

Abstract
Nucleotide synthesis is essential to proliferating cells, but the preferred precursors for de novo biosynthesis are not defined in human cancer tissues. We have employed multiplexed stable isotope-resolved metabolomics to track the metabolism of [13C6]glucose, D2-glycine, [13C2]glycine, and D3-serine into purine nucleotides in freshly resected cancerous and matched noncancerous lung tissues from nonsmall cell lung cancer (NSCLC) patients, and we compared the metabolism with established NSCLC PC9 and A549 cell lines in vitro. Surprisingly, [13C6]glucose was the best carbon source for purine synthesis in human NSCLC tissues, in contrast to the noncancerous lung tissues from the same patient, which showed lower mitotic indices and MYC expression. We also observed that D3-Ser was preferentially incorporated into purine rings over D2-glycine in both tissues and cell lines. MYC suppression attenuated [13C6]glucose, D3-serine, and [13C2]glycine incorporation into purines and reduced proliferation in PC9 but not in A549 cells. Using detailed kinetic modeling, we showed that the preferred use of glucose as a carbon source for purine ring synthesis in NSCLC tissues involves cytoplasmic activation/compartmentation of the glucose–to–serine pathway and enhanced reversed one-carbon fluxes that attenuate exogenous serine incorporation into purines. Our findings also indicate that the substrate for de novo nucleotide synthesis differs profoundly between cancer cell lines and fresh human lung cancer tissues; the latter preferred glucose to exogenous serine or glycine but not the former. This distinction in substrate utilization in purine synthesis in human cancer tissues should be considered when targeting one-carbon metabolism for cancer therapy.
Keywords: metabolic tracer, metabolism, nucleoside/nucleotide biosynthesis, lung cancer, one-carbon metabolism, dynamic compartmentation, ex vivo human lung tissue slice cultures, multiplexed Stable Isotope-resolved Metabolomics (mSIRM)
Introduction
De novo nucleotide biosynthesis is required to meet the demand for maintaining energy, nucleotide levels, and new nucleic acids in dividing cells (1, 2). Synthesis of pyrimidine nucleotides in cancer cells utilizes glucose and glutamine for the carbon of the uracil and cytosine rings, which then condense with the ribose subunit primarily derived from glucose via the pentose-phosphate pathway (1, 3). The purine nucleotides are synthesized by building the purine ring directly onto phosphoribosyl pyrophosphate (PRPP)6 using glycine (direct route), CO2, and glycine-derived N10-formyl tetrahydrofolate (CHO-THF, indirect route) as the carbon sources for C4,5, C6, and C2,8, respectively (cf. Fig. 1). Cellular glycine may derive from glucose via de novo synthesis from serine (1, 4), via protein degradation, and/or via uptake from external sources, i.e. common cell culture media (1, 2) or the circulating blood (5). Serine derived from glucose or external sources is converted (through one-carbon metabolism) into Gly and 5,10-methylene THF (CH2-THF) via the serine hydroxymethyltransferase (SHMT) activity (Fig. 1). CH2-THF is oxidized to CHO-THF via the methylene tetrahydrofolate cyclohydrolase activity, prior to incorporation into purine rings.
Figure 1.

[13C6]Glucose is more efficiently incorporated into purine rings than D2-Gly regardless of the MYC status. PC9 (a and c) and A549 (b and d) cells under control (CTL) or MYC suppression (siMYC) conditions were cultured in media containing [13C6]glucose and 2H2 (D2)-Gly and then analyzed by UHR-FTMS for 13C and D fractional enrichment in nucleotides and amino acids. The fractional 13C enrichment in the purine rings (Ring) of ATP and GTP was calculated by summing the 13C fraction of the 13C1–4 and 13C6–8 or 13C6–9 isotopologues, whereas the 13C and D fractional enrichment for Gly was obtained by summing the fraction of all labeled species. Schematics of the contributions of metabolic pathways to purine biosynthesis are shown in the middle panel. Black circle, 12C; red circle, 13C; and gray circle, 14N, D 2H in green; encircled 1 and encircled 2 refer, respectively, to direct and indirect routes of glycine incorporation into purines. a and b, fractional enrichment of 13C and D in purine rings (ATP and GTP). c and d, fractional enrichment of Gly isotopologues. Values are means ± S.E., n = 3. [13C6]Glucose was more efficiently incorporated into ATP and GTP than D2-Gly under both control and siMYC conditions. In addition, glucose-13C enrichment in ATP and GTP was strongly attenuated by MYC suppression in PC9 cells (**, t = 7.603, p = 0.006 for ATP, and t = 6.718, p = 0.0026 for GTP) but much less so in A549 cells (*, t = 3.162, p = 0.034 for ATP, and t = 1.109, p = 0.33 for GTP). MYC suppression had insignificant effects on [13C2]Gly and D-Gly isotopologue distribution (c and d; p > 0.05).
There has been considerable interest in the nucleotide synthesis pathway, which can be exploited for cancer therapy (6–11). In particular, the relative importance of exogenous glucose, glycogen, and serine in fueling purine biosynthesis and proliferation has been investigated in a variety of cell lines, with different conclusions. Some studies indicate that exogenous glycine does not enter purine biosynthesis nor support proliferation, whereas serine does (4). However, other reports indicate that exogenous glycine is incorporated into purines in some cell lines (12, 13). The importance of exogenous serine, formate, and the glycine decarboxylase (GLDC) system in purine biosynthesis has also been implicated in different cancer cell lines (14, 15). Thus, the fuels for purine synthesis appear to be cell line–dependent. With the NCI 60 cell panel, serine and glycine uptake rates varied widely; in some cases, glycine uptake was essential for proliferation, and in others a net efflux of glycine occurred (16). Moreover, in some breast and lung cancer cell lines, glucose is an important carbon source for serine and glycine biosynthesis (17–19). Notably, none of these studies included human cancer and paired benign tissues.
Here we report using freshly resected paired human non-cancer and lung cancer tissues, which have distinct architectures and microenvironments compared with pure 2D cell cultures. As the conclusions depended on cell types and culture conditions, we investigated nucleotide synthesis in human NSCLC tissues ex vivo for comparison with in vitro cell models. We have shown previously that these tissues are metabolically active while maintaining all of the cell types and architecture of the original tissue (20, 21). To trace the fate of individual atoms from source molecules into nucleotides and relevant metabolites for rigorous metabolic pathway reconstruction, we employed stable isotope-resolved metabolomics (SIRM) with NMR and MS detection. Both single (13C) and dual (i.e. multiplexed 13C and 2H or D) tracer SIRM analyses (20, 22–25) were employed to track the fate of individual tracer atoms from [13C6]Glc (glucose), [13C2]Gly, D2-Gly, and D3-Ser into the metabolites of one-carbon and purine nucleotide biosynthetic pathways in human lung tissues ex vivo and lung cancer cells in vitro. These include two NSCLC adenocarcinoma cell lines (PC9 and A549) and seven matched pairs of ex vivo cultured cancerous (CA) and noncancerous (NC) lung tissue slices freshly resected from NSCLC patients. In addition, as MYC is known to activate many genes in the one-carbon and nucleotide biosynthesis pathways to promote proliferation (15, 26, 27), we suppressed MYC expression in the two lung cancer cells to track their response in purine nucleotide biosynthesis and growth.
We found that [13C6]glucose was much more efficient than [13C2]Gly or D3-Ser in fueling purine synthesis ex vivo in human CA compared with their matched NC lung tissues, which had a much lower mitotic index and MYC expression. In contrast, the two cancer cell studies revealed D3-Ser to be the best substrate, although D2-Gly was poorly incorporated into purines. In addition, incorporation of [13C6]glucose, [13C2]Gly, and D3-Ser into purines was attenuated by growth inhibition via MYC suppression in PC9 but not in A549 cells. Altogether, these results indicate that cancer cells and tissues differ in their utilization of nutrient sources for purine biosynthesis via the one-carbon pathway. Lung cancer tissues used de novo–synthesized Ser and Gly far more than exogenous supplies for purine synthesis, which can be attributed to activation of de novo Ser synthesis, dynamic compartmentation of de novo Ser synthesis products in the cytoplasm, efficient access of these products to the purine synthesis machinery, and reversal of exogenously derived serine to one carbon fluxes.
Results
Efficiency in incorporation of exogenous nutrients into purine rings and its regulation by MYC differ in human lung cancer cells in vitro
To compare the efficiency of glucose and Gly utilization for purine ring synthesis, we performed dual-tracer experiments by incubating control and siRNA MYC–suppressed PC9 and A549 cells in media containing [13C6]glucose ± D2-Gly. We then tracked the fate(s) of de novo–synthesized [13C]glucose-derived Gly and exogenous Gly (D-Gly) into purine nucleotides using ultrahigh resolution Fourier transform MS (UHR-FTMS) analysis, which distinguishes 13C from D in the same molecule (23). Fig. 1 shows the incorporation of the heavy atoms (red circles) into the purine ring subunits of nucleotides in both A549 and PC9 cells. Glucose-derived 13C (∼3–8%) was incorporated to a much greater extent than Gly-derived D (∼0–0.8%) into the purine rings of ATP and GTP regardless of the cell type or MYC status (Fig. 1, a and b). In contrast, the de novo–synthesized Gly ([13C2]Gly) pool was at most 1% of the total (unlabeled Gly ([12C]Gly) + [13C]Gly) pool compared with D-glycine (D1 + D2), which accounted for 10–20% of the total pool (Fig. 1, c and d). Also, the % enrichment in D2-Gly and its SHMT-scrambled product D1-Gly was much higher than that in the D-purine products of ATP and GTP for both cell types (Fig. 1, a versus c; and b versus d). These results suggest that exogenous glycine is much less efficient in fueling purine synthesis than de novo–synthesized glycine. Moreover, MYC suppression attenuated MYC protein levels in both cell lines (Fig. S1), but only PC9 cells showed a substantial reduction in purine synthesis from glucose (Fig. 1, a and b) as well as growth and lactate production (Table S1).
