

Abstract
The peptide hormone adropin regulates energy metabolism in skeletal muscle and plays important roles in the regulation of metabolic homeostasis. Besides muscle, the liver has an essential role in regulating glucose homeostasis. Previous studies have reported that treatment of diet-induced obese (DIO) male mice with adropin34–76 (the putative secreted domain) reduces fasting blood glucose independently of body weight changes, suggesting that adropin suppresses glucose production in the liver. Here, we explored the molecular mechanisms underlying adropin's effects on hepatic glucose metabolism in DIO mice. Male DIO B6 mice maintained on a high-fat diet received five intraperitoneal injections of adropin34–76 (450 nmol/kg/injection) over a 48-h period. We found that adropin34–76 enhances major intracellular signaling activities in the liver that are involved in insulin-mediated regulation of glucose homeostasis. Moreover, treatment with adropin34–76 alleviated endoplasmic reticulum stress responses and reduced activity of c-Jun N-terminal kinase in the liver, explaining the enhanced activities of hepatic insulin signaling pathways observed with adropin34–76 treatment. Furthermore, adropin34–76 suppressed cAMP activated protein kinase A (PKA) activities, resulting in reduced phosphorylation of inositol trisphosphate receptor, which mediates endoplasmic reticulum calcium efflux, and of cAMP-responsive element–binding protein, a key transcription factor in hepatic regulation of glucose metabolism. Adropin34–76 directly affected liver metabolism, decreasing glucose production and reducing PKA-mediated phosphorylation in primary mouse hepatocytes in vitro. Our findings indicate that major hepatic signaling pathways contribute to the improved glycemic control achieved with adropin34–76 treatment in situations of obesity.
Keywords: type 2 diabetes, insulin resistance, c-Jun N-terminal kinase (JNK), endoplasmic reticulum stress (ER stress), lipogenesis, insulin receptor substrate 1 (IRS1), energy metabolism, gluconeogenesis, protein kinase A (PKA), adropin
Introduction
Adropin is a small peptide that is implicated in the physiological regulation of metabolic homeostasis (1–3). In mice and humans, adropin is abundantly expressed in the brain as well as the liver (3). Although the source and the mechanism of secretion are elusive, circulating adropin is readily detected in both mice and humans (2–5).
Mounting evidence indicates that adropin may act as a hormone in regulating metabolic homeostasis, in part by controlling substrate (glucose and fatty acid) metabolism in skeletal muscle (1–3). Using male C57BL/6J (B6)8 mice, our previous studies identified a therapeutic potential for adropin in treating impaired glycemic control that is frequently observed with obesity (1, 3). Adropin knockout (AdrKO) mice are insulin-resistant, whereas transgenic overexpression of adropin improves glycemic control of the mice maintained on either chow or high-fat diet (HFD) (1, 3). Moreover, treatment of diet-induced obese (DIO) B6 mice with the putative secreted domain of adropin, adropin34–76, improves glucose tolerance as well as whole-body insulin sensitivity (3, 6). These responses are accounted for at least in part by adropin's enhancement of the insulin intracellular signaling pathway in skeletal muscle (6).
Adropin is expressed in the liver (3) and appears to be co-regulated with genes involved in hepatic glucose and lipid metabolism (5). The liver is an important target organ for insulin, and insulin's metabolic actions in the liver are essential for glucose homeostasis (7, 8). Our previous studies demonstrate that AdrKO mice display an impaired suppression of hepatic glucose production under a hyperinsulinemic-euglycemic clamp condition, indicating that adropin deficiency associates with insulin resistance in the liver (1). In addition, fasting hyperinsulinemia and hyperglycemia are observed in AdrKO mice (1). As liver glucose production is a major determinant of fasting blood glucose level and the suppression of liver glucose production has a central role in insulin's glucose-lowering effect (9), these observations suggest that adropin influences insulin action and glucose metabolism in the liver (1, 10). Moreover, we have reported that adropin34–76 treatment reduced fasting hyperglycemia and hyperinsulinemia in diabetic DIO mice (3), indicating that adropin treatment improves hepatic glucose metabolism and may enhance insulin's hepatic intracellular signaling actions in obesity.
In obesity, insulin resistance and the aberrant hepatic glucose metabolism involve multiple mechanisms (7, 8). Among them, obesity-associated endoplasmic reticulum (ER) stress leads to cellular insulin resistance in part by activating c-Jun N-terminal kinase (JNK), the activation of which plays a prominent role in impairing the insulin intracellular signaling pathway (11, 12).
Insulin signaling interacts with cAMP-dependent pathways to coordinately regulate glucose metabolism in the liver (13). cAMP is a second messenger in the G protein–coupled receptor (GPCR) signaling pathway, and cAMP-dependent pathways play a central role in mediating the hepatic actions of glucagon, another key hormone in controlling glucose homeostasis (13). Of relevance, recent studies suggest that GPCR mediates adropin's intracellular signaling pathways (14, 15).
Here we report studies that address the effects of adropin34–76 treatment on key signaling pathways underlying insulin's effect on hepatic glucose metabolism in DIO mice. We further investigated adropin's actions on ER stress and JNK activity. In addition, we explored the effect of adropin on cAMP-dependent signaling pathways in the liver.
Results
Adropin34–76 treatment enhances intracellular signaling actions underlying insulin's effect on hepatic glucose metabolism
In the current report, we employ an adropin34–76 treatment protocol (five injections over 48 h with 450 nmol/kg for each injection) previously assessed in a dose-response study investigating effects on glucose homeostasis (3). The specific dosage and time period of treatment robustly enhance glucose tolerance as well as whole-body insulin sensitivity without altering body weight in the DIO mice (3, 6). In the current studies, we first confirmed adropin's glucose-lowering effect by showing that adropin34–76 treatment reduced fasting hyperglycemia as compared with the vehicle treatment in the DIO mice (Fig. S1).
Insulin plays an essential role in controlling hepatic glucose production in part by modulating liver metabolism (7–9). We then assessed hepatic intracellular signaling pathways that are employed by insulin to regulate glucose metabolism. Analysis of key mediators of insulin signaling showed marked differences between adropin34–76 treatment and vehicle control groups (Figs. 1 and 2). Increased Ser307 phosphorylation of insulin receptor substrate 1 (IRS1) that is frequently observed in B6 mice fed HFD (Fig. S2A) (7, 16) was markedly reduced by adropin34–76 treatment (Fig. 1A). Ser307 phosphorylation inhibits IRS1 signaling by antagonizing its tyrosine phosphorylation by insulin receptor (7, 16). Here we showed that the phosphorylation of IRS1 on Tyr608 that was reduced in mice on HFD (Fig. S2A) was increased with adropin34–76 treatment (Fig. 1A). Hepatic expression of IRS2 was reduced in mice fed HFD (Fig. S2A) (7, 16), but this level in DIO mice was increased with adropin34–76 treatment (Fig. 1B).
Figure 1.

Adropin34–76 treatment enhanced IRS signaling in the liver. A, the phosphorylation levels of Ser307 in IRS1 (n = 3–5) and total IRS1 protein levels (n = 4–5) as well as the phosphorylation levels of Tyr608 in IRS1 immunoprecipitates (IP) (n = 4) were measured by Western blotting (IB). The Western blotting of the phosphorylation levels of Ser307 in IRS1 were repeated (n = 4–5), and similar changes were detected. α-Tubulin was used as the loading control for pIRS1 (Ser307) and total IRS1. The same α-tubulin band serving as the loading control for total IRS1 was used as the loading control for the blots of pAKT (Ser473) and total AKT (Fig. 2A) and the blots of pIKK (α/β) (Ser176/180) and total IKK (α/β) (Fig. S6). B, IRS2 protein levels (n = 4–5) and message levels (Irs2) (n = 5–6) were determined by Western blotting and RT-PCR, respectively. In Western blotting, GAPDH was used as the loading control for IRS2. The same GAPDH band was used as the loading control for the blots of p-c-Jun (Ser63) and total c-Jun (Fig. 4E) and the blots of pCREB (Ser133) and total CREB (Fig. 8B). *, p ≤ 0.05; ***, p < 0.0005, adropin versus vehicle. Error bars, S.E.
