

Abstract
Dynamic control of thioredoxin (Trx) oxidoreductase activity is essential for balancing the need of cells to rapidly respond to oxidative/nitrosative stress and to temporally regulate thiol-based redox signaling. We have previously shown that cytokine stimulation of the respiratory epithelium induces a precipitous decline in cell S-nitrosothiol, which depends upon enhanced Trx activity and proteasome-mediated degradation of Txnip (thioredoxin-interacting protein). We now show that tumor necrosis factor-α–induced Txnip degradation in A549 respiratory epithelial cells is regulated by the extracellular signal-regulated protein kinase (ERK) mitogen-activated protein kinase pathway and that ERK inhibition augments both intracellular reactive oxygen species and S-nitrosothiol. ERK-dependent Txnip ubiquitination and proteasome degradation depended upon phosphorylation of a PXTP motif threonine (Thr349) located within the C-terminal α-arrestin domain and proximal to a previously characterized E3 ubiquitin ligase–binding site. Collectively, these findings demonstrate the ERK mitogen-activated protein kinase pathway to be integrally involved in regulating Trx oxidoreductase activity and that the regulation of Txnip lifetime via ERK-dependent phosphorylation is an important mediator of this effect.
Keywords: thioredoxin, extracellular-signal-regulated kinase (ERK), proteasome, ubiquitylation (ubiquitination), nitrosative stress, oxidative stress, epithelial cell
Introduction
Txnip (thioredoxin-interacting protein) is an α-arrestin family protein which functions in the regulation of thioredoxin (Trx)2 activity. Txnip inhibits Trx oxidoreductase activity through an intermolecular disulfide bond formation involving the Trx dithiol active site Cys32 (1). Increased cell Txnip expression results in oxidative stress, and augmented Txnip expression has been demonstrated in a number of pathologic conditions (e.g. diabetes, sepsis) in which redox homeostasis is known to be altered (2). Furthermore, the NO-dependent decrease in Txnip expression protects cytokine-stimulated macrophages from nitrosative injury via an increase in Trx denitrosylase activity (3). On the other hand, Txnip has been suggested to be a tumor suppressor and enhancer of apoptosis (4, 5), and reduced expression of Txnip is associated with poor prognosis in liver and bladder cancers, among others (6, 7).
The Txnip gene promoter contains antioxidant-response elements, which respond to oxidative stress (via the Keap1–Nrf2 pathway) and negatively regulate Txnip transcription (8). However, intracellular Txnip levels are also controlled by ubiquitination and proteasome degradation, which allows for expediency in augmenting Trx oxidoreductase activity (9). Txnip ubiquitination is mediated by the E3 ligase ITCH, which interacts with two PPXY motifs in the C-terminal region of the protein (5). Interestingly, tyrosine phosphorylation within the PPXY motif inhibits rather than promotes ITCH binding, implicating another signaling mechanism by which Txnip is targeted for ubiquitination and proteasome degradation (10).
The extracellular signal-regulated protein kinase (ERK) pathway has been shown to play a role in protecting the cell from oxidative injury with proposed impact on several different molecular pathways (11, 12). For example, ERK has also been shown to enhance NF-κB activation by targeting substrates in both the cytoplasm and nucleus (13, 14). In the respiratory epithelium, we have previously shown that Txnip undergoes rapid proteasome-dependent degradation in response to TNFα stimulation (15). We now show that cytokine-induced Txnip degradation depends upon the ERK kinase pathway, which facilitates Txnip ubiquitination via phosphorylation of a PXTP motif threonine located in the C-terminal region of the protein. The decline in cell Txnip results in an increase in Trx oxidoreductase activity and subsequent decrease in cell ROS and SNO.
Results
TNFα-induced Txnip degradation is ERK-dependent
We have previously shown that TNFα stimulation of A549 respiratory epithelial cells induces a rapid decrease in cell Txnip levels with concomitant increase in Trx denitrosylase activity (15). Given that the ERK kinase pathway has been shown to regulate redox-sensitive pathways (11, 12), we investigated whether ERK activity functions to mediate TNFα-induced Txnip degradation in the respiratory epithelium and thus serves as a regulator of Trx oxidoreductase activity. Pretreatment of A549 cells with the MEK (ERK 1/2) inhibitor U0126 prevented TNFα-induced Txnip degradation 1 h poststimulation (Fig. 1, A and B). Interestingly, A549 cells showed significant ERK activity at baseline (as demonstrated by phosphorylated ERK substrate) and U0126 treatment induced a slight increase in Txnip levels above baseline. Treatment with the proteasome inhibitor MG132 prevented TNFα-induced Txnip degradation, consistent with our previous observations (15), and abrogated the effect of ERK inhibition. These data indicate that the ERK pathway plays a crucial role in regulating Txnip expression with the process dependent upon the proteasome.
Figure 1.
