitial cohorts. α1-M/Cr ≥ 120 mg/g (> 13.6 mg/mmol) had the highest sensitivity and specificity when differentiating DD1 and studied CKiD populations.
Keywords: Dent disease, FSGS, CLCN5, proteinuria, low-molecular weight proteinuria, α1-microglobulin
Introduction
Dent disease is a rare X-linked inherited disorder, commonly characterized by low molecular weight proteinuria (LMWP), hypercalciuria and nephrocalcinosis [1, 2]. Approximately 60 % of patients have mutations in the CLCN5 gene (Dent disease 1, OMIM300009); 15 % have mutations in OCRL1 gene (Dent disease 2, OMIM300555) and approximately 25 % of patients lack a molecular genetic characterization [3, 4]. Patients often develop chronic kidney disease (CKD) and progress to end-stage renal disease, and may develop nephrocalcinosis and kidney stones. However, the phenotype is variable and the correct diagnosis is often challenging [4–6]. A characteristic feature of the disease is increased LMWP including retinol binding protein (RBP), α1 microglobulin (α1-M), and β2 microglobulin (β2M), typically in excess of 10 times normal [3]. Nephrotic range proteinuria (defined as protein/creatinine ratio > 1800 mg/g or timed protein excretion > 40 mg/m2/h) was found in 46 % of a recent Dent cohort [7]. However, since screening for LMWP is not routinely performed, Dent patients with nephrotic range proteinuria often get a kidney biopsy. The typical histologic feature seen on Dent disease renal biopsies is focal global glomerulosclerosis (FGGS) and may be easily interpreted as focal segmental glomerulosclerosis (FSGS) [8–11]. Thus, the disease is sometimes only suspected after a patient receives and fails inappropriate treatment with immunosuppressive medications, and the diagnosis might be significantly delayed, or patients may not be diagnosed at all. This creates missed opportunities for patients and for research to identify the most appropriate treatments for these patients.
Dent disease is considered a disease of renal tubule [12], even though glomerular sclerosis is commonly present [9, 10, 13]. Given the overlap of phenotypes and histology, it is unknown what percentage of patients diagnosed as FSGS might instead have unrecognized Dent disease. In addition, based on observation of our large Dent disease registry, a significant number of patients are diagnosed late and have other family members entirely undiagnosed. Therefore, in this study we sought undiagnosed Dent disease cases by evaluating three cohorts of kidney disease patients who we hypothesized might harbor undiagnosed Dent patients: Chronic Kidney Disease in Children (CKiD); Multicenter FSGS-Clinical Trial (FSGS-CT) and Novel Therapies for Resistant FSGS Trial (FONT). We tested biobanked specimens from these cohorts for CLCN5 gene mutations. Dent disease 1 patients (DD1) and Dent disease 1 carriers (DC) were selected from the Rare Kidney Stone Consortium (RKSC) Dent Registry with available biobanked urinary specimens. Our goal was to establish more accessible criteria for Dent Disease screening in a routine nephrology practice in order to facilitate screening and ultimately early diagnosis of Dent disease. Specifically, we were interested in performance of A/TP (total protein) ratio as a simple and readily available screening test for Dent disease and other tubular diseases.
Methods
Study sample
This study was approved by the Mayo Clinic Institutional Review Board. Ancillary studies were approved and biospecimens were obtained from the Chronic Kidney Disease in Children (CKiD; n=112); Multicenter FSGS-Clinical Trial (FSGS-CT; n=96) and Novel therapies for resistant FSGS Trial (FONT; n=30) studies [14–16].
General information and laboratory results, including serum creatinine and age, were abstracted from the RKSC Registry data and were obtained from the CKiD study. Renal function was assessed by serum creatinine values to estimate GFR using the CKD-EPI formula in the Dent disease cohort. Values obtained nearest to and within 1 year of the urinary testing were reported. The updated Schwartz formula [17] for bedside estimating equation (bedGFR = 41.3 [height (m) / SCr]) was used to estimate GFR in the CKiD cohort.
CLCN5 genetic screening
DNA (stored at −70°C) was obtained from the CKiD and FONT biobanks. Sanger sequencing was performed on all coding exons and exon/intron boundaries of the Dent gene CLCN5, NM_000084.2 using M13-tailed primers (Beckman Coulter). Primer sequences can be obtained by request. PCR products were purified and sequenced by Beckman Coulter Genomics Agencourt single-pass sequencing services and resulting sequencing chromatograms were analyzed using Mutation Surveyor version 4.06 (Softgenetics) software.
Urine measurements
Whole urine aliquots frozen to −70°C in the CKiD and Rare Kidney Stone Consortium biobanks were used for the current measurements. At the time of assay, samples were thawed to 37°C and spun (1000g × 12 min) to remove cellular debris. All urine measurements were performed in a centralized laboratory at the Mayo Clinic, Rochester, MN using a Cobas C311 autoanalyzer (Roche, Indianapolis, IN). Creatinine was measured using the standardized (isotope dilution mass spectrometry traceable) enzymatic creatinine assay (Roche). Total urine protein was measured using a pyrogallol red colorimetric assay (Wako Diagnostics, Richmond, VA). Urine αM was assayed using the K-ASSAYAlpha-1 microglobulin kit, Kamya Biomedical Company, Seattle, WA.
Statistical methods
We compared available urine specimens from DD1, DC, CKiD patients with glomerular disease (CKiD G) and CKiD patients with tubulointerstitial disease (CKiD TI) using nonparametric methods. The median, first, and third quartile were used as summary statistics because of outlier variability. A priori two-way comparisons utilizing the Wilcoxon rank sum test compared DD1 with each other group, while CKiD TI was further compared with CKiD G to assess within-group differences.