Figure 2.

Adropin34–76 treatment enhanced AKT signaling in the liver. A and B, the phosphorylation levels of Ser473 in AKT and total AKT protein levels (n = 4) (A) and the phosphorylation levels of Ser9 in GSK 3β and total GSK 3β protein levels (n = 4–5) (B) were determined by Western blotting. In A, α-tubulin was used as the loading control. The same α-tubulin band was used as the loading control for the blot of total IRS1 (Fig. 1A) and the blots of pIKK (α/β) (Ser176/180) and total IKK (α/β) (Fig. S6). In B, GAPDH was used as the loading control. C, glycogen levels were determined and were normalized to tissue masses (n = 8). D, nuclear levels (n = 4–5) and whole-tissue level (n = 4) of FoxO1 were measured by Western blotting. Histone H3 was used as the loading control in the blot of nuclear lysates. The same histone H3 band was used as the loading control for the blots of (n)SREBP1c (Fig. 6A), (n)CRTC2 (Fig. 8B), and (n)NF-κB p65 (Fig. S6). GAPDH was used as the loading control in the blot of whole-tissue lysates. *, p ≤ 0.05, adropin versus vehicle. Error bars, S.E.
AKT is a critical mediator of IRS1/2 signaling (7), and Ser473 phosphorylation is frequently used as a surrogate marker of AKT activity (6). In our studies, we showed that AKT Ser473 phosphorylation was increased with adropin34–76 treatment (Fig. 2A), indicating an activation of AKT (6). Activated AKT phosphorylates glycogen synthase kinase-3 (GSK-3) and members of the Forkhead box O (FoxO) family (7). We found that adropin34–76 treatment increased the phosphorylation level of Ser9 in GSK-3β (Fig. 2B), indicating an inhibition of GSK activity (7). The inhibition of GSK activity is expected to promote glycogen synthesis (7), and consistent with this prediction, liver glycogen content was increased following adropin34–76 treatment (Fig. 2C). FoxO1 phosphorylation by AKT results in its nuclear exclusion and degradation, leading to inhibition of FoxO1-dependent transcription (7). Here we found that adropin34–76 treatment reduced the nuclear level of FoxO1 as well as its whole-tissue level (Fig. 2D), which is expected to lead to an inhibition of FoxO1 transcription activity.
FoxO1 down-regulates the expression of glucokinase (Gck), a key enzyme facilitating glucose uptake, and up-regulates the expressions of G6Pase (G6pc) and phosphoenolpyruvate carboxykinase (PEPCK) (Pck1), enzymes involved in hepatic glucose production (17). Consistent with these effects, we found that adropin34–76 treatment increased Gck expression (Fig. 3A), whereas it down-regulated the expressions of G6pc and Pck1 (Fig. 3B). Pyruvate carboxylase (PC) is another enzyme playing a key role in hepatic gluconeogenesis (8, 18). However, adropin34–76 treatment altered neither its expression level (percentage of vehicle: adropin, 101 ± 5.5%; vehicle, 100 ± 1.7%) nor the level of acetyl-CoA (Fig. S3A), an allosteric regulator of PC activity (8, 18). The gene expression level of liver pyruvate kinase (Pklr), the last rate-limiting enzyme in the hepatic glycolysis pathway, was also not altered with adropin34–76 treatment (Fig. 3A).
Figure 3.

Adropin34–76 treatment altered the expression of glucose metabolic genes in the liver. The message levels of genes in glycolysis (A), including glucokinase (Gck) (n = 6) and liver pyruvate kinase (Pklr) (n = 6), and genes in glucose production (B), including G6Pase (G6pc) (n = 6) and PEPCK-1 (Pck1) (n = 6–7) were determined by real-time RT-PCR. *, p ≤ 0.05, adropin versus vehicle. Error bars, S.E.
Taken together, our data suggest that adropin34–76 treatment rapidly enhances intracellular signaling actions that are employed by insulin. It should be noted that the DIO mice displayed no significant difference in circulating insulin levels between the adropin-treated and vehicle-treated group (2.27 ± 0.20 versus 2.38 ± 0.14 ng/ml), which is consistent with the previous report (3). Therefore, the observed effects of insulin-signaling action are not accounted for by changes in circulating insulin concentrations.
Adropin34–76 treatment alleviates hepatic ER stress responses
ER stress triggers unfolded protein responses through pathways mediated by three classical signal transducers: PKR-like ER kinase (PERK), inositol-requiring enzyme 1α (IRE1α), and activating transcription factor 6 (ATF6) (11, 19). PERK activation results in phosphorylation level of eukaryotic initiation factor 2α (eIF2α) (19). In our studies, we found that adropin34–76 treatment of DIO mice decreased phosphorylation of eIF2α (Fig. 4A) that was increased by high fat diet feeding (Fig. S2B). The result shows that adropin treatment may attenuate PERK activation. Activation of IRE1α leads to increases in the spliced form of X-box–binding protein 1 (XBP1s) and nuclear translocation of XBP1s protein (19). Our data showed that adropin34–76 treatment reduced the nuclear level as well as the whole-tissue level of XBP1s protein (Fig. 4B), thus suggesting an inhibition of adropin on the signaling actions of IRE1α branch. Activation of ATF6 in response to ER stress induces proteolytic cleavage and nuclear translocation of the cleaved form (19); however, we did not detect changes in the nuclear level of ATF6 (the cleaved form) with adropin34–76 treatment (percentage of vehicle: adropin, 96 ± 9.2%; vehicle, 100 ± 18%).
Figure 4.

Adropin34–76 treatment alleviated ER stress responses and diminished JNK signaling in the liver. A and B, the phosphorylation levels of Ser51 in eIF2α and total eIF2α levels in whole-tissue lysates (A) and the levels of XBP-1s in nuclear lysates (n = 4–5) and whole-tissue lysates (n = 4) (B) were determined by Western blotting (n = 4–5). In A, α-tubulin was used as the loading control. In B, histone H3 was used as the loading control for nuclear XBP1s, and GAPDH was used as the loading control for whole-tissue XBP1s. C, BiP message (Hspa5) levels (n = 6) and protein levels in whole-tissue lysates (n = 4–5) were determined by real-time RT-PCR and Western blotting, respectively. In Western blotting, GAPDH was used as the loading control for BiP. D and E, the phosphorylation levels of Thr183/Tyr185 in JNK and total JNK levels (n = 4–5) (arrows indicating JNK splice isoforms) (D) and the phosphorylation levels of Ser63 in c-Jun and total c-Jun levels (n = 4–5) in whole-tissue lysates (E) were determined by Western blotting (n = 4–5). In D, α-tubulin was used as the loading control. The same α-tubulin band was used as the loading control for the blot of whole-tissue IP3R1 (Fig. 7). In E, GAPDH was used as the loading control. The same GAPDH band was used as the loading control for the blot of total IRS2 (Fig. 1B) and the blots of pCREB (Ser133) and total CREB (Fig. 8B). *, p ≤ 0.05; ****, p < 0.0001, adropin versus vehicle. Error bars, S.E.