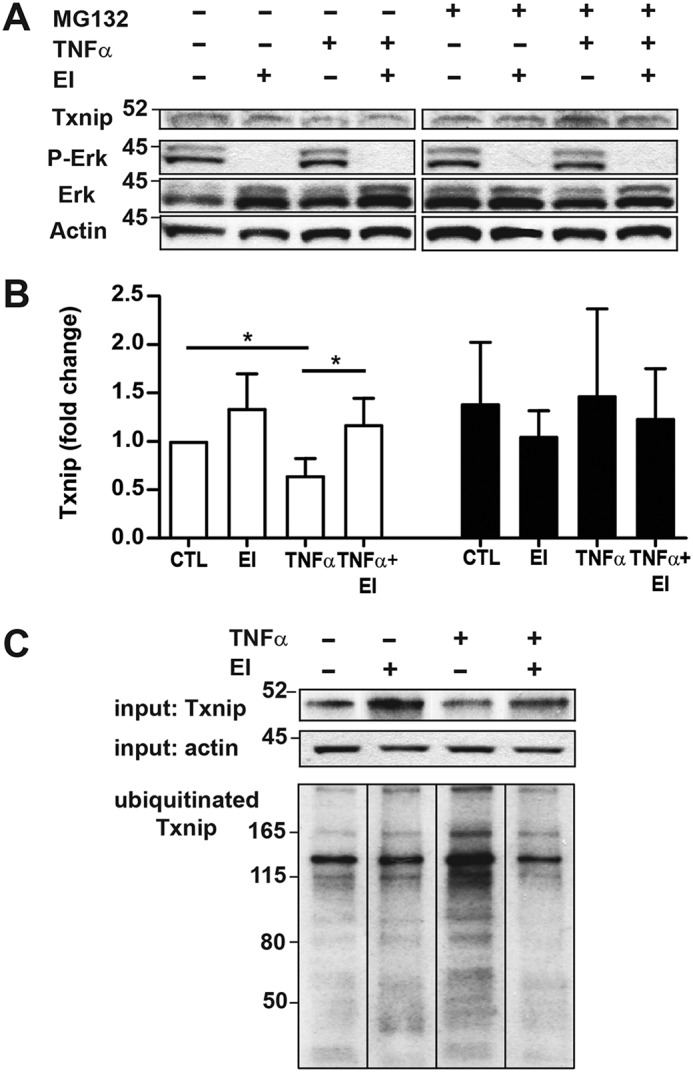
Cytokine-induced degradation of Txnip is ERK- and proteasome-dependent. A549 cells were treated with the ERK1/2 MAP kinase inhibitor (EI) (U0126, 10 μm, 60 min) and the proteasome inhibitor MG132 (10 μm) and stimulated with TNFα (10 ng/ml, 60 min) followed by cell lysate preparation. A, immunoblots of Txnip, phospho-ERK (P-ERK), ERK, and β-actin. B, densitometric analysis of Txnip blots shown in A. Open bars, −MG132; filled bars, +MG132 (n = 3 per condition, one-sample t test). *, p < 0.05. C, A549 cells were treated with the proteasome inhibitor MG132 (10 μm) prior to ERK inhibition and TNFα stimulation. Ubiquitinated proteins were then isolated from protein lysates by IP, and Txnip immunoblots were prepared from IP eluates. CTL, control.
The stimulus-regulated degradation of Txnip protein likely reflects a need for cells to rapidly augment Trx oxidoreductase activity as necessitated by stress-induced redox-signaling and as a means to protect the cell from oxidative and nitrosative stress. In this regard, Txnip has been shown to have a short half-life in vivo that is primarily governed by proteasome degradation (5). We have shown that the acute decline in A549 cell Txnip levels after TNFα stimulation depends upon enhanced Txnip ubiquitination and subsequent proteasome degradation (15). Treatment of A549 cells with the ERK kinase inhibitor U0126 prevented the TNFα-induced increase in Txnip ubiquitination (Fig. 1C). Moreover, ERK inhibition attenuated Txnip ubiquitination even under nonstressed, basal conditions that correlated with changes in Txnip expression (Fig. 1, A and B). Collectively, these data indicate ERK to be a principal regulator of Txnip proteasome degradation under both tonic- and stimulus-mediated conditions.
Trx oxidoreductase activity is regulated by ERK
Txnip inhibits Trx oxidoreductase activity by binding to the active site dithiol (1), and we have previously shown TNFα-induced Txnip degradation coincides with an increase in Trx denitrosylase activity in A549 cells with a concomitant decrease in intracellular SNO (15). Given the demonstrated role of ERK in regulating cell Txnip levels, we sought to determine whether there was a resultant impact on Trx activity. Using photolysis–chemiluminescence to quantify intracellular SNO, we found SNO levels in A549 cells to be decreased after TNFα stimulation with inhibition of the ERK pathway reversing this effect (Fig. 2A). Although intracellular ROS were unchanged in A549 cells after TNFα stimulation (likely because of concomitant increase in ROS production) (16), treatment with the ERK kinase inhibitor U0126 did increase ROS levels both at baseline and after TNFα stimulation (Fig. 2B). Moreover, Trx disulfide reductase activity was augmented with TNFα stimulation with the effect attenuated by ERK kinase inhibition (Fig. 2C). Importantly, we saw no change in Trx1 protein expression with either ERK inhibition or TNFα stimulation (Fig. 2D and Fig. S1). These data indicate that the ERK kinase pathway functions to regulate Trx activity primarily by controlling degradation of Txnip.
Figure 2.

ERK regulation of Trx activity. A, total SNO quantified by photolysis–chemiluminescence in lysates prepared from A549 cells after TNFα stimulation (10 ng/ml, 60 min) and treatment with ERK1/2 MAP kinase inhibitor (EI) (U0126, 10 μm; n = 5, means ± S.D.). B, intracellular ROS quantified in A549 cells using a fluorescence dye assay (Cellular ROS assay kit; Abcam). The cells were treated with TNFα (10 ng/ml) ± EI (10 μm) for 60 min followed by quantification on a fluorescence microplate reader (relative fluorescence units (RFU); n = 5, means ± S.D.). C, Trx reductase activity quantified by fluorescence assay (Cayman Chemical) in lysates prepared from A549 cells after TNFα stimulation (10 ng/ml, 60 min) and EI treatment (10 μm; n = 3, means ± S.D.). *, p < 0.05; **, p < 0.01. D, Trx1 immunoblots from A549 cells treated with EI and stimulated with TNFα as above. CTL, control.
