

Summary
When the Lyme disease spirochete, Borrelia burgdorferi, transfers from a feeding tick into a human or other vertebrate host, the bacterium produces vertebrate‐specific proteins and represses factors needed for arthropod colonization. Previous studies determined that the B. burgdorferi BpuR protein binds to its own mRNA and autoregulates its translation, and also serves as co‐repressor of erp transcription. Here, we demonstrate that B. burgdorferi controls transcription of bpuR, expressing high levels of bpuR during tick colonization but significantly less during mammalian infection. The master regulator of chromosomal replication, DnaA, was found to bind specifically to a DNA sequence that overlaps the bpuR promoter. Cultured B. burgdorferi that were genetically manipulated to produce elevated levels of BpuR exhibited altered levels of several proteins, although BpuR did not impact mRNA levels. Among these was the SodA superoxide dismutase, which is essential for mammalian infection. BpuR bound to sodA mRNA in live B. burgdorferi, and a specific BpuR‐binding site was mapped 5′ of the sodA open reading frame. Recognition of posttranscriptional regulation of protein levels by BpuR adds another layer to our understanding of the B. burgdorferi regulome, and provides further evidence that bacterial protein levels do not always correlate directly with mRNA levels.
Introduction
Borrelia burgdorferi, the spirochetal agent of Lyme disease, has evolved mechanisms to infect both vertebrates and ticks, and to cycle between those two very different types of animals. This complex cycle requires that B. burgdorferi accurately control production of the factors that are required for each step, and disruption of those regulatory networks can prevent the bacterium from colonizing one or both hosts (Samuels, 2011; Radolf et al., 2012; Stevenson and Seshu, 2018). From a public health standpoint, characterizing B. burgdorferi's regulatory mechanisms can identify critical targets for development of novel therapies to combat Lyme disease.
A number of proteins and small signaling molecules have been identified that control transcription in B. burgdorferi (Stevenson and Seshu, 2018). Recently, it has become obvious that the Lyme spirochete also uses posttranscriptional mechanisms to regulate protein levels. These mechanisms include RNA‐binding proteins and noncoding RNAs that alter translation of mRNAs, enzymatic modifications of proteins and factors that change the stability of target proteins (Lybecker and Samuels, 2007; Karna et al., 2011; Salman‐Dilgimen et al., 2011; Sze et al., 2011; Dulebohn et al., 2014; Lybecker and Samuels, 2017; Popitsch et al., 2017; Bontemps‐Gallo et al., 2018; Savage et al., 2018).
Among these posttranscription regulatory factors is a small protein named BpuR (Jutras et al., 2013a). It is a homodimer of two 122 residue polypeptides, which fold into a symmetric ‘PUR’ domain (Graebsch et al., 2009; 2010). That motif is conserved in prokaryotic and eukaryotic nucleic acid‐binding proteins, and is so‐named because PUR domain proteins exhibit high affinity for purine‐rich stretches of nucleic acids (Bergemann et al., 1992; Daniel and Johnson, 2018). BpuR was first identified as a DNA‐binding protein that functions as a co‐repressor of B. burgdorferi erp operons, enhancing the activity of the BpaB repressor protein (Burns et al., 2010; Jutras et al., 2012b; 2013a). However, in vitro analyses demonstrated that BpuR did not affect erp transcriptional initiation in the absence of BpaB. Our subsequent studies of BpuR revealed that it has a substantially greater affinity for RNA than it does for either double‐ or single‐stranded DNAs (Jutras et al., 2013a). Moreover, BpuR binds with high specificity and affinity to the 5′ end of its own transcript, which inhibits translation of bpuR mRNA (Jutras et al., 2013d).
The BpuR posttranscriptional mechanism of autoregulation prevents B. burgdorferi from producing this protein above a specific threshold (Jutras et al., 2013d). However, cultured B. burgdorferi can reduce cellular levels of BpuR, implying the existence of a mechanism(s) that inhibits protein production (Jutras et al., 2013a). This repression mechanism is linked to the rate of bacterial replication: bacteria cultured under conditions that enable rapid division produce substantially less BpuR than do bacteria cultured under growth‐inhibiting conditions. This is hypothesized to reflect conditions encountered during B. burgdorferi's infectious cycle, where bacterial replication rates vary from slow (or none) during survival in the unfed tick, to rapid growth and division as the tick ingests nutritious blood (Piesman et al., 1990; 2001; de Silva and Fikrig, 1995; Piesman and Schneider, 2002; Dunham‐Ems et al., 2009; Radolf et al., 2012; Jutras et al., 2013a; 2013c).
This report presents results of studies that address several key questions about BpuR, including an additional mechanism by which BpuR levels are controlled by the bacterium, identification of a connection between bacterial replication and BpuR production, and identification of a virulence‐associated protein that is posttranscriptionally regulated by BpuR.
Results
Regulation of bpuR transcription and translation
During the natural mammal–tick infectious cycle, B. burgdorferi undergoes periods of both rapid and slow growth. Our previous studies demonstrated that B. burgdorferi controls cellular levels of BpuR protein in response to changes in medium composition or incubation temperature (Jutras et al., 2013a; 2013d). In culture, concentrations of the protein decreased under conditions that enhance bacterial replication, and increased under slow‐growth conditions (Jutras et al., 2013a). We hypothesized that regulation of bpuR transcription could facilitate such reductions in BpuR levels.
We previously constructed a chimeric plasmid, pGJ1, in which the bpuR promoter drives transcription of gfp, yet lacks the BpuR‐binding sequence necessary for posttranscriptional control (Jutras et al., 2013d). Thus, GFP protein levels are dependent upon transcriptional initiation. B. burgdorferi strain JB26, which carries pGJ1, was cultured under conditions that yielded optimal growth (complete medium at 35°C) or three‐ to fourfold reduced division rates (incomplete medium at 35°C) (Jutras et al., 2013c). Individual bacteria were not detectably different in size when grown in the different culture media. The GFP content of individual bacteria of those cultures was measured by flow cytometry, revealing that the bacteria cultured under slow‐replication conditions produced greater levels of the reporter protein (Fig. 1). Reduction of rabbit serum content in medium resulted in significantly increased peak, mean and median GFP content per cell. Reduction of other medium contents increased the peak level, although this did not increase either mean or median GFP to statistically significant extents. Altogether, these results indicate that B. burgdorferi regulates initiation of bpuR transcription.
Figure 1.
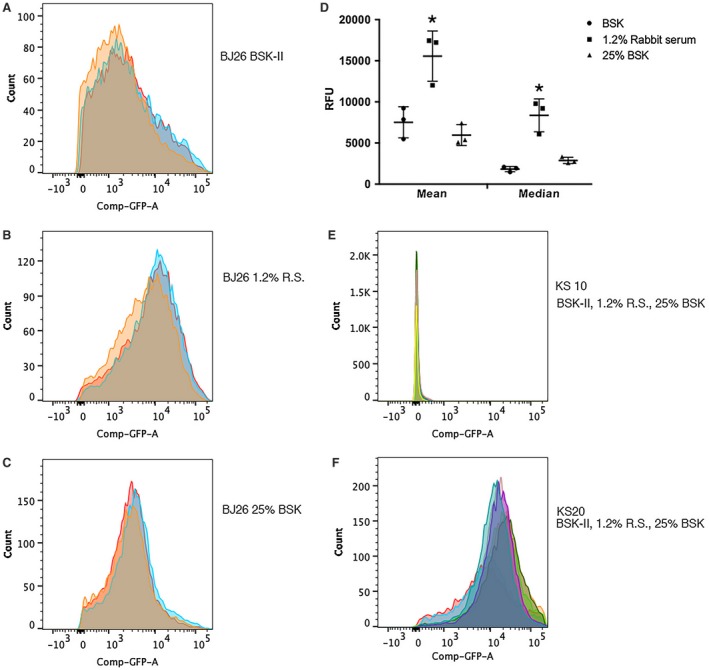
Transcriptional regulation of the bpuR promoter in response to changing culture conditions. B. burgdorferi strain BJ26 carries pGJ1, a transcriptional fusion between the bpuR promoter and gfp (Jutras et al., 2013d). That strain was incubated in either complete BSK‐II medium, or in one of two incomplete formulations that reduce bacterial division rates 3–4‐fold (Jutras et al., 2013c). Bacteria from mid‐exponential phase cultures (approximately 107 bacteria/ml) were collected, resuspended in PBS and level of GFP per bacterium was assayed by flow cytometry. Biological triplicates were assayed for all strains and culture conditions. Results from replicate experiments are illustrated as overlapping graphs in each panel, each indicated by a different colored outline and fill. The X‐axes indicate the relative level of green fluorescence per bacterium, which is proportional to production of GFP (Carroll et al., 2003; Babb et al., 2004). The Y‐axes indicate the number of bacterial cells with a particular level of GFP. A. Results of analyses of three distinct cultures of BJ26 in complete BSK‐II medium. B. Results of analyses of three distinct cultures of BJ26 in BSK‐II + 1.2% rabbit serum. C. Results of analyses of three distinct cultures of BJ26 in 25% BSK‐II + the normal concentration of rabbit serum (6%). D. Statistical analyses comparing GFP content of BJ26 cultured in incomplete medium relative to complete medium. Asterisks (*) indicate values that are statistically different from results of BJ26 in complete medium (P < 0.05 by two‐way ANOVA). E. Control strain KS20, which carries a promoterless gfp (Babb et al., 2004), cultured under all three growth conditions (triplicate cultures of each condition were assayed, one representative of each is illustrated here). F. Control strain KS20, which carries a fusion between a minimal B. burgdorferi promoter and gfp (Babb et al., 2004), cultured under all three growth conditions (triplicate cultures of each condition were assayed, one representative of each is illustrated here).
Extending investigations on regulation of the bpuR transcript, levels of its mRNA were determined during tick and mammal stages of the infectious cycle. To begin, Ixodes scapularis larvae were colonized through feeding on B. burgdorferi‐infected mice. Those ticks were allowed to digest their blood meal and molt into nymphs. B. burgdorferi within larvae that had completed blood‐feeding produced levels of bpuR mRNA that were comparable to that of the constitutively expressed flaB transcript (Fig. 2). Similar levels were observed in ticks that had molted into nymphs. During nymphal feeding, B. burgdorferi initiates expression of genes and proteins important for transmission and vertebrate infection, while also repressing tick‐specific transcripts (Schwan et al., 1995; Narasimhan et al., 2002; Miller et al., 2003; Bykowski et al., 2006; Iyer et al., 2015). Relative levels of bpuR transcript dropped significantly as nymphs began feeding on naïve mice, which is a time of increased bacterial replication (Fig. 2, 48 and 72 h fed nymphs). Upon completion of feeding, relative levels of bpuR mRNA began to increase (Fig. 2, detached and 1 week postfed nymphs). In contrast, bpuR transcripts were undetectable in mice that had been infected for 1 month, although the infecting bacteria produced flaB mRNA at readily detectable levels (Fig. 2, 1 month mice).
Figure 2.
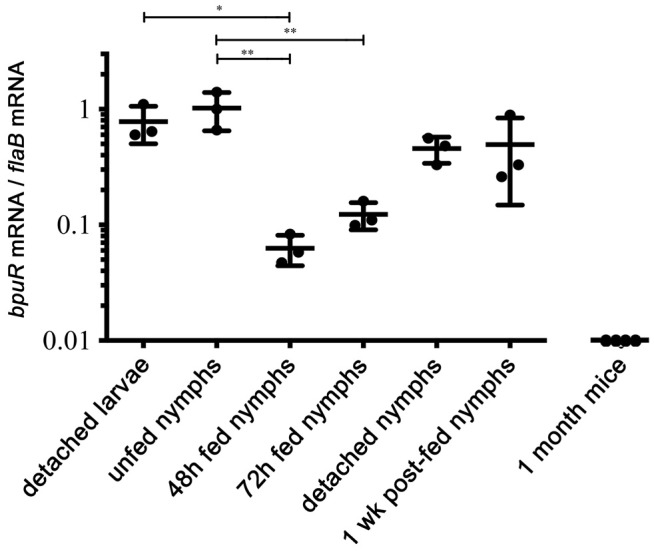
Borrelia burgdorferi differentially expresses bpuR during the tick–mammal infectious cycle. Quantitative RT‐PCR (qRT‐PCR) analyses of bpuR mRNA levels in ticks or mice, expressed as ratios of bpuR vs. transcript of the flagellin‐encoding flaB. Examined tick stages and conditions were as follows: larvae examined after completion of feeding on infected mice (detached larvae), infected ticks immediately after molting from larvae to nymphs (unfed nymphs), infected nymphs that were interrupted from feeding 48 or 72 h after attachment to naïve mice (‘48 h’ and ‘72 h fed nymphs’), infected nymphs that had completed feeding and detached mouse hosts (detached nymphs), and infected nymphs 1 week after completion of feeding (1 week postfed nymphs). Urinary bladders from mice were also analyzed, collected 1 month after the mice were infected by inoculation with cultured B. burgdorferi. Mean and standard error are shown for each sample set. One‐way analysis of variance with Tukey’s Multiple Comparison Test was used to determine significant differences between samples (*, P ≤ 0.05; **P ≤ 0.01). None of the mouse tissues yielded a positive signal for bpuR transcript, although all produced an amplicon from flaB primers.