As Fig. 1 shows, [13C6]glucose-derived glycine contributes 13C atoms to the purine ring both directly (1) and indirectly (2). In contrast, D2-Gly contributes D atoms to the purine ring only via the indirect route (2), which requires mitochondrial GLDC activity (Fig. 1). To measure glycine incorporation into purines via both routes, we carried out separate experiments with [13C2]Gly as sole tracer, [13C6]glucose as sole tracer, and dual [13C6]glucose ± D3-Ser tracer on PC9 and A549 cells with or without MYC suppression. We found that exogenous D3-Ser (DSer + [13C]Glc, green bars, Fig. S2, a and b) was a better substrate for adenine and guanine synthesis than either exogenous [13C6]glucose (no D + [13C]Glc, red bars, Fig. S2, a and b) or [13C2]Gly (no D + [13C]Gly, blue bars, Fig. S2, a and b) in PC9 cells. This preference for exogenous D-Ser was even more marked in A549 cells (Fig. S2, d and e). Exogenous [13C2]Gly was comparable with [13C6]glucose as substrates for purine synthesis in PC9 cells, but in A549 cells exogenous [13C2]Gly was preferred over [13C6]glucose (Fig. S2, a–d). MYC suppression significantly attenuated the incorporation of all three tracers into purines in PC9 cells but not at all or only to a small extent in A549 cells (Fig. S2, a–d), as in Fig. 1. Altogether, these results indicate the following in lung cancer cell lines in vitro: 1) exogenous Ser is the most efficient substrate for purine synthesis; 2) Gly is as efficient or is more efficient than glucose as a substrate for purine synthesis via direct incorporation but not via the indirect or GLDC-mediated route; and 3) regulation of purine synthesis and growth by MYC varies among cell lines. Although our results are consistent with the literature for some cell types in vitro, whether they reflect the substrate preferences in human CA lung tissues ex vivo has not been investigated.
De novo–synthesized serine and glycine preferentially enter purine rings in human lung tissues ex vivo
To determine the substrate preference for purine ring synthesis in human lung tumors, we used our ex vivo human tissue slice system that recapitulates metabolic reprogramming in vivo (20). We procured thin slices of matched CA and NC lung tissue freshly resected from six NSCLC patients (see “Experimental procedures”) and cultured them in media containing [13C6]Glc only, [13C6]Glc + D3-Ser (DSer), or [13C6]Glc + D2-Gly (DGly) for 24 h. These conditions paralleled those used for the 2D cell culture studies described above. We found that Ser and Gly de novo–synthesized from glucose were the dominant precursors to the purine ring of both ATP and GTP in CA lung tissue slices, as illustrated in Fig. 2, a and b, for patient UK022. In contrast, there was minimal incorporation of exogenous DGly or DSer into the purine rings in the CA lung tissue slices. In the matched NC tissues, no DGly incorporation was observed but exogenous DSer was a significant substrate for purine synthesis in addition to [13C6]Glc (Fig. 2, a and b). Similar results were consistently observed in five other pairs of CA and NC lung tissue slices from NSCLC patients (Fig. 3, a–e) that had distinct clinical attributes (Table S2). These subjects were diagnosed with adenocarcinoma or squamous cell carcinoma and differed in tumor stage, age, gender, and comorbidities. These results indicated that glucose, rather than Ser, was by far the preferred precursor for purine synthesis in CA lung tissues, in contrast to the case for lung cancer cells above and other mammalian cells (12). However, similar to the case in vitro, GLDC-mediated Gly incorporation into purines (route 2) did not occur in lung tissues. Altogether, these findings suggest that substrate preference for de novo nucleotide synthesis is context-dependent and differs substantially between cancer cell lines and fresh human cancer tissues.
Figure 2.

Glucose is a preferred substrate over exogenous serine and glycine for fueling purine synthesis in human cancerous lung tissues ex vivo. Freshly resected and thinly cut lung tissue slices (in 2–3 replicates) from NSCLC patient UK022 were treated with [13C6]glucose (13C Glc), [13C6]Glc + D3-Ser (DSer), or [13C6]Glc + D2-Gly (DGly) for 24 h. The tissue slices were then harvested, processed, and analyzed for 13C and D enrichment in ATP (a), GTP (b), and GSH disulfides (GSSG) (c) by direct infusion UHR-FTMS, and for nuclear localization of PCNA (d) and MYC (e) by immunohistochemical staining, as described under “Experimental procedures.” Also shown in the middle panel is the 13C (red circle) and D (green circle) atom tracking from the [13C6]Glc, D2-Gly (DGly), or D3-Ser (DSer) precursors to the adenine ring in ATP via glycolysis, Ser synthesis, and one-carbon metabolism. 13C enrichments from [13C]Glc in a and b (n = 2–3) were 10–30-fold higher than D enrichment from DSer in CA tissues (**, t = 15.55, p < 0.0001 for ATP; t = 5.466, p = 0.0054), and 2–3.5-fold in matched NC tissues (*, t = 4.466, p = 0.0012 for ATP; t = 5.665, p = 0.0002 for GTP). For GSSG, the 13C/D enrichment ratio in CA tissues was 2–3-fold, respectively, for DGly and DSer (**, t = 15.556, p < 0.0001, and t = 11.314, p < 0.0001), whereas in the NC tissues, this ratio for DGly and DSer was less than 1 (*, t = 4.025, p = 0.0024; t = 3.969, p = 0.0026, respectively). PCNA expression was highest in CA tissues (*, Nprox versus CA t = 2.803, p = 0.049; **, Ndist versus CA t = 3.463, p = 0.026) (d). Similarly, MYC expression was highest in CA tissues (*, Nprox versus CA t = 5.68, p = 0.0047; **, Ndist versus CA t = 7.943, p = 0.0014) (e). Data are means ± S.E., n = 6. Ndist, non-cancerous lung tissue distal (>5 cm) from the tumor margin; Nprox, non-cancerous lung tissue proximal (<1 cm) from the tumor margin.
Figure 3.

Consistent trend of preference for glucose-derived Ser/Gly over exogenous Gly or Ser for purine ring synthesis in human lung CA tissues ex vivo. Human tissue sample preparation, treatment, processing, extraction, and analysis were as described in Fig. 2, except that the lung tissue slices of UK055 patient (f) were treated with [13C6]Glc (13CGlc and no D), [13C6]Glc + D3-Ser (DSer), or [13C2]Gly (13CGly and no D) for 24 h. Similar patterns of 13C and D incorporation into ATP, GTP, and GSSG as those in Fig. 2 were observed for five other human CA and NC lung tissue pairs (UK018, UK024, UK025, UK028, and UK043, a–e: the 13C/D ratio in ATP was 5.6 in CA and 1.26 in NC (paired t test, n = 5, t = 5.55, p = 0.0052); in GTP it was 27.6 in CA and 4.8 in NC (paired t test, n = 5, t = 2.421, p = 0.0073); and in GSSG it was 1.66 in CA and 0.3 in NC for DSer (paired t test, n = 5, t = 3.562, p = 0.0235) and 2.056 in CA and 0.256 in NC for DGly (paired t test, n = 5, t = 8.209, p = 0.0012). f, for ATP, *, p = 0.019, t = 2.928; for GTP, **, p = 0.006, t = 5.66; and for [13C]Glc versus [13C]Gly comparison (two-tailed t test).
To determine the efficiency of direct glycine incorporation into the C4,5-positions of the purine rings (cf. route 1, Fig. 1), we performed [13C2]Gly tracer experiments on three replicate pairs of matched CA and NC lung tissue slices procured from a seventh (UK055) patient. We found that [13C]glucose-derived Gly was preferred over exogenous [13C2]Gly in fueling adenine and guanine ring synthesis in CA tissue slices, but the opposite trend was evident for adenine ring synthesis in NC tissue slices (Fig. 3f). Similar to six other patient tissue slices (Fig. 3, a–e), [13C6]glucose was preferred over D3-Ser as precursor for purine synthesis in CA tissue slices (Fig. 3f). Moreover, in all seven cases, exogenous D3-Ser, D2-Gly, or [13C2]Gly was a more efficient substrate for GSH synthesis in NC tissue slices but not or less so in CA tissue slices (Figs. 2 and 3).
We further noted that for all seven patient tissues, the mitotic index (cf. PCNA expression, Fig. 2d and Fig. S3, a and c) decreased from CA tissues to NC tissues proximal (≤1 cm) and distal (≥2–3 cm) to the CA tissues, which paralleled changes in the nuclear MYC expression (Fig. 2e and Fig. S3, b and d). This result is consistent with MYC as a general driver of proliferation and metabolism in CA tissues.
Serine–glycine metabolism is distinctly compartmentalized in cancerous lung tissues for GSH versus purine synthesis
We then asked whether the high demand for de novo–synthesized Ser and Gly as purine precursors in CA lung tissues could be related to compartmentation of Ser and Gly metabolism. We used UHR-FTMS to determine the total abundance and fractional enrichment of the 13C and D isotopologues of Ser and Gly extracted from the UK022 tissue slices. First, endogenous glucose-derived Ser (e.g. [13C3]Ser) or Gly (e.g. [13C2]Gly) was barely detected in any of the tissues (Fig. 4, a–d), despite their substantial incorporation into purines and GSSG (Figs. 2, a–c, and 3). In addition, the only isotopologue detected was the fully labeled [13C3]Ser and [13C2]Gly. These results suggest that the majority of de novo–synthesized Ser and Gly was consumed for purine/GSH synthesis and did not undergo exchange reactions in lung tissues (cf. Fig. 5). Second, CA lung tissues converted almost all exogenous D2-Gly to the “scrambled” D1-Gly product, whereas the ratio of D2/D1-Gly was nearly equal in matched NC lung tissues (Fig. 4, b and d). Likewise, CA lung tissues scrambled exogenous D3-Ser to a much greater extent than the NC counterparts (Fig. 4, a and c). Such differential D label scrambling patterns of Gly and Ser implicated a more extensive exchange in the mitochondrial Ser–Gly one-carbon pathway (cf. Fig. 5) in CA than in NC lung tissues, as GLDC-driven Gly to one-carbon interconversions that scramble Ds in D2-Gly or D3-Ser occur only in mitochondria. Such an exchange process may reduce the pool of cytoplasmic Gly and Ser available for incorporation into purines in CA lung tissues (cf. Fig. 5). Third, significantly higher enrichment of D in GSSG (Fig. 2c) than in the precursor D-Gly was evident in DGly-treated CA but not in NC lung tissue (Fig. 4d). Together with the dominance of unlabeled (12C) Gly in the total Gly pool in CA lung tissues (Fig. 4d), these data suggest that Gly in CA tissues was derived primarily from internal (unlabeled) sources, whereas labeled exogenous Gly was more efficiently utilized for GSH synthesis in CA than NC lung tissues. In contrast to D-Gly, D-Ser represented a major Ser pool in DSer-treated lung tissues (Fig. 4, a and c), which could reflect efficient uptake of Ser (28) and/or less efficient utilization. The 13C- and D-labeling patterns for Ser and Gly were qualitatively similar in other pairs of lung tissue slices (Fig. S4 and data not shown).