Three different markers (α1-M/Cr, α1M/TP and A/TP) were selected to further assess their ability to distinguish DD1 from CKiD (G and TI) in diagnostic testing using each biomarker as a continuous variable in a logistic regression. The best marker was selected based upon the lowest p-values and highest C statistics for distinguishing DD1 from CKiD G and CKiD TI; however, subsequent analyses are shown for all markers to allow readers to assess each separately. The C statistic is equivalent to the area under the ROC curve. Following selection of the best marker, a cutoff value was selected for each marker; in order to utilize a single cutoff for comparison with both CKiD G and CKiD TI, their combined data was used in a logistic regression in comparison to DD1 separately for each marker. The cutoff for each marker was identified by selecting the cutoff with the highest sensitivity and specificity, with the goal that sensitivity and specificity both exceeded 80 % (0.80) if possible. These cutoffs were then used to identify separate sensitivity and specificity for DD1 vs. CKiD G and DD1 vs. CKiD T1 for each marker.
Results
A total of 112 DNA specimens and 104 urine specimens were available from the CKiD cohort (8 of 104 specimens sent to us had no DNA sample available and 14 DNA specimens had no available urine). The median age of all CKiD patients was 13 years, the serum creatinine was 1.4 mg/dl (123.8 μmol/L), and estimated glomerular filtration rate (eGFR) calculated by updated Schwartz formula or bedside estimating equation (bedGFR = 41.3 [height (m) / SCr])) was 44.2 ml/min/1.73m2 (Table 1) [17]. CKiD cases had the following diagnoses and were divided into two groups: 1) CKiD Glomerular (CKiD G): focal segmental glomerulosclerosis (FSGS; n=33), familial nephritis (Alport syndrome or hereditary nephritis) (n=6), chronic glomerulonephritis (not further classified) (n=5), congenital nephrotic syndrome (n=2); and 2) CKiD tubulointerstitial (CKiD TI): medullary cystic kidney disease (n=4), and reflux nephropathy (hypodysplasia and reflux) (n=54). The cohort of DD1 (n=14) patients had a median age of 20.6 years, the serum creatinine was 1.8 mg/dl (159 μmol/L), and eGFR (calculated by CKD-EPI formula) was 45.3 ml/min/1.73m2. DC (n=8) had a median age of 40.5 years (Table 1). Serum creatinine values were not available for DCs, but kidney function was reported to be normal.
Table 1.
Characteristics of 112 patients from CKiD cohort, 14 patients from Dent disease 1 cohort and 8 Dent disease 1 carriers
| N | Age (min, max) | Serum creatinine mg/dl | eGFR ml/min/1.73m2 | |
|---|---|---|---|---|
| CKiD | 112 | 13 (3.5, 17.4) | 1.4 (0.6, 2.8) | 44.2 (18.2, 113.6)b |
| DD1 | 14 | 20.6 (2.5, 55.8) | 1.8 (0.3, 9.4) | 45.3 (6.5, 211.9)c |
| DC | 8 | 40.5 (11.8, 56.5) | NA | NA |
Abbreviations: CKiD = Chronic Kidney Disease in Children; DD1 = Dent disease 1; DC = Dent disease 1 carriers; eGFR =estimated glomerular filtration rate
Continuous data are presented as median (minimum, maximum).
bedGFR
CKD-EPI
No pathogenic mutations were identified in CLCN5, the most common genetic cause of Dent disease, among patients in the CKiD (n=112), FSGS-CT (n=96) and FONT (n=30) cohorts.
Available urinary specimens from CKiD subjects (n=104), DD1 patients (n=14) and DC (n=8) were also tested for α1-M, A and TP. Median TP/Cr (mg/g) was similar in DD1 and CKiD G, however DD1 had a significantly greater TP/Cr (mg/g) than both CKiD TI and DC (Table 2). Median A/Cr (mg/g) was significantly higher in CKiD G compared with DD1 and CKiD TI (Table 2).
Table 2.
Comparison of urinary proteins measured from CKiD cohort with tubulointerstitial disease, CKiD cohort with glomerular disease, Dent disease 1 patients and Dent disease 1 carriers. Logistic regression analysis was used to determine the best marker to discriminate between groups based upon the lowest p-values and highest C statistics
| DD1 (n=14) | DC (n=8) | p-value1 | CKiD TI (TI) (n=58) | p-value2 C | CKiD G (G) (n=46) | p-value3 C | p-value4 C | |
|---|---|---|---|---|---|---|---|---|
| A (mg/L) | 68.2 (57.4-124.6) | 5.7 (3-15) | <0.001 | 35.8 (5.1-131) | 0.14 0.81 | 1098.0 (148-2048) | <0.001 0.63 | <0.001 0.82 |
| TP (mg/L) | 443.0 (369-675) | 63.5 (10.5-100.5) | <0.001 | 147.0 (977-255) | 0.002 0.72 | 1625.0 (306-3040) | 0.012 0.77 | <0.001 0.82 |
| TP/Cr (mg/g) | 1581.5 (1157-1814) | 33.0 (23.5-75.5) | <0.001 | 315.0 (125-875) | 0.002 0.58 | 1625.0 (595-6429) | 0.35 0.77 | <0.001 0.78 |
| A/Cr (mg/g) | 230.0 (156-295) | 10.5 (6.5-13) | <0.001 | 90.0 (14-502) | 0.064 0.78 | 1084.0 (343-4012) | 0.002 0.66 | <0.001 0.79 |
| A/TP | 0.18 (0.13-0.20) | 0.19 (0.14-0.49) | 0.43 | 0.32 (0.08-0.59) | 0.22 0.84 | 0.67 (0.55-0.72) | <0.001 0.61 | <0.001 0.61 |
| α1M (mg/L) | 60.2 (47.4-89) | 4.4 (0.6,7.8) | <0.001 | 9.4 (4.4-17.7) | <0.001 0.95 | 15.2 (5.5-37.6) | <0.001 0.87 | 0.050 0.61 |
| α1M/Cr (mg/g) | 204.6 (140.4-245) | 3 (2.4, 6.7) | <0.001 | 16.6 (9.1-44.7) | <0.001 0.95 | 20.6 (8.4-49.2) | <0.001 0.94 | 0.56 0.53 |
| α1M/TP | 0.14 (0.12-0.17) | 0.11 (0.07, 0.15) | 0.23 | 0.06 (0.03-0.11) | <0.001 0.86 | 0.01 (0.01-0.02) | <0.001 0.95 | <0.001 0.82 |
| α1M/A | 0.81 (0.70-0.91) | 0.42 (0.16, 0.97) | 0.17 | 0.32 (0.05-0.73) | 0.003 0.75 | 0.01 (0.01-0.03) | <0.001 0.91 | <0.001 0.82 |
Abbreviations: CKiD = Chronic Kidney Disease in Children; CKiD TI = CKiD tubulointerstitial disease; CKiD G = CKiD glomerular disease; DD1 = Dent disease 1; DC = Dent disease 1 carriers; A = urinary albumin; TP = urinary total protein; α1M = urinary α1-microglobulin; Cr = urinary creatinine Note: A, TP, Cr and α1M were measured from one spot urinary sample.