Transcriptional regulation is often accomplished through one or more proteins that bind to a specific DNA sequence and influence interactions between the promoter and RNA polymerase. Pursuing that line, electrophoretic mobility shift assays (EMSAs) were employed using a labeled DNA probe that spanned the intergenic region from the bpuR ribosome binding site to the beginning of the upstream gene (Fig. 3A). This probe lacked the previously identified BpuR‐binding site (Jutras et al., 2013d). Cell extract from cultured B. burgdorferi contained a protein(s) that bound to the bpuR probe (Fig. 3B). That same fragment of bpuR 5′ noncoding DNA was then used as bait to pull down proteins from the B. burgdorferi lysate, followed by elution with increasing concentrations of NaCl (Fig. 3C) (Jutras et al., 2012c). The 0.5 M NaCl elution contained two proteins that were identified by mass spectrometry as the outer surface proteins OspA and OspC. Neither of those proteins would be able to interact with cytoplasmic DNA in intact bacteria, and so were not considered further. However, a ca. 58kD protein eluted at 0.75M NaCl, and was tentatively identified by mass spectrometry as being the B. burgdorferi homologue of DnaA. In other bacterial species, DnaA binds to specific DNA sites and serves as a transcriptional regulatory protein, in addition to controlling chromosomal replication (Messer and Weigel, 1997; Smith and Grossman, 2015). To assess whether DnaA truly has an affinity for the bpuR locus, recombinant B. burgdorferi DnaA was purified and found to bind to the bpuR promoter region in a dose‐dependent manner (Fig. 3D, lanes 1–4). Unlabeled bpuR promoter DNA competed for DnaA binding, whereas a 100‐fold excess of an unrelated DNA sequence did not (Fig. 3D, lanes 9 and 8, respectively). Three unlabeled DNAs that tile along the bpuR 5′ noncoding region were assessed for competition; only fragment 6 competed with the full‐length labeled probe (Fig. 3D, lanes 5‐7). We conclude that DnaA binds specifically to a sequence that is included in competitor fragment 6, but not in fragments 5 or 7. That region overlaps the bpuR transcriptional promoter (Fig. 3A). This region contains a sequence, 5′‐TTTTTAAA‐3′, that is similar to the relaxed consensus DnaA box sequence that was compiled from studies of other eubacterial species, 5′‐T(T/C)(T/A)T(A/C)CA(A/C)A‐3′ (Fuller et al., 1984; Moriya et al., 1988).
Figure 3.
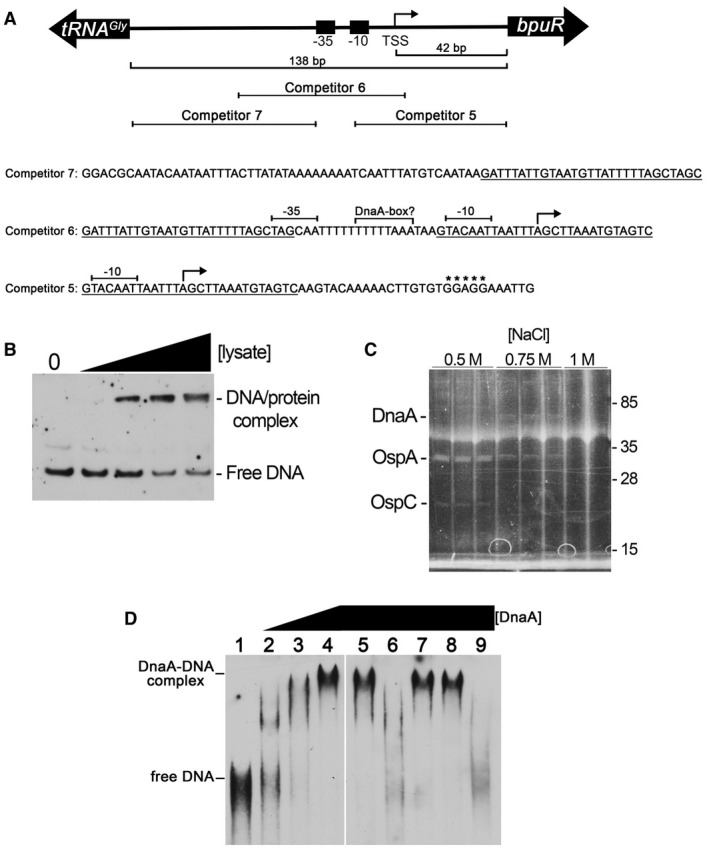
DnaA binds specifically to the bpuR promoter. A. Diagram illustrating the orientation of the bpuR and tRNA genes, with sequences and locations of relevant features. The −10, −35 and TSS of bpuR have previously been mapped (Jutras et al., 2013d). Sequences are provided for the three unlabeled competitor DNAs used in the EMSAs that are shown in panel D: overlapping sequences of the competitors are indicated by underlining, the TSS is indicated by an angled arrow, and the bpuR ribosome‐binding site is indicated by asterisks. A sequence within competitor 6, between the −10 and −35 elements, is similar to the relaxed consensus DnaA‐box sequence of other eubacterial species (Fuller et al., 1984; Moriya et al., 1988). B. EMSA using a labeled probe that consisted of the DNA sequence between the bpuR ribosome‐binding site and the upstream tRNA locus. The first lane contains DNA alone. The next four lanes contain increasing concentrations of B. burgdorferi extract. C. A DNA probe consisting of the sequence between the bpuR ribosome‐binding site and the upstream tRNA gene was incubated with B. burgdorferi cellular extract at a physiological NaCl concentration (150 mM), then adhered proteins eluted with successively greater concentrations of NaCl. Eluates were separated by SDS‐PAGE and proteins visualized by Sypro Ruby stain. Two proteins in the 500 mM NaCl elution were identified as the abundant outer surface proteins OspA and OspC, and were disregarded as irrelevant contaminants. The ca. 58kDa protein that eluted at 750 mM NaCl was identified as DnaA. The haze across all lanes at ca. 40kDa is an electrophoresis artifact. D. Electrophoretic mobility shift assays (EMSAs) using a labeled DNA probe that spans the bpuR‐tRNA intergenic region, recombinant B. burgdorferi DnaA, and unlabeled competitor DNAs. Lane 1: labeled probe alone. Lanes 2–4, labeled probe plus successively doubled concentrations of DnaA (0.35, 0.70 and 1.4 µM DnaA in lanes 2, 3 and 4, respectively). Lanes 5–7, labeled probe plus 1.4 µM DnaA and 100x excesses of unlabeled DNA competitors 5, 6 or 7, respectively. Lane 8, labeled probe plus 1.4 µM DnaA and 100x excess unlabeled competitor based on the B. burgdorferi flaB promoter sequence. Lane 9, labeled probe plus 1.4 µM DnaA and 100x excess unlabeled competitor with the exact sequence as the labeled probe. The unlabeled DNAs in lanes 6 and 9 evidently destabilized the DnaA–probe complex during electrophoresis, resulting in signal smears as the probe was released (Fried, 1989; Hellman and Fried, 2007). The illustrated data are from a single EMSA gel, with irrelevant lanes removed, as indicated by a vertical white line.
BpuR exerts posttranscriptional impacts on the B. burgdorferi proteome
An unsaturated transposon insertion library of B. burgdorferi does not include a bpuR mutant (Lin et al., 2012). Our repeated attempts to delete the bpuR locus have not been successful. It is possible that BpuR may be essential for B. burgdorferi survival in culture, or, since the bpuR gene is adjacent to an essential tRNA gene, it may be that disruption of bpuR impacts upon production of that tRNA (Fraser et al., 1997; Jutras et al., 2013d). However, since B. burgdorferi cultured at 35°C do not produce maximal levels of the protein, effects of BpuR can be evaluated by studying bacteria engineered to produce elevated levels of BpuR (Jutras et al., 2013a; 2013d). BpuR's posttranscriptional autoregulation mechanism prevents the bacteria from accumulating nonphysiological levels of the protein (Jutras et al., 2013d).
Wild‐type B. burgdorferi was transformed with a plasmid that contains bpuR under the control of an IPTG‐inducible promoter (Blevins et al., 2007). Bacteria were cultured in complete BSK‐II medium at 35°C, with or without added IPTG, then total RNA was purified and assayed by RNA‐Seq. Levels of all transcripts were analyzed, including coding and noncoding RNAs (Arnold et al., 2016; 2018). Under those culture conditions, we found only two transcripts that met the commonly used criteria for differential expression (2X expression and padj <0.05) (Supplemental Fig. S1 and Table S1). As expected, the transcript for bpuR was increased approximately 4.3‐fold upon IPTG induction. The only other transcript that was significantly affected was hslU (ORF BB0295) (Fraser et al., 1997), which was increased 2.6‐fold. Independently grown and induced cultures were examined by quantitative reverse transcription PCR (qRT‐PCR) for levels of bpuR, hslU, hslV, sodA and flaB transcripts. Consistent with the RNA‐Seq results, bpuR transcript abundance was always significantly increased. However, levels of hslU transcript were not reproducibly altered to significant extents by induction of BpuR, nor were any other assayed transcripts. As an additional control, the BpuR expression plasmid was isolated from B. burgdorferi and sequenced, which showed that no mutations had occurred. We conclude that, under the examined conditions, enhanced production of BpuR did not reproducibly affect levels of borrelial transcripts to significant extents.
BpuR exhibits an approximately 10‐fold greater affinity for RNA than it does for DNA, and regulates its own translation by binding to its mRNA (Jutras et al., 2013a; 2013d). Those data raised the possibility that BpuR might exert posttranscriptional control on additional proteins. To examine that hypothesis, two‐dimensional electrophoresis was used to separate total proteins of mid‐exponential cultures (107 bacteria/ml) of a B. burgdorferi strain that constitutively expresses elevated levels of BpuR, and an isogenic strain carrying an empty expression vector. Several protein spots were appreciably darker or fainter in the elevated BpuR strain, suggestive of differences in protein levels (Fig. 4). Several proteins that appeared to be differentially expressed were extracted and subjected to mass spectrometric analysis. Protein spots with the prefix ‘U’ appear to have been upregulated by elevated BpuR concentration, and those with ‘D’ appear to have been downregulated. Table 1 lists the tentative identifications of proteins. Assays of the protein indicated as D‐1 in Fig. 4 yielded a significant probability of being the SodA superoxide dismutase, with a Mascot score of 160.7. SodA as a target of regulation is particularly important, as this protein is essential for B. burgdorferi to survive during mammalian infection (Esteve‐Gassent et al., 2009; Troxell et al., 2012; Aguirre et al., 2013; Esteve‐Gassent et al., 2015). For that reason, initial focus was placed on SodA. Characterization of the effects of BpuR on the other proteins that appear to have been differentially expressed is ongoing.
Figure 4.