The activation of unfolded protein response signaling pathways up-regulates the expression of molecular chaperones facilitating protein folding, including binding immunoglobulin protein (BiP), to counter ER stress (20). We observed a decrease in BiP message level following adropin treatment, which is in line with the attenuated ER stress resulting in a reduced demand of molecular chaperones. However, the level of BiP protein was not altered by adropin (Fig. 4C), which might be accounted for by the long half-life of the protein (21).
Adropin34–76 treatment diminishes JNK signaling in the liver
ER stress can lead to the activations of JNK signaling as well as inhibitor κB kinase (IKK) (7, 19, 22, 23). Consistent with the previous finding (22), in parallel with inducing ER stress in liver, high fat diet feeding led to increases in the phosphorylation levels of JNK and c-Jun (the canonical JNK substrate) (Fig. S2B), which together indicates JNK activation (22). These phosphorylation levels were reduced following adropin34–76 treatment (Fig. 4, D and E), which indicates a suppression of JNK activity by adropin. JNK regulates expression of pro-inflammatory cytokines that can antagonize insulin receptor signaling (23). However, the expression levels of three key cytokines, including tumor necrosis factor (Tnf), interleukin-1b (Il1b), and interleukin-6 (Il6), were not changed with adropin34–76 treatment (Fig. S4). Inhibition of JNK may promote expression of fibroblast growth factor-21 (FGF21), which could contribute to an improved insulin action (23), whereas in our studies, adropin34–76 treatment did not affect Fgf21 expression (Fig. S5). In addition, we found that adropin treatment did not alter either the phosphorylation levels of IKK or the nuclear level of NF-κB that translocates into the nucleus upon IKK activation (Fig. S6). Taken together, the results demonstrate that adropin exerts a selective effect on liver JNK in the DIO mice.
Adropin34–76 treatment down-regulates hepatic lipogenic genes
Our previous studies demonstrated that 14 days of adropin34–76 treatment reduced hepatic steatosis in DIO mice, and transgenic overexpression of adropin markedly reduced plasma triacylglycerol (TAG) level (3). Our current studies showed that short-term treatment of adropin resulted in a trend for the reduction of hepatic TAG content (Fig. 5A), which confirms the previous finding employing the same treatment protocol (3). This short-term treatment did not alter plasma TAG levels (adropin, 59 ± 15 mg/dl; vehicle, 71 ± 7.8 mg/dl).The shorter time period (2 days) of treatment in the current studies may underlie the lack of significant changes in the TAG levels. Despite this, we observed that adropin34–76 treatment significantly reduced or induced strong trends of decrease in the expression of the enzymes involved in de novo fatty acid synthesis (Fig. 5B) and TAG synthesis (Fig. 5C). The expression of acetyl-CoA carboxylase-β that plays a key role in fatty acid oxidation (24, 25) was reduced by adropin (Fig. 5D). The adropin-induced down-regulation of acetyl-CoA carboxylase expression is consistent with our previous finding that adropin34–76 treatment reduced the level of hepatic malonyl-CoA, the product of acetyl-CoA carboxylase and a negative regulator of fatty acid oxidation (6).
Figure 5.

Adropin34–76 treatment reduced the expressions of lipogenic genes in the liver. A, triacylglycerol contents were measured and were normalized to tissue masses (n = 8). Real-time RT-PCR was performed to determine the message levels of genes in de novo fatty acid synthesis, including acetyl-CoA carboxylase-α (Acaca) (n = 6), fatty acid synthase (Fasn) (n = 5–6), stearoyl-CoA desaturase (Scd1) (n = 6), and Elovl6 (elongase) (n = 6) (B); de novo TAG synthesis, including mitochondrial glycerol-3-phosphate acyltransferase (Gpam) (n = 6) and diacylglycerol acyltransferase-2 (Dgat2) (n = 6) (C); and acetyl-CoA carboxylase-β (Acacb) (n = 5) (D). *, p ≤ 0.05, adropin versus vehicle Error bars, S.E.
The expression of lipogenic enzymes is partly controlled by sterol regulatory element–binding protein 1c (SREBP1c) located in the ER membrane. Post-translational processing of nascent (precursor) SREBP1c results in the release of the short-form SREBP1C that translocates into the nucleus to regulate gene transcription (24). Here, we observed that adropin34–76 treatment reduced nuclear (short form) SREBP1c levels without altering the levels of its precursor (Fig. 6A), which indicates that adropin suppresses post-translational processing of SREBP1c.
Figure 6.

Adropin34–76 treatment reduced the nuclear level of SREBP1c in the liver. A, the nuclear levels of SREBP1c (n = 4–5) and the levels of precursor SREBP1c in whole-tissue lysates (n = 4–5) were measured by Western blotting. GAPDH and histone H3 were used as the loading control in the blot of whole-tissue lysates and nuclear lysates, respectively. The same histone H3 band was used as the loading control for the blots of (n)FoxO1 (Fig. 2D), (n)CRTC2 (Fig. 8B), and (n)NF-κB p65 (Fig. S6). B, BiP protein levels in the immunoprecipitates (IP) of precursor SREBP1c from microsomal fractions were determined by Western blotting (IB) (n = 4–5). The blotting was repeated twice, and the blot with 3 samples/treatment was presented. *, p ≤ 0.05; **, p < 0.01, adropin versus vehicle. Error bars, S.E.
Under normal conditions, BiP interacts with precursor SREBP1c to sequester it at the ER membrane (26). However, ER stress disrupts this interaction and thus promotes the post-translational processing and nuclear translocation of SREBP1c (26). We observed that adropin34–76 treatment increased the level of BiP in the SREBP1c immunoprecipitates from microsomal fraction (Fig. 6B), as indicated by the elevated ratio of BiP to SREBP1c. The result suggests that adropin promotes the interaction between BiP and SREBP1c, which would contribute to the reduction of precursor SREBP1c processing and subsequent nuclear translocation of the short form.
Lipid intermediates impact cellular insulin signaling actions (8), and we performed lipidomic profiling to determine the levels of several lipid species that are known to modulate insulin pathways. Adropin34–76 treatment did not alter the levels of major long-chain acyl-CoAs, although reduced stearoyl-CoA (18:0) was observed (Fig. S3B), which might be accounted for by the reduced expression of elongase (Elovl6) (Fig. 5B). Further analysis of the ratio of saturated acyl-CoA (the sum of 16:0 and 18:0) to unsaturated acyl-CoA (the sum of 16:1 and 18:1) reveals a trend of decrease in adropin-treated mice compared with vehicle-treated ones (Fig. S3C). Adropin34–76 treatment also did not alter the levels of either ceramide (Fig. S3D) or diacylglycerol (adropin/vehicle ratio: 1,2-dipalmitoylglycerol, 0.8; 1,3-dipalmitoylglycerol, 1.0). Moreover, the treatment did not affect the phosphorylation level of Thr172 in AMP-activated protein kinase (Fig. S7), an enzyme involved in nontranscriptional regulation of lipid metabolism (27), which indicates that adropin does not alter AMP-activated protein kinase activity in the DIO liver.
Adropin34–76 treatment coordinately alters the phosphorylation levels of inositol-1,4,5-triphosphate receptor (IP3R) in the liver
Disruption of ER calcium homeostasis leads to ER stress, and the impairment of ER calcium retention underpins the development of hepatic ER stress in obesity (28). IP3R is the major channel mediating calcium efflux from ER, and its phosphorylation state that impacts channel activity is modulated by kinases such as PKA and AKT (29, 30). In our studies, obese mice (on a high fat diet) displayed an increased phosphorylation level of PKA substrate sites in IP3R as compared with the mice on a low fat diet (Fig. S8), which indicates a potential activation of the channel in mediating calcium efflux from ER (30). Adropin34–76 treatment of the obese mice reduced this level, suggesting the attenuation of the activation in the DIO mice (Fig. 7).