Thr349 is a target of ERK kinase
The role of phosphorylation in modulating ubiquitin-dependent proteaseomal degradation is well-known, and tyrosine phosphorylation of conserved PPXY motifs in Txnip have been shown to disrupt binding of the E3 ubiquitin ligase ITCH (5, 10). We used MS to identify potential sites of ERK-mediated phosphorylation. N-terminal FLAG-tagged Txnip (TxnipWT) was expressed in A549 cells, and Txnip immunoprecipitates (IPs) were separated by SDS-PAGE and analyzed by LC/tandem MS (GeLC–MS/MS), with or without enrichment for phosphopeptides using titanium dioxide. Despite achieving >70% sequence coverage of Txnip, including the first PPXY motif (Fig. 3A), we detected phosphorylation of only a single peptide, spanning residues 344–371 of Txnip. Although this peptide had a total of six potential acceptor Ser, Thr, and Tyr sites, there was a 95% probability of localization on Thr349 based on Ascore analysis of the MS/MS spectrum (Fig. 3B) (17). In addition, examination of the extracted ion chromatogram for this peptide suggested that there was a single species (Fig. 3C), whereas phosphopeptides containing differing sites of localization can often be resolved by LC (18). We also noted that among sites of phosphorylation that have identified in unbiased MS analyses (as compiled in the Phosphosite database; www.phosphosite.org; Ref. 33),3 pThr349 (annotated in 10 studies) was second only to the PPXY motif target pTyr378 (Fig. 3D; data not shown).
Figure 3.

Analysis of Txnip phosphorylation. Txnip IPs were analyzed by GeLC–MS/MS with or without TiO2 enrichment for phosphopeptides. A, sequence coverage Txnip as determined by GeLC–MS/MS of unenriched Txnip IPs. Identified residues are highlighted in yellow, with green indicating variable modification on Met (oxidation) or Asn/Gln (deamidation). The location of the phosphopeptide spanning residues 344–371 is underlined. B, EThcD MS/MS spectrum of the phosphorylated peptide containing pThr349 from TiO2-enriched Txnip IPs. The data are the results of Mascot search, and the spectrum was assigned >95% probability of pThr349 site localization using the Ascore algorithm (Ascore value of 18.44). C, extracted ion chromatogram of the Txnip phosphopeptide (107.4746 m/z; 3+ charge) spanning amino acids 344–371. Two peaks were detected (23.5 and 42.9 min), but only the peak at 42.9 min was assigned to the Txnip phosphopeptide based on accurate mass (−0.4 ppm). D, domain structure of Txnip indicating known and purported sites of dynamic phosphorylation. The data are representative of n = 2 replicates.
Thr349 is located within a canonical PX(S/T)P motif preferred by MAP kinases (including ERK; Fig. 3A). To confirm the phosphorylation at this site, we immunoprecipitated TxnipWT and T349V Txnip (Txnip349V) protein expressed in A549 cells followed by immunoblotting with PXpTP-specific antibody. We found that TxnipWT had basal PXTP phosphorylation that was attenuated by treatment with ERK inhibitor and modestly enhanced by TNFα stimulation (Fig. 4A). On the other hand, PXTP phosphorylation was not detectable in the Txnip349V IP at baseline or after TNFα stimulation, indicating Thr349 to be the kinase effector site. To determine whether Txnip Thr349 is phosphorylated by direct interaction with ERK, we performed an in vitro ERK kinase assay with TxnipWT and Txnip349V expressed in A549 cells and purified by IP. ERK-mediated PXTP phosphorylation was observed only with WT substrate and not the T349V mutant substrate (Fig. 4, B and C). Thus, our results establish that Thr349 is the only PXpTP motif site in Txnip and that this site is directly targeted by ERK for phosphorylation.
Figure 4.