Figure 4.

Analysis of 13C and D isotopologues of Ser and Gly reveals reduced Ser/Gly uptake but enhanced Ser–Gly one-carbon exchanges in human CA versus matched NC lung tissues ex vivo. UK022 patient sample treatment, processing, extraction, and ECF derivatization were as described under “Experimental procedures.” The respective level (a and b) and fractional enrichment (c and d) in 13C and d isotopologues of Ser or Gly in tissues and uptake or release of these Ser or Gly species in the medium (e and f) were obtained from UHR-FTMS analysis, as described under “Experimental procedures.” D1, D2-Ser in a, c, and e and D1-Gly in b, d, and f represented D-scrambled Ser and Gly, respectively. no D, [13C6]Glc only; DSer, D3-Ser + [13C6]Glc; DGly, D2-Gly + [13C6]Glc. Using the two-tailed unpaired t test, the absolute levels of D1-, D2-, and D3-serine differed between CA versus NC tissues (n = 3 each) (a) 2-fold for D1 (p = 0.01, t = 4.472), 3-fold for D2 (p = 0.002, t = 70.7), and 0.27 for D3 (p = 0.0032, t = 6.31). The fractional enrichment differed for CA versus NC tissues with (c) 7-fold for D1 (p = 0.0042, t = 5.884), 3.5-fold for D2 (p = 0.001, t = 8.575), and 0.27-fold for D3 (p = 0.0001, t = 19.677). For D1- and D2-glycine, the fractional enrichments differed between CA versus NC tissues (d) 0.3-fold for D1 (p = 0.0001, t = 24.748) and 0.07-fold for D2 (p = 0.0029, t = 6.492). The absolute levels in b were too low for reliable statistical testing. Because of large standard errors, the difference (4.4-fold) in the uptake of D3-serine from the medium did not reach statistical significance between CA versus NC tissues (e), but the differential uptakes (4.2–5.2-fold) of unlabeled (12C) Ser between the two tissue types were statistically significant (*, p = 0.018, t = 4.695). The uptake of D2-Gly decreased 23-fold in CA versus NC tissues (f) (*, p = 0.02, t = 6.856).
Figure 5.
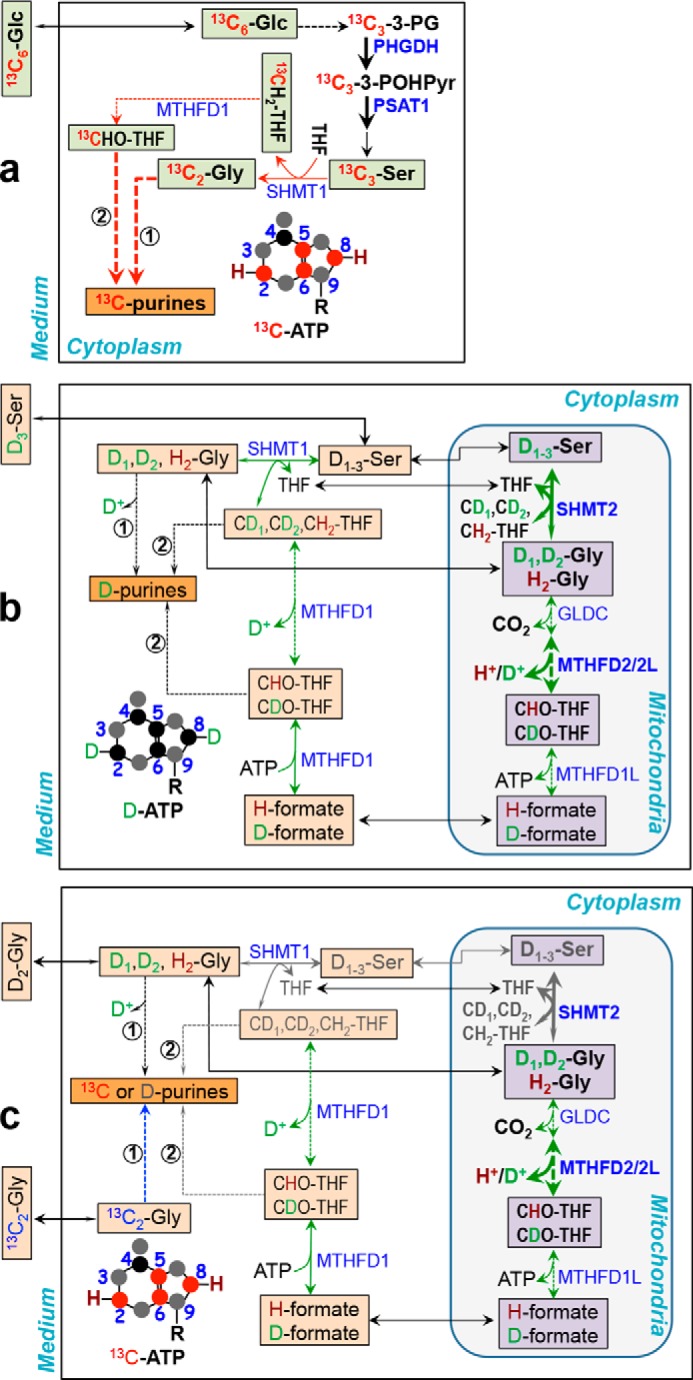
Activation of Ser/Gly synthesis from glucose, their efficient incorporation into purines, and reverse flux of the Ser–Gly one-carbon pathway underlie preference for glucose as substrate for purine synthesis in lung cancer tissues ex vivo. The three schemes depict our key findings on the metabolism of exogenous glucose (a), Ser (b), and Gly (c) into purines in CA lung tissues. These were modified from the mammalian cell literature (4, 32, 40–43). a, 1) net [13C3]glucose (Glc) uptake, enhanced conversion to [13C]Ser/[13C2]Gly in the cytoplasm (pool 1, light green square), and efficient incorporation (thick red arrow) into purine carbons (red circle) (e.g. adenine of ATP) via the action of cytoplasmic SHMT1 (route 1), MTHFD1 (route 2), and other enzymes (not shown) in the purine nucleotide synthesis pathway; 2) lack of cytoplasmic and mitochondrial exchange for [13C3]Ser/[13C2]Gly. b, 3) net uptake of exogenous D3-Ser into the cytoplasm (pool 2, peach square), which does not readily exchange with pool 1 but exchanges with the mitochondrial pool (lavender square) and interconverts with Gly and one-carbon metabolites (green double-headed arrow); 4) less access of D3-Ser-derived Gly and one carbon metabolites (peach square pools) to purine synthesis machinery (e.g. orange square purinosomes (44)); 5) enhanced mitochondrial Gly to Ser fluxes (uneven green double-headed arrow) possibly driven by activation of SHMT2 and MTHFD2 (thick green double-headed arrow). b and c, 6) loss of D (or gain of H) in one carbon metabolites via mitochondrial MTHFD2/2L and cytoplasmic MTHFD1 exchange reactions (green double-headed arrow); 7) loss of D via direct incorporation of D-Gly into C5,6 of purines (thin arrow); 8) negligible incorporation of Gly-derived one carbon metabolites into purines (gray arrow). c, 9) net Gly efflux (uneven arrow); 9) less favored direct (route 1, thin blue arrow) (c) and indirect (route 2, not depicted) incorporation of [13C2]Gly into purines. PSAT1, phosphoserine aminotransferase; MTHFD1, cytoplasmic NADP+-dependent methylene tetrahydrofolate (CH2-THF) dehydrogenase/methylene THF; cyclohydrolase/formyl THF (CHO-THF) synthetase; MTHFD2, mitochondrial NAD+-dependent methylene THF dehydrogenase/methylene THF cyclohydrolase; MTHFD2L, mitochondrial NADP+-dependent methylene THF dehydrogenase; MTHFD1L, mitochondrial formyl THF synthetase; 3-PG, 3-phosphoglycerate; 3-POHPyr, 3-phosphohydroxypyruvate. Solid and dashed arrows: one- and multistep reactions, respectively.
We further noted that CA tissue slices consumed less Ser from the medium than NC tissue slices (Fig. 4e), which is consistent with less D-Ser incorporation into purines and glutathiones (Fig. 2, a–c). Furthermore, both tissue types showed a net efflux of unlabeled Gly into the medium (Fig. 4f), which has also been observed in a panel of cancer cells (16). However, D2-Gly was still consumed from the medium by lung tissues, and more so by NC than by CA lung tissues (red boxes, Fig. 4f). This is consistent with the higher D enrichment of GSSG in the DGly-treated NC than CA lung tissues (Fig. 2C).
Altogether, these results pointed to the presence of at least three intracellular pools of Ser and Gly with distinct metabolic fates in human lung tissues. The smallest pool (pool 1) is derived from glucose as the dominant substrate for purine synthesis in CA tissues but less so in NC tissues. The second (larger than the first) pool (pool 2) is acquired externally as more efficient substrates for GSH synthesis in NC than CA tissues. The third (often the dominant) pool (pool 3) is derived from unlabeled sources such as protein turnover, which is much less utilized for purine or GSH synthesis in human lung tissues. To illuminate this finding further, we employed kinetic modeling of purine synthesis.