Continuous data are presented as median (interquartile range)
p-values from the Wilcoxon rank sum test comparing DC vs DD1
p-values from the Wilcoxon rank sum test comparing TI vs DD1
p-values from the Wilcoxon rank sum test comparing G vs DD1
p-values from the Wilcoxon rank sum test comparing G vs TI
When the median A/TP was compared between groups, the highest median A/TP was found in CKiD G, as expected, and was significantly higher than in DD1 and CKiD TI, whereas A/TP in DD1, CKiD TI and DC were similar (Table 2).
Median urinary α1M/Cr (mg/g) was substantially and significantly higher in DD1, compared to all three groups CKiD G, CKiD TI and DC (Table 2). The difference between CKiD G and CKiD TI was not statistically significant, however median α1M/TP was significantly higher in CKiD G compared to CKiD TI.
A scatter plot demonstrates separation of the DD1 patients when A/TP is plotted as a function of TP/Cr (Figure 1). We have found that using the combination of TP/Cr > 600 mg/g (> 68 mg/mmol) and A/TP < 0.3, we can distinguish DD1 from CKiD (CKiD total, as well as CKiD TI and CKiD G) with a sensitivity of 0.86 and a specificity of 0.98 (Figure 1).
Fig. 1.
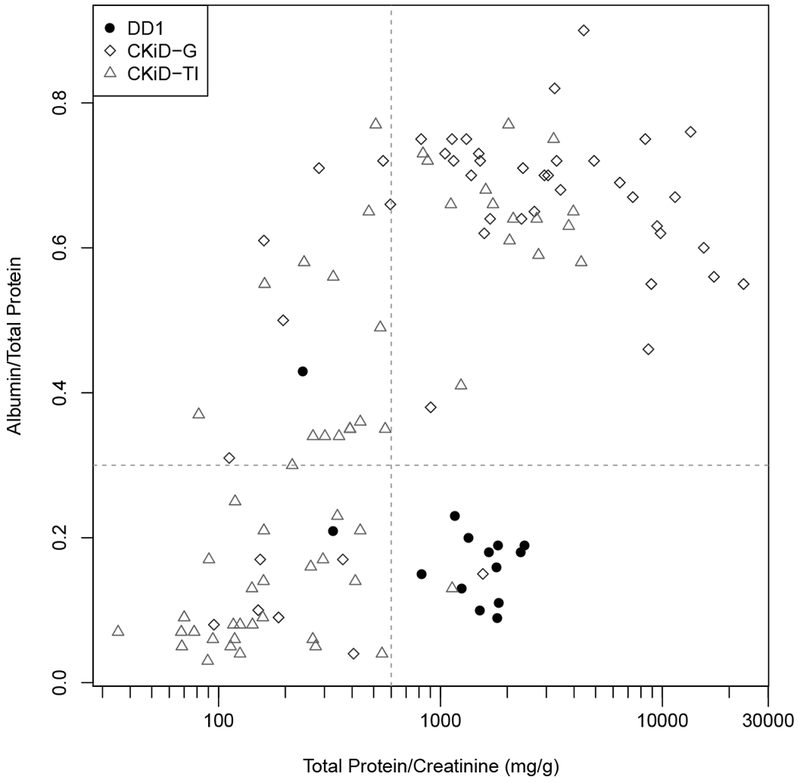
Relationship between Total Protein/Creatinine (mg/g) versus Albumin/Total Protein for Dent disease (DD1) (filled circles), CKiD with glomerular disease (CKiD G) (unfilled rhombs) and CKiD with tubulointerstitial disease (CKiD TI) (unfilled triangles) cohorts.
Next, markers α1M/Cr, α1M /TP and A/TP were chosen to compare DD1, CKiD T1 and CKiD G (using C-statistics or area under the ROC curves), to determine which marker could best discriminate these groups. α1M /Cr was best able to identify DD1 patients (p < 0.001, C = 0.95 in comparison to CKiD T1 and p < 0.001, C = 0.94 in comparison to CKiD G; results shown in Table 3).
Table 3.