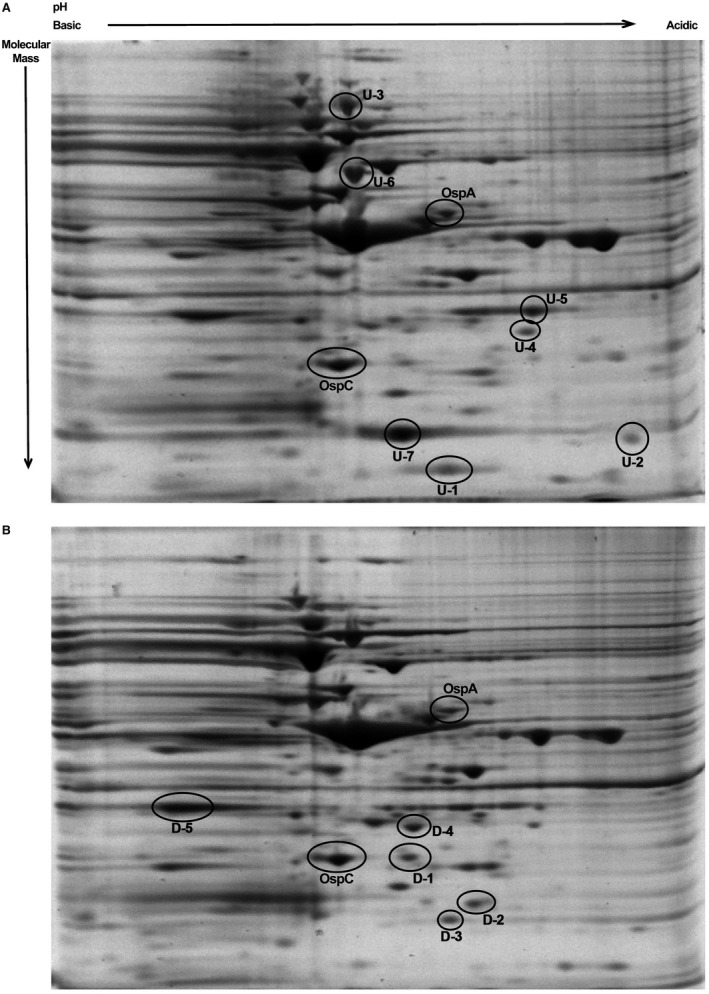
Enhanced expression of BpuR in cultured B. burgdorferi results in changes of the proteome. Two‐dimensional gel analyses of (A) B. burgdorferi that were genetically manipulated to produce elevated levels of BpuR or (B) B. burgdorferi carrying the empty vector. Several protein spots were extracted and analyzed by mass spectrometry. Proteins that appear to have been upregulated by increased levels of BpuR are prefixed ‘U’, while those that appear to have been downregulated are prefixed ‘D’. The differentially abundant spot marked D‐1 was identified through LC‐MS/MS as being SodA. Other proteins that appear to be affected by BpuR are listed in Table 1, and are currently under investigation.
Table 1.
Borrelia burgdorferi proteins identified by LC‐MS/MS analysis of spots on two‐dimensional polyacrylamide gels such as those shown in Fig. 4.
| Spot ID | Protein description | Annotation | MASCOT Score | Isoelectric point (pI) | Theoretical molecular mass (Daltons) |
|---|---|---|---|---|---|
| U‐1 | Putative Outer membrane protein | BBA03 | 97.4 | 5.27 | 19,222 |
| U‐2 | OmpH | BB0796 | 381.4 | 7.88 | 20,513 |
| U‐3 | Outer surface protein P66 | BB0603 | 2286.3 | 6.04 | 68,172 |
| U‐4 | Pfs, 5′‐methylthioadenosine/S‐adenosylhomocysteine nucleosidase | BB0375 | 348.9 | 6.98 | 26,586 |
| U‐5 | OspD | BBJ09 | 130.3 | 5.37 | 28,434 |
| U‐6 | FlaB, flagellin | BB0147 | 2390.6 | 5.53 | 35,765 |
| U‐7 | 50S ribosomal protein L25/Ctc, general stress protein | BB0786 | 270.5 | 6.75 | 20,450 |
| D‐1 | SodA, superoxide dismutase | BB0153 | 160.7 | 6.05 | 23,523 |
| D‐2 | LA‐7, lipoprotein | BB0365 | 128.6 | 5.53 | 21,866 |
| D‐3 | Putative transcription factor/CarD like | BB0355 | 108.9 | 6.12 | 19,020 |
| D‐4 | OMS28, porin | BBA74 | 1065.9 | 6.05 | 27,948 |
| D‐5 | SpoIIIJ‐associated protein | BB0443 | 107.5 | 9.46 | 28,267 |
Each identified protein that appeared to be differentially expressed as given an identification code (Spot ID) with prefix U or D, depending upon whether enhanced expression of BpuR resulted in the apparent protein level going up or down, respectively. Note that only protein D‐1 (SodA) was investigated further, and the apparent effects of BpuR on the other proteins remain to be clarified. For example, spot U‐6 was identified as the flagellin, FlaB, the major component of the borrelial flagella. Both strains appeared to be equally motile, raising the possibility that BpuR may have influenced a posttranslational modification of FlaB that caused a shift in motility on two‐dimensional polyacrylamide gels.
Reproducibility of the effect of elevated BpuR on SodA content was independently examined by analysis of the above‐described BpuR‐inducible strain. Bacteria were cultured either with or without IPTG, an aliquot of each removed for RNA extraction, and the remainder used for protein analysis. As before, qRT‐PCR demonstrated significant increases in bpuR transcript levels, but no significant changes to sodA mRNA levels (Fig. 5A). Immunoblot analyses of total proteins from each culture, using specific antibodies, showed that BpuR protein levels increased in each of three IPTG‐induced cultures, while levels of SodA protein decreased (Fig. 5B). Densiometric analyses of the three sets of immunoblots illustrated in Fig. 5B indicated that BpuR signals changed +2.6, +3.1 and + 4.3 and SodA signals changed −1.4, −1.4 and −1.9, respectively. Control immunoblots using antibody to the constitutively expressed FlaB protein demonstrated equal loading of lysate in each lane.
Figure 5.
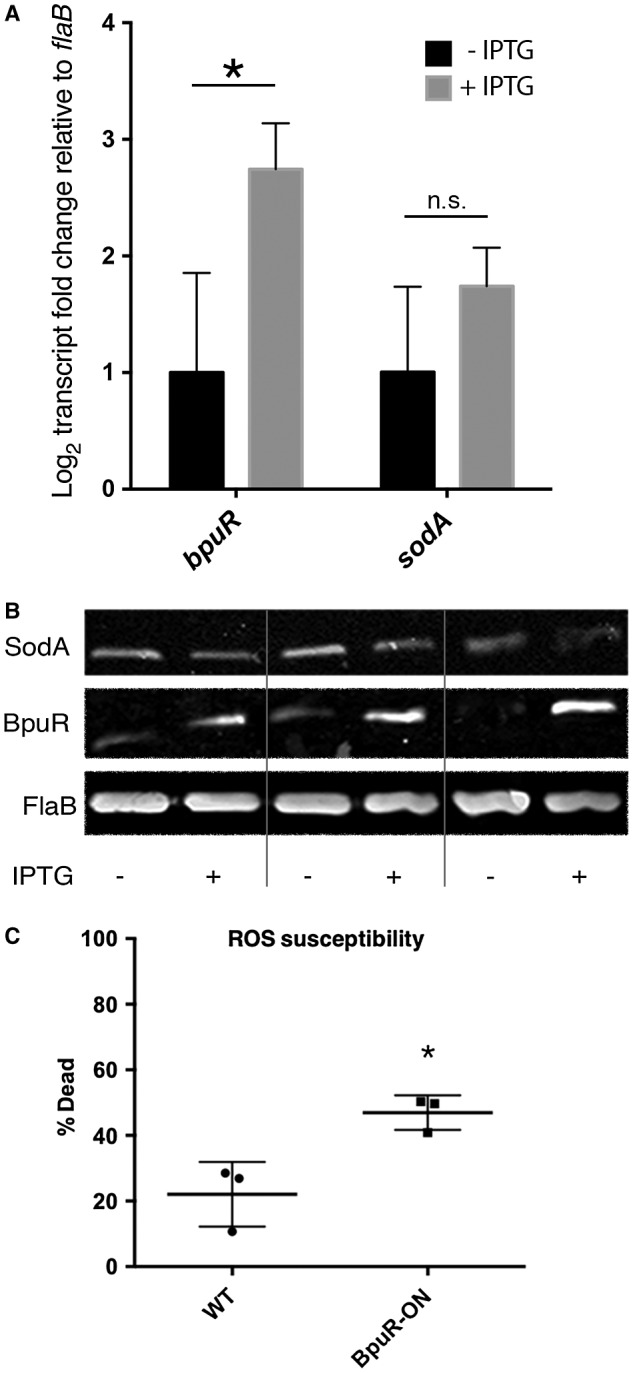
Increased levels of BpuR reduce SodA protein content, but do not affect sodA mRNA levels. Cultures of B. burgdorferi that contain an inducible bpuR construct were grown with or without IPTG inducer. A. qRT‐PCR analyses of bpuR and sodA transcripts in uninduced and induced cultures (−IPTG and +IPTG, respectively), relative to the flaB transcript. Addition of IPTG yielded significant increases in bpuR transcripts (*, P = 0.033), but did not have significant effects on sodA levels (N.S, P = 0.187), by t‐test of ∆Ct values. B. Immunoblot analyses of SodA, BpuR and FlaB proteins in three paired uninduced and induced cultures (− and +, respectively). Densiometric analyses of these images showed that BpuR signals changed +2.6, +3.1 and +4.3, and SodA signals changed −1.4, −1.4 and −1.9, for cultures 1, 2 and 3, respectively. C. B. burgdorferi carrying an inducible bpuR construct were incubated without or with IPTG, then incubated with methyl viologen (paraquat). Alive and dead bacteria were then identified by LIVE/DEAD staining and flow cytometry. BpuR‐induced bacteria were significantly more susceptible to reactive oxygen than were the uninduced bacteria (*, analyzed by unpaired t‐test, P = 0.0296).
B. burgdorferi requires SodA for mammalian infection, and for survival in culture when exposed to reactive oxygen species (ROS) (Esteve‐Gassent et al., 2009; 2015; Troxell et al., 2012; Aguirre et al., 2013). The impact of altering BpuR expression level on ROS susceptibility was assessed by incubating BpuR‐induced and uninduced B. burgdorferi in methyl viologen (paraquat). Bacteria induced to increase cellular concentrations of BpuR, and therefore reduced SodA, were significantly more sensitive to killing by ROS than were uninduced borreliae (Fig. 5C).
Finding that BpuR negatively impacted SodA protein levels but did not affect sodA transcript led us to hypothesize that BpuR binds to sodA mRNA. Targeted RNA immunoprecipitation (RIP) analyses previously demonstrated that BpuR binds to its own mRNA in live B. burgdorferi (Jutras et al., 2013d). That method was again used to examine whether BpuR binds to sodA transcript in vivo. After cross‐linking and immunoprecipitation with BpuR‐specific antibodies, purified RNA was subjected to RT‐PCR using transcript‐specific oligonucleotide primers. This yielded an amplicon with sodA‐specific primers, but not with primers specific for two other borrelial genes: gap and dnaK (Fig. 6, lanes 1). Control chromatin immunoprecipitation (ChIP) demonstrated that BpuR did not bind to sodA or other tested DNAs in vivo (Fig. 6, lanes 2). Other controls using irrelevant IgG‐coated beads, or beads alone, demonstrated that neither condition pulled down any of the tested transcripts (Fig. 6, lanes 3 and 4, respectively). As a final control, all three primer pairs produced PCR amplicons from purified genomic DNA template (Fig. 6, lanes 5). We are currently assessing whether BpuR binds to additional RNAs in live B. burgdorferi, and quantifying site occupation, through use of RIP‐Seq (Li et al., 2018).
Figure 6.
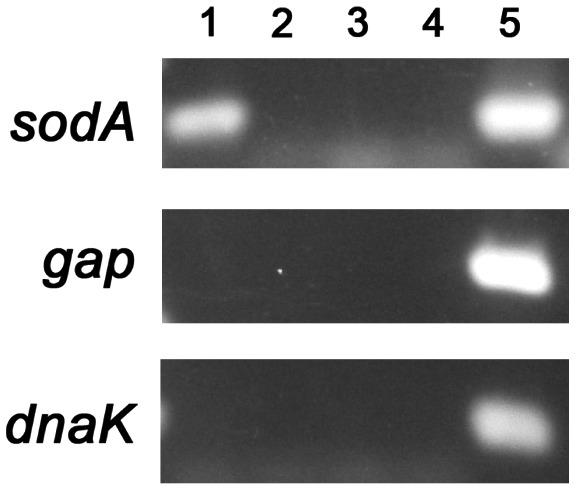
BpuR binds sodA mRNA in live B. burgdorferi. RNA immunoprecipitation (RIP) using BpuR‐specific antibodies pulled down sodA mRNA, but not other transcripts (dnaK and gap). Lanes 1; PCR of cDNAs from BpuR‐RIP. Lanes 2; PCR of cDNAs from RIP with an irrelevant antibody. Lanes 3; PCR of cDNAs from RIP using beads alone. Lanes 4; PCR of DNAs pulled‐down by BpuR‐chromatin immunoprecipitation (ChIP). Lanes 5; PCR of purified genomic DNA template. Primer dimers are visible of some lanes.
Those results were explored further by EMSA with purified BpuR and RNAs derived from the sodA locus. The initiation codon of B. burgdorferi sodA is 14bp downstream of the termination codon of secA, and there are no indications of a transcriptional terminator between the two genes (Fraser et al., 1997; Arnold et al., 2016). Two transcriptional start sites (TSS) have been mapped 5′ of sodA, one within the secA open reading frame and one directly 5′ of secA (Adams et al., 2017) (Fig. 7A). Both promoters are predicted to produce sodA mRNAs, so we focused on sequences shared by both transcripts. For use in RNA EMSA, two 59–60 nt labeled RNA probes were produced: probe sodA1 includes sequences 5′ of the sodA ribosome‐binding site (RBS), and sodA2 spans the sodA RBS and extends into the open reading frame. BpuR bound to probe sodA1, but did not bind probe sodA2 at the tested concentrations (Fig. 7B). Control EMSAs using the BpuR‐binding sequence of the bpuR locus also demonstrated binding by the recombinant protein.