Flux modeling of metabolic compartmentation reveals distinct differences in CA versus NC human lung tissues ex vivo
To obtain an integrative and quantitative view of purine metabolism in human lung tissue slices, we developed kinetic models (see “Experimental procedures”) that utilized available 13C and D enrichment data in different metabolites (Table S3) to estimate fluxes of the purine synthetic pathway in both CA and paired NC tissue slices of patient UK022. The best-fit model required three distinct pools of cytoplasmic Ser/Gly, with one pool derived from [13C6]Glc, a second from exogenous D3-Ser or D2-Gly, and the third from unlabeled sources. The model showed that the rates of Glc, Ser, and Gly influx and efflux as well as Glc conversion to Ser (but not to PRPP, the 1st intermediate of purine synthesis) were higher in CA than NC lung tissues during 24 h of tracer treatments (Fig. S5a). The enhancement of the glycine efflux over the influx rate for CA relative to NC tissue slices was consistent with the greater release of unlabeled (12C) Gly into the CA than NC tissue medium (cf. red boxes, Fig. 4f). Our findings are in contrast to a study showing a correlation of Gly influx and proliferation of the NCI 60 cancer cell lines (13) but are consistent with other reports on net Gly efflux from cancer cells in culture (16, 28). Our model also suggested a lower net rate of Ser consumption by CA than by NC lung tissues (Fig. S5b).
The kinetic model further revealed a larger apparent rate constant (k) for the forward SHMT action, i.e. the production of the purine precursor CH2-THF from glucose-derived Ser (pool 1) than from exogenous (pool 2) or unlabeled Ser sources (pool 3) in CA but not in matched NC tissue slices (Fig. S5c). In contrast, the apparent rate constant for the reverse reaction (CH2-THF to Ser) (k−1) was much greater for pools 2 and 3 than pool 1 in CA tissues and negligible in their NC counterparts (Fig. S5d). These results indicate that the forward path of glucose → Ser → CH2-THF and the reverse path of CH2-THF → Ser for nonglucose sources was much favored in CA tissue slices, which can at least in part account for the higher glucose preference for fueling purine synthesis in CA tissues than in their NC counterparts. The above modeling outcomes were consistently obtained under all three different tracer treatments, i.e. [13C6]Glc only, [13C6]Glc + D3-Ser, and [13C6]Glc + D2-Gly. Altogether, our kinetic models support the notion that the preference for glucose over Ser and Gly for purine synthesis in CA lung tissues is mediated by the activation of de novo Ser synthesis from glucose and reversal of Ser to one carbon fluxes from nonglucose sources.
Cell line–specific MYC enhancement of growth-related de novo serine synthesis from glucose and serine one-carbon exchanges
The ex vivo tissue experiments described above showed that the serine one-carbon exchanges were more extensive in the more proliferative, higher MYC-expressing CA lung tissues and may contribute to their preference for glucose over exogenous Ser in purine synthesis. We therefore asked whether MYC expression governs such a preference in lung cancer cells in vitro. We compared Ser metabolism in PC9 versus A549 cells in response to siRNA-mediated MYC suppression. We found that in PC9 cells MYC suppression (siMYC) attenuated the fractional enrichment of [13C]Ser (derived from [13C6]Glc) and D-scrambled (D1 + 2)-Ser (derived from D3-Ser) relative to control (siNT) treatment (Fig. S6a), whereas that of parent D3-Ser was enhanced by siMYC, which was not the case for A549 cells (Fig. S6c). D-Ser scrambling results from Ser one-carbon exchange processes (cf. Fig. 5). Likewise, in PC9 cells, MYC suppression reduced the ratios of de novo–synthesized (13C-labeled) to exogenously derived (D-labeled) Ser and Gly (Fig. S6b) but not in A549 cells (Fig. S6d). These data indicated that MYC enhanced de novo Ser and Gly synthesis from glucose and serine one-carbon exchange reactions for exogenously derived Ser in PC9 cells. This paralleled the MYC-induced increase in efficiency of [13C]Glc (relative to exogenous D-Ser) incorporation into purines (Fig. S2c), glucose consumption, and growth (Table S1). Similarly, MYC overexpression in lung CA versus NC tissues ex vivo could be related to increased mitotic index (Fig. 2 and Fig. S3c) and preference for glucose as substrate for purine synthesis (Figs. 2 and 3).
MYC regulation of key genes in the Ser synthesis and mitochondrial one-carbon pathways relates to substrate preference for purine synthesis and growth
To determine whether reduced serine one-carbon exchanges induced by MYC suppression is driven at the transcriptional level, we measured key gene expression events in the glucose–Ser–Gly one-carbon pathway (cf. Fig. 5) in lung cancer cells in response to MYC suppression. In PC9 cells, MYC suppression attenuated the expression of serine hydroxymethyltransferase (SHMT2), phosphoserine aminotransferase (PSAT), phosphoglycerate dehydrogenase (PHGDH), and methylenetetrahydrofolate dehydrogenase (MTHFD2), as shown in Fig. S7a. PHGDH and PSAT are key enzymes in the de novo Ser biosynthesis pathway, whereas SHMT2 and MTHFD2 catalyze the interconversions of mitochondrial Ser to Gly and the one-carbon metabolite (cf. Fig. 5). These gene expression changes in PC9 cells are consistent with the reduced 13C or D incorporation from [13C6]Glc, D3-Ser, or [13C2]Gly into ATP and GTP following MYC suppression in PC9 cells (Fig. S2, a and b). However, MYC abrogation also enhanced the SHMT1 and GLDC expression in PC9 cells, which was not evident in A549 cells. Furthermore, the attenuating effect on MTHFD2 expression was smaller in A549 than in PC9 cells (Fig. S7a). These differences can be related to the reduced response of glucose consumption, growth (Table S1), and ATP/GTP synthesis from the three precursors as well as the lack of preference for glucose as substrate for purine synthesis in A549 versus PC9 cells (Fig. S2). MYC's up-regulation of MTHFD2 and down-regulation of SHMT1 and GLDC in PC9 cells could restrict the supply of cytoplasmic Gly and one-carbon metabolites derived from exogenous Ser and Gly (Fig. 5, b and c), thereby decreasing their use for purine synthesis. This is in contrast to MYC's activation of de novo Ser synthesis and its enhanced use in purine synthesis in PC9 cells (Fig. S2).
Up-regulation of Ser synthesis and one-carbon exchange pathways relates to preference of glucose for fueling purine synthesis in lung cancer tissues
The same set of genes up-regulated by MYC in PC9 cells was highly overexpressed in human CA versus NC lung tissues, as illustrated for patient UK018 (Fig. S8a). A similar trend was observed for other patients except for UK024, where only MTHFD2 was overexpressed (data not shown). To determine whether the gene expression changes result in protein level changes, we performed reverse-phase protein array (RPPA) analysis on the protein extracts of seven sets of paired patient tissues (cf. Figs. 2 and 3). This assay is quantitative and well-suited for multiplexed protein profiling with very limited patient tissue samples (31, 32). We confirmed the overexpression of key enzymes in the Ser synthesis (PSAT and PHGDH) and mitochondrial one-carbon pathways (SHMT2 and MTHFD2) in CA versus NC tissues (Fig. S8b). Thus, all of the distinct metabolic properties of CA tissues defined above are consistent with glucose being the preferred substrate for purine synthesis due to the following: 1) enhanced de novo Ser synthesis; 2) efficient access of de novo–synthesized Ser to the purine synthesis machinery; and 3) blunted supply of one-carbon metabolites derived from exogenous Ser and Gly via increased mitochondrial Ser one-carbon exchanges. Furthermore, they were related to the overexpression of nuclear MYC and increased mitotic index (Fig. 2 and Fig. S3), suggesting a possible role of MYC in stimulating the use of glucose for purine synthesis in lung cancer tissues and to a smaller extent in PC9 cells.
Discussion
De novo nucleotide synthesis is at the core of cancer metabolism and provides a vulnerability that has long been exploited for chemotherapy by targeting one-carbon metabolism. Two separate signaling pathways involving mammalian target of rapamycin–activating transcription factor 4 (ATF4) (12) or nuclear factor E2–related factor 2 (NRF2)–ATF4 (17) axes have recently been shown to activate purine nucleotide synthesis. MYC has also been documented to transcriptionally regulate the expression of many nucleotide synthesis genes required for cell proliferation (2, 28, 34, 35). Despite these recent advances in elucidating the signaling networks for regulating purine nucleotide synthesis, little is known about nutrient preferences for purine synthesis in human cancer tissues as compared with cell lines. Herein, we document profound differences in the use of substrates by freshly explanted human lung cancer tissues as compared with paired noncancerous tissues or lung cancer cell lines. These findings are important not only for the fundamental understanding of nucleotide metabolism but also for its exploitation as target(s), such as Ser/Gly starvation (10), in human cancer therapy.
By tracking in detail the fate of [13C6]glucose, D3-Ser, D2-Gly, and [13C2]Gly, we found that serine–glycine metabolism is distinctly compartmentalized in cancerous lung tissues for GSH versus purine synthesis. Glucose was the preferred substrate for purine synthesis in CA over NC lung tissues, which could be related to the activation of glucose to Ser one-carbon metabolism, preferred access of de novo-synthesized Ser/Gly by the cytoplasmic purine synthesis machinery, and enhanced mitochondrial Ser/Gly one-carbon exchange reactions. In contrast, D2-Gly was consumed from the medium by NC more than by CA lung tissues with a higher deuterium enrichment of GSSG in the NC than CA lung tissues.