Cutoffs for each marker based on the highest sensitivity and specificity. Sensitivity and specificity for Dent disease 1 vs. CKiD glomerular, Dent disease 1 vs. CKiD tubulointerstitial and Dent disease 1 vs. all CKiD for each identified marker
| Sensitivity (95% CI) | Specificity (95% CI) | ||
|---|---|---|---|
| DD1 vs CKiD G | α1M/Cr ≥ 120 mg/g | 0.86 (0.57, 0.98) | 0.96 (0.85, 0.99) |
| α1M/TP ≥ 0.11 | 0.86 (0.57, 0.98) | 0.93 (0.82, 0.99) | |
| A/TP ≤ 0.21 | 0.86 (0.57, 0.98) | 0.85 (0.71, 0.94) | |
| DD1 vs CKiD TI | α1M/Cr ≥ 120 mg/g | 0.86 (0.57, 0.98) | 0.95 (0.86, 0.99) |
| α1M /TP ≥ 0.11 | 0.86 (0.57, 0.98) | 0.74 (0.61, 0.85) | |
| A/TP ≤ 0.21 | 0.86 (0.57, 0.98) | 0.55 (0.42, 0.68) | |
| DD1 vs CKiD (all) | α1M/Cr ≥ 120 mg/g | 0.86 (0.57, 0.98) | 0.95 (0.89, 0.98) |
| α1M/TP ≥ 0.11 | 0.86 (0.57, 0.98) | 0.83 (0.74, 0.89) | |
| A/TP ≤ 0.21 | 0.86 (0.57, 0.98) | 0.68 (0.58, 0.77) |
Abbreviations: CKiD TI = CKiD tubulointerstitial disease; CKiD G = CKiD glomerular disease; DD1 = Dent disease 1; A = urinary albumin; TP = urinary total protein; α1M = urinary α1-microglobulin; Cr = urinary creatinine
We analyzed α1-M/Cr, α1M /TP and A/TP using logistic regression analysis to establish the cut off values most useful to screen for patients with Dent disease. The cutoff for each marker was identified by selecting the value with the highest sensitivity and specificity. Using this approach, we identified α1M/Cr cut off ≥ 120 mg/g (> 13.6 mg/mmol), α1M/TP ≥ 0.1 and A/TP ≤ 0.21. All three ratios had good sensitivity; the specificity differed. α1M/Cr ≥ 120 mg/g (> 13.6 mg/mmol) had the highest sensitivity and specificity when differentiating DD1 and CKiD total population (Table 3). It also had most value in differentiating DD1 and CKiD G, and was good in differentiating DD1 and CKiD TI. The performance of α1M/TP ≥ 0.11 was inferior in comparing DD1 to the total CKiD group, DD1 to CKiD G and DD1 to CKiD TI. The most available test A/TP ≤ 0.21 was not as good at differentiating DD1 from the whole group of CKiD patients analyzed, but performed well differentiating DD1 from CKiD G (Table 3). This can also be reviewed on corresponding ROC curves where cut-off points for each marker, chosen based on highest sensitivity and specificity, are represented with circles (Fig 2, 3 and 4).
Fig. 2. ROC curves comparing the predictive performance of urinary α1-microglobulin/Creatinine (α1M/Cr), urinary α1-microglobulin/Total Protein (α1M/TP), and Albumin to Total Protein (A/TP) for distinguishing Dent disease 1 (DD1) from the total CKiD cohort.
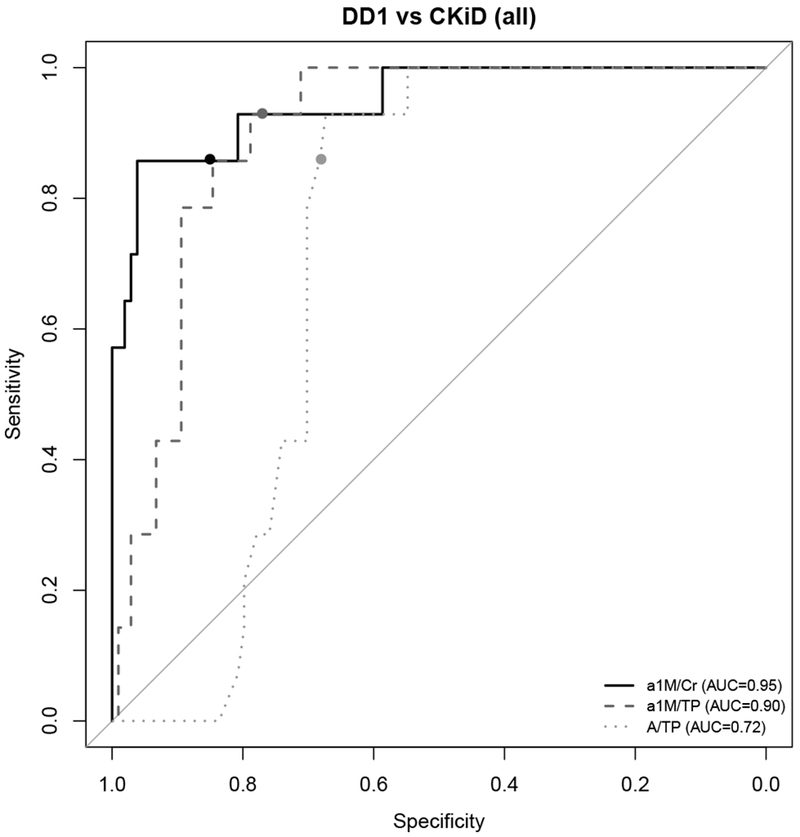
Circles on each ROC curve represent the cutoff for each marker with the highest sensitivity and specificity.
Fig. 3. ROC curves comparing the predictive performance of α1-microglobulin/Creatinine (α1M/Cr), urinary α1-microglobulin/Total Protein (α1M/TP), and Albumin to Total Protein (A/TP) for distinguishing Dent disease 1 (DD1) and CKiD with glomerular disease (CKiD G) cohort.