Figure 7.
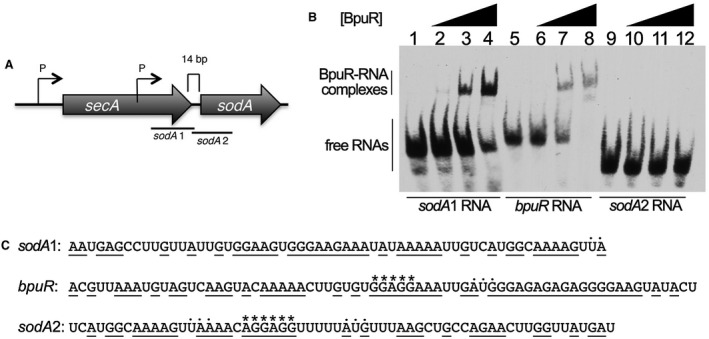
BpuR binds to a sequence within sodA mRNA. A. Diagram of the orientation of sodA in the B. burgdorferi main chromosome. Two promoters have been identified, one 5′ of the upstream secA gene, and another within secA (Adams et al., 2017). There are 14bp between the two open reading frames. Relative locations of RNA probes sodA1 and sodA2 are illustrated. B. EMSAs of interactions between recombinant BpuR protein and labeled RNA probes. Lanes 1–4 contain probe sodA1. Lanes 5–8 contain a control probe derived from the bpuR mRNA sequence, to which BpuR was previously shown to bind (Jutras et al., 2013a). Lanes 9–12 contain probe sodA2. Lanes 1, 5 and 9 lack BpuR, while lanes 2–4, 6–8 and 10–12 contain increasing concentrations of BpuR: Lanes 2, 6 and 10 contain 50 nM BpuR; Lanes 3, 7 and 11 contain 1.3 µM BpuR; and Lanes 4, 8 and 12 contain 5.0 µM BpuR. C. Sequences of the three assayed RNA probes. Translational start and stop codons (AUG and UAA, respectively) are indicated by dots above the nucleotides. Ribosomal‐binding sites are indicated by asterisks above the nucleotides. Purine nucleotides are underlined.
‘PUR’ domains, such as that in BpuR, exhibit affinity to purine‐rich nucleic acid sequences (Bergemann et al., 1992; Graebsch et al., 2010; Jutras et al., 2013a; 2013d; Daniel and Johnson, 2018). The two BpuR‐binding sites that have been identified conform to that prediction: the sodA1 and bpuR RNA probes both contain extensive stretches of purine nucleotides (Fig. 7C). In contrast, the sodA2 RNA probe contains fewer contiguous purines. These data indicate that the purine‐rich RBS, by itself, is not sufficient for BpuR to bind an mRNA.
Discussion
B. burgdorferi regulates levels of numerous proteins during the many stages of its tick–mammal infectious cycle. For the most part, previous research has focused on identifying mechanisms by which the spirochete controls transcription throughout the cycle. Results of our studies indicate that posttranscriptional regulation can also have significant effects on B. burgdorferi protein levels. We previously demonstrated that BpuR regulates its own protein levels via binding to bpuR mRNA, and the current report indicates that it also controls levels of at least one virulence‐associated protein, SodA (Fig. 8). Comparisons of proteomic and transcriptomic data of BpuR‐induced versus uninduced B. burgdorferi suggest that BpuR influences levels of numerous other borrelial proteins (Fig. 4). The evident global effects of BpuR on borrelial protein levels are likely to be complex: protein binding to target RNAs and DNAs will be influenced not only by their relative affinities for BpuR, but also by the cellular concentrations of both BpuR protein and bpuR mRNA, since bpuR transcript could act as a sink to affect levels of free protein. If other BpuR‐binding RNAs are also differentially transcribed, then variations in concentration of those targets could likewise alter levels of available BpuR in the bacterial cell. Studies are under way to identify additional BpuR‐binding RNAs and differentially expressed proteins, to address those questions and increase understanding of the functions of this nucleic acid‐binding protein.
Figure 8.
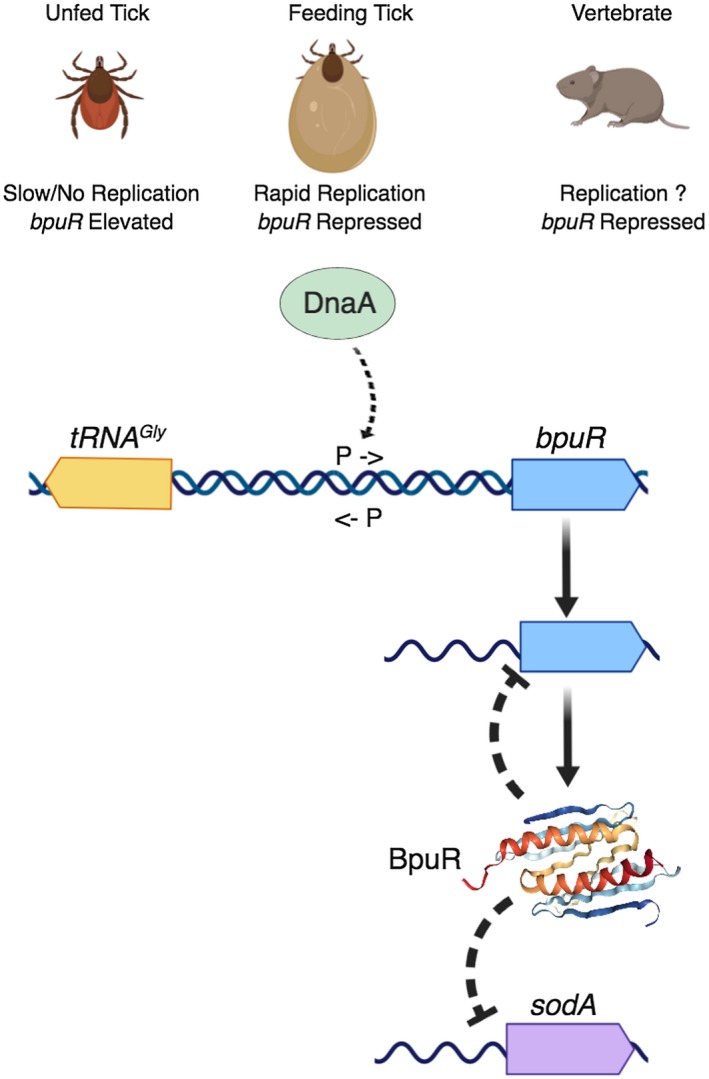
Diagram summarizing results of these and previous studies of B. burgdorferi BpuR. B. burgdorferi within unfed ticks produced high levels of bpuR transcript. As ticks feed, levels of bpuR transcript were reduced. B. burgdorferi within infected vertebrates did not produce detectable levels of bpuR mRNA. The extent to which B. burgdorferi replicates during vertebrate infection is not currently known. The DnaA protein binds to a DNA site that is adjacent to the bpuR transcriptional promoter. The promoter of the divergently transcribed tRNAGly has not been precisely mapped, but spacing indicates that the −10 and −35 sequences must either overlap the bpuR promoter or be located 3′ of the bpuR promoter. The BpuR homodimer was previously found to bind its own mRNA and repress translation (Jutras et al., 2013d). BpuR also binds to sodA mRNA 5′ of the translational start site. Results from the current studies indicate that BpuR inhibits SodA translation. The three‐dimensional image of the BpuR homodimer is adapted from its solved structure, www.rcsb.org/3d-view/3NM7 (Graebsch et al., 2010). Image produced with BioRender (biorender.com).
B. burgdorferi controls production of BpuR on at least two levels: initiation of transcription and posttranscriptional inhibition of mRNA translation. Translational autoregulation prevents the spirochete from overproducing BpuR, suggesting that cellular levels beyond a threshold might be deleterious to the bacterium (Jutras et al., 2013d). The evident impacts on the proteome from genetically manipulating BpuR concentrations support that possibility. Levels of bpuR mRNA change during the borrelial infection cycle, being considerably higher during tick colonization than during mammalian infection. Those differences are consistent with known functions of BpuR. The current studies found that elevated levels of BpuR coincided with reduced levels of SodA, an enzyme that is critical for mammalian infection. By virtue of its ability to also bind DNA, BpuR serves as co‐repressor of erp transcription, and expression of erp genes is repressed during tick colonization and enhanced during infection of mammals (Hefty et al., 2001; Stevenson et al., 2001; Miller et al., 2003; Jutras et al., 2013a). Presumably, induction of BpuR during the current studies did not significantly change erp transcription because cellular levels of the BpaB repressor are reduced during culture at 35°C, while levels of the EbfC anti‐repressor are elevated (Jutras et al., 2012b; 2013c). It is possible that some of the effects seen, or not seen, in these studies of cultured B. burgdorferi may be counteracted or amplified by differences in other proteins and transcripts during certain stages of the natural vertebrate–tick infection cycle. That is a caveat of any study of B. burgdorferi or any other bacterium.
Transcription of bpuR can be modulated by altering culture conditions: temperature or media formulations that reduce bacterial division rate will result in elevated levels of bpuR mRNA (Fig. 1 and (Jutras et al., 2013a). That characteristic is hypothesized to mimic an environmental change that occurs during the borrelial infectious cycle. The midgut of an unfed tick is relatively nutrient‐poor, and B. burgdorferi do not appear to grow or divide to any great extent (Dunham‐Ems et al., 2009). But, as the tick begins to feed on a vertebrate host, ingested blood provides nutrients that allow the bacteria to divide very rapidly (Piesman et al., 1990; 2001; de Silva and Fikrig, 1995; Piesman and Schneider, 2002; Dunham‐Ems et al., 2009). While the vertebrate–tick infectious cycle involves many steps, with numerous different interactions between the bacteria and host components, B. burgdorferi has been following the same tick‐to‐vertebrate‐to‐tick cycle for millennia. This consistent routine appears to have resulted in B. burgdorferi evolving mechanisms to respond to conditions that are indicative of specific points in the cycle. The division rate of B. burgdorferi changes from slow to rapid at only one point: when the tick begins to ingest blood. The discovery that DnaA, the master regulator of chromosomal replication, binds to the bpuR promoter provides a plausible mechanistic link between B. burgdorferi replication and bpuR transcription (Fig. 8).
In addition to serving as the master regulator of chromosomal replication, DnaA proteins of other bacterial species are known to bind multiple sites throughout the genome and affect transcription levels of diverse genes (Messer and Weigel, 1997; Smith and Grossman, 2015; Washington et al., 2017) This study was the first to investigate the B. burgdorferi DnaA protein, and thus the first to identify a DNA segment to which DnaA binds. This region contains a sequence that is similar to the relaxed consensus DnaA box sequence of other bacterial species (Fuller et al., 1984; Moriya et al., 1988). The B. burgdorferi oriC region contains three sites that meet similar criteria (Picardeau et al., 1999). Details of the impacts of DnaA on bpuR transcription remain to be determined. The putative DnaA box is situated between the bpuR −10 and −35 promoter elements (Jutras et al., 2013d), suggesting a promoter occlusion model. Complicating matters, bpuR is divergently transcribed from a tRNA gene, there are 138 bp between the bpuR initiation codon and the first nucleotide that encodes the mature tRNA, and the bpuR TSS is 42 bp 3′ of its initiation codon (Fraser et al., 1997; Jutras et al., 2013d). Those data indicate that the transcriptional promoter of the tRNA is directly adjacent to or overlaps the bpuR promoter (Figs 3A and 8). Elucidation of the impacts of DnaA on bpuR transcription will necessarily include characterization of DnaA's effects on the tRNA locus.
The SodA superoxide dismutase is required for B. burgdorferi to infect vertebrate hosts (Esteve‐Gassent et al., 2009; 2015; Troxell et al., 2012; Aguirre et al., 2013). One hypothesized function of SodA is to detoxify superoxides derived from oxygen in the blood that is ingested by the feeding tick. The evident posttranscriptional regulation of SodA production could enable rapid accumulation of SodA protein, with preformed mRNA becoming accessible for translation upon depletion of the BpuR repressor. BpuR bound specifically to a segment of RNA that is 5′ of the sodA open reading frame. The BpuR‐binding sequence does not include the sodA RBS. Whether BpuR alters mRNA conformation, interacts with additional factors, or has some other effect is currently under investigation. As discussed above, the sodA gene is evidently transcribed from two distinct promoters, producing either a bicistronic mRNA of secA–sodA, or a monocistronic message that encodes only sodA. Both of those transcripts include the sequence that bound BpuR. While altering cellular BpuR concentrations did not significantly affect levels of any borrelial transcript, two‐dimensional electrophoresis indicated that BpuR affects levels of numerous other borrelial proteins (Fig. 4). Presumably, those changes were also mediated through interactions between BpuR and mRNAs. Studies are under way to identify and confirm additional RNA targets of BpuR.