Figure 7.

Adropin34–76 treatment decreased PKA phosphorylation and increased AKT phosphorylation of IP3R in the liver. A, the phosphorylation levels of PKA substrate sites (n = 4) and the phosphorylation levels of AKT substrate sites in IP3R1 following immunoprecipitation (IP) of IP3R1 as well as total IP3R1 levels in whole-tissue lysates (n = 4–5) were determined by Western blotting (IB). α-Tubulin was used as the loading control for whole-tissue IP3R1. The same α-tubulin band was used as the loading control for the blots of pJNK (Thr183/Tyr185) and total JNK (Fig. 4D). **, p < 0.01, adropin versus vehicle. Error bars, S.E.
In parallel to the enhanced AKT action, adropin34–76 treatment increased the phosphorylation level of the AKT substrate site in IP3R (Fig. 7), indicating an inhibition of the channel activity (30). The concerted effects by adropin on IP3R phosphorylation state are expected to lead to a suppression of IP3R channel activity resulting in a reduced calcium efflux from ER.
Adropin34–76 treatment inhibits PKA signaling actions in the liver
In addition to AKT, PKA plays a key role in regulating liver glucose metabolism (13). Here, we demonstrate that adropin34–76 treatment decreased PKA activity in liver crude cytosolic extracts (percentage of vehicle: adropin, 74 ± 8.4%; vehicle, 100 ± 3.6%; p = 0.05) as well as reduced the level of cAMP (Fig. 8A), the canonical second messenger activating PKA (31). These changes are consistent with the observed reduction of PKA-mediated IP3R phosphorylation following adropin treatment (Fig. 7). Besides IP3R, the cAMP-responsive element-binding protein (CREB) is a well-established PKA substrate and a central transcription factor mediating cAMP-dependent gene transcription (31). Here, we demonstrate that adropin34–76 treatment reduced the phosphorylation level of Ser133 in CREB (Fig. 8B), indicating a potential reduction of CREB transcriptional activity (31). Moreover, adropin treatment reduced the nuclear level of CREB-regulated transcription co-activator 2 (CRTC2) (Fig. 8B), a key co-activator of CREB in cAMP-dependent gene transcription (32). Together, these results suggest that adropin actions suppress the cAMP-PKA signaling pathway in the liver of DIO mice.
Figure 8.

Adropin34–76 treatment decreased cAMP level and the phosphorylation level of CREB in the liver. A, cAMP contents were measured and were normalized to tissue masses (n = 8). B, the phosphorylation levels of Ser133 in CREB and total CREB levels in whole-tissue lysates (n = 4–5) as well as the nuclear levels of CRTC2 (n = 4–5) were measured by Western blotting. GAPDH and histone H3 were used as the loading control in whole-tissue lysates and nuclear lysates, respectively. The same GAPDH band was used as the loading control for the blot of total IRS2 (Fig. 1B) and the blots of p-c-Jun (Ser63) and total c-Jun (Fig. 4E). The same histone H3 band was used as the loading control for the blots of (n)FoxO1 (Fig. 2D), (n)SREBP1c (Fig. 6A), and (n)NF-κB p65 (Fig. S6). *, p ≤ 0.05, adropin versus vehicle. Error bars, S.E.
Adropin34–76 directly suppresses glucose production in cultured hepatocytes
Primary cultured mouse hepatocytes were used to explore whether adropin34–76 would exert a direct effect on liver glucose production. Endogenous glucose production was induced in serum-starved primary cultured hepatocytes following the addition of glucagon and pyruvate (33). We found that adropin34–76 treatment attenuated glucose production (Fig. 9A), which demonstrates that adropin directly inhibits glucose production in hepatocytes. To explore the underlying mechanisms, we assessed cAMP-PKA signaling. In our experimental settings, we found that the cAMP level in primary hepatocytes was too low, which would prevent a potential decrease in response to adropin34–76 from being detected. We then measured cAMP level in HepG2 liver cells treated with the same amount of adropin34–76 as in primary hepatocytes and found decreases in this level, as compared with vehicle-treated cells (Fig. 9B). Consistent with the in vivo findings, adropin34–76 reduced the phosphorylation levels of CREB and multiple other PKA substrates in the primary hepatocytes (Fig. 9C). Expression levels of G6pc and Pck1 in the primary hepatocytes were also suppressed by adropin34–76 treatment (Fig. 9D).
Figure 9.

Adropin34–76 treatment suppresses glucose production in primary mouse hepatocyte. A, glucose production from the hepatocytes was determined by quantifying glucose levels in culture media. The assay was performed from three hepatocyte preparations, and the data were pooled and presented as a percentage of the vehicle-treated values (n = 10). The levels of glucose production in the vehicle-treated group were around 0.1 mg/mg of protein/h. B, cAMP levels in HEPG2 liver cells were measured in the presence of increasing levels of forskolin (an activator of adenylate cyclase) in the culture media. The experiments were repeated three times. C, the phosphorylation levels of Ser133 in CREB and total CREB levels, and the phosphorylation levels of PKA substrates in the hepatocytes were determined by Western blotting (n = 2). Heat shock protein 90 (Hsp90) was used as the loading control. D, the message levels of glucose production genes, including G6Pase (G6pc) (n = 5) and PEPCK (Pck1) (n = 2–3), in the hepatocytes were determined by real-time PCR. The quantitation of Pck1 was repeated in another experiment (n = 3), and the levels of Pck1 in the adropin-treated group were below the detection limit. Hypoxanthine guanine phosphoribosyltransferase was used as the reference gene. *, p ≤ 0.05, adropin versus vehicle. Error bars, S.E.
Discussion
The major finding of this report is that adropin34–76 treatment enhances hepatic IRS-AKT signaling actions in DIO mice. These data suggest that adropin sensitizes the insulin intracellular signaling pathway, leading to reduced fasting hyperglycemia. The finding is in line with our previous study showing that adropin34–76 treatment sensitizes insulin intracellular signaling pathways in skeletal muscle in DIO mice (6) as well as the report demonstrating that adropin augments AKT signaling actions in endothelial cells (34). Furthermore, consistent with our current results, recent data reveal that adropin34–76 treatment enhances IRS and AKT signaling actions in the heart (35). In the current studies, despite the enhanced intracellular signaling actions, the serum insulin level was not altered following adropin treatment. We believe the lack of changes is likely due to the short time period of the treatment because our previous studies demonstrate a marked reduction of serum insulin in the mice with transgenic overexpression of adropin (3).
Through enhancing AKT signaling, adropin suppresses the action of FoxO1, which can up-regulate the transcription of Gck, the enzyme catalyzing glucose influx (9, 17). Along with this, adropin suppresses GSK3 (7), the activation of which inhibits glycogen synthesis. These changes are expected to promote glycogen synthesis and lead to the observed increase in glycogen content. Moreover, the suppression of FoxO1 action would also contribute to the down-regulation of Pck1 and G6pc, two key enzymes involved in hepatic glucose production (9, 17). Together, the concerted changes in the molecular machinery mediating glucose flux would ultimately result in the net reduction of hepatic glucose output, which underlies adropin's effect on fasting blood glucose level. In support of our findings, overexpression of GK in the liver of Zucker diabetic fatty rats has been shown to correct hepatic glucose flux and normalize plasma glucose level (36). Moreover, liver-specific ablation of FoxO, which reduces the G6Pase/GK ratio, increased glucose uptake and utilization and consequently suppressed hepatic glucose production (17). Of interest, our studies provide further support for GK as a target of novel anti-hyperglycemic drugs (36). One concern with targeting GK is that its activation may promote de novo lipogenesis (17), thus leading to hepatic steatosis and offsetting the beneficial effects of lowering blood glucose (36). Importantly, our studies indicate that short-term adropin34–76 treatment promotes GK action, whereas it reduces lipogenic gene expression in DIO mice. Indeed, long-term treatment (14 days) with adropin34–76 enhances glucose tolerance and ameliorates insulin resistance while markedly attenuating the development of hepatic steatosis in DIO mice (3).