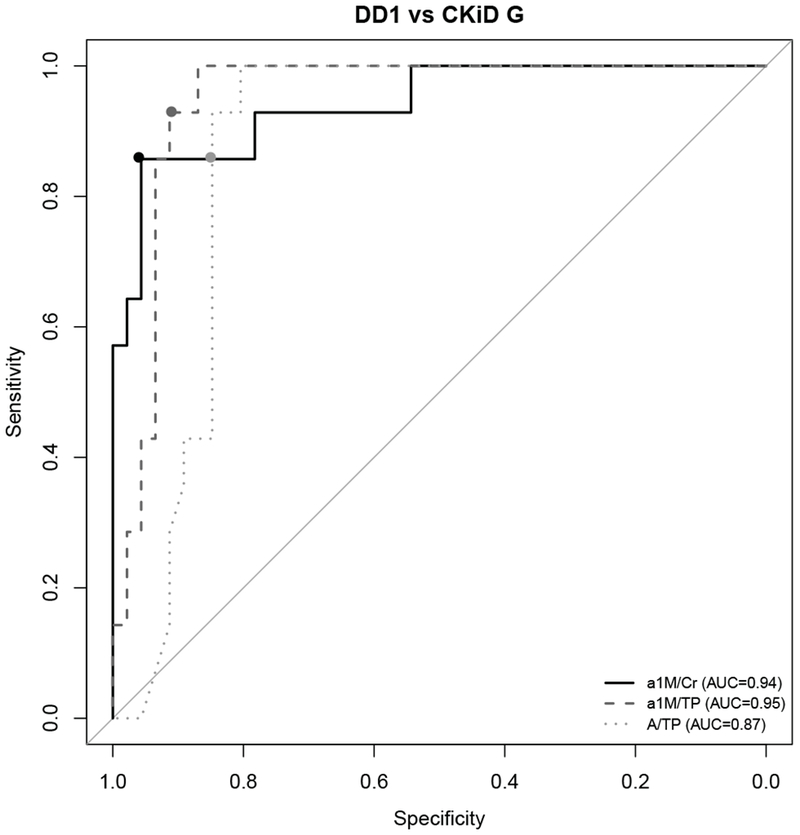
Circles on each ROC curve represent the cutoff for each marker with the highest sensitivity and specificity.
Fig. 4. ROC curves comparing the predictive performance of α1-microglobulin/Creatinine (α1M/Cr), urinary α1-microglobulin/Total Protein (α1M/TP), and Albumin to Total Protein (A/TP) for distinguishing Dent disease 1 (DD1) and tubulointerstitial disease (CKiD TI) cohort.
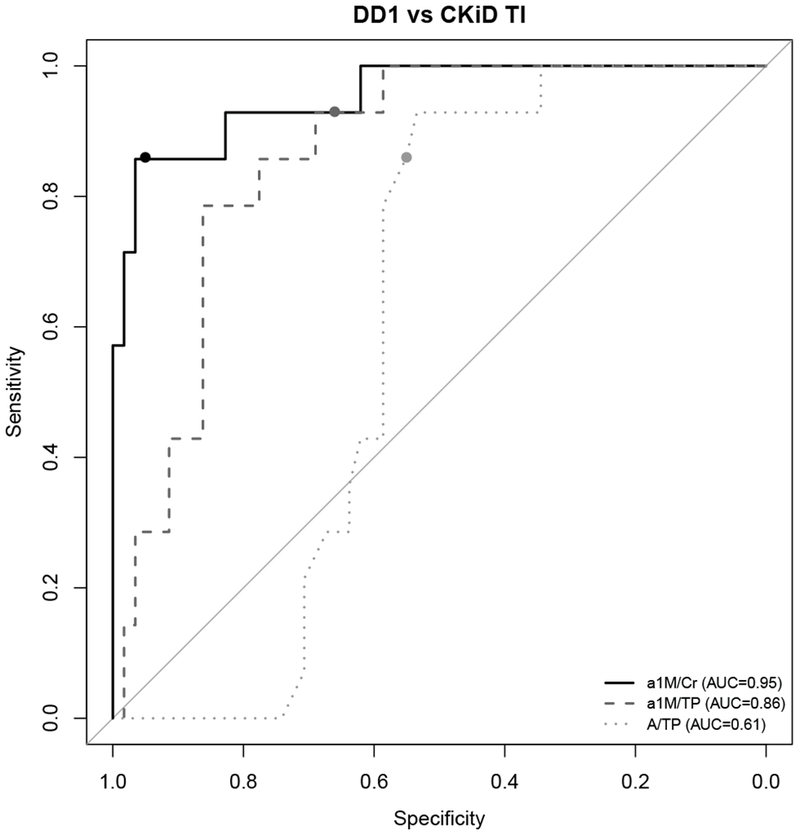
Circles on each ROC curve represent the cutoff for each marker with the highest sensitivity and specificity.
Discussion
In the current study, we confirmed that DD1, caused by CLCN5 mutations, is rare in pediatric populations with FSGS, as well as in other selected diagnoses that could be confused clinically with Dent disease. Based on this finding, genetic testing of all pediatric FSGS patents for DD1 does not seem warranted. Therefore, we also analyzed the proteinuria pattern for comparison with other patient groups in order to discern a practical screening approach.
Dent disease is often diagnosed late and sometimes not diagnosed at all. As we and others have reported, Dent disease patients may undergo a kidney biopsy and a trial of steroid and immunosuppressive treatment, as well as repeated kidney biopsy for steroid-resistant proteinuria [10, 11, 13]. These Dent patients often had proteinuria without significant nephrocalcinosis or a history of kidney stones, and the biopsy only revealed focal global sclerosis and milder tubulointerstitial findings. Therefore, there is a need for a better screening tool in order to increase awareness of Dent disease as a possible diagnosis.