Our findings support context-dependent differences in Ser/Gly metabolism and the metabolic schemes for CA tissues in Fig. 5, which were revised from known membrane-delineated compartmentation of Ser/Gly one-carbon metabolism (36–39). In Fig. 5a, increased PHGDH and PSAT1 expression enhances Ser synthesis from glucose in CA as compared with NC lung tissues. A lack of 13C scrambling in [13C6]Glc-derived Ser and Gly indicates a favored forward SHMT1 reaction (→) and lack of mitochondrial exchange activity. Minimal in [13C]Ser or [13C]Gly (Fig. 4, a–d) and much higher in 13C enrichment in purines than in the buildup to their Ser/Gly precursors (Figs. 2 and 3 versus 4, a–d) point to the highly-efficient use of de novo–synthesized Ser/Gly for purine synthesis (→). Less incorporation of exogenous D3-Ser (→, Fig. 5b; Figs. 2 and 3) or [13C2]Gly (→, Fig. 5c; Fig. 3f) into purines suggests less efficient access of the exogenously-derived Gly and one-carbon pools to the purine synthesis machinery (possibly organized as purinosomes (44)) (Fig. S5d) and revealed a favorable reverse SHMT1/2 reaction from CH2-THF to Ser (→, Fig. 5, b and c). Such pathway reversal has been shown to block the incorporation of exogenous Ser into purines in cancer cells, which involves restricting the supply of formate from mitochondria to the cytoplasm (4). Overexpression of SHMT2/MTHFD2 (Fig. S8) can facilitate such reverse flow from Gly to Ser to reduce the formate supply (Fig. 5, b and c). Restricted formate supply is also consistent with total or nearly total lack of D labeling in purines with D2-Gly as tracer in lung tissues (Figs. 2 and 3) and PC9 cells (Fig. 1). Total loss of D occurs from direct D2-Gly incorporation into C4,5 of purines (route 1, Fig. 5, b and c) but exchange reactions catalyzed by mitochondrial SHMT2/GLDC/MTHFD2/2L and cytoplasmic SHMT1 and MTHFD1 (route 2, Fig. 5, b and c) should still lead to D incorporation into purines unless D-formate is in short supply.
Although the transcriptional regulation for such substrate preference in vivo is unclear, we surmise that MYC-mediated alterations in growth and purine metabolism is a plausible mechanism, based on studies of PC9 cells in vitro. We also postulate that such preference for glucose in purine synthesis in human cancer lung tissues offers metabolic advantages. By linking purine synthesis to ribose 5-phosphate production via glucose metabolism would help avoid nonproductive purine synthesis fueled by Ser and Gly (e.g. from proteolysis) in poorly vascularized tumor tissues where glucose is in short supply.
In conclusion, by tracing the fate of different purine substrates with 13C and/or D labels, we discovered a high preference for glucose or de novo–synthesized Ser/Gly in fueling purine synthesis in active human lung cancer tissues, which contrasted with the preferred use of exogenous Ser for purine synthesis in noncancerous lung tissues or cancer cell lines in vitro (4). Our results and those of others clearly indicate that substrate utilization for purine biosynthesis varies greatly according to cell type and environment. This lung CA tissue preference for glucose could be attributed to the following: 1) activation of glucose to Ser one-carbon pathways; 2) compartmentation of cytoplasmic Ser/Gly one-carbon pathways and preferred access of de novo–synthesized Ser/Gly by the purine synthesis machinery; and 3) enhanced reverse mitochondrial Ser to Gly fluxes. Our findings suggest that blocking the glucose to the Ser pathway may be more efficacious than dietary Ser/Gly starvation (10) for lung cancer therapy.
Experimental procedures
PC9 and A549 cell growth and MYC suppression
A549 and PC9 cells were obtained from the American Type Culture Collection (ATCC) (Manassas, VA) with authentification, and they were checked for mycoplasma contamination monthly. Cells were cultured in DMEM (Sigma) or RPMI 1640 medium (Life Technologies, Inc.) containing 2 mm glutamine, 0.2% glucose, with 10% (v/v) FBS and 100 units/ml penicillin and 100 μg/ml streptomycin (Thermo Fisher Scientific, Waltham, MA). 24 h before siRNA transfection, cells were seeded at a density of 1.8 × 104/cm2 or 2.7 × 104/cm2 for siNT (control vector) or siMYC, respectively. 100 nm siRNA was transfected using Dharmafect 1, as per the manufacturer's protocol. 48 h after siRNA addition, cells were placed in labeled tracer media (see below). 72 h post-transfection, one plate was harvested for Western blotting to confirm knockdown and the remaining three plates from each siRNA transfection were harvested with metabolic quenching in cold acetonitrile.
All siRNAs used for study were ordered from Dharmacon (ON-TARGET plus): nontargeting siRNA (siNT), UAAGGCUAUGAAGAGAUACAA; MYC siRNA (siMYC), GGACUAUCCUGCUGCCAAGUU. Knockdown efficiencies are shown in Figs. S1 and S8c.
Stable isotope tracer treatments of lung cancer cells
All 13C and 2H (D) tracers were purchased from Sigma or Cambridge Isotope Laboratories (Tewksbury, MA). For the tracer experiments, A549 or PC9 cells were grown for 24 h, respectively, in DMEM lacking glucose, glycine, and serine supplemented with 10% dialyzed FBS (Life Technologies, Inc.), 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mm Gln, plus one of the following tracer cocktails: 10 mm [13C6]Glc + 0.4 mm each unlabeled Gly and Ser; 10 mm [13C6]Glc + 0.4 mm D2-Gly + 0.4 mm unlabeled Ser; 10 mm [13C6]Glc + unlabeled Gly + 0.4 mm D3-Ser; and 10 mm unlabeled Glc + 0.4 mm [13C2]Gly + 0.4 mm unlabeled Ser.
Stable isotope tracer treatments of lung tissues ex vivo
All NSCLC patients investigated were recruited for lung tissue collection with written informed consent in accordance with HIPAA regulations, and all experiments were carried out via a protocol approved by the University of Kentucky Institutional Review Board (IRB, 14-0288-F6A). These studies abide by the Declaration of Helsinki principles.
Fresh CA and surrounding NC lung tissues were collected in the operating room within 5 min of surgical resection. The pair of tissues were thinly sliced by the surgeon at ∼0.7 to 1-mm thickness as described previously (20, 21, 25, 41, 42) and incubated in glucose, Ser, and Gly-free DMEM supplemented with dialyzed FBS, penicillin, streptomycin, Gln, plus the same set of tracer cocktails as described above for the cell experiments for 24 h at 37 °C, 5% CO2 with gentle rocking to facilitate nutrient supplies and waste product mixing (21, 24). Culture media were sampled at 0 and 24 h of incubation. Tissue slices were then quickly rinsed in cold PBS three times to remove medium components, blotted dry, weighed on a four-place balance for wet weight, and flash-frozen in liquid N2.
Extractions and derivatization of cell and tissue extracts
At the end of the treatments, A549 and PC9 cells were washed with cold PBS, quenched with acetonitrile/H2O (2:1.5), and then extracted using the acetonitrile/H2O/CHCl3 (2:1.5:1) partitioning method (43). Medium samples were extracted in cold 10% TCA (44) or 80% acetone (see below).
The frozen tissue slices were homogenized in 60% cold CH3CN in a ball mill (Precellys-24, Bertin Technologies, Washington, D. C.) for denaturing proteins and optimizing extraction. Polar metabolites were extracted by the solvent partitioning method with a final CH3CN/H2O/CHCl3 (2:1.5:1, v/v) ratio (48). The polar extracts were lyophilized in aliquots before reconstitution in 50% D2O for NMR analysis or in H2O for direct infusion UHR-FTMS analysis. The culture media were deproteinized in 80% acetone at −20 °C for 0.5 h before centrifugation, lyophilization, and reconstitution in 50% D2O for 1H NMR analysis.
A separate aliquot of lyophilized cell, tissue, or medium extracts was spiked with 5 μl of 0.4 mg/ml uniformly 15N-labeled amino acids mixture (NLM-6695, Cambridge Isotope Laboratories, Cambridge, MA) as internal standards, dissolved in 100 μl of water/ethanol/pyridine (6:3:1) solution, followed by addition of 5 μl of ethyl chloroformate (ECF) for chemoselective derivatization of amino- and carboxylate-containing metabolites. The mixture was extracted twice with 100 μl of chloroform with vortexing for 30 s The pooled chloroform extract was diluted 10-fold with 90% acetonitrile plus 20 μm NaCl to sodiate the ions (50). The samples were then directly infused into an UHR-FTMS instrument (Thermo Fusion Orbitrap Tribrid MS) using nano-electrospray ionization. The Triversa Nanomate was operated at 1.5 kV and 0.5 p.s.i. head pressure for positive mode. The maximum ion time for the automatic gain control was set to 100 ms, and microscans were set to 5. Each spectrum was acquired for 10 min with m/z range of 100–1000 at a resolution target set to 500,000 (achieving >370,000 at 400 m/z). The concentration of 15N-labeled amino acids in the internal standard mixture was verified by adding a known amount of unlabeled amino acid mixture (amino acids mixture A6407 and A6282 plus glutamine, Sigma), followed by ECF derivatization and UHR-FTMS analysis.
NMR and MS analysis of cell, tissue, and medium metabolites
Metabolites were analyzed by NMR at 14.1 T, as described previously (44, 46). The metabolites were identified using in-house databases (47–49) and quantified using MNova software (Mestrelab Research, SL, Santiago de Compostela, Spain).
For nucleotide analysis by direct-infusion UHR-FTMS, samples were prepared using a slightly modified procedure, as described previously (23). Briefly, lyophilized polar extracts were first reconstituted in 50 μl of 5 mm aqueous hexylamine, pH 6.3, with acetic acid (solvent A). The samples were then loaded onto a 100-μl capacity C18 tip (Pierce-Thermo Fisher Scientific, Rockford, IL) via four slow aspirations followed by washing twice with 50 μl of solvent A. The metabolites were eluted with two 50-μl 70% solvent A plus 30% of 1 mm ammonium acetate in 90% methanol, pH 8.5 (solvent B) by aspirating 10 times. UHR-FTMS analyses were performed on an Orbitrap Fusion Tribrid (Thermo Fisher Scientific, San Jose, CA) equipped with a TriVersa NanoMate (Advion Biosciences, Ithaca, NY). Nanoelectrospray ionization was initiated from the nozzle by applying 1.5 kV with a 0.5 p.s.i. head pressure in negative ion mode. All UHR-FTMS data were recorded in profile mode using a maximum injection time of 100 ms, automated gain control at 2.0 × 105, 10 microscans, and a target resolution set to 500,000 (achieving >370,000 at 400 m/z) (49). A sample run was completed in 15 min.
Results from both NMR and MS measurements were normalized to the protein weight for A549 and PC9 cells, except for the fractional enrichment of 13C or D, where no normalization was necessary. 13C and D isotopologues were identified based on their accurate masses acquired from UHR-FT-MS, and their fractional enrichments quantified after natural isotope abundance contributions of each of the isotopologues in the MS data were corrected as described previously (50, 51).