Homologues of BpuR are encoded by many other spirochetes, including members of the genera Treponema and Spirochaeta, and by some other genera, such as Bacteroides and Butyricimonas. Intriguingly, PUR domain proteins are widespread throughout eukaryotes, where they often perform critical regulatory functions through interactions with nucleic acids (Daniel and Johnson, 2018). Results of these and other studies on BpuR suggest that its functions are distinct from the well‐characterized bacterial RNA‐binding protein Hfq (Lybecker et al., 2010; Woodson et al., 2018). There are similarities in the apparent effects of BpuR and SpoVG, another bacterial RNA‐ and DNA‐binding proteins that are produced by all firmicutes and spirochetes (Rosenbluh et al., 1981; Jutras et al., 2013b; Burke and Portnoy, 2016; Savage et al., 2018). Thus, studies of BpuR will influence understanding of the functions of dual RNA/DNA‐binding proteins across much of the Prokaryota.
To summarize, B. burgdorferi controls levels of BpuR by regulating initiation of both transcription and translation (Fig. 8). The master regulator of chromosomal replication, DnaA, binds to a specific sequence in the bpuR transcriptional promoter. This likely ties bpuR transcription to B. burgdorferi replication, consistent with high levels of bpuR transcript in bacteria that divide slowly, and low levels of bpuR under states of rapid replication. Under the tested culture conditions, changes in BpuR levels did not have any significant effects on any borrelial transcript. In contrast, alterations in bacterial BpuR content influenced levels of numerous proteins, one of which was identified as the virulence factor SodA. BpuR binds to sodA mRNA in live B. burgdorferi, 5′ of the RBS. Studies are ongoing to identify other targets of BpuR and define the mechanisms through which it affects borrelial protein levels. Perhaps of greatest importance, these results further emphasize that B. burgdorferi controls its protein content through multiple mechanisms, and transcript levels do not necessarily reflect protein levels.
Experimental procedures
Bacteria, plasmids and culture conditions
B. burgdorferi strain B31‐e2 is a readily‐transformable clone of type strain B31 (Casjens et al., 1997; Babb et al., 2004). Strain KS10 was previously produced by transforming B31‐e2 with plasmid pBLS590, which contains a promoterless gfp locus (Babb et al., 2004). Strain KS20 was similarly produced by transformation with pBLS599, which contains a minimal promoter element fused to gfp (Babb et al., 2004). Strain BJ26 was produced by transformation of B31‐e2 with pGJ1, which contains a transcriptional fusion between the bpuR promoter and gfp (Jutras et al., 2013d).
All other studies used the B31 culture that has been completely sequenced, B31‐MI (Fraser et al., 1997; Casjens et al., 2000). Strain B31‐MI contains 21 distinct, naturally occurring small DNA replicons (plasmids), lacking the ancestral cp32‐2, cp32‐5 and cp9‐2 (Casjens et al., 1997; 2000; Miller et al., 2000). It is fully infectious to both mammals and ticks (Miller et al., 2003).
The effects of increased BpuR levels on the B. burgdorferi transcriptome and proteome were examined through use of an inducible bpuR locus. Plasmid pWA10 was produced from pJSB268 (Blevins et al., 2007), by replacing the luc gene with bpuR. This placed bpuR under the transcriptional control of the IPTG‐inducible lac promoter. B. burgdorferi B31‐MI clone A3 was stably transformed with pWA10 (Samuels, 1995; Elias et al., 2002).
The initial studies on the impacts of elevated BpuR concentrations were performed using a plasmid construct that puts bpuR under control of a constitutive promoter. Plasmid pBLS715 was derived from cloning vector pBSV2‐G by removing the multiple cloning sequence region, then inserting an RBS and restriction endonuclease cleavage sites immediately 3′ of aac1, the gentamicin resistance‐encoding gene (Elias et al., 2003; Byram et al., 2015). Open reading frames cloned into those sites are transcribed by RNA polymerase initiating from the constitutive PflgB promoter that is 5′ of aac1. The bpuR gene was inserted into pBLS715 to produce pBLJ307. That chimeric plasmid was stably introduced into B. burgdorferi strain B31‐MI clone 5A4NP1 (Kawabata et al., 2004).
Unless otherwise indicated, all B. burgdorferi were cultivated at 35°C in complete Barbour–Stoenner–Kelly II medium (BSK‐II) with 6% (vol/vol) rabbit serum (Zückert, 2007). Two formulations of incomplete BSK‐II were used for nutrient shift studies: fivefold reduced serum (the standard 6% rabbit serum was replaced with 1.2% rabbit serum), and fourfold diluted medium (BSK‐II was diluted fourfold with PBS, while rabbit serum was added to the standard 6%) (Jutras et al., 2013c). Culture densities were determined by counting bacteria with a Petroff–Hausser chamber and darkfield microscopy.
Flow cytometry
B. burgdorferi strain BJ26, which contains the PbpuR::gfp transcriptional fusion, was cultured to mid‐exponential phase (107 bacteria/ml) in either complete BSK‐II or one of the two incomplete formulations. B. burgdorferi strains KS10 and KS20 were similarly grown in complete BSK‐II and used as negative and positive controls, respectively. Bacteria were collected by centrifugation, washed and then resuspended in PBS. GFP levels in individual bacteria were detected by use of a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA), with excitation at 488 nm and detection at 530 nm (Babb et al., 2004). Analyses were performed using FlowJo10. Results were compared by two‐way ANOVA. At least 10,000 events were counted for each sample.
Quantitative Reverse Transcription PCR (qRT‐PCR) analyses of mRNA expression during mammalian and tick colonization
Relative expression levels of bpuR during tick and mouse infection were determined using the oligonucleotide primers listed in Supplemental Table S2.
For analyses of transcript levels during tick colonization, naive I. scapularis larvae were colonized by feeding to repletion on mice that were infected with B. burgdorferi B31‐MI clone A3 (Elias et al., 2002). Engorged ticks were allowed to molt to nymphs, then fed upon naïve mice. Ticks were collected at specific time points, snap‐frozen in liquid nitrogen and stored at −80°C until use. RNA was isolated from pools of ticks, with each pool containing the following numbers of ticks: 10 fed larvae/pool; 10–20 unfed nymphs/pool; 2–5 fed nymphs/pool; and 10 nymphs 1 week postfeeding/pool. RNA was isolated from pools of frozen ticks by mechanical disruption as follows: ticks were placed in 100 µl PBS in a 1.5 ml tube and crushed with a disposable pestle. RNA was purified using Nucleospin kits (Macherey‐Nagel Co., Bethlehem, PA) per manufacturer's recommendations, or TRIzol reagent (Invitrogen, Carlsbad, CA). After DNase treatment, 500 ng of RNA was converted to cDNA using High‐Capacity cDNA Reverse Transcription kits (Applied Biosystems, Foster City, CA) as previously described (Stewart et al., 2012). The cDNA samples were diluted 1:20 with water and 5 µl of each was used in the subsequent quantitative PCR reactions. Three biological replicates (independent pools of ticks) were assessed in triplicate technical replicates for gene expression by qRT‐PCR as previously described (Stewart et al., 2008). A standard curve was generated from genomic DNA, representing a range of 106 cells to 10 cells (in 10‐fold serial dilutions) using the Ct values from the flaB primer/probe set. This standard curve was then used to interpolate the bpuR transcript copy number from the Ct values generated by the bpuR primer/probe set. Transcript levels of bpuR were normalized to copies of flaB transcript. Negative controls lacking reverse transcriptase confirmed that all genomic DNA had been degraded and did not contribute to the signal.
For analyses of transcript levels during mammalian infection, four female 4–6 week old C3H/HeN mice were infected by subcutaneous injection of 1 x 106 B31‐MI clone 16 (Miller et al., 2003), from a mid‐exponential‐phase culture. Two weeks post infection, mouse blood was drawn from the saphenous vein, processed to serum and presence of B. burgdorferi‐specific antibodies were determined by enzyme‐linked immunosorbent assay (ELISA) as previously described (Floden et al., 2013). Mice were euthanized after 4 weeks of infection, urinary bladders collected, then flash frozen in liquid nitrogen and stored at −80°C until use. Bladders were chosen because they are regularly colonized by B. burgdorferi (Schwan et al., 1988). Frozen tissues were ground with mortars and pestles, followed by homogenization (PRO Scientific) in TRIzol reagent on ice. RNA was resuspended in RNAsecure reagent (Ambion) and treated with DNase I (Ambion) to remove contaminating DNA. DNase was inactivated using DNase Inactivation Reagent (Ambion). Aliquots of each RNA preparation was reverse transcribed using First Strand cDNA synthesis kits (Life Technologies) with random hexamers. Quantitative PCR was performed using a BioRad myIQ2 Real‐Time PCR instrument. Briefly, cDNA or diluted genomic DNA (see below) was added to an 20 μl master mixture containing 2x SybrGreen Supermix (BioRad) and oligonucleotide primers (0.5 µM final concentration), and nuclease‐free water (Ambion). All cDNA samples were analyzed in triplicate. Each qRT‐PCR run included negative controls of RNA processed without RTase to test for DNA contamination of each RNA preparation and samples that lacked template to test for DNA contamination of reagents. Tenfold serial dilutions of B31‐MI‐16 genomic DNA (100 ng–100 fg) were included in every assay for each primer set. This generated standard curves from which the amount of transcript present in each cDNA sample could be calculated. Average expression values obtained from triplicate runs of each cDNA sample for bpuR transcript were calculated relative to the average triplicate value for the constitutively expressed B. burgdorferi flaB transcript from the same cDNA preparation (Miller, 2005).
All animal studies were performed with approval from, and under supervision of, Institutional Animal Care and Use Committees of the University of Kentucky, University of North Dakota and Rocky Mountain Laboratories, NIH.
Electrophoretic mobility shift assays
Nucleic acids utilized as probes or competitors are described in Supplemental Tables S2 and S3. Double‐stranded DNA probes were generated by PCR of cloned genomic DNAs, by use of Q5 polymerase (New England Biolabs), a 5′‐end biotinylated primer and an unlabeled primer (IDT, Coralville, IA). Each PCR amplicon was purified by agarose gel electrophoresis, the amplicon extracted and purified, then re‐amplified before use as an EMSA probe.
DNA and RNA EMSAs were performed essentially as described previously (Babb et al., 2006; Riley et al., 2009; Jutras et al., 2013d; Savage et al., 2018). Binding reactions took place in EMSA binding buffer (50 mM Tris, 1 mM EDTA, 1 mM DTT, 10% Glycerol, 2.4% Protease inhibitor cocktail and 0.6% phosphatase inhibitor cocktail) with 1nM biotin‐labeled DNA probe and either cellular extract or purified protein. Where appropriate, competitor DNAs were included to a final concentration of 100nM (100‐fold excess). After incubation at room temperature for 20 min, 2.5 µl of loading dye (15% w/v Ficol, 0.04% w/v Orange G) was mixed with the reaction, and 5.5 µl of this was electrophoresed through 6% TBE acrylamide gels (ThermoFisher) for 60 min at 100V. DNAs were then transferred to nylon Biodyne B membranes (ThermoFisher), then crosslinked to the membrane using a Stratagene UV Crosslinker (Stratagene, San Diego, CA). Biotin‐labeled DNAs were visualized using Chemiluminescent Nucleic Acid Detection Modules (ThermoFisher) and autoradiography.
DNA affinity chromatography
DNA affinity chromatography was performed essentially as previously described (Jutras et al., 2012c). The bpuR promoter and open reading frame, consisting of nucleotides 46,019–46,483 of the B. burgdorferi B31 main chromosome (Fraser et al., 1997) was PCR amplified using primers BiobpuRP‐1 and bpuRP‐2 (Table 1). Purified probe was adhered to streptavidin‐coated Dynal magnetic beads (ThermoFisher). Cell extracts from mid‐exponential phase (1 × 107 cells/ml) cultures of B. burgdorferi were prepared and mixed with the DNA‐bound magnetic beads. Poly‐dI‐dC was included at a final concentration of 10 µg/ml, to compete for nonspecific DNA‐binding factors. The beads were then washed repeatedly in buffer BS/THES with 10 µg/ml poly‐dI‐dC. Proteins were eluted from DNA by washing in buffer with increasing concentrations of NaCl. Eluted fractions were separated by SDS‐polyacrylamide gel electrophoresis, and proteins visualized by staining with Sypro Ruby (Sigma, St. Louis, MO). Protein bands were excised and submitted to the University of Kentucky Mass Spectrometry core for identification.