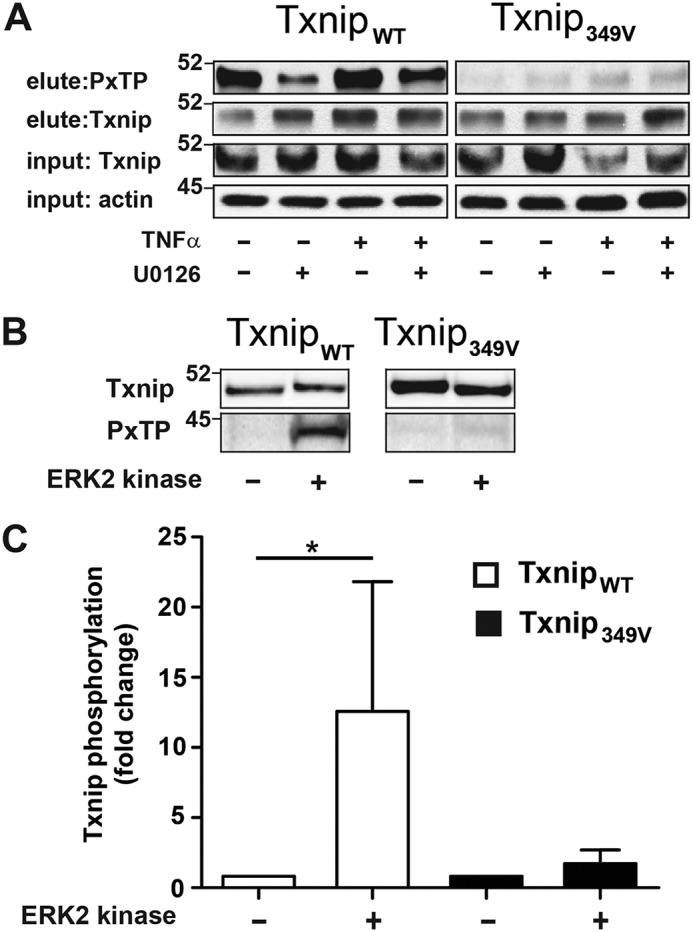
Cytokine-induced phosphorylation of Txnip threonine 349 (Thr349) by ERK. A, A549 cells were infected with lentivirus that express either C-terminally FLAG-tagged WT (TxnipWT) or T349V (Txnip349V) Txnip. The cells were treated with ERK1/2 MAP kinase inhibitor (U0126, 10 μm) and stimulated with TNFα (10 ng/ml, 60 min), and protein lysates were prepared. Txnip was immunoprecipitated with anti–FLAG-agarose, and immunoblots of eluate were probed with a phospho-MAPK (PXTP) and Txnip antibody. B, protein lysates were prepared from A549 cells expressing either TxnipWT or Txnip349V. Txnip was immunoprecipitated from cell lysate with anti-FLAG agarose and treated with ERK2 kinase (5 ng/μl), 60 min, 30 °C. PXTP and Txnip immunoblots were prepared from IP eluate. C, densitometric analysis of PXTP blots shown in B (n = 4 per condition, one-sample t test). *, p < 0.01.
ERK-dependent degradation of Txnip is modulated by Txnip phosphorylation
To determine whether phosphorylation of Thr349 facilitates Txnip protein degradation, we performed cyclohexamide (CHX) chase assays on A549 cells expressing either TxnipWT or Txnip349V. Degradation of TxnipWT was observed at 30 and 60 min post-CHX, with TNF enhancing and ERK inhibition attenuating the process, respectively (Fig. 5, A and B). On the other hand, the degradation of Txnip349V was not significantly altered by either TNFα stimulation or treatment with ERK inhibitor.
Figure 5.

ERK phosphorylation of Txnip Thr349 regulates cytokine-induced degradation. A549 cells were infected with lentivirus expressing either FLAG-tagged TxnipWT or Txnip349V. The cells were treated with CHX (50 μm) and ERK1/2 MAP kinase inhibitor (EI) (U0126, 10 μm) for 10 min prior to stimulation with TNFα (10 ng/ml). A, immunoblots of Txnip at indicated time post-CHX treatment. 0-min blots for the Txnip349V CTL/EI group and TNFα/TNFα-EI group are duplicates, which serve as the respective controls for those groups because they were run on the same gel. B, densitometric analysis of Txnip blots shown in A. The results are relative to CHX-only control (CTL) at 0-min treatment time (n = 3 per condition). **, p < 0.01 EI + TNFα compared with TNFα alone.
We next evaluated the effects of Thr349 mutation on Txnip ubiquitination. Consistent with the effects on Txnip protein degradation, TNFα stimulation significantly enhanced ubiquitination of TxnipWT protein but induced no change in Txnip349V ubiquitination (Fig. 6, A and B). Collectively, these findings establish the importance of the ERK kinase pathway in regulating cell Txnip expression and identify a specific molecular signaling mechanism (phosphorylation of Txnip Thr349) by which ERK targets Txnip for ubiquitination and proteasome degradation with the resultant downstream effect being augmentation of Trx oxidoreductase activity.
Figure 6.
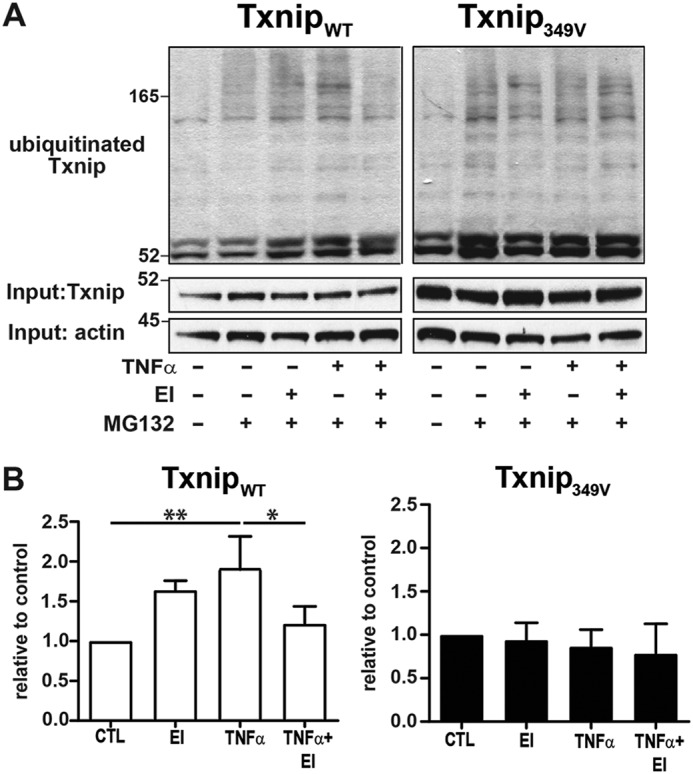
Phosphorylation of Txnip Thr349 regulates cytokine-induced ubiquitination. A, A549 cells were infected with lentivirus expressing either TxnipWT or Txnip349V, treated with the proteasome inhibitor MG132 (10 μm), and stimulated with TNFα (10 ng/ml, 60 min) ± ERK1/2 MAP kinase inhibitor (EI) (U0126, 10 μm). Ubiquitinated proteins were isolated from cell lysates by IP, and Txnip immunoblots were prepared from IP eluates. B, densitometric analysis of ubiquitinated Txnip blots shown in A. The results are relative to MG132-only control (CTL; n = 3 per condition, one-sample t test). **, p < 0.01; *, p < 0.05.