ER stress plays a causal role in the development of hepatic insulin resistance and hepatic steatosis in obesity (37, 38). Our data show that adropin's actions diminish ER stress responses in the liver of DIO mice, which can underlie both the enhancement of hepatic insulin signaling actions and the attenuation of hepatic lipogenesis by adropin. Chronic ER stress promotes sustained activation of JNK in obesity (7, 19), and JNK activation further antagonizes IRS's signaling, which leads to insulin resistance (7). Adropin34–76 treatment suppressed hepatic JNK activity in DIO mice, which could be in part accounted for by the alleviated ER stress. Our data are consistent with numerous studies showing that the suppression of JNK activity enhances insulin sensitivity in obesity (23). Among a variety of the distinct mechanisms underlying JNK's effect on insulin signaling pathway (23), our data favor the classical model (12) in which JNK activation phosphorylates the Ser residue in IRS blocking Tyr phosphorylation in this protein that is essential in the activation of the downstream signaling cascade. It is noteworthy that the hepatic expression levels of pro-inflammatory cytokines were not altered along with the changes in JNK, and this is likely due to the potential cell type–specific effect of adropin treatment on hepatocytes. Recent evidence demonstrates that the mice with hepatocyte-specific JNK deficiency display no defect in the development of hepatic inflammation, and these mice display a similar level of LPS-induced up-regulation of Tnf as the WT control mice (39), indicating that JNK may not be a major mediator in the expression of the pro-inflammatory cytokines in hepatocytes.
In addition to the effect on insulin signaling, ER stress is implicated in regulating SREBP1c activity and lipogenic gene expression impacting hepatic steatosis (11, 37, 38). ER stress activates SREBP1c by promoting the dissociation of BiP from precursor SREBP1c in the ER membrane, resulting in increased expression of lipogenic enzymes (26). Our data show that adropin34–76 treatment promotes the sequestration of precursor SREBP1c in the ER, thus preventing nuclear localization of the mature form and abrogating the activation of its target lipogenic gene transcription. In addition, SREBP1c represses Irs2 transcription, thereby inhibiting hepatic insulin signaling (40). Thus, the inactivation of SREBP1c by adropin could make an additional contribution to the enhanced insulin-signaling pathway through up-regulating IRS2. It deserves mention that our studies did not support a role of lipid metabolites in modulating insulin sensitivity, as no changes in the levels of a variety of fatty acid intermediates were detected despite the enhanced actions of insulin-signaling mediators following adropin treatment.
Calcium plays a critical role in the ER protein folding process, and the depletion of ER calcium level underlies the development of ER stress in obesity (28, 29). Furthermore, the calcium channel activity of IP3R in the liver is enhanced, and the cytosolic calcium concentration increases in both genetically and diet-induced obese mouse models (30, 41). Our studies suggest that adropin treatment inhibits the channel activity of IP3R by the concerted actions of PKA and AKT, which would attenuate ER calcium efflux, thus alleviating ER stress. In support of this prediction, it has been demonstrated that blocking the channel activity of IP3R, resulting in suppression of ER calcium release, attenuates ER stress (42). Alternatively, the alleviation of ER stress by adropin may be caused by the potential reduction of ER membrane lipid saturation (43), as we observed a trend of decrease in the degree of saturation of major cellular fatty acyl-CoAs. However, the analysis of lipid saturation degree specifically in ER membrane is warranted to assess this hypothesis.
As with the IP3R, the reduced phosphorylation of CREB (Ser133) following adropin treatment likely results from the effects on cAMP level and PKA activity. In parallel, the nuclear level of CRTC2 (co-activator of CREB) that translocates into the nucleus upon PKA activation (32) was reduced following adropin treatment. The activation of the insulin signaling pathway can dissociate CRTC from CREB, excluding CRTC from the nucleus (32). Thus, adropin can reduce the nuclear level of CRTC by both preventing it from entering the nucleus as a result of the suppressed PKA activity and promoting nuclear exclusion (of CRTC) as a consequence of the enhanced insulin signaling action. Adropin's effects on CREB and CRTC strongly suggest that CREB transcriptional activity is reduced, which then makes an additional contribution to the decreased expression of G6pc and Pck1.
cAMP-PKA signaling pathway plays a central role in mediating the effect of glucagon on hepatic glucose metabolism (13, 44). Glucagon enhances hepatic glucose production by activating the cAMP/PKA signaling pathway, which leads to up-regulation of CREB-dependent gene expression, including G6pc and Pck1 (13, 44). Of relevance, diabetes is frequently associated with hyperglucagonemia, and augmented hepatic glucagon signaling actions, including activation of CREB, have been observed in diabetic DIO mice (45). The current studies indicate that in addition to sensitizing insulin intracellular signaling, adropin may antagonize the glucagon signaling pathway in reducing hyperglycemia. In this regard, adropin34–76 appears to share aspects of the molecular mechanisms underlying metformin's actions on reducing hepatic glucose production. A recent report shows that metformin treatment inhibits adenylate cyclase, resulting in reduction of cAMP level and phosphorylation of PKA substrates including IP3R, which leads to suppression of hepatic glucagon signaling (46).
Our in vitro data demonstrate that adropin suppresses glucose production in primary hepatocytes, which shows a direct effect of adropin on hepatic glucose metabolism. The underlying mechanisms appear to involve adropin's suppression of the phosphorylations of CREB (Ser133) and other PKA substrates. The observed direct effect on hepatocytes suggests that liver cells express a receptor that mediates adropin's action on glucose metabolism in an autocrine/paracrine manner. Furthermore, recent studies have shown that adropin likely acts through GPCRs (14, 15). The observed effect of adropin on cAMP-PKA, a major signaling pathway downstream from GPCR (47), is indeed in line with these reports. As the activation of inhibitory G protein (Gi) induces the decrease in cAMP level (by suppressing adenylate cyclase) (48), the potential adropin receptor might be coupled to Gi protein. Thus, adropin might activate Gi protein, leading to the decrease in cAMP level and the attenuation of PKA-mediated signaling actions. Interestingly, deficiency of the Gi subunit has been shown to impair insulin actions in liver, leading to insulin resistance (48).
Low circulating adropin level may be causally linked to the impaired glycemic control in obesity. The circulating adropin levels are low in diabetic DIO mice (3) as well as in obese subjects (4). Recent evidence also shows that nonhuman primates with low plasma adropin level display enhanced sensitivity to high-sugar diet–induced obesity and hyperglycemia (5). In light of these findings, the current report, together with previous studies (3, 6), has provided strong support for the potential of adropin therapy in improving glycemic control in obesity. Regarding the prospect of therapeutic potential, it also deserves mention that the high-dose adropin treatment appears to exert a liver-protective role. A recent report demonstrates that acute treatment of adropin with a similar dose (500 nmol/kg intraperitoneally) significantly reduces serum levels of alanine aminotransferase and aspartate aminotransferase in a mouse model of nonalcoholic steatohepatitis (49).
In conclusion, our data show that adropin34–76 treatment modulates major intracellular signaling pathways in the liver to reduce hyperglycemia in diet-induced obesity (Fig. 10). These signaling actions appear to underlie peptide adropin's therapeutic potential of suppressing fasting hyperglycemia and improving glycemic control in obesity-associated type-2 diabetes.