Our analyses demonstrate that the median TP/Cr was not statistically different in DD1 and in CKiD G, however, it was significantly higher compared to both CKiD TI and DC, confirming the potential for Dent disease to be easily confused with glomerular diseases. Our results confirm urinary a-|M to creatinine ratio (α-ιΜ/Cr) mg/g as the best marker to differentiate Dent disease both from tubulointerstitial and glomerular disease, especially a value of α1M /Cr ≥ 120 mg/g (> 13.6 mg/mmol). α1M/TP ≥0.11 was not as specific at differentiating DD1 cohort from CKiD TI cohort. Interestingly, although α-ιΜ/Cr was not significantly different between CKiD G and CKiD TI, α1M/TP was significantly higher in CKiD TI compared with CKiD G. We have measured α1M as a screening low molecular weight protein for Dent disease, however retinol binding protein (RBP) is also an excellent marker, whereas β2 microglobulin (β2M) is not recommended as it is unstable in acidic urine [18–20]. Norden et al. suggested the use of a combination of RBP and a formula for the upper limit of albumin secretion for the diagnosis of tubular proteinuria, however this formula has not been widely used, perhaps because of its complexity [21].
Since urinary α1M as well as other low molecular weight proteins, are not always readily available, and not commonly ordered by nephrologists, we also sought a more accessible Dent disease screening test. Such a test would be of particular importance in the population of patients with nephrotic range proteinuria and without apparent nephrocalcinosis and kidney stones, the usual findings suggestive of Dent disease. In the Dent Registry of the RKSC, which numbered 167 patients in July 2017, nephrocalcinosis was present in 65 % and kidney stones only in 30 %, corresponding to previously reported data [6, 22, 23]. Thus a significant percentage lack obvious pathologic consequences of hypercalciuria.
The median A/Cr was significantly higher in the CKiD G cohort compared to DD1 and CKiD TI. Moreover, the A/TP clearly separated DD1 from CKiD G. DD1 patients had a median A/TP of 0.18 compared with 0.67 in the CKiD G population. CKiD TI at A/TP of 0.32 was not significantly different from DD1; however, DD1 did have significantly higher TP/Cr and α-ιΜ/Cr levels compared with the CKiD TI group, which can therefore be used to separate them. According to our scatter plot, if P/Cr was higher than 600 mg/g creatinine (67.8 mg/mmol) and A/TP < 0.3, we were able to differentiate DD1 from both CKD TI and CKD G with sensitivity of 0.86 and specificity of 0.98 (Fig 1).
The CKiD TI group included a majority of patients who had reflux nephropathy (54 out of 58), which is known to cause tubular proteinuria, but may also cause FSGS with unknown exact mechanism. The podocyte injury might be caused by the lengthening of the glomerular capillaries due to hyperfiltration [24]. In our population of reflux patients, the median A/TP was still significantly lower compared with CKiD G, however some patients did have A/TP > 0.5 (Fig 1). Interestingly, DC also had a very low and not significantly different A/TP comparable to DD1, and some of the carriers had increased TP (up to 371 mg/gm Cr). This, along with similar α1M/TP, supports a similar mechanism of proteinuria in DC.
We have identified cut off values for three different markers (α1M/Cr, a-|M/TP and A/TP) to further assess their ability to distinguish DD1 from CKiD (G and TI) based on the highest sensitivity and specificity. α-ιΜ/Cr ≥ 120 mg/g had the highest sensitivity and specificity when differentiating DD1 and CKiD. The most readily available test A/TP ≤ 0.21 differentiated DD1 from CKiD G well, with high sensitivity and specificity, supporting leaning away from kidney biopsy when finding a patient with such low A/TP ratio, at least not before DD1 is ruled out. In addition, normal serum A level and absence of edema will support the diagnosis of a tubular disease. When comparing DD1 to CKiD TI, A/TP ≤ 0.21 had high sensitivity for Dent disease, but lower specificity. Sensitivity and specificity increases significantly when proteinuria is higher than 600 mg/g (67.8 mg/mmol) creatinine. Supportive X-linked recessive family history, additional laboratory findings including α-ιΜ/Cr in Dent disease and the genetic test will aid in the definite diagnosis.
We suggest the following approach to diagnosis of Dent disease for patients with proteinuria of unknown origin. We propose quantifying and qualifying proteinuria with TP and A/TP ratio. If the patients have higher P/Cr 600 mg/g (67.8 mg/mmol) and the proportion of A/TP is ≤ 0.3, it is indicated by our study that the sensitivity and specificity for DD1 is higher than 0.85. In patients with nephrotic range proteinuria, normal serum albumin and absence of edema additionally suggest that primary glomerular disease is not a likely cause of proteinuria. To further clarify the diagnosis, we suggest measuring urinary α1M, with a-|M/Cr ≥ 120 mg/g being the most specific indicator of DD1. The clinical diagnosis of Dent disease is then established by the additional presence of at least two to three of the secondary findings: hypercalciuria, nephrocalcinosis, nephrolithiasis, hematuria, hypophosphatemia, CKD, and family history consistent with X-linked recessive inheritance. The diagnosis can be confirmed by genetic testing, however about 25 % of Dent patients lack explanatory genetic characterization, and the diagnosis is established by the above clinical and laboratory criteria.
The majority of urinary protein in Dent patients is LMWP; however, patients commonly have significant albuminuria. In our previous biopsy review, we found a significant FGGS prevalence among Dent patients, even more prevalent than tubulointerstitial disease. We reviewed 30 biopsies of Dent patients, 83 % of whom were found to have FGGS, with the low median (25th, 75th percentile) percentage of globally sclerotic glomeruli 9.5 % (2.2, 24.3). FSGS was documented in only 7 % of patients. Foot process effacement was noted in 57 % of patients; it was segmental in all patients and mild in the majority of patients [13]. A recent literature review of proteinuria in Dent patients by van Berkel et al. has similarly noted glomerulosclerosis in almost two thirds of the biopsies with a median of 17 % sclerosed glomeruli. In contrast with the report by van Berkel et al., we did find a significant association of lower eGFR at biopsy with higher 24-hour urine protein excretion and with a higher percentage of globally sclerotic glomeruli on biopsy [25]. We were able to follow up 18 patients for a median of 3 (1, 7) years. On multivariate analysis, only foot process effacement and not tubulointerstitial changes or percentage of globally sclerotic glomeruli, were significantly associated with decline of eGFR [13].