Discussion
In the context of redox signaling and under conditions of acute oxidative or nitrosative stress, it is essential that cells have the capability to expeditiously enhance Trx oxidoreductase activity. In this regard, Txnip, an inhibitor of Trx activity and a protein with a half-life tightly regulated by the proteasome, is an ideal mediator. We have previously shown that in the respiratory epithelium, which expresses both constitutive and inducible nitric-oxide synthase (19, 20), TNFα induces rapid ubiquitination and proteasome degradation of Txnip concurrent with an increase in Trx denitrosylase activity (15). We now show this response to be mediated by the ERK signaling pathway and that phosphorylation of a single threonine residue in Txnip is critical for the effect. The ERK pathway has long been known to play a protective role in both oxidative and nitrosative cell injury (21, 22). In the respiratory epithelium, oxidant-induced Nrf2 nuclear translocation and antioxidant-response element–dependent gene transcription has been shown to be dependent on ERK activity (12). Although Trx expression is regulated, in part, by Nrf2, in the current study we saw no change in Trx expression in A549 cells with either TNFα stimulation or ERK inhibition (Fig. 2D). Rather, the change in Trx activity occurred in conjunction with modification in cellular levels of the Trx inhibitor Txnip.
Regulation of intracellular Txnip levels occurs predominantly through enhanced proteasome degradation. The E3 ubiquitin protein ligase ITCH targets Txnip for ubiquitination and proteasomal degradation with two PPXY motifs located in the C-terminal region of Txnip instrumental in ITCH–Txnip protein interaction (5, 10). Phosphorylation of these tyrosine residues inhibits ITCH–Txnip interaction, thus prolonging the half-life of the protein (10). Interestingly, we did not detect phosphorylation of either tyrosine in our phosphoprotein analysis nor did mutation of the ERK-targeted PXTP phosphorylation site (Thr349) alter ITCH binding to Txnip (Fig. S2). It is possible that the observed increase in Txnip ubiquitination may be the result of Thr349 phosphorylation enhancing transfer of ubiquitin from the E2-conjugating enzyme irrespective of attenuation in ITCH–Txnip interaction. Txnip now joins a growing list of proteins, including c-Myc, STAT1, and the tumor suppressor protein FBW7, in which ubiquitination and proteasomal degradation are regulated by ERK-dependent phosphorylation (23–25).
Our results further emphasize the importance of Txnip interaction with Trx in regulating cell ROS and SNOs. Under conditions of oxidative stress, Txnip interaction with Trx-2 in the mitochondria results in activation of ASK1 (Apoptosis signal-regulating kinase 1) and the induction of apoptosis (26). Txnip-induced increase in ROS has also been demonstrated to augment mitophagy and autophagy with signaling, at least in part, via the mTOR pathway (27, 28). In turn, ROS have been shown to induce Txnip dissociation from Trx with subsequent binding to NLRP3 and activation of the inflammasome (29). Although we and others have shown Txnip to inhibit the pro-inflammatory, anti-apoptotic NF-κB pathway by attenuating Trx activity (6, 15), in the present study we saw no change in TNFα-induced NF-κB activity despite the observed decrease in Txnip degradation with ERK inhibition (Fig. S3). ERK inhibition has been shown to enhance NF-κB activity by increasing phosphorylation of the p65 subunit, which could explain these findings (30). Nevertheless, it is clear that Txnip–Trx interaction is important in regulating the immune response, with manipulation in this interaction holding promise as a therapeutic maneuver in the treatment of inflammatory diseases.
Experimental procedures
Reagents
Antibodies were purchased from the following vendors: Txnip rabbit polyclonal (Invitrogen); Txnip mouse monoclonal (MBL International); ERK1/2, phospho-ERK1/2, and phospho-MAPK substrate (PXTP) rabbit polyclonal antibodies (Cell Signaling); IκBα (C-21), NF-κB p65 (C-20), α-tubulin (B-7), Lamin A (H-102), and β-actin (C-11) (Santa Cruz Biotechnology). Other reagents were purchased from the following vendors: MEK1/2 inhibitor U0126 and MG132 (Cell Signaling), cycloheximide (Cayman Chemical), ERK2 kinase (Millipore Sigma), phosphatase inhibitors (Abcam), and TNFα (Peprotech). All other reagents were purchased from Sigma unless otherwise indicated.
Cell culture
A549 (CCL-185) cells were grown in F12K medium (Invitrogen) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. HEK 293-T cells were grown in Dulbecco's modified Eagle's medium with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. All cultures were maintained in 95% air, 5% CO2 at 37 °C. For the Txnip overexpression experiments, the cells were infected with lentivirus Txnip expression vectors, and positive transduction was confirmed by puromycin resistance (5.0 μg/ml concentration). After puromycin selection, cell stocks were prepared and stored in liquid nitrogen for later use. Whole cell, cytoplasmic, and nuclear lysates were prepared as previously described (15). Protein concentration of cell extracts was determined by BCA (Pierce).