Immunohistochemical analysis of human tissues
The human specimens studied were formalin-fixed and paraffin-embedded before 4-μm tissue sections were prepared. Immunohistochemistry (IHC) was performed with the Dako EnVision + System-HRP (DAB) kit (K4010, Dako). Primary antibody used was anti-c-Myc (Y69) (1:100, ab32072, Abcam). The antibody dilution buffer was 1% BSA and 0.3% Triton X-100 in 1× PBS. Antigen retrieval was carried out using Tris-EDTA buffer (pH 9.0) for c-Myc in a microwave oven for 20 min. Any endogenous peroxidase activity was quenched by incubating the specimen for 5 min with Peroxidase Block, followed by incubation with Protein Block Serum-Free (Dako, X0909) for 15 min to inhibit nonspecific background staining. The specimen was then incubated with the primary antibody, followed by incubation with the labeled polymer-HRP anti-rabbit, using two sequential 30-min incubations. Staining was completed by a 5–10-min incubation with 3,3′-diaminobenzidine (DAB+) substrate-chromogen and then hematoxylin counterstain.
Digital images of IHC slides were obtained at ×40 magnification using a whole slide scanner (ScanScope XT, Aperio) and viewed by the ImageScope software (version 11.2, Aperio). The optimally tuned Nuclear version 9 and Cytoplasmic version 2 algorithms were used to quantify nuclear staining. Staining intensity was graded as 0 (negative), 1 (weak), 2 (moderate), and 3 (strong). The nuclear H-score was obtained by the formula: (3× percentage of strongly staining nuclei) + (2× percentage of moderately staining nuclei) + (1× percentage of weakly staining nuclei), giving a range of 0 to 300. For each sample, three regions of interest were annotated, analyzed, and averaged.
Immunofluorescence staining was performed with the PCNA (D3H8P) XP® rabbit mAb (1:800, 13110, Cell Signaling). Antigen retrieval was carried out using sodium citrate buffer (pH 6.0) in a microwave oven for 20 min. Tissue sections were then incubated with ice-cold 100% methanol for 10 min at −20 °C for permeabilization and blocked with 5% normal goat serum for 1 h at room temperature to prevent nonspecific binding. Slides were incubated with primary antibody overnight at 4 °C, followed by incubation with the goat anti-rabbit Alexa Fluor® 488 secondary antibody (1:200, A-11008, Thermo Fisher Scientific) for 1 h at room temperature in the dark. Slides were then mounted in ProLong® Gold Antifade Mountant with DAPI (P-36931, Thermo Fisher Scientific).
Fluorescence images were acquired using a laser-scanning confocal microscope (FluoViewTM FV1000, Olympus) with a ×60 oil immersion lens (UPLSAPO 60XO) under the same setting. Fluorescence intensity was measured using Fiji (ImageJ, National Institutes of Health, Bethesda, MD) with a constant threshold. The percent PCNA positivity was calculated by normalizing the PCNA-positive area against the DAPI-stained area. For each sample, three representative fields were acquired, analyzed, and averaged.
Gene expression analysis
A549 and PC9 cells were harvested 72 h following siRNA transfection. Total RNA was extracted using the Norgen total RNA purification kit, and RNA was subjected to on-column DNase I (Omega Biotek) treatment before elution. cDNA was synthesized using the Maxima First Strand cDNA synthesis kit (Thermo Fisher Scientific). Quantitative real-time PCR was performed using the Maxima SYBR Green Mastermix reagent (Thermo Fisher Scientific). The primer sequences used were as follows: MTHFD2 (F, GCTGCGACTTCTCTAATGTCTGC, and R, ATCTGCTGGGCCAGTTTCCTT); MTHFD2L (F, TCATACGCAGCTGGCAGATA, and R, TCCTGTCACTGGATCGTGGA); PSAT (F, GTCCTCAAACTTCCTGTCCAA, and R, GCAGGTCATCACGGACAATC); PHGDH (F, GCAAATCTGCGGAAAGTGCT, and R, AATAAGGCCTTCACAGTCCTGC); GLDC (F, CAGGGGTGCAAGAGGTTATGT, and R, GTCTCTTGGCCACATCCACA); MTHFD1 (F, GTCTACACGAAGCAGGGCTTT, and R, ATGTCGCGAATGGGCAGAAT); MTHFD1L (F, GGCTGGTCTGAACATCACTC, and R, GGCCATGTACTCTGGTATCTTC); SHMT1 (F, GTACCCGGGCCAGAGATACT, and R, GCACTGTGGGTCCAGCTTAT); SHMT2 (F, CTGACTGCTCGACTTTTCCG, and R, GCTTTGACTTCATCACACACC); MYC (F, GTAGTGGAAAACCAGCAGCCTC, and R, GTTCTCCTCCTCGTCGCAGTA); and 18 S rRNA (F, AACGGCTACCACATCCAA, and R, GACTCATTCCAATTACAGGGC).
For qRT-PCR analysis of CA/NC tissue slices, RNA was extracted from ∼10 mg of pulverized frozen tissue and subsequent cDNA synthesis, and qRT-PCR was carried out using the same reagents and primers as with the A549/PC9 cell analysis.
Protein analysis by RPPA
Protein extracts (at 0.5 and 0.3 mg/ml) from bulk CA paired NC lung tissues were spotted as two drops per spot onto a slide coated with 16 nitrocellulose membrane pads using a microarray printer (ArrayJet, Ltd., Roslin, UK). Membranes were incubated in a 1: 5 × 106 dilution of 12.3 nm stock of FastGreen protein stain for 5 min, dried, and scanned with InnoScan 710 AL Microarray Scanner (Innopsys, Inc. Carbonne France) to determine the amount of proteins deposited per sample spot. Slides were then rinsed briefly in nanopure H2O and washed three times at 10 min each in TBST with each pad sealed under ProPlate slide chambers (Grace Bio-Labs). After blocking for 30 min in 5% FBS, each pad was incubated in a primary antibody (at 1:100 dilution; see below for vendor information) against a selected protein for 2 h at 20 °C, followed by rinsing three times at 5 min each in TBST, and incubation with fluorescent secondary antibody (LICOR-IRDye 800) at 1:1000 dilution in 5% FBS for 1 h at 20 °C. Slides were rinsed three times for 5 min each in TBST and spun dry before scanning with InnoScan 710 AL. Fluorescence image analysis of spots was done using Innopsys's Mapix software. Background fluorescence for each spot was subtracted from the fluorescence signal for that spot followed by normalization to the FastGreen signal. Normalized signals were averaged across replicates, where feasible. Protein expression in each CA tissue was shown as fold change in normalized fluorescence relative to that of the NC counterpart. The primary antibodies used were obtained from ProteinTech Group with the following catalogue numbers: GAPDH, 60004-1-Ig; PSAT1, 10501-1-AP; MTHFD2, 12270-1-AP; SHMT2, 11099-1-AP; and PHGDH, 14719-1-AP; GLDC, 24827-1-AP.
Development of kinetic models based on SIRM data
The nonsteady-state kinetic models of purine de novo synthesis were developed to describe the transformation dynamics of metabolites, which included glucose, PRPP, serine, glycine, THF, N5,10-methylene-THF, N10-formyl-THF, formate, glycinamide ribonucleotide, formylglycinamide ribonucleotide, 5′-IMP, and 5′-AMP. The kinetic modeling used the established approach of ordinary differential equations (52, 53) and accounted for isotopomers and isotopologues of metabolites using an isotopic mapping algorithm. This algorithm was developed to trace the transfer of atoms (both labeled and unlabeled) from given tracers to various product(s) based on relevant enzyme reaction mechanisms. For example, the conversion of D3-Ser to D1-Gly in the cytoplasm by SHMT1 transfers one each of carboxyl carbon, methine carbon, and attached deuterium atoms from Ser to Gly as well as one methylene carbon and two attached deuterium atoms from Ser to N5,10-methylene-THF. The constructed isotopic atom maps were then used in the kinetic models to track individual isotopes in metabolites of various pathways.
Specifically, our models dynamically simulated the processes in the synthesis of AMP from glucose, serine, or glycine via glycolysis, pentose-phosphate pathway, one-carbon metabolism, and/or de novo purine synthesis pathway. The transformations of three different tracer configurations were simulated, including [13C6]glucose only, [13C6]glucose + D3-serine, and [13C6]glucose + D2-glycine. Both types of labeled atoms (13C and D) as well as 12C and 1H atoms were dynamically and simultaneously tracked to account for different isotopomer/isotopologue distributions of relevant labeled metabolites. Multiple compartments, including medium, cytoplasm, mitochondria, and subcompartments (i.e. three cytoplasmic pools of Ser/Gly), were modeled.
To best model the SIRM data, we used machine-learning methods, including the genetic algorithm described by Conn et al. (54, 55) and Powell's gradient-based algorithm (56). Specifically, for the paired CA and NC tissue slice data of patient UK022, we generated three sets of models: each assumed one, two, or three cytoplasmic Ser/Gly pools. These amounted to a total of six models (three hypotheses × two types of tissue slices) developed to explain the SIRM data. For these models, each machine-learning algorithm (54–57) was applied for least-squares fitting of kinetic models to SIRM data (29), and results from these algorithms were compared with finding the best fit for each combination of hypotheses and types of tissue slices. Then, the optimal models were chosen according to the Akaike information criterion (30) (−144.78, −175.57, and −195.42 for one- two-, and three-pool CA models, respectively). The multiple-pool CA model was also consistent with the observed lack of [13C]Ser release into the medium, in contrast to such release predicted by the one-pool CA model. Moreover, flux dynamics or reaction rate constants for CA versus NC tissue slices were compared in terms of the uptake and usage of glucose, serine, and glycine.
Statistical analyses
The data were analyzed using Student's two-tailed t test and significance was defined as p < 0.05. The n, t, and p values are given for specific data in the figure legends. The t tests were carried out using GraphPad or Excel. For RPPA and quantitative PCR analysis of paired tissue samples, the raw intensities for cancer and proximal samples were normalized to those of the lung tissue distal to the cancer, and the t test was calculated using the single group with a test value of 1 in Kaleidagraph (Synergy Software). RPPA data normalized to protein concentrations were also compared using the paired Wilcoxon test. For multiple testing, q values and the false discovery rates were calculated (33). Simulations and regression analyses were carried out using Kaleidagraph, which reports the best estimate of the parameters and the estimated standard deviations, as well as the χ2 and an overall correlation coefficient (R2).
Author contributions
T. W. M. F., P. P. S., L. J. B., Z. Q., A. L. M., R. M. H., C. V. D., and A. N. L. conceptualization; T. W. M. F., R. M. H., C. V. D., and A. N. L. resources; T. W. M. F., R. C. B., Y. Y., H. S., Y. C., P. D., Y. Z., P. P. S., L. J. B., Z. Q., A. L. M., R. M. H., C. V. D., and A. N. L. data curation; T. W. M. F., L. J. B., Z. Q., A. L. M., C. V. D., and A. N. L. software; T. W. M. F., R. C. B., Y. Y., H. S., Y. C., P. D., Y. Z., P. P. S., L. J. B., Z. Q., A. L. M., R. M. H., C. V. D., and A. N. L. formal analysis; T. W. M. F., A. L. M., R. M. H., C. V. D., and A. N. L. supervision; T. W. M. F., A. L. M., R. M. H., C. V. D., and A. N. L. funding acquisition; T. W. M. F. and A. N. L. validation; T. W. M. F., R. C. B., Y. Y., H. S., Y. C., P. D., Y. Z., P. P. S., L. J. B., Z. Q., A. L. M., R. M. H., C. V. D., and A. N. L. investigation; T. W. M. F., R. C. B., Y. Y., H. S., A. L. M., R. M. H., C. V. D., and A. N. L. methodology; T. W. M. F., R. C. B., Y. Y., H. S., Y. C., P. D., Y. Z., P. P. S., L. J. B., Z. Q., A. L. M., R. M. H., C. V. D., and A. N. L. writing-original draft; T. W. M. F., C. V. D., and A. N. L. project administration; T. W. M. F., C. V. D., and A. N. L. writing-review and editing.
Supplementary Material
Acknowledgment
We thank Jin Lian Tan for excellent technical assistance.
This work was supported National Institutes of Health Grants 1R01CA118434-01, 3R01CA118434-02, and 1R01ES022191-01 (to T. W. M. F.); 1P01CA163223-01A1 (to A. N. L. and T. W. M. F.); 1U24DK097215-01A1 (to R. M. H., T. W. M. F., and A. N. L.); Redox Metabolism Shared Resource of the University of Kentucky Markey Cancer Center National Institutes of Health Grant P30CA177558; the Lustgarten Fund 90049125; National Institutes of Health Grant T32 CA165990 (to R. C. B.); National Institutes of Health Grants 5R01CA051497 and 5R01CA057341; Leukemia Lymphoma Society Grant LLS-6363-11; and Stand-Up-to-Cancer/American Association for Cancer Research translational grant (to C. V. D.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S8, Tables S1–S3 and supporting Refs. 1–3.
- PRPP
- phosphoribosyl pyrophosphate
- CA
- cancer
- ECF
- ethyl chloroformate
- FBS
- fetal bovine serum
- mSIRM
- multiplexed stable isotope-resolved metabolomics
- NC
- noncancer
- NSCLC
- nonsmall-cell lung cancer
- THF
- tetrahydrofolate
- UHR-FTMS
- ultrahigh resolution Fourier transform mass spectrometry
- RPPA
- reverse-phase protein array
- SHMT
- serine hydroxymethyltransferase
- F
- forward
- R
- reverse
- CHO-THF
- N10-formyl tetrahydrofolate
- CH2-THF
- 5,10-methylene tetrahydrofolate
- GLDC
- glycine decarboxylase
- PSAT
- phosphoserine aminotransferase
- PHGDH
- phosphoglycerate dehydrogenase
- siNT
- nontargeting siRNA
- DMEM
- Dulbecco's modified Eagle's medium
- IHC
- immunohistochemistry
- PCNA
- proliferating cell nuclear antigen
- DAPI
- 4′,6-diamidino-2-phenylindole.
References
- 1. Lane A. N., and Fan T. W. (2015) Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 43, 2466–2485 10.1093/nar/gkv047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liu Y.-C., Li F., Handler J., Huang C. R., Xiang Y., Neretti N., Sedivy J. M., Zeller K. I., and Dang C. V. (2008) Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS One 3, e2722 10.1371/journal.pone.0002722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fan T. W., Tan J., McKinney M. M., and Lane A. N. (2012) Stable isotope resolved metabolomics analysis of ribonucleotide and RNA metabolism in human lung cancer cells. Metabolomics 8, 517–527 10.1007/s11306-011-0337-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Labuschagne C. F., van den Broek N. J., Mackay G. M., Vousden K. H., and Maddocks O. D. (2014) Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 7, 1248–1258 10.1016/j.celrep.2014.04.045 [DOI] [PubMed] [Google Scholar]
- 5. Wishart D. S., Jewison T., Guo A. C., Wilson M., Knox C., Liu Y., Djoumbou Y., Mandal R., Aziat F., Dong E., Bouatra S., Sinelnikov I., Arndt D., Xia J., Liu P., et al. (2013) HMDB 3.0-The human metabolome database in 2013. Nucleic Acids Res. 41, D801–D807 10.1093/nar/gks1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hori H., Tran P., Carrera C. J., Hori Y., Rosenbach M. D., Carson D. A., and Nobori T. (1996) Methylthioadenosine phosphorylase cDNA transfection alters sensitivity to depletion of purine and methionine in A549 lung cancer cells. Cancer Res. 56, 5653–5658 [PubMed] [Google Scholar]
- 7. Hayes J. D., and Dinkova-Kostova A. T. (2014) The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 39, 199–218 10.1016/j.tibs.2014.02.002 [DOI] [PubMed] [Google Scholar]
- 8. Wikoff W., Grapov D., Fahrmann J., DeFelice B., Rom W., Pass H., Kim K., Nguyen U., Taylor S. L., Kelly K., Fiehn O., and Miyamoto S. (2015) Metabolomic markers of altered nucleotide metabolism in early stage adenocarcinoma. Cancer Prevent. Res. 8, 410–418 10.1158/1940-6207.CAPR-14-0329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tedeschi P. M., Vazquez A., Kerrigan J. E., and Bertino J. R. (2015) Mitochondrial methylenetetrahydrofolate dehydrogenase (MTHFD2) overexpression is associated with tumor cell proliferation and is a novel target for drug development. Mol. Cancer Res. 13, 1361–1366 10.1158/1541-7786.MCR-15-0117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maddocks O. D. K., Athineos D., Cheung E. C., Lee P., Zhang T., van den Broek N. J. F., Mackay G. M., Labuschagne C. F., Gay D., Kruiswijk F., Blagih J., Vincent D. F., Campbell K. J., Ceteci F., Sansom O. J., et al. (2017) Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544, 372–376 10.1038/nature22056 [DOI] [PubMed] [Google Scholar]
- 11. Paone A., Marani M., Fiascarelli A., Rinaldo S., Giardina G., Contestabile R., Paiardini A., and Cutruzzolà F. (2014) SHMT1 knockdown induces apoptosis in lung cancer cells by causing uracil misincorporation. Cell Death Dis. 5, e1525 10.1038/cddis.2014.482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ben-Sahra I., Hoxhaj G., Ricoult S. J. H., Asara J. M., and Manning B. D. (2016) mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351, 728–733 10.1126/science.aad0489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jain M., Nilsson R., Sharma S., Madhusudhan N., Kitami T., Souza A. L., Kafri R., Kirschner M. W., Clish C. B., and Mootha V. K. (2012) Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336, 1040–1044 10.1126/science.1218595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang W. C., Shyh-Chang N., Yang H., Rai A., Umashankar S., Ma S., Soh B. S., Sun L. L., Tai B. C., Nga M. E., Bhakoo K. K., Jayapal S. R., Nichane M., Yu Q., Ahmed D. A., et al. (2012) Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 148, 259–272 10.1016/j.cell.2011.11.050 [DOI] [PubMed] [Google Scholar]
- 15. Tedeschi P. M., Markert E. K., Gounder M., Lin H., Dvorzhinski D., Dolfi S. C., Chan L. L., Qiu J., DiPaola R. S., Hirshfield K. M., Boros L. G., Bertino J. R., Oltvai Z. N., and Vazquez A. (2013) Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell Death Dis. 4, e877 10.1038/cddis.2013.393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dolfi S. C., Chan L. L., Qiu J., Tedeschi P. M., Bertino J. R., Hirshfield K. M., Oltvai Z. N., and Vazquez A. (2013) The metabolic demands of cancer cells are coupled to their size and protein synthesis rates. Cancer Metab. 1, 20 10.1186/2049-3002-1-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. DeNicola G. M., Chen P.-H., Mullarky E., Sudderth J. A., Hu Z., Wu D., Tang H., Xie Y., Asara J. M., Huffman K. E., Wistuba I. I., Minna J. D., DeBerardinis R. J., and Cantley L. C. (2015) NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 47, 1475–1481 10.1038/ng.3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Possemato R., Marks K. M., Shaul Y. D., Pacold M. E., Kim D., Birsoy K., Sethumadhavan S., Woo H.-K., Jang H. G., Jha A. K., Chen W. W., Barrett F. G., Stransky N., Tsun Z.-Y., Cowley G. S., et al. (2011) Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350 10.1038/nature10350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Locasale J. W., Grassian A. R., Melman T., Lyssiotis C. A., Mattaini K. R., Bass A. J., Heffron G., Metallo C. M., Muranen T., Sharfi H., Sasaki A. T., Anastasiou D., Mullarky E., Vokes N. I., Sasaki M., et al. (2011) Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 43, 869–874 10.1038/ng.890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sellers K., Fox M. P., Bousamra M., Slone S. P., Higashi R. M., Miller D. M., Wang Y., Yan J., Yuneva M. O., Deshpande R., Lane A. N., and Fan T. W. (2015) Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J. Clin. Invest. 125, 687–698 10.1172/JCI72873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fan T. W., Lane A. N., and Higashi R. M. (2016) Stable isotope resolved metabolomics studies in ex vivo tissue slices. Bio Protoc. 6, e1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Le A., Lane A. N., Hamaker M., Bose S., Gouw A., Barbi J., Tsukamoto T., Rojas C. J., Slusher B. S., Zhang H., Zimmerman L. J., Liebler D. C., Slebos R. J., Lorkiewicz P. K., Higashi R. M., et al. (2012) Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 15, 110–121 10.1016/j.cmet.2011.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lorkiewicz P., Higashi R. M., Lane A. N., and Fan T. W. (2012) High information throughput analysis of nucleotides and their isotopically enriched isotopologues by direct-infusion FTICR-MS. Metabolomics 8, 930–939 10.1007/s11306-011-0388-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fan T. W., Lorkiewicz P. K., Sellers K., Moseley H. N., Higashi R. M., and Lane A. N. (2012) Stable isotope-resolved metabolomics and applications for drug development. Pharmacol. Ther. 133, 366–391 10.1016/j.pharmthera.2011.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fan T. W., Warmoes M. O., Sun Q., Song H., Turchan-Cholewo J., Martin J. T., Mahan A., Higashi R. M., and Lane A. N. (2016) Distinctly perturbed metabolic networks underlie differential tumor tissue damages induced by immune modulator β-glucan in a two-case ex vivo non-small-cell lung cancer study. Cold Spring Harb. Mol. Case Stud. 2, a000893 10.1101/mcs.a000893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morrish F., Neretti N., Sedivy J. M., and Hockenbery D. M. (2008) The oncogene c-Myc coordinates regulation of metabolic networks to enable rapid cell cycle entry. Cell Cycle 7, 1054–1066 10.4161/cc.7.8.5739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu Y. C., Li F., Handler J., Huang C. R., Xiang Y., Neretti N., Sedivy J. M., Zeller K. I., and Dang C. V. (2008) Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS ONE 3, e2722 10.1371/journal.pone.0002722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murphy T. A., Dang C. V., and Young J. D. (2013) Isotopically nonstationary 13C flux analysis of Myc-induced metabolic reprogramming in B-cells. Metab. Eng. 15, 206–217 10.1016/j.ymben.2012.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Johnson M. (2008) in Biophysical Tools for Biologists (Detrich J. J., ed) pp. 781–807, Academic Press, San Diego, CA [Google Scholar]
- 30. Akaike H. (1974) A new look at the statistical model identification. IEEE Transactions on Automatic Control 19, 716–723 10.1109/TAC.1974.1100705 [DOI] [Google Scholar]
- 31. Boellner S., and Becker K.-F. (2015) Reverse phase protein arrays—quantitative assessment of multiple biomarkers in biopsies for clinical use. Microarrays 4, 98–114 10.3390/microarrays4020098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Akbani R., Becker K. F., Carragher N., Goldstein T., de Koning L., Korf U., Liotta L., Mills G. B., Nishizuka S. S., Pawlak M., Petricoin E. F. 3rd., Pollard H. B., Serrels B., and Zhu J. (2014) Realizing the promise of reverse-phase protein arrays for clinical, translational, and basic research: a workshop report: the RPPA (reverse phase protein array) society. Mol. Cell. Proteomics 13, 1625–1643 10.1074/mcp.O113.034918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rosner B. (2006) Fundamentals of Biostatistics, 6th Ed., pp. 573–581, Thomson, Belmont, CA [Google Scholar]
- 34. Kim J.-W., Zeller K. I., Wang Y., Jegga A. G., Aronow B. J., O'Donnell K. A., and Dang C. V. (2004) Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol. Cell. Biol. 24, 5923–5936 10.1128/MCB.24.13.5923-5936.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wise D. R., DeBerardinis R. J., Mancuso A., Sayed N., Zhang X.-Y., Pfeiffer H. K., Nissim I., Daikhin E., Yudkoff M., McMahon S. B., and Thompson C. B. (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. U.S.A. 105, 18782–18787 10.1073/pnas.0810199105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Momb J., Lewandowski J. P., Bryant J. D., Fitch R., Surman D. R., Vokes S. A., and Appling D. R. (2013) Deletion of Mthfd1l causes embryonic lethality and neural tube and craniofacial defects in mice. Proc. Natl. Acad. Sci. U.S.A. 110, 549–554 10.1073/pnas.1211199110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tedeschi P. M., Markert E. K., Gounder M., Lin H., Dvorzhinski D., Dolfi S. C., Chan L. L., Qiu J., DiPaola R. S., Hirshfield K. M., Boros L. G., Bertino J. R., Oltvai Z. N., and Vazquez A. (2013) Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell Death Dis. 4, e877 10.1038/cddis.2013.393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tibbetts A. S., and Appling D. R. (2010) Compartmentalization of mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 30, 57–81 10.1146/annurev.nutr.012809.104810 [DOI] [PubMed] [Google Scholar]
- 39. Pike S. T., Rajendra R., Artzt K., and Appling D. R. (2010) Mitochondrial C1-tetrahydrofolate synthase (MTHFD1L) supports the flow of mitochondrial one-carbon units into the methyl cycle in embryos. J. Biol. Chem. 285, 4612–4620 10.1074/jbc.M109.079855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. French J. B., Jones S. A., Deng H., Pedley A. M., Kim D., Chan C. Y., Hu H., Pugh R. J., Zhao H., Zhang Y., Huang T. J., Fang Y., Zhuang X., and Benkovic S. J. (2016) Spatial colocalization and functional link of purinosomes with mitochondria. Science 351, 733–737 10.1126/science.aac6054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xie H., Hanai J., Ren J.-G., Kats L., Burgess K., Bhargava P., Signoretti S., Billiard J., Duffy K. J., Grant A., Wang X., Lorkiewicz P. K., Schatzman S., Bousamra M. 2nd., Lane A. N., et al. (2014) Targeting lactate dehydrogenase-A (LDH-A) inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor initiating cells. Cell Metab. 19, 795–809 10.1016/j.cmet.2014.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fan T. W., Lane A. N., and Higashi R. M. (2016) Stable isotope resolved metabolomics studies in ex vivo tissue slices. Bio Protoc. 6, e1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fan T. W.-M. (2012) in The Handbook of Metabolomics: Pathway and Flux Analysis, Methods in Pharmacology and Toxicology (Fan T. W.-M., Lane A. N., and Higashi R. M., eds) pp. 7–27, Springer Science, New York [Google Scholar]
- 44. Fan T. W.-M., Bandura L., Higashi R. M., and Lane A. N. (2005) Metabolomics–edited transcriptomics analysis of Se anticancer action in human lung cancer cells. Metabolomics 1, 325–339 [Google Scholar]
- 45. Yang Y., Fan T. W., Lane A. N., and Higashi R. M. (2017) Chloroformate derivatization for tracing the fate of amino acids in cells by multiple stable isotope resolved metabolomics (mSIRM). Anal. Chim. Acta 976, 63–73 10.1016/j.aca.2017.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fan T. W., Kucia M., Jankowski K., Higashi R. M., Rataczjak J., Rataczjak M. Z., and Lane A. N. (2008) Proliferating rhabdomyosarcoma cells shows an energy producing anabolic metabolic phenotype compared with primary myocytes. Mol. Cancer 7, 79 10.1186/1476-4598-7-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fan T. W., and Lane A. N. (2011) in Methodologies for Metabolomics: Experimental Strategies and Techniques (Lutz N., Sweedler J. V., and Weavers A. R., eds), pp. 525–584, Cambridge University Press, New York [Google Scholar]
- 48. Fan T. W., and Lane A. N. (2008) Structure-based profiling of metabolites and isotopomers by NMR. Progr. NMR Spectrosc. 52, 69–117 10.1016/j.pnmrs.2007.03.002 [DOI] [Google Scholar]
- 49. Fan T. W.-M. (1996) Metabolite profiling by one- and two-dimensional NMR analysis of complex mixtures. Progr. Nuclear Magnetic Reson. Spectrosc. 28, 161–219 10.1016/0079-6565(95)01017-3 [DOI] [Google Scholar]
- 50. Lane A. N., Fan T. W., Xie Z., Moseley H. N., and Higashi R. M. (2009) Stable isotope analysis of lipid biosynthesis by high resolution mass spectrometry and NMR. Anal. Chim. Acta 651, 201–208 10.1016/j.aca.2009.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Moseley H. (2010) Correcting for the effects of natural abundance in stable isotope resolved metabolomics experiments involving ultra-high resolution mass spectrometry. BMC Bioinformatics 11, 139 10.1186/1471-2105-11-139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Voit E. O., Qi Z., and Miller G. W. (2008) Steps of modeling complex biological systems. Pharmacopsychiatry 41, Suppl. 1, S78–84 10.1055/s-2008-1080911 [DOI] [PubMed] [Google Scholar]
- 53. Qi Z., Miller G. W., and Voit E. O. (2008) A mathematical model of presynaptic dopamine homeostasis: implications for schizophrenia. Pharmacopsychiatry 41, Suppl. 1, S89–S98 10.1055/s-2008-1080936 [DOI] [PubMed] [Google Scholar]
- 54. Conn A. R., Gould N. I. M., and Toint P. L. (1997) A globally convergent augmented Lagrangian barrier algorithm for optimization with general inequality constraints and simple bounds. Mathemat. Comput. 66, 261–288 10.1090/S0025-5718-97-00777-1 [DOI] [Google Scholar]
- 55. Conn A. R., Gould N. I. M., and Toint P. L. (1991) A globally convergent augmented Lagrangian algorithm for optimization with general constraints and simple bounds. SIAM J. Numer. Anal. 28, 545–572 10.1137/0728030 [DOI] [Google Scholar]
- 56. Powell M. J. D. (1977) in Numerical Analysis (Watson G. A., ed), pp. 144–157, Springer Verlag, New York [Google Scholar]
- 57. Marler R. T., and Arora J. S. (2004) Survey of multi-objective optimization methods for engineering. Structural and Multidisciplinary Optimization 26, 369–395 10.1007/s00158-003-0368-6 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.