Both CLCN5 and OCRL1, as well as megalin, and cubulin, are expressed in the glomerulus [26–29]. Podocytes of proteinuric patients overexpress CLC-5 at both mRNA and protein level, suggesting a link between CLC-5 and protein absorption [26].
Limitations of our study are attributable to the rare nature of Dent disease, including the relatively low number of Dent patients and separate specimen collection from Dent and CKiD patients. In addition, there was a wider age range for the Dent patients compared with the CKiD cohort. However, the median proteinuria in the Dent cohort was representative of proteinuria in Dent patients reported in literature. Although we have only studied DD1 patients, we can speculate that DD2 patients and DD patients with unknown genetic mutation have a similar pattern of proteinuria, which was also suggested in the recent data review of van Berkel et al. [25].
We also included a limited number of diagnoses with tubulointerstitial disease from the CKiD cohort, including patients with reflux disease with a possible component of FSGS, however the reflux group selected had a low median proteinuria and albuminuria and other tubulointerstitial diseases might also have secondary glomerular sclerosis.
In conclusion, we suggest that patients with proteinuric kidney diseases of unclear origin be screened for tubulointerstitial diseases, including DD, by looking at the TP and A/TP ratio. In our study, a cut off of TP/Cr > 600 mg/g (> 68 mg/mmol) and A/TP < 0.3 had a high sensitivity and specificity to distinguish DD1 from both CKiD glomerular and tubulointerstitial cohorts. The most specific confirmatory test for Dent disease was α1M/Cr ≥120. Prudent qualitative analysis and knowledge in interpretation of proteinuria, as well as the awareness of Dent disease, may avoid unnecessary biopsy and treatments and improve timeliness of diagnosis.
ACKNOWLEDGMENTS
We thank Alicia Meek for her assistance in data collection.
This study was supported by Rare Kidney Stone Consortium grant U54KD083908, a part of the Rare Diseases Clinical Research Network, an initiative of the Office of Rare Diseases Research, the National Center for Advancing Translational Sciences (NCATS).
This consortium is funded through a collaboration between the NCATS and the National Institute of Diabetes and Digestive and Kidney Diseases.
The study sponsor had no role in study design; collection, analysis, and interpretation of data; writing the report; and the decision to submit the report for publication.
The Chronic Kidney Disease in Children Cohort Study (CKiD) was conducted by the CKiD Investigators and supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), with additional funding from the National Institute of Child Health and Human Development, and the National Heart, Lung, and Blood Institute (U01-DK-66143, U01-DK-66174, U01DK-082194, U01-DK-66116). The data and samples from the CKiD study reported here were supplied by the NIDDK Central Repositories. This manuscript does not necessarily reflect the opinions or views of the CKiD study, the NIDDK Central Repositories, or the NIDDK.
The Multicenter FSGS-Clinical Trial (FSGS-CT) and Novel therapies for resistant FSGS Trial (FONT) was conducted by the FSGS-CT and FONT investigators and supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The data and samples from the FSGS-CT and FONT reported here were supplied by the NIDDK Central Repositories. This manuscript does not necessarily reflect the opinions or views of the FSGS-CT and FONT study, the NIDDK Central Repositories, or the NIDDK.
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Footnotes
COMPLIANCE WITH ETHICAL STANDARDS
This study was approved by the Mayo Clinic Institutional Review Board. Ancillary studies were approved and biospecimens were obtained from CKiD, FSGS-CT and FONT studies.
DISCLOSURE
The authors declare that they have no conflicts of interest.
REFERENCES
- 1.Dent CE, Friedman M (1964) Flypercalciuric Rickets associated with renal tubular damage. Arch Dis Child 39:240–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scheinman SJ (1998) X-linked hypercalciuric nephrolithiasis: clinical syndromes and chloride channel mutations. Kidney Int 53:3–17 [DOI] [PubMed] [Google Scholar]
- 3.Lieske JC, Milliner DS, Beara-Lasic L, Harris P, Flopp K, Cogal A, Mattison K (1993) Dent Disease. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K (eds) GeneReviews(R), Seattle (WA) [PubMed] [Google Scholar]
- 4.Ludwig M, Utsch B, Monnens LA (2006) Recent advances in understanding the clinical and genetic heterogeneity of Dent’s disease. Nephrol Dial Transplant 21:2708–2717 [DOI] [PubMed] [Google Scholar]
- 5.Frymoyer PA, Scheinman SJ, Dunham PB, Jones DB, Hueber P, Schroeder ET (1991) X-linked recessive nephrolithiasis with renal failure. N Engl J Med 325:681–686 [DOI] [PubMed] [Google Scholar]
- 6.Devuyst 0, Thakker RV (2010) Dent’s disease. Orphanet J Rare Dis 5:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Berkel Y, Ludwig M, van Wijk JAE, Bokenkamp A (2017) Proteinuria in Dent disease: a review of the literature. Pediatr Nephrol 32:1851–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gambaro G, Vezzoli G, Casari G, Rampoldi L, D’Angelo A, Borghi L (2004) Genetics of hypercalciuria and calcium nephrolithiasis: from the rare monogenic to the common polygenic forms. Am J Kidney Dis 44:963–986 [DOI] [PubMed] [Google Scholar]