Figure 10.
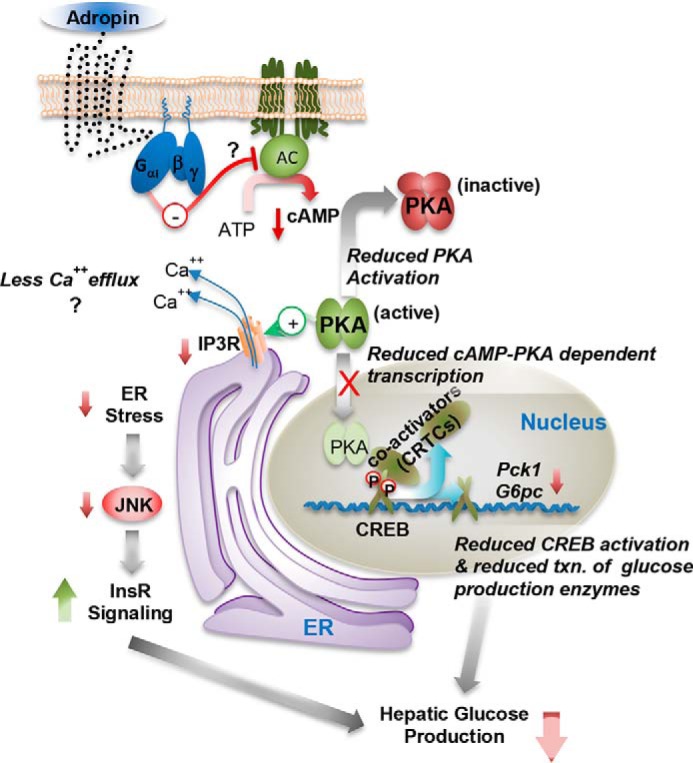
A model of adropin actions in regulating hepatic glucose metabolism. AC, adenylate cyclase.
Experimental procedures
Animals
Mouse experiments were approved by the Institutional Animal Care and Use Committees of the Scripps Research Institute (Jupiter, FL) and the University of Alberta Health Sciences Animal Welfare Committee.
Male DIO B6 mice that were weaned on an HFD (60% kcal fat; New Brunswick, NJ) were purchased from the Jackson Laboratory (Bar Harbor, ME) and were housed in a light/dark (12-h/12-h) cycle-controlled room at a temperature of 26 °C. The mice were maintained on the same HFD until they were used. In a separate experiment, age-matched C57BL/6 WT mice maintained on a laboratory chow diet (low-fat diet) and DIO B6 mice maintained on an HFD were used as the control.
Mice were monitored and handled daily for habituation and were subject to repeated mock injections to minimize the experimental stress. Injections of adropin34–76 were administered after the animals had become fully habituated. The mice subject to the experimental procedures were around 24 weeks old. The animals were maintained under ad libitum fed conditions throughout the injection process.
Treatment with adropin34–76
Adropin34–76 purchased from ChinaPeptides (Shanghai, China) (2, 3, 6) was dissolved in 0.1% BSA/PBS solution, and administered as five intraperitoneal injections (450 nmol/kg for each injection) over a 48-h period to DIO mice fed ad libitum (6). Body weight was measured right before the first injection and right after the last injection, and the data were published in the previous studies (6). Food was removed after the last injection, which took place 4 h before the dark onset, and the mice were euthanized 2 h after the last injection. The livers were freeze-clamped and flash-frozen in liquid nitrogen for subsequent analysis.
Blood glucose and insulin determination
Blood glucose levels were determined by a OneTouch blood glucose meter (LifeScan Europe, Zug, Switzerland). Insulin levels (serum and plasma) were determined by ELISA (Crystal Chem, Downers Grove, IL).
Glycogen and triacylglycerol assays
The levels of glycogen and triacylglycerol in the liver were measured by the Glycogen Assay Kit from Abcam (Cambridge, MA) and the Triglyceride Colorimetric Assay Kit from Cayman (Ann Arbor, MI) according to the manufacturers' instructions, which were described previously (6).
Measurement of lipid metabolites
Lipid metabolites were analyzed with HPLC as described before (6) and by Metabolon (Durham, NC).
Western blotting
Whole-cell lysate of liver was prepared by use of the protocol from Cell Signaling Technology. Nuclear extract was prepared with the Nuclear Extraction Kit from Active Motif (Carlsbad, CA). Microsomal membranes were isolated as described previously (26).
Immunoprecipitation and immunoblotting procedures, based on the protocols detailed by Cell Signaling Technology and Invitrogen (Carlsbad, CA), were described in the previous studies (2, 6). Co-immunoprecipitation was performed as described previously (26). In general, the protein extracts were heated at 70 ºC for 10 min before gel loading.
The antibodies against phospho-IRS1 (Ser307), phospho-IRS1 (Tyr608), and histone H3 were from EMD Millipore (Mahopac, NY). The antibodies against XBP-1, SREBP1c, NF-κB-p65, and β-actin were from Santa Cruz Biotechnology, Inc. (Dallas, TX). The antibody against BiP was from Abcam. The antibody against CRTC2 was from Bethyl Laboratories (Montgomery, TX). All of the other primary antibodies were from Cell Signaling Technology (Danvers, MA). GAPDH, β-actin, and α-tubulin were used as the loading control in whole-liver cell lysate analysis. Histone H3 was used as the loading control in liver nuclear extract analysis.
Proteins were visualized using enhanced chemiluminescence (Waltham, MA), and the signals were exposed on film. In general, the same membrane was used for detecting different proteins. Whenever possible, the membranes were stripped with OneMinute Stripping Buffer (GM Bioscience, Fredrick, MD) and reprobed with primary antibodies for other proteins of interest. Loading controls from the same membranes were shared. Four to six contiguous gel bands with two to three from each group are presented. Densitometry was performed by use of Image J software (National Institutes of Health).
Quantitative PCR
The extraction of total RNA and cDNA synthesis were performed as described previously (2, 6). PCR was conducted using the ABI 7900 real-time PCR system, based on the instructions from TaqMan Gene Expression Assays (Thermo Fisher Scientific). Gapdh was used as the reference gene (6).
Primary hepatocytes
Primary hepatocytes were isolated from the WT B6 mouse as described previously (33). 12–16 h after isolation, the hepatocytes were washed twice with PBS and starved in Hanks' balanced salt solution for 2 h. Hepatocytes were then treated with Hanks' balanced salt solution containing 100 ng/ml glucagon and 5 mm pyruvate to induce glucose production (33) with or without 100 nm adropin (or vehicle) for 3 h. Glucose levels in the culture media were measured with a glucose assay kit from Sigma and were normalized to cellular protein levels.
Statistical analysis
Data values are expressed as mean ± S.E. Comparisons between the means were performed by unpaired Student's t test. A value of p ≤ 0.05 is defined as statistically significant.
Author contributions
S. Gao and A. A. B. conceptualization; S. Gao, S. Ghoshal, L. Z., and J. R. S. data curation; S. Gao, S. Ghoshal, J. R. S., G. D. L., and A. A. B. formal analysis; S. Gao, S. Ghoshal, L. Z., K. S. M., G. D. L., and A. A. B. investigation; S. Gao visualization; S. Gao, S. Ghoshal, L. Z., K. S. M., and B. N. F. methodology; S. Gao writing-original draft; S. Gao, S. Ghoshal, K. S. M., G. D. L., and A. A. B. writing-review and editing; K. S. M., B. N. F., G. D. L., and A. A. B. resources; B. N. F., G. D. L., and A. A. B. supervision; G. D. L. and A. A. B. funding acquisition; A. A. B. project administration.