Mass spectrometry
Proteins in polyacrylamide gels were identified by previously described methods (Babb et al., 2006; Burns et al., 2010; Jutras et al., 2013a; 2013b). Briefly, a piece of gel containing the protein in question was excised, subjected to dithiothreitol reduction, iodoacetamide alkylation and in‐gel trypsin digestion using a standard protocol. The tryptic peptides were subjected to shot‐gun proteomics analysis (Yang et al., 2014). Liquid chromatography MS/MS analysis was performed using an LTQ‐Orbitrap mass spectrometer (ThermoFisher) coupled with an Eksigent Nanoflex cHiPLC system (Eksigent, Dublin, CA) through a nanoelectrospray ionization source. The peptide samples were separated with a reversed phase cHiPLC column. The mass analysis method consisted of one segment with eight scan events. The first scan event was an Orbitrap MS scan (300‐1800 m/z) with 60,000 resolution for parent ions followed by data dependent MS/MS for fragmentation of the seven most intense multiple charged ions with collision‐induced dissociation (CID) method.
LC‐MS/MS data were submitted to a local Mascot server for MS/MS protein identification via Proteome Discoverer 1.3 (ThermoFisher) against a custom database containing 990 reviewed proteins of Borrelia burgdorferi strain B31 (downloaded 08/19/2015) (http://www.matrixscience.com). Typical parameters used in the Mascot MS/MS ion search were: trypsin digest with maximum of two mis‐cleavages, cysteine carbamidomethylation, methionine oxidation, a maximum of 10 ppm MS error tolerance, and a maximum of 0.8 Da MS/MS error tolerance. A decoy database was built and searched. Filter settings that determine false discovery rates (FDR) are used to distribute the confidence indicators for the peptide matches. Peptide matches that pass the filter associated with the FDR rate of 1% and 5% are assigned as high and medium confident peptides, respectively.
Recombinant protein expression and purification
Recombinant BpuR was produced from the previously described construct (Jutras et al., 2013a). Using oligonucleotide primers that are listed in Supplemental Table S2, the B. burgdorferi dnaA gene was PCR amplified using Q5 polymerase (New England Biolabs, Ipswitch, MA), then cloned into pET200 (ThermoFisher). The insert of the resulting plasmid was sequenced to ensure that no mutations had been introduced. E. coli Rosetta 2 DE3 pLysS (Millipore, Billerica, MA) were transformed with either a BpuR‐ or DnaA‐producing plasmid.
Cultures were induced with 0.5 mM IPTG, bacteria harvested by centrifugation and pellets frozen at −80°C. Bacteria were thawed and resuspended in 20 ml of ice‐cold washing/binding buffer (100 mM HEPES, 10 mM imidazole, pH = 7.5). Pierce protease inhibitor mini tablets, EDTA free (ThermoFisher) were added at a ratio of 1 tablet per 10 ml of cell suspension. Bacterial suspensions were lysed by sonication with a Branson 102C sonicator (Emerson, St. Louis, MO) at amplitude of 20% for five cycles of 15 s. Cellular debris was pelleted by centrifugation at 4°C for 30 min at 27,000× g. Soluble fractions were incubated with MagneHis beads (Promega, Madison, WI) at 4°C for 1 h. Beads were pulled down by magnet, the supernatant was decanted and then the beads were washed with fresh washing/binding buffer. This was repeated 5 times. Remaining bound proteins were eluted with 100 mM HEPES, 500 mM imidazole, pH = 7.5. Eluted protein suspensions were dialyzed overnight in 10K MWCO (ThermoFisher) cassettes against EMSA dialysis buffer (10% v/v Glycerol, 0.5 mM EDTA, 50 mM Tris, 0.1% PMSF, 0.01% Tween‐20, 50µM KCl, 1 mM DTT, pH = 8.0). Dialyzed samples were concentrated using Amicon 50K Ultra Centrifugal Units (Millipore). Concentrated protein was aliquoted, assessed for purity by SDS‐PAGE with Coomassie brilliant blue staining and then frozen at −80°C.
RNA‐sequencing, analysis and validation
B. burgdorferi harboring the BpuR‐inducible construct pWA10 were inoculated from thawed −80°C stocks were inoculated into BSK‐II, then grown at 34°C until reaching mid‐exponential phase (~1 × 107 bacteria/ml). Equal aliquots were passaged into six bottles of 20 ml fresh BSK‐II, to a final density of 1 × 105 cells/ml, and placed at 34°C. Upon reaching ~1 × 106 cells/ml, IPTG was added to three of the cultures, to a final concentration of 0.5 mM. All cultures were further incubated at 34°C until bacterial densities reached 1 × 107 cells/ml (approximately 24 h). Bacteria were harvested by centrifugation, pellets were suspended in 60°C TRIzol (Ambion, Foster City, CA), then frozen at −80°C. Bacteria‐TRIzol suspensions were thawed at room temperature. RNA was isolated using Zymo RNA Direct‐Zol miniprep kits (Zymo, Irvine, CA) and eluted into RNase‐free water. RNA integrity was assayed by microfluidic analysis using an Agilent 2100 Bioanalyzer and Agilent RNA 6000 Nano chips (Agilent, CA USA). All samples assayed were of high quality with RIN score >9. RNA concentration was determined using a Nanodrop 2000 (ThermoFisher). Purified RNAs were stored at −80°C.
Illumina cDNA libraries were generated using the RNAtag‐Seq protocol (Shishkin et al., 2015; Arnold et al., 2016; 2018), with minor modifications. Briefly, 840 ng of total RNA was fragmented, depleted of genomic DNA, dephosphorylated, then ligated to DNA adapter barcodes. Barcoded RNAs were pooled and depleted of rRNA using RiboZero (Illumina, San Diego, CA). Pools of barcoded RNAs were converted to Illumina cDNA libraries in three main steps: (i) reverse transcription of the RNA by priming a constant region of the barcoded adaptor; (ii) degradation of the RNA followed by template switching addition of a second poly G priming site; and (iii) PCR amplification using primers that target the constant regions of the 3′ ligated adapter and 5′ poly‐G tract and contain the full sequence of the Illumina sequencing adaptors. cDNA libraries were sequenced on an Illumina Nextseq 500 (Illumina, San Diego, CA) in the paired end configuration for 75 cycles.
For the analyses of RNAtag‐Seq data, reads from each sample in the pool were identified and deconvoluted, based on their associated barcode, using custom scripts (Shishkin et al., 2015). Up to one mismatch in the barcode was allowed, with the caveat that it did not allow assignment to more than one barcode. Quality of reads was assessed prior to quality trimming using FastQC (v0.11.5) (Andrews, 2010). Paired FASTQ files were trimmed of low‐quality sequences and reads before being filtered to remove reads which contained less than 25 bases using Trimmomatic (v0.36) (Bolger et al., 2014). A custom transcriptome file described previously was indexed using the Salmon‐index function set for quasi mapping with default settings and auto library detection (v0.8.2) (Patro et al., 2017). Mapping and counting was conducted using Salmon (v.0.8.2) in quasi mode with seqBias and GCbias flags activated. Data mapping indicated that the analyzed cultures contained all of the naturally occurring plasmids of the parental strain.
All intermediary data and code used in these studies are available on Figshare at 10.6084/m9.figshare.5594488. Raw Illumina sequencing reads were deposited to SRA under BioProject PRJNA422408.
Quantitative RT‐PCR
Independently grown cultures of the above‐described B. burgdorferi that carry the BpuR‐inducible BpuR construct were similarly incubated with or without IPTG. Bacteria were harvested by centrifugation and then resuspended in 60°C TRIzol (ThermoFisher). Suspensions were processed with Zymo Mini‐zol RNA isolation kits (Zymo), then eluted into 35 µl of nuclease free H2O. RNA was depleted of genomic DNA using Ambion Turbo DNase (ThermoFisher), using the ‘high genomic DNA contamination’ protocol. The qRT‐PCR method was performed essentially as described (Miller, 2005; Arnold et al., 2016). Briefly, purified RNAs were converted to cDNA using iScript cDNA synthesis kits (BioRad, Hercules, CA). Then, cDNAs were diluted 1:20 with nuclease free H2O. qPCR was performed using iTaq Syber Green Supermix (BioRad) on a BioRad CFX96 Touch Real‐time PCR detection platform (BioRad). Primers for qPCR were designed for gene‐specific targets using the IDT Primer Quest tool on default settings (IDT, Coralville, IA) (Table 1). Technical triplicates were performed for all assays. Cycling was performed as follows; 94°C for 3 min, 40 cycles at 94°C for 10 s, followed by 30 s at 60°C. Melt curves were performed for all assays to validate the amplification of single specific products. Each assay was performed with RNA treated identically but lacking reverse transcriptase, to ensure the total depletion of genomic DNA. Ct values were normalized to ftsK transcript using the ∆Ct method and fold changes between conditions were calculated using the ∆∆Ct method (Schmittgen and Livak, 2008).
Two‐dimensional gel electrophoresis
Two‐dimensional gel electrophoresis was performed essentially as described previously (Babb et al., 2005). Briefly, B. burgdorferi were resuspended in equilibration buffer and loaded on to precast IPG strips (BioRad) (pH 4–7). Strips were subject to isoelectric focusing for 3000 V‐h. Strips were equilibrated and loaded across the tops of 12.5% SDS‐polyacrylamide gels, and proteins separated by electrophoresis. Gels were stained with Sypro Ruby (Sigma) and imaged using transmitted ultraviolet light. Select spots were excised and submitted to the University of Kentucky Mass Spectrometry core for identification (see above).
Immunoblot analyses
Immunoblots were performed essentially as described previously (Stevenson et al., 1995). For evaluation of the effects of elevated production of BpuR, B. burgdorferi were incubated with or without IPTG, as described above. Before cell harvesting, cultures were split, with one aliquot prepared for qRT‐PCR and the other for immunoblot analyses. Primary antibodies consisted of rabbit polyclonal anti‐BpuR serum (Jutras et al., 2013a) (1:100), mouse polyclonal anti‐SodA serum (Esteve‐Gassent et al., 2009) or mouse monoclonal anti‐FlaB (Barbour et al., 1986). Bound antibodies were detected with fluorescence‐conjugated secondary antibodies and an Odyssey CLx imaging system (LiCor, Lincoln, NE). Band intensities were analyzed with Image J (Schneider et al., 2012).
Bacterial sensitivity to reactive oxygen
B. burgdorferi carrying the inducible bpuR construct were cultured to mid‐exponential phase (approximately 107 bacteria/ml) either with or without inclusion of 0.5 mM IPTG. Bacteria were harvested by centrifugation and washed 3 times with PBS. Aliquots of each culture were resuspended in PBS + 20 mM methyl viologen and incubated for 2 h at room temperature (Esteve‐Gassent et al., 2009). Control aliquots of each culture were incubated in PBS alone. Bacteria were then washed in PBS, and viability assessed by bacterial LIVE/DEAD stain (ThermoFisher). Cells were analyzed by flow cytometry using a CytoFLEX LX (Beckman Coulter) with 488 nm excitation. Optical filters were set up such that red fluorescence was measured above 630 nm and green fluorescence was measured at 520 nm. For each aliquot, 100,000 events were analyzed. The live and dead cells were counted, and proportion of dead cells calculated. The proportions were graphed from three independent cultures. Results were compared by unpaired t‐test.