Thioredoxin activity assays
Total cell SNOs were quantified in A549 protein lysates using photolysis–chemiluminescence as previously described (15). Quantification of intracellular ROS was determined using a cell-permeable fluorescent dye (cellular ROS detection assay kit; Abcam). A549 cells were grown on 96-well plates and loaded with dye followed by the addition of MEK1/2 inhibitor and TNFα. Fluorescence was then measured using a multiwall fluorimeter (excitation, 520; emission, 605 nm).
Trx oxidoreductase activity was measured using a fluorescence-labeled insulin substrate kit (Cayman Chemical). Briefly, cell lysates were preincubated in a 96-well plate with 0.9 mm NADPH for 10 min at 37 °C, after which eosin-labeled insulin was added. Fluorescence intensity was then quantified on a multiwall fluorimeter (excitation, 485; emission, 520 nm) with measurements recorded every 5 min for 50 min to determine the rate of insulin disulfide reduction.
ERK kinase assay
A549 cells were infected with lentivirus that expressed either C-terminally FLAG-tagged TxnipWT or Txnip349V. Txnip was immunoprecipitated from cell lysate with anti–FLAG-agarose, resuspended in kinase buffer (20 mm HEPES, 10 mm MG132, 0.1% Nonidet P-40, 2 mm DTT, protease and phosphatase inhibitors, 500 μm ATP), and treated with ERK2 kinase (5 ng/μL, 60 min, 30 °C). Proteins were then eluted from agarose beads and separated by SDS-PAGE, and PXTP were immunoblots prepared from IP eluate.
Immunoprecipitation
A549 cells were lysed in IP buffer (0.1% Nonidet P-40, 150 mm NaCl, 10 mm Na2HPO4, 2 mm EDTA, protease inhibitors). Anti-DYKDDDDK affinity resin (BioLegend) and anti-ubiquitin TUBE resin (LifeSensor) was used to isolate FLAG-tagged Txnip and ubiquitinated proteins, respectively. Resin was washed extensively with IP buffer, and proteins were eluted by boiling in Laemmli buffer and separated by SDS-PAGE followed by Western blotting.
Txnip lentivirus construction
N-terminal FLAG-tagged Txnip was amplified from human Txnip cDNA (IMAGE clone 7939547) by PCR using the primers: 5′-NheI_FLAG_TXNIP (5′-GCC GTC GCT AGC ACC ATG GAC TAC AAA GAC GAT GAC GAC AAG ATG GTG ATG TTCA AGA AG-3′) and 3′-TXNIP_NotI (5′-GCT TAA GCG GCC GCT CAC TGC ACA TTG TTG TTG AGG-3′) and ligated into the NheI and NotI endonuclease sites of the pCDH-CMV-MCS-EF1-puro vector (System Biosciences). Mutation of Txnip threonine 349 (to valine) was performed using QuikChange site-directed mutagenesis kits (Agilent Technologies) using the following forward and reverse primers: 5′-CGA TTG GAG AGC CCA ACC GTT CCT CTG CTA GAT GAC AT-3′ and 5′-ATG TCA TCT AGC AGA GGA ACG GTT GGG CTC TCC AAT CG-3′. Lentivirus expressing FLAG-tagged WT and T349V Txnip lentivirus were produced in HEK-293T cells by co-transfection with VSV-G and deltaNRF plasmids as previously described (31). Supernatants containing lentivirus were collected 48 h post-transfection, filtered with a 0.45-μm filter, and stored at −80 °C. Viral titers were determined by p24 ELISA (Cell Biolabs).
Mass spectrometry
Txnip IPs were separated by SDS-PAGE (NuPage 4–20% Bis-Tris) followed by Colloidal Blue staining. The ∼50-kDa region containing Txnip was excised and subjected to in-gel digestion with trypsin as previously described (32). Phosphopeptides were enriched using titanium oxide and enriched samples analyzing by LC–MS/MS using a Waters NanoACQUITY interfaced to a Thermo Q-Exactive Plus MS or Thermo Fusion Lumos as previously described (18), followed by database searching with Mascot 2.4 (Matrix Sciences) and annotation at a 1% peptide false discovery in Scaffold (Proteome Software). Confidence of site localization was evaluated using the Ascore algorithm in Scaffold PTM (Proteome Software).
Data analysis
The data are expressed as means ± S.D. Significant differences between groups were identified by Student's and one-sample t tests.
Author contributions
Z. T. K., M. W. F., and H. E. M. data curation; Z. T. K., C. W., and M. W. F. formal analysis; Z. T. K., C. W., and M. W. F. validation; Z. T. K., C. W., M. W. F., and H. E. M. investigation; Z. T. K., M. T. F., M. W. F., and H. E. M. methodology; Z. T. K., M. W. F., and H. E. M. writing-review and editing; M. T. F., M. W. F., and H. E. M. conceptualization; M. T. F., M. W. F., and H. E. M. visualization; M. W. F. software; H. E. M. resources; H. E. M. supervision; H. E. M. funding acquisition; H. E. M. writing-original draft; H. E. M. project administration.
Supplementary Material
This work was supported by National Institutes of Health Grant HL092994 (to H. E. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- Trx
- thioredoxin
- SNO
- S-nitrosothiol
- Txnip
- thioredoxin-interacting protein
- ERK
- extracellular signal-regulated protein kinase
- ROS
- reactive oxygen species
- IP
- immunoprecipitate/immunoprecipitation
- CHX
- cycloheximide
- TNF
- tumor necrosis factor
- MAP
- mitogen-activated protein
- MEK
- MAP kinase/ERK kinase.
References
- 1. Patwari P., Higgins L. J., Chutkow W. A., Yoshioka J., and Lee R. T. (2006) The interaction of thioredoxin with Txnip: evidence for formation of a mixed disulfide by disulfide exchange. J. Biol. Chem. 281, 21884–21891 10.1074/jbc.M600427200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yoshihara E., Masaki S., Matsuo Y., Chen Z., Tian H., and Yodoi J. (2014) Thioredoxin/Txnip: redoxisome, as a redox switch for the pathogenesis of diseases. Front. Immunol. 4, 514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Forrester M. T., Seth D., Hausladen A., Eyler C. E., Foster M. W., Matsumoto A., Benhar M., Marshall H. E., and Stamler J. S. (2009) Thioredoxin-interacting protein (Txnip) is a feedback regulator of S-nitrosylation. J. Biol. Chem. 284, 36160–36166 10.1074/jbc.M109.057729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Frullanti E., Colombo F., Falvella F. S., Galvan A., Noci S., De Cecco L., Incarbone M., Alloisio M., Santambrogio L., Nosotti M., Tosi D., Pastorino U., and Dragani T. A. (2012) Association of lung adenocarcinoma clinical stage with gene expression pattern in noninvolved lung tissue. Int. J. Cancer 131, E643–E648 10.1002/ijc.27426 [DOI] [PubMed] [Google Scholar]
- 5. Zhang P., Wang C., Gao K., Wang D., Mao J., An J., Xu C., Wu D., Yu H., Liu J. O., and Yu L. (2010) The ubiquitin ligase Itch regulates apoptosis by targeting thioredoxin-interacting protein for ubiquitin-dependent degradation. J. Biol. Chem. 285, 8869–8879 10.1074/jbc.M109.063321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kwon H. J., Won Y. S., Suh H. W., Jeon J. H., Shao Y., Yoon S. R., Chung J. W., Kim T. D., Kim H. M., Nam K. H., Yoon W. K., Kim D. G., Kim J. H., Kim Y. S., Kim D. Y., et al. (2010) Vitamin D3 upregulated protein 1 suppresses TNF-α–induced NF-κB activation in hepatocarcinogenesis. J. Immunol. 185, 3980–3989 10.4049/jimmunol.1000990 [DOI] [PubMed] [Google Scholar]
- 7. Nishizawa K., Nishiyama H., Matsui Y., Kobayashi T., Saito R., Kotani H., Masutani H., Oishi S., Toda Y., Fujii N., Yodoi J., and Ogawa O. (2011) Thioredoxin-interacting protein suppresses bladder carcinogenesis. Carcinogenesis 32, 1459–1466 10.1093/carcin/bgr137 [DOI] [PubMed] [Google Scholar]
- 8. He X., and Ma Q. (2012) Redox regulation by nuclear factor erythroid 2-related factor 2: gatekeeping for the basal and diabetes-induced expression of thioredoxin-interacting protein. Mol. Pharmacol. 82, 887–897 10.1124/mol.112.081133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Otaki Y., Takahashi H., Watanabe T., Funayama A., Netsu S., Honda Y., Narumi T., Kadowaki S., Hasegawa H., Honda S., Arimoto T., Shishido T., Miyamoto T., Kamata H., Nakajima O., et al. (2016) HECT-type ubiquitin E3 ligase ITCH interacts with thioredoxin-interacting protein and ameliorates reactive oxygen species–induced cardiotoxicity. J. Am. Heart Assoc. 5, e002485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu Y., Lau J., Li W., Tempel W., Li L., Dong A., Narula A., Qin S., and Min J. (2016) Structural basis for the regulatory role of the PPxY motifs in the thioredoxin-interacting protein TXNIP. Biochem. J. 473, 179–187 10.1042/BJ20150830 [DOI] [PubMed] [Google Scholar]
- 11. Hung C. C., Ichimura T., Stevens J. L., and Bonventre J. V. (2003) Protection of renal epithelial cells against oxidative injury by endoplasmic reticulum stress preconditioning is mediated by ERK1/2 activation. J. Biol. Chem. 278, 29317–29326 10.1074/jbc.M302368200 [DOI] [PubMed] [Google Scholar]
- 12. Papaiahgari S., Kleeberger S. R., Cho H. Y., Kalvakolanu D. V., and Reddy S. P. (2004) NADPH oxidase and ERK signaling regulates hyperoxia-induced Nrf2–ARE transcriptional response in pulmonary epithelial cells. J. Biol. Chem. 279, 42302–42312 10.1074/jbc.M408275200 [DOI] [PubMed] [Google Scholar]
- 13. Chen B., Liu J., Ho T. T., Ding X., and Mo Y. Y. (2016) ERK-mediated NF-B activation through ASIC1 in response to acidosis. Oncogenesis 5, e279 10.1038/oncsis.2016.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jiang B., Xu S., Hou X., Pimentel D. R., Brecher P., and Cohen R. A. (2004) Temporal control of NF-κB activation by ERK differentially regulates interleukin-1β–induced gene expression. J. Biol. Chem. 279, 1323–1329 10.1074/jbc.M307521200 [DOI] [PubMed] [Google Scholar]
- 15. Kelleher Z. T., Sha Y., Foster M. W., Foster W. M., Forrester M. T., and Marshall H. E. (2014) Thioredoxin-mediated denitrosylation regulates cytokine-induced nuclear factor κB (NF-κB) activation. J. Biol. Chem. 289, 3066–3072 10.1074/jbc.M113.503938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim H., Hwang J. S., Woo C. H., Kim E. Y., Kim T. H., Cho K. J., Kim J. H., Seo J. M., and Lee S. S. (2008) TNF-α–induced up-regulation of intercellular adhesion molecule-1 is regulated by a Rac-ROS-dependent cascade in human airway epithelial cells. Exp. Mol. Med. 40, 167–175 10.3858/emm.2008.40.2.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beausoleil S. A., Villén J., Gerber S. A., Rush J., and Gygi S. P. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 10.1038/nbt1240 [DOI] [PubMed] [Google Scholar]
- 18. Foster M. W., Gwinn W. M., Kelly F. L., Brass D. M., Valente A. M., Moseley M. A., Thompson J. W., Morgan D. L., and Palmer S. M. (2017) Proteomic analysis of primary human airway epithelial cells exposed to the respiratory toxicant diacetyl. J. Proteome Res. 16, 538–549 10.1021/acs.jproteome.6b00672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Asano K., Chee C. B., Gaston B., Lilly C. M., Gerard C., Drazen J. M., and Stamler J. S. (1994) Constitutive and inducible nitric oxide synthase gene expression, regulation, and activity in human lung epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 91, 10089–10093 10.1073/pnas.91.21.10089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brindicci C., Kharitonov S. A., Ito M., Elliott M. W., Hogg J. C., Barnes P. J., and Ito K. (2010) Nitric oxide synthase isoenzyme expression and activity in peripheral lung tissue of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 181, 21–30 10.1164/rccm.200904-0493OC [DOI] [PubMed] [Google Scholar]
- 21. Buckley S., Driscoll B., Barsky L., Weinberg K., Anderson K., and Warburton D. (1999) ERK activation protects against DNA damage and apoptosis in hyperoxic rat AEC2. Am. J. Physiol. 277, L159–L166 10.1152/ajplung.1999.277.1.L159 [DOI] [PubMed] [Google Scholar]
- 22. Kim S.-J., Ju J.-W., Oh C.-D., Yoon Y.-M., Song W. K., Kim J.-H., Yoo Y. J., Bang O.-S., Kang S.-S., and Chun J.-S. (2002) ERK-1/2 and p38 kinase oppositely regulate nitric oxide–induced apoptosis of chondrocytes in association with p53, caspase-3, and differentiation status. J. Biol. Chem. 277, 1332–1339 10.1074/jbc.M107231200 [DOI] [PubMed] [Google Scholar]
- 23. Ji S., Qin Y., Shi S., Liu X., Hu H., Zhou H., Gao J., Zhang B., Xu W., Liu J., Liang D., Liu L., Liu C., Long J., Zhou H., et al. (2015) ERK kinase phosphorylates and destabilizes the tumor suppressor FBW7 in pancreatic cancer. Cell Res 25, 561–573 10.1038/cr.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee S. H., Hu L.-L., Gonzalez-Navajas J., Seo G. S., Shen C., Brick J., Herdman S., Varki N., Corr M., Lee J., and Raz E. (2010) ERK activation drives intestinal tumorigenesis in Apcmin/+ mice. Nat. Med. 16, 665–670 10.1038/nm.2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deleted in proof.
- 26. Saxena G., Chen J., and Shalev A. (2010) Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J. Biol. Chem. 285, 3997–4005 10.1074/jbc.M109.034421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang C., Zhang Y., Kelly D. J., Tan C. Y., Gill A., Cheng D., Braet F., Park J.-S., Sue C. M., Pollock C. A., and Chen X.-M. (2016) Thioredoxin interacting protein (TXNIP) regulates tubular autophagy and mitophagy in diabetic nephropathy through the mTOR signaling pathway. Sci. Rep. 6, 29196 10.1038/srep29196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Qiao S., Dennis M., Song X., Vadysirisack D. D., Salunke D., Nash Z., Yang Z., Liesa M., Yoshioka J., Matsuzawa S., Shirihai O. S., Lee R. T., Reed J. C., and Ellisen L. W. (2015) A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to control stress-induced autophagy and sustain exercise capacity. Nat. Commun. 6, 7014 10.1038/ncomms8014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou R., Tardivel A., Thorens B., Choi I., and Tschopp J. (2010) Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 11, 136–140 10.1038/ni.1831 [DOI] [PubMed] [Google Scholar]
- 30. Yeh P. Y., Yeh K.-H., Chuang S.-E., Song Y. C., and Cheng A.-L. (2004) Suppression of MEK/ERK signaling pathway enhances cisplatin-induced NF-κB activation by protein phosphatase 4-mediated NF-κB p65 Thr dephosphorylation. J. Biol. Chem. 279, 26143–26148 10.1074/jbc.M402362200 [DOI] [PubMed] [Google Scholar]
- 31. Cockrell A. S., and Kafri T. (2007) Gene delivery by lentivirus vectors. Mol. Biotechnol. 36, 184–204 10.1007/s12033-007-0010-8 [DOI] [PubMed] [Google Scholar]
- 32. Foster M. W., Thompson J. W., Ledford J. G., Dubois L. G., Hollingsworth J. W., Francisco D., Tanyaratsrisakul S., Voelker D. R., Kraft M., Moseley M. A., and Foster W. M. (2014) Identification and quantitation of coding variants and isoforms of pulmonary surfactant protein A. J. Proteome Res. 13, 3722–3732 10.1021/pr500307f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hornbeck P. V., Zhang B., Murray B., Kornhauser J. M., Latham V., and Skrzypek E. (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 10.1093/nar/gku1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.