- 9.Copelovitch L, Nash MA, Kaplan BS (2007) Hypothesis: Dent disease is an underrecognized cause of focal glomerulosclerosis. Clin J Am Soc Nephrol 2:914–918 [DOI] [PubMed] [Google Scholar]
- 10.Frishberg Y, Dinour D, Belostotsky R, Becker-Cohen R, Rinat C, Feinstein S, Navon-Elkan P, Ben-Shalom E (2009) Dent’s disease manifesting as focal glomerulosclerosis: Is it the tip of the iceberg? Pediatr Nephrol 24:2369–2373 [DOI] [PubMed] [Google Scholar]
- 11.Fervenza FC (2013) A patient with nephrotic-range proteinuria and focal global glomerulosclerosis. Clin J Am Soc Nephrol 8:1979–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wrong OM, Norden AG, Feest TG (1994) Dent’s disease; a familial proximal renal tubular syndrome with low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and a marked male predominance. QJM 87:473–493 [PubMed] [Google Scholar]
- 13.Wang X, Anglani F, Beara-Lasic L, Mehta AJ, Vaughan LE, Herrera Hernandez L, Cogal A, Scheinman SJ, Ariceta G, Isom R, Copelovitch L, Enders FT, Del Prete D, Vezzoli G, Paglialonga F, Harris PC, Lieske JC; Investigators of the Rare Kidney Stone Consortium (2016) Glomerular Pathology in Dent Disease and Its Association with Kidney Function. Clin J Am Soc Nephrol 11:2168–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong CJ, Moxey-Mims M, Jerry-Fluker J, Warady BA, Furth SL (2012) CKiD (CKD in children) prospective cohort study: a review of current findings. Am J Kidney Dis 60:1002–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joy MS, Gipson DS, Dike M, Powell L, Thompson A, Vento S, Eddy A, Fogo AB, Kopp JB, Cattran D, Trachtman FI (2009) Phase I trial of rosiglitazone in FSGS: I. Report of the FONT Study Group. Clin J Am Soc Nephrol 4:39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferris M, Norwood V, Radeva M, Gassman JJ, Al-Uzri A, Askenazi D, Matoo T, Pinsk M, Sharma A, Smoyer W, Stults J, Vyas S, Weiss R, Gipson D, Kaskel F, Friedman A, Moxey-Mims M, Trachtman H (2013) Patient recruitment into a multicenter randomized clinical trial for kidney disease: report of the focal segmental glomerulosclerosis clinical trial (FSGS CT). Clin Transl Sci 6:13–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwartz GJ, Munoz A, Schneider MF, Mak RH, Kaskel F, Warady BA, Furth SL (2009) New equations to estimate GFR in children with CKD. J Am Soc Nephrol 20:629–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bernard AM, Moreau D, Lauwerys R (1982) Comparison of retinol-binding protein and beta 2-microglobulin determination in urine for the early detection of tubular proteinuria. Clin Chim Acta 126:1–7 [DOI] [PubMed] [Google Scholar]
- 19.Blumsohn A, Morris BW, Griffiths H, Ramsey CF (1991) Stability of beta 2-microglobulin and retinol binding protein at different values of pH and temperature in normal and pathological urine. Clin Chim Acta 195:133–137 [DOI] [PubMed] [Google Scholar]
- 20.Guder WG, Hofmann W (2008) Clinical role of urinary low molecular weight proteins: their diagnostic and prognostic implications. Scand J Clin Lab Invest Suppl 241:95–98 [DOI] [PubMed] [Google Scholar]
- 21.Norden AG, Scheinman SJ, Deschodt-Lanckman MM, Lapsley M, Nortier JL, Thakker RV, Unwin RJ, Wrong 0 (2000) Tubular proteinuria defined by a study of Dent’s (CLCN5 mutation) and other tubular diseases. Kidney Int 57:240–249 [DOI] [PubMed] [Google Scholar]
- 22.Bokenkamp A, Bockenhauer D, Cheong HI, Hoppe B, Tasic V, Unwin R, Ludwig M (2009) Dent-2 disease: a mild variant of Lowe syndrome. J Pediatr 155:94–99 [DOI] [PubMed] [Google Scholar]
- 23.Lloyd SE, Pearce SHS, Guenther W, Kawaguchi H, Igarashi T, Jentsch TJ, Thakker RV (1997) Idiopathic low molecular weight proteinuria associated with hypercalciuric nephroclacinosis in Japanese children is due to mutations of teh renal chloride channel (CLCN5). J Clin Invest 99:967–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tada M, Jimi S, Hisano S, Sasatomi Y, Oshima K, Matsuoka H, Takebayashi S (2001) Histopathological evidence of poor prognosis in patients with vesicoureteral reflux. Pediatr Nephrol 16:482–487 [DOI] [PubMed] [Google Scholar]
- 25.van Berkel Y, Ludwig M, van Wijk JA, Bokenkamp A (2016) Proteinuria in Dent disease: a review of the literature. Pediatr Nephrol 32:1851–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ceol M, Tiralongo E, Baelde HJ, Vianello D, Betto G, Marangelli A, Bonfante L, Valente M, Della Barbera M, D’Angelo A, Anglani F, Del Prete D (2012) Involvement of the tubular CIC-type exchanger CIC-5 in glomeruli of human proteinuric nephropathies. PLoS ONE 7:e45605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erb BC, Velazquez H, Gisser M, Shugrue CA, Reilly RF (1997) cDNA cloning and localization of OCRL-1 in rabbit kidney. Am J Physiol 273:F790–795 [DOI] [PubMed] [Google Scholar]
- 28.Prabakaran T, Christensen El, Nielsen R, Verroust PJ (2012) Cubilin is expressed in rat and human glomerular podocytes. Nephrol Dial Transplant 27:3156–3159 [DOI] [PubMed] [Google Scholar]
- 29.Prabakaran T, Nielsen R, Larsen JV, Sorensen SS, Feldt-Rasmussen U, Saleem MA, Petersen CM, Verroust PJ, Christensen El (2011) Receptor-mediated endocytosis of alpha-galactosidase A in human podocytes in Fabry disease. PLoS ONE 6:e25065. [DOI] [PMC free article] [PubMed] [Google Scholar]