RNA immunoprecipitation (RIP)
Targeted RNA immunoprecipitation was performed essentially as described previously (Jutras et al., 2012a). B. burgdorferi were cultured at 34°C to mid‐exponential phase (approximately 5 × 107 bacteria/ml). Formaldehyde was added to the cultures at a final concentration of 1% and then incubated for 8 min at room temperature while shaking. Crosslinking was stopped by addition of glycine to a final concentration of 0.3 M. Bacteria were pelleted by centrifugation and washed twice with Tris‐buffered saline (20 mM Tris [pH7.5], 150 mM NaCl). Cell pellets were frozen at −80°C and resuspended in a 1:4 ratio of lysis buffer:immunoprecipitation (IP) buffer. Lysozyme was added to a final concentration of 10 mg/ml. To shear bacterial RNA, lysates were sonicated using a Branson 102C sonicator (Branson Ultrasonics) to shear the RNA, with 10 pulses of 15 s each at 15% amplitude. RIP was performed using Protein G magnetic beads following manufacturer's recommended procedures (Active Motif, Carlsbad CA). Following shearing and centrifugation to remove cellular debris, 20 µl of BpuR‐specific antiserum was added directly to 800 µl of cleared supernatant and rocked overnight at 4°C. As controls, BpuR‐specific antiserum, anti‐IgG control antibodies (Santa Cruz) or PBS alone (bead control) were individually incubated with cleared lysate under the same conditions. To immunoprecipitate antibody complexes, 50 µl of Protein G magnetic beads were added and rocked for 2 h at room temperature. The formaldehyde crosslink was reversed in RNase‐free Tris‐EDTA buffer, by heating at 75°C for 10 min. Eluted nucleic acids were treated for 30 min with Turbo DNase I (Ambion, Waltham, MA), and purified using Epicenter RNA MasterPure kits (Illumina). The DNase and RNA purification steps were repeated for a total of 2 times. Complementary DNA synthesis was performed using Transcript First‐Strand cDNA synthesis (Roche, Madison, WI). Enriched fractions were subjected to qPCR as described above. All materials were treated with DEPC prior to use and each step included 0.5 U/mL of RiboGuard (ThermoFisher).
Targeted RIP is a simple and efficient technique to determine whether a particular RNA of interest is included among the immunoprecipitate (Jutras et al., 2012a). Specificity of RIP is ascertained by PCR of cDNA using primers specific for additional transcripts. The possibility of immunoprecipitation of protein–DNA complexes (i.e. chromosomal immunoprecipitation, ChIP) is assessed by PCR of precipitate that was not subjected to reverse transcription. For the current studies, oligonucleotide primers that are specific for the sodA, dnaK or gap open reading frames were used in separate PCR reactions with the BpuR RIP eluate, RIP control cDNAs, ChIP DNAs or purified B. burgdorferi strain B31 genomic DNA. Amplicons were separated by agarose gel electrophoresis, stained with ethidium bromide and imaged.
Conflicts of interest
The authors declare that they do not have any conflicts of interest.
Supporting information
Acknowledgements
The authors thank Paula Schlax for funding assistance; Ashutosh Verma, Walter Finch, Amy Bowman, Alicia Chenail, Alyssa Antonicello and Timothy Saylor for experimental assistance; Jonathan Livny and Jessica Alexander for their assistance with RNA‐Seq library construction and sequencing; Michael Fried for assistance in design and interpretation of electrophoretic mobility shift assays; Jing Chen and the University of Kentucky Proteomics Core Facility for proteomic identification; Jon Blevins for providing plasmid pJSB268; and Timothy Casselli, Timothy Saylor and Paula Schlax for helpful comments on the manuscript.
These studies were funded by grants from the National Institutes of Heath: R21AI120602 (to B. Stevenson), R21AI139956 (to B. Stevenson, C.A. Brissette, W.R. Zückert, and P. Schlax), R03AI113648 (to B. Stevenson, J. Seshu, and J. Livny) and P20GM113123 (to C.A. Brissette); and by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health (to P.E. Stewart, K. Tilly, A. Bestor, and P.A Rosa). The Orbitrap mass spectrometer was acquired by High‐End Instrumentation Grant S10RR029127 (to H. Zhu). The University of Kentucky Flow Cytometry & Immune Function core facility is supported in part by the Office of the Vice President for Research, the Markey Cancer Center and an NCI Center Core Support Grant (P30 CA177558) to the University of Kentucky Markey Cancer Center.
References
- Adams, P.P. , Flores Avile, C. , Popitsch, N. , Bilusic, I. , Schroeder, R. , Lybecker, M. , et al (2017) In vivo expression technology and 5′ end mapping of the Borrelia burgdorferi transcriptome identify novel RNAs expressed during mammalian infection. Nucleic Acids Research, 45, 775–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre, J.D. , Clark, H.M. , McIlvin, M. , Vazquez, C. , Palmere, S.L. , Grab, D.J. , et al (2013) A manganese‐rich environment supports superoxide dismutase activity in a Lyme disease pathogen, Borrelia burgdorferi . Journal of Biological Chemistry, 288, 8468–8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews, S. (2010) FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- Arnold, W.K. , Savage, C.R. , Brissette, C.A. , Seshu, J. , Livny, J. and Stevenson, B. (2016) RNA‐Seq of Borrelia burgdorferi in multiple phases of growth reveals insights into the dynamics of gene expression, transcriptome architecture, and noncoding RNAs. PLoS ONE, 11, e0164165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold, W.K. , Savage, C.R. , Lethbridge, K.G. , Smith, T.C. , Brissette, C.A. , Seshu, J. , et al (2018) Transcriptomic insights on the virulence‐controlling CsrA, BadR, RpoN, and RpoS regulatory networks in the Lyme disease spirochete. PLoS ONE, 13, e0203286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babb, K. , Bykowski, T. , Riley, S.P. , Miller, M.C. , DeMoll, E. and Stevenson, B. (2006) Borrelia burgdorferi EbfC, a novel, chromosomally‐encoded protein, binds specific DNA sequences adjacent to erp loci on the spirochete's resident cp32 prophages. Journal of Bacteriology, 188, 4331–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babb, K. , von Lackum, K. , Wattier, R.L. , Riley, S.P. and Stevenson, B. (2005) Synthesis of autoinducer 2 by the Lyme disease spirochete, Borrelia burgdorferi . Journal of Bacteriology, 187, 3079–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babb, K. , McAlister, J.D. , Miller, J.C. and Stevenson, B. (2004) Molecular characterization of Borrelia burgdorferi erp promoter/operator elements. Journal of Bacteriology, 186, 2745–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour, A.G. , Hayes, S.F. , Heiland, R.A. , Schrumpf, M.E. and Tessier, S.L. (1986) A Borrelia‐specific monoclonal antibody binds to a flagellar epitope. Infection and Immunity, 52, 549–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergemann, A.D. , Ma, Z.W. and Johnson, E.M. (1992) Sequence of cDNA comprising the human pur gene and sequence‐specific single‐stranded‐DNA‐binding properties of the encoded protein. Molecular and Cellular Biology, 12, 5673–5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blevins, J.S. , Revel, A.T. , Smith, A.H. , Bachlani, G.N. and Norgard, M.V. (2007) Adaptation of a luciferase gene reporter and lac expression system to Borrelia burgdorferi . Applied and Environmental Microbiology, 73, 1501–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bontemps‐Gallo, S. , Gaviard, C. , Richards, C.L. , Kentache, T. , Raffel, S.J. , Lawrence, K.A , et al. (2018) Global profiling of lysine acetylation in Borrelia burgdorferi B31 reveals Its role in central metabolism. Frontiers in Microbiology, 9 Available at: 10.3389/fmicb.2018.02036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke, T.P. and Portnoy, D.A. (2016) SpoVG is a conserved RNA‐binding protein that regulates Listeria monocytogenes lysozyme resistance, virulence, and swarming motility. mBio, 7, e00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns, L.H. , Adams, C.A. , Riley, S.P. , Jutras, B.L. , Bowman, A. , Chenail, A.M. , et al (2010) BpaB, a novel protein encoded by the Lyme disease spirochete's cp32 prophages, binds to erp Operator 2 DNA. Nucleic Acids Research, 38, 5443–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bykowski, T. , Babb, K. , von Lackum, K. , Riley, S.P. , Norris, S.J. and Stevenson, B. (2006) Transcriptional regulation of the Borrelia burgdorferi antigenically variable VlsE surface protein. Journal of Bacteriology, 188, 4879–4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byram, R. , Gaultney, R.A. , Floden, A.M. , Hellekson, C. , Stone, B.L. , Bowman, A. , et al (2015) Borrelia burgdorferi RevA significantly affects pathogenicity and host response in the mouse model of Lyme disease. Infection and Immunity, 83, 3675–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll, J.A. , Stewart, P.E. , Rosa, P. , Elias, A.F. and Garon, C.F. (2003) An enhanced GFP reporter system to monitor gene expression in Borrelia burgdorferi . Microbiology, 149, 1819–1828. [DOI] [PubMed] [Google Scholar]
- Casjens, S. , Palmer, N. , van Vugt, R. , Huang, W.M. , Stevenson, B. , Rosa, P. , et al (2000) A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs of an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi . Molecular Microbiology, 35, 490–516. [DOI] [PubMed] [Google Scholar]
- Casjens, S. , van Vugt, R. , Tilly, K. , Rosa, P.A. and Stevenson, B. (1997) Homology throughout the multiple 32‐kilobase circular plasmids present in Lyme disease spirochetes. Journal of Bacteriology, 179, 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel, D.C. and Johnson, E.M. (2018) PURA, the gene encoding Pur‐alpha, member of an ancient nucleic acid‐binding protein family with mammalian neurological functions. Gene, 643, 133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulebohn, D.P. , Hayes, B.M. and Rosa, P.A. (2014) Global repression of host‐associated genes of the Lyme disease spirochete through post‐transcriptional modulation of the alternative sigma factor RpoS. PLoS ONE, 9, e93141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham‐Ems, S.M. , Caimano, M.J. , Pal, U. , Wolgemuth, C.W. , Eggers, C.H. , Balic, A. , et al (2009) Live imaging reveals a biphasic mode of dissemination of Borrelia burgdorferi within ticks. Journal of Clinical Investigation, 119, 3652–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias, A.F. , Bono, J.L. , Kupko, J.J. , Stewart, P.E. , Krum, J.G. and Rosa, P.A. (2003) New antibiotic resistance cassettes suitable for genetic studies in Borrelia burgdorferi . Journal of Molecular Microbiology and Biotechnology, 6, 29–40. [DOI] [PubMed] [Google Scholar]
- Elias, A.F. , Stewart, P.E. , Grimm, D. , Caimano, M.J. , Eggers, C.H. , Tilly, K. , et al (2002) Clonal polymorphism of Borrelia burgdorferi strain B31 MI: implications for mutagenesis in an infectious strain background. Infection and Immunity, 70, 2139–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteve‐Gassent, M.D. , Elliott, N.L. and Seshu, J. (2009) sodA is essential for virulence of Borrelia burgdorferi in the murine model of Lyme disease. Molecular Microbiology, 71, 594–612. [DOI] [PubMed] [Google Scholar]
- Esteve‐Gassent, M.D. , Smith, T.C. , Small, C.M. , Thomas, D.P. and Seshu, J. (2015) Absence of sodA increases the levels of oxidation of key metabolic determinants of Borrelia burgdorferi . PLoS ONE, 10, e0136707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floden, A.M. , Gonzalez, T. , Gaultney, R.A. and Brissette, C.A. (2013) Evaluation of RevA, a fibronectin‐binding protein of Borrelia burgdorferi, as a potential vaccine candidate for Lyme disease. Clinical and Vaccine Immunology, 20, 892–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser, C.M. , Casjens, S. , Huang, W.M. , Sutton, G.G. , Clayton, R. , Lathigra, R. , et al (1997) Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi . Nature, 390, 580–586. [DOI] [PubMed] [Google Scholar]
- Fried, M.G. (1989) Measurement of protein‐DNA interaction parameters by electrophoresis mobility shift assay. Electrophoresis, 10, 366–376. [DOI] [PubMed] [Google Scholar]
- Fuller, R.S. , Funnel, B.E. and Kornberg, A. (1984) The DnaA protein complex with the E. coli chromosomal origin (oriC) and other sites. Cell, 38, 889–900. [DOI] [PubMed] [Google Scholar]