Supplementary Material
This work was supported by a Proof of Principle Award from Novo Nordisk's Diabetes Innovations Award Program (to A. A. B.), by American Diabetes Association Grant 7-08-RA16 (to A. A. B.), and in part by a grant from the Canadian Institutes of Health Research (to G. D. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S8.
- B6
- C57BL/6J
- PKA
- protein kinase A
- IP3R
- inositol-1,4,5-triphosphate receptor
- JNK
- c-Jun N-terminal kinase
- G6Pase
- glucose-6-phosphatase
- PEPCK
- phosphoenolpyruvate carboxykinase
- ER
- endoplasmic reticulum
- AdrKO
- adropin knockout
- HFD
- high fat diet
- DIO
- diet-induced obese
- GPCR
- G protein–coupled receptor
- IRS
- insulin receptor substrate
- GSK
- glycogen synthase kinase
- PC
- pyruvate carboxylase
- PERK
- PKR-like ER kinase
- IRE
- inositol-requiring enzyme
- ATF
- activating transcription factor
- eIF
- eukaryotic initiation factor
- BiP
- binding immunoglobulin protein
- IKK
- inhibitor κB kinase
- TAG
- triacylglycerol
- SREBP
- sterol regulatory element–binding protein
- CREB
- cAMP-responsive element-binding protein
- GK
- glucokinase
- CRTC
- CREB-regulated transcription co-activator
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- XBP1s
- spliced form of X-box–binding protein 1.
References
- 1. Ganesh Kumar K., Zhang J., Gao S., Rossi J., McGuinness O. P., Halem H. H., Culler M. D., Mynatt R. L., and Butler A. A. (2012) Adropin deficiency is associated with increased adiposity and insulin resistance. Obesity 20, 1394–1402 10.1038/oby.2012.31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gao S., McMillan R. P., Jacas J., Zhu Q., Li X., Kumar G. K., Casals N., Hegardt F. G., Robbins P. D., Lopaschuk G. D., Hulver M. W., and Butler A. A. (2014) Regulation of substrate oxidation preferences in muscle by the peptide hormone adropin. Diabetes 63, 3242–3252 10.2337/db14-0388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kumar K. G., Trevaskis J. L., Lam D. D., Sutton G. M., Koza R. A., Chouljenko V. N., Kousoulas K. G., Rogers P. M., Kesterson R. A., Thearle M., Ferrante A. W. Jr., Mynatt R. L., Burris T. P., Dong J. Z., Halem H. A., et al. (2008) Identification of adropin as a secreted factor linking dietary macronutrient intake with energy homeostasis and lipid metabolism. Cell Metab. 8, 468–481 10.1016/j.cmet.2008.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Butler A. A., Tam C. S., Stanhope K. L., Wolfe B. M., Ali M. R., O'Keeffe M., St-Onge M. P., Ravussin E., and Havel P. J. (2012) Low circulating adropin concentrations with obesity and aging correlate with risk factors for metabolic disease and increase after gastric bypass surgery in humans. J. Clin. Endocrinol. Metab. 97, 3783–3791 10.1210/jc.2012-2194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Butler A. A., Zhang J., Price C. A., Stevens J. R., Graham J. L., Stanhope K. L., King S., Krauss R. M., Bremer A. A., and Havel P. J. (2019) Low plasma adropin concentrations increase risks of weight gain and metabolic dysregulation in response to a high-sugar diet in male nonhuman primates. J. Biol. Chem. 294, 9706–9719 10.1074/jbc.RA119.007528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gao S., McMillan R. P., Zhu Q., Lopaschuk G. D., Hulver M. W., and Butler A. A. (2015) Therapeutic effects of adropin on glucose tolerance and substrate utilization in diet-induced obese mice with insulin resistance. Mol. Metab. 4, 310–324 10.1016/j.molmet.2015.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boucher J., Kleinridders A., and Kahn C. R. (2014) Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 6, a009191 10.1101/cshperspect.a009191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Samuel V. T., and Shulman G. I. (2016) The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J. Clin. Invest. 126, 12–22 10.1172/JCI77812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Han H.-S., Kang G., Kim J. S., Choi B. H., and Koo S.-H. (2016) Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 48, e218 10.1038/emm.2015.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen S., Zeng K., Liu Q.-C., Guo Z., Zhang S., Chen X.-R., Lin J.-H., Wen J.-P., Zhao C.-F., Lin X.-H., and Gao F. (2017) Adropin deficiency worsens HFD-induced metabolic defects. Cell Death Dis. 8, e3008 10.1038/cddis.2017.362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dara L., Ji C., and Kaplowitz N. (2011) The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 53, 1752–1763 10.1002/hep.24279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Win S., Than T. A., Zhang J., Oo C., Min R. W. M., and Kaplowitz N. (2018) New insights into the role and mechanism of c-Jun-N-terminal kinase signaling in the pathobiology of liver diseases. Hepatology 67, 2013–2024 10.1002/hep.29689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hædersdal S., Lund A., Knop F. K., and Vilsbøll T. (2018) The role of glucagon in the pathophysiology and treatment of type 2 diabetes. Mayo Clin. Proc. 93, 217–239 10.1016/j.mayocp.2017.12.003 [DOI] [PubMed] [Google Scholar]
- 14. Stein L. M., Yosten G. L., and Samson W. K. (2016) Adropin acts in brain to inhibit water drinking: potential interaction with the orphan G protein-coupled receptor, GPR19. Am. J. Physiol. Regul. Integr. Comp. Physiol. 310, R476–R480 10.1152/ajpregu.00511.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thapa D., Stoner M. W., Zhang M., Xie B., Manning J. R., Guimaraes D., Shiva S., Jurczak M. J., and Scott I. (2018) Adropin regulates pyruvate dehydrogenase in cardiac cells via a novel GPCR-MAPK-PDK4 signaling pathway. Redox Biol. 18, 25–32 10.1016/j.redox.2018.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Copps K. D., and White M. F. (2012) Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 55, 2565–2582 10.1007/s00125-012-2644-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haeusler R. A., Hartil K., Vaitheesvaran B., Arrieta-Cruz I., Knight C. M., Cook J. R., Kammoun H. L., Febbraio M. A., Gutierrez-Juarez R., Kurland I. J., and Accili D. (2014) Integrated control of hepatic lipogenesis versus glucose production requires FoxO transcription factors. Nat. Commun. 5, 5190 10.1038/ncomms6190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kumashiro N., Beddow S. A., Vatner D. F., Majumdar S. K., Cantley J. L., Guebre-Egziabher F., Fat I., Guigni B., Jurczak M. J., Birkenfeld A. L., Kahn M., Perler B. K., Puchowicz M. A., Manchem V. P., Bhanot S., et al. (2013) Targeting pyruvate carboxylase reduces gluconeogenesis and adiposity and improves insulin resistance. Diabetes 62, 2183–2194 10.2337/db12-1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Malhi H., and Kaufman R. J. (2011) Endoplasmic reticulum stress in liver disease. J. Hepatol. 54, 795–809 10.1016/j.jhep.2010.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yoshida H., Matsui T., Yamamoto A., Okada T., and Mori K. (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 10.1016/S0092-8674(01)00611-0 [DOI] [PubMed] [Google Scholar]
- 21. Rutkowski D. T., Arnold S. M., Miller C. N., Wu J., Li J., Gunnison K. M., Mori K., Sadighi Akha A. A., Raden D., and Kaufman R. J. (2006) Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 4, e374 10.1371/journal.pbio.0040374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ozcan U., Cao Q., Yilmaz E., Lee A. H., Iwakoshi N. N., Ozdelen E., Tuncman G., Görgün C., Glimcher L. H., and Hotamisligil G. S. (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461 10.1126/science.1103160 [DOI] [PubMed] [Google Scholar]
- 23. Solinas G., and Becattini B. (2017) JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol. Metab. 6, 174–184 10.1016/j.molmet.2016.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sanders F. W. B., and Griffin J. L. (2016) De novo lipogenesis in the liver in health and disease: more than just a shunting yard for glucose. Biol. Rev. Camb. Philos. Soc. 91, 452–468 10.1111/brv.12178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oh S.-Y., Park S.-K., Kim J.-W., Ahn Y.-H., Park S.-W., and Kim K.-S. (2003) Acetyl-CoA carboxylase β gene is regulated by sterol regulatory element-binding protein-1 in liver. J. Biol. Chem. 278, 28410–28417 10.1074/jbc.M300553200 [DOI] [PubMed] [Google Scholar]
- 26. Kammoun H. L., Chabanon H., Hainault I., Luquet S., Magnan C., Koike T., Ferré P., and Foufelle F. (2009) GRP78 expression inhibits insulin and ER stress–induced SREBP-1c activation and reduces hepatic steatosis in mice. J. Clin. Invest. 119, 1201–1215 10.1172/JCI37007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boudaba N., Marion A., Huet C., Pierre R., Viollet B., and Foretz M. (2018) AMPK re-activation suppresses hepatic steatosis but its downregulation does not promote fatty liver development. EBioMedicine 28, 194–209 10.1016/j.ebiom.2018.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fu S., Yang L., Li P., Hofmann O., Dicker L., Hide W., Lin X., Watkins S. M., Ivanov A. R., and Hotamisligil G. S. (2011) Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473, 528–531 10.1038/nature09968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Arruda A. P., and Hotamisligil G. S. (2015) Calcium homeostasis and organelle function in the pathogenesis of obesity and diabetes. Cell Metab. 22, 381–397 10.1016/j.cmet.2015.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang Y., Li G., Goode J., Paz J. C., Ouyang K., Screaton R., Fischer W. H., Chen J., Tabas I., and Montminy M. (2012) InsP3 receptor regulates hepatic gluconeogenesis in fasting and diabetes. Nature 485, 128–132 10.1038/nature10988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hagiwara M., Brindle P., Harootunian A., Armstrong R., Rivier J., Vale W., Tsien R., and Montminy M. R. (1993) Coupling of hormonal stimulation and transcription via the cyclic AMP-responsive factor CREB is rate limited by nuclear entry of protein kinase A. Mol. Cell Biol. 13, 4852–4859 10.1128/MCB.13.8.4852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Altarejos J. Y., and Montminy M. (2011) CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 12, 141–151 10.1038/nrm3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McCommis K. S., Chen Z., Fu X., McDonald W. G., Colca J. R., Kletzien R. F., Burgess S. C., and Finck B. N. (2015) Loss of mitochondrial pyruvate carrier 2 in the liver leads to defects in gluconeogenesis and compensation via pyruvate-alanine cycling. Cell Metab. 22, 682–694 10.1016/j.cmet.2015.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lovren F., Pan Y., Quan A., Singh K. K., Shukla P. C., Gupta M., Al-Omran M., Teoh H., and Verma S. (2010) Adropin is a novel regulator of endothelial function. Circulation 122, S185–S192 10.1161/CIRCULATIONAHA.109.931782 [DOI] [PubMed] [Google Scholar]
- 35. Altamimi T. R., Gao S., Karwi Q. G., Fukushima A., Rawat S., Wagg C. S., Zhang L., and Lopaschuk G. D. (2019) Adropin regulates cardiac energy metabolism and improves cardiac function and efficiency. Metabolism 98, 37–48 10.1016/j.metabol.2019.06.005 [DOI] [PubMed] [Google Scholar]
- 36. Agius L. (2009) Targeting hepatic glucokinase in type 2 diabetes: weighing the benefits and risks. Diabetes 58, 18–20 10.2337/db08-1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Farese R. V. Jr., Zechner R., Newgard C. B., and Walther T. C. (2012) The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metab. 15, 570–573 10.1016/j.cmet.2012.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Baiceanu A., Mesdom P., Lagouge M., and Foufelle F. (2016) Endoplasmic reticulum proteostasis in hepatic steatosis. Nat. Rev. Endocrinol. 12, 710–722 10.1038/nrendo.2016.124 [DOI] [PubMed] [Google Scholar]
- 39. Das M., Sabio G., Jiang F., Rincón M., Flavell R. A., and Davis R. J. (2009) Induction of hepatitis by JNK-mediated expression of TNF-α. Cell 136, 249–260 10.1016/j.cell.2008.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ide T., Shimano H., Yahagi N., Matsuzaka T., Nakakuki M., Yamamoto T., Nakagawa Y., Takahashi A., Suzuki H., Sone H., Toyoshima H., Fukamizu A., and Yamada N. (2004) SREBPs suppress IRS-2-mediated insulin signalling in the liver. Nat. Cell Biol. 6, 351–357 10.1038/ncb1111 [DOI] [PubMed] [Google Scholar]
- 41. Kang J. K., Kim O.-H., Hur J., Yu S. H., Lamichhane S., Lee J. W., Ojha U., Hong J. H., Lee C. S., Cha J.-Y., Lee Y. J., Im S.-S., Park Y. J., Choi C. S., Lee D. H., et al. (2017) Increased intracellular Ca2+ concentrations prevent membrane localization of PH domains through the formation of Ca2+-phosphoinositides. Proc. Natl. Acad. Sci. U.S.A. 114, 11926–11931 10.1073/pnas.1706489114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Luciani D. S., Gwiazda K. S., Yang T.-L. B., Kalynyak T. B., Bychkivska Y., Frey M. H. Z., Jeffrey K. D., Sampaio A. V., Underhill T. M., and Johnson J. D. (2009) Roles of IP(3)R and RyR Ca2+ channels in endoplasmic reticulum stress and β-cell death. Diabetes 58, 422–432 10.2337/db07-1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Volmer R., van der Ploeg K., and Ron D. (2013) Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. U.S.A. 110, 4628–4633 10.1073/pnas.1217611110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Han H. S., Kang G., Kim J. S., Choi B. H., and Koo S. H. (2016) Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 48, e218 10.1038/emm.2015.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Song W.-J., Mondal P., Wolfe A., Alonso L. C., Stamateris R., Ong B. W. T., Lim O. C., Yang K. S., Radovick S., Novaira H. J., Farber E. A., Farber C. R., Turner S. D., and Hussain M. A. (2014) Glucagon regulates hepatic kisspeptin to impair insulin secretion. Cell Metab. 19, 667–681 10.1016/j.cmet.2014.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Miller R. A., Chu Q., Xie J., Foretz M., Viollet B., and Birnbaum M. J. (2013) Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 494, 256–260 10.1038/nature11808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sassone-Corsi P. (2012) The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol. 4, a011148 10.1101/cshperspect.a011148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Moxham C. M., and Malbon C. C. (1996) Insulin action impaired by deficiency of the G-protein subunit Giα2. Nature 379, 840–844 10.1038/379840a0 [DOI] [PubMed] [Google Scholar]
- 49. Chen X., Xue H., Fang W., Chen K., Chen S., Yang W., Shen T., Chen X., Zhang P., and Ling W. (2019) Adropin protects against liver injury in nonalcoholic steatohepatitis via the Nrf2 mediated antioxidant capacity. Redox Biol. 21, 101068 10.1016/j.redox.2018.101068 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.