- Graebsch, A. , Roche, S. , Kostrewa, D. , Söding, J. and Niessing, D. (2010) Of bits and bugs ‐ on the use of bioinformatics and a bacterial crystal structure to solve a eukaryotic repeat‐protein structure. PLoS ONE, 5, e13402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graebsch, A. , Roche, S. and Niessing, D. (2009) X‐ray structure of Pur‐α reveals a Whirly‐like fold and an unusual nucleic acid binding surface. Proceedings of the National Academy of Sciences of the United States of America, 106, 18521–18526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefty, P.S. , Jolliff, S.E. , Caimano, M.J. , Wikel, S.K. , Radolf, J.D. and Akins, D.R. (2001) Regulation of OspE‐related, OspF‐related, and Elp lipoproteins of Borrelia burgdorferi strain 297 by mammalian host‐specific signals. Infection and Immunity, 69, 3618–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellman, L.M. and Fried, M.G. (2007) Electrophoretic mobility shift assay (EMSA) for detecting protein‐nucleic acid interactions. Nature Protocols, 2, 1849–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer, R. , Caimano, M.J. , Luthra, A. , Axline, D. , Corona, A. , Iacobas, D.A. , et al (2015) Stage‐specific global alterations in the transcriptomes of Lyme disease spirochetes during tick feeding and following mammalian host‐adaptation. Molecular Microbiology, 95, 509–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutras, B.L. , Bowman, A. , Brissette, C.A. , Adams, C.A. , Verma, A. , Chenail, A.M. , et al (2012a) EbfC (YbaB) is a new type of bacterial nucleoid‐associated protein, and a global regulator of gene expression in the Lyme disease spirochete. Journal of Bacteriology, 194, 3395–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutras, B.L. , Chenail, A.M. , Carroll, D.W. , Miller, M.C. , Zhu, H. , Bowman, A. , et al (2013a) Bpur, the Lyme disease spirochete's PUR‐domain protein: identification as a transcriptional modulator and characterization of nucleic acid interactions. Journal of Biological Chemistry, 288, 26220–26234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutras, B.L. , Chenail, A.M. , Rowland, C.L. , Carroll, D. , Miller, M.C. , Bykowski, T. , et al (2013b) Eubacterial SpoVG homologs constitute a new family of site‐specific DNA‐binding proteins. PLoS ONE, 8, e66683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutras, B.L. , Chenail, A.M. and Stevenson, B. (2013c) Changes in bacterial growth rate govern expression of the Borrelia burgdorferi OspC and Erp infection‐associated surface proteins. Journal of Bacteriology, 195, 757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutras, B.L. , Jones, G. , Verma, A. , Brown, N.A. , Antonicello, A.D. , Chenail, A.M. , et al (2013d) Post‐transcriptional autoregulation of the Lyme disease bacterium's BpuR DNA/RNA‐binding protein. Journal of Bacteriology, 195, 4915–4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutras, B.L. , Verma, A. , Adams, C.A. , Brissette, C.A. , Burns, L.H. , Whetstine, C.R. , et al (2012b) BpaB and EbfC DNA‐binding proteins regulate production of the Lyme disease spirochete’s infection‐associated Erp surface proteins. Journal of Bacteriology, 194, 778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutras, B.L. , Verma, A. and Stevenson, B. (2012c) Identification of novel DNA‐binding proteins using DNA‐affinity chromatography/pull down. Current Protocols in Microbiology, 1F, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karna, S.L.R. , Sanjuan, E. , Esteve‐Gassent, M.D. , Miller, C.L. , Maruskova, M. and Seshu, J. (2011) CsrA modulates levels of lipoproteins and key regulators of gene expression critical for pathogenic mechianisms of Borrelia burgdorferi . Infection and Immunity, 79, 732–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata, H. , Norris, S.J. and Watanabe, H. (2004) BBE02 disruption mutants of Borrelia burgdorferi B31 have a highly transformable, infectious phenotype. Infection and Immunity, 72, 7147–7154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Förstner, K. and Chao, Y. (2018) Computational analysis of RNA‐protein interactions via deep sequencing. Methods in Molecular Biology, 1751, 171–182. [DOI] [PubMed] [Google Scholar]
- Lin, T. , Gao, L. , Zhang, C. , Odeh, E. , Jacobs, M.B. , Coutte, L. , et al (2012) Analysis of an ordered, comprehensive STM mutant library in infectious Borrelia burgdorferi: insights into the genes required for mouse infectivity. PLoS ONE, 7, e47532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lybecker, M.C. , Abel, C.A. , Feig, A.L. and Samuels, D.S. (2010) Identification and function of the RNA chaperone Hfq in the Lyme disease spirochete Borrelia burgdorferi . Molecular Microbiology, 78, 622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lybecker, M.C. and Samuels, D.S. (2007) Temperature‐induced regulation of RpoS by a small RNA in Borrelia burgdorferi . Molecular Microbiology, 64, 1075–1089. [DOI] [PubMed] [Google Scholar]
- Lybecker, M.C. and Samuels, D.S. (2017) Small RNAs of Borrelia burgdorferi: characterizing functional regulators in a sea of sRNAs. Yale Journal of Biology and Medicine, 90, 317–323. [PMC free article] [PubMed] [Google Scholar]
- Messer, W. and Weigel, C. (1997) DnaA initiator ‐ also a transcription factor. Molecular Microbiology, 24, 1–6. [DOI] [PubMed] [Google Scholar]
- Miller, J.C. (2005) Example of real‐time quantitative reverse transcription‐PCR (Q‐RT‐PCR) analysis of bacterial gene expression during mammalian infection: Borrelia burgdorferi in mouse tissues. Current Protocols in Microbiology, 1D.3. [DOI] [PubMed] [Google Scholar]
- Miller, J.C. , Bono, J.L. , Babb, K. , El‐Hage, N. , Casjens, S. and Stevenson, B. (2000) A second allele of eppA in Borrelia burgdorferi strain B31 is located on the previously undetected circular plasmid cp9‐2. Journal of Bacteriology, 182, 6254–6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, J.C. , von Lackum, K. , Babb, K. , McAlister, J.D. and Stevenson, B. (2003) Temporal analysis of Borrelia burgdorferi Erp protein expression throughout the mammal‐tick infectious cycle. Infection and Immunity, 71, 6943–6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya, S. , Fukuoka, T. , Ogasawara, N. and Yoshikawa, H. (1988) Regulation of initiation of the chromosomal replication by DnaA‐boxes in the origin of the Bacillus subtilis chromosome. EMBO Journal, 7, 2911–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan, S. , Santiago, F. , Koski, R.A. , Brei, B. , Anderson, J.F. , Fish, D. , et al (2002) Examination of the Borrelia burgdorferi transcriptome in Ixodes scapularis during feeding. Journal of Bacteriology, 184, 3122–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro, R. , Duggal, G. , Love, M.I. , Irizarry, R.A. and Kingsford, C. (2017) Salmon provides fast and bias‐aware quantification of transcript expression. Nature Methods, 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picardeau, M. , Lobry, J.R. and Hinnebusch, B.J. (1999) Physical mapping of an origin of bidirectional replication at the centre of the Borrelia burgdorferi linear chromosome. Molecular Microbiology, 32, 437–445. [DOI] [PubMed] [Google Scholar]
- Piesman, J. , Oliver, J.R. and Sinsky, R.J. (1990) Growth kinetics of the Lyme disease spirochete (Borrelia burgdorferi) in vector ticks (Ixodes dammini). American Journal of Tropical Medicine and Hygiene, 42, 352–357. [DOI] [PubMed] [Google Scholar]
- Piesman, J. and Schneider, B.S. (2002) Dynamic changes in Lyme disease spirochetes during transmission by nymphal ticks. Experimental and Applied Acarology, 28, 141–145. [DOI] [PubMed] [Google Scholar]
- Piesman, J. , Schneider, B.S. and Zeidner, N.S. (2001) Use of quantitative PCR to measure density of Borrelia burgdorferi in the midgut and salivary glands of feeding tick vectors. Journal of Clinical Microbiology, 39, 4145–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popitsch, N. , Bilusic, I. , Rescheneder, P. , Schroeder, R. and Lybecker, M. (2017) Temperature‐dependent sRNA transcriptome of the Lyme disease spirochete. BMC Genomics, 18, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radolf, J.D. , Caimano, M.J. , Stevenson, B. and Hu, L.T. (2012) Of ticks, mice, and men: understanding the dual‐host lifestyle of Lyme disease spirochaetes. Nature Reviews Microbiology, 10, 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley, S.P. , Bykowski, T.T. , Cooley, A.E. , Burns, L.H. , Babb, K. , Brissette, C.A. , et al (2009) Borrelia burgdorferi EbfC defines a newly‐identified, widespread family of bacterial DNA‐binding proteins. Nucleic Acids Research, 37, 1973–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluh, A. , Banner, C.D. , Losick, R. and Fitz‐James, P.C. (1981) Identification of a new developmental locus in Bacillus subtilis by construction of a deletion mutation in a cloned gene under sporulation control. Journal of Bacteriology, 148, 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salman‐Dilgimen, A. , Hardy, P.O. , Dresser, A.R. and Chaconas, G. (2011) HrpA, a DEAH‐box RNA helicase, is involved in global gene regulation in the Lyme disease spirochete. PLoS ONE, 6, e22168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels, D.S. (1995) Electrotransformation of the spirochete Borrelia burgdorferi . Methods in Molecular Biology, 47, 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels, D.S. (2011) Gene regulation in Borrelia burgdorferi . Annual Review of Microbiology, 65, 479–499. [DOI] [PubMed] [Google Scholar]
- Savage, C.R. , Jutras, B.L. , Bestor, A. , Tilly, K. , Rosa, P.A. , Tourand, Y. , et al (2018) Borrelia burgdorferi SpoVG DNA‐ and RNA‐binding protein modulates the physiology of the Lyme disease spirochete. Journal of Bacteriology, 200, e00033–00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen, T.D. and Livak, K.J. (2008) Analyzing real‐time PCR data by the comparative CT method. Nature Protocols, 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Schneider, C.A. , Rasband, W.S. and Eliceiri, K.W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nature Methods, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwan, T.G. , Burgdorfer, W. , Schrumpf, M.E. and Karstens, R.H. (1988) The urinary bladder, a consistent source of Borrelia burgdorferi in experimentally infected white‐footed mice (Peromyscus leucopus). Journal of Clinical Microbiology, 26, 893–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwan, T.G. , Piesman, J. , Golde, W.T. , Dolan, M.C. and Rosa, P.A. (1995) Induction of an outer surface protein on Borrelia burgdorferi during tick feeding. Proceedings of the National Academy of Sciences of the United States of America, 92, 2909–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishkin, A.A. , Giannoukos, G. , Kucukural, A. , Ciulla, D. , Busby, M. , Surka, C. , et al (2015) Simultaneous generation of many RNA‐seq libraries in a single reaction. Nature Methods, 12, 323–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Silva, A.M. and Fikrig, E. (1995) Growth and migration of Borrelia burgdorferi in Ixodes ticks during blood feeding. American Journal of Tropical Medicine and Hygiene, 53, 397–404. [DOI] [PubMed] [Google Scholar]
- Smith, J.L. and Grossman, A.D. (2015) In vitro whole genome DNA binding analysis of the bacterial replication initiator and transcription factor DnaA. PLoS Genetics, 11, e1005258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson, B. , Schwan, T.G. and Rosa, P.A. (1995) Temperature‐related differential expression of antigens in the Lyme disease spirochete, Borrelia burgdorferi . Infection and Immunity, 63, 4535–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson, B. and Seshu, J. (2018) Regulation of gene and protein expression in the Lyme disease spirochete In: Adler B. (Ed.) Spirochete Biology: The Post Genomic Era. Heidelberg: Springer‐Nature, pp. 83–112. [DOI] [PubMed] [Google Scholar]
- Stevenson, B. , Zückert, W.R. and Akins, D.R. (2001) Repetition, conservation, and variation: the multiple cp32 plasmids of Borrelia species In: Saier M.H. and García‐Lara J. (Eds.) The Spirochetes: Molecular and Cellular Biology. Oxford: Horizon Press, pp. 87–100. [Google Scholar]
- Stewart, P.E. , Bestor, A. , Cullen, J.N. and Rosa, P.A. (2008) A tightly regulated surface protein of Borrelia burgdorferi is not essential to the mouse‐tick infectious cycle. Infection and Immunity, 76, 1970–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, P.E. , Carroll, J.A. , Dorward, D.W. , Stone, H.H. , Sarkar, A. , Picardeau, M. , et al (2012) Characterization of the Bat proteins in the oxidative stress response of Leptospira biflexa . BMC Microbiology, 12, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze, C.W. , Morado, D.R. , Liu, J. , Charon, N.W. , Xu, H. and Li, C. (2011) Carbon storage regulator A (CsrA(Bb)) is a repressor of Borrelia burgdorferi flagellin protein FlaB. Molecular Microbiology, 82, 851–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troxell, B. , Xu, H. and Yang, X.F. (2012) Borrelia burgdorferi, a pathogen that lacks iron, encodes manganese‐dependent superoxide dismutase essential for resistance to streptonigrin. Journal of Biological Chemistry, 287, 19284–19293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington, T.A. , Smith, J.L. and Grossman, A.D. (2017) Genetic networks controlled by the bacterial replication initiator and transcription factor DnaA in Bacillus subtilis . Molecular Microbiology, 106, 109–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodson, S.A. , Panja, S. and Santiago‐Frangos, A. (2018) Proteins that chaperone RNA regulation. Microbiology Spectrum, 6 Available at: 10.1128/microbiolspec.RWR-0026-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, L. , Gal, J. , Chen, J. and Zhu, H. (2014) Self‐assembled FUS binds active chromatin and regulates gene transcription. Proceedings of the National Academy of Sciences, 111, 17809–17814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zückert, W.R. (2007) Laboratory maintenance of Borrelia burgdorferi . Current Protocols in Microbiology, 12C, 1–10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials